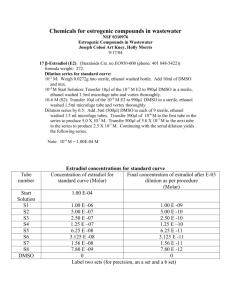
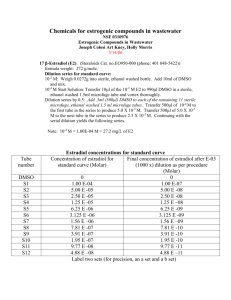
Cell Culture technique

BASIC CELL CULTURE TECHNIQUES
STERILE TECHNIQUES
Aseptic or sterile technique is the execution of tissue culture procedures without introducing contaminating microorganisms from the environment. In doing tissue culture work, 70% of the problems are due to a lack of good sterile technique. Microorganisms causing the contamination problems exist everywhere, on the surface of all objects and in the air. A conscious effort must be made to keep them out of a sterile environment. Because many and sometimes awkward manipulations are required for various techniques, tissue culture media used are often supplemented with antibiotics. Antibiotics do not eliminate problems of gross contamination which result from poor sterile technique or antibioticresistant mutants. Autoclaving renders pipettes, glassware, and solutions sterile.
Nutrient medium cannot be autoclaved. The compounds in nutrient medium are destroyed by the heat of autoclaving. Medium must therefore be sterilized by passing it through a sterile filter small enough in pore size to hold back bacteria and mycoplasmas (Millipore Sterivex - GS 0.22u disposable filter units).
Here are some rules of thumb to follow to keep your medium, cultures, and glassware from becoming contaminated:
1. Wipe your work area and hands with 70% ethanol before starting. Spray any container you plan to put into the sterile workspace with 70% ethanol.
2. Never uncover a sterile flask, bottle, petri dish, etc., until the instant you are ready to use it. Return the cover as soon as you are finished. Never leave it open to the environment.
3. Sterile pipettes should never be taken from the wrapper until they are to be used. Keep your pipettes at your work area. Sterile pipettes do not have to be flamed. Pipetting your cells with a hot pipette will kill them.
4. When removing the cap from a bottle, flask, etc., do not place the cap with the open end upright on the lab bench. Do not hold the opening straight up into the air. If possible, tilt the container so that any falling microorganisms fall onto the lip.
5. Be careful not to talk, sing, or whistle when you are performing these sterile procedures, and never tissue culture when you are sick with a cold or cough.
6. Do not draw from different bottles with the same pipette. Because such a pipette has been exposed; the chance for contamination is too great; use sterile pipette for each bottle -- especially when pipetting medium. Avoid forming bubbles as these are more likely to gather microbes if present.
7. Techniques should be performed as rapidly as possible to minimize contamination.
8. When you finish in the culture hood, spray the deck with 70% ethanol and wipe down thoroughly.
Turn on the vacuum and vacuum 70% ethanol through the tubing and into the waste flask. Never leave a dirty hood, throw all cell culture materials into the autoclave bag, and generally make sure there is a sterile workspace left behind.
You may find yourself involved in a procedure which these sterile technique "rules-of-thumb" do not cover. Therefore, you must constantly be aware that microorganisms are everywhere and take proper steps to keep them out of your cultures. When first developing your aseptic technique you must always be thinking of sterility. Eventually it will become second nature to you. Mastering good aseptic technique will save you considerable frustration in the labs to follow. Furthermore, the same principles for good aseptic technique also minimize biohazard risk to the investigator when infectious organisms or dangerous chemicals are used.
THAWING CELLS
1. Cells generally do not like cold media. Before you begin, put your media into the 37 o water bath to warm for about 30 min prior to the start of this process. Once it is warm, spray the bottle with 70% ethanol and place it on the deck of the hood.
Cells are generally stored in medium that contains DMSO. DMSO is added so that ice crystals do not form in the cells as they are freezing. Unfortunately, you don’t want to culture your cells in medium containing DMSO, so part of this process dilutes out the DMSO in the freezing medium.
2.
Thaw cells as quickly as possible. Generally you can put them in the 37 o C water bath next to the hood.
3.
Once the cells are thawed completely spray the cell tube with 70% ethanol and put it into the hood.
Transfer the cells from the freezer tube to a sterile 15 ml screw top tube.
4.
Add 6 ml of growth medium, shake gently, and spin for 2-3 min at level 5. The cells will form a small flocculent pellet in the tip of the tube.
5.
Using the vacuum flask with an unplugged pipet tip in the end of the tubing, vacuum most of the medium off of your cells (this is slightly contaminated with the DMSO the cells were frozen in).
6.
Resuspend the cells in about 1 ml of new medium. This will be the suspension that you will “seed” your growth plates with.
SEEDING CELLS INTO CULTURE PLATES
Generally cells are seeded into a container that is specific for the needs of that particular cell type and/or the type of experiment you are going to do. Adherent cells are generally maintained in sterile 6-well plates. That is what we will use for seeding our cells.
1.
Without touching the packaging to the surface of the hood deck, unwrap a sterile 6-well plate, put it onto the deck, and throw away the packaging without ever letting it touch the deck.
