Hemoglobinopathies
advertisement

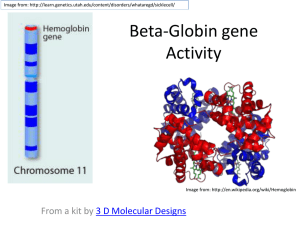
Hemoglobinopathies Globin Hemoglobin is the molecule that carries oxygen in the blood. It is contained in red blood cells. Heme (porphyrin ring, red) Hemoglobin globin Chains:、、、、、 -type chains: , ----141 amino acids -type chains: 、、、 (G、A) -----146 amino acids Adult Hb A Hb A2 Hb F Fetal Hb F Embryonic Hb Gower I (12 weeks) Hb Gower II Hb Portland 22 97~98% 22 2~3% 22 <1% 22 2 2 22 22 -type chain gene group : 16 p12 2 1 Normal: / -type chain gene group : 11p12 2 G Normal: A 1 / 22 22 22 GowerⅠ GowerⅡ Portland Embryonic Hb G A 1 22 22 22 F Fetal Hb A2 A Adult Hb Hemoglobinopathy is a group of rare, inherited disorders involving inherited mutation of the globin genes leading to a qualitative or quantitative abnormality of globin synthesis. Structural hemoglobinopathy (qualitative) Amino acid substitution in the globin chain 471 variants ( 144, 259, other 59, two chains 9) The Thalassemias (quantitative) Syndromes in which the rate of synthesis of a globin chain is reduced. Beta thalassemia - reduced beta chain synthesis Alpha thalassemia – reduced alpha chain synthesis Gene Mutations 1. Substitution (different amino acid) HbS (Sickle cell anemia) 6 GAA → GUA 2. Substitution (termination) Hb Mckees-Rock 144AAGUAUCACUAA→AAGUAACACUAA Terminator 3. Addition Hb Constant Spring --141CGUUAA→--CGUCAAGCU---- UAA 173 Terminator 4. Shift Hb Wayne 142 137ACCUCCAAAUACCGUUAA ACCUCAAAUACCGUUAAG-----CGGUAG 5. Missing Hb Gum Hiu 147 91 95 98 89AGUGAGCUGCACUGUGACAAGCUGCAC GUG 89 90 96 97 98 AGUGAGCUGCACGUG 6. Fusion Hb Lepore G A G G A A Classification & Terminology Alpha Thalassemia • Normal • Silent carrier • Minor • Hb H disease • Barts hydrops fetalis - : Indicates a gene deletion: / - / -/- --/ --/- --/-- Classification & Terminology Beta Thalassemia • Normal • Minor • Intermedia • Major 0 / /0 /+ 0/+ 0/0 +/+ :Indicates no production of globin chain by gene +: Indicates diminished, but some production of globin chain by gene Lab Tests CBC (complete blood count) •the number, size and shape of the red blood cells present. •how much hemoglobin is in them •MCV (mean corpuscular volume) is a measurement of the size of the red blood cells. A low MCV is often the first indication of thalassemia. If the MCV is low and iron-deficiency has been ruled out, the person may be a thalassemia trait carrier or have one of the hemoglobin variants that cause microcytosis (for example, Hgb E). Blood smear In this test, a trained laboratorian looks at a thin stained layer of blood, on a slide, under a microscope. •The number and type of white blood cells, red blood cells, and platelets •The red blood cells may be: Microcytic (smaller than normal) Hypochromic (pale –indicating less hemoglobin) Varying in size (anisocytosis) and shape (poikilocytosis) Nucleated (not normal in a mature RBC) Have uneven hemoglobin distribution (producing “target cells” that look like a bull’s-eye under the microscope). Detection of hemoglobin variants These tests identify the type and measure the relative amount of hemoglobins. hemoglobin electrophoresis, isoelectric focusing, or high performance liquid chromatography. These techniques separate different hemoglobins based on their charge. Most of the common variants can be identified using one of these methods or a combination. The relative amounts of any variant hemoglobin detected can help to diagnose combinations of hemoglobin variants and thalassemia. DNA analysis This test is used to investigate deletions and mutations in the alpha and beta globin producing genes. Family studies can be done to evaluate carrier status and the types of mutations present in other family members. DNA testing is not routinely done but can be used to help diagnose hemoglobin variants, thalassemia, and to determine carrier status. Methods Specimens were drawn into tubes containing dipotassium EDTA. All specimens were analyzed on the Bio-Rad Variant II HPLC system. Over a 32-month period, 60,293 samples were analyzed in the Special Hematology Laboratory at Bellevue Hospital Center for quantification of hemoglobin fractions and screening for hemoglobin variants. Alkaline and acid hemoglobin electrophoresis, and in certain cases globin chain electrophoresis, isoelectric focusing, and DNA analysis, were performed to document the identities of the hemoglobin variants. (A), a normal patient. C), Hb Twin Peak elution demonstrating the characteristic hump on the downward slope of the Hb A0 elution peak at 2.34 min. (B), Hb Ko¨ln elutes at a retention time of 2.26 min and the characteristic denatured Hb Ko¨ln at retention time 4.87 min. (D), Hb Lepore elution demonstrating the characteristic increased proportion of Hb A2 (11.6%) and hump on the downward slope of the Hb A2 peak at 3.34 min. (E), Hb A2’ (4.56 min) elution (F), Hb G-Philadelphia elutes the characteristic lower proportion of Hb A2 (0.8%) at 3.63 min, the Hb G-Philadelphia elution peak at 4.24 min, and the presence of the characteristic minor peak (2G-Philadelphia22 ) at 4.63 min. (G), Hb Hasharon elutes the characteristic decreased proportion of Hb A2 (1.5%) at 3.60 min, a characteristic minor peak at 4.27 min, and the characteristic minor peaks preceding and succeeding (2Hasharon2) the Hb Hasharon elution peak at 4.85 min. (H), Hb O-Arab elution demonstrating the characteristic minor peak at 3.98 min, which is absent in Hb C samples. Results: The mean (SD) imprecision (CV) of the retention time was 1.0 (0.7)% . Among 60 293 samples tested, we encountered 34 unique hemoglobin variants and 2 tetramers. 18 variants and 2 tetramers could be identified solely by retention time 3 variants by retention time and proportion of total hemoglobin 4 variants could be identified by retention time and peak characteristics 8 variants by retention time and electrophoretic mobility. 1 variant (Hb New York) was missed on HPLC. Retention time on HPLC was superior to electrophoresis for the differentiation and identification of 6 members of the Hb J family, 4 members of the Hb D family, and 3 variants with electrophoretic mobilities identical or similar to that of Hb C. 6 variants with electrophoretic mobilities identical or similar to that of Hb S could be differentiated and identified by retention time and proportion of total hemoglobin. HPLC detected two variants (Hb Ty Gard and Hb Twin Peaks) missed on electrophoresis. Hb A1C eluted in the P2 (1.24 –1.40 min) window. When the elution peak was >7% of the total hemoglobin, the patient records were checked for indication of diabetes and Hb A1C quantification. If no quantification was available, the Hb A1C was quantified in the hospital’s chemistry laboratory by phenol-borate affinity HPLC (Primus Corpora-tion). If the %Hb in the P2 window and the Hb A1C values were concordant, i.e., within 15% of each other, no further studies were performed. The only hemoglobin variant found to elute in this window was Hb Hope, which had a mean (SD) %Hb [45.9 (2.2)%] much greater than would be expected for Hb A1C .