PPT
advertisement

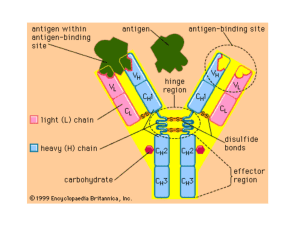
Microarray Gene Expression Analysis Differential expression, clustering, networks, and functional enrichment STEMREM 201 Fall 2012 Aaron Newman, Ph.D. 10/17/12 A genomics approach to biology involves… Finding significant patterns in high-throughput data Interpreting these patterns in the context of prior knowledge Generating new hypotheses and predictions • A plethora of techniques and tools exist… • My goal is to introduce some practical, powerful, and freely available methods for gene expression analysis Example workflow Gene expression profiling DEGs Cluster analysis Functional enrichment Network analysis Biological meaning GSEA Materials and Methods GSE22651 ESCs and differentiated cells Excel & GenePattern Hierarchical & AutoSOME DAVID Toppfun STRING & Cytoscape Biological meaning GSEA Microarray normalization • Probe/array-level normalization (reduce inter-array technical errors and noise) – Raw CEL files merged and normalized text file • Robust Multi-Chip Averaging (RMA) for Affymetrix arrays – Affymetrix Gene Expression Console software • Quantile Normalization for Agilent, Illumina, and others. – Sets all arrays to the same distribution (mean, median, sd, etc.) • Analysis-level normalization – Log2 Transformation • Improves statistical properties for analysis (log-normal) – Median/mean centering • Useful for reducing impact of transcript abundance on identifying/visualizing co-expressed genes center – Unit variance • Standardize each column (array) to mean of 0 and standard deviation of 1 Differentially expressed genes (DEGs) • Goal: given known phenotypes, determine which genes exhibit significant differential expression. • Many genes will be tested; multiple hypothesis testing should be performed to control the false discovery rate (FDR). – Bonferroni (α / n) – Benjamini-Hochberg (largest k s.t. P(k) ≤ (αk) / n) – Storey Q-value (p-value specific FDR) • Q-value software - http://genomics.princeton.edu/storeylab/qvalue/ • Sample permutations can improve p-value accuracy – *Only apply with ≥ 10 samples per class – Implemented in GenePattern (ComparativeMarkerSelection module), SAM Supervised DEG identification tools (i.e., classes are known) • Likely familiar – Excel (T-test) coupled with FDR assessment • Watch for conversion to dates! (e.g., MARCH6 3/6) • Basic to Intermediate – Statistical Analysis of Microarrays (Windows only) – GenePattern (Broad Institute) – p.adjust (R) • Advanced – Bioconductor packages in R (e.g., limma) Tutorial 1: Identifying DEGs in Excel using FDR cutoff of 1% (BH method) Open “DEG_example_large.txt” in Excel (2 classes, 25151 genes). In column L, use T-test function to test for significant differential expression between ESCs and non-ESCs. 1) 2) • • 3) 4) 5) =TTEST(“ESCs”, “non-ESCs”, 2, 3) 2-tailed, unpaired with unequal variance Sort p-values in column L in ascending order. In column M, input p-value rank, going from 1,2,3…n. Input following formula into column N to test for FDR of 1% • • • 6) In general: = (0.01 * rank) / n In our case: = (0.01 * M2) / 25151 Autocomplete column N In column O, test for p-values that do not exceed FDR of 1% • = if(L2 <= N2, 1, 0) That’s it! All genes with a 1 in column O are significant at an FDR of 1%. 