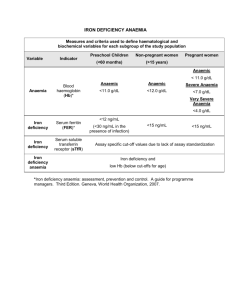
Table 2.3 Normal haemoglobins in adult blood. HbA Hb F Hb A2 Structu re α2β2 α2γ2 α2δ2 Normal (%) 96–98 0.5–0.8 1.5–3.2 Anaemia Key facts and checkpoints • • • • • • • In Australia, most people with anaemia will have iron deficiency 5% for children to 20% for menstruating females. Elderly 30-45% Anaemia of chronic disorders. 15-30% IDA The incidence of haemoglobinopathy traits, especially thalassaemia, is increasing in multicultural Western societies. If a patient presents with precipitation or aggravation of myocardial ischaemia, heart failure or intermittent claudication, consider the possibility of anaemia. The serum ferritin level, which is low in cases of iron-deficiency anaemia, is probably the best test to monitor iron-deficiency anaemia as its level reflects the amount of stored iron. Normal reference values for peripheral blood: adults male female Haemoglobin (g/L) 130–180 115–165 Red cells (× 1012/L) 4.5–6 4–5.5 PCV (haematocrit) 40–53 35–47 MCV (fL) 80–100 Platelets (× 109/L) 150–400 White cell count (× 109/L) 4–11 Neutrophils 2.5–7.5 Lymphocytes 1.5–4 Monocytes 0.2–1 Eosinophils <0.5 Reticulocytes (%) 0.5–2 ESR (mm/hour) <20 mm <35 mm if >70 years Haemoglobin (g/L) Red cells (erythrocytes) (× 1012/L) PCV (haematocrit) (%) Mean cell volume (MCV) (fL) Mean cell haemoglobin (MCH) (pg) Reticulocyte count (×109/L) Males Females 135.0–175.0 4.5–6.5 40–52 80–95 115.0–155.0 3.9–5.6 36–48 27–34 50–150 White cells (leucocytes) Total (×109/L) Neutrophils (×109/L) Lymphocytes (×109/L) Monocytes (×109/L) Eosinophils (×109/L) Basophils (×109/L) Platelets (×109/L) Serum iron (μmol/L) 4.0–11.0 1.8–7.5 1.5–3.5 0.2–0.8 0.04–0.44 0.01–0.1 150–400 10–30 anaemia DxT→ fatigue + palpitations + exertional dyspnoea • • • • • • • • • • Clinical features Patients with anaemia may be asymptomatic. When symptoms develop they are usually non- specific. Symptoms: tiredness/fatigue muscle weakness headache and tinnitus lack of concentration faintness/dizziness dyspnoea on exertion palpitations angina on effort intermittent claudication pica—usually brittle and crunchy food, e.g. ice (iron-deficiency anaemia) Signs Non-specific signs include pallor, tachycardia, systolic flow murmur and angular cheilosis. If severe, signs can include ankle oedema and cardiac failure. • Specific signs include • jaundice—haemolytic anaemia • koilonychias (spoon-shaped nails)—iron-deficiency anaemia. History • The history may indicate the nature of the problem: • iron deficiency: inadequate diet, pregnancy, GIT loss, menorrhagia, NSAID and anticoagulant ingestion • folate deficiency: inadequate diet especially with pregnancy and alcoholism, small bowel disease • vitamin B12 deficiency: previous gastric surgery, ileal disease or surgery, pernicious anaemia, selective diets (e.g. vegetarian, fad) • haemolysis: abrupt onset anaemia with mild jaundice possibly lead toxicity, especially in children Classification of anaemia • The various types of anaemia are classified in terms of the red cell size— the mean corpuscular volume (MCV): • microcytic—MCV ≤ 80 fL • macrocytic—MCV >100 fL • normocytic—MCV 80–100 fL • anaemia of chronic disorders (chronic infection, inflammation and malignancy) can occasionally be microcytic as well as normocytic; • the anaemia of hypothyroidism can be macrocytic in addition to the more likely normocytic; • the anaemia of bone marrow disorder or infiltration can also be occasionally macrocytic. Classification of anaemia by mean RBC volume (MCV) with selected causes Microcytic (MCV < 80 fL) Microcytic (MCV > 100 fL) Normocytic (MCV 80–100 fL) Iron deficiency Thalassaemia Anaemia of chronic disease Sideroblastic anaemia Vitamin B12 deficiency Folate deficiency Myelodysplastic disorders Cytotoxic drugs Liver disease/alcoholism Kidney disease Anaemia of chronic disease Endocrine failure/hypothyroidism Haemolysis Aplastic anaemia Classification of anaemia. Microcytic, hypochromic Normocytic, normochromic Macrocytic MCV <80fL MCV 80–95fL MCV >95fL MCH <27pg MCH ≥27pg Megaloblastic: vitamin B12 or folate deficiency Non‐megaloblastic: alcohol, liver disease, myelodysplasia, aplastic anaemia, etc. (see Table 5.10) Iron deficiency Many haemolytic anaemias Thalassaemia Anaemia of chronic disease (some cases) Lead poisoning Sideroblastic anaemia (some cases) Anaemia of chronic disease (some cases) After acute blood loss Factors impairing the normal reticulocyte response to anaemia. Marrow diseases, e.g. hypoplasia, infiltration by carcinoma, lymphoma, myeloma, acute leukaemia, tuberculosis Deficiency of iron, vitamin B12 or folate Lack of erythropoietin, e.g. renal disease Reduced tissue O2 consumption, e.g. myxoedema, protein deficiency Ineffective erythropoiesis, e.g. thalassaemia major, megaloblastic anaemia, myelodysplasia, myelofibrosis Chronic inflammatory or malignant disease Untitled 2 IRON DEFICIENCY ANAEMIA Microcytic anaemia—MCV ≤80 FL • The main causes of microcytic anaemia are iron deficiency and haemoglobulinopathy, particularly thalassaemia. Consider lead poisoning. • Iron-deficiency anaemia • Iron deficiency is the most common cause of anaemia worldwide. It is the biggest cause of microcytic anaemia, with the main differential diagnosis of microcytic anaemia being a haemoglobinopathy such as thalassaemia. However, it is caused by bleeding until proved otherwise. • Clinical and laboratory features An understanding of the interpretation of iron studies is important in management. • Microcytic anaemia • Serum ferritin level low (NR: F 15–200 mcg/L: M 30–300 mcg/L) <15 • Serum iron level low • Increased transferrin level • Microcytic hypochromic red cells • MCV ↓, MCH ↓, MCHC ↓ • Reduced transferrin saturation • Response to iron therapy Non-haematological effects of chronic iron deficiency • Angular cheilosis/stomatitis • Glossitis • Oesophageal webs • Atrophic gastritis • Brittle nails and koilonychias Causes • Blood loss • Menorrhagia • Gastrointestinal bleeding (e.g. carcinoma, haemorrhoids, peptic ulcer, hiatus hernia, GORD, NSAID