2.
In the corners on the lid of your plate, write your initials, what type of cell you are growing, the date, and the “passage number” of the cells.
The “passage number” indicates the number of times the cells have been lifted from the plate, and recultured into a new set of wells. This is also called “splitting”. Even though most cells used in cell cuture are hybridomas and are said to have the ability to divide infinitely, most take on odd morphologies and unwanted characteristics over time. You should always know how many times the cells you are using have been lifted and re-seeded into new plates.
3.
Each group should fill three wells of a plate with 5 ml of medium containing serum, and antibiotics.
4.
Using the cells from #6 above, seed the wells with 1 drop, 2 drops, and 3 drops respectively in each of your three wells.
5.
Examine the wells under the inverted microscope. You should see small shiny balls of cells floating across the medium. The well that you placed one drop of cells into should have fewer cell balls than the well that you put three drops of cells into. Once you are sure there are cells in every well, place the plate in a humidified incubator. Plan on checking the cells to see if they are adhering and starting to grow on the bottom of your wells within 24 hours.
"EYEBALLING" THE CULTURES
Before doing anything with a culture, its general "health" and appearance should be evaluated. This can be done quickly and quantitatively by making the following observations:
1. Check the pH of the culture medium by looking at the color of the indicator, phenol red. As a culture becomes more acid the indicator shifts from red to yellow-red to yellow. As the culture becomes more alkaline the color shifts from red to fuchsia (red with a purple tinge). As a generalization, cells can tolerate slight acidity better than they can tolerate shifts in pH above pH 7.6.
2. Cell attachment. Are most of the cells well attached and spread out? Are the floating cells dividing cells or dying cells which may have an irregular appearance?
3. Percent confluency. The growth of a culture can be estimated by following it toward the development of a full cell sheet (confluent culture). By comparing the amount of space covered by cells with the unoccupied spaces you can estimate percent confluency.
4. Cell shape is an important guide. Round cells in an uncrowded culture is not a good sign unless these happen to be dividing cells. Look for doublets or dividing cells. Get to know the effect of crowding on cell shape.
5. Look for giant cells. The number of giant cells will increase as a culture ages or declines in "wellbeing." The frequency of giant cells should be relatively low and constant under uniform culture conditions.
6. One of the most valuable guides in assessing the success of a "culture split" is the rate at which the cells in the newly established cultures attach and spread out. Attachment within an hour or two suggests that the cells have not been traumatized and that the in vitro environment is not grossly abnormal.
Longer attachment times are suggestive of problems. Nevertheless, good cultures may result even if attachment does not occur for four hours.
7. Keep in mind that some cells will show oriented growth patterns under some circumstances while many transformed cells, because of a lack of contact inhibition may "pile up" especially when the culture becomes crowded. Get to recognize the range of cells shapes and growth patterns exhibited by each cell line.
SUBCULTURING PROTOCOL FOR ANCHORAGE-DEPENDENT CELLS :
(Use Sterile Technique Throughout!!!)
1. Vacuum supernatant fluid from culture into a waste collection jar, applying the principles of sterile technique.
2. Now add 3 ml of cold trypsin and place in the incubator for about 5-10 min. At the end of that period, examine cells under low magnification for 3-7 minutes with the inverted scope. When it appears that most of the cells have rounded up, but have not yet completely detached, gently slosh the liquid back and forth. Then immediately return to the hood and pipette the cells up and down several times to help break up the clumps of cells.
5. Remove the cells in trypsin and place in sterile 15 ml tube. Spin for 2-3 min at level 5. Vacuum off as much trypsin as possible. Add 5 ml media and resuspend cells. Spin again, remove as much media as possible. Add about 500 µl of medium, resuspend cells.
6. Add 5 ml fresh media to the wells of a 6-well plate (or other plate as needed). Add variable drops of cells to the 6 wells (eg. 1 drop in well 1, 2 drops in well 2, 3 drops in well 3, and so on). Add the drops to the outer area of the well as cells tend to migrate toward the center. Before incubating, look at the wells under the inverted microscope. Cells will appear as bright round balls floating on the surface of the medium. If you don’t see any of these, there is a problem.
7. Incubate at 34 o C in a humidified incubator with 5% CO2. Monitor periodically, beginning 30-45 minutes after inoculation. Rapid attachment (within 1 hour or so) is indicative, but not proof, that all went well.
FREEZING BACK CELLS
It is general protocol to “freeze back” a portion of your first passage of cells. This is so you always have a set of low passage number cells to begin experiments with. The medium for freezing back is slightly different than that used to culture cells. In addition to the normal ingredients, the medium also generally contains 10% DMSO.
1.
Prepare 1ml of medium containing 10% DMSO. Find a sterile cryotube and write the cell type, passage number, and date on the cryotube.
2.
Pass cells as described above, but when resuspending as in step 5, resuspend in the medium containing DMSO.
3.
Place the tube of cells into the -80freezer in the special freezing container. Be sure to place them where they won’t be bumped, as this causes ice crystals to form more easily. After they have been in the -80 freezer overnight, place the tube on a cane in the liquid-nitrogen storage tank.