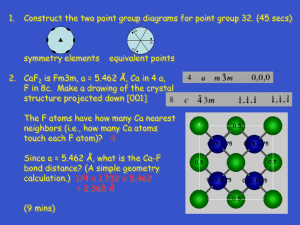
Characteristics of crystalline state
advertisement

Minerals • A mineral is a naturally occurring solid chemical substance formed through biogeochemical processes, having characteristic chemical composition, highly ordered atomic structure, and specific physical properties. • By comparison, a rock is an aggregate of minerals and does not have a specific chemical composition. • International Mineralogical Association (IMA, http://www.ima-mineralogy.org/) approved the following definition in 1995: "A mineral is an element or chemical compound that is normally crystalline and that has been formed as a result of geological processes." • According to this definition and classification scheme, biogenic materials were excluded from the mineral kingdom: – • "Biogenic substances are chemical compounds produced entirely by biological processes without a geological component (e.g., urinary calculi, oxalate crystals in plant tissues, shells of marine molluscs, etc.) and are not regarded as minerals. However, if geological processes were involved in the genesis of the compound, then the product can be accepted as a mineral." There are currently more than 5,000 known minerals, according to the IMA, which is responsible for the approval of and naming of new mineral species found in nature. Of these, perhaps 100 can be called "common", 50 are "occasional", and the rest are "rare" to "extremely rare". RRUFF database: http://rruff.info/ American Mineralogist Database: http://rruff.geo.arizona.edu/AMS/amcsd.php Webmineral database: http://webmineral.com/ Types of chemical bonds and their general characteristics • Because of the nature of ionic and covalent bonds, the materials produced by those bonds tend to have quite different macroscopic properties. The atoms of covalent materials are bound tightly to each other in stable molecules, but those molecules are generally not very strongly attracted to other molecules in the material. On the other hand, the atoms (ions) in ionic materials show strong attractions to other ions in their vicinity. This generally leads to low melting points for covalent solids, and high melting points for ionic solids. • Ionic Compounds – – – – • Crystalline solids (made of ions) High melting and boiling points Conduct electricity when melted Many soluble in water but not in nonpolar liquid Why is diamond an exception? Covalent Compounds – – – – Gases, liquids, or solids (made of molecules) Low melting and boiling points Poor electrical conductors in all phases Many soluble in nonpolar liquids but not in water • Elements from opposite ends of the periodic table will generally form ionic bonds. They will have large differences in electronegativity and will usually form positive and negative ions. The elements with the largest electronegativities are in the upper right of the periodic table, and the elements with the smallest electronegativities are on the bottom left. If these extremes are combined, such as in RbF, the dissociation energy is large. Hydrogen is an exception to that rule, forming covalent bonds. • Elements which are close together in electronegativity tend to form covalent bonds and can exist as stable free molecules. Carbon dioxide is a common example. Oxidation state of atoms Oxidation state is an indicator of the degree of oxidation of an atom in a chemical compound. The formal oxidation state is the hypothetical charge that an atom would have if all bonds to atoms of different elements were 100% ionic. Oxidation states are typically represented by integers, which can be positive, negative, or zero. In some cases the average oxidation state of an element is a fraction, such as 8/3 for iron in magnetite. The increase in oxidation state of an atom through a chemical reaction is known as an oxidation; a decrease in oxidation state is known as a reduction. Such reactions involve the formal transfer of electrons, a net gain in electrons being a reduction and a net loss of electrons being an oxidation. For pure elements, the oxidation state is zero. Oxidation state General rules for simple compounds without structural formulae: • • • • Any pure element (even if it forms diatomic molecules like chlorine, Cl2) has an oxidation state (OS) of zero. Examples of this would be Cu or O2. For monatomic ions, the OS is the same as the charge of the ion. For example S2- has an OS of 2, whereas Li+ has an OS of +1. The sum of OSs for all atoms in a molecule or polyatomic ion is equal to the charge of the molecule or ion, so that the OS of one element can be calculated from the OS of the other elements. For example, in (SO3)2- (sulfite ion), the total charge of the ion is -2, and each oxygen is assumed to have its usual oxidation state of -2. The sum of OSs is then OS(S) + 3(-2) = -2, so that OS(S) = +4. Do not confuse the formal charge on an atom with its formal oxidation state, as these may be different, and often are different, in polyatomic ions. For example, the charge on the nitrogen atom in ammonium ion NH4+ is +1, but the formal oxidation state is -3, the same as it is for nitrogen in ammonia. In this case, the charge on the atom changed, but its oxidation state did not. • • • • Ionic radius, \is