by gene by genomic segment
advertisement

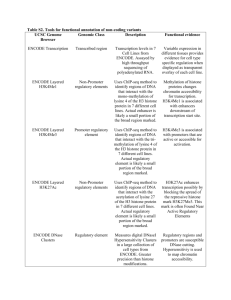
Monica C. Sleumer (苏漠) 2012-09-19 Human Genome • • • • • 3,101,804,739 base pairs 22 chromosomes plus X and Y 21,224 protein-coding genes 15,952 ncRNA genes 3–8% of bases are under selection – From comparative genomic studies • Question: What is the genome doing? Objectives • Find all functional elements – – – – Bound by specific proteins Transcribed Histone modifications DNA methylation • Use this information to annotate functional regions – – – – – – – Genes (coding and non-coding) Promoters Enhancers Specific transcription factor binding sites Silencers Insulators Chromatin states • Cross-reference data from other studies – Comparative genomics – 1000 Genomes Project – Genome-wide association studies (GWAS) ENCODE projects • ENCODE pilot project: 1% of the genome 2003-2007 • modENCODE: Drosophila and C. elegans • ENCODE main project 2007-2012 – – – – – 1649 dataset-generating experiments 147 cell types 235 antibodies and assay protocols 450 authors 32 institutes • 31 publications 2012-09-06 – – – – 6 in Nature 18 in Genome Research 6 in Genome Biology 1 in BMC Genetics www.nature.com/encode/category/research-papers Materials • 147 types of human cell lines, 3 priority levels • Tier 1 cell lines: top priority for all experiments Name Description Lineage Tissue Karyotype GM12878 B-lymphocyte, lymphoblastoid, Epstein-Barr Virus, mesoderm 1000 Genomes Project blood normal H1-hESC embryonic stem cells inner cell mass embryonic stem cell normal K562 leukemia, 53-year-old female with chronic myelogenous leukemia mesoderm blood cancer • Tier 2 cell lines to be done after Tier 1 (next slide) • Tier 3: any other cell lines Tier 2 Cell Lines Name Description Lineage lung carcinoma epithelium, 58-yearendoderm old caucasian male donor B cells: RO01778 and CD20+ mesoderm RO01794 CD20+_RO01778 B cells, caucasian mesoderm CD20+_RO01794 B cells, African American mesoderm neurons derived from H1 H1-neurons ectoderm embryonic stem cells HeLa-S3 cervical carcinoma ectoderm HepG2 hepatocellular carcinoma endoderm HUVEC umbilical vein endothelial cells mesoderm IMR90 fetal lung fibroblasts endoderm skeletal myoblasts from pectoralis LHCN-M2 mesoderm major muscle, 41 year old caucasian MCF-7 mammary gland, adenocarcinoma ectoderm MonocytesMonocytes-CD14+, leukapheresis mesoderm CD14+ from RO 01746 and RO 01826 SK-N-SH neuroblastoma, 4 year old ectoderm A549 Tissue Karyotype epithelium cancer blood normal blood blood normal normal neurons normal cervix liver blood vessel lung skeletal muscle myoblast breast cancer cancer normal normal monocytes normal brain cancer http://encodeproject.org/ENCODE/cellTypes.html cancer Methods RNA-Seq Different fractions of RNA -> sequencing CAGE 5’ Capped RNA sequencing RNA-PET Sequencing 5’ Cap plus poly-A tail ChIP-seq Chromatin immunoprecipitation of a DNA binding protein -> sequencing DNase-seq Cut exposed DNA with DNase I -> sequencing FAIRE-seq Nucleosome-depleted DNA -> sequencing RRBS Bisulphite treatment: unmethylated C->U -> sequencing 3C,5C, ChIA-PET Chromatin interactions -> sequencing Results: RNA Sequencing • 62% of the genome is transcribed into sequences >200 bp long – 5.5% of this is exon – 31% is intergenic – no annotated gene – Remaining: intronic • CAGE-seq: 62,403 TSS – 44% within 100bp of the 5’ end of a GENCODE gene – Others: exons and 3’ UTRs, significance unknown • Lots of short ncRNAs: tRNA, miRNA, snRNA etc. • Further description: Wu Dingming, 9:30 Results: Transcribed and protein-coding regions • GENCODE reference gene set – 20,687 Protein-coding • • • • 6.3 alternatively spliced transcripts on average 3.9 protein isoforms on average Protein-coding exons: 1.22% of the genome Still more to come: unidentified peptides in mass-spec – 18,441 ncRNA genes • 8801 short ncRNA • 9640 long nc RNA – 11,224 pseudogenes • 863 transcribed ChIP-Seq Acronym Description ChromRem ATP-dependent chromatin complexes DNA repair DNARep HISase Other Pol2 Factors analysed 5 3 Histone acetylation, 8 deacetylation