therapy) • Frequent blood donations • Malignancy • Hookworm (common in tropics) • Increased physiological requirements • Prematurity, infant growth • Adolescent growth • Pregnancy • Malabsorption • Coeliac disease • Postgastrectomy • Dietary • Inadequate intake Special diets (e.g. fad, vegetarianism) • Pica—eating unnatural food, e.g. dirt, ashes Investigations • Investigations are based on the • history • physical examination, including the rectal examination. • If GIT bleeding is suspected, the faecal occult blood test is not considered very valuable but appropriate investigations include gastroscopy and colonoscopy, small bowel biopsy and small bowel enema. Haematological investigations: typical findings • • • • • • Microcytic, hypochromic red cells Anisocytosis (variation in size), poikilocytosis (shape)—pencil-shaped rods Low serum iron level Raised iron-binding capacity Serum ferritin level low (the most useful index) Soluble transferrin receptor factor—this factor is increased in iron deficiency, but not in chronic disease. Therefore, it is very helpful in differentiating iron deficiency from other forms. It is an indirect marker of what is happening in the bone marrow. The state of the iron stores is assessed by considering the serum iron, the serum ferritin and the serum transferrin levels in combination • Typically, in • iron deficiency, the serum iron and ferritin levels are low and the transferrin high, • but the serum iron level is also low in all infections— severe, mild and even subclinical—as well as in inflammatory states, malignancy and other chronic conditions. • Serum ferritin estimations are spuriously raised in liver disease of all types, chronic inflammatory conditions and malignancy; • transferrin is normally raised in pregnancy. Since each of these estimations can be altered in conditions other than iron deficiency, all three quantities have to be considered together to establish the iron status The interpretation of iron studies Condition Serum Fe TIBC % Transferrin Ferritin saturation Iron deficiency ↓ N or ↑ ↓ <10% ↓↓ β-thalassaemia N or ↑ N N or ↑ N or ↑ Anaemia of chronic disease ↓ N or ↓ ↓ N or ↑ Sideroblastic anaemia N or ↑ N N or ↑ ↑ ↓ ↑↑ ↑↑ Haemochromatosi ↑ s Treatment • Correct the identified cause • Diet—iron-rich foods, vitamin C-rich foods (Iron is present in meat and legumes as Fe+ + + and therefore requires gastric acid for conversion to Fe++. ) • Elemental iron supplements 100–200 mg daily (adults). • Iron preparations: oral iron (ferrous sulphate 1–2 tablets daily between meals for 6 months), e.g. Ferro- Gradumet or Ferro-grad C (avoid taking with milk) with orange juice or ascorbic acid until Hb is normal parenteral iron preferably by IV infusion is probably best reserved for special circumstances such as a failed trial of oral iron for symptomatic iron-deficiency anaemia (there is a risk of an allergic reaction, a serum sickness-like illness for 48 hours and post- infusion skin staining around the cannula site). Cover with an antihistamine or IV hydrocortisone 30 minutes beforehand. Infusion is best with ferric carboxymaltose in 0.9% (N) saline. Avoid blood transfusions if possible. IM iron is not recommended. Optimal adult diet for iron deficiency Adults should limit milk intake to 500 mL a day while on iron tablets Avoid excess caffeine, faT diets and excess processed bread Eat ample iron-rich foods (especially protein) Protein foods Meats—beef (especially), veal, pork, liver, poultry Fish and shellfish (e.g. oysters, sardines, tuna) Seeds (e.g. sesame, pumpkin) Eggs, especially egg yolk Fruits Dried fruit (e.g. prunes, figs, raisins, currants, peaches) Juices (e.g. prune, blackberry) Most fresh fruit Vegetables Greens (e.g. spinach, silver beet, lettuce) Dried peas and beans (e.g. kidney beans) Pumpkin, sweet potatoes Grains Iron-fortified breads and dry cereals Oatmeal cereal or better iron absorption, add foods rich in vitamin C (e.g. citrus fruits, cantaloupe, Brussels sprouts, broccoli, cauliflower) Response • Anaemia responds after about 2 weeks and is usually corrected after 2 months (if underlying cause addressed). • Oral iron is continued for 3 to 6 months to replenish stores. • Monitor progress with regular serum ferritin levels. • A serum ferritin level >50 mcg/L generally indicates adequate stores. Failure of iron therapy • poor compliance • continuing blood loss • malabsorption (e.g. severe coeliac disease) • incorrect diagnosis (e.g. thalassaemia minor, chronic disease) • bone marrow infiltration Anaemia of chronic disorders • One of the most common anaemias occurs in patients with a variety of chronic inflammatory and malignant diseases (Table 3.6). The characteristic features are: • 1 Normochromic, normocytic or mildly hypochromic (MCV rarely <75 fL) indices and red cell morphology. • 2 Mild and non‐progressive anaemia (haemoglobin rarely <90g/L) – the severity being related to the severity of the disease. • 3 Both the serum iron and TIBC are reduced. 4 The serum ferritin is normal or raised. 5 Bone marrow storage (reticuloendothelial) iron is normal • but erythroblast iron is reduced • pathogenesis of this anaemia appears to be related to decreased release of iron from macrophages to plasma because of raised serum hepcidin levels, reduced red cell lifespan and an inadequate erythropoietin response to anaemia caused by the effects of cytokines such as IL‐1 and tumour necrosis factor (TNF) on erythropoiesis. Causes of the anaemia of chronic disorders. Chronic inflammatory diseases Infections (e.g. pulmonary abscess, tuberculosis, osteomyelitis, pneumonia, bacterial endocarditis) Non‐infectious (e.g. rheumatoid arthritis, systemic lupus erythematosus and other connective tissue diseases, sarcoidosis, inflammatory bowel disease, liver disease) Malignant diseases Carcinoma, lymphoma, sarcoma Laboratory diagnosis of a hypochromic anaemia. Iron deficiency Chronic inflammation or malignancy Thalassaemia trait Sideroblastic (α or β) anaemia MCV/ MCH Reduced in relation Normal or mild to severity of