Practice Problem
advertisement

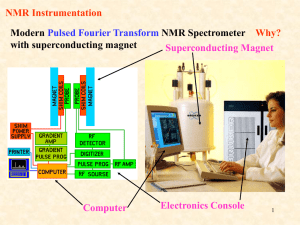
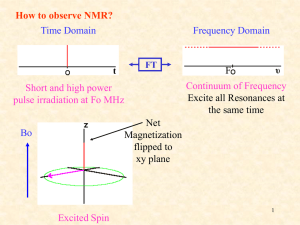
Chapter 13 Introduction • Mass Spectrometry (MS) – determines the size and formula • Infrared (IR) Spectroscopy – determines the kinds of functional groups present • Nuclear Magnetic Resonance Spectroscopy (NMR) – – determines the carbonhydrogen framework • Ultraviolet Spectroscopy (UV) – determines if a conjugated p electron system is present The Use of NMR Spectroscopy • It is used to determine relative location of atoms within a molecule • It is the most helpful spectroscopic technique in organic chemistry • It is related to Magnetic Resonance Imaging (MRI) in medicine • It maps carbon-hydrogen framework of molecules • It depends on very strong magnetic fields 1. Nuclear Magnetic Resonance Spectroscopy (NMR) • Nuclear Magnetic Resonance Spectroscopy (NMR) – is a technique used to map the carbon-hydrogen framework – It detects the energy absorption accompanying the transition between nuclear spin states that occurs when a molecule is placed in a strong magnetic field and irradiated with radiofrequency waves. • The 1H and 13C nuclei: – have spins (i.e behave as if they were spinning about an axis) – are magnetic (i.e interact with an external magnetic field, Bo) • Other magnetic nuclei include: – all nuclei with an odd number of protons (1H, 2H, 14N, 19F, 31P,...) – all nuclei with an odd number of neutrons (13C) • Nonmagnetic nuclei include: – nuclei with even numbers of protons and neutrons (12C, 16O,…) • In the absence of an external magnetic field, magnetic nuclear spins are oriented randomly • In the presence of an external field, Bo, magnetic nuclear spins orient with (parallel to) or against (antiparallel to) the external field. parallel antiparallel • The parallel spin state (orientation) is slightly lower in energy and therefore favored (more populated). parallel antiparallel • When the oriented nuclei are irradiated with proper electromagnetic radiation frequency, energy is absorbed and the nuclei “spin-flip” from the lower-energy state (parallel) to the higher-energy state (antiparallel) Magnetic nuclei are said to be in resonance with the applied radiation (hence the name of nuclear magnetic resonance). The absorption of energy is detected, amplified and displayed as a nuclear magnetic resonance spectrum • The exact frequency necessary for resonance depends on: – the strength of the external magnetic field – the identity of the nuclei • The exact frequency necessary for resonance is proportional to field strength, Bo. – In the absence of an applied magnetic field, spin states have equal energies. In the presence of a magnetic field (Bo), spin states have unequal energies – If a very strong magnetic field is applied, the energy difference between the two spin states (DE) is larger, and higher-frequency (higher-energy) radiation is required for a spin-flip. Practice Problem: The amount of energy required to spin-flip a nucleus depends both on the strength of the external magnetic field and on the nucleus. At a field strength of 4.7 T, rf energy of 200 MHz is required to bring a 1H nucleus into resonance , but energy of only 187 MHz will bring a 19F nucleus into resonance. Use the equation below to calculate the amount of energy required to spin-flip a 19F nucleus. Is this amount greater or less than that required to spin-flip a 1H nucleus? E= 1.20 X 10-4 kJ/mol . l (m) and n= c . l Practice Problem: Calculate the amount of energy required to spin-flip a proton in a spectrometer operating at 300 MHz. Does increasing the spectrometer frequency from 200 to 300 MHz increase or decrease the amount of energy necessary for resonance? 2. The Nature of NMR Absorptions • The absorption frequency is not the same for all 1H or 13C nuclei in a molecule. • All nuclei in molecules are surrounded by electrons with their own magnetic fields Blocal • When an external field is applied to a molecule, the effective field (Beffective) felt by the nucleus is a bit smaller than the applied field (Bapplied) : Beffective = Bapplied - Blocal • Electrons setting up local magnetic fields shield nearby nuclei from the full effect of the applied field Nuclei are shielded from the full effect of the applied field • Since each specific nucleus in a molecule is shielded to a slightly different extent, the effective magnetic field is not the same for each nucleus: A distinct NMR signal for each chemically distinct nucleus in a molecule can be detected. 