Experiments - p61-142
advertisement

EXPERIMENTS Biotechnology Protocols 61 Experiments SEPARATING MICRO-ORGANISMS Source: NQ Curriculum Support Intermediate 2 Biotechnology (Unit 2 Student Materials) When professional microbiologists isolate micro-organisms from the environment or an infected person, it is extremely rare to obtain a pure culture. It is therefore necessary to separate micro-organisms. Plating or streaking can be used to achieve this. Using isolated single colonies as inocula for further streak plates, pure cultures can be obtained. Streaking out a mixed broth culture on an agar plate and incubating it to obtain single colonies of different types of bacteria or yeasts can simulate this. Streak plate showing individual colonies Biotechnology Protocols 62 Experiments Instructions These instructions are for right-handed people. If you are left handed, please reverse the instructions accordingly. 1 Wear a lab coat. 2 Prepare your work space on the bench, collect the materials and set them out correctly on the bench. 3 Label the bases of the Petri dishes containing the appropriate sterile agar with initials, date and name of culture. 4 Using the mixed broth culture as the inoculum, streak out applying the method you have used previously. 5 Incubate the plates at room temperature for 48 hours. If you have obtained well isolated single colonies, use these as inocula for further streak plates using the method you have used previously to obtain pure cultures. Biotechnology Protocols 63 Experiments CONJUGATION IN MUCOR (ZYGOSPORE PRODUCTION) Source: James Watt College Label an agar plate as shown here: + - Sterilise a platinum stab wire or a scalpel blade. Transfer a piece of mycelium from a Mucor (+) strain to one half of the agar plate. Resterilise the wire/blade. Transfer a piece of mycelium from a Mucor (-) strain to the other half of the agar plate Use sellotape tabs to secure lid to base of plate Carefully invert the plate and store at 25C for 3 – 7 days. Note the growth which is found after this period of incubation. Biotechnology Protocols 64 Experiments EFFECTS OF TEMPERATURE ON MICROBIAL GROWTH Source: James Watt College Stock cultures (24 hours) of the following microbes: Bacillus subtilus Escherichia coli Bacillus stearothermophilus Saccharomyces cerevisiae 5 nutrient agar plates. Divide each plate into four quadrants and label appropriately with microbes name or initials. Inoculate each quadrant with a different microbe by means of a single line of inoculation of each microbe in its appropriately labelled section. Secure lids to plates. Incubate in inverted position at appropriate temperature (10C, 20C, 37C, 50C and 60C) for 24 hours. Record amount of growth: (-) (1+) (2+) (3+) = = = = Absence of growth Scant growth Moderate growth Abundant growth Comment on the temperature ranges the 4 microbes grow best at. Biotechnology Protocols 65 Experiments ANTIBIOTIC SENSITIVITY: KIRBY-BAUER METHOD Source: James Watt College This technique is used to determine the sensitivity of various microbes to antibiotics present on discs. Inoculate agar plates with 0.1 ml of test microbe: E. coli Klebsiella pneumoniae Staphylococcus epidermis albus i.e. set up spread plates Allow culture to soak in for 10 minutes. Add antibiotic disks using sterile forceps. Do not move the disk once it is placed on the agar Secure lids to plates. Incubate at 30C for 24 hours. Do not invert. Take a note of the specific antibiotics used here. Determine whether the bacteria are resistant, intermediate or susceptible to each antibiotic by comparing ‘zones of inhibition’ ie areas of clearing around the antibiotic discs. Deduce which antibiotic(s) would be best to treat each individual species of bacteria. Biotechnology Protocols 66 Experiments BIOCHEMICAL TESTS Source: James Watt College Oxidase Test This identifies cytochrome c oxidase, an enzyme found in obligate aerobic bacteria. Soak a small piece of filter paper in a fresh solution of 1% (w/v) NN-N1-N1-tetramethyl-p-phenylenediamine dihydrochloride on a microscope slide. Rub a small amount from the surface of a young, active colony onto the filter paper using a glass rod or plastic loop: a purple-blue colour within 10 s is a positive result Note: Performing the oxidase test – never use a nichrome wire loop, as this will react with the reagent, giving a false positive result. To avoid false negatives ensure you use sufficient material during oxidase and catalase testing, otherwise you may obtain a false negative result: a clearly visible ‘clump’ of bacteria should be used. Catalase Test This identifies catalase, an enzyme found in obligate aerobes and in most facultative anaerobes, which catalyses the breakdown of hydrogen peroxide into water and oxygen. Transfer a small sample of your unknown bacterium onto a coverslip using a disposable plastic loop or glass rod. Invert onto a drop of hydrogen peroxide on a slide: the appearance of bubbles within 30 s is a positive reaction. This method minimizes the dangers from aerosols formed when gas bubbles burst. The oxidase and catalase tests effectively allow us to sub-divide bacteria on the basis of their oxygen requirements. Obligate aerobes will be oxidase and catalase positive; Facultative anaerobes will be oxidase negative and catalase positive; Microaerophilic bacteria, aerotolerant anaerobes and strict (obligate) anaerobes will be oxidase and catalase negative – the latter group will grow only under anaerobic conditions. Once you have reached this stage (colony characteristics, shape, Gram reaction, oxidase and catalase status) it may be possible to make a tentative identification, at least for certain Gram-positive bacteria, at the generic level. To identify Gram-negative bacteria further tests are usually required. Biotechnology Protocols 67 Experiments SERIAL DILUTION Source: HSDU Biology and Biotechnology Microbiological Techniques Intermediate 1-Advanced Higher Folder Materials Lab coat Eye protection Benchkote if necessary Disinfectant and paper towels Discard jar with disinfectant Bunsen burner Labels 7 sterile 1 cm3 syringes or pipette tips 7 sterile test tubes or bottles each containing 9 cm3 diluent Broth culture of yeast or bacteria to be diluted Instructions 1 Wear a lab coat and use eye protection. 2 Label the test tubes or bottles with the appropriate dilution, 10 -1 – 10-7. 3 Make sure that the lid of the culture tube is firmly attached then shake the culture vigorously to separate clumps of cells and to distribute the organisms evenly throughout the liquid. 4 Remove a sterile syringe or pipette tip from its pack/container. Do not touch the parts which will come in contact with organism. If using a pipette tip, carefully attach to the dispenser. 5 Using aseptic technique (i.e. flaming the neck of the tube or bottle after removal and before replacement of its lid), remove exactly 1 cm 3 of the fluid and transfer to the dilution tube next in the series. 6 Place syringe or pipette tips into discard jar. 7 Mix the dilution well. 8 Repeat steps 4 – 7 until the last dilution tube is reached. This process dilutes the organisms in the original sample to a countable number. Biotechnology Protocols 68 Experiments COUNTING YEAST CELLS USING A HAEMOCYTOMETER Source: HSDU Biology and Biotechnology Microbiological Techniques Intermediate 1-Advanced Higher Folder Materials Haemocytometer and coverslip Tissue Alcohol Lens tissue Water Suspension of yeast cells Capillary tube Petri dish with moist tissue Microscope Instructions – setting up the slide 1 Clean the haemocytometer with alcohol, then wipe with lens tissue. 2 Using a damp tissue, moisten the slide as shown in diagram. 3 Push the special coverslip on to the slide as shown in diagram, pressing down on the outside edges of the coverslip at the same time until you can see Newton’s rings (see diagram). If you push the centre of the coverslip, it is likely to break. Biotechnology Protocols 69 Experiments Instructions – loading the haemocytometer 4 Shake the cell suspension gently. 5 Insert the end of the capillary tube into the suspension. The liquid will rise into the tube. 6 Run the end of the capillary tube along the edge of the coverslip between the arms of the ‘H’. The suspension should fill the area between the coverslip and the top half of the ‘H’ (shaded in diagram below). If the suspension flows into the troughs (the lines of the ‘H’), clean the slide and start again. 7 Turn the slide through 180 and repeat for the opposite edge of the coverslip. 8 Place the haemocytometer on a damp tissue in a Petri dish for at least two minutes to equilibrate. Biotechnology Protocols 70 Experiments Instructions – counting the cells The haemocytometer has two grids situated as shown in the diagram: 1 Place the haemocytometer on the microscope stage 2 Using the instructions for use of the microscope, examine the haemocytometer using the 4x objective lens. You should be able to view one whole grid as shown in the diagram. 3 Increase the magnification to the 10x objective lens. You should be able to see the 25 central squares, each of which is divided into 16 smaller squares (see diagram at instruction 4). 4 Increase the magnification to the 40x objective lens. You will see one of the 25 central square made up of 16 small squares. 5 Count the cells in each of the four corner squares and the central square; (see shaded squares in instruction 3). Note that you will count five groups of 16. Include in the count those cells touching the top or right side of the square; do not count those cells touching the bottom or left side. This takes account of cells which are half in and half out the square. Biotechnology Protocols 71 Experiments Instructions – calculation Length of side of grid = 1 mm Area of grid = 1 mm2 Depth between coverslip and slide = 0.1 mm Volume under squared area (25 squares) of grid = 1 mm2 x 0.1 mm = 0.1mm3 Volume under 5 squares (the number counted) = 0.1/ 5 mm3 = 0.02mm3 You have therefore counted the number of cells in 0.02 mm 3 and can use the following calculation to estimate the cell concentration of your original suspension. Number of cells (total in 5 squares) in 0.02 mm3 = n Number of cells in 1 mm3 = n x 50 Number of cells in 1 cm3 = (n x 50) x 1000 If the cell suspension counted has been diluted, then the above result must be multiplied by the appropriate dilution factor to give the concentration of the original culture. Biotechnology Protocols 72 Experiments VIABLE COUNT: POUR PLATE METHOD Source: HSDU Biology and Biotechnology Microbiological Techniques Intermediate 1-Advanced Higher Folder Materials Lab coat Eye protection Benchkote if necessary Disinfectant and paper towels Discard jar with disinfectant Bunsen burner Dilution series of organism Sterile pipettes or syringes (0.1 cm3) Sterile Petri dishes 20 cm3 volumes of sterile molten nutrient agar at 45C Glass spreader Alcohol Instructions 1 Wear a lab coat and use eye protection. 