Chapter 8
advertisement

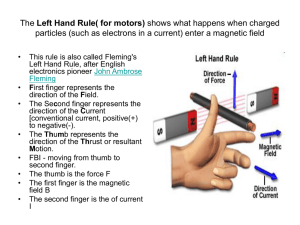
Chapter 8 Magnetic and optical properties Magnetic Properties Railgun Magnetization and Magnetic Susceptibility If a body is placed in a homogeneous magnetic field Ho, the magnetic field with the body varies from free-space value. That is, B = Ho + 4p M B : magnetic induction (the field within the body) M : intensity of magnetization B/Ho = 1 + 4p (M/Ho) = 1 + 4p cv M/Ho : dimensionless cv : magnetic susceptibility per volume Other definition of magnetic susceptibility Gram Susceptibility: cg = cv/d (unit: cm3/g) d = density Molar Susceptibility: cm = cg.(MW) (unit: cm3/mol, or emu) MW: molecular weight Macroscopic Point of View Magnetic moment, M, can be related to the rate the energy change of a body in the magnetic field, Ho. -E M= Ho The sign of the induced magnetic moment, M, defines two types of magnetic behaviors: Diamagnetism and Paramagnetism -E M= Ho M (and cv, cg, cm) is negative: diamagnetism Materials with only paired electrons (In fact, all materials exhibit diamagnetism) M (and cv, cg, cm) is positive: paramagnetism Materials with unpaired electrons (In fact, these materials exhibit both paramagnetism and diamagnetism) Ferromagnetism, antiferromagnetism and ferrimagnetism Ferromagnetism, antiferromagnetism and ferrimagnetism can be considered as special forms of paramagnetism. Diamagnetism a property of all materials It arises from the interactions of electron pairs with magnetic field, generating a small magnetic field opposing the applied magnetic field, Ho. Diamagnetism N S N B = Ho + 4p M S Diamagnetism Diamagnetic materials move to the region of lower field strength (repelled by Ho) M= -E Ho For diamagnetic materials, M < 0, ∴ E >0 Ho Ho That is, the energy of the system increases with the applied magnetic field, Ho. ∴ the body moves in the direction of lower energy (i.e. lower field). The process is exothermic. Superconductors are perfect diamagnetic materials How to obtain diamagnetism (cdia)? cT = cpara + cdia To study the paramagnetic behaviors of materials, cdia must be subtracted from cT Pascal’s constants Calculation of cdia from Pascal’s constants: cdia = i cA + i cB i A: atoms B: bonds i Table of Selected Pascal’s Constants atom cA (x10-6 emu) atom cA (x10-6 emu) atom cA (x10-6 emu) H -2.93 F -63 Na+ -6.8 C -6.00 Cl -20.1 K+ -14.9 C(aromatic) -6.24 Br -30.6 bond cB (x10-6 emu) N -5.57 I -44.6 C=C +5.5 N(aromatic) -4.61 Mg2+ -5 C≡C +0.8 N(monamide) -1.54 Zn2+ -15 C=N +8.2 N(diamide, imide) -2.11 Pb2+ -32.0 C≡N +0.8 O -4.61 Ca2+ -10.4 N=N +1.8 O2(carboxylate) -7.95 Fe2+ -12.8 N=O +1.7 S -15.0 Cu2+ -12.8 C=O +6.3 P -26.3 Co2+ -12.8 anions c (x10-6 emu) Hg2+ -40.0 Ni2+ -12.8 C≡N- -13.0 Example 1: 5 x C (ring) = 5 x (-6.24) = -31.2 5 x H = 5 x (-2.93) = -14.6 1 x N (ring) = 1 x (=4.61) = -4.61 H H H N H H pyridine i cA i = -31.2 + (-14.6) + (-4.61) = -50.3 x 10-6 cm3/mol (or emu) In this case, cBi is zero, because the “ring values” of C and N are used. i Example 