7) • Should be 3237 significant genes. Cleaning up result and creating a heat map 1) Calculate log2 fold change in column P • • = AVERAGE(B2:F2) – AVERAGE(G2:K2) Autocomplete Use filter to isolate significant genes with absolute fold change ≥ 5 Copy and paste into new tab Sort by fold change in descending order Save as new file (e.g., DEG_example_sorted_fdr01_fold5.txt) Center genes in Cluster 3.0 2) 3) 4) 5) 6) • • • • 7) Open file Go to “Adjust Data” Check “Center genes” and select “Median” Press “Apply”, then save file (e.g. DEG_example_sorted_fdr01_fold5_medcen.txt) Open in Java TreeView. • • To customize heat map display and text, use “Settings>Pixel Settings”, and “Settings>Font Settings”. Export heat map image using “Export>Save Tree Image”. Expected result FDR ≤ 1% Fold change ≥ abs(25) = 79 genes Tutorial 2: Identifying DEGs in GenePattern 1) 2) 3) 4) Create account (if you have not already) and log in to GenePattern Select “Differential Expression Analysis”. Skip to ComparativeMarkerSelection (step 2) and click “Open module”. Enter following fields: • • • 5) 6) 7) 8) 9) input* file = GSEA_example_expression_large.gct cls* file = GSEA_example_classes.cls number of permutations* = 0 Select “Run” When processing is finished, select “ComparativeMarkerSelectionViewer” from the pulldown menu next to “GSEA_example_expression_large.comp.marker.odf” Select “Run” Select “Open Visualizer” and “Allow” A table will appear showing all genes and various statistics, including: 1) 2) 3) 4) Benjamini_Hochberg corrected p-value; FDR(BH) Storey q-value; Q Value Bonferroni p-value Fold change Extracting data and displaying as a heat map 1) 2) 3) 4) 5) Pipe .odf file to “ExtractComparativeMarkerResults” module. Select genes with BH FDR ≤ 1%. Filtered data are available as a new .gct file. To display a heat map, pipe .gct file to “HeatMapViewer” module. Select “Run” Example workflow Gene expression profiling DEGs Cluster analysis Functional enrichment Network analysis Biological meaning GSEA The “Clustering Problem” for Large Data Sets Widespread Importance – Genomics – Phylogenetics – Disease – Galaxy Clusters – etc., etc., etc. Common clustering methods Toy data set K-means Hierarchical (Eisen) SOM Figure 1. D’haeseleer, Nat Biotechnol. 2005 Feature Comparison Method Handle Diverse Large Cluster Datasets Shapes √ Hierarchical K-Means √ SelfOrganizing Map (SOM) √ Detect Cluster Number Identify Outliers Low Output Variance √* √ √* Tutorial 3: Hierarchical Clustering 1) Open “GSE22651_filtered.txt” in Cluster 3.0 1) Normalize data (Adjust Data tab) • Check “Log transform data” • Check “Center genes” and select “Median” • Press “Apply Button” 2) Cluster data (Hierarchical tab) • Check “Cluster” under Genes and under Arrays • Leave “Similarity Metric” at Uncentered Correlation Press “Centroid linkage” under “Cluster method” 3) • • • • 4) Centroid = distance between cluster centers Single = closest distance between clusters Complete = farthest distance between clusters Average = mean of all pairwise distances between clusters Open clustered data table file (*.cdt) in Java TreeView. Java TreeView Navigate the cluster tree to highlight genes with distinct expression patterns in particular samples Export>”Save List” to copy or save gene lists of interest for further analysis. Expected result Genes Samples Automatic clustering of Self-Organizing Map Ensembles AutoSOME Serial application of - SOM - Density Equalization - Minimum Spanning Tree - Ensemble Averaging Newman and Cooper (2010) BMC Bioinformatics, 11:117 AutoSOME Method Handle Diverse Large Cluster Datasets Shapes Detect Cluster Number √ Hierarchical Identify Outliers Low Output Variance √* √ K-Means √ √* SelfOrganizing Map (SOM) √ Affinity Propagation √* Spectral Clustering √* √ √ nNMF √ √ √ AutoSOME √ √ √ √ √ √ √ √ AutoSOME Webstart http://jimcooperlab.mcdb.ucsb.edu/autosome Tutorial 4: Identifying discrete clusters without prior knowledge of cluster number 1) 2) 3) 4) Launch the AutoSOME GUI via the large launch button. Open “GSE22651_filtered.txt” Skip filtering Show “Basic Fields” • Set p-value = 0.05 Show “Input Adjustment” 5) • Check Log2 Scaling, Unit Variance, Median Center, and Sum Squares = 1 Press the large “Run” button on the left. From the menubar, select View>heat map> green red. Select cluster 1 in the cluster list. The data are rendered as a normalized heat map. To change the display, go to View>settings>image settings. 