reduction anaemia Reduced; very low for degree of anaemia Usually low in congenital type but MCV usually raised in acquired type Serum iron Reduced Reduced Normal Raised TIBC Raised Reduced Normal Normal Serum ferritin Reduced Normal or raised Normal Raised Bone marrow iron stores Absent Present Present Present Erythroblast iron Absent Absent Present Ring forms Haemoglobin electrophoresis Normal Normal Hb A2 raised in β form Normal Sideroblastic anaemia Classification of sideroblastic anaemia. Hereditary X chromosome linked ALA‐S mutation or rarely with spinocerebellar degeneration and ataxia Usually occurs in males, transmitted by females; also occurs rarely in females Other rare types (see text) Acquired Primary Myelodysplasia (refractory anaemia with ring sideroblasts) (see p. 178) N.B. Ring sideroblast formation (<15% of erythroblasts) may also occur in the bone marrow in: other malignant diseases of the marrow (e.g. other types of myelodysplasia, myelofibrosis, myeloid leukaemia, myeloma) drugs, e.g. antituberculous (isoniazid, cycloserine), alcohol, lead other benign conditions (e.g. haemolytic anaemia, megaloblastic anaemia, malabsorption, rheumatoid arthritis) ALA‐S, δ‐aminolaevulinic acid synthase. Lead poisoning • Lead inhibits both haem and globin synthesis at a number of points. In addition it interferes with the breakdown of RNA by inhibiting the enzyme pyrimidine 5′ nucleotidase, causing accumulation of denatured RNA in red cells, the RNA giving an appearance called basophilic stippling on the ordinary (Romanowsky) stain (see Fig. 2.17). The anaemia may be hypochromic or predominantly haemolytic, and the bone marrow may show ring sideroblasts. Free erythrocyte protopor phyrin is raised. Haemochromatosis • Hereditary haemochromatosis (HHC), which is a disorder of iron overload, is the most • • • • • • • • common serious single gene genetic disorder in our population. It is a common condition in which the total body iron concentration is increased to 20– 60 g (normal 4 g). The excess iron is deposited in and can damage several organs: liver—cirrhosis (10% develop cancer) pancreas—‘bronze’ diabetes skin—bronze or leaden grey colour heart—restrictive cardiomyopathy, pituitary—hypogonadism, impotence joints—arthralgia (especially hands), chondrocalcinosis It is usually hereditary (autosomal recessive = AR) or may be secondary to chronic haemolysis and multiple transfusions. Note: Hereditary haemochromatosis is the genetic condition; haemosiderosis is the secondary condition. Genetic profile • Being an autosomal recessive disorder, the patient must inherit two altered (mutated) copies of the gene. It is a problem mainly affecting Caucasians, usually from middle age onwards. • About 1 in 10 people are silent carriers of one mutated gene, while 1 in 200 are homozygous and are at risk of developing haemochromatosis. • These people can have it to a variable extent (the penetrance factor), and some are asymptomatic while others have a serious problem. It is rare for symptoms to manifest before the third decade. • The two common identified specific mutations in the HFE gene are C282Y and H63D (another is S65C): • homozygous C282Y—high risk for HHC • homozygous H63D—unlikely to develop clinical HHC • heterozygous C282Y and H63D—milder form of HHC Clinical features • Most patients are asymptomatic but may have extreme lethargy, abdominal discomfort, signs of chronic liver disease, polyuria and polydipsia, arthralgia, erectile dysfunction, loss of libido and joint signs. • Signs: look for hepatomegaly, very tanned skin, cardiac arrhythmias, joint swelling, testicular atrophy. The key diagnostic sensitive markers are serum transferrin saturation and the serum ferritin level. The serum iron level is not a good indicator. An elevated ferritin level is not diagnostic of HHC but is the best serum marker of iron overload. Diagnosis • Increased serum transferrin saturation: >50% (F); >60% (M) • Increased serum ferritin level: >200 mcg/L (F); >300 mcg/L (M) CT, MRI or FerriScan—increased iron deposition in liver • Liver biopsy (if liver function test enzymes are abnormal or ferritin >1000 mcg/L or hepatomegaly)—FerriScan now preferred • Genetic studies: HFE gene—a C282Y and/or H63D mutation • Screen first-degree relatives (serum ferritin levels and serum transferrin saturation in older relatives and genetic testing in younger ones). No need to screen before adulthood. HbEPG gene for pregnant patient and partner. • Routine screening not recommended Note: Full blood count (FBE) and erythrocyte sedimentation rate are normal. Management • Refer for specialist care • Weekly venesection 500 mL (250 mg iron) until serum iron stores are normal (may take at least 2 years), then every 3–4 months to keep serum ferritin level <100 mcg/L (usually 40–80 mcg/L), serum transferrin saturation <50% and iron levels normal • Desferrioxamine can be used but not as effective as venesection • Normal, healthy low-iron diet • Avoid or limit alcohol • Avoid iron tablets and vitamin C • Life expectancy is normal if treated before cirrhosis or diabetes Megaloblastic anaemias and other macrocytic anaemias •MEGALOBLASTIC ANAEMIAS Causes of megaloblastic anaemia. Vitamin B12 deficiency Folate deficiency Abnormalities of vitamin B12 or folate metabolism (e.g. transcobalamin deficiency, nitrous oxide, antifolate drugs) Other defects of DNA synthesis Congenital enzyme deficiencies (e.g. orotic aciduria) Acquired enzyme deficiencies (e.g. alcohol, therapy with hydroxyurea, cytosine arabinoside) Vitamin B12 and folate: nutritional aspects. Vitamin B12 Folate Normal daily dietary intake 7–30 μg 200–250 μg Main foods Animal produce only Most, especially liver, greens and yeast Cooking Little effect Easily destroyed Minimal adult daily requirement 1–2 μg 100–150 μg Body stores 2–3mg (sufficient for 2–4 years) 10–12mg (sufficient for 4 months) Absorption Site Mechanism Limit Ileum Intrinsic factor 2–3 μg/day Duodenum and jejunum Conversion to methyltetrahydrofolate 50–80% of dietary content Enterohepatic