2 Label the underside of the plates with initials, date, sample and dilution. For greatest reliability/precision, each dilution should be plated in triplicate and the average of the three counts used. 3 Remove a sterile 0.1 cm3 syringe or pipette tip from its pack/container. Do not touch the parts which will come in contact with organism. If using a pipette tip, carefully attach to the dispenser. 4 Start with the highest dilution (i.e. 10-7). 5 Using aseptic technique (i.e. flaming the neck of the tube or bottle after removal and before replacement of its lid), remove exactly 0.1 cm 3 of the sample and transfer to the base of a sterile Petri dish. 6 Using aseptic technique, pour 20 cm3 sterile nutrient agar over the sample and mix gently. 7 Place syringe or pipette tip into discard jar. 8 Repeat steps 5 – 7 for dilutions 10-6, 10-5 and 10-4. 9 When the plates are solidified and dry, incubate upside down at the appropriate temperature for the appropriate time. 10 After incubation, select plates for counting that contain 30 – 300 colonies (samples which contain <30 colonies/0.1 cm3 diluent are subject to large fluctuations in numbers or sampling errors, plates which contain >300 colonies are likely to have overlapping colonies). 11 Count accurately and record the number of colonies on each plate. 12 Calculate the concentration of viable cells or colony forming units (cfu) in the original suspension. Biotechnology Protocols 73 Experiments VIABLE COUNT : SPREAD PLATE METHOD Source: HSDU Biology and Biotechnology Microbiological Techniques Intermediate 1-Advanced Higher Folder Materials Lab coat Eye protection Benchkote if necessary Disinfectant and paper towels Discard jar with disinfectant Bunsen burner Labels Dilution scrics of organism Sterile pipettes or syringes (0.1 cm3) Sterile nutrient agar plates Glass spreader Alcohol Instructions 1 Wear a lab coat and use eye protection. 2 Label the underside of the plates with initials, date, sample and dilution. For greatest reliability/precision, each dilution should be plated in triplicate and the average of the three counts used. 3 Remove a sterile 0.1 cm3 syringe or pipette tip from its pack/container. Do not touch the parts which will come in contact with organism. If using a pipette, carefully attach to the dispenser. 4 Start with the highest dilution (i.e. 10-7). 5 Using aseptic technique (i.e. flaming the neck of the tube or bottle after removal and before replacement of its lid), remove exactly 0.1 cm 3 of the sample and transfer to the surface of an appropriately labelled sterile nutrient agar plate. Note: Keep remainder of all samples 6 Using aseptic technique, spread the sample evenly across the plate with the glass spreader. 7 Place syringe or pipette tip into discard jar. 8 Repeat steps 5 – 7 for dilutions 10-6, 10-5 and 10-4. 9 When the plates are dry, incubate upside down at the appropriate temperature for the appropriate time. 10 After incubation, select plates for counting that contain 30 – 300 colonies (samples which contain <30 colonies/0.1 cm3 diluent are subject to large fluctuations in numbers or sampling errors, plates which contain >300 colonies are likely to have overlapping colonies). 11 Count accurately and record the number of colonies on each plate. 12 Calculate the concentration of viable cells or colony forming units (cfu) in the original suspension. Biotechnology Protocols 74 Experiments YEAST GROWTH CURVE Source: Adapted from SAPS ‘Growth Curve: Determination of Doubling Time’ http://www-saps.plantsci.cam.ac.uk/worksheets/scotland/double.htm Technical guides are also available from the same source. Read through the Student Activity Guide and consider the following questions. Analysis of Activity What is the aim of the activity? What measurements are you going to make? How will you record these measurements? How will you determine the information you require to make the final calculation? What constant will you calculate? Getting organised for experimental work In your group decide how the activity will be managed by allocating tasks to each member. It is very important that samples are removed at least three times per day: ideally early-morning, lunchtime and late afternoon. This will happen over 3 days. [A rota for removing samples may help]. Recording of data Prepare tables and a graph to record your group results. You should use a ruler, correct headings and appropriate units. Evaluation How effective were the methods which you used? What were the limitations of the equipment? What were the sources of error? What possible improvements could be made to the experiment? What ideas do you have for further work? What is the economic importance of the process which you are studying and the calculations which you will make? Biotechnology Protocols 75 Experiments STUDENT ACTIVITY GUIDE Introduction Stages of Growth Growth is the process during which living organisms synthesise new chemical components for the cell and as a result they usually increase in size. In unicellular organisms, such as bacteria and yeast, growth leads to cell division and consequently an increase in population size. The growth of a population of single-celled micro-organisms grown in a closed environment typically shows four stages: lag phase; exponential phase; stationary phase; death phase. The lengths and characteristics of these phases will depend upon factors such as the nature of the growth medium and temperature of incubation. In industry, it is important to understand the factors which affect the growth rate of a given micro-organism in order to generate maximum product by the most economic means. For example, if the desired product is a secondary metabolite such as an antibiotic which is produced when the organism has stopped growing, the manufacturer will want to provide optimum conditions for the culture to reach maximum numbers in stationary phase in the shortest time possible. In some cases, the product is the organism itself e.g. the production of yeast biomass to be used as starter cultures for brewing or baking, or as the starting point for autolysis which produces a huge variety of food flavourings. Growth of a population can be measured using the following methods: Cell counts: total numbers of cells are counted directly using a microscope and a special slide called a haemocytometer. Biotechnology Protocols 76 Experiments Dilution plating: the culture is serially diluted and a known volume of each dilution plated out and incubated. Resulting colonies are counted giving a measure of viable numbers of cells in the original population. Turbidometric methods: Cell density is measured using a colorimeter. This is a photometric method which measures the light scattered by the cells in suspension. Increase in cell density is an extremely accurate method of measuring cell growth rates. Biotechnology Protocols 77 Experiments In this practical, you will produce a growth curve of absorbance against time for a culture inoculated with a known dry mass of Saccharomyces cerevisiae (bakers’ yeast) then grown over several days. From this you will be able to calculate generation time and a growth rate constant. Equipment and materials Materials required Materials required by each student/group: 1 x 5 cm3 sterile yeast glucose broth as blank 99 cm3 sterile yeast glucose broth in flask dried yeast (not fast acting) weighing boat spatula 10 cm3 sterile water (if balance is accurate to 0.01g) 100 cm3 sterile water sterile 1 cm3 pipette discard jar containing 1% Virkon semi-log graph paper Materials to be shared: water bath or incubator at 30C balance (accurate to 0.001 g (preferably) or 0.01 g) colorimeter (440 nm) Instructions 1 Start this experiment late afternoon at the start of a week. 2 Draw a table showing date, time, hours of incubation and absorbance. 3 Using aseptic technique, add 0.025 g dried yeast to 100 cm 3 sterile distilled water at 30C. Shake gently to ensure that the cells are evenly distributed and suspended. 4 Using aseptic technique, dilute 100 times by adding 1 cm 3 to 99 cm3 sterile broth in a flask. This should give a starting concentration of 0.0025 g/l for your growth curve. 5 Using sterile medium as the reference, calibrate the colorimeter (i.e. set it to zero). Note: keep this reference medium (the blank) in the refrigerator throughout experiment. 6 Shake the flask containing the yeast culture gently to distribute the cells evenly. Using aseptic technique, withdraw a 5 cm 3 sample. 7 Measure the absorbance of the sample you have just withdrawn. Record date, time and absorbance in the table. 