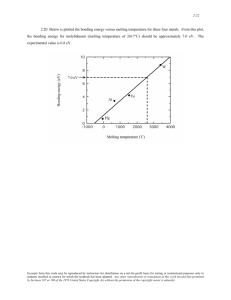
Assignment 5 1. (4 marks) Explain what is understood by re-entrant melting and why this phenomenon can occur. Give an example of at least one other material where this phenomenon occurs (besides Na, Mg and Al discussed in this paper)? Re-entrant melting is generally understood to be a phenomenon which occurs when a substance’s melting point starts to decrease below a certain pressure. A melting curve, which generally increases as pressure increases, may eventually invert its trend such that the melting point decreases with pressure after this point. As a result of this, when a material is compressed at a constant temperature (which is smaller than its maximum melting temperature, or the turning point of the melting curve), one will observe the liquid-solid-liquid sequence of phases which characterize re-entrant melting (shown in the sketch below). This is an especially surprising result as this implies that metals could have a liquid phase which is denser than its solid phase. Other materials for which this phenomenon has been observed so far include other alkali metals such as lithium (Rousseau et al., 2011). 2. (6 marks) Describe the melting simulation method used, in particular include a short description on how the potential energy of the system is computed and what method is used to simulate the melting process itself. In this paper, the authors used the coexistence method to simulate melting of the relevant systems. This method is commonly used to investigate the thermodynamic properties of a system at a phase transition point, and the information it provides can then be extrapolated to find the melting point of a material. Within this method, the simulation is first initialized within a cell containing a region with the material in its solid phase and a region with the material in its liquid phase, given a desired set of conditions. Then, the simulation is run at a specific temperature for enough time that the system reaches an equilibrium between the solid and liquid phases. While equilibration occurs, data is collected periodically on the properties of the system. The simulation is then run in the same way for different temperatures, and statistical analysis is undertaken on the resulting data set to estimate the temperature at which the solidliquid coexistence is the most prominent- this corresponds to the approximate melting temperature of the material under the specified conditions. During each step of the simulation, the potential energy of the system needs to be calculated in order to determine its behavior. This computation is undertaken by VASP, which uses the exchange-correlation energy within density functional theory (DFT) to account for the contribution of electron-electron interactions towards the potential energy, and pseudopotentials to capture the effect of electron-ion interactions. To describe the electron-electron interactions, DFT first makes an initial guess for the electron density of the system (typically using atomic positions or other approximations) and then undergoes several iterations (solving the KohnSham equations each time) to find the electron density which minimizes the total energy of the system. The interactions are then calculated using the exchange-correlation energy, which is estimated using the generalized-gradient approximation. The electron-ion interactions are then calculated using pseudopotentials, which reduce the computational effort as they essentially replace the full electron-ion potential with an approximate potential which only acts on the valence electrons (with the core electron interactions being implicitly included within the pseudopotentials). In the case of this study, the pseudopotentials are generated using the projector-augmented wave (PAW) method and verified by comparison with code that calculates the electron-ion interactions more rigorously. Because of the specialized conditions of this study (the system being under high pressures), a correction to the pseudopotentials is also made treating the semi-core s and p states as valence states. 3. (6 marks) Describe the information contained in the phase diagram of sodium shown in Fig. 1. Include sketches of the conventional unit cells of the three solid phases of sodium found up to about 110 GPa. You will have to do a bit of literature search for the solid phase encountered above about 105 GPa. ® The melting curve of sodium has been experimentally observed by Gregoryanz et al. to increase at low pressures towards a peak at about 30GPa where it decreases until reaching a plateau at around 65-80GPa, and then decreases again at a higher rate, with the melting point eventually reaching room temperature at about 120GPa. ® However, comparison of Gregoryanz’s work with its predecessors at low pressures shows significant discrepancies, with Gregoryanz’s work showing larger melting temperature and a larger gradient for the melting curve. ® Several studies have been conducted using DFT in combination with different computational methods to calculate the melting curve of sodium. The melting curves from these studies agree with each other, and with early experimental results at low pressures, but differ vastly from the melting