here - Purdue University Cytometry Laboratories
advertisement

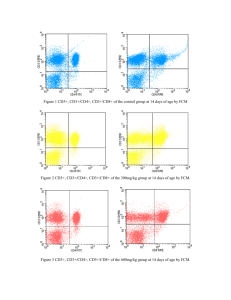
MULTIPARAMETRIC INVESTIGATION OF THE IMMUNE SYSTEM DURING MAJOR SURGERY BY LASER SCANNING CYTOMETRY A. GERSTNER1, W. LAFFERS1, F. BOOTZ1, A. TÁRNOK2 1 Department of Otorhinolaryngology; 2Department of Pediatric Cardiology, Cardiac Centre; University of Leipzig, Leipzig, Germany; Address requests to: Dr. Andreas Gerstner, gersta@medizin.uni-leipzig.de SUMMARY Investigations of the immune system in the fast majority are based on flow cytometry (FCM). The drawback of most of the FCM assays is that cells cannot be analysed a second time and that the morphology of the cells cannot be studied. This however is possible by using laser scanning cytometry (LSC). We developed an assay for immunophenotyping of peripheral blood leukocytes (PBLs) by LSC. The percentage of cells in different subsets determined by LSC correlates very well with those determined by FCM but only 25% of material is needed. We expect LSC to play a major role in further investigations on the role of specific cell subsets in systemic immune responses according to its unique facility to analyse the same cells repeatedly by different assay. INTRODUCTION Many clinical situations demand repeated analyses of blood parameters but permit only minimal amounts of peripheral blood to be taken. This includes neonates with low birth weight, during extensive operations on young children, or patients with restricted bone marrow function. In order to determine the function of the cellular immune system, LSC is the ideal tool to determine the distribution of different subsets of the PBLs in these cases. A major advantage of LSC as compared to FCM is the fact that the cells are fixed on the slide. The instrument is build around a standard research fluorescence microscope. The cells on the glass slide are scanned by two lasers (Ar + HeNe) and the emitted fluorescence is measured on a pixel-topixel basis; the analysis itself is performed on the resulting bit-map similar to image analysis (1, 2). This process leaves the cells unchanged. The immobilisation of the cells on the slide allows the direct morphological control of the measured cells by relocating them on the slide. Additionally, it reduces the amount of both, peripheral blood and reagents needed. Furthermore, slides can be stored and re-analyzed later. Existing assays (3) for immunophenotyping with the LSC are based on triggering the analysis on the forward light scatter (FSC) of the cells. We experienced substantial difficulties in achieving data of sufficient quality and reliability by this strategy: on the one hand any particle or air bubble on the slide could produce a FSC-signal indistinguishable of a cell. On the other hand some cells might have virtually invisible scattering features and consequently could be lost from analysis. We were therefore looking for an alternative assay for immunophenotyping of PBLs by LSC and developed a strategy where analysis is triggered on nuclear fluorescence. MATERIALS AND METHODS Blood was taken from patients undergoing resection of oro- and hypopharyngeal cancer in advanced metastatic stages with reconstruction by free microvascular reanastomosed flaps (forearm flap, latissimus dorsi flap, lateral arm flap, scapular flap) 60-90min after onset of anaesthesia. For analysis, full peripheral EDTA blood is aliquoted to 10µl and incubated with different arrays of CD-antibodies (2,5µl each). After lysis of the erythrocytes cells are fixed with paraformaldehyd and dispersed on a conventional microscopic glass slide to pre-defined areas; this makes the analysis easier and more standardised. The slides are allowed to air-dry and are incubated with acetone. Cells are finally stained with the DNA-stain 7-aminoactinomycin-D (7-AAD; 5µg/ml) and are coverslipped. For analysis, pre-set scan areas on the microscope slide are used. The fluorescence of the nuclei is taken as the contouring triggering signal. This ensures that all nucleated cells are included in the analysis. Furthermore, staining with 7-AAD allows the differentiation between PMNs and mononuclear cells (not shown). The FSC is used to define the eosinophils (and basophils): eosinophils are characterised by strongly light-scattering granula and high autofluorescence and therefore would otherwise interfere with the specific immunofluorescence. Further discrimination of monocytes and