13C or 1H Different signals appear for nuclei in different environments Nuclear Magnetic Resonance (NMR) Spectrum • NMR spectrum – plots the effective field strength felt by the nuclei (x-axis) versus the magnitude or intensity of NMR resonance signals (corresponding to the intensity of rf energy) (y-axis) – The intensity of NMR resonance signals is proportional to the molar concentration of the sample. – Each peak in the NMR spectrum corresponds to a chemically distinct 1H or 13C nucleus in the molecule – 1H or 13C spectra cannot be observed on the same spectrometer because of the difference in energy required to spin-flip the nuclei 1H NMR Spectrum 13C NMR Spectrum • Chemically equivalent nuclei – are shielded to the same extent (i.e they have the same electronic environment) – always show a single absorption • Example: Methyl Acetate The 3H’s on –OCH3 are equivalent The 3H’s on CH3C=O are equivalent NMR Spectrometer • The Operation of a typical NMR spectrometer is illustrated: The NMR Measurement • The sample is dissolved in a solvent that does not have a signal itself and placed in a long thin tube • The tube is placed within the gap of a magnet and spun • Radiofrequency energy is transmitted and absorption is detected • If two species interconverting faster than 103 times per second are present in a sample, NMR records only a single, averaged spectrum, rather than separate spectra of the two distinct species: “blurring effect” • Species that interconvert give an averaged signal that can be analyzed to find the rate of conversion • Example: Cyclohexane Practice Problem: 2-Chloropropene shows signals for three kinds of protons in its 1H NMR spectrum. Explain 3. Chemical Shifts • Chemical shift - is the relative energy of resonance of a particular nucleus resulting from its local environment - is the position on the NMR chart where a nucleus absorbs • NMR spectra show the applied field strength increasing from left to right: – Left part is downfield (or low-field) – Right part is upfield (or high-field) • NMR spectra show the applied field strength increasing from left to right: – Nuclei that absorb on the downfield side are weakly shielded – Nuclei that absorb on the upfield side are strongly shielded • NMR chart is calibrated versus a reference point, set as 0, tetramethylsilane [TMS] – TMS is used as a reference for both 1H and 13C Measuring Chemical Shift • Numeric value of chemical shift – is the difference between strength of magnetic field at which the observed nucleus resonates and field strength for resonance of a reference – Difference is very small but can be accurately measured – Taken as a ratio to the total field and multiplied by 106 so the shift is in parts per million (ppm) • Absorptions normally occur downfield of TMS, to the left on the chart Measuring Chemical Shift • NMR charts are calibrated using an arbitrary scale called the delta scale (d), where 1d = 1 ppm (part per million) of spectrometer frequency – d is the number of parts per million (ppm) of the magnetic field expressed as the spectrometer’s operating frequency (it is a ratio and not a unit) d= Observed chemical shift (number of Hz away from TMS) Spectrometer frequency in MHz Measuring Chemical Shift • The chemical shift of an NMR absorption in d units is constant, regardless of the operating frequency of the spectrometer – It is independent of instrument’s field strength – Example: A 1H nucleus at d = 2.0 ppm on a 60 MHz instrument also absorbs at d = 2.0 ppm on a 300 MHz. Measuring Chemical Shift • Interpretation of a spectrum is easier with higher operating spectrometer frequency (less likelihood of signal overlap) For 60 MHz instrument, 1d = 60 Hz For 300 MHz instrument, 1d = 300 Hz • Absorptions normally occur downfield of TMS: – Almost all 1H NMR absorptions occur 0-10 ppm from TMS – Almost all 13C NMR absorptions occur 0-220 ppm from TMS. Practice Problem: When the 1H NMR spectrum of acetone, CH3COCH3, is recorded on an instrument operating at 200 MHz, a single sharp resonance at 2.1 d is seen. a) How many hertz downfield from TMS does the acetone resonance correspond to? b) If the 1H NMR spectrum of acetone were recorded at 500 MHz, what would be the position of the absorption in d units? c) How many hertz downfield from TMS does this 500 MHz resonance correspond to? Practice Problem: The following 1H NMR peaks were recorded on a spectrometer operating at 200 MHz. Convert each into d units. a) CHCl3; 1454 Hz b) CH3Cl; 610 Hz c) CH3OH; 693 Hz d) CH2Cl2; 1060 Hz] 4. 