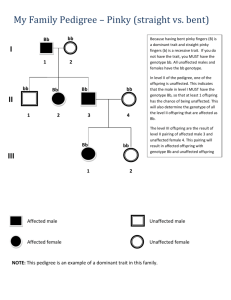
Hemorrhagic diseases in children Haemorrhagic disease (diathesis) hematological syndromes, various etiology and pathogenesis , main feature is pathological increased bleeding. Hemostasis - process which causes bleeding to stop, (the opposite of hemostasis is hemorrhage). • I. Vascular spasm (Vasoconstriction) • II. Platelet plug formation (primary hemostasis) • III. Clot formation (secondary hemostasis) • activated partial thromboplastin time (30s - 50s) speed of clotting by means of the intrinsic pathway. Normal PTT times require the presence of the following coagulation factors: I, II, V, VIII, IX, X, XI and XII. Used to monitor the treatment effects with heparin • Prothrombin time speed of clotting by means of the extrinsic pathway determine the clotting tendency of blood, in the measure of warfarin dosage, liver damage, and vitamin K status. PT measures factors I, II, V, VII, and X • Bleeding time (2-7 minutes) medical test done to assess their platelets function. It involves making a patient bleed then timing how long it takes for them to stop bleeding. • Platelet component of blood whose function (along with the coagulation factors) is to stop bleeding by clumping and clotting blood vessel injuries.150–400 IVY method • A standard-sized incision is made around 10 mm long and 1 mm deep. • The time from when the incision is made until all bleeding has stopped is measured and is called the bleeding time. • Every 30 seconds, filter paper or a paper towel is used to draw off the blood. • The test is finished when bleeding has stopped. • Normal values fall between 3 – 10 minutes Duke method • The patient is pricked with a special needle or lancet, preferably on the earlobe or fingertip. • The patient then wipes the blood every 15 seconds with a filter paper. • The test ceases when bleeding ceases. • The usual time is about 2–5 minutes. Laboratory findings in various platelet and coagulation disorders Partial Prothrombin Condition thromboplastin Bleeding time time time Platelet count Prolonged Normal or mildly prolonged Unaffected Unaffected Disseminated intravascular coagulation Prolonged Prolonged Prolonged Decreased Von Willebrand disease Unaffected Prolonged Prolonged Hemophilia Aspirin Unaffected Unaffected Prolonged Unaffected Unaffected Prolonged Decreased and/or Rejected Unaffected Unaffected Thrombocytopenia Unaffected Unaffected Prolonged Decreased Liver failure, early Prolonged Unaffected Unaffected Unaffected Liver failure, end-stage Prolonged Prolonged Prolonged Decreased Uremia Unaffected Unaffected Prolonged Unaffected Congenital afibrinogenemia Prolonged Prolonged Prolonged Unaffected Factor V deficiency Prolonged Prolonged Unaffected Unaffected Factor X deficiency as seen in amyloid purpura Prolonged Prolonged Unaffected Unaffected Glanzmann's thrombasthenia Unaffected Unaffected Prolonged Unaffected Bernard-Soulier syndrome Unaffected Unaffected Prolonged Decreased or unaffected Factor XII deficiency Unaffected Prolonged Unaffected Unaffected Vitamin K deficiency or warfarin Coagulopathy Hereditary : • • • • Hemophilia A (VIII factor) Hemophilia B (IX factor) Von Willebrand disease Hemophilia C (XI factor ) Acquired: • Vit. K deficiency • DIC • Hemostatic disorders caused by non-specific immune inhibitors - such as lupus anticoagulant Haemorrhagic disease of the newborn • Also known as vitamin K deficiency bleeding (VKDB), is a coagulation disturbance in newborn infants. • As a consequence of vitamin K deficiency there is an impaired production of coagulation factors II, VII, IX, X, protein C and protein S by the liver, resulting in excessive bleeding (hemorrhage). • The disease causes an increased risk of bleeding. The most common sites of bleeding are the umbilicus, mucous membranes, gastrointestinal tract, circumcision and venepunctures. • Precise diagnosis by measuring proteins induced by vitamin k absence (PIVKA). But this is usually not required. • Treatment consists of vitamin K supplementation.This is often given prophylactically to newborns shortly after birth HAEMOPHILIA Haemophilia is a bleeding disorder that slows the blood clotting process. The major types of this condition are hemophilia A (also known as classic hemophilia or factor VIII deficiency) and hemophilia B (also known as Christmas disease or factor IX deficiency). Although the two types have very similar signs and symptoms, they are caused by mutations in different genes. Epidemiology •The two major forms of hemophilia occur