circulation 5–10 μg/day 90 μg/day Transport in plasma Most bound to haptocorrin; TC essential for cell uptake Weakly bound to albumin Major intracellular physiological forms Methyl‐ and deoxyadenosylcobalamin Reduced polyglutamate derivatives Causes of severe vitamin B12 deficiency. Nutritional Especially vegans Malabsorption Gastric causes Pernicious anaemia Congenital lack or abnormality of intrinsic factor Total or partial gastrectomy Intestinal causes Intestinal stagnant loop syndrome – jejunal diverticulosis, blind‐loop, stricture, etc. Chronic tropical sprue Ileal resection and Crohn’s disease Congenital selective malabsorption with proteinuria (autosomal recessive megaloblastic anaemia) Fish tapeworm Causes of mild vitamin B12 deficiency include poor diet and other causes of malabsorption of vitamin B12 (malabsorption of food B12 caused by atrophic gastritis particularly in the elderly, therapy with proton pump inhibitors or metformin), severe pancreatitis, gluten‐induced enteropathy and HIV infection. These • Vitamin B12 neuropathy (subacute combined degeneration of the cord) • Severe B12 deficiency can cause a progressive neuropathy affect- ing the peripheral sensory nerves and posterior and lateral columns (Fig. 5.9). The neuropathy is symmetrical and affects the lower limbs more than the upper limbs. The patient notices tingling in the feet, difficulty in walking and may fall over in the • dark. Rarely, optic atrophy or severe psychiatric symptoms are present. Anaemia may be severe, mild or even absent, but the blood film and bone marrow appearances are always abnormal. The peripheral neuropathy is usually reversible with B12 therapy but spinal cord recovery is incomplete, especially if the neu- ropathy has been present for more than a few weeks or months. • The cause of the neuropathy is likely to be related to the accumulation of S‐adenosyl homocysteine and reduced levels of S‐adenosyl methionine in nervous tissue resulting in defec- tive methylation of myelin and other substrates. Folate and B12 deficiency has been associated with reduced cognitive function and Alzheimer’s disease but no benefit has been shown for pro- phylactic folic acid or B12. Causes of folate deficiency. Nutritional Especially old age, institutions, poverty, famine, special diets, goat’s milk anaemia, etc. Malabsorption Tropical sprue, gluten‐induced enteropathy (adult or child). Possible contributory factor to folate deficiency in some patients with partial gastrectomy, extensive jejunal resection or Crohn’s disease Excess utilization Physiological Pregnancy and lactation, prematurity Pathological Haematological diseases: haemolytic anaemias, myelofibrosis Malignant disease: carcinoma, lymphoma, myeloma Inflammatory diseases: Crohn’s disease, tuberculosis, rheumatoid arthritis, psoriasis, exfoliative dermatitis, malaria Excess urinary folate loss Active liver disease, congestive heart failure Drugs Anticonvulsants, sulfasalazine Mixed Treatment of megaloblastic anaemia. Vitamin B12 deficiency Folate deficiency Compound Hydroxocobalamin Folic acid Route Intramuscular* Oral Dose 1000μg 5mg Initial dose 6 × 1000μg over 2–3 weeks Daily for 4 months Maintenance Prophylactic 1000μg every 3 months Depends on underlying disease; life‐long therapy may be needed in chronic inherited haemolytic anaemias, myelofibrosis, renal dialysis Total gastrectomy Ileal Pregnancy, severe haemolytic anaemias, • Macrocytic anaemias show an increased size of circulating red cells (MCV >98 fL). Causes include vitamin B12 (B12, cobalamin) or folate deficiency, alcohol, liver disease, hypothyroidism, myelodysplasia, paraproteinaemia, cytotoxic drugs, aplastic anaemia, pregnancy and the neonatal period. • B12 or folate deficiency cause megaloblastic anaemia, in which the bone marrow erythroblasts have a typical abnormal appearance. • Folates take part in biochemical reactions in DNA synthesis. B12 has an indirect role by its involvement in folate metabolism. • B12 deficiency may also cause a neuropathy due to damage to the spinal cord and peripheral nerves. • B12 deficiency is usually caused by B12 malabsorption brought about by pernicious anaemia in which there is autoimmune gastritis, resulting in severe deficiency of intrinsic factor, a glycoprotein made in the stomach which facilitates B12 absorption by the ileum. • ■ Other gastrointestinal diseases as well as a vegan diet may cause B12 deficiency. • ■ Folate deficiency may be caused by a poor diet, malabsorption (e.g. gluten‐induced enteropathy) or excess cell turnover (e.g. pregnancy, haemolytic anaemias, malignancy). • ■ Treatment of B12 deficiency is usually with injections of hydroxocobalamin and of folate deficiency with oral folic (pteroylglutamic) acid. • ■ Rare causes of megaloblastic anaemia include inborn errors of B12 or folate transport or metabolism, and defects of DNA synthesis not related to B12 or folate. Pernicious anaemia (PA) • This is caused by autoimmune attack on the gastric mucosa leading to atrophy of the stomach. The wall of the stomach becomes thin, with a plasma cell and lymphoid infiltrate of the lamina propria. Intestinal metaplasia may occur. There is achlorhydria and secretion of IF is absent or almost absent. Serum gastrin levels are raised. Helicobater pylori infection may initiate an autoimmune gastritis which presents in younger subjects as iron deficiency and in the elderly as PA. Pernicious anaemia: associations. Female Vitiligo Blue eyes Myxoedema Early greying Hashimoto’s disease Northern European Thyrotoxicosis Familial Addison’s disease Blood group A Hypoparathyroidism Hypogammaglobulinaemia Carcinoma of the stomach Causes of macrocytosis other than megaloblastic anaemia. Alcohol Liver disease Myxoedema Myelodysplastic syndromes Antimetabolite drugs, e.g. hydroxycarbamide Aplastic anaemia Pregnancy Smoking Reticulocytosis Myeloma and paraproteinaemia •Hemolytic anaemia Intravascular and extravascular haemolysis • Hemolytic anemias are characterized by an excessive breakdown of red blood cells (RBCs). They can be classified according to the cause of hemolysis (intrinsic or extrinsic) and by the location of hemolysis (intravascular or extravascular) • There are two mechanisms whereby red cells are destroyed in haemolytic anaemia. There may be excessive removal of red cells by cells of the RE system (extravascular haemolysis) or they may be broken down directly in the circulation (intravascular haemolysis) Laboratory findings • The laboratory findings are conveniently divided into three groups. • 1 Features of increased red cell breakdown: (a) serum bilirubin raised, unconjugated and bound to albumin; (b) urine urinobilinogen increased; (c) serum haptoglobins absent because the haptoglobins become saturated with haemoglobin and the complex is removed by RE cells. 