HNO 3
advertisement

Simulating materials with atomic detail at
IBM: From biophysical to high-tech
applications
Application Team
Glenn J. Martyna,
Dennis M. Newns,
Jason Crain,
Andrew Jones,
Razvan Nistor,
Ahmed Maarouf,
Marcelo Kuroda,
Physical Sciences Division, IBM Research
Physical Sciences Division, IBM Research
School of Physics, Edinburgh University
School of Physics, Edinburgh University
Physical Sciences Division, IBM Research
Physical Sciences Division, IBM Research and EGNC
Physical Sciences Division, IBM and CS, UIUC
Methods/Software Development Team
Glenn J. Martyna,
Physical Sciences Division, IBM Research
Mark E. Tuckerman,
Department of Chemistry, NYU.
Laxmikant Kale,
Computer Science Department, UIUC
Ramkumar Vadali,
Computer Science Department, UIUC
Sameer Kumar,
Computer Science, IBM Research
Eric Bohm,
Computer Science Department, UIUC
Abhinav Bhatele*,
Computer Science Department, UIUC
Ramprasad Venkataraman, Computer Science Department, UIUC
*George Michael Memorial High Performance Computing Fellows for 2009
Funding : NSF, IBM Research, ONRL
IBM’s Blue Gene/L network torus
supercomputer
Our fancy apparatus.
Goal : The accurate treatment of complex heterogeneous
systems to gain physical insight.
Characteristics of current models
Empirical Models: Fixed charge,
non-polarizable,
pair dispersion.
Ab Initio Models: GGA-DFT,
Self interaction present,
Dispersion absent.
Problems with current models (empirical)
Dipole Polarizability : Including dipole polarizability changes
solvation shells of ions and drives them to the surface.
Higher Polarizabilities: Quadrupolar and octapolar
polarizabilities are NOT SMALL.
All Manybody Dispersion terms: Surface tensions and bulk
properties determined using accurate pair potentials are
incorrect. Surface tensions and bulk properties are both
recovered using manybody dispersion and an accurate pair
potential. An effective pair potential destroys surface
properties but reproduces the bulk.
Force fields cannot treat chemical reactions:
Problems with current models (DFT)
• Incorrect treatment of self-interaction/exchange:
Errors in electron affinity, band gaps …
• Incorrect treatment of correlation: Problematic
treatment of spin states. The ground state of transition
metals (Ti, V, Co) and spin splitting in Ni are in error.
Ni oxide incorrectly predicted to be metallic when
magnetic long-range order is absent.
• Incorrect treatment of dispersion : Both exchange
and correlation contribute.
• KS states are NOT physical objects : The bands of the
exact DFT are problematic. TDDFT with a frequency
dependent functional (exact) is required to treat
excitations even within the Born-Oppenheimer
approximation.
Conclusion : Current Models
•
•
Simulations are likely to provide semi-quantitative
accuracy/agreement with experiment.
Simulations are best used to obtain insight and examine
physics .e.g. to promote understanding.
Nonetheless, in order to provide truthful solutions of the
models, simulations must be performed to long time scales!
Limitations of ab initio MD
(despite our efforts/improvements!)
Limited to small systems (100-1000 atoms)*.
Limited to short time dynamics and/or sampling
times.
Parallel scaling only achieved for
# processors <= # electronic states
until recent efforts by ourselves and others.
Improving this will allow us to sample longer
and learn new physics.
*The methodology employed herein scales as O(N3) with
system size due to the orthogonality constraint, only.
Solution: Fine grained parallelization of CPAIMD.
Scale small systems to 105 processors!!
Study long time scale phenomena!!
(The charm++ QM/MM application is work in progress.)
IBM’s Blue Gene/L network torus
supercomputer
The worlds fastest supercomputer!
Its low power architecture requires fine grain parallel
algorithms/software to achieve optimal performance.
Density Functional Theory : DFT
Electronic states/orbitals of water
Removed by introducing a non-local electron-ion interaction.
Plane Wave Basis Set:
The # of states/orbital ~ N where N is # of atoms.
The # of pts in g-space ~N.
Plane Wave Basis Set:
Two Spherical cutoffs in G-space
gz
y(g)
gz
n(g)
gy
gx
y(g) : radius gcut
gx
gy
n(g) : radius 2gcut
g-space is a discrete regular grid due to finite size of sys
Plane Wave Basis Set:
The dense discrete real space mesh.
z
z
y(r)
x
y
y(r) = 3D-FFT{ y(g)}
n(r)
y
x
n(r) = Sk|yk(r)|2
n(g) = 3D-IFFT{n(r)} exactly
Although r-space is a discrete dense mesh, n(g) is generated exactly!
