Ji`s Awesome Powerpoint slides 3 - Bio 5068
advertisement

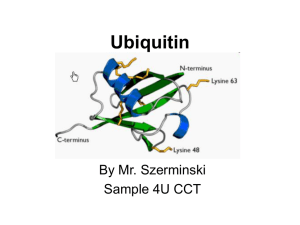
MCB Exam III Review Ji Woong Park December 13th, 2014 Material Coverage • My review covers 45 points. It’s lectures by Stewart, Huettner, Weihl, Amarasinghe, and Fremont. • Due to the diverse range of topics (cancer to crystallography), you may need to use slides not in the review to help your understanding. • But, the exam questions will be based on the review slides. There will be, however, some questions that require solid understanding/application to answer them (hence, the above point) • Previous years’ exams will be helpful but it’s incorrect to assume they will be like Exam II. Terms To Know for “Application” Questions • • • • • • • • RT-qPCR/RNAi/Knockout Gene or Mice Western Blot/IP/Co-IP Transfection/Infection Electrophysiology Mass Spec/NMR/Crystallography Site-Directed Mutagenesis Inhibitors/Dominant Negatives Fluorescence/Viability/Toxicity Assays Telomere Function distinguishes between the chromosome end and a double strand break protects the chromosome from end-to-end fusions An emerging paradigm: the telomere complex does not exclude DNA surveillance, repair and replication machinery; rather it directs, modulates and specializes the activities of these proteins to ensure high fidelity replication and telomere stability Old view TIN2 TPP1 POT1POT1 RAP1 DNA repair machinery New view DNA repair/replication machinery The “telomere” hypothesis Telomere Length Population Doubling Senescence 1 2 3 4 Time 5 6 The telomere hypothesis p53 Rb Telomere Length Stop Time Senescence The telomere hypothesis Telomere Length p53 Rb Continued proliferation Crisis telomere dysfunction genetic catastrophe cell death Time Telomerase adds telomeric repeats to the 3’ termini of the chromosome -only the catalytic (hTERT) and RNA (hTR) components are required for activity in vitro hTERT hTR CAAUCCCAAUC The telomere hypothesis Telomere Length ALT Stop Senescence p53 Rb Crisis hTERT 1 in ~107 Stable telomere maintenance Time Telomere Length The telomere hypothesis ALT Stop Crisis hTERT 1 in ~107 Time ALT Telomerase + Telomerase allows ALT cells to form tumors hTERT H-ras TUMORS GM847 Immortal Expresses SV40 Early region Lacks telomerase Stewart et al 2002 Extra-telomeric functions of hTERT Overexpression of mTERT in murine models increases tumor rates in aged mice Overexpression of hTERT results in resistance to apoptosis Ectopic expression of mTERT in skin results “hairy” mice, increased stem cell pool Telomerase is favored over ALT in human tumors hTERT expression is required in normal fibroblasts to avoid senescence Time/Stress Senescence Senescence evasion Stromal Promotion Normal ECM Epithelial cell Premalignant Altered ECM Endothelial cell Preneoplastic cell Tumor Young fibroblast Senescent fibroblast Cancer cell Immune cell Senescent epithelial cell For additional reading (highly recommend) Types of Stem Cells Embryonic – from the inner cell mass of preimplantation embryos, prior to formation of the 3 germ layers (ectoderm, mesoderm, endoderm) Somatic – undifferentiated cells found in specific locations in “mature” tissues iPS cells – induced pluripotent stem cells generated by reprogramming differentiated cells (or cell nuclei, i.e. therapeutic cloning) Reprogramming • SCNT – somatic cell nuclear transfer (reproductive and therapeutic cloning) – deterministic and fairly rapid • iPS – induced pluripotent stem cells – slow and stochastic (until recently) • Transdifferentiation – conversion of one terminally differentiated cell type into another without dedifferentiation to an immature phenotype. Must rule out cell fusion or other explanations. Generating iPS cells • Express transcription factors: Oct3/4, Sox2, Klf4 and c-Myc (OSKM) Oct3/4, Sox2, Nanog and Lin28 • Initial de-differentiation and proliferation (day 1-3, enhanced by Myc); histone modification and chromatin reorganization • 2nd wave of gene expression - stem cell and development related genes (day 9-12); DNA demethylation and X reactivation OR Transdifferentiation • Conversion from one differentiated cell type to another without evident de-differentiation and re-differentiation • Must not be confused by cell fusion or selection for rare pluripotent cells in the source material. • Induced by expression of transcription factors and microRNAs Protein Degradation in the Cell Ub Autophagy Nucleus Aggresome Ub UPS Ub Ub Endocytosis