8 Incubate at 30C. 9 Repeat instructions 5 – 8 three times per day for the next three days (early morning, lunch-time and late afternoon if possible). If it is not possible to measure the absorbance at the time of taking the sample, place it in a sterile container, label with initials, date and time and refrigerate until Biotechnology Protocols 78 Experiments 10 11 12 13 convenient to do so, preferably within 24 hours. Make sure that the yeast is fully suspended before reading the absorbance. Plot absorbance vs time. Identify on your graph the lag, log and exponential phases of the growth curve. From the exponential (log) phase of growth curve, work out the time in hours taken for the absorbance and hence the population size to double. Calculate growth rate constant. SUPPLEMENTARY STUDENT INFORMATION Calculation of growth rate constant Growth rate constant, k, is a measure of the number of generations (the number of doublings) that occur per unit of time in an exponentially growing culture. k= n 2 g where n 2 is the natural log of 2 (determine this from your calculator) and g is the time in hours taken for the population to double during the exponential phase of growth. Biotechnology Protocols 79 Experiments DETERMINING DRY WEIGHT USING TURBIDITY: VERSION 1 Source: Adapted from SAPS ‘Growth Curve: Determination of Dry Weight from Standard Curve’. http://www-saps.plantsci.cam.ac.uk/worksheets/scotland/curve.htm Technical guides are also available from the same source. PREPARING FOR THE ACTIVITY Read Through the Student Activity Guide and consider the following questions. Analysis of activity What is the aim of the activity? What measurements are you going to make? How will you record these measurements? How will you determine concentration of yeast cells in a growing culture? Getting organised for experimental work In your group decide how the activity will be managed by allocating tasks to each member. Recording of data Prepare tables and graph paper to record your group results. You should use a ruler, correct headings and appropriate units. Evaluation How effective were the methods which you used? What is the significance of using dry mass as the measurement of concentration? What were the limitations of the equipment? What were the sources of error? What possible improvements could be made to the experiment? What ideas do you have for further work? What is the economic importance of the process which you are studying and the calculations which you will make? Biotechnology Protocols 80 Experiments STUDENT ACTIVITY GUIDE Introduction In industry it is often important to determine the actual concentration of cells in a growing culture. This may involve counting total numbers of cells, numbers of viable cells or cell concentration in terms of dry mass. In this experiment you will use commercially available dried yeast to produce a standard curve of absorbance at 440 nm against concentration (dry mass in grams/litre). You will then use your standard curve to determine the concentration of yeast in each of a series of diluted samples. [These can be from a previous experiment. Equipment and materials Materials required Materials required by each student/group: 100 cm3 sterile yeast glucose broth in flask or bottle 11 large test tubes (or universals) test tube rack dried yeast (not fast acting) weighing boat spatula 4 x 10 cm3 syringes (or pipettors with tips) 1 cm3 pipette cuvettes discard jar containing 1% Virkon graph paper marker pen Materials to be shared: Balance (accurate to 0.01 g) Colorimeter (440 nm) Crushed ice (optional) Instructions In order to make up the appropriate concentrations of yeast you will weight out 0.2 g dried yeast and add it to 20 cm3 sterile broth to give a concentration of dry mass of 10 grams/litre. You will then use this suspension (the standard dilution) to make further dilutions giving you yeast concentrations in g/l of 5, 4, 3, 2.5, 2, 1.5, 1.0, 0.5, 0.25, 0.05. Look at the table carefully and make sure you understand it before you start. Biotechnology Protocols 81 Experiments You may need crushed ice to keep your tubes cool while you carry out the experiment. 1 Withdraw 20 cm3 broth from a flask of sterile yeast glucose broth and add to a test tube (or universal). 2 Add 0.2 g dried yeast to the broth in the test tube. This will give you a concentration of 10 g/litre. Shake gently occasionally until the cells are fully resuspended. This is your standard dilution. Use it to make further dilutions. 3 Draw a table with test tube number, yeast concentration in g/l and absorbance at 440 nm. 4 Label clean test tubes 1 – 10. Add the volume of sterile broth shown in Row A in the table below to each tube. Pay close attention as you are required to add different volumes to the tubes. 5 From your standard dilution add the volume shown in Row B to tubes 1 – 5. Shake the standard dilution tube gently each time before removing the sample. Make sure that you understand how the yeast concentration in g/l is worked out. 6 Add the volumes from the numbered tubes shown in Row B to tubes 6 – 10. Note: Pay careful attention to tube numbers and volumes. Remember to suspend cells by shaking gently before taking a sample and use a fresh syringe for each tube. Dispose of used syringes and pipettes in the discard jar. Make sure that you understand how the yeast concentration in g/l is worked out. A B Tube no 1 Volume 5 sterile broth (cm3) Volume 5 yeast suspension (cm3) 2 6 3 7 4 7.5 5 8 6 5 7 5 8 5 9 5 10 9 4 3 2.5 2 yeast conc. 5 (g/l) 4 3 2.5 2 5 cm3 from tube 3 1.5 5 cm3 from tube 5 1 5 cm3 from tube 7 0.5 5 cm3 from tube 8 0.25 1 cm3 from tube 8 0.05 7 Using sterile broth as the reference, calibrate the colorimeter (i.e. set it to zero) at 440 nm. 8 Starting with tube 10 shake the test tube to distribute the cells evenly and use a pipette to transfer about 3 cm3 into a cuvette. Biotechnology Protocols 82 Experiments 9 Measure the absorbance at 440 nm of the sample you have just withdrawn. Record it in the table. 10 Return the sample to its original tube and repeat steps 7 and 8, using the same pipette and cuvette, to obtain readings for tubes 9 – 1. 11 Draw a graph of absorbance at 440 nm vs. yeast concentration (grams/litre). This is the standard curve which you will use to determine yeast concentration from absorbances of serially diluted samples in earlier experiments e.g. viable count experiment. Biotechnology Protocols 83 Experiments Using the standard curve to determine concentration in grams/litre from known absorbance 1 Use the absorbances measured in the previous experiment. 2 Draw a table showing time of sample, absorbance and concentration. 3 Fill in the time and absorbance rows. 4 Carry out the following for each absorbance: Mark the value on the absorbance (vertical) axis of your standard curve Draw a horizontal line till it meets the standard curve (a). Mark the point Draw a vertical line from there to meet the yeast concentration axis (b) Read the value and complete the concentration row on the table 5 Determine the dry weight of yeast (in g/litre) present in the original sample. 6 Write a report on your practical placing particular emphasis on evaluation of the equipment and methods used with respect to the resulting accuracy and reliability. Biotechnology Protocols 84 Experiments DETERMINING DRY WEIGHT USING TURBIDITY: VERSION 2 Source: James Watt College Take stock culture of micro-organism & serial dilute to 10-7. e.g. 1 ml 1 ml 1 ml 9 ml diluent 9 ml diluent 9 ml diluent = 10-1 = 10-2 = 10-3 1.0 ml stock 10-7 This will give 8 samples in total (including original stock). Measure the turbidity of each of these samples using a spectrophotometer set to 600 nm. Plot graph of absorbance against dilution factor: is there a direct relationship between dilution and turbidity? Given the following piece of reference information, convert your absorbance readings to quantities of actual micro-organism (a sample calculation is provided to help). Absorbance at 600nm value of 0.5 is equivalent to a total cell mass of 1 mg dry cells per ml. Example of calculation A600 = 0.298 from machine for 10-1 dilution 0.298 x 1.0 mg/ml = 0.596 mg/ml for 10-1 dilution 0.500 Correcting for dilution factor, this becomes 0.596 x 10 5.96 mg/ml. Biotechnology Protocols 85 Experiments CARBOHYDRATE FERMENTATION Source: James Watt College Carbohydrate utilization tests Some bacteria can use a particular carbohydrate as a carbon and energy source. Acid end-products can be identified using a pH indicator dye while CO2 is detected in liquid culture using a Durham tube (inverted small test tube). Aerobic breakdown is termed oxidation while anaerobic breakdown is known as fermentation. Identification tables usually incorporate tests for several different carbohydrates. Durham tube in carbohydrate utilization broth. Air within the Durham tube is replaced by broth during the autoclaving procedure. Collect the following materials: 24 hour broth cultures of E. coli, Staphylococcus epidermis albus, Erwinia carotovora and Micrococcus luteus. Phenol red dextrose, lactose and sucrose peptone broths in test tubes also containing Durham tubes. Label one tube of each type of medium and inoculate with E. coli. Label one tube of each type of medium and inoculate with Staphylococcus epidermis albus. Label one tube of each type of medium and inoculate with Erwinia carotovora. Label one tube of each type of medium and inoculate with Micrococcus luteus. Label one tube of each type of medium and do not incubate i.e. control tubes. During incubation do not tip the fermentation tube as this may accidentally force a bubble of air into the Durham tube to give a false positive result. Mix tubes by rolling them back and forth between the palms of the hands. Incubate tubes at 30C for 24 – 48 hours. Examine all cultures for evidence of acid (pH change), and/or acid and gas production for each sugar. Determine type of fermentation occurring. Biotechnology Protocols 86 Experiments DIGESTION OF CELLULOSE SOURCE: “Microbial Friends & Allies” BBSRC publication (originally developed by NCBE http://www.ncbe.reading.ac.uk/ ) Cellumonas bacteria secrete cellulase enzyme so could potentially be used to utilise cellulose waste (eg paper) as feedstock for their fermentation process, converting low cost start materials into products of greater value. 1 Use sterile forceps to place a disc of sterile filter paper on the surface of a nutrient agar plate. Use the tips of the forceps to smooth the paper onto the agar, to ensure that there is a good contact. 2 Take the lid off the Cellulomonas culture bottle. Keep the bottle top in your hand – do not place it on the bench. Briefly pass the neck of the bottle through a Bunsen burner flame. 3 Dip a sterile cotton wool bud into the culture. Do not allow the culture to drip on the bench. Flame the neck of the bottle again and replace the lid. 4 Quickly ‘paint’ a message or picture on the filter paper with the culture (see diagram below). Dispose of the cotton wool bud into a beaker of disinfectant. 5 Seal the Petri dish diagonally with a small amount of tape. Label the base of the Petri dish with your initials, the date, and the name ‘Cellulomonas’. 