2 Features of increased red cell production: a)reticulocytosis; b)bone marrow erythroid hyperplasia; the normal marrow myeloid:erythoid ratio of 2:1 to 12:1 is reduced to 1 : 1 or reversed. 3 Damaged red cells: a) morphology (e.g. microspherocytes, elliptocytes, fragments); (b) osmotic fragility; (c) specific enzyme, protein or DNA tests. Causes of intravascular haemolysis. Mismatched blood transfusion (usually ABO) G6PD deficiency with oxidant stress Red cell fragmentation syndromes Some severe autoimmune haemolytic anaemias Some drug‐ and infection‐induced haemolytic anaemias Paroxysmal nocturnal haemoglobinuria March haemoglobinuria Unstable haemoglobin G6PD, glucose‐6‐phosphate dehydrogenase • The main laboratory features of intravascular haemolysis therefore are (Fig. 6.3): • 1 Haemoglobinaemia and haemoglobinuria. 2 Haemosiderinuria. 3 Methaemalbuminaemia (detected spectrophotometrically). Classification of haemolytic anaemias. Hereditary Acquired Membrane Hereditary spherocytosis, hereditary elliptocytosis Metabolism G6PD deficiency, pyruvate kinase deficiency Haemoglobin Genetic abnormalities (Hb S, Hb C, unstable); see Chapter 7 Immune Autoimmune Warm antibody type (see Table 6.5) Cold antibody type Alloimmune Haemolytic transfusion reactions Haemolytic disease of the newborn Allografts, especially marrow transplantation Drug associated Red cell fragmentation syndromes See Table 6.6 March haemoglobinuria Infections Malaria, clostridia Chemical and physical agents Especially drugs, industrial/domestic substances, burns Secondary Liver and renal disease Paroxysmal nocturnal haemoglobinuria (see Chapter 22) • Hereditary haemolytic anaemias Hereditary spherocytosis • This is the commonest cause of inherited haemolytic anaemia in northern Europeans. • It is an autosomal dominant disorder of variable severity, although in 25% of patients neither parent is affected, suggesting spontaneous mutation in some instances. • Jaundice may present at birth or be delayed or occur not at all. • Splenomegaly is a feature and • splenectomy is considered to be the treatment of choice in severe cases. • Maintenance of folic acid levels is important. Glucose-6-phosphate dehydrogenase deficiency • G6PD deficiency is a common disorder affecting over 400 million people worldwide. • It is the most common red cell enzyme defect that causes episodic haemolytic anaemia because of the decreased ability of red blood cells to cope with oxidative stresses. • It is an X-linked recessive inherited disorder with a high prevalence among people of African, Mediterranean or Asian ancestry. • In some countries such as Malaysia there is a national screening program. The important clinical features are: • asymptomatic in many • neonatal jaundice—infants at risk should be observed after delivery (at least 5 days) • episodic acute haemolytic anaemia—triggered by antioxidants and infections, and drugs, especially antimalarials, sulfonamides, nitrofurantoin, quinolones, traditional medicines, vitamins C and K, high dose aspirin, fava (broad) beans and naphthalene (e.g. moth balls) • There is no specific treatment. Known precipitants should be avoided. Avoid penicillin and probenecid. • Diagnosis is by G6PD assay and a blood film during an attack. Agents that may cause haemolytic anaemia in glucose‐6‐phosphate dehydrogenase (G6PD) deficiency. Infections and other acute illnesses (e.g. diabetic ketoacidosis) Drugs Antimalarials (e.g. primaquine, pamaquine, chloroquine, Fansidar, Maloprim) Sulphonamides and sulphones (e.g. co‐trimoxazole, sulfanilamide, dapsone, sufasalazine) Other antibacterial agents (e.g. nitrofurans, chloramphenicol) Analgesics (e.g. aspirin), moderate doses are safe Antihelminths (e.g. β‐naphthol, stibophen) Miscellaneous (e.g. vitamin K analogues, naphthalene (mothballs), probenecid) Fava beans (possibly other vegetables) N.B. Many common drugs have been reported to precipitate haemolysis in G6PD deficiency in some patients (e.g. aspirin, quinine and penicillin) but not at conventional dosage. Acquired haemolytic anaemias • Immune haemolytic anaemias • Autoimmune haemolytic anaemias Immune haemolytic anaemias: classification. Warm type Cold type Autoimmune Idiopathic Secondary SLE, other ‘autoimmune’ diseases CLL, lymphomas Drugs (e.g. methyldopa) Idiopathic Secondary Infections – Mycoplasma pneumonia, infectious mononucleosis Lymphoma Paroxysmal cold haemoglobinuria (rare, sometimes associated with infections, e.g. syphilis) Alloimmune Induced by red cell antigens Haemolytic transfusion reactions Haemolytic disease of the newborn post stem cell grafts Drug induced Drug–red cell membrane complex Immune Hemolytic anaemia • hemolytic anaemia are those with a red cell membrane defect and include • hereditary spherocytosis, • hereditary elliptocytosis and • hereditary stomatocytosis. • Haemolytic anaemia is caused by shortening of the red cell life. The red cells may break down in the reticuloendothelial system (extravascular) or in the circulation (intravascular). • Haemolytic anaemia may be caused by inherited red cell defects, which are usually intrinsic to the red cell, or to acquired causes, which are usually caused by an abnormality of the red cell environment. • Features of extravascular haemolysis include