6) 7) 8) 9) • • Under the Normalization tab, check “Display Original Data”, “Log2 Scaling”, and “Median Center”. Check “Manually adjust range for contrast” and set minimum to -2 and maximum to 2. Press “Update” (lower left corner). AutoSOME continued… 1) 2) 3) Select clusters 1 to 5 (hold down shift). Right click mouse in heat map window to resize. Set “Zoom Factor” to 40 and Press “Save”. See website for further tutorials and documentation. Representative heat map View>heat map>rainbow Example workflow Gene expression profiling DEGs Cluster analysis Functional enrichment Network analysis Biological meaning GSEA How to interpret clustering results • Gene set functional annotation – DAVID – Toppfun – MSigDB http://david.abcc.ncifcrf.gov/ http://toppgene.cchmc.org/enrichment.jsp http://www.broadinstitute.org/gsea/msigdb/ • Network analysis – STRING http://string-db.org/ Gene sets: where do they come from? • Gene ontology – An attempt to semantically organize genes and their functional relationships. – Data are arranged in a graph structure, from broad to specific – Ontologies: • Biological process (BP): series of ordered events • Molecular function (MF): activities that occur at the molecular level • Cellular component (CC): part of a cell • Biocarta/KEGG pathways (curated wiring diagrams) • High-throughput studies e.g., MSigDB Tutorial 5: Functional annotation of a large cluster using DAVID http://david.abcc.ncifcrf.gov/ 1) In AutoSOME, find the cluster of genes with higher expression in stem cells. 2) Go to View>raw data. Highlight all genes in the cluster and copy the list. 3) Open the DAVID homepage and press “Start Analysis” Select “Upload” tab and paste in gene list. Under “Select Identifier,” select “OFFICIAL_GENE_SYMBOL”. Select “Gene List” for “List Type”. Submit the list. Press OK at multi-species warning message. Select “Homo sapiens” and press “Select Species”. Select “Functional Annotation Tool” Press “Functional Annotation Clustering” button. 4) 5) 6) 7) 8) 9) 10) 11) DAVID Output Similar gene sets are clustered together, eliminating redundancy and facilitating interpretation Gene sets Pathways Tutorial 6: Protein-protein associations with STRING Top 50 non-ESC genes Protein-protein association network among top 50 non-ESC genes Evidence types Top 100 non-ESC genes Example workflow Gene expression profiling DEGs Cluster analysis Functional enrichment Network analysis Biological meaning GSEA Gene set enrichment in a ranked list • Gene Set Enrichment Analysis (GSEA) • “Threshold-less” – Arbitrary DEG cutoffs are avoided • Two modes of operation to rank input genes: – Rank by differential expression between phenotypes • Default metric is signal to noise, defined as: (avg[class 1] – avg[class 2]) / (sd[class 1] + sd[class 2]) – Pre-ranked according to user-defined criteria • Evaluates statistical bias in the distribution of each defined gene set over the list of ranked input genes. Input format • Input: – Expression data (gene cluster text file format, *.gct) – Classes (*.cls) • File extensions matter! – If using Notepad in Windows, set Save as type: to “All Files (*.*) – If using TextEdit in Mac, go to Preferences > Open and Save > and uncheck ‘Add “.txt” extension to plain text files’. • Formatting instructions: http://www.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_for mats Tutorial 7: GSEA Input files: GSEA_example_expression_large.gct, GSEA_example_classes.cls Load data Run GSEA Output Summary Gene expression profiling DEGs Cluster analysis Functional enrichment Network analysis Biological meaning GSEA