6 Incubate the Petri dish at 25 - 30C in an inverted position: the filter paper should stick to the surface of the nutrient agar. 2 or 3 weeks later After incubation, the sealed plates may be examined for digestion of the filter paper under a binocular microscope or using a hand lens. You should be able to read the message/see the picture as the paper should have been digested where the Cellulomonas bacteria were painted. Biotechnology Protocols 87 Experiments Biotechnology Protocols 88 Experiments TESTING VIABILITY OF YEAST AT DIFFERENT STAGES OF THE AUTOLYSIS PROCESS SOURCE: SAPS http://www-saps.plantsci.cam.ac.uk/worksheets/scotland/yeast.htm Teacher and technical guides can also be found at this site. Preparing for the Activity Read through the Student Activity Guide and consider the following questions. Analysis of activity What is the aim of the activity? What is being varied in the activity? What measurements are you going to make? Getting organised for experimental work What precautions you must be taken to prevent contamination of the agar plates? Can you successfully examine material under a microscope at x400 magnification? In your group decide how the activity will be managed by allocating tasks to each member. It is important that you play an active part in setting up the experiment and in collecting results. Recording of data Prepare summary tables to record your group results. You should use a ruler, correct headings and appropriate units. Evaluation How effective were the tests which you used? What were the limitations of the equipment? Were there any possible sources of error? What possible improvements could be made to the experiment? What is the biological importance of the process which you are investigating? Biotechnology Protocols 89 Experiments Student Activity Guide Introduction Yeasts are versatile micro-organisms which have been used for centuries by man to produce bread and alcoholic drinks. In more recent years they have been used to produce flavourings for the food industry. The yeast is grown in huge fermenters to produce biomass – Upstream Processing and then it is treated in different ways to produce different flavourings. These flavourings are found in most of the savoury snack foods which we eat – crisps, soups, snacks etc. The yeast goes through a series of different treatments to develop the huge variety of different end products. All of these treatments involve the process of autolysis and are examples of what is known as Downstream Processing. The yeast products may be powders, granules or pastes and they are then incorporated into processed foods to provide natural flavourings. This process of AUTOLYSIS (auto-self; lysis-splitting) involves killing the yeast and encouraging the breakdown of the cells by enzymes. These may be the cells own endogenous enzymes or enzymes may be added. It is these products of enzyme degradation which produce the specific flavour molecules. Autolysis usually begins with the addition of salt to the cells, causing water to leave the cells by osmosis and beginning the process of cell breakdown. The cells are then heated encouraging further breakdown of the cells. In this practical you are going to carry out the process of autolysis and try to find at what point in the process the cells actually die. You will salt and heat yeast and then test the viability by plating out the treated yeast to see if it will grow. You will also test the autolysed product to see if the dehydrogenase enzymes are active and to see if the cells take up methylene blue dye. Biotechnology Protocols 90 Experiments Yeast Autolysis Equipment and materials Materials required by each student/group: 10 g fresh yeast salt 5x 200 cm3 beakers stirring rod Materials to be shared: Ovens at different temperatures Instructions 1 Autolyse the yeast in 4 different ways. You could alter the temperature at which the yeast is autolysed or the amount of salt which is added to the yeast. One possible regime is suggested below: 2 g yeast 1 g salt at room temp overnight 2 g yeast 1 g salt at 40C overnight 2 g yeast 1 g salt at 60C overnight 2 g yeast 1 g salt at 80C overnight 2 Collect dried yeast samples and rehydrate by slowly adding water to the dried sample, stirring constantly. Continue to add water to the samples to make them up to 100 cm3. 3 Autolyse a fresh sample of yeast. Mix 2 g yeast with 1 g salt and add water to make this sample up to 100 cm3. You can now test the viability of these yeast samples by 3 different methods. Biotechnology Protocols 91 Experiments Method 1: Plating Out Autolysed Yeast Samples Streaking an agar plate Equipment and materials Materials required by each student/group: Disinfectant and cloth 3 Yeast agar plates Metal inoculating loop Rehydrated yeast samples Marker pen Sellotape Materials to be shared Incubator at 30C Instructions 1 Make sure that you are working in an area which has been swabbed with disinfectant. 2 Turn your petri dish upside down and use a pen to mark the base, as shown, and label it with the yeast samples to be used, the date and your initial. 1 2 3 Open your petri dish and using a sterile inoculating loop, place your sample of yeast onto one side of the petri dish. Make a shape like this. Biotechnology Protocols 92 Experiments 4 Repeat with other yeast samples and plates. 5 Seal your plates with tabs of sellotape and place it in an incubator at 30C. 6 Record any growth of yeast over the next 5 days. Biotechnology Protocols 93 Experiments Method 2: Comparing the activity of the dehydrogenase enzymes present in autolysed yeast samples. Background Information During a metabolic pathway such as aerobic respiration glucose is gradually broken down and energy is released. Hydrogen is released from the glucose in a process knows as oxidation. This hydrogen binds to a co-enzyme and each reaction is catalysed by an enzyme known as a dehydrogenase. Although it would not be possible to detect this reaction in a test-tube some chemicals such as resazurin dye change colour when they gain hydrogen. It changes colour in the following ways: BLUE LILAC Unreduced MAUVE PINK Partially reduced COLOURLESS Reduced You can use this reaction to compare the activity of the dehydrogenase enzymes present in each of your autolysed yeast samples. The time it takes for the dye to change colour will indicate the activity – the faster the colour change takes place the greater the activity of the dehydrogenase enzymes. Activity of enzymes such as dehydrogenases would indicate that the yeast is likely to be viable. Biotechnology Protocols 94 Experiments Equipment and materials Materials required by each student/group: Rehydrated yeast samples 1 test-tube rack with 5 test tubes 5 labels 1 stop clock 1 pair safety spectacles 1 syringe/measuring cylinder 16 cm3 Resazurin dye 20 cm3 5% glucose solution colour chart Materials to be shared: Waterbath at 35C Instructions 1 Collect the materials indicated above. 2 Label 5 test-tubes and add 3 cm3 of resazurin dye to each tube. 3 Add 3 cm3 of the appropiate yeast suspension to the labelled tubes. 4 Shake each tube and place in a water bath at 35C. 5 Using the colour chart, record the colour of each tube every 2 mins for 20 mins. If you do not get a reaction in any of the tubes after 10 minutes add 3 cm 3 of 5% glucose solution to each test-tube and shake. 6 Record the results in a table with suitable headings. Biotechnology Protocols 95 Experiments Method 3: Testing the yeast samples with methylene blue Methylene blue dye will diffuse into the yeast cells. If the cells are living they will pump the blue dye out but if they are dead they will remain blue. Equipment and materials Materials required by each student/group: rehydrated yeast samples microscope microscope slides and coverslips dropper 0.1% methylene blue solution 50 cm3 beaker stirring rod distilled water Instructions 1 Place a drop of one of the diluted yeasts onto the microscope slide and add a drop of methylene blue dye and wait 5 minutes. 2 Place the slide onto the stage of your microscope and focus. 3 Count all the blue cells and clear cells in your field of view. 4 Repeat steps 1-3 for each of the yeast samples. You may find that there are too many cells to count, if this is the case then you can dilute your samples and start again. 5 Record your results in a suitable table. 6 Calculate the percentage of viable ie unstained cells. Biotechnology Protocols 96 Experiments TISSUE CULTURE: EFFECT OF M&S SALTS ON EXPLANT GROWTH SOURCE: LTScotland National Curriculum Support Materials Intermediate 2 Biotechnology, Unit 3 Biotechnological Processes, Student Materials Background information This tissue culture technique uses (cotyledon) explants taken from the top of a seedling. Micro-organisms grow faster than plant cells. It is important to keep seeds, medium and equipment sterile. Follow aseptic technique throughout. Equipment and materials required by each student/group: Part 1 Eye protection Disinfectant and paper towels 10 mustard seeds 30 cm3 10% bleach in lidded container forceps clingfilm new, clean, block shaped sponge germination vessel, half filled with sterile water Part 2 (after 2 – 3 days) All equipment must be very clean Disinfectant and paper towels Bunsen burner Eye protection Forceps Scissors 6 small glass containers: 2 half filled with 1% plain agar 2 half filled with 1% plain agar plus 2.2 g litre M&S salts 2 half filled with 1% plain agar plus 4.4 g litre M&S salts clingfilm Materials to be shared Light bank Hazard bags for disposal of plates Biotechnology Protocols 97 Experiments Instructions Part 1: Sterilising seeds for germination 1 Prepare work space: clear area, swab bench with disinfectant. 2 Collect materials and equipment for part 1. 3 Place 10 mustard seeds in lidded container of bleach. Swirl seeds and leave for 10 minutes in the bleach. Remember your seeds are now sterile. Do not touch with your hands. 4 Pour off bleach. 5 Rinse three times with fresh, sterile water, leaving them covered with a little water. 6 Sprinkle the seeds onto the clean sponge, making sure the seeds are spaced out. 7 Place sponge in germination vessel, with water level half way up sponge. 8 Label vessel with your name date and seed type. 9 Place under a light bank in a warm place (ideally 20 - 26C), for 2 to 3 days. 10 Leave until germinated and cotyledons (young leaves) have just started to unfold. Biotechnology Protocols 98 Experiments Instructions Part 2 (after 2 – 3 days) 1 Collect germination vessel and equipment for part 2 (see Student Guide). 2 Select the 6 straightest and longest seedlings for your experiment. 3 Label six glass containers with date and check there are 2 each of three medium types: You should have 6 containers: 2 with agar and no M&S salts 2 with agar and 2.2 g/l M&S salts 2 with agar and 4.4 g/l M&S salts 4 Cut off six cotyledons as shown below. These are your explants. 