Supplementary Information (doc 4087K)
advertisement
")
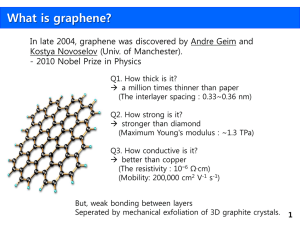
SUPPLEMENTARY INFORMATION Electrical control of nanoscale chemical modification in graphene Ik-Su Byun1†, Wondong Kim2†, Danil W. Boukhvalov3†, Inrok Hwang1,4, Jong Wan Son1, Gwangtaek Oh1, Jin Sik Choi1,5, Duhee Yoon6,7, Hyeonsik Cheong7, Jaeyoon Baik8, HyunJoon Shin8, Hung Wei Shiu9, Chia-Hao Chen9, Young-Woo Son3* and Bae Ho Park1* 1 Division of Quantum Phases & Devices, Department of Physics, Konkuk University, Seoul, 143701 Korea 2 Division of Industrial Metrology, Korea Research Institute of Standards and Science, Daejeon 305340, Korea 3 School of Computational Sciences, Korea Institute for Advanced Study, Seoul 130-722, Korea 4 Electronic Materials Research Center, Korea Institute of Science and Technology, Seoul 136-791, Korea 5 Creative Research Center for Graphene Electronics, Electronics and Telecommunications Research Institute, Daejeon, 305-700, Korea 6 Department of Physics, Sogang University, Seoul 121-742, Korea 7 Electrical Engineering, University of Cambridge, Cambridge CB3 0FA, United Kingdom 8 Pohang Accelerator Laboratory, Pohang, Kyungbuk 790-784, Korea 9 National Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu 30076, Taiwan † These authors contributed equally to this work. *email: hand@kias.re.kr and baehpark@konkuk.ac.kr 1 SPEM image of oxidized graphenes Figure S1. The 16-channel SPEM images obtained near the boundary between the graphene and SiO2 substrate. The red dotted line and white dotted areas denote the boundary and oxidized areas on graphene, respectively. Figure S1 displays a series of 16 SPEM images obtained near the boundary between graphene and SiO2 with a BE bandwidth of 1 eV and center BE ranging from 278 eV to 293 eV. The images were taken at a photon energy of 380 eV with an image size of 200 pixels × 200 pixels, pixel width of 300 nm, dwelling time of 60 ms, pass energy of 23.5 eV, and total acquisition time of approximately 30 minutes. These graphenes (light yellow area) and graphene oxides (white dotted rectangular area) on SiO2 can be identified in 9 or 10 channels, clearly. Because of the distinguished boundary (red dotted line) image, a center BE of 284 eV or 285 eV was used for further characterization. 2 Effect of pre-annealing treatment We can identify the three areas of 1LG oxidized with bias voltages of +6 V, +8 V, and +10 V using a friction force microscope (FFM) image, as shown in Figure S2(a). Figures S2(b) and S2(c) show the SPEM images near the boundary between graphene and SiO2, which are obtained at room temperature before and after pre-annealing treatment at 500 K, respectively. Although the areas of oxidized graphene are not clearly discriminated from that of SiO2 in Figure S2(b) because of the residual carbon contamination, they become distinguishable after pre-annealing treatment at 500 K and 10-9 Torr over 2 hours, as shown in Figure S2(c). Figure S2. FFM (a) and SPEM (b and c) images of pristine and oxidized graphene on SiO2 substrate before (a and b) and after (c) pre-annealing treatment at 500 K. By combining spatially resolved elemental and chemical mapping with XPS, we are able to measure a piece of local information with high lateral and energy resolution instead of average information, and distinguish bonds other than those from pristine graphene after pre-annealing treatment at 500 K and 700 K, as shown in Figure S3. Before the quantitative survey using by micro-XPS system, we should find the exact locations of graphene that those SPEM images 3 obtained from oxidized graphene areas after pre-annealing treatment at 500 K, as shown in Figure S3. After additional annealing at 700 K, graphene oxide appears to be partly reduced from boundaries between graphene and oxidized graphene samples, compared to the sample preannealed at 500 K. Furthermore, we can observe that these oxidized areas of 3LG are reduced less than those of 1LG and 2LG, implying that such reduction depends on the number of graphene layers. Figure S3. SPEM images of oxidized 1LG (a and b) or 2LG and 3LG (c and d) after preannealing treatment at 500 K (a and c) and 700 K (b and d). White dotted rectangular areas denote the oxidized graphene, and the red lines delineate the boundaries between 2LG and 3LG. 4 XPS data analysis for oxidized graphene We introduced a fitting procedure in which an XPS peak was decomposed with a Doniac and Sunjic35 line shape using a Lorentzian linewidth of 165 meV and an asymmetry parameter of 0.0636. The C 1s spectra of pristine 1LG consists of two peaks: the main peak at a binding energy (BE) of 