jaundice, gallstones and splenomegaly with raised reticulocytes, unconjugated bilirubin and absent haptoglobins. • In intravascular haemolysis (e.g. caused by ABO mismatched blood transfusion), there is haemoglobinaemia, methaemalbuminaemia, haemoglobinuria and haemosiderinuria. • ■ Genetic defects include those of the red cell membrane (e.g. hereditary spherocytosis), enzyme deficiencies (e.g. glucose‐6‐phosphate dehydrogenase or pyruvate kinase deficiency) or haemoglobin defects (e.g. sickle cell anaemia, see Chapter 7). • ■ Acquired causes of haemolytic anaemia include warm or cold, auto‐ or allo‐antibodies to red cells, red cell fragmentation syndromes, infections, toxins and paroxysmal nocturnal haemoglobinuria Microangiopathic hemolytic anemia • MAHA • A type of hemolytic anemia that is the result of mechanical damage to erythrocytes by microthrombi in small blood vessels. • Characterized by schistocytes on the peripheral blood smear. • Common causes include, oThrombotic thrombocytopenic purpura oHemolytic uremic syndrome oDisseminated intravascular coagulation oHELLP syndrome. • If the patient has severe symptoms of anemia or a lifethreatening cause is suspected (i.e., TTP/HUS, disseminated intravascular coagulation, HELLP syndrome, acute hemolytic transfusion reaction), proceed directly to treatment in parallel with diagnostic evaluation. Hereditary haemoglobinopathies • The commonest haemoglobinopathies are the thalassaemias, which are caused by a deficiency in the quality of globin chains, whereas other haemoglobinopathies are caused by structural variations in the globin chain. These conditions include HbS (sickle cell), HbC, HbD, HbE, HbO and HbLepore. Thalassaemia Four gene deletion α‐thalassaemia β0-Thalassaemia trait Thalassaemia major β+‐Thalassaemia trait Transfusion dependent, homozygous β0‐thalassaemia or other combinations of β‐thalassaemia trait Thalassaemia intermedia (non‐transfusion dependent thalassaemia) α0‐Thalassaemia trait See Table 7.3 α+‐Thalassaemia trait Genetic Type Haplotype Heterozygous Homozygous thalassaemia trait (minor)* – –/ MCV, MCH low Hydrops fetalis MCV, MCH minimally reduced As heterozygous α0‐thalassaemia† Compound heterozygote α0α+ (– –/–α) is haemoglobin H disease α‐Thalassaemias† α0 α+ –α/ Thalassaemia • This inherited condition is seen mainly (although not exclusively) in people from the Mediterranean basin, the Middle East, north and central India and South-East Asia, including south China. • The heterozygous form is usually asymptomatic; patients show little if any anaemia and require no treatment. The condition is relatively common in people from these areas. • The homozygous form is a very severe congenital anaemia needing lifelong transfusional support but is comparatively rare, even among the populations prone to thalassaemia • The key to the diagnosis of heterozygous thalassaemia minor is significant microcytosis quite out of proportion to the normal Hb or slight anaemia, and confirmed by finding a raised HbA2 on Hb electrophoresis. • DNA screening analysis is now available. • The importance of recognising the condition lies in distinguishing it from iron-deficiency anaemia, for iron does not help people with thalassaemia and is theoretically contraindicated. Even more importantly, it lies in recognising the risk that, if both parents have thalassaemia minor, they run a one in four chance of having a baby with thalassaemia major in every pregnancy, with devastating consequences for both the affected child and the whole family. • Treatment of thalassaemia major is transfusion to a high normal Hb with packed cells plus desferrioxamine. Thalassemia • the most common human single-gene disorders in the world, are a group of hereditary disorders characterised by a defect in the synthesis of one or more of the globin chains (α or β)—there are two of each (α2, β2). • This causes defective haemoglobin synthesis leading to hypochromic microcytic anaemia. • α-thalassaemia is usually seen in people of Asian origin. • β-thalassaemia is seen in certain ethnic groups from the Mediterranean, the Middle East, South- East Asia and the Indian subcontinent. • However, in our multicultural communities one cannot assume a person’s origins. • It is recommended that all women of child-bearing age be screened for thalassaemia. • The thalassaemias are described as ‘trait’ when there are laboratory features without clinical expression. Genetic profile • α-thalassaemia is usually due to the deletion of one or more of the four genes for α-globin, • the severity depending on the number of genes deleted: • deletion of all four genes—α-thalassaemia (hydrops fetalis); • of three genes—haemoglobin H disease, which results in lifelong anaemia of mildto-moderate degree; • of one or two genes—a symptomless carrier. • In β-thalassaemia, the β-chains are produced in decreased quantity rather than having large deletions. People who have two mutations (one in each β-globin gene) have β-thalassaemia major. • β-thalassaemia minor—a single mutation (heterozygous)—the carrier or trait state • β-thalassaemia major—two mutations (homozygous)—the person who has the disorder • If both parents are carriers, there is a 1 in 4 chance that their child will have the disorder. Clinical features • Carriers are clinically asymptomatic and do not need treatment apart from counselling. • Patients with thalassaemia major present with symptoms of severe anaemia (haemolytic anaemia). • Without treatment, children with thalassaemia major are lethargic and inactive, show a failure to thrive or to grow normally, and delayed puberty, hepatosplenomegaly and jaundice. • Signs usually appear after 6 months and death from cardiac failure used to be common but with regular blood transfusions and ironchelating treatment people can now live in good health. DxT pallor + jaundice + hepatosplenomegaly → thalassaemia major • Diagnosis • FBE: in most carriers the mean corpuscular haemoglobin/mean corpuscular volume is low but can be normal. There is usually mild hypochromic microcytic