Simple Flow Chart : Scalar Ops
Memory penalty
Object
States
Density
Orthonormality
: Comp : Mem
: N2 log N : N2
: N log N : N
: N3
: N2.33
Flow Chart : Data Structures
Parallelization under charm++
Transpose
Transpose
Transpose
Transpose
RhoR
Transpose
Effective Parallel Strategy:
The problem must be finely discretized.
The discretizations must be deftly chosen to
–Minimize the communication between
processors.
–Maximize the computational load on the
processors.
NOTE , PROCESSOR AND DISCRETIZATION
ARE
Ineffective Parallel Strategy
The discretization size is controlled by the
number of physical processors.
The size of data to be communicated at a given
step is controlled by the number of physical
processors.
For the above paradigm :
–Parallel scaling is limited to #
processors=coarse grained parameter in the
model.
THIS APPROACH IS TOO LIMITED TO
ACHIEVE FINE GRAINED PARALLEL
Virtualization and Charm++
Discretize the problem into a large number of
very fine grained parts.
Each discretization is associated with some
amount of computational work and
communication.
Each discretization is assigned to a light weight
thread or a ``virtual processor'' (VP).
VPs are rolled into and out of physical processors
as physical processors become available
(Interleaving!)
Mapping of VPs to processors can be changed
easily.
The Charm++ middleware provides the data
Parallelization by over partitioning of parallel objects :
The charm++ middleware choreography!
Decomposition of work
Charm++ middleware maps work to processors dynamically
Available Processors
On a torus architecture, load balance is not enough!
The choreography must be ``topologically aware’’.
Challenges to scaling:
Multiple concurrent 3D-FFTs to generate the states in real space
require AllToAll communication patterns. Communicate N2 data pts.
Reduction of states (~N2 data pts) to the density (~N data pts)
in real space.
Multicast of the KS potential computed from the density (~N pts)
back to the states in real space (~N copies to make N2 data).
Applying the orthogonality constraint requires N3 operations.
Mapping the chare arrays/VPs to BG/L processors in a topologically
aware fashion.
Scaling bottlenecks due to non-local and local electron-ion interactions
removed by the introduction of new methods!
Topologically aware mapping for CPAIMD
Density
N1/12
States
~N1/2
Gspace
~N1/2
(~N1/3)
•The states are confined to rectangular prisms cut from the torus to
minimize 3D-FFT communication.
•The density placement is optimized to reduced its 3D-FFT
communication and the multicast/reduction operations.
Topologically aware mapping for
CPAIMD : Details
Distinguished Paper Award at Euro-Par 2009
Improvements wrought by topological aware mapping
on the network torus architecture
Density (R) reduction and multicast to State (R) improved.
State (G) communication to/from orthogonality chares improved.
‘’Operator calculus for parallel computing”, Martyna and Deng (2009) in preparation.
Parallel scaling of liquid water* as a function of system size
on the Blue Gene/L installation at YKT:
*Liquid water has 4 states per molecule.
•Weak scaling is observed!
•Strong scaling on processor numbers up to ~60x the number of states!
Scaling Water on Blue Gene/L
Software : Summary
Fine grained parallelization of the Car-Parrinello ab
initio MD method demonstrated on thousands of
processors :
# processors >> # electronic states.
Long time simulations of small systems are now
possible on large massively parallel
supercomputers.
Instance parallelization
• Many simulation types require fairly uncoupled instances
of existing chare arrays.
• Simulation types is this class include:
1) Path Integral MD (PIMD) for nuclear quantum effects.
2) k-point sampling for metallic systems.
3) Spin DFT for magnetic systems.
4) Replica exchange for improved atomic phase space
sampling.
• A full combination of all 4 simulation is both physical and
interesting
Replica Exchange : M classical subsystems each at a different
temperature acting indepently
Replica exchange uber index active for all chares.
Nearest neighbor communication required to exchange temperatures
and energies
PIMD : P classical subsystems connect by harmonic bonds
Classical particle
Quantum particle
PIMD uber index active for all chares.
Uber communication required to compute harmonic interactions
K-points : N-states are replicated
and given a different phase.
k0
k1
Atoms are assumed to be part of a periodic structure and
are shared between the k-points (crystal momenta).
The k-point uber index is not active for atoms and electron density.
Uber reduction communication require to form the e-density and
atom forces.
Spin DFT : States and electron density are given a
spin-up and spin-down index.
Spin up
Spin dn
The spin uber index is not active for atoms.
Uber reduction communication require to form the atom forces
``Uber’’ charm++ indices
• Chare arrays in OpenAtom now posses
4 uber ``instance’’ indices.