or methylation complexes Cyclin kinase associated with 1 transcription Pol II subunit 1 (2 forms) Pol3 Pol III-associated 6 TFNS General Pol II-associated factor, not site-specific Pol II transcription factor with sequence-specific DNA binding 8 TFSS Total 87 119 www.illumina.com/technology/chip_seq_assay.ilmn ChIP-Seq: Histone modifications Histone modification or Signal variant characteristics H2A.Z Peak Association dynamic chromatin H3K4me1 Peak/region H3K4me2 Peak enhancers and other distal elements, also downstream of transcription starts promoters and enhancers H3K4me3 Peak promoters/transcription starts H3K9ac Peak promoters H3K9me1 Region 5′ end of genes H3K9me3 Peak/region H3K27ac Peak Gene repression, constitutive heterochromatin and repetitive elements Gene expression, active enhancers and promoters H3K27me3 Region H3K36me3 Region H3K79me2 Region polycomb complex, repressive domains and silent developmental genes Elongation, transcribed portions of genes, 3′ regions after intron 1 Transcription, 5′ end of genes H4K20me1 Region 5′ end of genes Results: ChIP-Seq • 636,336 binding regions • 8.1% of the genome • Sequence-specific TF ChIP-seq: – 86% of the DNA segments occupied by sequencespecific transcription factors contained a strong DNA-binding motif – 55% cases contained the expected motif • Further description: Qin Zhiyi & Ma Xiaopeng, 13:30 DNase I hypersensitivity • • • • • 2,890,000 unique hypersensitive sites (DHSs) 4,800,000 sites across 25 cell types Tier 1 and tier 2 cell types: 205,109 DHSs per cell type 98.5% of ChIP-seq TFBS within DHSs Further description: Guo Weilong 12:30, He Chao 14:30 https://www.nationaldiagnostics.com/electrophoresis/article/dnase-i-footprinting FAIRE-seq • Like the opposite of ChIP-seq • Cross-link the nucleosomes to the DNA – But not the sequence-specific TFs • Shear the DNA into small pieces • Remove the protein-bound DNA • Sequence the non-bound DNA Gaulton KJ et al, Nature Genetics 42, 255–259 (2010) doi:10.1038/ng.530 DNA methylation • CpG methylation: regulates gene expression – In promoters: gene repression – In genes: gene transcription • 1,200,000 methylated CpGs in 82 cell lines and tissues – 96% differentially methylated, especially those in genes • Unmethylated genic CpG islands associated with P300 binding , an enhancer-related histone acetyltransferase • Allele-specific methylation: genomic imprinting • Aberrant methylation in cancer cell lines • Reproducible methylation outside CpG dinucleotides http://www.diagenode.com/en/applications/bisulfite-conversion.php Chromosome conformation capture Montavon and Duboule, Trends in Cell Biology (2012) 22:7, 347–354 Results: Chromosome interactions • Chromosome conformation capture (3C) : – 5C: 3C-carbon copy – ChIA-PET • Identified 127,417 promoter-centred chromatin interactions using ChIA-PET – 98% intra-chromosomal • 2,324 promoters involved in ‘single-gene’ enhancer– promoter interactions • 19,813 promoters were involved in ‘multi-gene’ interaction complexes spanning up to several megabases • 50–60% of long-range interactions occurred in only one of the four cell lines • Further discussion: Li Yanjian, 10:40 Primary Findings • 80.4% of the human genome is doing at least one of the following: – Bound by a transcription factor – Transcribed – Modified histone • 99% is within 1.7 kb of at least one of the biochemical events • 95% within 8 kb of a DNA–protein interaction or DNase I footprint • 7 chromatin states: – 399,124 enhancer-like regions – 70,292 promoter-like regions • Correlation between transcription, chromatin marks, and TF binding • Functional regions contain lots of SNPs – Disease-associated SNPs in non-coding regions tend to be in functional elements End of Introduction Summary of ENCODE elements • 80.4% of the human genome is covered by at least one ENCODE-identified element • 62% of the genome is transcribed • 56% of the genome associated with histone modifications • Excluding RNA elements and broad histone elements, 44.2% of the genome is covered – open chromatin (15.2%) – transcription factor