functional_enrichment_new - Baliga Lab at Institute for Systems
advertisement

Introduction to Systems Biology 2012
Functional Enrichment Analysis:
making sense of big data
Aaron Brooks & Fang Yin Lo
8/21/2012
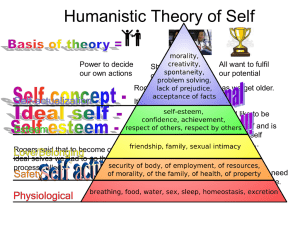
Insight
Experimental
Design
Data
analysis
Data
collection
Mischel et al, 2004
From knots to knowledge
• What is functional enrichment?
- Tools and caveats (e.g. DAVID and pvals)
• How can you apply these tools to large,
complex analysis problems (i.e. automation)?
Why functional enrichment analysis?
Interesting glioblastoma study
Common
Pathways
Gene
e.g. Glycolysis
Gene
Urn problem:
Gene
Gene Gene
Gene Gene
Function
Common
Functions
e.g. sugar metabolism
Draw 6 marbles (k)
How likely am I to draw 3 or more marbles?
N m
m
( )( )
P(X q)
()
q
Total: 15 marbles (N):
5 red (m)
10 black (N-m)
kq
N
k
N: population size
m: # of positives in the population
k: # of draws
q: # of positives
The workflow of typical enrichment tools
e.g. Gene Ontology
(GO), KEGG Pathway,
etc
e.g. GO Terms that are
enriched in the input
gene list
Nucleic Acids Research, 2009, Vol. 37, No. 1 1–13
(the database for annotation, visualization and
integrated discovery)
• Diverse, web-based functional analysis tool
• Integrates a suite of databases and statistical tools
(GO, KEGG, Interpro, Disease)
• User-friendly,
• Problematic for large analysis problems (many
independent sets)
staRt your engines
http://baliga.systemsbiology.net/events/sysbio/content/bicluster-307
http://baliga.systemsbiology.net/events/sysbio/content/bicluster-353
What if you had
many sets to
analyze?
AUTOMATION!!!
topGO
• An R package that facilitates semi-automated enrichment
analysis for Gene Ontology
• Three main Steps:
1. Prepare data
- create a topGO object with list of genes identiers, gene-to-GO
annotations)
2. Run enrichment tests
3. Display the results
Structured controlled vocabularies (ontologies) that describe relationships
between gene products and their associated biological roles
• cellular components : the parts of a cell or its extracellular
environment
• molecular functions: activities, such as catalytic or binding
activities, that occur at the molecular level (e.g. catalytic activity, Toll
receptor binding)
• biological processes: series of events accomplished by one or
more ordered assemblies of molecular functions (e.g. signal
transduction, pyrimidine metabolic process )
GO structure
• Directed Acyclic Graph(DAG)
• Child terms are more specialized
• Child can have more than one parent
Data preparation
# Install topGO and Affymetrix Human Genome U133 Plus 2.0 Array
annotation data
> source("http://bioconductor.org/biocLite.R")
> biocLite("topGO")
> biocLite("hgu133plus2.db")
> geneSets # Input a list a genes
#Boot the gaggle
> library(gaggle)
> gaggleInit()
#load the library
> library(topGO)
> library(hgu133plus2.db)
Data preparation
### Initializing the analysis ###
# hgu133plus2ACCNUM: an R object that contains mappings
between the manufacturers identifiers and gene names of
Affymetrix Human Genome U133 Plus 2.0 Array
# all.genes: all background genes ( gene universe )
> all.genes <- ls(hgu133plus2ACCNUM)
Other annotation packages at
Bioconductor
Other annotation packages can be found at:
http://www.bioconductor.org/packages/release/data/annotation/
Data preparation: Input gene lists
### Make gene lists ###
# We will make a list that includes two sets of genes of interest
# Initialize the list:
>glioblastoma.genes = list()
http://baliga.systemsbiology.net/events/sysbio/content/bicluster-307
# broadcast 'bicluster 307 genes' to R
>glioblastoma.genes[["bc307"]] = sapply(getNameList(),tolower)
Do the same for the other gene list:
http://baliga.systemsbiology.net/events/sysbio/content/bicluster-353
>glioblastoma.genes[["bc353"]] = sapply(getNameList(),tolower)
Data preparation: make topGO object
## Analyze genes in bc353 first
> relevant.genes <- factor(as.integer(all.genes %in% glioblastoma.genes[["bc353"]]))
> names(relevant.genes) <- all.genes
# Construct the topGOdata object for automated analysis
>GOdata.BP <- new("topGOdata", ontology='BP', allGenes = relevant.genes,
annotationFun = annFUN.db, affyLib = 'hgu133plus2.db')
# ontology:'BP','MF; or 'CC'
# allGenes: named vector of type numeric or factor. The names attribute
contains the genes identifiers. The genes listed in this object define the gene
universe.