2: O 3 x C = 3 x (-6) = -18 6 x H = 6 x (-2.93) = -17.6 1 x O = -4.61 H3C CH3 acetone cA = -18 + (-17.6) + (-4.61) = -40.21 x 10-6 emu i cB = 1 x (C=O) = +6.3 x 10-6 emu i i i cdia = i cA + i cB i i = -33.9 x 10-6 emu Example 3: K4Fe(CN)6 Transition metal complex 4 x K+ = 4 x (-14.9) = -59.6 1 x Fe2+ = -12.8 6 x (C≡N-) = 6 x (-13.0) = -78.0 cdia = i cA + i cB i i = -150.4 x 10-6 emu How to obtain cdia of Cr(acac)3? Cr(acac)3 is paramagnetic so it is difficult to measure its diamagnetism directly. Method I. Calculate cdia from Pascal’s constants. Method II. Synthesize Co(acac)3, Co3+: d6 low spin. Use the cdia value of Co(acac)3 as that of Cr(acac)3. Method III. Measure the cdia value of Na(acac), to obtain the cdia value of acac-. Then include this value in Pascal’s constant calculation. H H Expt’l Agreement -50.4 x 10-6 -49 x 10-6 good H N H Calculated H AsMe2 -147 x 10-6 -194 x 10-6 poor AsMe2 However, since cpara >> cdia , the disagreement usually does not cause too much problem. Disagreement of cdia Paramagnetism Paramagnetism arises from the interaction of Ho with the magnetic field of the unpaired electron due to the spin and orbital angular momentum. Derivation of M and cm from microscopic point of view For S = 1/2 system (Spin only, no orbital contribution) = -gS : magnetic moment S : spin angular momentum ge : electron g-factor (ge = 2.0037 for free e-) ( or B) : Bohr magneton of the electron = 9.3 x 10-21 erg/Gauss Interaction energy between magnetic moment and applied field The Hamiltonian describing the interaction energy of this moment with the applied magnetic field, Ho, is: H = -.H = gS.H The energies for S = ½ system The energies (eigen values) for S = 1/2 (ms = +1/2, -1/2) system are: E = msgHo E1/2 = (1/2)gHo and E-1/2 = –(1/2)gHo ms=1/2 Energy ms=1/2 E = gHo ms=-1/2 Zeeman effect ms=-1/2 Ho Relative populations of ½ and –½ states When Ho = 25 kG E ~ 2.3 cm-1 At 300 K, kT ~ 200 cm-1 Boltzmann distribution P1/2 e-E/kT ~ 1 = P-1/2 The populations of ms = 1/2 and –1/2 states are almost equal with only a very slight excess in the –1/2 state. That means that even under very large applied field, the net magnetic moment is still very small. To obtain M ( or cm), we need to consider all the energy states that are populated. ∵ H = -.H = gS.H ∴ The magnetic moment, n with the direction // Ho, of an electron in a quantum state n can be obtained by: n = -(∂En)/(∂H) = -msg So we consider: (1) The magnetic moment of each energy state. (2) The population of each energy state. That is, M = Nn.Pn N : Avogadro’s number Pn : probability in state n. M = Nn.Pn H = -.H = gS.H n = -(∂En)/(∂H) = -msg Pn = Nn NT = -En/kT e -E /kT e n Nn: population of state n NT: population of all the states Energy ms=1/2 ms=-1/2 ms=1/2 n 1/2gH -1/2g E = gHo ms=-1/2 E = msgHo En -1/2gH 1/2g Ho n = -(∂En)/(∂H) = -msg M = Nn.Pn n e N ms -En/kT M= Since gH<<kT when Ho ~ 5kG e -En/kT ms ±x e g/2kT = N [ g/2 e -g/2kT -g/2e g/2kT -g/2kT [e +e N g [ 1 + g/2kT (1 = 2 1 + g/2kT + (1 = N