the radius ascribed to an atom's ion. Although neither atoms nor ions have sharp boundaries, it is useful to treat them as if they are hard spheres with radii such that the sum of ionic radii of the cation and anion gives the distance between the ions in a crystal lattice. Ions may be larger or smaller than the neutral atom, depending on the ion's charge. When an atom loses an electron to form a cation, the lost electron no longer contributes to shielding the other electrons from the charge of the nucleus; consequently, the other electrons are more strongly attracted to the nucleus, and the radius of the atom gets smaller. Similarly, when an electron is added to an atom, forming an anion, the added electron shields the other electrons from the nucleus, with the result that the size of the atom increases. The ionic radius is not a fixed property of a given ion, but varies with coordination number, spin state and other parameters. Nevertheless, ionic radius values are sufficiently transferable to allow periodic trends to be recognized. As with other types of atomic radius, ionic radii increase on descending a group. Ionic size (for the same ion) also increases with increasing coordination number, and an ion in a high-spin state will be larger than the same ion in a low-spin state. In general, ionic radius decreases with increasing positive charge and increases with increasing negative charge. An "anomalous" ionic radius in a crystal is often a sign of significant covalent character in the bonding. No bond is completely ionic, and some supposedly "ionic" compounds, especially of the transition metals, are particularly covalent in character. Ionic radius Bond valence method • The bond valence method (or bond valence sum) is a popular method in coordination chemistry to estimate the oxidation states of atoms and expected bond lengths. • The basic idea is that the valence V of an atom is the sum of the individual bond valences vi surrounding the atom: • The individual bond valences in turn are calculated from the observed bond lengths. • Ri is the observed bond length, R0 is a tabulated parameter expressing the (ideal) bond length when the element i has exactly valence 1, and b is an empirical constant, typically 0.37 Å. • Bond Valence wizard: http://orlov.ch/bondval/index.html#4 Characteristics of crystalline state • Great majority of minerals are crystals • The two most important characteristics of a crystal are: – Periodicity – Symmetry Description of crystal structure and direct space • • Unit cell parameters Crystal system Metric symmetry of the unit cell does not determine the crystal system – the symmetry does • • • • Space group Wyckoff positions and fractional atomic coordinates Site occupancy factors Atomic displacement parameters (anisotropic or isotropic) Crystal data Formula sum Z Crystal system Space group Unit cell dimensions SiO2 3 trigonal P 31 2 1 (no. 152) a = 4.8815(7) Å c = 5.3816(15) Å 111.06(4) Å3 2.695 g/cm3 Cell volume Density, calculated Atomic coordinates Atom SI1 O1 Wyck. 3a 6c Anisotropic displacement parameters (in Å 2) Atom SI1 O1 U11 0.00618 0.01193 U22 0.00551 0.01158 U33 0.01209 0.02161 U12 0.00309 0.00913 U13 0.00051 -0.00280 U23 0.00026 -0.00316 x 0 0.27246 y 0.46621 0.41287 z 2/3 0.78002 • • • • • • Unit cell is the simplest element of the crystal that is repeated by translations to form infinite crystal lattice. Geometrically, the unit cell is fully characterized by 6 parameters describing the lengths of the edges and the inter-edge angles. Alternatively, a set of principal translation vectors a1, a2, a3 can be described, to form a basis of direct space reference system. The content of the unit cell (atomic positions) are usually described using fractional atomic coordinates (x,y,z)f, which are coordinates in the direct space reference system. Alternatively, atoms can be described in Cartesian coordinates of laboratory reference system. A conventional choice of the Cartesian reference system to describe unit cell contents is given by matrix A: a b cos A 0 b sin 0 0 c cos c(cos cos cos ) / sin c sin 2 cos2 cos2 2 cos cos cos 1/ 2 / sin Unit cell and direct space xyzC Axyzf Metric tensor • Metric tensor (symmetric, positive definite) of direct space can be calculated as follows: T GA A • Matrix A is a Cholesky decomposition of the metric tensor. • Determinant of the Metric tensor is equal to the square of the unit cell volume: detG V 2 • Unit cell volume can also be calculated from vector equation: V a1 a2 a3 • Inverse of the metric tensor, Gr=G-1 is the reciprocal space metric tensor Calculation of density • • • • • • Unit cell of SiO2 quartz has a volume of 111.06 A3 The Z-number is 3 Atomic mass unit: 1u = 1.660 10-24 g Avogadro’s number NA=6.022 1023 1/mol What is the density of quartz? What is the molar volume of quartz? • • • • • All properties of a crystal remain invariant under lattice translation, therefore only angular dependence of the properties is of importance. From the point of view of diffraction it is important to consider families of parallel planes of lattice points. These families of planes can be characterized by their vectors normal. It is convenient to introduce another reference frame basis, built