13C NMR Spectroscopy: Signal Averaging and FT-NMR • Carbon-13, 13C – is the only naturally occurring carbon isotope with a nuclear spin – Natural abundance is only 1.1% of C’s in molecules – Sample is thus very dilute in this isotope • Two techniques have been developed to detect the 13C isotope in an organic sample by NMR: – Signal averaging (increases instrument sensitivity) – Fourier-transform NMR (increases instrument speed) • Signal averaging: increases instrument sensitivity – Any individual 13C NMR spectrum is extremely “noisy”, but when hundreds of individual runs are added together by computer and then averaged, a greatly improved spectrum results. • Fourier-transform NMR: increases instrument speed – In the FT-NMR technique, all signals are recorded simultaneously – The sample placed in a magnetic field of constant strength is irradiated with a short burst of rf energy. – All 1H and 13C in the sample resonate at once, and the complex composite signal is manipulated using so-called Fourier transforms before it can be displayed. Large amount of noise A single run A average of 200 runs 5. Characteristics of 13C NMR Spectroscopy • The carbon NMR spectrum of a compound provides the number of different types of electronic environments of carbon atoms in a molecule – It displays a single sharp signal for each chemically distinct 13C nucleus in the molecule. – Most 13C resonances are between 0 to 220 ppm downfield from TMS – 13C NMR spectrum displays a single sharp signal for each chemically distinct 13C nucleus in the molecule. • Example: Methyl Acetate The C on –OCH3 The C of C=O The C on –CH3C=O Correlation of Environment with Chemical Shift • Most 13C resonances are between 0 to 220 ppm downfield from TMS • The exact chemical shift of each 13C depends on its electronic environment: – Electronegativity of nearby atoms – Hybridization of 13C atom – Electronegativity of nearby atoms: C bonded to O, N, or halogen absorb downfield because O, N, or halogen pull electrons away from nearby 13C atoms, decreasing their electron density and “deshielding” them. – Hybridization of 13C atom: – sp3 C signal is in the range 0-90 d – sp2 C signal is in the range 110-220 d – C=O signal is at the low-field end, in the range 160-220 d C=O signal is at the low-field end, in the range 160-220 d 2-butanone para-bromo acetophenone Example: Only six C absorptions with different peak sizes Practice Problem: Identify the different 13C NMR absorptions Five absorptions: 14.1, 60.5, 128.5, 130.3, and 166.0 d Practice Problem: Predict the number of carbon resonance lines you would expect in the 13C NMR spectra of the following compounds: a) Methyl cyclopentane b) 1-Methylcyclohexene c) 1,2-Dimethylbenzene d) 2-Methyl-2-butene Practice Problem: Propose structures for compounds that fit the following descriptions: a) A hydrocarbon with seven lines in its 13C NMR spectrum b) A six-carbon compound with only five lines in its 13C NMR spectrum c) A four-carbon compound with three lines in its 13C NMR spectrum Practice Problem: Assign the resonances in 13C NMR spectrum of methyl propanoate, CH3CH2CO2CH3 1 2 3 4 6. DEPT 13C NMR Spectroscopy • DEPT-NMR (distortionless enhancement by polarization transfer) – is one of the improved pulsing and computational methods that give additional information – can help distinguish among signals due to CH3, CH2, CH, and C – determines the number of H’s bonded to each C A DEPT experiment is done in three stages: • First stage: is to run an ordinary spectrum (called a broadband-decoupled spectrum): – It locates the chemical shifts of all C’s • Second stage: is called a DEPT- 90 run: – only signals due to CH carbons appear • Third stage: is called DEPT- 135 or INEPT (insensitive nuclear enhancement by polarization transfer) run: – positive signals for CH3 and CH carbons – negative signals for CH2 carbons. – zero signal for carbons having no hydrogen (C) • Information from all three spectra can be used to determine the number of protons attached to each carbon. Example: DEPT-NMR spectrum for 6-methyl-5-hepten-2-ol CH3 CH3 CH3 Broadband-decoupled spectrum C 6 DEPT- 90 spectrum 5 2 CH CH 4 8371 DEPT- 135 spectrum CH2 CH2 Practice Problem: Assign a chemical shift to each carbon in 6methyl-5-hepten-2-ol Practice Problem: Estimate the