# annotationFun: function that maps gene identifiers to GO terms.
# annFUN.db extracts the gene-to-GO mappings from the affyLib object
# affyLib: character string containing the name of the Bioconductor annotaion
package for a specific microarray chip.
Run Enrichment Analysis
> results <- runTest(GOdata.BP, algorithm = 'classic', statistic = 'fisher’)
Analysis of results: summary
# generate a summary of the enrichment analysis
> results.table <- GenTable(GOdata.BP, results, topNodes =
length(results@score))
# How many GO terms were tested?
> dim(results.table)[1]
# reduce results to GO terms passing Benjamini-Hochberg multiple hypothesis
corrected pval <= 0.05, FDR <= 5%
>results.table.bh <results.table[which(p.adjust(results.table[,"result1"],method="BH")<=0.05),]
Analysis of results: get significant GO terms
# reduce results to GO terms passing Benjamini-Hochberg multiple hypothesis
corrected pval <= 0.05, FDR <= 5%
>results.table.bh <results.table[which(p.adjust(results.table[,"result1"],method="BH")<=0.05),]
# How many terms are enriched?
>dim(results.table.bh)[1]
# What are the top ten terms?
>results.table.bh[1:10,]
Analysis of results: get genes in top GO terms
# Get all the genes annotated to a specific GO term of interest:
>GOid.of.interest = results.table.bh[1,"GO.ID"]
>all.term.genes = genesInTerm(GOdata.BP,GOid.of.interest)[[1]]
# Which of these genes is in the bicluster?
>genes.of.interest <- intersect(glioblastoma.genes[["bc353"]],all.term.genes)
# print table with probe ID and gene symbol
>gene.symbol= toTable(hgu133plus2SYMBOL[genes.of.interest])
# print table with probe ID and gene names
>gene.name= toTable(hgu133plus2GENENAME[genes.of.interest])
# Combine the information of the genes, output to csvfile:
>cbind(gene.symbol,gene.name[,2])
>write.csv(cbind(gene.symbol,gene.name[,2]), file =
"glioblastoma.genes_bc353_in_immune response.csv“)
Automation
results <- list()
for( bc in names(glioblastoma.genes) ) {
cat(paste("Computing functional enrichment for...",bc,"\n"))
relevant.genes <- factor(as.integer(all.genes %in%
glioblastoma.genes[[bc]]))
names(relevant.genes) <- all.genes
GOdata.BP <- new("topGOdata", ontology='BP', allGenes =
relevant.genes, annotationFun = annFUN.db, affyLib =
'hgu133plus2.db')
results[[bc]] <- GenTable(GOdata.BP,runTest(GOdata.BP, algorithm =
'classic', statistic = 'fisher'),topNodes = length(results@score))
}
Questions?
Other algorithms supported by topGO
• Standard implementations of GO testing compute the
significance of a node independent of the significance of the
neighboring nodes (‘classic’)
• Other algorithms take into considerations of the GO structure
and try to find more specific GO terms (e.g.’elim’, ‘weight’,
Alexa et al. (2006). Bioinformatics (2006) 22 (13): 1600-1607.
‘weight01’,etc)
Other algorithms supported by topGO
# Try running other algorithms and compare the results:
>r1.BP.elim = runTest(GOdata.BP, algorithm = 'elim', statistic = 'fisher')
>r1.BP.weight = runTest(GOdata.BP, algorithm = 'weight', statistic = 'fisher‘)
# This will take a while…
# After the runs are done, visually compare resulting p values from different algorithms:
>pValue.classic <- score(r1.BP)
>pValue.elim <- score(r1.BP.elim)[names(pValue.classic)]
>pValue.weight <- score(r1.BP.weight)[names(pValue.classic)]
>gstat <- termStat(GOdata.BP, names(pValue.classic))
>gSize <- gstat$Annotated / max(gstat$Annotated) * 4
>colMap <- function(x) {
.col <- rep(rev(heat.colors(length(unique(x)))), time = table(x))
return(.col[match(1:length(x), order(x))])
}
>gCol <- colMap(gstat$Significant)
>plot(pValue.classic, pValue.elim, xlab = "p-value classic", ylab = "p-value elim",pch = 19,
cex = gSize, col = gCol)
Broadcasting gene list to DAVID
1. In R:
> broadcast(geneSetGenes)
2.