on vectors “reciprocal” to the direct space basis vectors: This space is called reciprocal space. Reciprocal space coordinates of vectors are always integer and are called Miller indices. The length of the reciprocal space vectors is defined as inverse of the interlayer spacing in direct space. The Brillouin zone is a primitive unit cell of the reciprocal lattice. By analogy to Cholesky decomposition of the direct space metric tensor, a reciprocal space Cartesian basis matrix B can be defined. B=A-1 Reciprocal space Reciprocal space • • • • z • hkl X-ray x y • • Direct space relates to atoms in the unit cell, while reciprocal space relates to peaks in diffraction experiment. Vectors in reciprocal space correspond to families of planes in the crystal (the direction of the rec. vector is normal to the family of planes). In a conventional crystal (as opposed to incommensurately modulated crystal or quasi-crystal) vectors in reciprocal space can occupy only points on a 3-dimensional grid. Grid coordinates of reciprocal vectors are known as Miller indices hkl Geometry of the reciprocal space grid is determined by the unit cell of the crystal. And can be directly measured in diffraction experiment. D-spacing is equal to inverse of the reciprocal vector length. Coordinates of vectors in reciprocal space are described in laboratory (instrument-related) reference coordinate system. Seven crystal systems Crystal System Characteristic Symmetry Triclinic 1× 1-fold -1 Monoclinic 1× 2-fold 2/m Orthorhombic 3× 2-fold Tetragonal Syngony Unit-Cell Parameters Indep. Parameters a ≠ b ≠ c; α ≠ β ≠ γ 6 a ≠ b ≠ c; α = γ = 90°; β ≠ 90° 4 mmm a ≠ b ≠ c; α = β = γ = 90° 3 1× 4-fold 4/mmm a = b ≠ c; α = β = γ = 90° 2 Trigonal (see note) 1× 3-fold 6/mmm -3m(R) a = b ≠ c; α = β = 90°; γ = 120° 2 Hexagonal 1× 6-fold 6/mmm a = b ≠ c; α = β = 90°; γ = 120° 2 Cubic 4× 3-fold m-3m a = b = c; α = β = γ = 90° 1 Scalar and vector equations to calculate d-spacing d 1 Bhkl • UB matrix relates the Miller indices of reciprocal vector (hkl) with its Cartesian coordinates in lab reference system (xyz) at zero goniometer position. Orientation matrix xyz=UB hkl • • • • Columns in the orientation matrix are coordinates of the principal vectors in reciprocal space 1 0 0 , 0 1 0 and 0 0 1. UB matrix is composed of two sub-matrices, U and B U describes the orientation of the crystal axes with respect to the laboratory reference system, while B stores information about the unit cell parameters. By inverting the above equation one can calculate what are the Miller indices of a measured reciprocal vector xyz hkl=UB-1 xyz z hkl X-ray x y Symmetry operations in crystals • • • • • • Simple symmetry operations is crystals include: Identity 1 Inversion -1 Proper rotation axes 2, 3, 4, 6 Improper rotation (rotoinversions) -3, -4, -6 Mirror plane m Only symmetry operation that allow infinite tiling of space are allowed Simple symmetry operations inversion center 2-fold axis Axis is always described by direction parallel (e.g. along a), whereas plane is described by direction normal (e.g. perpendicular to b) Symmetry element can be described by its type and general equation of its invariant points: mirror plane normal to b, located at y=1/2 is (x,1/2, z) 3-fold axis along to c, located at x=1/2, y=0 is (1/2,0, z) Transforming atom coordinates with symmetry operations Mirror plane All symmetry operations in crystals can be expressed by combination of rotation, translation and inversion. For example, a matrix describing a reflection in mirror plane perpendicular to a, located at x=0 (0,y,z) is: 1 0 0 0 1 0 0 0 1 (x,y,z) → (-x,y,z) Write transformation matrices and transformation formulae for a 2-fold and 4-fold axis, parallel to c, located at 1/2, 0, 0 (1/2, 0, z)? 1/2 +X,1/2 -Y, -Z Point groups (describe external morphology of crystals) Crystal System 32 Crystallographic Point Groups Triclinic 1 -1 Monoclinic 2 m 2/m Orthorhombic 222 mm2 mmm Tetragonal 4 -4 4/m 422 4mm Trigonal 3 -3 32 3m -3m Hexagonal 6 -6 6/m 622 6mm Cubic 23 m-3 432 -43m m-3m -42m 4/mmm -62m 6/mmm • A lattice is a regular array of points. Each point must have the same number of neighbors as every other point and the neighbors must always be found at the same distances and directions. • All points are in the same environment. • A Bravais Lattice is a three dimensional lattice, characterized by translation symmetry, which tiles space without any gaps or holes. • There are 14 ways in which this can be accomplished. Bravais Lattices contain seven crystal systems and four lattice centering types. Bravais lattice types Symmetry operations involving translation • • Screw axes always have translation along the direction parallel. Subscript denotes fraction of the translation period (e.g. 42 has a translation period of ½, 41 and 43 have a period of ¼, 62 has a period of 2/6). Translation period is never more than ½. Glide planes can have one (a, b, c) or two (n, d) translation directions. Translation is never along the direction perpendicular. The letter describing the plane type denotes the directions of translation (e.g. a has translation along x, b along y, c along z, n along two directions). All glide translations have a period of ¼, except