A Test of Improved Force Field Parameters for Urea
advertisement

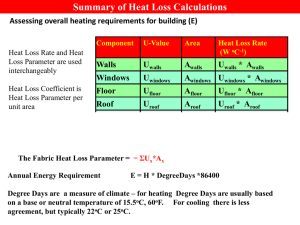
A Test of Improved Force Field Parameters for Urea: Molecular-Dynamics Simulations of Urea Crystals Gül Altınbaş Özpınar,a,b,c,# Frank R. Beierlein,b,c,# Wolfgang Peukert,c,d Dirk Zahn,b,c and Timothy Clark*,b,c a Department of Chemistry, Natural Sciences, Architecture and Engineering Faculty, Bursa Technical University, Osmangazi, 16000 Bursa, Turkey. b Computer-Chemie-Centrum and Interdisciplinary Center for Molecular Materials, Friedrich-Alexander Universität Erlangen-Nürnberg, Nägelsbachstr. 25, 91052 Erlangen, Germany. c Excellence Cluster Engineering of Advanced Materials, Friedrich-Alexander-Universität Erlangen-Nürnberg, Nägelsbachstr. 49b 91052 Erlangen, Germany. d Lehrstuhl für Feststoff- und Grenzflächenverfahrenstechnik, Friedrich-AlexanderUniversität Erlangen-Nürnberg, Cauerstraße 4, 91058 Erlangen, Germany. # These authors contributed equally to this work Table of Contents 1. Force field parameters for urea used in this study........................................... 2. RMSD, RMSF, energies and temperature during the initial MD simulations of cubic, rectangular prismatic, and sheet urea crystals at 300 K in vacuo..... 3. S3 S4 Average total energies of urea monomer and crystals in cubic, rectangular prismatic, and sheet shapes obtained from the initial MD simulations in vacuo at 300 K................................................................................................. 4. Total and potential energies obtained from the “instantaneous heating” MD simulation at 382 K.......................................................................................... 5. S9 RMSD and orientational order parameter obtained from the “instantaneous heating” MD simulations at 300 and 350 K..................................................... 6. S8 S10 RMSF and orientational order parameter mapped on the initial coordinates of all C atoms in the system, as obtained from the “instantaneous heating” MD simulation at 382 K.................................................................................. S12 S1 7. Temperature and energy plots obtained from the “gradual heating” simulations....................................................................................................... 8. Snapshots from the MD simulation using the “gradual heating” protocol and a heating rate of 20 K ns1........................................................................ 9. S14 S21 RMSD and orientational order parameter from the MD simulation using the “gradual heating” protocol and a heating rate of 5 K ns-1............................... S22 S2 1. Force field parameters for urea used in this study Table S1. Improved general AMBER force field parameters from the previous study. [14] Bond Parameters Kra rb 656 1.250 424 1.383 434 1.010 Angle Parametres angle K θc θd n –c –o 80 120.9 c –n –hn 30 120.0 hn –n –hn 35 120.0 n –c –n 70 118.6 Dihedral Parameters torsion no. of pathse Vn/2f γg nh hn –n –c –o 1 2.5 180 -2 hn –n –c –o 1 2.0 0 1 Improper Dihedral Parameters torsion Vn/2f γg nh n –n –c –o 10.5 180 2 c –hn –n –hn 1.1 180 2 Nonbonding Parameters atom type R*i εj hn 0.6000 0.0157 o 1.6612 0.2100 c 1.9080 0.0860 n 1.8240 0.1700 a 1 2 b Force constant (kcal mol Å ). Bond distance (Å). c Force constant (kcal mol1 radian2). d Angle (deg.). e Number of bond paths that the total Vn/2 is divided into. f Magnitude of torsion (kcal mol1). g Phase offset (°). h The periodicity of the torsion. A negative value is not used in the computation but signifies more than one component around a given bond. i (Å). j (kcal mol1). bond c –o c -n hn-n Table S2. RESP charges used. [14] Atoms RESP 0.884 C -0.660 O -0.888 N 0.388 H S3 2. RMSD, RMSF, energies and temperature during the initial MD simulations of cubic, rectangular, and flat urea crystals at 300 K in vacuo 2.5 330 Cubic Cubic Rectengular Rectengular Flat Flat 320 2 Temperature (K) 310 RMSD 1.5 1 300 290 280 270 0.5 260 250 0 0 1000 2000 3000 4000 5000 6000 7000 8000 -150000 9000 0 10000 1000 2000 3000 4000 5000 6000 7000 8000 -150000 Cubic Flat -170000 Potential Energy (kcal/mol) Total Energy (kcal/mol) Rectengular -160000 Flat -170000 10000 Cubic Rectengular -160000 9000 -180000 -190000 -200000 -210000 -220000 -180000 -190000 -200000 -210000 -220000 -230000 -230000 -240000 -240000 -250000 -250000 0 1000 2000 3000 4000 5000 6000 7000 8000 9000 10000 0 1000 2000 3000 4000 5000 6000 7000 8000 9000 10000 Figure S1. RMSD (top left), total energy (bottom left), potential energy (bottom right) and temperature (top right) during the initial simulations of cubic, rectangular prismatic, and sheet urea crystals in vacuo at 300 K. S4 Figure S2. Plots of the root-mean-square fluctuation [Å] (RMSF, top left, calculated for the heavy atoms of each urea molecule), mean van der Waals (bottom left), electrostatic (top right) and total interaction energies (bottom right) [kcal mol-1] of each urea molecule with the rest of the system, as obtained from the initial simulations of the cubic urea crystal at 300 K in vacuo. Values were mapped on the geometric centers of the average coordinates of the heavy atoms in each urea molecule. The [001] surface corresponds to the “upper” and “lower” crystal boundaries (plane with constant c-values). S5 Figure S3. Plots of the root-mean-square fluctuation [Å] (RMSF, top, calculated for the heavy atoms of each urea molecule), mean van der Waals, electrostatic and total interaction energies (bottom) [kcal mol-1] of each urea molecule with the rest of the system, as obtained from the initial simulations of the rectangular prismatic urea crystal at 300 K in vacuo. Values were mapped on the geometric centers of the average coordinates of the heavy atoms in each urea molecule. S6 FigureS4 Plots of the root-mean-square fluctuation [Å] (RMSF, top left, calculated for the heavy atoms of each urea molecule), mean electrostatic (top right), van der Waals (bottom left) and total interaction energies (bottom right) [kcal mol-1] of each urea molecule with the rest of the system, as obtained from the initial simulations of the sheet urea crystal at 300 K in vacuo. The b coordinate axis was scaled by a factor of 10 to improve clarity. Values were mapped on the geometric centers of the average coordinates of the heavy atoms in each urea molecule. S7 3. Average total energies of urea monomer and crystals in cubic, rectangular prismatic, and sheet shapes obtained from the initial MD simulations in vacuo at 300 K Table S3. Average total energies [kcal mol-1] of urea monomer and crystals in cubic, rectangular prismatic, and sheet shapes obtained from the MD simulations in vacuo at 300K Number of urea molecules Sheet Rectangular prismatic Cubic Monomer Average Etotal 1220 1125 -201353.9636 -196506.9957 1152 1 -203462.7163 -153.7973 S8 4. Total and potential energies obtained from the “instantaneous heating” MD simulation at 382 K Figure S5. Plot of total (green) and potential (black) energies obtained from the “instantaneous heating” MD simulation at 382 K. S9 5. RMSD and orientational order parameter obtained from the “instantaneous heating” MD simulations at 300 and 350 K. Figure S6. RMSD (left) and orientational order parameter (right) calculated for all C atoms obtained from the “instantaneous heating” protocol MD simulation at 300 K. The first 500 ps (restrained MD, heat-up) were omitted for the analyses. S10 Figure S7. RMSD (left) and orientational order parameter (right) calculated for all C atoms obtained from the “instantaneous heating” protocol MD simulation at 350 K. The first 500 ps (restrained MD, heat-up) were omitted for the analyses. S11 6. RMSF and orientational order parameter mapped on the initial coordinates of all C atoms in the system, as obtained from the “instantaneous heating” MD simulation at 382 K. Figure S8. RMSF (left) and orientational order parameter (right) calculated for all C atoms mapped on the initial coordinates of all C atoms in the system, as obtained from the “instantaneous heating” protocol MD simulation at 382 K. The viewing angle corresponds to “side view” in Figure 4 in the main text. The first 500 ps (restrained MD, heat-up) were omitted for the analyses. S12 Figure S9. RMSF (left) and orientational order parameter (right) calculated for all C atoms mapped on the initial coordinates of all C atoms in the system, as obtained from the “instantaneous heating” protocol MD simulation at 382 K. The viewing angle corresponds to “side view rotated by 45°” in Figure 4 in the main text. The first 500 ps (restrained MD, heatup) were omitted for the analyses. S13 7. Temperature and energy plots from the “gradual heating” simulations gradual heating (30K/ns) Figure S10. Temperature vs. time in the “gradual heating” simulation with a heating rate of 30 K/ns. S14 gradual heating (20K/ns) Figure S11. Temperature vs. time in the “gradual heating” simulation with a heating rate of 20 K/ns. S15 gradual heating (10K/ns) Figure S12. Temperature vs. time in the “gradual heating” simulation with a heating rate of 10 K/ns. S16 gradual heating (5K/ns) Figure S13. Temperature vs. time in the “gradual heating” simulation with a heating rate of 5 K/ns. S17 melting interval 7.4 ns 405.5 K 7.85 ns 419 K gradual heating (30 K/ns) Figure S14. Total (green) and potential energies (black) in the “gradual heating” simulation with a heating rate of 30 K/ns. S18 melting interval 9.5 ns 403 K 10.0 ns 413 K gradual heating (20 K/ns) Figure S15. Total (green) and potential energies (black) in the “gradual heating” simulation with a heating rate of 20 K/ns. S19 melting interval 15.9 ns 403 K 16.45 ns 408.5 K gradual heating (10 K/ns) Figure S16. Total and potential energies (black) in the “gradual heating” simulation with a heating rate of 10 K/ns. S20 8. Snapshots from the MD simulation using the “gradual heating” protocol and a heating rate of 20 K ns1 Figure S17. Snapshots from the MD simulation using the “gradual heating” protocol and a heating rate of 20 K ns1. S21 9. RMSD and orientational order parameter from the MD simulation using the “gradual heating” protocol and a heating rate of 5 K ns-1 Figure S18. RMSD (left) and orientational order parameter (right) calculated for all C atoms obtained from the “gradual heating” protocol MD simulation and a heating rate of 5 K ns-1. The first 500 ps (restrained MD, heat-up) were omitted for the analyses. S22