Structure Name Exp. Vol - Electron poor materials
advertisement

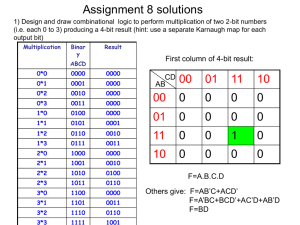
Electron poor materials research group Group meeting Dec 16, 2010 Theory- PAW_PBE psuedo potentials. EOS relaxations and bader analysis of resultant structures. System’s under study Electron Poor Materials ZnSb ZnAs Li2Sb Mg3Sb2 Tetrahedral, sp3 systems InSb GaSb ZnSe ZnTe GaAs Si Procedure Very accurate EOS relaxations were preformed. The system cell shape and volume was relaxed while the volume was kept fixed to give a point in the EOS curve. A final relaxation was performed to bring the system to the target volume given by the BirchMurnaghan EOS. From the final relaxation a static calculation was performed to get the charge densities for Bader analysis. Bader analysis was performed on the charge densities from the static run. Example INCAR for EOS relaxation. It should be noted that not all relaxations have the same VASP setup as I had difficulties with some of the systems crashing in VASP with very accurate relaxation parameters. This was machine independent. System = InSb NSW = 5 | number of ionic steps ISIF = 4 | ISIF=4 relax cellshape and ions. NOT volume IBRION = 1 | ionic relaxation algorithm EDIFF = 1E-5 | break condition for elec. SCF loop EDIFFG = -1E-4 | break condition for ionic relaxation loop MAXMIX = 80 NELMIN = 6 NFREE = 15 | keep dielectric function between ionic movements | minimum number of electronic steps | number of degrees of freedom (don't go above 20) #RECOMMENDED MINIMUM SETUP #GGA= #xchange-correlation #VOSKOWN= #=1 if GGA=91; else = 0 PREC = ACCURATE #PRECISION, sets fft grid ENCUT = 225 #energy cutoff, commented to use enmax in potcar LREAL = .FALSE. #.FALSE. MEANS USE RECIPROCAL LATTICE ISMEAR = 0 #0 means use gaussian smearing Example INCAR for EOS static. It should be noted that not all relaxations have the same VASP setup as I had difficulties with some of the systems crashing in VASP with very accurate relaxation parameters. This was machine independent. System = InSb #ISTART = 0 # startjob: no WAVECAR file #ICHARGE = 2 # charge: from atoms #INIWAV = 1 # random initialization for wf. NELM = 40 # maximum of 40 electronic steps NELMIN = 4 # minimum of 2 steps NELMDL = -5 # no update of charge for 3 steps EDIFF = 1E-5 # accuracy for electronic minimization #RECOMMENDED MINIMUM SETUP #GGA= #xchange-correlation #VOSKOWN= #=1 if GGA=91; else = 0 PREC = ACCURATE #PRECISION, sets fft grid ENCUT = 225 #energy cutoff, commented to use enmax in potcar LREAL = .FALSE. #.FALSE. MEANS USE RECIPROCAL LATTICE ISMEAR = -5 #-5 means use tetrahedral with blochl INCAR For final relaxation calculations System = InSb relaxsetup.sh NSW = 20 | number of ionic steps ISIF = 4 | (ISIF=2 Relax ions only, ISIF=3 Relax everything) IBRION = 1 | ionic relaxation algorithm EDIFF = 1E-9 | break condition for elec. SCF loop EDIFFG = -1E-8 | break condition for ionic relaxation loop MAXMIX = 80 NELMIN = 8 NFREE = 20 | keep dielectric function between ionic movements | minimum number of electronic steps | number of degrees of freedom (don't go above 20) #RECOMMENDED MINIMUM SETUP #GGA= #xchange-correlation #VOSKOWN= #=1 if GGA=91; else = 0 PREC = ACCURATE #PRECISION, sets fft grid ENCUT = 225 #energy cutoff, determines number of lattice vectors LREAL = .FALSE. #.FALSE. MEANS USE RECIPROCAL LATTICE ISMEAR = 0 #determines how partial occupancies