Protein Degradation Ubiquitin/Proteasome Pathway 80-90% Most intracellular proteins • Lysosomal processes 10-20% Extracellular proteins Cell organelles Some intracellular proteins UBIQUITIN Small peptide that is a “TAG” 76 amino acids C-terminal glycine - isopeptide bond with the e-amino group of lysine residues on the substrate Attached as monoubiquitin or polyubiquitin chains G K Ubiquitination of proteins is a FOUR-step process First, Ubiquitin is activated by forming a link to “enzyme 1” (E1). Then, ubiquitin is transferred to one of several types of “enzyme 2” (E2). Then, “enzyme 3” (E3) catalizes the transfer of ubiquitin from E2 to a Lys e-amino group of the “condemned” protein. Lastly, molecules of Ubiquitin are commonly conjugated to the protein to be degraded by E3s & E4s AMP The proteasomal DUB Usp14 impairs protein degradation Lee, BH et al Nature 467:179-84 2010 Autophagy • Lysosomal degradation of proteins and organelles • Occurs via three routes – Macroautophagy – Microautophagy (direct uptake of cellular debris via the lysosome) – Chaperone mediated autophagy (selective import of substrates via Hsc70 and Lamp2a) Selective Autophagy • • • • • • Aggregaphagy– p62/SQSTM1, Nbr1 Mitophagy – Parkin, Nix Reticulophagy – endoplasmic reticulum Ribophagy – translating ribosomes Xenophagy – e.g. Salmonella via optineurin Lipophagy – autophagy mediated lipolysis • Performed by an expanding group of ubiquitin adaptors Rapamycin as an inducer of autophagy Immunosuppressant used to treat transplant rejection Inhibits the mTOR pathway mTOR integrates extrinsic growth signals and cellular nutrient status and energy state Active mTOR Protein synthesis and cell growth Inactive mTOR (or rapamycin treatment) Inhibition of protein synthesis and increased autophagic degradation of protein Protein Structures from an NMR Perspective Analyzing NMR Data is a Non-Trivial Task! there is an abundance of data that needs to be interpreted X Not A Direct Path! Interpreting NMR Data Requires Making Informed “Guesses” to Move Toward the “Correct” Fold Distance from Correct Structure Initial rapid convergence to approximate correct fold Correct structure NMR Data Analysis Iterative “guesses” allow “correct” fold to emerge Protein Structure Determination by NMR •Stage I—Sequence specific resonance assignment •State II – Conformational restraints •Stage III – Calculate and refine structure Why use deuteration? • What are the advantages? • What are the disadvantages? NMR Structure Determination NOE NOE - a through space correlation (<5Å) - distance constraint 4.1Å 2.9Å Coupling Constant (J) - through bond correlation J NH CaH - dihedral angle constraint Chemical Shift - very sensitive to local changes in environment - dihedral angle constraint Dipolar coupling constants (D) - bond vector orientation relative to magnetic field - alignment with bicelles or viruses CaH D NH Analysis of the Quality of NMR Protein Structures Is the “Average” NMR Structure a Real Structure? • No-it is a distorted structure level of distortions depends on the similarity between the structures in the ensemble provides a means to measure the variability in atom positions between an ensemble of structures Expanded View of an “Average” Structure Some very long, stretched bonds Position of atoms are so scrambled the graphics program does not know which atoms to draw bonds between Some regions of the structure can appear relatively normal An 7-step program for protein structure determination by x-ray crystallography 1. Produce monodisperse protein either alone or as relevant complexes 2. Grow and characterize crystals 3. Collect X-ray diffraction data 4. Solve the phase problem either experimentally or computationally 5. Build and refine an atomic model using the electron density map 6. Validation: How do you know if a crystal structure is right? 