chemical shift of each carbon in the following molecule. Predict which carbons will appear in the DEPT-90 spectrum, which will give positive peaks in the DEPT-135 spectrum, and which will give negative peaks in the DEPT-135 spectrum. Practice Problem: Propose a structure for an aromatic hydrocarbon, C11H16, that has the following 13C NMR spectral data. Broadband decoupled 13C NMR: 29.5, 31.8, 50.2, 125.5, 127.5, 130.3, 139.8 d DEPT-90: 125.5, 127.5, 130.3 d DEPT-135: positive peaks at 29.5, 125.5, 127.5, 130.3 d negative peak at 50.2 d 7. • 13C Uses of 13C NMR Spectroscopy NMR spectroscopy - provides useful information for structure determination which cannot be provided by infrared spectroscopy or mass spectroscopy • Example: Does the E2 elimination of alkyl halide, 1-chloromethyl cyclohexane, with strong base give the more highly substituted alkene, 1-methylcyclohexene (Zaitsev’s product) or methylenecyclohexane? Five sp3 C peaks Two sp2 C peaks Three sp3 C peaks Two sp2 C peaks • Example: Difference in symmetry of products is directly observed in the spectrum. The spectrum of the reaction product clearly identifies 1-methylcyclohexene as the product formed in the E2 reaction. Five sp3 C peaks (d 20-50) Two sp2 C peaks (d 100-150) Practice Problem: Addition of HBr to a terminal alkyne leads to the Markovnikov addition product, with the Br bonding to the more highly substituted carbon. How could you use 13C NMR to identify the product of the addition of 1 equivalent of HBr to 1-hexyne? 8. • 1H 1H NMR Spectroscopy and Proton Equivalence NMR spectroscopy – is much more sensitive than 13C because the active nucleus (1H) is nearly 100 % of the natural abundance – shows how many kinds of nonequivalent hydrogens are present in a compound • A quick look at the structure is usually enough to decide how many kinds of nonequivalent protons are present in a molecule. • Example: Methyl Acetate The 3H’s on –OCH3 are equivalent The 3H’s on CH3C=O are equivalent • The nonequivalence of two H’s can be determined by seeing if replacing each H with “X” gives the same or different structure – Equivalent H’s have the same signal while nonequivalent H’s have different signals – There are degrees of nonequivalence Nonequivalent H’s • Replacement of each H with “X” gives a different constitutional isomer – The H’s are in constitutionally heterotopic environments – They have different NMR absorption (d) – They are nonequivalent under all circumstances Equivalent H’s • Replacement of either H with “X” gives the same structure – The H’s are in homotopic (identical) environments – They have same NMR absorption (d) – They are equivalent Enantiotopic Distinctions • Replacement of the pro-R or pro-S H with “X” gives a different enantiomer – The H’s are in enantiotopic (mirror images of each other) environments – They have same NMR absorption (d) – They are electronically equivalent Diastereotopic Distinctions • In chiral molecules, replacement of the pro-R or pro-S H with “X” gives a different diastereomer – The H’s are in diastereotopic environments – They will have different NMR absorption (d) – They are chemically and electronically nonequivalent Practice Problem: Identify the indicated sets of 1H’s as unrelated, homotopic, enantiopic, or diasterotopic (a) Enantiotopic (d) Diastereotopic (b) Diastereotopic (e) Diastereotopic (c) Diastereotopic (f) Homotopic Practice Problem: How many kinds of electronically nonequivalent protons are present in each of the following compounds, and thus how many NMR absorptions might you expect in each? a) CH3CH2Br b) CH3OCH2CH(CH3)2 c) CH3CH2CH2NO2 d) Methylbenzene e) 2-Methyl-1-butene f) cis-3-Hexene Practice Problem: How many absorptions would you expect the following compound to have in its 1H NMR spectrum? 9. Chemical Shifts in 1H NMR Spectroscopy • Chemical shifts (d) are due to Blocal : – Nuclei that are strongly shielded require higher Bapplied and thus absorb on the upfield side – Nuclei that are weakly shielded require lower Bapplied and thus absorb on the downfield side • Most 1H chemical shifts fall within the range 0-10 d. • Most 1H chemical shifts fall within the range 0-10 d. – H’s attached to sp3 C have higher field signals – H’s attached to sp2 C have lower field signals – H’s on C bonded to electronegative atoms (O, N, X) absorb at the low-field end Saturated d 0-1.5 Allylic d 1.5-2.5 Y = O, N, X d 2.5-4.5 Cl Vinylic d 4.5-6.5 Aromatic d 6.5-8.0 Practice