for d, for which the period is ¼. Glide plane Screw axis What symmetry operation is described by the following transformation? (x,y,z)→(1/2 +x,1/2 -y, -z ) 21 (X, ¼, 0) • • • • • • • • All simple symmetry operations have invariant points (points that transform into themselves as a result of the operation). Examples: • mirror plane - all points on the plane • Rotation axis – all points along the axis • Inversion – the point where the inversion center is located An atom located on a symmetry element invariant point has special properties. Such position is referred to as “special position”. There are less independent parameters needed to describe an atom located on a special position. A position within unit cell which is not located on any symmetry element invariant point is called general position. Multiplicity of a position is the number of equivalent repetitions of this position within the unit cell. General positions have higher multiplicity than special positions. Symmetry elements involving translations do not have invariant points and do not generate special positions. General and special atomic positions Understanding space group symbol • Thre are 230 space groups in 3 dimensions. They were first enumerated by Fyodorov (1891), Barlow (1894) and Schönflies (1891). • Space group symbol consists of Capital letter describing the Bravais lattice type, and three groups of lower case letters/numbers describing symmetry elements in different symmetry independent directions Explain space group symbols: I-43m P-421c Pmc21 P3112 Crystal System Symmetry Direction Primary Secondary Tertiary Triclinic None Monoclinic [010] Orthorhombi c [100] [010] [001] Tetragonal [001] [100]/[010] [110] Hexagonal/ Trigonal [001] [100]/[010] [120]/[1 `1 0] [100]/[010]/ [001] [111] [110] Cubic Centrosymmetric, polar and chiral space groups • Crystals with centrosymmetric space groups (including inversion centers) exhibit some common special properties: do not have piezoelectric effect and do not cause second harmonic generation (frequency doubling) • Symmetry element that include inversion symmetry are: – – – -1 2k/m rotation axis+perpendicular plane -3 • Acentric space groups are also called polar. • The point groups that possess no improper (-3,-4,-6) rotations and no inversion centers are called enantiomorphic. Enantiomorphic molecules have right-handed and left-handed forms, although in nature usually one form strongly predominates over the other. An example of enantiomorphic mineral is quartz. Location of symmetry elements in the unit cell • • • • • Space group symbol equivocally defines the relative location of the symmetry elements. Symmetry elements repeat every half unit cell. There are certain conventions regarding the choice of origin in the unit cell (e.g. usually on an inversion center, if present). Most space groups can be represented in multiple settings (e.g. P21/c=P21/n). Only primary symmetry element are described in the space group symbol. There usually are additional symmetry elements that are consequences of the presence of the primary elements. Find coordinates of all equivalent general positions in the unit cell Thermal vibrations and atomic displacement parameters • • • • • • Atoms in crystals vibrate about their equilibrium positions. These vibrations are thermally activated and their amplitude increases with temperature. To account for “blurring” of electron density around atoms resulting from thermal vibrations crystallographers use atomic displacement parameters (ADP) describing radius of a sphere or shape and size of ellipsoid of highest probability of finding given atom. Because of anisotropy of interatomic interactions (e.g. bonding) the vibration amplitudes might be anisotropic as well. Isotropic ADP require one parameter per atom, anisotropic ADP require up to 6 parameters per atom (3 axial lengths and 3 parameter describing orientation of the ellipsoid). Number of independent anisotropic ADPs is modified by crystal and site symmetry. Atomic vibrations in the presence of rigid bonds can cause errors in determination of bond lengths using crystallographic methods. To account for this effect a TSL modeling (accounting for libraton) is used. Chemical substitution and site occupancy factors • • • • Atoms and ions of similar size (radius) and similar charge often substitute for each other in crystal structures, without changing crystal symmetry. This effect is know as solid solutions. It is also possible for atoms in the crystal lattice to be missing from their crystallographic sites, leading to formation of vacancy defects. If defects or substitutions show long-range order, the crystal symmetry changes. In the description of the crystal structure site occupancy factor account for the degree of filling of the given crystallographic site by the given atom type (1=full, 0=completely empty) averaged over the whole crystal. Software for visualization and analysis of crystal structures XtalDraw (free) http://www.geo.arizona.edu/xtal/group/software.htm PowderCell (free) http://www.ccp14.ac.uk/ccp/web-mirrors/powdcell/a_v/v_1/powder/e_cell.html Endeavour (commercial, 60 day free fully functional demo) http://www.crystalimpact.com/endeavour/download.htm