g2 4kT ] ~ 1±x when x << 1 ] g/2kT) ] g/2kT) = M M for S=1/2 system Curie Law of paramagnetic materials cm = cm M = slope = C N g2 4kT Curie Law: cm = C/T N g2 ∴ 1/T C= 4k Curie-Weiss Law is called Weiss constant. If is positive, then the magnets tend to align parallelly. If is negative, then the magnets tend to align antiparallelly. cm 1 cm = C/(T-) T If the system is not magnetically dilute (pure paramagnetic), the neighboring magnetic moments may exhibit an overall tendency of parallel alignment or antiparallel alignment. (still considered as paramagnetic, not ferromagnetic or antiferromagnetic) For general S values (not only S = 1/2) En = msgbH ms = -S, -S+1, …. , S-1, S N m=-S (-msg)e S M= -msgH/kT = s e -msgH/kT N g2H S(S+1) 3kT ms cm = M = N g2 S(S+1) 3kT spin only cm = M = N g2 S(S+1) 3kT S=3/2 For S=1/2 cm = N g2 For S=1 cm = 2N g2 3kT For S=3/2 cm = 5Ng2 4kT S=1 cm S=1/2 slope = Curie const. 4kT 1/T c for S = 1/2, 1, 3/2 Definition of eff cm = eff N g2 S(S+1) 3kT =( 3k N ) ( cmT ) ~ 2.828( cmT ) 1/2 1/2 1/2 eff = g[S(S+1)]1/2 (BM, Bohr Magneton) or eff = [n(n+2)]1/2 where n= number of unpaired e- Saturation of Magnetization The Curie-Wiess law does not hold where the system is approaching saturation. An approximation has been made: gH << kT so that ±x e ~ 1±x If the applied magnetic field is very large, Curie-Weiss law is not valid. (M is not proportional to H) cm = M = N g2 S(S+1) 3kT Saturation of Magnetization follow Curie -Weiss law S=2 4 S=3/2 M/ 3 mol-1 S=1 2 Energy ms=1/2 ms=-1/2 ms=1/2 E = gHo ms=-1/2 S=1/2 1 0 1 H/kT 2 M = -msg M = for 1 magnet. If H is large enough, the probability of ms= -1/2 populated is close to 100%. Ho Plot of eff vs Temperature 3.87 eff S=3/2 eff = 2[S(S+1)]1/2 (BM) eff = 2.828(cT)1/2 temperature All the molecules are in the S=3/2 state at all temperatures. Plot of eff vs Temperature 3.87 eff S=3/2 (BM) 1.7 S=1/2 temperature The eff value of the system gradually decreases from hightemperature value of 3.87 BM (S=3/2) to low-temperature value 1.7 BM (S=1/2) Spin equilibrium and Spin Crossover 3.87 eff S=3/2 (BM) 3.87 eff S=3/2 (BM) 1.7 S=1/2 temperature 1.7 S=1/2 temperature Calculation of eff for f-block elements Now, we consider spin-only cases. For f-block elements, spin-orbit coupling is very large eff = g[J(J+1)]1/2 S(S+1)-L(L+1)+J(J+1) g = 1+ 2J(J+1) g-value for free ions Example: calculate the eff of Nd3+ (4f3) ml +3 +2 +1 0 -1 -2 -3 g = 1+ S(S+1)-L(L+1)+J(J+1) 2J(J+1) Lmax = 3+2+1 = 6 Smax = 3 x 1/2 = 3/2 2S+1= 2 x 3/2 + 1 = 4 Ground state J = L-S = 6-3/2 = 9/2 Ground state term symbol: 4I9/2 3/2(3/2+1)-6(6+1)+(9/2)(9/2+1) g = 1+ = 0.727 2x(9/2)(9/2+1) eff = g[J(J+1)]1/2 = 0.727[(9/2)(9/2+1)]=3.62 BM eff values of d-block elements For d-block elements, spin-orbit coupling is less important. In many cases, eff = g[S(S+1)]1/2 is valid. The orbital angular momentum is often “quenched” by special electronic configuration, especially when the symmetry is low. Spin-Orbit Coupling z y y ex dx2-y2 x dxy For