curve recorded by Gregoryanz et al. ® The melting curve calculated within this study agrees closely with two other DFT calculations (and likely establish the DFT melting curve of sodium) and with the early experimental results, but is still very different from that recorded by Gregoryanz et al. ® The existence of reentrant melting within sodium at about 750 K and 35 GPa is confirmed by this study. ® At low pressures (up to about 65GPa), the solid phase of sodium is a b.c.c. lattice with the following conventional unit cell (from textbook): ® At higher pressures (around the range 65-105GPa), the solid phase of sodium is an f.c.c. lattice with the conventional unit cell shown on the right (also from textbook): ® At very high pressures (between about 105GPa and 120GPa), the solid phase of sodium enters the more complex cI16 phase (McMahon et al., 2007). 4. (1 bonus mark) Can you find a possible explanation why this less symmetric structure is adopted under these high pressures? As the pressure increases, the lattice becomes more squished together, meaning the interatomic distances between the sodium ions decrease. This change in interatomic distance could somehow change the interactions between either the ions themselves or the electrons, such that the lowest energy configuration of the system is now given within this structure. 5. (4 marks) Describe how the extremely high pressures are achieved in experiments. (You should find some more information in the experimental work by Gregoryanz et al. mentioned in the paper.) Gregoryanz et al. state that their measurements were carried out using modified hightemperature piston-cylinder Mao-Bell diamond anvil cells equipped with double heaters and thermocouples. Diamond anvil cells are an experimental apparatus which allow for extremely high pressures to be applied to a material, by compressing a small sample between two anvils made of synthetic diamond. Diamond is incredibly strong and hard, which means that very large pressures can be applied without shattering the anvils, and the truncated cone shape of the diamonds allow for multiplication of the input force applied. In this experiment, the sodium sample was placed inside the cell within an Ar atmosphere, contained by the diamond anvils and stainless steel gaskets. Pressure is then applied to the anvils using a piston-cylinder assembly, which is then transferred to the Ar gas and then to the sodium sample. The pistoncylinder set up allows for much larger pressures to be applied with less mechanical effort, and the Mao-Bell configuration adds increased stability and ensures the sample does not escape its containment under large pressures. The double heaters and thermocouples are then used to more precisely control the temperature of the experimental set-up. These experimental techniques combined allow for extremely high pressures to be applied to the sodium sample within a controlled environment. 6. (4 marks) Summarize the possible explanations for the discrepancies observed between experimental and computational data given in the paper. The paper states that both the experimental and computational data have shortcomings which may be causing this discrepancy. On the experimental side, they cite challenging experimental conditions, such as the high reactivity of sodium (which could cause it to react with the air, impurities within the cell, or the diamond anvils at high pressures) which may be causing inaccuracies in the experimentally determined melting curve. From the computational side, the exchange-correlation energy within the DFT model which is calculated using the generalized- gradient approximation is just an estimate and may not accurately describe the properties of sodium. This may lead to flaws in the calculated melting curve which mean that it does not match up with experimental results. 7. (6 marks) State two possible explanations for a volume reduction in the liquid phase with respect to the solid phase. Which of these two explanations is favoured by the authors in this publication and what are the reasons given. The authors state that volume can be composed into two factors: the coordination number (which describes the number of other atoms surrounding a given atom) and the pairwise distance between nearest neighbors (which describes how close atoms are to each other). Therefore, the volume reduction for the liquid phase could be explained by a change in either of these factors- if the structure of the liquid phase has a larger coordination number, or the interatomic distance within the liquid phase is smaller, these would both make the liquid phase more compact, leading to a smaller volume. The authors also cite experimental results which support each of these two explanations- on one hand, there is evidence that the solid structures formed by sodium and magnesium have low coordination numbers, which could lead to an increase in coordination number when melting; however, the work by Raty et al. shows that there is a softening of intermolecular interactions which occurs at a faster rate in liquids for growing pressures, which causes decreased atomic repulsion and smaller interatomic distances. Of these two explanations, the authors support the latter. To refute the