lymphocytes is performed on the basis of their CD45-fluorescence. In order to compare the analysis by LSC with FCM data the same staining is run but starting with 40µl of peripheral full EDTA blood instead of 10µl. The sample is split after fixation with paraformaldehyd: at that point three quarters of the material are taken for FCM analysis in a Becton-Dickinson FACSCalibur. Following the analysis in the LSC the coverslips are removed, the slides are placed in PBS, and a conventional H&E staining is performed. Slides are coverslipped again, placed in the microscope stage, and single cells of interest are localised to confirm and document the morphology of the cells (see figures 1&2). Slides can be stored afterwards without any special requirements. RESULTS The protocol described above yields reliable and consistent data. Processing of the material must start within 60min after taking the specimen, the total time of the protocol is 4h 15min. The specimens can be left overnight after fixation in paraformaldehyd. The measurement in the LSC should be carried out within 48h because of significant loss of fluorescence with storage. With this method we can determine the percentage of eosinophils (and basophils), neutrophils, monocytes, and lymphocytes, and can divide the lymphocytes into CD3+CD4+-, CD3+CD4–-, CD3–CD19+-, and CD3–CD19–-cells in a single scan using 10µl whole blood (see fig. 2). The percentage of the subsets correlates very well with those determined by FCM on the same material (see tab. 1). The ratio of the integrated fluorescence values of distinct cell subsets (CD3+CD4+ vs. CD3+CD4–, e.g.) on the LSC is optimised to the range yielded on FCM. This shows that the resolution of the fluorescence intensities by LSC is equivalent to that by FCM. Relocating single cells of interest after H&E staining worked very reliably. CONCLUSIONS We expect the LSC to prove its capacity for multi-parameter analysis of PBLs in minimal amounts of peripheral blood. Its unique feature of relocating cells allows to perform additional investigations on the same cells. This will be especially helpful in investigating the role of specific cell subsets of the immune system in systemic responses post-operatively e.g. after cardiopulmonary-bypass: we are working on an assay that allows consecutive immunophenotyping, intra-cellular chemokine detection, and TUNEL-assay. REFERENCES 1 2 3 KAMENTSKY et al.: Slide-based laser scanning cytometer. Acta Cytol 41:123-143, 1997 REEVE et al.: New technology in the analytical cell sciences: the laser scanning cytometer. Eur J Surg Oncology 23:445-450, 1997 CLATCH et al.: Simplified immunophenotypic analysis by laser scanning cytometry. Cytometry (Clin. Comm.) 34:3-16, 1998 ACKNOWLEDGEMENT Reprint permission kindly provided by Monduzzi Editore, Bologna, Italy, 2000. Eosinophils (100x objective) 1 2 Fig. 1: PBLs stained with CD45-APC, CD3-FITC, CD4-PE, CD14-PE, CD19-PE, and 7-AAD; dot plot in the inset shows area versus FSC Max Pixel for all measured events. Single cells are gated; region 1 is gated on eosinophils, region 2 is set on the remaining single cells. Cells in region 1 were relocalised after H&E staining and correspond to eosinophils (photographs taken with 100x objective). Monocytes (40x objective) 3 Neutrophils 4 Fig. 2: Same material as in fig. 1 but additionally gated on region 2. No compensation is set. Dot plot of PE (CD4-CD14-CD19) vs. APC (CD45). Upper line: cells within the region 3 correspond to monocytes (see photographs). Lower line: cells within the region 4 correspond to neutrophils (see photographs). Patient 1 Patient 2 LSC FCM LSC FCM 57,8% 59,6% 79,4% 79,2% Eosinophils + Basophils 2,5% 2,1% 1,4% 1,7% Monocytes 7,0% 5,8% 3,0% 3,9% 32,3% 29,3% Neutrophils Lymphocytes all + CD3 - CD3 70,2% 29,7% 16,1% 15,8% + 13,8% 15,7% - 86,2% 84,3% 68,8% CD19 31,1% CD19 Tab. 1: Comparison of the percentages of cells in different cell subsets as determined by LSC and by FCM (left and right column, resp.) in blood of two different patients (Pat.1 and Pat.2). CD3+CD4+ Lymphocytes 5 6 CD3+CD4– (40x objective) Fig. 3: Same material as in fig. 1, but additionally gated on cells with bright CD45fluorescence (not shown). Compensation is set. Dot plot showing CD3 vs. CD4-CD19. Cells within region 5 correspond to CD3 +CD4+ lymphocytes (see photographs in upper line), cells within region 6 correspond to CD3+CD4– lymphocytes (see photographs in lower line).