much more commonly in males than in females. •Hemophilia A is the most common type of the condition; 1 in 4,000 to 1 in 5,000 males worldwide are born with this disorder. •Hemophilia B occurs in approximately 1 in 20,000 newborn males worldwide. Epidemiology •The two major forms of hemophilia occur much more commonly in males than in females. •Hemophilia A is the most common type of the condition; 1 in 4,000 to 1 in 5,000 males worldwide are born with this disorder. •Hemophilia B occurs in approximately 1 in 20,000 newborn males worldwide. Etiology • Changes in the F8 gene are responsible for hemophilia A, while mutations in the F9 gene cause hemophilia B. • The F8 gene provides instructions for making a protein called coagulation factor VIII. A related protein, coagulation factor IX, is produced from the F9 gene. • Mutations in the F8 or F9 gene lead to the production of an abnormal version of coagulation factor VIII or coagulation factor IX, or reduce the amount of one of these proteins. Etiology • Hemophilia A and hemophilia B are inherited in an Xlinked recessive pattern. • The genes associated with these conditions are located on the X chromosome, which is one of the two sex chromosomes. • In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. • In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Haemophilia A and B Both types haemophilia share the same symptoms and inheritance pattern - only blood tests can differentiate between the two. Important to know which factor is defective so that the correct treatment can be given. Except in very rare cases both haemophilia A and haemophilia B affect only males. DISEASE SEVERITY 50-200% 5-50% 2-5% <1% Degrees of Severity NORMAL RANGE 50 – 150% Clotting Factor Normal blood coagulation MILD HAEMOPHILIA 5-50% Clotting Factor Bleeding problems usually associated tooth extractions, surgery, severe accident. Often not diagnosed until later in life MODERATE HAEMOPHILIA 2-5% Clotting Factor prolonged or delayed bleeding usually associated with injury – knock/ deep cut. Can present like severe haemophilia SEVERE HAEMOPHILIA <1% Clotting Factor Bleeding is frequent and often spontaneous into joints, muscles, and any site including brain. Usually diagnosed in first year of life. Types of Bleeds • Joint bleeding - hemarthrosis • Muscle hemorrhage • Soft tissue • Life threatening-bleeding • Other Life-Threatening Bleeding • Head / Intracranial – Nausea, vomiting, headache, drowsiness, isual changes, loss of consciousness • Neck and Throat – Pain, swelling, difficulty breathing/swallowing • Abdominal / GI – Pain, tenderness, swelling, blood in the stool Diagnosis • genetic tests (Molecular, Chromosomal, Biochemical) • factors IX, VIII diagnosis - Before pregnancy Genetic testing are available to help determine the risk of passing the condition onto a child. This may involve testing a sample of tissue or blood to look for signs of the genetic mutation that causes haemophilia. - During pregnancy A pregnant woman with a history of haemophilia in her family can test for the haemophilia gene. Such tests include: chorionic villus sampling (CVS) – a small sample of the placenta is removed from the womb and tested for the haemophilia gene, usually during weeks 11-14 of pregnancy amniocentesis – a sample of amniotic fluid is taken for testing, usually during weeks 15-20 of pregnancy SURGERY AND HAEMOPHILIA Factor replacement should be given pre surgery and during post op period, In suture removal, drain removal Factor levels should be taken to confirm expected rise in levels Continuous infusion should never be switched off as levels will fall rapidly post op No IM injections No asprin or NSAID Treatment of Hemophilia • Replacement of missing clotting protein – On demand – Prophylaxis Factor VIII Concentrate Factor IX Concentrate history 34 Von Willebrand Disease • First described by Erik von Willlebrand in individuals living on the Aland Islands, an archipeloga between Sweden and Finland in 1926. • Characterized by mutations that lead to an impairment in the synthesis or function of vWF. Acquired forms are caused by different pathophysiologic mechanisms. • Manifests as mucocutaneuos bleeding (epistaxis, menorrhagia, GI bleed, ecchymosis). • Most common inherited bleeding disorder affecting up to 1% of population. • Autosomal inheritance Function of vWF 1. adhesion of platelets to collagen 2. Binding factor VIII Classification A- Quantitative deficiency of VWF Type 1: Partial quantitative deficiency of vWF Type 3: Virtually