first explanation, they state that the liquid and solid phases of magnesium do not have a significant difference in coordination number, even though within this explanation the volume reduction in the liquid phase would imply that the coordination number of the liquid is much larger than the low coordination number seen in the solid bcc structure of magnesium. Furthermore, in the regions of the sodium phase diagram where the solid fcc structure is stable (and therefore the coordination number is consistently at a maximum), a volume decrease is still observed, even though the first explanation would imply otherwise. Therefore, it is illogical to link changing coordination numbers to the decrease in volume of the liquid phase. To confirm the second explanation, the authors employ a computational method to change the radial distances (while keeping the coordination number constant), and confirm that the volume difference between the liquid and solid phases becomes negative once the radial distances have been sufficiently compressed. 8. (4 marks) Explain how the results in Fig. 4 were obtained and give their interpretation. The results are based on the computational method that the authors have used to scale the radial distances without changing the coordination number. This method involves taking a random “snapshot” of the system in both the solid and liquid phases, and then estimating the pressure-volume relation by uniformly scaling the lattice vectors in order to simulate expanding the structure (or increasing the volume of the material) and then observing the resultant effect on the pressure. Within this method, the coordination numbers are untouched as the atomic coordinates are not altered. The results in Fig. 4 were obtained by simulating expansion on one random solid and liquid snapshot of sodium, magnesium, and aluminum. We first have a graph for each material showing the pressure as the volume increases for both the solid and liquid, which shows the expected negative slope and a very small deviation of the behavior of pressure for increasing volume between the two phases. The second set of graphs shows a much more zoomed in picture of the difference between the pressure of the liquid phase and the pressure of the solid phase as the volume increases. We can see that initially, Pl – Ps is negative, but as the volume increases, this value becomes less negative until eventually Pl > Ps and the pressure of the liquid phase is larger than that of the solid phase. Using the relationship between pressure and volume, we can use this trend to infer that as the lattice vectors scale negatively (the pairwise distance decreases), we initially have that Vl > Vs , but for high pressures (where reentrant melting occurs) we get that Vs > Vl, which causes the negative volume change during reentrant melting as the phase transition from solid to liquid causes the volume to reduce. This confirms that the negative volume change is related to the radial distance, as the computational method was able to reproduce the desired volume difference between the solid and liquid phases by changing only the radial distances and not the coordination numbers of the phases. 9. (4 marks) State the two main conclusions of the paper. ® According to the DFT melting curves calculated by the authors, magnesium and aluminum also exhibit reentrant melting at very high pressures (it isn’t as rare as we thought!) ® The theory that the origin of reentrant melting lies in the softening of interatomic potentials (causing a decrease in pairwise distance between nearest neighbors), and thus a smaller volume in the liquid phase than the solid with increasing pressures, is confirmed. 10. (2 marks) Several computational studies have been performed and are summarized in the paper - what do all these studies have in common? They all use density functional theory to calculate potential energies, which are inputted into the various computational methods used to determine the melting curve for a material. 11. (3 bonus marks) Write a short proposal (one or two paragraphs) for a follow-up computational simulation study on sodium that could be performed to extend and thus check the simulations performed up to now. This isn’t really an answer to the question, but I would be way more curious to try to reproduce the experimental results by Gregoryanz et al. and see if I could get them closer to the results predicted by the various computational simulations- from what I can tell, no one has tried to confirm the experimental measurements at high pressures so it’s entirely possible that there are errors in the results which cause the large discrepancies with the simulations. References: Rousseau, B., Xie, Y., Ma, Y. et al. Exotic high pressure behavior of light alkali metals, lithium and sodium. Eur. Phys. J. B 81, 1–14 (2011). https://doi.org/10.1140/epjb/e2011-10972-9 McMahon, M., Gregoryanz, E., Lundegaard, L. F., Loa, I., Guillaume, C. L., Nelmes, R. J., Kleppe, A. K., M. Amboage, Wilhelm, H., & Jephcoat, A. P. (2007). Structure of sodium above 100 GPa by single-crystal x-ray diffraction. 104(44), 17297–17299. https://doi.org/10.1073/pnas.0709309104 Simon, S. H. (2013). The Oxford Solid State Basics. United Kingdom: OUP Oxford.