anaemia but this is severe with the homozygous type. • Haemoglobin electrophoresis: measures relative amounts of normal adult haemoglobin (HbA) and other variants (e.g. HbA2, HbF). This will detect most carriers. • Serum ferritin level: helps distinguish from iron deficiency, which has a similar blood film. • DNA analysis: for mutation detection (mainly used to detect or confirm carriers). Treatment for thalassaemia major • Treatment is based on a regular blood transfusion schedule for anaemia. • Avoid iron supplements. • Folate supplementation and a low-iron diet are advisable. • Excess iron is removed by iron chelation (e.g. desferrioxamine). • Allogeneic bone marrow transplantation has been used with success. • Splenectomy may be appropriate. Sickle-cell disorders • The most important abnormality in the haemoglobin (Hb) chain is sickle-cell haemoglobin (HbS), which results from a single base mutation of adenine to thymine, leading to a substitution of valine for glutamine at position 6 on the β-globin chain. • The defective Hb causes the red cells to become deformed in shape— ‘sickled’. • The sickled cells tend to flow poorly and clog the microcirculation, resulting in hypoxia, which compounds the sickling. Such attacks, which result in tissue infarction, are called ‘crises’. • Sickling is precipitated by infection, hypoxia, dehydration, cold and acidosis, and may complicate operations. • The autosomal recessive disorder occurs mainly in Africans (25% carry the gene), but it is also found in India, South-East Asia, the Middle East and southern Europe. • Heterozygous state for HbS = sickle-cell trait • Homozygous state = sickle-cell anaemia/disease • Sickle-cell anaemia • This varies from being mild or asymptomatic to a severe haemolytic anaemia and recurrent painful crises. • It may present in children with anaemia and mild jaundice. • Children may develop digits of varying lengths from the hand-and-foot syndrome due to infarcts of small bones. • Features of infarctive sickle crises include: • bone pain (usually limb bones) • abdominal pain • chest—pleuritic pain kidney—haematuria • spleen—painful infarcts • precipitated by cold, hypoxia, dehydration or infection • • • • • • Hb electrophoresis is needed to confirm the diagnosis. Long-term problems ; chronic leg ulcers, susceptibility to infection, aseptic necrosis of bone (especially head of femur), blindness and chronic kidney disease. T he prognosis is variable. Children in Africa often die within the first year of life. Infection is the commonest cause of death. • Sickle-cell trait • People with this usually have no symptoms unless they are exposed to prolonged hypoxia, such as anaesthesia and flying in non-pressurised aircraft. The disorder is protective against malaria. Classification of bleeding disorders Vascular disorders Inherited • Hereditary haemorrhagic telangiectasia • Connective tissue disease, e.g. Marfan syndrome • Easy bruising syndrome Acquired • Senile purpura • Infection, e.g. dengue, meningococcal • Henoch-Schonlein purpura • Corticosteroid purpura • Vitamin C deficiency (scurvy) • Painful bruising syndrome Platelet disorders Inherited Fanconi syndrome Glanzmann disease Acquired (immune) Idiopathic thrombocytopenic purpura Aplastic anaemia Drug induced thrombocytopenia, e.g. heparin Thrombotic thrombocytopenia purpura Post-transfusion purpura Non-immune Disseminated intravascular coagulation Myeloproliferative disorders Kidney failure/uraemia Bone marrow replacement (e.g. leukaemia) or failure Coagulation disorders • • • • • • • • • • Inherited Haemophilia A Haemophilia B von Willebrand disease (types 1, 2 and 3) Acquired Disseminated intravascular coagulation (DIC) Vitamin K deficiency Oral anticoagulation therapy or overdose Acquired haemophilia Liver disease The coagulation factors. Factor number Descriptive name Active form I Fibrinogen Fibrin subunit II Prothrombin Serine protease III Tissue factor Receptor/cofact or* V Labile factor Cofactor VII Proconvertin Serine protease VIII Antihaemophilic factor Cofactor Table 3.7 Laboratory diagnosis of a hypochromic anaemia. Iron deficiency Chronic inflammation or malignancy Thalassaemia trait (α or β) Sideroblastic anaemia MCV/ MCH Reduced in relation to severity of anaemia Normal or mild reduction Reduced; very low for degree of anaemia Usually low in congenital type but MCV usually raised in acquired type Serum iron Reduced Reduced Normal Raised TIBC Raised Reduced Normal Normal Serum ferritin Reduced Normal or raised Normal Raised Bone marrow iron stores Absent Present Present Present Erythroblast iron Absent Absent Present Ring forms Haemoglobin electrophoresis Normal Normal Hb A2 raised in β form Normal MCH, mean corpuscular haemoglobin; MCV, mean corpuscular volume; TIBC, total iron‐binding capacity. • Aplastic anaemia • Aplastic (hypoplastic) anaemia is defined as pancytopenia resulting from hypoplasia of the bone marrow (Fig 22.1). It is classified into primary (congenital or acquired) or secondary types Causes of pancytopenia. Decreased bone marrow function Aplasia (reduction of haemopoietic stem cells) Acute leukaemia, myelodysplasia, myeloma Infiltration with lymphoma, solid tumours, tuberculosis Megaloblastic anaemia Paroxysmal nocturnal haemoglobinuria Myelofibrosis Haemophagocytic syndrome Increased peripheral destruction Splenomegaly Causes of aplastic anaemia. Primary Secondary Congenital (Fanconi and non-Fanconi types) Ionizing radiation: accidental exposure (radiotherapy, radioactive isotopes,) Idiopathic acquired Chemicals: benzene, organophosphates and other organic solvents, DDT and other pesticides, recreational drugs (ecstasy) Drugs: Those that regularly cause marrow depression (e.g. busulfan, melphalan, cyclophosphamide, anthracyclines, nitrosoureas) Those that occasionally or rarely cause marrow depression (e.g. chloramphenicol, sulphonamides, gold, anti-inflammatory, antithyroid, psychotrophic, anticonvulsant/ antidepressant drugs) Viruses: viral hepatitis (non-A, non-B, non-C, non-G in most cases), EBV Autoimmune