AutoDock 4 and AutoDock Vina
advertisement

AutoDock 4 and AutoDock Vina
-Brief Intruction
Guanglin Kuang
2014-3-25
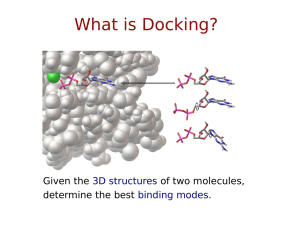
Docking
-wikipedia
• General definition:
In the field of molecular modeling, docking is a method which predicts the
preferred orientation of one molecule to a second when bound to each
other to form a stable complex. Knowledge of the preferred orientation in
turn may be used to predict the strength of association or binding affinity
between two molecules using for example scoring functions.
• Usage:
Docking is frequently used to predict the binding orientation of small
molecule drug candidates to their protein targets in order to in turn
predict the affinity and activity of the small molecule. Hence docking plays
an important role in the rational design of drugs, for example, hit
identification and lead optimization.
lock-and-key
Theoretical Background
Docking approaches:
Shape complementarity
Simulation
Search algorithm:
Systematic or Stochastic torsional searches about rotatable bonds
Genetic algorithms to "evolve" new low energy conformations
Molecular dynamics simulations
Structural flexibility:
Ligand flexibility
Receptor flexibility (Induced fit docking)
Scoring function:
Force field
Empirical
Knowledge -based
Popular Softwares:
DOCK, AUTODOCK, GOLD, Glide (Schrödinger), FlexX (Sybyl), MOE-Dock …
J. Med. Chem., 2004, 47 (12), pp 3032–3047
Nat Rev Drug Discov. 2004 Nov;3(11):935-49.
AutoDock 4.0
AutoDock is an automated procedure for
predicting the interaction of ligands with
biomacromolecular targets.
The current version of AutoDock, using the
Lamarckian Genetic Algorithm and empirical
free energy scoring function.
𝑽 = 𝑾𝒗𝒅𝒘
(
𝒊,𝒋
𝑨𝒊𝒋
𝒓𝟏𝟐
𝒊𝒋
−
𝑩𝒊𝒋
)+
𝒓𝟔𝒊𝒋
𝑾𝒉𝒃𝒐𝒏𝒅
𝒊,𝒋
𝒒𝒊 𝒒𝒋
+ 𝑾𝒆𝒍𝒆𝒄
𝒊,𝒋
𝜺 𝒓𝒊𝒋 𝒓𝒊𝒋
𝑪𝒊𝒋 𝑫𝒊𝒋
𝑬(𝒕)( 𝟏𝟐 − 𝟏𝟎 )
𝒓𝒊𝒋
𝒓𝒊𝒋
+ 𝑾𝒔𝒐𝒍
(𝑺𝒊 𝑽𝒋 + 𝑺𝒋 𝑽𝒊 )𝒆
𝒓𝟐𝒊𝒋
− 𝟐
𝟐𝝈
𝒊,𝒋
𝑳−𝑳
𝑷−𝑷
𝑷−𝑷
𝑷−𝑳
𝑷−𝑳
∆𝑮 = 𝑽𝑳−𝑳
𝒃𝒐𝒖𝒏𝒅 − 𝑽𝒖𝒏𝒃𝒐𝒖𝒏𝒅 + 𝑽𝒃𝒐𝒖𝒏𝒅 − 𝑽𝒖𝒏𝒃𝒐𝒖𝒏𝒅 + 𝑽𝒃𝒐𝒖𝒏𝒅 − 𝑽𝒖𝒏𝒃𝒐𝒖𝒏𝒅 + ∆𝑺𝒄𝒐𝒏𝒇
AutoDock calculations are performed in several
steps:
Preparation of coordinate files using
AutoDockTools
Recalculation of atomic affinities using AutoGrid
Docking of ligands using AutoDock,
Analysis of results using AutoDockTools.
Journal of Computational Chemistry, Vol. 19, No. 14, 1639]1662 (1998)
Journal of Computational Chemistry: Volume 30, Issue 16, pages 2785–2791, December 2009
http://autodock.scripps.edu/
AutoDock Vina
AutoDock 4 and AutoDock Vina were both
developed in the Molecular Graphics Lab at The
Scripps Research Institute. AutoDock Vina
inherits some of the ideas and approaches of
AutoDock 4 . They use the same type of structure
format (PDBQT) for maximum compatibility with
auxiliary software. However, the source code, the
scoring function and the actual algorithms used
are brand new.
Advantages (Compared to AutoDock 4.0):
Faster (can be used for virtual screening)
Ease of use
More accurate (arguably)
Disadvantages
The algorithm and scoring function is not as
straightforward and physical as that of AutoDock
4. Works more like a black-box
Journal of Computational Chemistry, Volume 31, Issue 2, pages 455–461, 30 January 2010
http://vina.scripps.edu/manual.html
Virtual Screening Using Vina
Step 1: Prepare the protein:
Use AutoDockTools
Step 2: Prepare the Ligands:
corina -d wh,rs,neu,r2d,errorfile=errors.sdf database.sdf database-3D.sdf
babel -isdf database-3D -opdb ligand.pdb -m
#! /bin/bash
for f in ligand*.pdb; do
b=`basename $f .pdb`
echo Processing ligand $b
mkdir -p $b
pythonsh $ADT_Utilities24/prepare_ligand4.py -l $f -o ${b}/${b}.pdbqt
done
conf.txt
receptor = protein.pdbqt
cpu = 8
Step 3: Virtual Screening
#! /bin/bash
for f in ligand*.pdb; do
b=`basename $f .pdb`
echo Processing ligand $b
vina --config conf.txt --ligand ${b}/${b}.pdbqt --out ${b}/${b}_out.pdbqt --log ${b}/${b}_log.txt
done
center_x = -2.0
center_y = 57.1
center_z = 52.3
size_x = 22.5
size_y = 22.5
size_z = 22.5
Step 4: Analysis
Find the top scored ligands: vina_screen_get_top.py
Analyze the binding modes with AutoDockTools
http://vina.scripps.edu/manual.html