284.5 eV (FWHM = 1.05 eV) is assigned to sp2 C-C; the other peak at higher BE with core-level shifts (CLS) of +0.7 eV is assigned to sp3 (C-C or C-H)33,34. The C 1s spectra of the three oxidized 1LG can be fitted with four peaks: one at 284.5 eV is assigned to sp2 C-C, and the others with CLS of +0.9, +2.1, and +3.1 eV are assigned typically for hydroxyl (C-OH)37, epoxy (C-O-C)38, and carbonyl (C=O)39, respectively. For quantitative approaches, the area percentage, normalized area ratio (area of each bond divided by the area of sp2 C-C)40, and oxygen coverage (sum of peak areas for the oxygen-containing bonds divided by that for all the carbon bonds) are also calculated from the fitting results for the corresponding XPS spectra, as shown in Figures S4 and S5. 5 Figure S4. The area percentage of sp2 C-C, C-OH, C-O-C, and C=O XPS peaks of oxidized areas on 1–3LG, which depend on number of graphene layers, applied bias, and annealing temperature. The pristine 2LG and 3LG also have two peaks assigned to sp2 C-C and sp3 (C-C or C-H)33,34. The peaks assigned to sp2 C-C bonds are located at 284.4 eV (2LG) and 284.3 eV (3LG) and have smaller FWHMs of 0.65 eV (2LG) and 0.60 eV (3LG) than that of pristine 1LG. The peak shift and narrowing may be due to the reduction of the charging effect from the silicon oxide substrate and the different electrical conductivity (1LG < 2LG < 3LG). The C 1s spectra of oxidized 2LG and 3LG were also fitted with four peaks: sp2 C-C peak located at 284.4 eV (2LG) and 284.3 eV (3LG); C-OH, C-O-C, and C=O bond peaks had CLSs of +0.9, +2.1 and +3.1 eV, respectively37-39. Similar to the oxidized 1LG, the normalized ratio of each bond enables the quantitative analysis for the oxidation of 2LG and 3LG, as shown in Figure S4. 6 Figure S5. The normalized area ratios of C-OH, C-O, and C=O XPS peaks with respect to the area of sp2 C-C XPS peak (a and b) and oxygen coverage of oxidized areas on graphene (c), which depend on the number of graphene layers, applied bias, and annealing temperature. Figures S5(a) and S5(b) show the normalized area ratio of each XPS peak depending on number of graphene layers, applied bias, and annealing temperature. Using these results, we can quantitatively analyze each chemical bond that has been induced on graphene by positive AFM lithography. We were able to confirm the high normalized ratio of C-O-C peaks that have the values of 4.8 (1LG), 14.7 (2LG), and 7.6 (3LG) with +10 V. Figure S5(c) shows the oxygen coverage of oxidized areas on graphene calculated using XPS spectra. After annealing at 700 K, the oxygen coverage of 1LG significantly decreases; however, the coverages of 2LG and 3LG are negligibly reduced, probably because of both the formation of irreversibly broken sp2 C-C bonds and the protection effect of the top-most graphene layer. 7 Figure S6. AFM (a), FFM (b, and conductive-AFM image (c) on recovered 1LG through UHV annealing at 700K. Figure S6 shows AFM, FFM, and conductive-AFM images (obtained at +2 V dc bias) of oxidized 1LG after UHV annealing at 700K. Partial change in AFM and conductive-AFM images of 1LG oxidized with +5 V and +7 V are observed after annealing while FFM images remain nearly unchanged. Figure S7. Optical microscope images (a), SPEM images at a BE of 284.5 eV (b), and XPS C 1s spectra (c) of 1LG oxidized with +10 V before and after irradiation of photons with energy of 380 eV for 1 hour. 8 Irradiation of photons with energy of 380 eV for 1 hour can change the chemical bonding characteristics of 1LG oxidized with +10 V, as shown in Figure S7. Spectral weight transfer from the C=O peak to C-OH peak is observed, and the sp2 C-C peak is unchanged. Thus, we can argue that the combination method of thermal40 and photon-irradiation treatments on oxidized graphene could allow for further reduction and recovery to graphene. Graphene hydrogenated using negative AFM lithography Figure S8. Schematic diagram of negative AFM lithography (a). Optical microscope (b), topographic AFM (c), and FFM images (d) of 1LG including areas hydrogenated with negative AFM lithography using -6 V, -8 V, and -10 V. 9 We have hydrogenated graphene in the nanoscale using negative AFM lithography where a negative dc bias is applied to the graphene, as shown in Figure S8(a). Figures S8(b), S8(c), and S8(D) respectively exhibit optical microscope, topographic AFM, and FFM images of three areas on 1LG, which were hydrogenated using -6 V, -8 V, and -10 V. The hydrogenated areas are indistinguishable in the optical microscope and topographic AFM images, whereas they are clearly observable in the FFM image because of the higher friction value of hydrogenated graphene compared to pristine graphene15. Figure S9. The Raman spectra of pristine and hydrogenated 1LG. The hydrogenation was performed by negative AFM lithography using -6 V, -8 V, and -10 V biases applied to graphene. Figure S9 shows the measured Raman spectra of hydrogenated and pristine 1LG. The Raman spectrum of pristine 1LG reveals sharp G and 2D peaks, whereas defect-related D, D, and D+D peaks appear on the hydrogenated 1LG irrespective of the external bias applied during negative AFM lithography. 