5 Use cooled, sterile forceps to transfer one explant into each of the six containers. Push cut end of explant into the agar, making sure the cotyledons are not touching the agar’s surface. 6 Cover containers with cling film and place in a warm area under a light bank for 1 to 2 weeks. 7 Observe and note the number of explants with roots after 3, 6 and 9 days approximately. 8 Collect class results and calculate % explants with roots for the three different media on the three different days. 9 Present your results as three lines on a single graph (one line for each medium) with suitable scales and axes labelled with quantities and units. Biotechnology Protocols 99 Experiments CLONING CAULIFLOWER Source: NCBE Practical Biotechnology at http://www.ncbe.reading.ac.uk/NCBE/PROTOCOLS/PRACBIOTECH/PDF/ cauli.pdf A similar protocol, which includes colour photographs, can be found at http://www-saps.plantsci.cam.ac.uk/docs/tissue.pdf Materials required for each student or group of students: Fresh cauliflower curd (the white part) Sterile distilled water (100 cm3) 70% ethanol (50 cm3) 20% Domestos solution (100 cm3) Boiling tubes, each containing 2-3 cm3 of plant tissue growth medium Sterile Petri dish Metal forceps and scalpel Non-absorbent cotton wool and aluminium foil White tile or suitable cutting surface Practical details 1 Swab the working area with 70% ethanol. Keep the ethanol away from exposed flames! 2 Cut out a small piece of cauliflower curd; roughly the size of a cherry. Working on a clean Petri dish, divide the curd into three. 3 Place the pieces (explants) in bleach e.g. Domestos solution for 10 minutes to surface sterilize the tissue. From this point on, quick, aseptic operations are important to prevent contamination. 4 Rinse the explants in three successive beakers of sterile distilled water. Use flamed, cooled forceps to do this – the correct way to flame forceps and other instruments is to dip them in alcohol, then to pass them briefly through a flame to ignite the ethanol. As the ethanol burns off, it heats the surface of the instruments to 70C, killing any contaminating organisms. Do not heat forceps and scalpels until red hot, and remember to keep ethanol away from exposed flames. 5 The explants can be left in the final beaker of sterile water (covered with a Petri dish lid) until required. 6 Take the first tube of growth medium withdraw the cotton wool plug, then briefly flame the neck. Use flamed, cooled forceps to pick up an explant and quickly drop it into the tube. Return the forceps to the ethanol beaker. Flame the neck of the tube once more, then replace the plug. 7 Repeat Step 6 with the two remaining explants and two fresh tubes of growth medium. 8 The tubes should be kept in a warm, light place. Growth should be visible within 10 days. Contamination, if it has occurred, should also be visible by this time. Failure of anything to grow usually indicates that the bleach has not been rinsed from the plant tissue. Biotechnology Protocols 100 Experiments Safety Students should wear safety goggles when using bleach solution. Ethanol used for sterilizing working surfaces should be kept away from naked flames. Results Observe periodically throughout the 10 day incubation process and describe growth. Biotechnology Protocols 101 Experiments Biotechnology Protocols 102 Experiments ENZYMES & FRUIT JUICE PRODUCTION Source: Adapted from NCBE Practical Biotechnology http://www.ncbe.reading.ac.uk/NCBE/PROTOCOLS/PRACBIOTECH/PDF/ juice.pdf Extracting Fruit Juice The enzyme pectinase can break down the pectin found in cell walls. Fruit juice companies use pectinase to improve the juice extraction and to produce a clearer juice. Read the following procedure and think carefully about what apparatus you will need and what information you will need to record. Remember to label your beakers and measuring cylinders Experiment 1 Practical details Collect a piece of apple and grate it. Place an equal weight of grated apple into two beakers To one beaker add 2 ml of pectinase and to the other add 2 ml of distilled water. Stir each beaker and leave for 5 minutes. Filter the juice from the apples. Record the volume of juice every 2 minutes. Apparatus Biotechnology Protocols 103 Experiments Results Use the following grid and design your results table Conclusion Biotechnology Protocols 104 Experiments ENZYMES Extracting Fruit Juice Questions 1 What effect did the enzyme pectinase have on the volume of juice produced? ___________________________________________________________ 2 What effect did the enzyme have on the clarity of the juice? ___________________________________________________________ 3 Why did you set up a beaker which had the apples and distilled water in it? ___________________________________________________________ 4 What other factors might affect the volume of juice produced? ___________________________________________________________ ___________________________________________________________ You will investigate some of these factors when you know a little more about juice extraction. Biotechnology Protocols 105 Experiments ENZYMES Clarifying Fruit Juice You have already discovered some effects of the enzyme pectinase. Pectinase was first used by industry to clarify juice. In this experiment you are going to look at the effects of pectinase and amylase on the clarity of juice. What does the enzyme amylase do? ______________________________________________________________ Experiment 2 Practical Details Set up the following test tubes Biotechnology Protocols 106 Experiments ENZYMES Clarifying Fruit Juice Add 10 ml of cloudy apple juice to each tube and stir Incubate for 30 minutes at 40C Observe the tubes every 5 minutes and record their appearance Results: Make a results table to record your results Questions 1 What effect did each enzyme have on the clarity of the juice? AMYLASE ___________________________________________________________ PECTINASE ___________________________________________________________ 2 What was the effect of using both enzymes? ___________________________________________________________ ___________________________________________________________ Biotechnology Protocols 107 Experiments DEHYDROGENASE ACTIVITY IN YEAST Source: NCBE Practical Fermentation http://www.ncbe.reading.ac.uk/ncbe/protocols/fermentation.html Yeasts are living organisms. They belong to the group of microbes known as FUNGI. Yeasts are single celled organisms which reproduce by splitting into two. This process in yeast is known as budding. Yeast has been used for thousands of years to make beer, bread and wine. These technologies have developed because yeast cells can produce carbondioxide and alcohol when they grow. The process is known as fermentation. Nowadays yeast is still used in the brewing and baking industries but it is also used to produce new products such as flavourings for foods, fizzy drinks and the enzymes needed to make cheese. Biotechnology Protocols 108 Experiments Dehydrogenase Enzymes in Yeast During aerobic respiration glucose is gradually broken down and energy is released. glucose + oxygen energy + carbon dioxide + water In a metabolic pathway such as this, it is usually the removal of hydrogen from the glucose which allows the energy to be released. The removal of hydrogen is called oxidation. As the glucose is oxidised, hydrogen is released at various stages along the pathway. This hydrogen binds to a chemical called a co-enzyme and each reaction is catalysed by an enzyme known as a dehydrogenase. Therefore dehydrogenase enzymes catalyse the oxidation of substrates by transferring hydrogen to co-enzymes such as NAD. These co-enzymes carry the hydrogen as they become reduced. AH2 substrate e.g. glucose + 2 NAD co-enzyme A oxidised substrate + 2 NADH reduced co-enzyme Although it would not be possible to detect this reaction in a test-tube some chemicals such as resazurin dye change colour when they gain hydrogen. We say the chemical has become reduced. It changes colour in the following ways: blue (unreduced) lilac mauve (partially reduced) pink colourless (reduced) This reaction enables you to test to see if respiration is taking place. You can also use it to investigate respiration rates as you alter variables. Yeast is a suitable organism to use for these investigations. Biotechnology Protocols 109 Experiments Experiment – Testing respiration rate in yeast Apparatus 3 test tubes 3 labels 5 ml or 10 ml syringe resazurin dye fresh yeast solution boiled and cooled yeast solution glucose solution water bath timer Method 1 Label 3 test-tubes A, B & C 2 Add 3 ml of resazurin dye to each tube 3 Add 3 ml of glucose solution to A & C Add 3 ml of water to B 4 Add 3 ml of fresh yeast suspension to A & B Add 3 ml of boiled and cooled yeast suspension to C 5 Shake each tube and place in a water bath at 35C Result Record the colour in each tube in the table below Time in minutes A B C 0 Blue 3 Lilac 6 Mauve 9 Pink 12 Colourless 15 18 21 24 Biotechnology Protocols 110 Experiments QUESTIONS 1 Give an example of a metabolic pathway __________________________ ___________________________________________________________ 2 What happens to the hydrogen when it is first released from the glucose? ___________________________________________________________ ___________________________________________________________ 3 What do dehydrogenase enzymes do? ___________________________________________________________ ___________________________________________________________ 4 What is a reduced coenzyme? ___________________________________________________________ 5 What happens to resazurin dye when hydrogen is added to it? ___________________________________________________________ 6 In which test-tube did the yeast respire most rapidly? ___________________________________________________________ 7 What was the purpose of test-tube C? ___________________________________________________________ Biotechnology Protocols 111 Experiments DNA FROM KIWI FRUIT Source: Adapted from NCBE Illuminating DNA http://www.ncbe.reading.ac.uk/NCBE/PROTOCOLS/PRACBIOTECH/PDF/ onion.pdf In this practical you are going to isolate DNA from plant cells (i.e. not from micro-organisms). Detergent is used to degrade the cell and nuclear membranes. Cell fragments are separated by filtration leaving the DNA and the soluble proteins in the filtrate. A protease enzyme called neutrase breaks down the protein and then the DNA is precipitated using ice cold ethanol. QUESTIONS 1 Which part of the cell and nuclear membrane is the detergent degrading? ___________________________________________________________ 2 What will the neutrase enzyme break its substrate into? ___________________________________________________________ APPARATUS A balance Weigh boat Sodium chloride Washing up liquid 5 ml syringe 2 x 250 ml beaker 50 g of kiwi fruit stirring rod coffee filter paper measuring cylinder boiling tube ice cold ethanol A liquidizer, an ice bath and a water bath at 60C must be available in the lab. METHOD Part 1 Preparing the Fruit extract 1 Add 1.5 g of sodium chloride and 5 ml of washing up liquid to a 250 ml beaker. 