• Appropriate section reductions and
broadcasts across the ‘’Ubers’’ have
been enabled.
• We expect full results for July 2010.
Goal : The accurate treatment of complex heterogeneous
systems to gain physical insight.
Transparent Conducting Electrodes (TCEs)
for (inexpensive) amorphous silicon solar cells
Conventional TCEs:
• Indium Tin Oxide (ITO)
• Zinc Oxide (ZnO)
Graphene TCEs:
• 1 – 8 atomic layers
Performance:
• Transparency
95%
• Sheet resistance 10W
Performance:
• Transparency
85%
• Sheet resistance 100W
Manufacturing:
Manufacturing:
cm X cm size sheets
Science, 324, p. 1312 (2009).
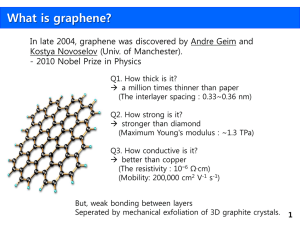
Graphene – single atomic layer of carbon
Chemical Doping:
http://www.rsc.org/
Transparent – 2% loss per layer
High Mobility – 2 x 105 (cm2 / Vs)
Engineering goal
Goal
# layers
Experimental collaborators G. Tulevski (IBM), A. Kasry (EGNC), A. Boll (IBM)
aSi + Graphene on BG/L
Plane wave based DFT CODES:
1) OpenAtom
2) Quantum Espresso
3) Abinit
PZ functional
800 eV (60 Ry.) Ecut
100ps quench aSi
10ps relaxation
Graphene + intercalates:
Explore various n and p dopants on layers
Orientation of Layers – Marcelo, Ahmed talks
Graphene + substrate:
Explore interface and transport from
semiconducting layer to graphene
Entire system:
Calculate electronic properties of entire +500
atom systems using full ab initio
Graphene on aSi:H – Structural results
Ideal single layer:
Relaxed single layer:
0.2 eV
Relaxed multi layer:
1% strain
0.1eV gap
See also Zhen Hua Ni et al.
ACS Nano, 2008, 2 (11), pp 2301
Graphene Intercalates
Electron donors:
Hole donors:
Alkali group elements – Li, K
HNO3
EFermi
AlCl3, FeCl3,
SbCl5
Graphene Intercalates – HNO3
EFermi
Graphene Intercalates – HNO3
EFermi
No shift – no doping
Graphene Intercalates – HNO3
EFermi
No shift – no doping
Graphene Intercalates – HNO3
HNO3 decomposition
Graphene layers facilitate decomposition
Graphene Intercalates – HNO3
HNO3 decomposition
(movie) - Top view with invisible graphene
Decomposition product - NO3 anion
EFermi
Decomposition product - NO3 anion
EFermi
Old EFermi
Integrate 1 hole per molecule
HNO3 decomposition products
From graphene
NO3-
1/3
O1
N
s
O2
1/3
1/3
O3
p
See also:
Wehling et. al, Nano Letters, 8, 173 (2008)
Gierz et. al, Nano Letters, 8, 4603 (2008)
e from graphene in
oxygen p-orbitals
AlCl3 intercalate (1 unit per 6.3 C)
Al
Ionic compound
Cl
s
p
AlCl3 intercalate (1 unit per 6.3 C)
EFermi
No shift – no doping
AlCl3 intercalate (1 unit per 6.3 C) – with Defects
Induce defect by removing Al atom:
Cl
From graphene
Cl
s
p
SbCl6 intercalate (1 unit per 14 C)
EFermi
Old EFermi
Integrate 1 hole per defect
Review of p – dopant studies
HNO3 intercalate:
- Charge transfer complexes: NO2, NO3
AlCl3 crystals:
- Charge transfer complexes: AlCl4 or AlCl2.3
SbCl5 crystals:
- Relaxed pseudo-crystal configuration with density (1unit / 14C).
- Charge transfer complexes: SbCl4, SbCl6
Summary
Structure:
Graphene layers on aSi:H
p dopants:
~1 hole per defect or decomposition product:
NO2, NO3, AlCl4, SbCl4, SbCl6
doping:
Classic rigid band shifts observed from various intercalate compounds
Future Work
- Quantify transparency of intercalate layers
- Investigate charge transfer to graphene layers from substrate
- Large scale electronic structure calculations
Conclusions
• Important physical insight can be gleaned from
high quality, large scale computer simulation
studies.
• The parallel algorithm development required
necessitates cutting edge computer science.
• New methods must be developed hand-in-hand
with new parallel paradigms.
• Using clever hardware with better methods and
parallel algorithms shows great promise to impact
both science and technology.