diseases: systemic lupus erythematosus Transfusion associated GVHD (see p. 342) Screening tests used in the diagnosis of coagulation disorders (see also Fig. 24.10) Screening tests Abnormalities indicated by prolongation Most common cause of coagulation disorder Thrombin time (TT) Deficiency or abnormality of fibrinogen or inhibition of thrombin DIC Heparin therapy by heparin or FDPs Prothrombin time (PT) Deficiency or inhibition of one or more of the following coagulation factors: VII, X, V, II, fibrinogen Liver disease Warfarin therapy DIC Activated partial thromboplastin time (APTT or PTTK) Deficiency or inhibition of one or more of the following coagulation factors: XII, XI, IX (Christmas disease), VIII (haemophilia), X, V, II, fibrinogen Haemophilia, Christmas disease (+ conditions above) Fibrinogen quantitation Fibrinogen deficiency DIC, liver disease DIC, disseminated intravascular coagulation; FDPs, fibrin degradation products. N.B. Platelet count and the tests of platelet function are also used in screening patients with a bleeding disorder ( • Normal haemostasis requires vasoconstriction, platelet aggregation and blood coagulation. The intact endothelial cell separates collagen and other subendothelial connective tissues that would stimulate platelet aggregation from circulating blood. The endothelial cells also produce prostacyclin, nitric oxide and an ectonucleotidase, which inhibit platelet aggregation. • Platelets are produced from megakaryocytes in the bone marrow stimulated by thrombopoietin. They have surface glycoproteins which facilitate direct adherence to subendothelial tissues and also, via von Willebrand factor, to collagen, to other platelets (aggregation) and to fibrinogen. Platelets contain different types of storage granules which are released after platelet activation. • ■ Blood coagulation in vivo in response to vascular injury commences with tissue factor binding to clotting factor VII and this initiates a cascade which results in thrombin generation. Thrombin then activates cofactors VIII and V and factor XI which greatly amplify the coagulation pathway resulting in a fibrin clot. • ■ Coagulation factor inhibitors include antithrombin, protein C and protein S. • ■ Dissolution of fibrin clots (fibrinolysis) occurs by activation of plasminogen to plasmin. • ■ Tests of haemostatic function include the thrombin time (TT), prothrombin time (PT), activated partial thromboplastin time (APTT) as well as individual coagulation factor assays and assay of von Willebrand factor. Tests of platelet function include the PFA‐100 and platelet aggregation tests. Causes of thrombocytopenia. Failure of platelet production Selective megakaryocyte depression rare congenital defects (see text) drugs, chemicals, viral infections Part of general bone marrow failure cytotoxic drugs radiotherapy aplastic anaemia leukaemia myelodysplastic syndromes myelofibrosis marrow infiltration (e.g. carcinoma, lymphoma, Gaucher’s disease) multiple myeloma megaloblastic anaemia HIV infection Increased consumption of platelets Immune autoimmune idiopathic associated with systemic lupus erythematosus, chronic lymphocytic leukaemia or lymphoma; infections: Helicobacter pylori, HIV, other viruses, malaria drug‐induced, e.g. heparin post‐transfusional purpura feto‐maternal alloimmune thrombocytopenia Thrombocytopenia as a result of drugs or toxins. Bone marrow suppression Predictable (dose‐related) ionizing radiation, cytotoxic drugs, ethanol Occasional chloramphenicol, co‐trimoxazole, idoxuridine, penicillamine, organic arsenicals, benzene, etc. Immune mechanisms (proven or probable) Analgesics, anti‐inflammatory drugs gold salts Antimicrobials penicillins, rifamycin, sulphonamides, trimethoprim, para‐ aminosalicylate Sedatives, anticonvulsants diazepam, sodium valproate, carbamazepine Diuretics acetazolamide, chlorathiazides, furosemide Antidiabetics chlorpropamide, tolbutamide Others digitoxin, heparin, methyldopa, oxyprenolol, quinine, quinidine Thrombotic thrombocytopenic purpura (TTP) • TTP has traditionally been described as a pentad of • thrombocytopenia, microangiopathic haemolytic anaemia, neurological abnormalities, renal failure and fever. Main clinical and laboratory findings in haemophilia A, factor IX deficiency (haemophilia B, Christmas disease) and von Willebrand disease. Inheritance Haemophilia A Factor IX deficiency von Willebrand disease Sex‐linked Sex‐linked Dominant (incomplete) Main sites of haemorrhage Muscle, joints, post‐ trauma or postoperative Muscle, joints, post‐trauma or postoperative Mucous membranes, skin cuts, post‐trauma or postoperative Platelet count Normal Normal Normal PFA‐100 Normal Normal Prolonged Prothrombin time Normal Normal Normal Partial thromboplastin time Prolonged Prolonged Prolonged or normal Haemostasis tests: typical results in acquired bleeding disorders. Platelet count Prothromb in time Activated partial thrombopl astin time T ti Liver disease Low Prolonged Prolonged N (r pr DIC Low Prolonged Prolonged G pr Massive transfusion Low Prolonged Prolonged N Coumarin anticoagula nts Normal Grossly prolonged Prolonged N Heparin Normal (rarely low) Mildly prolonged Prolonged P Circulating Indications for the use of fresh frozen plasma (National Institutes of Health Consensus Guidelines). Coagulation factor deficiency (PCC where specific or combined factor concentrate is not available) Reversal of warfarin effect (only if PCC is unavailable) Multiple coagulation defects (e.g. in patients with liver disease, DIC) (PCC are much better, plasma is virtually useless) Massive blood transfusion with coagulopathy and clinical bleeding Thrombotic thrombocytopenic purpura Some patients with immunodeficiency syndromes DIC, disseminated intravascular coagulation; PCC, prothrombin complex concentrates. Venous thrombosis Pathogenesis and risk factors • Virchow’s triad suggests that there are three components that are important in thrombus formation: 1 Slowing down of blood flow; 2 Hypercoagulability of the blood; 3 Vessel wall damage.