2 Make up to 50 ml with distilled water. 3 Now, collect 50 g of fruit and chop it up, then add it to the salt/detergent solution. Biotechnology Protocols 112 Experiments 4 Stir the mixture and incubate for 13 minutes at 60C 5 Cool the mixture in an ice bath for 4 minutes. 6 Pour the mixture into a liquidizer and blend for 5 seconds at high speed. 7 Filter this mixture into a second beaker. Separating the DNA 1 Add 4 ml of fruit extract to a boiling tube 2 Add 2 drops of neutrase enzyme to the extract 3 Slowly trickle 6 ml of ice-cold ethanol very slowly down the side of the test-tube. Two layers of liquid should form. 4 Leave the tube for 2-3 minutes without disturbing it. 5 Gently rock the liquids taking care not to let the two layers mix too much. RESULT Describe the DNA which you have precipitated Biotechnology Protocols 113 Experiments DNA FROM CAVIAR Source: online Bioscience journal http://www.bioscience-explained.org/EN1.1/pdf/CaviarEN.pdf AIM This simple practical procedure allows the isolation of impure DNA from ‘caviar’ or fish eggs. The result is a pellet of thread-like material, which includes DNA but will still be contaminated with lipids, carbohydrates and proteins. EQUIPMENT AND MATERIALS Needed by each person or group 20-30 g caviar (about 2 heaped teaspoonsful) e.g. roe from capelin (Mallotus villosus) or lumpsucker (Cyclopteris lumpus). Note: such roe is sold under the Abba® brand name. The yellow or ‘natural’ variety works best. 15 ml washing-up liquid e.g. Fairy Liquid, diluted 1:10 with distilled water 1 teaspoon (about 6 g) of table salt 2 ml ethanol. This must be ice-cold and at least 80% pure. 3-4 drops of protease, e.g. Novozymes Neutrase ® Glass rod Coffee filter paper Funnel Small test tube Dropper or pipette for dispensing the enzyme Pasteur pipette, the tip of which has been melted and curved to form a small hook Biotechnology Protocols 114 Experiments PROCEDURE 1 Add the caviar and salt to a mortar, then crush the eggs using a pestle. The shells of the eggs have to be broken. Proteins are precipitated by the salt. 2 Add the washing-up liquid solution to the mortar. The liquid should cover the caviar completely. The detergent dissolves lipids from the membranes of the roe. 3 Add 3-4 drops of protease to the mixture and stir vigorously. The enzyme will partially degrade any soluble proteins. 4 Filter the mixture through the coffee filter and collect the filtrate in a clean test tube. 5 Add the ice-cold ethanol by carefully pouring it along the wall of the tube or use a pipette and add it at the bottom of the test tube. DNA precipitates as long threads in cold ethanol and can be found at the interface between the detergent solution and the ethanol. 6 Collect the DNA with the help of a Pasteur pipette with a hooked tip. The DNA may be transferred to a microcentrifuge tube and stored, frozen, for later use e.g. for gel electrophoresis or staining of the DNA. Biotechnology Protocols 115 Experiments RESTRICTION ENZYME DIGESTS & DNA GEL ELECTROPHORESIS Source: Adapted from NCBE ‘The Lambda DNA Protocol’ at http://www.ncbe.reading.ac.uk/NCBE/PROTOCOLS/PDF/LambdaSG.pdf A technical/teachers guide to this activity can be found at http://www.ncbe.reading.ac.uk/NCBE/PROTOCOLS/PDF/LambdaTG.pdf 1 CUTTING THE LAMBDA DNA WITH RESTRICTION ENZYMES The restriction enzymes are in the following tubes: Yellow – empty Green – HindIII Blue – Bam H1 Pink – EcoR1 Wear gloves (to protect the DNA from digestion by enzymes in your sweat). Add a fresh tip to the Gilson pipette. Practice aspirating and ejecting 10l of blue dye. [Volume errors in pipetting when using such small volumes can make big errors in your results]. Add a fresh tip to the pipette. Put 20 l of your chosen DNA solution into an enzyme tube of your choice. Mix the liquid with the dried enzyme by carefully drawing the liquid up and down in the tip a few times. The liquid in the enzyme tube should have a distinct blue colour and there should be no concentration of dye at the bottom of the tube. Cap the tube containing the enzyme and DNA, then incubate it at 37C in a waterbath for 30-45 minutes. Repeat with the other enzyme tubes using a fresh tip each time. It helps to flick the tubes occasionally during incubation, to ensure that their contents are thoroughly mixed. 2 LOADING THE GELS After the incubation, add 2 l of loading dye to each enzyme tube using a fresh tip. [The loading dye contains sucrose so the sample will sink to the bottom of the well in the gel and not float away in the buffer in the tank. It also is blue so you can see your samples]. Mix the loading dye and the DNA sample very thoroughly by drawing the mixture up and down the pipette tip. Put the gel and tank over a piece of black paper or card to make the wells more visible. Pour 10 ml of TBE buffer onto the gel. Load 10l of the mixture of loading dye and DNA into one of the wells, holding the tip above the well Biotechnology Protocols 116 Experiments but under the buffer solution. Take great care not to puncture the bottom of the well with the pipette tip. Depress the pipette plunger gently and the DNA and dye mixture should sink into the well as it leaves the pipette tip. Make a note of which DNA you have put in each well. Repeat these steps with the other DNA samples using a fresh tip for each sample. Load dye markers (5 l) in one well. 3 RUNNING THE GEL Fit a piece of carbon fibre tissue to each end of the tank so one end of it is in the buffer. Check the power pack is turned OFF. Put a lid on your tank. Using the red and black leads, connect the tissue to the power source. The red lead should go to the positive and be at the end of the tank furthest from the wells. The black lead should go to the negative and be at the end nearest to the wells. [The reason it is this way round is that DNA in this running buffer, has a negative charge and will be attracted to the positive terminal (anode) when a current is applied]. After a few minutes you should see bubbles forming at the cathode (negative end of the tank). After a few more minutes, the DNA and dye mixture should start migrating through the gel. If it is going the wrong way, or not moving at all, you have connected your leads up incorrectly. Try again. The gel should run until the blue dye (which runs in front of any DNA fragments) reaches the end of the gel. This will probably take a few hours. Switch off the power and remove the leads. If you let it run too long, the DNA will come off the end of the gel and be lost in the buffer!!! Biotechnology Protocols 117 Experiments 4 STAINING THE GEL Wear plastic gloves. Pour off the buffer solution and pour the stain onto the surface of the gel. Leave it for exactly 4 minutes, then return stain to the beaker/stain bottle. Very carefully wash the gel surface with cold distilled water to remove excess stain. Repeat 3-4 times, finally pouring off all the water from the gel. The remaining stain will gradually move down through the gel, staining the DNA as it does so. Faint bands should start to appear after 10 minutes. Better results are seen if the gel is left to develop over night. Leave the gel in a plastic bag in the fridge, to prevent the gel from drying out. Biotechnology Protocols 118 Experiments -GALACTOSIDASE INDUCTION Source: Adapted from NCBE Illuminating DNA http://www.ncbe.reading.ac.uk/ncbe/PROTOCOLS/DNA/PDF/DNA06.pdf The lac operon is the classic example of gene regulation, in which the production of -galactosidase (lactase) is induced by the presence of lactose in the growth medium. In this practical task, ONPG, rather than lactose, is used as a substrate for the enzyme. Aim To induce and measure the production of the enzyme -galactosidase (lactase) by E.coli. Day 1: Preparation You will need cultures of E.coli from a strain that possesses the lacZ (galactosidase) gene. These can be grown on solid agar 24-48 hours in advance. To induce the production of -galactosidase, lactose must be present in the growth medium. From stock E.coli plate prepare the following two streak plates: E.coli on nutrient agar E.coli on nutrient agar and lactose. Incubate at 30C for 24-48 hours. Day 2: Timing This activity takes about 60 minutes, including an incubation period of 10 minutes. Materials and equipment needed by each person or group Cultures of E.coli ONPG (ortho-nitrophenyl--D-galactoside) solution (2 cm3 per test sample) Methylbenzene; 1 drop per test sample Test tubes, caps, rack and marker pen Inoculation loop Pasteur pipettes 5 cm3 syringe, for transferring ONPG solution Waste container with disinfectant Stopclock Safety spectacles Biotechnology Protocols 119 Experiments Procedure Quick qualitative method 1 Using a syringe, transfer 2 cm3 of ONPG solution into each of two test tubes. One of these tubes will be a ‘control’; the other will be for the test culture. 2 Label the tubes appropriately. 3 Use a flamed wire loop to aseptically transfer a large colony of E.coli from the nutrient agar plate into the test solution. Suspend the microorganisms by agitating the loop, then sterilise it by flaming. Take care to introduce the loop into the flame slowly, to avoid sputtering! Repeat the process, this time transferring an E.coli colony from the plate where nutrient agar contains lactose, into the test solution. 4 Add a drop of methylbenzene to each tube, cap the tubes and shake well to mix. Methylbenzene kills the cells and partially disrupts the cell membranes, allowing the ONPG to diffuse into the cells. 5 Let the test tubes stand on the bench, until a strong yellow colour develops. This generally takes 5-20 minutes. The reaction can be speeded up by incubating the tubes at 37C. The colourless ONPG is broken down by -galactosidase to produce galactose and orthonitrophenyl (ONP). ONP is bright yellow in alkaline conditions. 