a set. INCAR For final static calculations System = InSb SIGMA = 0.01 #RECOMMENDED MINIMUM SETUP PREC = ACCURATE #PRECISION ENCUT = 225 LREAL = .FALSE. #.FALSE. MEANS USE RECIPROCAL LATTICE ISMEAR = 0 #USE GAUSSIAN SMEARING #FOR GW CALCULATIONS #LOPTICS = .TRUE. #NBANDS = 96 #FOR BADER ANALYSIS LAECHG=.TRUE. NGXF = 126 #USE 6X NGX for bader analysis NGYF = 126 NGZF = 126 Equations of State The equations of state for the 10 materials are below. % differences are caluclated via: Exp Vol: 67.97 PAW_PBE Vol: 73.48 Exp Vol: 56.63 PAW_PBE Vol: 60.31 Exp Vol: 45.98 PAW_PBE Vol: 47.35 Exp Vol: 56.83 PAW_PBE Vol: 59.17 Exp Vol: 45.17 lat constant: 5.6533 PAW_PBE Vol: 47.86 Exp Vol: 40.03 PAW_PBE Vol: 40.89 Exp Vol: 388.93 PAW_PBE Vol: 404.82 Exp Vol: 312.38 PAW_PBE Vol: 323.40 Exp Vol: 130.64 PAW_PBE Vol: 133.17 Exp Vol: 356.90 PAW_PBE Vol: 356.65 Li2Sb EOS troubles. The above graph is the ugliest out of the set which makes me worry about the results for this structure. I have tried changing psuedo-potentials to include more core electrons (currently there is only 1 valence electron in the calculation). this however has crashed in the relaxation step I believe due to the large number of plane waves needed. ENMAX in POTCAR is 650 eV for Li_pv. It is not uncommon for VASP to in relaxation on these larger systems if the plane waves gets too big or the precision flag is set too high. This is machine independent. Equation of State Results. Structure Name InSb GaSb ZnSe ZnTe GaAs Si ZnSb ZnAs Mg3Sb2 Li2Sb Exp. Vol (Ang^3) 67.97 56.63 45.98 56.83 45.17 40.03 388.93 312.38 130.64 356.90 VASP Vol (Ang^3) 73.48 60.31 47.35 59.17 47.86 40.89 404.82 323.40 133.17 356.65 %diff 8.11 6.50 2.98 4.12 5.96 2.15 4.09 3.53 1.94 0.07 Bader analysis bader analysis of the structures that were relaxed from the target volume of the EOS calculations above. InSb ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 0.0000 0.0000 0.0000 2.6002 1.2908 29.8637 2 1.6622 1.6622 1.6622 5.3998 1.4395 43.6185 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 8.0000 Charge Transfer: 0.3998 GaSb ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 0.0000 0.0000 0.0000 2.7022 1.1483 22.2941 2 1.5563 1.5563 1.5563 5.2978 1.4227 38.0197 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 8.0000 Charge Transfer: 0.2978 ZnSe ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 0.0000 0.0000 0.0000 11.2714 1.0616 15.9986 2 1.4358 1.4358 1.4358 6.7286 1.3224 31.3555 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 18.0000 Charge Transfer: 0.7286 ZnTe ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 0.0000 0.0000 0.0000 11.4898 1.1104 18.5303 2 1.5464 1.5464 1.5464 6.5102 1.4456 40.6387 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 18.0000 Charge Transfer: 0.5102 GaAs ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 0.0000 0.0000 0.0000 2.3848 1.0766 18.1258 2 1.4409 1.4409 1.4409 5.6152 1.2941 29.7357 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 8.0000 Charge Transfer: 0.6152 Si ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 0.0000 0.0000 0.0000 3.9740 1.1403 20.3153 2 1.3672 1.3672 1.3672 4.0260 1.1183 20.5746 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 8.0000 Charge Transfer: 0.0260 ZnSb ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 2.8910 0.8296 7.1852 11.7353 1.1719 18.5852 2 0.2526 6.9946 3.0706 11.7353 1.1719 18.5852 3 3.3962 