Problem: Each of the following compounds has a single 1H NMR peak. Approximately where would you expect each compound to absorb? a) Cyclohexane b) CH3COCH3 c) Benzene d) Glyoxal e) CH2Cl2 f) (CH3)3N O O II II H—C—C—H Practice Problem: Identify the different kinds of nonequivalent protons in the following molecule, and tell where you would expect each to absorb: 10. Integration of 1H NMR Absorptions: Proton Counting • In the 1H NMR spectrum, the relative intensity of a signal (integrated area) is proportional to the number of protons causing the signal – This information is used to deduce the structure – Integrated areas are presented as a “stair-step” line – For narrow peaks, the heights are the same as the areas and can be measured with a ruler • Example: In ethanol (CH3CH2OH), the signals have the integrated ratio 3:2:1 • Example: In methyl 2,2-dimethylpropanoate, the signals have the integrated ratio 1:3 Practice Problem: How many peaks would you expect in the 1H NMR spectrum of p-xylene? What ratio of peak areas would you expect on integration of the spectrum? Refer to Table 13.3 for approximate chemical shifts, and sketch what the spectrum would look like. Note: Aromatic rings have two resonance forms. Practice Problem: Identify the number of peaks and the ratio of peak areas on integration of the spectrum 2.4-2.7 d (6H’s) 6.5-8.0 d (4H’s) Two peaks 3:2 11. Spin-Spin Splitting in 1H NMR Spectra • Spin-spin splitting is when peaks often split into multiple peaks due to interactions between nonequivalent protons on adjacent carbons – The set of peaks is called a multiplet (2 = doublet, 3 = triplet, 4 = quartet) – Coupling is the interaction of the spins of nearby nuclei. • Example: Bromoethane (CH3CH2Br) The -CH2Br protons have four peaks (quartet) at 3.42 d The -CH3 protons have three peaks (triplet) at 1.68 d • Coupling is the interaction of the spins of nearby nuclei. – The tiny magnetic field produced by one nucleus (Bnucleus 1) affects the magnetic field felt by neighboring nuclei (Beffective 2) – Nuclear spins can have two possible alignments with respect to the magnetic field (can align either with or against the applied field) Example: The origin of spin-spin splitting in bromoethane • The -CH3 protons in bromoethane are neighbored by two magnetic -CH2Br protons • Each -CH2Br proton has its own nuclear spin which can align either with or against the applied field, affecting the -CH3 protons • These spin states are represented as 1:2:1 triplet Example: The origin of spin-spin splitting in bromoethane • The three different spin alignments of the two -CH2Br protons are: a. Both proton spins align with Bapplied, Beffective is larger; Bapplied is reduced (downfield) b. One proton spin aligns with Bapplied, and one proton spin aligns against Bapplied, no effect c. Both proton spins align against Bapplied, Beffective is smaller; Bapplied is increased (upfield) a b c Example: The origin of spin-spin splitting in bromoethane • The -CH2Br absorption is split into a 1:3:3:1 quartet • There are four possible combinations of the three spins of -CH3 protons • The “n + 1 rule” states that: – Protons that have n equivalent neighboring protons show n + 1 peaks in their NMR spectrum – The splitting is into one more peak than the number of H’s on the adjacent carbon • Example: 2-bromopropane The -CHBr proton is split by 6 equivalent -CH3 protons (n = 6): a septet The 6 equivalent -CH3 protons are split by 1 -CHBr proton (n = 1): a doublet n = 6 leads to 6+1 = 7 peaks n = 1 leads to 1+1 = 2 peaks Integration 6:1 • The distance between peaks in a multiplet is called the coupling constant, denoted J – It is measured in Hz and is in the 0-18 Hz range – It is dependent on the molecular geometry but independent of the spectrometer field strength • Some coupling patterns and relative intensities of lines: – If two multiplets have the same coupling constant (J), the H’s causing those multiplets are adjacent in the molecule – The relative intensities are in proportion of a binomial distribution Rules for Spin-Spin Splitting1 1. Chemically equivalent protons do not show spin-spin splitting. The equivalent protons may be on the same carbon or on different carbons Rules for Spin-Spin Splitting2 2. The signal of a proton that has n equivalent neighboring protons is split into a multiplet of n+1 peaks with coupling constant J. Protons that are farther than two carbon atoms apart don’t usually couple, although they sometimes show small coupling when they are separated by a p bond. Rules for Spin-Spin Splitting3 3. Two groups of protons coupled to each other have the same coupling constant J. Rules for Spin-Spin Splitting 1. Chemically equivalent protons do not show spinspin splitting. 