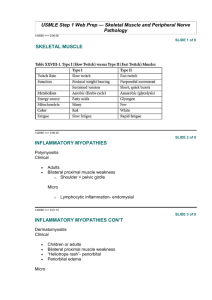
MYASTHENIA GRAVIS Neuromuscular junction disorder An autoimmune diseases caused by autoantibodies. Target is neuromuscular junction with fluctuating muscle weakness. Inherited o Dystrophinopathies 1. Duchenne Muscular Dystrophy, DMD 2. Becker Muscular Dystrophy, BMD Acquired o Inflammatory Myopathies 1. Polymyositis 2. Dermatomyositis 3. Inclusion body myositis AETIOLOGY Autoantibodies block the function of postsynaptic acetylcholine receptors at motor end plates. Results in degradation and depletion of the receptors. 30 percent of patient, young, have thymic hyperplasia meanwhile 10 percent of patients have thymoma. These thymic lesions may perturb tolerance to self-antigens, thereby setting the stage for the generation of autoreactive T and B cells. Thymic abnormalities are usually absent in cases of MG that occur in Older patients or that are not associated with anti-acetylcholine receptor autoantibodies. Other pathogenic Antibodies: Muscle specific kinase, MuSK Low density lipoprotein receptor related protein, LRP4 PATHOGENESIS Antibody; anti acetylcholine receptor, AChR antibodies blocks the receptor and degrade the receptors at the postsynaptic membrane and induce damage by complement fixation. Net result; depleted of acetylcholine receptors and postsynaptic membrane alteration in morphology. Sudden onset of the disorder In early stage, patient may feel fine in the morning develops diplopia and speech slurs later in the day. Always begins with ocular muscle weakness Muscle weakness degree varies greatly among individuals. Muscle weakness severity fluctuates dramatically; sometimes over only few minute periods. Hallmark; repetitive use of muscles makes the weakness more severe (diagnostically useful) After 30-60s reading aloud facial weakness; inability to hold the mouth closed; ‘hanging jaw sign’ Myasthenia-Fatigue Recovery Test ‘Simpson plus’ Upward gaze test Antibody test: Anti-acetylcholine receptor (Ach R) antibodies ice pack test Diplopia and ptosis due to extraocular muscle involvement; common Fascial weakness; inability to hold the mouth closed hanging jaw sign can be unilateral or bilateral. Myasthenic crisis: Complication of myasthenia gravis characterized by worsening of muscle weakness resulting in respiratory failure. Long-term immunomodulatory therapy: Steroid use may cause or aggravate osteoporosis, cataracts, hyperglycemia, weight gain, avascular necrosis pf hip, hypertension, opportunistic infection and other complications, gastritis or peptic ulcer disease. 1. INHERITED Dystrophinopathies Duchenne Muscular Dystrophy (DMD) Becker Muscular Dystrophy (BMD) 2. ACQUIRED Inflammatory Myopathies Polymyositis Dermatomyositis Inclusion body myositis o Links the cytoskeleton actin to the extracellular matrix. o Dystrophin provides mechanical stability to the myofibril and its cell membrane during muscle contraction. o Involve in signaling pathways; interacts with nitric oxide synthase, which generates NO. DYSTROPHINOPATHIES x-linked recessive disease DMD is more common than BMD. Aetiology: dystrophin gene mutations Gene; Located on the short arm of X chromosome, Xp21 One of the largest human genes, hence vulnerability to sporadic mutations. Most common mutations are deletion, point mutations. Protein; Found in skeletal, cardiac muscle, brain and peripheral nerves. Part of dystrophin-glycoprotein complex, DGC o Large multi-protein complex consists of dystrophin and other proteins; dystroglycans, sarcoglycans, syntrophins, dystrobrevins. Pathogenesis Mutations in the dystrophin gene on the X chromosome causing defect in DGC. Results in; Transient membrane tear o Constant Calcium influx; activate proteases increased on going myofibril damage and necrosis. o Regenerated myofibril either damaged again or replaced by fibrosis and fat. o Hence, produce muscle weakness and pseudohyperthropy. Enlargement of the muscles of the lower leg associated with weakness, termed pseudohyperthropy. Severity of the disease correlates with the degree of the dystrophin deficiency. Investigation Serum Creatine Kinase level; high Muscle biopsy; fibrosis, adipocytes, dystrophin in muscle cells. Clinical Features Duchenne; Walking is often delayed. The first indications of muscle weakness are clumsiness and inability to keep up with peers. Weakness begins in the pelvic girdle muscles. Extends to the shoulder girdle. Mutations in Duchenne dystrophy result in severe absence of dystrophin. Mutations in Becker dystrophy result in production of abnormal truncated dystrophin. Disrupt intracellular signaling, reduced NO. o Damaged and necrosis of myofibril o Muscle replaced by fibrosis and fat. o Muscle weakness and pseudohyperthropy. Normal at birth; very early motor milestones are met Why DMD is more severe than BMD? INCIDENCE DMD More common Early BMD Less common Late ONSET AGE OF ONSET, 2-5 8-10 Y/O AMBULATION, Till 9-11 Beyond 15 AGE CONTRACTURES Early Later INTELLIGENCE Subnormal Normal LIFE SPAN Shorter Normal Electromyography; shows weakness is caused by muscle destruction rather than by nerves damage. Genetic testing; looks for mutation of the dystrophin gene. Complications DMD; Contracture, scoliosis, respiratory insufficiency, pneumonia, cardia decompensation. BMD; Cardiac failure ACQUIRED; Inflammatory of Myopathies Polymyositis Aetiology Autoimmune disorder Pathogenesis Increased expression of MHC class I molecules on myofibrils and predominantly endomysia inflammatory infiltrates containing CD8 and cytotoxic T cells and leads to myofibril necrosis and subsequent. Clinical manifestation Weakness affects many different muscle in the body, especially the shoulder, hips and thigh muscle, but there is no skin rash. Dermatomyositis Aetiology Believed to have an autoimmune basis. Pathogenesis Type 1 interferon-induced gene products are strongly upregulated in affected muscles. Some patients have autoantibodies that are relatively specific for dermatomyosities; these include antibodies against Mi-2, a nuclear helicase and p155 and p140, proteins with uncertain functions. Clinical manifestation Affect the joints, esophagus, lungs and less commonly, the heart. Produce skin rash. Inclusion Body Myositis Aetiology Degenerative process with secondary inflammatory changes, unclear causes Pathogenesis Aggregates of the same proteins that accumulate in the brains of patients with neurodegenerative diseases; hyperphosphorylated, amyloid derived from b-amyloid precursor protein and TDP-43; lead to some speculate degenerative disorder of aging. Clinical manifestation Progressive muscle weakness; quadriceps, distal upper extremity muscles and dysphagia. Complications Aspiration pneumonia Hypoventilation Interstitial lung disease, ILD Dysphagia