3. Select target:
DAVID
4. Broadcast to
DAVID
Broadcasting gene list to DAVID
Broadcasting gene list to DAVID
Broadcasting gene list to DAVID
Gene list and population
background being analyzed
Clustering options and contingency
A group of terms having similar
biological meaning due to
sharing similar gene members
Original database/resources
where the terms orient
Enriched terms associate
with input gene list
Modified Fisher Corrected p-values
Exact p-values
Some remaining challenges
Realistically positioning the role of enrichment P-values in the current datamining environment:
•
•
•
•
•
•
•
high-throughput enrichment data-mining environment is extremely complicated
Variations of the user gene list size
deviation of the number of genes associated with each annotation
gene overlap between annotation
incompleteness of annotation content
strong connectivity/dependency among genes
unbalanced distributions of annotation content
Limitation of multiple testing correction on enrichment P-values
•
common multiple testing correction techniques maybe overly conservative approaches if there are thousands or
even more annotation terms involved in the analysis
Genome Inform. 2005;16:106-115.; Nucleic Acids Research, 2009, Vol. 37, No. 1 1–13
Cross-comparing enrichment analysis results derived from multiple gene lists
• the size of the gene list impacts the absolute enrichment P-values, therefore
difficult to directly compare the absolute enrichment P-values across gene lists
Some remaining challenges
some may treat the resulting enrichment P-values as a scoring system that
plays a advisory role
more of an exploratory procedure, with the aid of enrichment P-value, rather
than a pure statistical solution.
Nat. Protoc. 2008. doi: 10.1038/nprot.2008.211
the specificity of enrichment analysis is more impacted by non-statistical
layers than it is by statistical methods alone
Nucleic Acids Research, 2009, Vol. 37, No. 1 1–13
Working with topGO data object
# work with Godata.BP
# obtaining all genes
> a = genes(GOdata.BP)
> str(a)
chr [1:31777] "1007_s_at" "1053_at" "117_at" "121_at" "1255_g_at"
"1294_at" ...
# number of genes
> numGenes(GOdata.BP)
[1] 31777
# The list of significant genes can be accessed using the method sigGenes()
> sg = sigGenes(GOdata.BP)
> str(sg)
chr [1:22] "201291_s_at" "202095_s_at" "202589_at" "202705_at"
"203213_at" ...
Working with topGO data object
# accessing information related to GO and its structure
# which GO terms are available for analysis:
> ug = usedGO(GOdata.BP)
> str(ug)
chr [1:10921] "GO:0000002" "GO:0000003" "GO:0000012" "GO:0000018" "GO:0000019" ...
# select some random GO terms: (1). count the number of annotated genes and (2) obtain their annotation
> sel.terms <- sample(usedGO(GOdata.BP), 10)
> sel.terms
[1] "GO:0032913" "GO:0043372" "GO:0044259" "GO:0032700" "GO:0043122" "GO:2001141" "GO:0060587"
[8] "GO:0071352" "GO:0007256" "GO:0051343“
# Check what are the genes annotated to a specific GO term:
> genesInTerm(GOdata.BP, "GO:0032913" )
$`GO:0032913`
[1] "208650_s_at" "208651_x_at" "209771_x_at" "209772_s_at" "216379_x_at" "266_s_at"
# Number of genes annotated to the selected GO terms:
> num.ann.genes <- countGenesInTerm(GOdata.BP, sel.terms)
> num.ann.genes
GO:0032913 GO:0043372 GO:0044259 GO:0032700 GO:0043122 GO:2001141 GO:0060587 GO:0071352
GO:0007256
6
36
111
12
454
6809
2
5
11
GO:0051343
2
Working with topGO data object
> ann.genes <- genesInTerm(GOdata.BP, sel.terms)
> str(ann.genes)
List of 10
$ GO:0032913: chr [1:6] "208650_s_at" "208651_x_at" "209771_x_at" "209772_s_at" ...
$ GO:0043372: chr [1:36] "1554519_at" "1555689_at" "1565358_at" "1569748_at" ...
$ GO:0044259: chr [1:111] "1554383_a_at" "1555540_at" "1555896_a_at" "1556499_s_at" ...
$ GO:0032700: chr [1:12] "1552798_a_at" "1556190_s_at" "201300_s_at" "207160_at" ...
$ GO:0043122: chr [1:454] "1552360_a_at" "1552703_s_at" "1552798_a_at" "1552804_a_at" ...
$ GO:2001141: chr [1:6809] "121_at" "1316_at" "1405_i_at" "1487_at" ...
$ GO:0060587: chr [1:2] "201525_at" "207092_at"
$ GO:0071352: chr [1:5] "201940_at" "201941_at" "201942_s_at" "201943_s_at" ...
$ GO:0007256: chr [1:11] "1558984_at" "203652_at" "206362_x_at" "207347_at" ...
$ GO:0051343: chr [1:2] "207514_s_at" "214286_at"
Analysis of results: get genes in top GO terms
#We can also look at multiple GO terms at the same time:
> GOids.of.interest = results.table.bh[c(1:10),"GO.ID"]
> all.term.genes = genesInTerm(GOdata.BP,GOids.of.interest)
# Which of these genes is in the bicluster?
> genes.of.interest <sapply(names(all.term.genes),function(x){intersect(all.term.genes[[x]],gliobla
stoma.genes[["bc353"]])})
# print table with probe ID and gene symbol:
> geneSynmol.of.interest <lapply(names(genes.of.interest),function(x){toTable(hgu133plus2SYMBOL[ge
nes.of.interest[[x]]])})
> names(geneSynmol.of.interest)<- GOids.of.interest