10 Figure S10. XPS C 1s spectra of the three hydrogenated 1LG after UHV annealing for 2 hours at 500 K and 10-9 Torr. After annealing at 500 K, XPS C 1s spectra of 1LG hydrogenated with -6 V, -8 V, and -10 V become very similar to that of pristine 1LG, implying that hydrogen atoms can be completely dissociated by such thermal treatment, as shown in Figure S10. Transport properties of oxidized and hydrogenated graphene We fabricated the 1LG/oxidized-(or hydrogenated-)1LG/1LG junctions. Figure S11(a) shows the current-voltage curves of junctions with 1LG oxidized by +5 V, +7 V, and +10V, as measured with a voltage sweeping mode at room temperature (300K). The sweeping voltage was applied to 11 the conductive AFM tip on right side of 1LG while the left side of 1LG was grounded. More insulating behaviors are observed in the junction with 1LG oxidized by higher bias voltage. As shown in Figure S11(b), more insulating behaviors are observed in the junction with 1LG hydrogenated by a bias voltage with larger absolute value. These results demonstrate that higher bias voltage applied during AFM lithography induces chemically modified graphene with more insulating behaviors. Figure S11. Two terminal current-voltage characteristics of 1LG/oxidized-1LG/1LG (a) and 1LG/hydrogenated-1LG/1LG (b) junctions, which are plotted on semi-logarithmic scale. 12 First-principles modeling The modeling is carried out by the density functional theory realized in the pseudopotential code SIESTA41, as was done in our previous works42,43. All calculations are done in the local density approximation (LDA)41. This approximation provides significantly better results for interlayer distances and interlayer binding energies in such systems42,43. During the optimization, the electronic ground state was found self-consistently using norm-conserving pseudo-potentials for cores, a double-ζ plus polarization basis of localized orbitals for carbon and oxygen atoms, and a double-ζ basis for hydrogen. Optimization of the forces and total energies was performed with an accuracy of 0.04 eV/Å and 1 meV, respectively. All calculations were carried out for an energy mesh cut off of 360 Ry and an 8 × 8 × 2 k-point mesh in the Mokhorst-Park scheme45. For the modeling of the oxidation of graphene, a 3 × 3 graphene supercell (32 carbon atoms, also see Figures S12(a-d) was used. For the modeling of 3LG, three identical layers of graphene with a Bernal (AB) stacking order were employed. For the modeling of step-by-step oxidation, we have used the algorithm which that was previously employed for the description of nanographenes hydrogenation32. At each step, we optimized an atomic structure by selecting the structure with the lowest total energy among all reasonable structures for chemisorption of the next epoxy group on graphene. The energy required for oxidation is calculated with the standard formula: Eform = EG+On+1 – (EG+On + EO2/2), where EG+On is the total energy of graphene with chemisorbed n epoxy groups, and EO2 is the total energy of an oxygen molecule in a triplet (ground) state in an empty box. 13 For the modeling of vacancy formation on graphene, we removed one carbon atom and one of the nearest oxygen atoms from oxidized graphene at a distance of approximately 2 Å and added one more oxygen atom to this pair of atoms (to build a CO2 molecule). We optimized the atomic structure and calculated the vacancy formation energy by the following formula: Eform = EG – C + On-1 + CO2 – (EG + On + EO2/2), where EG-C+On-1+CO2 is the total energy of activated graphene and the CO2 molecule (Figures S12(b) and 3(d)). To check the graphene activation with the formation of a carbon monoxide (CO) molecule, we performed similar calculations and found that the energies required for this process are approximately 3 eV higher than for the case of vacancy and carbon dioxide formation. The energy required for transformation of epoxy groups to hydroxyl groups in the presence of water were calculated by the following formula: Eform = EG + On-1 + (OH)2 – EG +On +H2O, where EG + On-1 + (OH)2 is the total energy of graphene with n-1 epoxy groups and two hydroxyl groups (Figure S12(f)), and EG +On +H2O is the total energy of graphene with n epoxy groups and a water molecule over one of these groups (Figure S12(e)). The results of these step-by-step calculations are shown in Figure S12. We can divide the process of the oxidation of graphene (Figures S12(a) and S12(b)) into several stages. These stages can be correlated with the different applied voltages. We can see that the number of layers affects the oxidation level at each oxidation stage. The presence of second and third layers provides changes in the local distortions30 of the graphene sheet induced by the chemisorption of oxygen. These 14 distortions play a crucial role in the energetics of C-O-C to C-OH transformation in the presence of water (Figures S12(e) and S12(f)) because the distortion of the graphene sheet caused by the hydroxyl group is stronger than that caused by the epoxy group24. The second step of our modeling is the activation of a vacancy on graphene with the formation of a carbon dioxide molecule (Figures S12(c) and S12(d)). For 1LG, the energy required for activation is always higher than that for oxidation, in contrast to 2LG and 3LG. Another important feature is that passivation of the edges of vacancies by oxygen (Figures 3(g) and 3(j)) is always energetically favorable with formation energies (-2 – -4 eV). The calculation of the hydrogenation energy has been performed similarly to the calculation of the oxidation energy. The desorption energy is defined as the energy difference between hydrogenated graphene (Figures S13(a) and S13(c)) and the corresponding structure with a single hydrogen atom removed (Figures S13(b) and S13(d)). 15 Figure S12. Atomic structures of oxidized graphene: optimized atomic structures of 1LG with chemisorbed single oxygen atom (a) and maximal coverage of epoxy groups (c); activation of graphene with formation of carbon dioxide molecule at these concentrations (b and d); initial (e) and final (f) steps of the transformation of epoxy groups to hydroxyl groups in the presence of water. Energetics of oxidation (dashed blue line), activation of vacancies with formation of CO2 molecules (solid red line), and hydroxyl groups formation (dashed green line) as function of the oxidation of graphene surface for 1LG (g), 2LG (h), and 3LG (i). 16 Figure S13. Optimized atomic structures in the case of adsorption of the pairs of hydrogen atoms (a and c) and desorption of the single hydrogen atom from these structures (b and d) for the hydrogenation of the carbon surface with 6.25 at% (a and b) and 43.25 at% (c and d). 17 References 35. Doniach, S., Sunjic, M. Many-electron singularity in x-ray photoemission and x-ray line spectra from metals. J. Phys. C 3, 285 (1970). 36. Prince, K. C., Ulrych, I., Peloi, M., Ressel, B., Cha´b, V., Crotti, C., Comicioli, C. Core-level photoemission from graphite. Phys. Rev. B 62, 6866–6868 (2000). 37. Wang, P., Zhai, Y., Wang, D., Dong, S. Synthesis of reduced graphene oxide-anatase TiO2 nanocomposite and its improved photo-induced charge transfer properties. Nanoscale 3, 1640– 1645(2011). 38. Compton, O. C., Jain, B., Dikin, D. A., Abouimrane, A., Amine, K, Nguyen, S. T. Chemically active reduced graphene oxide with tunable C/O ratios. ACS Nano 5, 4380–4391 (2011). 39. Chen, J., Liu, S., Feng, W., Zhang, G. Q., Yang, F. L. Fabrication phosphomolybdic acid– reduced graphene oxide nanocomposite by UV photo-reduction and its electrochemical properties. Phys. Chem. Chem. Phys. 15, 5664–5669 (2013). 40. Yang, D., Velamakanni, A., Bozoklu, G., Park, S., Piner, R. D., Stankovich, S., Jung, I., Field, D. A., Ventrice, J. C. A., Ruoff, R. S. Chemical analysis of graphene oxide films after heat and chemical treatments by x-ray photoelectron and micro-raman spectroscopy. Carbon 47, 145–152 (2009). 41. Soler, J. M., Artacho, E., Gale, J. D., Garsia, A., Junquera, J., Orejon, P., Sanchez-Portal, D. The SIESTA method for ab initio order-n materials simulation. J. Phys.: Condens. Matter 14, 2745 (2002). 18 42. Bukhvalov, D. W., Katsnelson, M. I. A New route towards uniformly functionalized single layer graphene. J. Phys. D: Appl. Phys. 43, 1753302 (2010). 43. Bukhvalov, D. W., Katsnelson, M. I. Tuning the gap in bilayer graphene using chemical functionalization: density functional calculations. Phys. Rev. B 78, 085413 (2008). 44. Perdew, J. P., Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048 (1981). 45. Monkhorst, H. J., Park, J. D. Special points for brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976). 19