7. Develop structure-based hypothesis 1. Produce monodisperse protein either alone or as relevant complexes Methods to determine protein purity, heterogeneity, and monodispersity Gel electrophoresis (native, isoelectric focusing, and SDS-PAGE) Size exclusion chromatography Dynamic light scattering http://www.protein-solutions.com/ Circular Dichroism Spectroscopy http://www-structure.llnl.gov/cd/cdtutorial.htm Characterize your protein using a number of biophysical methods Establish the binding stoichiometry of interacting partners 2. Grow and characterize crystals Hanging Drop vapor diffusion Sitting drop, dialysis, or under oil Macro-seeding or micro-seeding Sparse matrix screening methods Random thinking processes, talisman, and luck The optimum conditions for crystal nucleation are not necessarily the optimum for diffraction-quality crystal growth Hanging Drop Sitting drop Commercial screening kits available from http://www.hamptonresearch.com; http://www.emeraldbiostructures.com Space Group P21 4 M3 /ASU diffraction >2.3Å 14.4% Peg6K NaCacodylate pH 7.0 200mM CaCl2 Space Group C2 2 M3 /ASU diffraction >2.1Å 18% Peg4K Malic Acid/Imidazole pH 5.1 100mM CaCl2 Space Group P3121 3 M3 + 3 MCP-1/ASU diffraction > 2.3Å 18% Peg4K NaAcetate pH 4.1 100mM MgCl2 No Xtals? Decrease protein heterogeneity Remove purification tags and other artifacts of protein production Remove carbohydrate residues or consensus sites (i.e., N-x-S/T) Determine domain boundaries by limited proteolysis followed by mass spectrometry or amino-terminal sequencing. Make new expression constructs if necessary. Think about the biochemistry of the system! Does your protein have cofactors, accessory proteins, or interacting partners to prepare as complexes? Is their an inhibitor available? Are kinases or phosphatases available that will allow for the preparation of a homogeneous sample? Get a better talisman 3. Collect X-ray diffraction data Initiate experiments using home-source x-ray generator and detector Determine liquid nitrogen cryo-protection conditions to reduce crystal decay While home x-rays are sufficient for some questions, synchrotron radiation is preferred Anywhere from one to hundreds of crystals and diffraction experiments may be required Argonne National Laboratory Structural Biology Center beamlineID19 at the Advanced Photon Source http://www.sbc.anl.gov 4. Solve the phase problem either experimentally or computationally Structure factor equation: By Fourier transform we can obtain the electron density. We know the structure factor amplitudes after successful data collection. Unfortunately, conventional x-ray diffraction doesn’t allow for direct phase measurement. This is know as the crystallographic phase problem. Luckily, there are a few tricks that can be used to obtain estimates of the phase a(h,k,l) Experimental Phasing Methods MIR - multiple isomorphous replacement - need heavy atom incorporation MAD - multiple anomalous dispersion- typically done with SeMet replacement MIRAS - multiple isomorphous replacement with anomalous signal SIRAS - single isomorphous replacement with anomalous signal Computational Methods MR - molecular replacement - need related structure Direct and Ab Initio methods - not yet useful for most protein crystals MAD phasing statistics for the AP-2 a-appendage 5. Build an atomic model using the electron density map Electron density for the AP-2 aappendage Initial bones trace for the AP-2 aappendage Final trace for the AP-2 aappendage The resolution of the electron-density map and the amount of detail that can be seen Resolution Structural Features Observed 5.0 Å 3.5 Å 3.0 Å 2.8 Å 2.5 Å Overall shape of the molecule Ca trace Side chains Carbonyl oxygens (bulges) Side chain well resolved, Peptide bond plane resolved Holes in Phe, Tyr rings Current limit for best protein crystals 1.5 Å 0.8 Å 6. Validation: How do you know if a crystal structure is right? The R-factor R = S(|Fo-Fc|)/S(Fo) where Fo is the observed structure factor amplitude and Fc is calculated using the atomic model. R-free An unbiased, cross-validation of the R-factor. The R-free value is calculated with typically 5-10% of the observed reflections which are set aside from atomic refinement calculations. Main-chain torsions: the Ramachandran plot Geometric Distortions in bond lengths and angles Favorable van der Waals packing interactions Chemical environment of individual amino acids Location of insertion and deletion positions in related sequences 7. Develop structure-based hypothesis Structure-Based Mutagenesis of the a-appendage Traub LM, Downs MA, Westrich JL, and Fremont DH: (1999) Crystal structure of the a-appendage of AP-2 reveals a recruitment platform for clathrin-coat assembly. Proc. Natl. Acad. Sci. U.S.A. 96:8907-8912. Selection of E16 specific epitope variants of DIII Yeast library of DIII variants created by error prone PCR E -DIII Pooled DIII mAbs E16 staining DIII mutations at Ser306, Lys307, ThrE330 and Thr332 significantly diminish E16 binding GOOD LUCK – Interview season is coming!