complete deficiency of vWF B- Qualitative deficiency of VWF Type 2A: Qualitative variants with decreased platelet dependent function associated with the absence of high and intermediate molecular weight vWF multimers Type 2B: Qualitative variants with increased affinity for platelet GPIb Type 2M: Qualitative variants with decreased platelet dependent function not caused by the absence of high-molecular weight vWF multimers Type 2N: Qualitative variants with markedly decreased affinity for factor VIII Given the autosomal dominant inheritance, genetic risk for offspring is 50% regardless of the sex of the fetus. The gene encoding the vWF activity is the twelfth chromosome. Von Willebrand disease type I and II, platelet-type inherited in an autosomal dominant pattern, while type III is inherited in an autosomal recessive manner. However, there are cases where two type also inherited recessively. Autosomal recessive (III type) Clinical manifestations • Heavy epistaxis 5% - 60% • Gingival bleeding 7% - 51% • Severe skin hemorrhages (ecchymosis, less bruising) 12% - 24% • Bleeding after tooth extraction 1% - 13% • Bleeding after tonsillectomy 2.4% - 11% • Postpartum haemorrhage 6% - 23% • Menorrhagia 23% - 44% • Hemarthrosis? • Intracranial hemorrhage? • After-and intra-operative bleeding? Laboratory findings in various platelet and coagulation disorders Condition Prothrombin time Partial thromboplastin time Bleeding time Platelet count Vitamin K deficiency or warfarin Prolonged Normal or mildly prolonged Disseminated intravascular coagulation Prolonged Prolonged Prolonged Decreased Von Willebrand disease Unaffected Prolonged Prolonged Decreased and/or Rejected Hemophilia Unaffected Prolonged Unaffected Unaffected Aspirin Unaffected Unaffected Prolonged Unaffected Thrombocytopenia Unaffected Unaffected Prolonged Decreased Liver failure, early Prolonged Unaffected Unaffected Unaffected Liver failure, end-stage Prolonged Prolonged Prolonged Decreased Uremia Unaffected Unaffected Prolonged Unaffected Congenital afibrinogenemia Prolonged Prolonged Prolonged Unaffected Factor V deficiency Prolonged Prolonged Unaffected Unaffected Factor X deficiency as seen in amyloid purpura Prolonged Prolonged Unaffected Unaffected Glanzmann's thrombasthenia Unaffected Unaffected Prolonged Unaffected Bernard-Soulier syndrome Unaffected Unaffected Prolonged Decreased or unaffected Factor XII deficiency Unaffected Prolonged Unaffected Unaffected Unaffected Unaffected Management • Cryoprocipitate fibrinogen, VIII, vWF, XIII (blood plasma) • Desmopressin for type 1 • antifibrinolytic agent for mucosal bleeds • factor 8 and vWF for surgery, trauma Management • Desmopressin for type 1 • For patients with vWD type 1 and vWD type 2A, desmopressin is recommended for use in cases of minor trauma, or in preparation for dental or minor surgical procedures. It stimulates the release of vWF. • Desmopressin is contraindicated in vWD type 2b because of the risk of aggravated thrombocytopenia and thrombotic complications. Platelet Response •Platelets adhere to vessel wall, then aggregate, leading to formation of a platelet plug Thrombocytopathies • Thrombocytopenia is a condition characterized by abnormally low levels of thrombocytes. • A normal human platelet count ranges from 150,000 to 450,000 platelets per microliter of blood • If the person's platelet count is between 30,000 and 50,000/mm3, bruising with minor trauma may be expected; if it is between 15,000 and 30,000/mm3, spontaneous bruising will be seen (mostly on the arms and legs) ThrombocytopeniaHow low is too low? • 150,000 - 50,000: no symptoms – No treatment generally required. • 50,000 - 20,000: first symptoms – Generally need to begin therapy • 20,000-10,000: life-threatening – Generally requires hospitalization • <10,000: risk for spontaneous intracranial hemorrhage Causes of Thrombocytopenia • Decreased production • Increased destruction • Medication-induced Decreased production • • • • • Vitamin B12 or folic acid deficiency Leukemia or myelodysplastic syndrome Decreased production of thrombopoietin by the liver in liver failure. Sepsis, systemic viral or bacterial infection Dengue fever can cause thrombocytopenia by direct infection of bone marrow megakaryocytes as well as immunological shortened platelet survival Hereditary syndromes – Congenital amegakaryocytic thrombocytopenia (CAMT) – Thrombocytopenia absent radius syndrome – Fanconi anemia – Bernard-Soulier syndrome, associated with large platelets – May-Hegglin