4.7417 5.1587 11.7353 1.1719 18.5852 4 6.0346 3.0825 1.0441 11.7353 1.1719 18.5852 5 3.3962 6.9946 1.0441 11.7353 1.1719 18.5852 6 6.0346 0.8296 5.1587 11.7353 1.1719 18.5852 7 2.8910 3.0825 3.0706 11.7353 1.1719 18.5852 8 0.2526 4.7417 7.1852 11.7353 1.1719 18.5852 9 0.8900 0.6496 0.9006 5.2645 1.3747 32.0161 10 2.2536 7.1746 5.0153 5.2650 1.3833 32.0181 11 5.3972 4.5617 3.2140 5.2650 1.3833 32.0181 12 4.0336 3.2625 7.3286 5.2645 1.3747 32.0161 13 5.3972 7.1746 7.3286 5.2650 1.3833 32.0181 14 4.0336 0.6496 3.2140 5.2645 1.3747 32.0161 15 0.8900 3.2625 5.0153 5.2645 1.3747 32.0161 16 2.2536 4.5617 0.9006 5.2650 1.3833 32.0181 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 136.0000 Charge Transfer: 0.2645 ZnAs ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 3.1025 4.5260 4.8534 11.5272 1.0976 15.9814 2 5.5241 2.8161 1.0239 11.5272 1.0976 15.9816 3 2.6485 0.8550 6.6352 11.5272 1.0976 15.9816 4 0.2270 6.4871 2.8056 11.5272 1.0976 15.9814 5 2.6485 2.8161 2.8056 11.5272 1.0976 15.9816 6 0.2270 4.5260 6.6352 11.5272 1.0976 15.9814 7 3.1025 6.4871 1.0239 11.5272 1.0976 15.9814 8 5.5241 0.8550 4.8534 11.5272 1.0976 15.9816 9 0.7864 0.5428 0.7776 5.4722 1.1688 24.4421 10 2.0891 6.7993 4.6072 5.4734 1.1816 24.4455 11 4.9647 4.2138 3.0519 5.4734 1.1816 24.4455 12 3.6619 3.1283 6.8814 5.4722 1.1688 24.4421 13 4.9647 6.7993 6.8814 5.4734 1.1816 24.4455 14 3.6619 0.5428 3.0519 5.4722 1.1688 24.4421 15 0.7864 3.1283 4.6072 5.4722 1.1688 24.4421 16 2.0891 4.2138 0.7776 5.4734 1.1816 24.4455 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 136.0000 Charge Transfer: 0.4722 Mg3Sb2 ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 0.0000 0.0000 0.0000 0.5183 1.0492 8.8431 2 0.0000 2.6546 4.5981 0.5550 0.9734 8.5351 3 2.2989 1.3273 2.6757 0.5550 0.9734 8.5351 4 0.0000 2.6546 1.6400 7.1859 1.7652 53.6294 5 2.2989 1.3273 5.6338 7.1857 1.7652 53.6263 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 16.0000 Charge Transfer: 2.1859 Li2Sb ACF.dat # X Y Z CHARGE MIN DIST ATOMIC VOL -------------------------------------------------------------------------------1 2.3289 0.0000 0.0000 0.1760 0.8271 4.1821 2 6.7906 2.0169 0.0000 0.1760 0.8271 4.1821 3 2.8130 4.8723 0.0000 0.1761 0.8331 4.1818 4 2.3289 0.0000 3.2538 0.1760 0.8271 4.1821 5 6.7906 2.0169 3.2538 0.1760 0.8271 4.1821 6 2.8130 4.8723 3.2538 0.1761 0.8331 4.1818 7 1.0465 6.8893 1.6269 0.1615 0.8513 4.2817 8 -2.5120 4.3509 1.6269 0.1615 0.8513 4.2817 9 1.4655 2.5383 1.6269 0.1616 0.8644 4.2829 10 5.4430 4.3509 4.8808 0.1615 0.8513 4.2817 11 5.0240 0.0000 4.8808 0.1615 0.8513 4.2817 12 1.4655 2.5383 4.8808 0.1616 0.8644 4.2829 13 0.0000 0.0000 1.6269 6.7396 1.5930 51.1225 14 0.0000 0.0000 4.8808 6.7396 1.5930 51.1225 15 0.0000 4.5928 6.5077 6.6271 1.5930 50.9728 16 0.0000 4.5928 3.2538 6.6271 1.5930 50.9728 17 3.9775 2.2964 0.0000 6.6206 1.5930 50.8370 18 3.9775 2.2964 3.2538 6.6206 1.5930 50.8370 -------------------------------------------------------------------------------VACUUM CHARGE: 0.0000 VACUUM VOLUME: 0.0000 NUMBER OF ELECTRONS: 42.0000 Charge Transfer: 1.7396 Bader Analysis results Crystal Name Bader charge transfer Pauling Electronegativity Philips Ionicity1 Si ZnSb GaSb InSb ZnAs ZnTe GaAs ZnSe Li2Sb Mg3Sb2 0.026 0.2645 0.2978 0.3998 0.4722 0.5102 0.6152 0.7286 1.7396 2.1859 0 0.4 0.24 0.27 0.53 0.45 0.37 0.9 0 1.) Phillips. Rev. Mod. Phys. 42, 3, 1970 0.261 0.321 0.546 0.31 0.676