binding (8.1%) – 19.4% DHS or transcription factor ChIP-seq peaks across all cell lines • 8.5% of bases are covered by either a transcription-factorbinding-site motif (4.6%) or a DHS footprint (5.7%) – 4.5x the amount of protein-coding exons (1.2%) – 2x the amount of conserved sequence between mammals • Estimate: 50% of DHS remain to be found – Based on saturation curves Diversity Diversity vs Conservation: Interactive Figure Conservation A high-resolution map of human evolutionary constraint using 29 mammals Nature 478, 476–482 (2011) Diversity Conservation in Bound Motifs vs Unbound Motifs Conservation http://www.nature.com/encode/interactive-figures/nature11247_F1 Model of gene expression – histone marks Model of gene expression – TF binding Transcription factor co-associations Seven major classes of genome states CTCF CTCF-enriched element CTCF signal , no histone modifications, open chromatin, may have insulator function, enriched for cohesin components RAD21 and SMC3 E Predicted enhancer Open chromatin, H3K4me1, other enhancer-associated marks, enriched for EP300, FOS, FOSL1, GATA2, HDAC8, JUNB, JUND, NFE2, SMARCA4, SMARCB1, SIRT6 and TAL1 sites, nuclear and whole-cell RNA poly(A) signal PF Predicted promoter flanking Regions that generally surround TSS segments R Predicted repressed H3K27me3 polycomb-enriched regions, REST, BRF2, CEBPB, MAFK, TRIM28, ZNF274 and SETDB1 sites or no signal at all TSS Predicted promoter including TSS H3K4me3, open chromatin, Pol II, Pol III, short RNAs, close to TSS sites T Predicted transcribed H3K36me3 transcriptional elongation signal., overlap with gene bodies, phosphorylated Pol II , cytoplasmic poly(A)+ RNA WE Weak enhancer Similar to the E state, but weaker signals and weaker enrichments. Data integration and genome segmentation Enhancer Transcribed Repressed TSS RNA expression Transcription factors Association between genome states and annotations Genome segment Genome segment Enhancer validation in mouse and fish Enhancer from K562 cell (leukemia) drives basal promoter with reporter gene in embryonic mouse blood cells and medaka fish Genome segment clustering 6 cell types Genome cluster function Genome state is related to gene function Allele-specific expression Pol II Txn Rpn Correlation of allele-specific signal by gene by genomic segment Genome-wide association studies Annotated diseasecausing SNPs Control SNPs Selected TFBS tracks Significant overlap Diseases No genes, but several TFBS near the disease-causing SNPs Conclusions • 80% of human genome annotated with at least one association – Protein-binding – Histone modification – Transcription • ENCODE data combination – Model gene expression – Genome segmented into 7 types • Different in each cell line • ENCODE data combined with other data – 1000 genomes: see influence of parental DNA – Genome-wide association studies Discussion • 147 types of cells, and the human body has a few thousand • 80% functional : controversial – 80% of the genome is being transcribed and/or has a protein bound to it some of the time – Heterochromatin: tightly packed repeat sequences – most of that activity isn’t particularly specific or interesting and may not have impact – Important not to overstate the findings – Ewan Birney: “cumulative occupation of 8% of the genome by TFs” • Reproducibility – In exactly the same cell lines, same conditions, different time or place – Same cell lines, different conditions – Same cell type, different people • Cell lines vs tissue • Cancer vs normal http://blogs.nature.com/news/2012/09/fighting-about-encode-and-junk.html http://blogs.discovermagazine.com/notrocketscience/2012/09/05/encode-the-rough-guide-to-the-human-genome/ Applications • Visible as genome tracks in UCSC • Mutation from – Cancer sequencing – GWAS – Find out what that part of the genome is doing • Compare with your cancer data (RNA-seq) • Comparative genome analysis • Gene or pathway of interest Online Resources • Interactive graphics in online version of paper • Interactive app on Nature ENCODE main page www.nature.com/encode/