example, if an electron can move back and forth between dx2-y2 and dxy orbitals, it can be considered as circulating about the z-axis, giving significant contribution to orbital angular momentum. Spin-Orbit Coupling dx2-y2 E dxy If dx2-y2 and dxy orbitals have different energies in a certain electron configuration, electrons cannot go back and forth between them. ∴ little contribution from orbital angular momentum. Spin-Orbit Coupling dx2-y2 dxy E Electrons have to change directions of spins to circulate Little contribution from orbital angular momentum. Spin-Orbit Coupling dx2-y2 dxy E Orbitals are filled. Little contribution from orbital angular momentum. Spin-Orbit Coupling dx2-y2 E dx2-y2 dxy dxy E Spin-orbit couplings are significant in the above two cases. Other orbital sets that may give spin-orbit coupling. dx2-y2/dxy dxz/dyz dxz/dxy dyz/dxy dz2/dxz dz2/dyz rotate about z-axis rotate about z-axis rotate about x-axis rotate about y-axis rotate about y-axis rotate about x-axis There are no spin-orbit coupling contribution for dz2/dx2-y2 and dz2/dxy Magic pentagon Related to “strength” of spin-orbit coupling dz2 6 2 dxz 2 dx2-y2 6 2 dyz 2 8 2 dxy Spin-orbit coupling influences g-value g = 2.0023 + n E1-E2 2.0023: g-value for free ion + sign for <1/2 filled subshell - sign for >1/2 filled subshell n: number of magic pentagon : free ion spin-orbit coupling constant For example: CuIIL4 system (Cu2+: d9) L dx2-y2 dxy L CuII dz2 dxz dyz g = 2.0023 + L n E1-E2 L D4h point group (axial symmetry) = -829 cm-1 E(dxy)-E(dx2-y2) = 15000 cm-1 (obtained by UV spectroscopy) gz = g// = 2.0023-8(-829)/15000 = 2.44 gx = gy = g⊥ = 2.0023 spin-orbit coupling has little contribution to x and y directions. Now, we can predict if the angular momentum will be quenched. Example: check all the electron configurations in an octahedral field. Which ones of the above electron configuration in Oh field have little spin-orbit contribution (with g ~ 2.0)? d3, d4(HS), d5(HS), d6(LS), d7(LS), d8, d9, d10 In low-symmetry field, spin-orbit coupling are quenched Remember that Oh is a high-symmetry field. If the symmetry is lowered, degeneracy will be destroyed and the orbital contribution will be quenched. Oh D4h D4h: all are quenched except d1 and d3 For low-symmetry field, all are quenched and therefore eff = g[S(S+1)]1/2 (spin-only) is valid. Van Vleck Equation In some cases, the paramagnetic behaviors are more complicated due to (1) mixing of low-lying excited state (2) zero field splitting (ZFS) (3) higher order Zeeman effect. These problems can be treated using Van Vleck Equation. Van Vleck Equation En : the energy of state n can be expressed as a power series of H. En = En(0) + H・En(1) + H2・En(2) + H3・En(3) + ….. En(0) : En of zero field; can be set to zero by convention. H : applied magnetic field En(1) : 1st order Zeeman coefficient En(2) : 2nd order Zeeman coefficient (related to mixing of low- lying excited state) En(3) : 3rd order Zeeman coefficient (very small and can be