6 Compare the colour of the two tubes. Safety Handling microorganisms and methylbenzene Good microbiological practice must be observed when handling microorganisms. Methylbenzene is flammable and produces harmful vapour. Large volumes should therefore be handled in a fume cupboard, although the small amounts used here can safely be handled at the bench (but keep away from flames). Skin and eye contact should be avoided. Eye protection must be worn. Biotechnology Protocols 120 Experiments Qualitative method STEPS 1&2 Transfer 2cm3 of ONPG solution into each of two test tubes. Label the tubes appropriately. 3. Aseptically transfer a colony from the plate to one of the tubes of ONPG solution. Twiddle to disperse the cells, then flame the loop to kill any remaining cells. 4-6. Add a drop of methylbenzene to each tube, cap the tubes and shake well to mix. Stand the tubes for 5-20 minutes until a yellow colour develops. Biotechnology Protocols 121 Experiments GENETIC CONTROL Source: Adapted from NCBE Practical Biotechnology http://www.ncbe.reading.ac.uk/NCBE/PROTOCOLS/PRACBIOTECH/PDF/cat milk.pdf The genes found in living cells control the production of proteins such as enzymes. Any one cell can contain many thousands of genes all coding for different proteins. In a multicellular organism only a small proportion of the genes available within the cell will be required. Scientists think that genes become switched on or off as they are required. We are going to look at the gene which codes for an enzyme which breaks down the sugar LACTOSE. The, so called, Jacob-Monod hypothesis suggests that the gene which produces the enzyme required to break down lactose becomes switched on only when the substrate is present. When there is no lactose present the gene would be switched off. Firstly you will set up an experiment to investigate the effect that the enzyme has on its substrate. Production of Lactose – free Milk The naturally occurring sugar found in milk is lactose. This disaccharide sugar is broken down to glucose and galactose by the enzyme galactosidase. Many people (75% of the world’s adult population) cannot digest lactose because they cannot produce this enzyme. They are known as lactose intolerant. These people cannot consume milk or milk products. In this experiment you are going to attempt to produce lactose free milk by immobilising the galactosidase enzyme and setting up a continuous flow system. APPARATUS galactosidase enzyme calcium chloride sodium alginate milk 2 x 10 ml syringe 250 ml beaker 2 x 100 ml beaker circle of gauze stirring rod tea strainer 3 way tap 2 clinistix paper towel Biotechnology Protocols 122 Experiments METHOD Step 1 Immobilising the galactosidase 1 Use a 10 ml syringe to draw up 2 ml of galactosidase enzyme 2 Collect a piece of paper towel and then add 8 ml of sodium alginate to the same syringe (use the paper towel to catch any alginate drips) 3 Rock the syringe until the two liquids mix completely (this may take about 5 minutes) 4 Collect a 250 ml beaker and add 100 ml of calcium chloride 5 Add the alginate/enzyme mixture to the calcium chloride one drop at a time. Leave the beads in the calcium chloride for 3 minutes to allow them to set. 6 Filter the immobilised enzyme beads from the calcium chloride solution and then rinse the beads in distilled water Step 2 Setting up a Continuous Flow System 7 Collect a 3 way tap and check that you know how to use it. Practise with water. 8 Place a piece of nylon gauze at the base of a 10 ml syringe and attach a 3 way tap to the end of the syringe. 9 Add the beads to the syringe. 10 Collect 25 ml of milk in a beaker. 11 Test the milk for the presence of glucose using a CLINISTIX (Clinistix changes from pink to purple if glucose is present). Biotechnology Protocols 123 Experiments 12 Slowly add the milk to the syringe and alter the 3 way tap to ensure that the milk passes through at a slow but steady rate. Collect the treated milk in a small beaker. 13 Test the milk produced for the presence of glucose RESULT: Clinistix colour Glucose presence (+ or -) Milk before enzyme treatment Milk after enzyme treatment CONCLUSION: QUESTIONS: 1 Name the enzyme which breaks down lactose ______________________ 2 What induces the production of this enzyme? ______________________ ___________________________________________________________ 3 Give 2 advantages of using a continuous flow system, such as this, rather than a batch process to produce lactose free milk. ___________________________________________________________ ___________________________________________________________ 4 Cheese making produces the waste product whey which is rich in lactose. The whey is usually dumped at sea. Describe a way in which this waste product might be upgraded into a useful product. ___________________________________________________________ ___________________________________________________________ Biotechnology Protocols 124 Experiments Lactose Metabolism in E. Coli The bacterium E. coli produces galactosidase only when lactose is present in it’s growing medium. The Jacob-Monod hypothesis suggests that the genes controlling the production of this enzyme work in the following way. There are 3 genes involved: The structural gene – codes for the enzyme The operator gene – switches on the structural gene The regulator gene – codes for a repressor molecule DNA chain Regulator gene Operator gene Structural gene DNA chain Repressor molecule The presence of lactose induces the production of the enzyme. Lactose is therefore known as the inducer. LACTOSE ABSENT When lactose is absent the repressor molecule combines with the operator, the structural gene is switched OFF and no galacosidase is produced. DNA chain Regulator gene Operator gene Structural gene OFF DNA chain repressor LACTOSE PRESENT When lactose is present some of it binds with the repressor and the operator gene is able to switch ON the structural gene and galactosidase is produced. DNA chain Regulator gene repressor Operator gene Structural gene ON DNA chain lactose When all the lactose has been digested the repressor molecule will bind again with the operator and the gene will be switched back off. Biotechnology Protocols 125 Experiments QUESTIONS 1 Name the 3 genes involved with this hypothesis ____________________ ___________________________________________________________ 2 Which gene codes for the enzyme? ______________________________ ___________________________________________________________ 3 What does the repressor molecule do? ___________________________ ___________________________________________________________ 4 Why is lactose known as the inducer? ____________________________ ___________________________________________________________ 5 Why might this system of gene control be described as energy-efficient? ___________________________________________________________ ___________________________________________________________ 6 Complete the blanks in the following passage. In E. coli a _________________________ gene codes for the production of an enzyme called ____________________. When lactose is absent a ___________________ molecule binds with the ___________________ gene and the structural gene is switched ____________________ . Biotechnology Protocols 126 Experiments ANTIBODY/ANTIGEN REACTIONS Source: James Watt College Suppliers of antibodies (antiserum) and antigens include Alba Bioscience (formerly the Scottish Antibody Production Unit) http://www.show.scot.nhs.uk/sapu/index.html and Sigma-Aldrich http://www.sigmaaldrich.com/Area_of_Interest/Europe_Home/UK.html This simple experiment utilizes (purified) agar plates with wells cut into them as depicted here: Antibody Ab1 Distance between central well & test wells must be equal Ab4 Ab2 Ab3 Antibody (Antiserum) is dropped in the central well and antigen (serum) from a variety of animals is placed in surrounding wells. If the antibodies meet recognizable antigens, the proteins react and precipitate, giving a visible line. Whether a reaction occurs depends on the evolutionary relatedness of the animals, so that sheep antiserum reacts with goat serum but not with mouse serum. This point illustrates the specificity of the antibody/antigen reaction. Biotechnology Protocols 127 Experiments Set up TWO plates as shown here: note which antibody you put in the central well 1 4 2 3 Note which Antigens you put in wells 1 8: Antigen 1 = Antigen 2 = Antigen 3 = Antigen 4 = 5 6 8 7 Antigen 5 = Antigen 6 = Antigen 7 = Antigen 8 = * Remember to include a control Biotechnology Protocols 128 Experiments THE EFFECT OF DISINFECTANTS AND ANTISEPTICS ON MICROBIAL GROWTH Source: Based on material from biology4all website http://www.biology4all.com/resources_library/source/2.doc AIMS Determine the effect of chemical agents on bacterial growth. 1 Describe the physical effect on the growth of bacteria on solid nutrient agar. 2 Evaluate the effectiveness of commonly available antiseptics and disinfectants. 3 Discuss the inaccuracies that are inherent in the filter paper disk method. INTRODUCTION You will study how certain commonly available disinfectants and antiseptics affect the growth of two common bacterial species. Disinfectants are described as antimicrobial agents used on inanimate objects such as instruments (medical or dental), plastics or surfaces such as kitchen worktops, toilets, washbasins etc). Disinfectants should remove pathogenic organisms. Antiseptics are antimicrobial agents that are used on living tissue such as skin. Disinfectants and antiseptics do not always sterilise because these compounds usually do not kill all fungal and bacterial spores and vegetative bacteria and fungi. Organic compounds e.g. dirt, grease … etc. can interfere with their action reducing the efficiency of the antimicrobial agent. This practical enables you to study some commonly available antiseptics and disinfectants and assess their efficiency against two common bacteria that can be isolated from kitchens, hospitals and from humans. You can use household products such as Dettol or you can vary the experiment to compare antibacterial soaps, skincare products such as facewashes etc, toothpastes, mouthwashes……. Useful references on the internet Lister and antiseptics – http://web.ukonline.co.uk/b.gardner/Lister.html E. coli – http://vm.cfsan.fda.gov/~mow/chap14.html Staphylococcus – http://www.niaid.nih.gov/dmid/staph.htm Biotechnology Protocols 129 Experiments MATERIALS four disinfectants or antiseptics (can bring from home) overnight broth cultures of: Escherichia coli Staphylococcus epidermis albus sterile nutrient agar plates (x2) sterile filter paper disks forceps glass spreader METHOD 1 Using a sterile pipette remove 0.1 ml from the broth culture of Escherichia coli, and inoculate one labelled nutrient agar plate. Spread evenly to obtain confluent growth after incubation. 