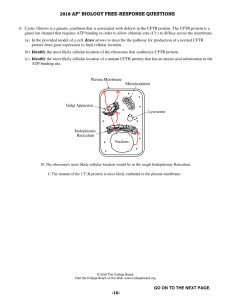
✅ Cystic fibrosis a genetic, multisystemic disorder characteristics: recurrent airway dysfunction pancreatic exocrine insufficiency intestinal dysfunction sweat gland dysfunction urogenital dysfunction pathogenesis autosomal recessive disease — involving gene is CFTR CF transmembrane conductance regulator gene - located on chromosome 7 function of CFTR - chloride channel and regulator of other ion channels location of CFTR - apical membrane of epithelium lining airway, sweat gland ducts, intestines, biliary duct, pancreatic ducts. dysfunctions due to CFTR loss in Sweat glands: loss of absorption of Cl- from sweat and thereby prevents absorption of Na+ ⇒ results into high Na and Cl concentration in sweat (baby tastes salty when kissed) in Exocrine Pancreas: absent CFTR causes lack of secretion of Na and HCO3 and Water into the duct ⇒ retention of enzymes in pancreas ⇒ destruction of pancreas in Liver: absent CFTR causes disruption in Na and Water balance in small biliary ducts ⇒ obstruction in the biliary tract Cystic fibrosis 1 in Genital tract (esp males): causes obstruction of small ducts ⇒ leads to atrophy, fibrosis of vas deferens, epididymis and seminal vesicles etc. in Ileum: disruption of salt and water secretion ⇒ thick dehydrated wastes in bowel ⇒ causes obstruction of bowel in Airways: high NaCl concentration in mucosa ⇒ reduced mucociliary clearance ⇒ impairment of local defence mechanisms (defective phagocytosis of pathogens) ⇒ repeated airway infections thick, viscous, purulent sputum ⇒ obstructs airways ⇒ leads to bronchiectasis clinical features: Newborn patients meconium ileus failure to thrive in infants - due to fat and protein malabsorption steatorrhea generalised edema - due to hypoproteinemia Adult patients recurrent LRTI recurrent cough, persisting longer thick, purulent, foul-smelling, blood tinged Sputum green-colored sputum → suspect Pseudomonas infection may develop presentation of bronchiectasis infertility cholelithiasis obstructive jaundice presentation investigations: Cystic fibrosis 2 Sweat Chloride levels if concentration of Cl- is ≥60 mEq/L ⇒ suggests CF Genetic testing for presence of CFTR gene Nasal transepithelial potential difference is raised Sputum examination, culture and AST Chest X-ray CT chest PFT treatment: Antibiotic therapy lasting for 2-3 weeks to be prescribed based on AST results of sputum cultures in case of Pseudomonas - Aminoglycoside + Beta-Lactam Chest physiotherapy chest percussion Postural drainage - helpful in bronchiectatic patient Bronchodilators prescribed only during exacerbations DEOXY-RIBO-NUCLEASE nebulised recombinant human DNase - Dornase alpha neutrophils in the lesions release DNA which forms long fibrils these fibrils cause increased viscosity of the sputum DNase can cleave DNA and thereby reduce sputum viscosity nebulised Hypertonic saline inhalation increases hydration of airway surface Cystic fibrosis 3 improves lung function reduces recurrent pulmonary exacerbations for patients with FEV1< 30% → LUNG TRANSPLANTATION oral Pancreatic enzymes and Vitamin supplements includes Lipase and trypsin given at meal times vitamin A, D, E are given along with it Vitamin K is given as per prothrombin time in case of development of diabetes, patients must be started on Insulin therapy Ivacaftor/ Lumacaftor/ Tezacaftor corrects the deletion mutations in the abnormal CFTR gene improves lung function reduced pulmonary exacerbations reduces concentration of sweat chloride Supportive measure: intake of salt is increased to make up for the NaCl lost in sweat immunise against influenza and pneumococcus avoid smoking complications: Cystic fibrosis 4