neglected) Van Vleck Equation En = En(0) + H・En(1) + H2・En(2) + H3・En(3) + ….. -En n = H =- En-2HEn o (1) (0) -En/kT e (1) (2) -En-HEn-H2En =e kT (2) (0) -En =e kT (1) (2) -HEn-H2En xe kT (0) (1) -En HEn) (1= kT x e kT ∵ when x is small, e-x ~ 1-x, and H2En(2) << kT M= N n n (0) -En/kT e e -En/kT n) x kT Nn (-En-2HEn) (1- HE e kT (1) = n -En (1) (2) (1- HE ) x e -E(0) n (1) n kT n kT For a paramagnetic system, when H = 0, M = 0 (0) ∴ En e = 0 (1) -En kT n ∴ cm = and (En(2))2 and (En(2)˙En(1)) are very small and can be neglected. cm = M/H N n [ (1) 2 (En) -En -2En]e -E e (2) kT (0) n n kT Van Vleck Equation (0) kT Application of Van Vleck Equation Example 1: S = 1/2, ms=+1/2, -1/2, no excited state En(2) = 0 En = En(0) + H・En(1) + H2・En(2) + ……. E = msgHo n = -(∂En)/(∂H) = -msg (0) Energy ms=1/2 ms=-1/2 ms=1/2 Energy En E(1)n 1/2g 0 1/2g -1/2g Ho 0 -1/2g E = gHo ms=-1/2 (0) cm = N n [ (1) 2 (En) -2En]e -E e (2) kT [ N n ms=1/2 Energy kT ms=1/2 n kT (1) 2 (En) kT e 0 ]e 0 = ms=-1/2 N[ 2 (g/2) kT E(1)n 1/2g 0 1/2g -1/2g Ho 0 -1/2g E = gHo ms=-1/2 (0) n cm = -E(0) n Energy En 2 + (-g/2) kT ] 1+1 n cm = Ng2 4kT cm = M = N g2 S(S+1) 3kT this result is the same as what we obtained from simple boltzmann distribution. Application of Van Vleck Equation S=1/2 Example 2: Cr3+, d3, S = 3/2 with zero field splitting (ZFS) = D (0) ms=+3/2 ms= ms= _ + 1/2 E=ZFS=D 0 Energy ms= +_ 1/2 0 D+3/2gH D 3/2g D-3/2gH D -3/2g ms=+1/2 1/2gH 0 1/2g ms=-1/2 -1/2gH 0 -1/2g _ 3/2 + ms= +_ 3/2 Energy En E(1)n ms=-3/2 H In this case, En(2) = 0. cm = N n [ (1) 2 (En) -En - ]e -E e kT (2) 2En (0) (0) ms=+3/2 kT (0) n ms= _ + 1/2 E=ZFS=D 0 Energy ms= +_ 1/2 0 2 2 2 (-g/2) [ e e cm = e e e 0 kT 0 cm = D+3/2gH D 3/2g D-3/2gH D -3/2g ms=+1/2 1/2gH 0 1/2g ms=-1/2 -1/2gH 0 -1/2g ms= +_ 3/2 ms= +_ 3/2 n kT Energy En E(1)n ms=-3/2 H 2 ] (g/2) 0 (3g/2) -D/kT (-3g/2) -D/kT N + kT + kT + kT 0 -D/kT -D/kT + + + e e e -D/kT Ng2 4kT [ 1 + 9e 1+ -D/kT e ] Example 2 cm = -D/kT Ng2 4kT [ 1 + 9e 1+ ] -D/kT e When ZFS is very small (D0) or at high temperature (kT >> D), e-D/kT 1 cm = Ng2 4kT x 10/2 = 5Ng2 4kT ms=+3/2 g2 cm = M = N3kT S(S+1) with S = 3/2 ms= +_ 3/2 ms=+1/2 ms= +_ 1/2 ms=-1/2 ms=-3/2 Example 2 when D 0 or at high temperature When D ∞ or at low temperature (kT<<D), e-D/kT 0. cm = -D/kT Ng2 4kT cm = [ 1 + 9e 1+ Ng2 4kT -D/kT e ] ms= +_ 3/2 ms= +_ 1/2 The system behaves like: cm = M N g2 S(S+1) = 3kT with S = 1/2 ms= +_ 3/2 E=ZFS=D If D is very large only ms = +1/2, -1/2 are populated ms= +_ 1/2 Value of ZFS can be obtained by curve-fitting 3.87 eff S=3/2 S=1/2 cm (BM) 1.7 S=1/2 temperature cm = S=3/2 HT 1/T LT -D/kT Ng2 4kT [ 1 + 9e 1+ -D/kT e ] ZFS (D) can be obtained by curve-fitting. Example 3. S =1 system (0) Energy En En _1 + ms=+1 E=ZFS=D ms=-1 ms= (1) D+gH D g D-gH D -g 0 0 ms= +_ 1 ms= 0 0 ms= 0 0 ms=0 Energy 0 cm = N [ n (1) 2 (En) H -E(0) n - ]e -E e kT (2) 2En (0) n n kT Van Vleck equation kT 2 (-g) 2 -D/kT (g) -D/kT + kT kT ] e e cm = -D/kT -D/kT e +e +e N[ 0 N[ 2 (-g) 2 -D/kT (g) -D/kT + kT kT e ] e cm = -D/kT -D/kT e +e +e 0 [ 2N g2 -D/kT e ] cm = -D/kT 1+2e kT At high temperature, or ZFS is very small (D<<kT) then e-D/kT 1 cm = 2Ng2 3kT The system appears to be like S = 1 with no ZFS. cm = M = N g2 S(S+1) 3kT At low temperature or very large ZFS (D>>kT), then e-D/kT 0. cm 0 The system appears to be diamagnetic because only ms = 0 state is populated. e.g. 3 Interactions of micromagnets Paramagnetic materials No interactions between magnets cm These magnets are oriented randomly under zero applied magnetic field. paramagnetic T If there are interactions between these micromagnets, these materials are ferromagnetic, antiferromagnetic or ferrimagnetic. With interactions among micromagnets Ferromagnetic, Antiferromagnetic and Ferrimagnetic ferromagnetic cm + Curie temperature ' temperature Neel cm + paramagnetic paramagnetic antiferromagnetic T T Magnetic domain H Average domain size: 20 ~200 nm Hysteresis curve of M vs H M Mr Ms Ms : saturation magnetization Mr: remanence magnetization Hc: coercive magnetic field Hc H Magnetic interaction of polynuclear clusters Cu2(OAc)4.(H2O)2 cm O O O H2O O Cu O Cu O paramagnetic O T OH2 ' temperature Neel O cm + paramagnetic antiferromagnetic T Antiferromagnetic coupling complexes Some terms to define: Magnetic orbital: orbital containing an unpaired electron Exchange interactions: magnetic interactions between metal centers HO 2 Exchange parameter: J, coupling constant O O O Cu O O Cu O O O OH2 Magnetic behavior of d1-d1 dimer O O O H2O O J: coupling constant J > 0 : antiferromagnetic coupling J < 0 : ferromagnetic coupling O S=1 S=0 J>0 J<0 S=1 J S=0 Cu S=0 -J S=1 Cu O O O OH2 The energy diagram of d1-d1 dimer system En = En(0) + H・En(1) + H2・En(2) + H3・En(3) + ….. (0) Energy En ms=+1 S=1 J 0 Energy S=0 0 E(1)n J+gH J g ms= 0 J J 0 ms=-1 J-gH J -g 0 0 0 ms=0 H Antiferromagnetic coupling system Van Vleck equation cm = [ N n (1) 2 (En) kT cm = cm = ms=+1 (0) -En - ]e -E e (2) 2En S=1 kT (0) n N[ (0) n kT J 0 Energy 2 (-g) 2 -J/kT (g) -J/kT + kT kT e e 0 + 3e -J/kT S=0 [3+ e ] J/kT J g ms= 0 J J 0 ms=-1 J-gH J -g 0 0 0 ms=0 H 0 2 kT ' temperature Neel + cm paramagnetic Bleany-Bowers Equation antiferromagnetic The value of J can be obtained by curve-fitting. E(1)n J+gH 2g -J/kT ] ] e = N[ e 1 + 3 e -J/kT 2Ng2 kT Energy En T Two extreme conditions of the d1-d1 system cm = 2Ng2 [3+ e ] kT J/kT 2g2 -J/kT N[ kT e = 1 + 3 e -J/kT ] O O O H2O O Cu O Cu O When J >> kT or T 0, cm 0, this system is diamagnetic or a Cu-Cu bond is formed. When J << kT or at high temperature, cm = Ng22/2kT Ng22/2kT can be considered as two Ng22/4kT, i.e., d1-d1 can be considered as two independent d1 system O O OH2 The relationship between J and TN ' temperature Neel cm cm = + paramagnetic antiferromagnetic T 2Ng2 [3+ e ] kT J/kT Solve (cm)/ (T) = 0 TN ≒ (5/8) J/K Direct couping through metal-metal bonding or through superexchange via ligands? Magnetic orbital dx2-y2 dz2 dxy dxz, dyz O O O H2O O Cu O Cu O O OH2 z-axis O Weak -bond is formed between the interaction of magnetic orbitals. Is the d1-d1 interaction through () -bonding ? (2) superexchange via ligands ? Goodgame’s experiment in 1969 [Cu2(O2CH)4(NCS)2]2d(Cu…Cu) = 2.716 Å [Cu2(O2CCH3)4(NCS)2]2d(Cu…Cu) = 2.643 Å S=1 J = 485 cm-1 S=1 J = 305 cm-1 S=0 S=0 This results indicate that the d1-d1 interaction O is not through metal-metal bonding only. HO Cu O Superexchange mechanism through ligands O may dominate. O O Cu 2 O O O OH2 Comparison of V4+-V4+ and Mo5+-Mo5+ dimers O L O L V L O L L V L L Mo L d(V…V) = 3.20 Å L z O L y L x Mo L d(Mo…Mo) = 2.78 Å S=1 dz2 S=1 J = 200 cm-1 S=0 Paramagnetic at HT Antiferromagnetically coupled At LT J = 3000 cm-1 S=0 Diamagnetic compound dx2-y2 dxz,dyz dxy Magnetic phenomena in 1D crystal Variation of flux density in diamagnetic and paramagnetic substances in a magnetic field Magnetic susceptibility Plot of reciprocal susceptibility against temperature Some properties of ferromagnetic materials Some Values of magnetic moments Antiferromagnetic coupling of spins of d electrons on Ni2+ ions through p electrons of oxide ions Ferromagnetic ordering in bcc Fe, fcc Ni and hcp Co Electronic constitution of iron, cobalt and nickel 4.8 + 2.6 = 7.4 4.8 up + 2.6 down 4.8 - 2.6 = 2.2 Occupied energy levels and density of states N(E) for 3d and 4s, 4p bands Pauli paramagnetism Electrons near Fermi level are paired. temperature independent H Excess unpaired electrons Induced by magnetic field Pauli paramagnetism is temperature independent but field dependent. Curie and Néel temperatures in lanthanides Magnetic structure of antiferromagnetic and ferromagnetic spinels Spinel [M2+]tet[Fe3+]2octO4 Inverse Spinel [Fe3+]tet[M2+, Fe3+]octO4 Partial inverse Spinel [Fe3+1-xZn2+x]tet[M2+1-xFe3+1+x]octO4 Variation in saturation magnetization with composition for ferrite solid solution M1-xZnxFe2O4 Partial inverse Spinel [Fe3+1-xZn2+x]tet[M2+1-xFe3+1+x]octO4 M M O When x is large, the antiferromagmnetic coupling is destroyed. Garnets Atomic coordinates for Y3Fe5O12 (YIG) x3 x3 x2 Variation of magnetic moment at 0 K of garnets Spontaneous Magnetization in Dy3Fe5O12 garnet Dy3Fe3Fe2O12 The spins on rare earth (Dy3+) sublattice randomize much rapidly than those on Fe3+ sublattice. Crystallographic data for ilmenite Schematic representation of the luminescence Schematic design a fluorescent lamp Luminescence spectra of activated ZnS phosphors after irradiation with UV light Various types of electronic transition in activator ions Ground state potential energy diagram for a luminescent center in an ionic host crystal Ground and excited state potential energy diagrams for a luminescent center Non-radiative energy transfer involved in operation of a sensitized phosphor Some lamp phosphor materials Schematic representation of anti-Stokes and normal luminescence phenomena Energy levels of the Cr3+ ion in ruby crystal and laser emission Design of a ruby laser Ga-As laser Energy levels of the Nd3+ ion in neodymium lasers