2 Do the same with the other agar plate, using the broth culture of Staphylococcus epidermis albus. 3 Label the four antiseptics or disinfectants that you will be testing A, B, C and D. 4 On the bottom of the agar plates, mark four sectors using a marker pen. Label the sectors A, B, C and D. These sectors correspond to the letters you put on the antiseptic or disinfectant containers. 5 Sterilise the tip of your forceps by passing it through the flame of the Bunsen burner two or three times. 6 Aseptically pick up a sterile filter paper disk with your sterile forceps and dip the disk into the disinfectant or antiseptic labelled A. Be sure that the excess disinfectant has drained off. 7 Place the disk in centre of Sector A of the Staphylococcus epidermis albus inoculated plate. 8 Using the same disinfectant, place another disk in Sector A of the E. coliinoculated plate. You are comparing the effectiveness of each disinfectant or antiseptic on both organisms. 9 Repeat steps 8 and 9, placing the other disinfectants in the other sectors. 10 Gently press the disks down with the tip of your flamed forceps to ensure contact with the nutrient agar. 11 When all four disinfectant-soaked disks have been placed in all four sectors of both plates, diametrically seal them with tabs of tape, invert the plates and incubate them at 30C for 48 hours. 12 Observe, measure, and compare the zone of no growth (inhibition), if any, around the disk for each disinfectant or antiseptic for both organisms. 13 Evaluate the effectiveness of each of the antiseptics/disinfectants at controlling the growth of the microbes used here. 14 Describe the usefulness of this experiment in terms of controls, reliability, & possible inaccuracies. Biotechnology Protocols 130 Experiments TRANSFORMING BACTERIA Source: Bio-Rad ‘pGLO Bacterial Transformation Quick Guide’ Available online at http://www.caam.rice.edu/~cox/lab1.pdf Transformation Procedure The aim of this experiment is to transform E.coli bacteria to express the green fluorescent protein. The pGLO plasmid is used as a vector in this experiment. 1 Label one closed micro test tube +pGLO and another -pGLO. Label both tubes with your group’s name. Place them in the foam tube rack. 2 Open the tubes and, using a sterile transfer pipette, transfer 250 l of transformation solution (CaCl2) into each tube. 3 Place the tubes on ice. Biotechnology Protocols 131 Experiments 4 Use a sterile loop to pick up a single colony of bacteria from your starter plate. Pick up the +pGLO tube and immerse the loop into the transformation solution at the bottom of the tube. Spin the loop between your index finger and thumb until the entire colony is dispersed in the transformation solution (with no floating chunks). Place the tube back in the tube rack in the ice. Using a new sterile loop, repeat for the –pGLO tube. 5 Examine the pGLO DNA solution with the UV lamp. Note your observations. Immerse a new sterile loop into the pGLO plasmid DNA stock tube. Withdraw a loopful. There should be a film of plasmid solution across the ring. This is similar to seeing a soapy film across a ring for blowing soap bubbles. Mix the loopful into the cell suspension of the +pGLO tube. Close the tube and return it to the rack on ice. Also close the –pGLO tube. Do not add plasmid DNA to the –pGLO tube. Why not? Biotechnology Protocols 132 Experiments 6 Incubate the tubes on ice for 10 minutes. Make sure to push the tubes all the way down in the rack so the bottom of the tubes stick out and make contact with the ice. 7 While the tubes are sitting on ice, label your four LB nutrient agar plates on the bottom (not the lid) as follows: Label one LB/amp plate: +pGLO Label the LB/amp/ara plate: +pGLO Label the other LB/amp plate: -pGLO Label the LB plate: -pGLO 8 Heat shock. Using the foam rack as a holder, transfer both the (+) pGLO and (-) pGLO tubes into the water bath, set at 42C, for exactly 50 seconds. Make sure to push the tubes all the way down in the rack so the bottom of the tubes stick out and make contact with the warm water. When the 50 seconds are done, place both tubes back on ice. For the best transformation results, the transfer from the ice (0C) to 42C and then back to the ice must be rapid. Incubate tubes on ice for 2 minutes. Biotechnology Protocols 133 Experiments 9 Remove the rack containing the tubes from the ice and place on the bench top. Open a tube and, using a new sterile pipette, add 250 l of LB nutrient broth to the tube and reclose it. Repeat with a new sterile pipette for the other tube. Incubate the tubes for 10 minutes at room temperature. 10 Tap the closed tubes with your finger to mix. Using a new sterile pipette for each tube, pipette 100 l of the transformation and control suspensions onto the appropriate nutrient agar plates. Biotechnology Protocols 134 Experiments Transformation plates Biotechnology Protocols Control plates 135 Experiments 11 Use a new sterile loop for each plate. Spread the suspensions evenly around the surface of the LB nutrient agar by quickly skating the flat surface of a new sterile loop back and forth across the plate surface. DO NOT PRESS TOO DEEP INTO THE AGAR. +pGLO LB/amp +pGLO -pGLO -pGLO LB/amp/ara LB/amp LB 12 Stack up your plates and tape them together. Put your group name on the bottom of the stack and place the stack of plates upside down in the 37C incubator until the next day. Biotechnology Protocols 136 Experiments MAKING PROTOPLASTS Source: Adapted from NCBE Practical Biotechnology http://www.ncbe.reading.ac.uk/NCBE/PROTOCOLS/PRACBIOTECH/PDF/pro to.pdf A similar protocol using lettuce leaves is available at http://www-saps.plantsci.cam.ac.uk/docs/protofusion.pdf Protoplasts are plant, fungal or bacterial cells which have had their cell walls removed; usually by enzymic digestion. The resultant ‘naked’ cells can be used in techniques such as the creation of transgenic plants. Materials 1 lettuce leaf (from a round lettuce) 0.1 cm3 21% sorbitol solution 20cm3 13% sorbitol solution 0.5cm3 Viscozyme (mixture of carbohydrase enzymes) Paper tissue 10ml syringe 2 x 1ml syringe Test tube Centrifuge tube Stirring rod Filter funnel Nylon gauze to plug filter funnel Microscope slide Cover slip Microscope with x40 objective Water bath set at 37oC Bench centrifuge Method Preparing the lettuce 1. Cut the lettuce leaf into pieces roughly 5mm x 5mm 2. Add 15-20 lettuce pieces to 9.5cm3 13% sorbitol solution in a test tube 3. Incubate the tube at 37oC for 5 minutes. Biotechnology Protocols 137 Experiments Digesting the cell walls 4. Gently stir 0.5cm3 of viscozyme into the sorbitol and lettuce preparation 5. Return the tube to the water bath for another 20 minutes. Gently stir the contents from time to time. Recovery of the protoplasts 6. Tightly pack the spout of the filter funnel with nylon gauze 7. Pour the digested lettuce suspension into the filter funnel 8. Wash any trapped protoplasts through the filter using 10cm3 of 13% sorbitol solution. Collect all the filtrates in a centrifuge tube. 9. Centrifuge the filtrate for approximately 5 minutes at 2000 rpm 10. Carefully pour off the supernatant, leaving a pellet of protoplasts at the bottom of the tube. 11. Resuspend the pellet in approximately 0.1cm3 of 21% sorbitol solution. Examining the protoplasts 12. Put some resuspended protoplasts on a slide and gently lower a coverslip into position. Protoplasts can easily be seen without staining using a x40 objective lense. Biotechnology Protocols 138 Experiments Biotechnology Protocols 139 Experiments PLAQUE ASSAY Source: James Watt College This technique is used to estimate the number of phage virus particles in a stock suspension. Plaques are clear areas in a lawn of bacteria caused by the lysis of a phage-infected bacterial cell. Materials 24 hour broth culture of E.coli B Suspension of phage virus (eg Philip Harris Bacteriophage T4B) 8 sterile nutrient broths (4.5 ml each) Gilson pipette Sterile blue tips 10 tubes sterile sloppy (0.7%) nutrient agar: 6mls each (NOTE: these must be kept at 45oC). 10 nutrient agar plates Water bath at 45oC Serial diluting the phage 1. Use sterile nutrient broth to prepare serial dilutions of the stock phage ranging from 10-1 to 10-8 (use 0.5ml phage and 4.5ml broth each time). Sterile nutrient broth (4.5ml per tube) Biotechnology Protocols 140 Experiments Combining the bacteria and virus NOTE: The following steps use sloppy agar. This will start to set if the temperature is allowed to drop below 450C. These tubes must be kept warm at all times. 2. Label each of the tubes of sloppy agar with a concentration ranging from 10-1 to 10-8. Also label a tube as ‘stock’ and another tube as ‘control’. 3. Add 0.4ml E.coli B to every tube. 4. To each tube add 0.2ml appropriate phage dilution: remember to add undiluted phage to the tube labelled ‘stock’ and to add nutrient broth to the tube labelled ‘control’. 5. Rapidly mix by rotating the tubes between the palms of your hands. Pouring the plates 6. Label the nutrient agar plates from 10-1 to 10-8, plus ‘stock’ and ‘control’ plates. 7. Pour the contents of the appropriate tube (see above) over the surface of a nutrient agar plate so it forms an even layer. 8. Allow to set and then invert & incubate for 28-24 hours at 30oC. Estimating the number of phage Some plates will have so many plaques they will overlap each other, whilst other plates (at higher dilutions) will have very few plaques. At least one plate will have plaques in countable numbers. 9. Find the plate or plates with plaques in countable numbers. 10.Count the number of plaques & multiply this figure by the dilution factor to obtain the concentration of the phage. Biotechnology Protocols 141 Experiments Biotechnology Protocols 142 Experiments






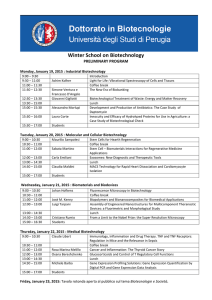

![Jefferson County, KY [Mission 5, Flight Experiment]](http://s2.studylib.net/store/data/005381659_1-6ff410f794c42188c46f63145dca8240-300x300.png)