2. The signal of a proton that has n equivalent neighboring protons is split into a multiplet of n+1 peaks with coupling constant J. 3. Two groups of protons coupled to each other have the same coupling constant J. • Example: para-methoxypropiophenone The -OCH3 protons are unsplit (n = 0): a singlet Each of two types of equivalent Ar-H protons is split by its neighbor (n = 1): a doublet The 2 equivalent O=CCH2 protons are split by 3 –CH3 protons (n = 3): a quartet and vice-versa, a triplet Why is there no splitting of carbon signals into mutiplets in 13C NMR? • No coupling of a 13C nucleus with nearby carbons and/or hydrogens is seen because: – The low natural abundance makes it unlikely that two 13C nuclei will be adjacent – The method used for 13C spectrum, broadband decoupling, cancels the 1H’s magnetic fields • Example: Propose a structure for C5H12O with the following 1H NMR data: – – – – 0.92 d (3H, triplet, J = 7Hz) 1.20 d (6H, singlet) 1.50 d (2H, quartet, J = 7Hz) 1.64 d (1H, broad singlet) Degree of unsaturation = ((2x5+2)-12)/2 = 0 The compound is saturated, either an alcohol or an ether Practice Problem: Predict the splitting patterns you would expect for each proton in the following molecules: Practice Problem: Draw structures for compounds that meet the following descriptions: a) C2H6O; one singlet b) C3H7Cl; one doublet and one septet c) C4H8Cl2O; two triplets d) C4H8O2; one singlet, one triplet, and one quartet Practice Problem: Propose a structure for a compound with a formula C4H10O consistent with the following 1H NMR spectrum Degree of unsaturation = ((2x4+2)-10)/2 = 0 The compound is saturated, either an alcohol or an ether CH3CH2OCH2CH3 One quartet 3.5d One triplet 1.2d 12. More Complex Spin-Spin Splitting Patterns • Spectra can be more complex due to: – overlapping signals – multiple nonequivalence • The n + 1 rule correctly predicts splitting caused by equivalent protons but not by nonequivalent protons – Splittings caused by nonequivalent protons are more complex. • Example: Toluene The 5 Ar-H protons are not equivalent but give a complex overlapping pattern • Example: Trans-cinnamaldehyde The 5 Ar-H proton signals overlap into a complex pattern The O=CH proton is split by its neighbor (n = 1): a doublet The =CH proton is split by its neighbor (n = 1): a doublet The =CH proton is split by two nonequivalent protons: a doublet of doublets • A tree diagram for the C2 proton of trans-cinnamaldehyde shows how it is coupled to the C1 and C3 protons with different coupling constants The peaks nearer the signal of the coupled partner are always larger; those farther from the signal are always smaller Practice Problem: 3-Bromo-1-phenyl-1-propene shows a complex NMR spectrum in which the vinylic proton at C2 is coupled with both the C1 vinylic proton (J = 16 Hz) and the C3 methylene protons (J = 8 Hz) Draw a tree diagram for the C2 proton signal. Explain the five-line multiplet observed. J 1-2 = 16 Hz J 2-3 = 8 Hz 13. Uses of 1H NMR Spectroscopy • 1H NMR spectroscopy- is a technique used to identify likely products in the laboratory quickly and easily • Example: Does the hydroboration/oxidation of methylenecyclohexane yield cyclohexylmethanol or 1-methylcyclohexanol? • Example: The 1H NMR spectrum clearly identifies the reaction product. 2-proton triplet (d 3.40) 3-proton singlet (d 1) Practice Problem: How could you use 1H NMR to determine the regiochemistry of electrophilic addition to alkenes? For example, does addition of HCl to 1-methylcyclohexene yield 1-chloro-1methylcyclohexane or 1-chloro-2methylcyclohexane? Chapter 13 Practice Problem: The compound whose 1H NMR spectrum is shown has the molecular formula C3H6Br2. Propose a structure. Practice Problem: The compound whose 1H NMR spectrum is shown has the molecular formula C4H7O2Cl has an I.R absorption peak at 1740 cm-1. Propose a structure. Practice Problem: Assign as many of the resonances as you can to specific carbon atoms in the 13C NMR spectrum of ethyl benzoate. Practice Problem: The 1H and 13C NMR spectra of compound A, C8H9Br, are shown. Propose a structure for A, and assign peaks in the spectra to your structure