anomaly, the combination of thrombocytopenia, pale-blue leuckocyte inclusions, and giant platelets – Grey platelet syndrome – Alport syndrome – Wiskott–Aldrich syndrome Increased destruction • • • • • • • • • • • Idiopathic thrombocytopenic purpura (ITP) Thrombotic thrombocytopenic purpura (TTP) Hemolytic-uremic syndrome (HUS) Disseminated intravascular coagulation (DIC) Paroxysmal nocturnal hemoglobinuria (PNH) Antiphospholipid syndrome Systemic lupus erythematosus (SLE) Post-transfusion purpura Neonatal alloimmune thrombocytopenia (NAITP) Splenic sequestration of platelets due to hypersplenism Dengue fever has been shown to cause shortened platelet survival and immunological platelet destruction • HIV-associated thrombocytopenia[4] Medication-induced • • Direct myelosuppression – Valproic acid – Methotrexate – Interferon – Other chemotherapy drugs – Singulair (montelukast sodium) – H2 blockers and Proton-pump inhibitors Immunological platelet destruction – A drug molecule binds to the Fab portion of an antibody. A classic example is the quinidine group of drugs. – A drug molecule binds to the Fc antibody - Heparin-induced thrombocytopenia (HIT) – Abciximab-induced thrombocytopenia. ITP - Immune/Idiopathic Thrombocytopenic Purpura • Definition: isolated thrombocytopenia with no clinically apparent associated conditions or other causes of thrombocytopenia. • Etiology: autoantibodies directed against platelets coat platelet surface. IgG-coated platelets are taken up by RE system. • Incidence: approximately 100 per million; half of these are children. ITP - Diagnosis • ITP is a Diagnosis of Exclusion • No laboratory test can diagnose ITP • Need to exclude other causes of thrombocytopenia Etiology: 1. viruses, rarer - a bacterial infection (the occurrence is usually 2-3 weeks after the onset of acute respiratory infections, measles, chicken pox, measles, influenza, and less of adenovirus infection, infectious mononucleosis) 2. mental and physical injuries 3. medications (salicylates, antibiotics, sulfonamides, digoxin, gold salts, hydrochlorothiazide) Pathogenesis: Increased destruction of platelets contain autoantibodies Reduced lifespan of platelets to several hours or even minutes. Classification Acute ITP Mostly children Male/Female = 1:1 Acute onset Plt. Count mostly <20,000/mm3 Spontaneous remission frequent Mortality : 0.5-1.5 % Chronic ITP Mostly adults Male/Female = 1:3-4 Usaully gradual onset Plt. Count 20 000 – 50 000/mm3 Spontaneous remission rare Chronic recurrent course Common Signs and Symptoms 1.Purpura 2.Menorrhagia 3.Epitaxis 4.Gingival bleeding 5.Recent viral illness (acute ITP) 6.Bruising tendency 55 Laboratory Findings 1.Isolated thrombocytopenia 2.No splenomegaly 3.Increase megakaryocytes in BM 4.No other cause of thrombocytopenia 5.Platelet auto-antibody found Laboratory findings in various platelet and coagulation disorders Partial Prothrombin Condition thromboplastin Bleeding time Platelet count time time Normal or Vitamin K Prolonged mildly Unaffected Unaffected deficiency or warfarin prolonged Disseminated intravascular Prolonged Prolonged Prolonged Decreased coagulation Decreased Von Willebrand disease Unaffected Prolonged Prolonged and/or Rejected Hemophilia Unaffected Prolonged Unaffected Unaffected Aspirin Unaffected Unaffected Prolonged Unaffected Thrombocytopenia Unaffected Unaffected Prolonged Decreased Liver failure, early Prolonged Unaffected Unaffected Unaffected Liver failure, end-stage Uremia Prolonged Unaffected Prolonged Unaffected Prolonged Prolonged Decreased Unaffected Congenital afibrinogenemia Prolonged Prolonged Prolonged Unaffected Factor V deficiency Prolonged Prolonged Unaffected Unaffected Factor X deficiency as seen in amyloid purpura Prolonged Prolonged Unaffected Unaffected Glanzmann's thrombasthenia Unaffected Unaffected Prolonged Unaffected Bernard-Soulier syndrome Unaffected Unaffected Prolonged Factor XII deficiency Unaffected Prolonged Unaffected Decreased or unaffected Unaffected Differential Diagnosis 1.Drugs induced thrombocytopenia -Drug use history -quinidine, quinine, sulfonamides, rifampin & heparin 2.Low platelet production -Bone marrow failure -Leukemia/Lymphoma 3.Over platelet destruction -Hypersplenism, TTP (The two best understood causes of TTP are due autoimmunity and an inherited deficiency of ADAMTS13 (known as the Upshaw-Schülman syndrome), SLE & DIC, Infection -HIV, DHF, Rubella, Infectious Mononucleosis -Leptospirosis -Malaria 4.Others : CLL, Hypogammaglobulinemia Mortality/Morbidity 1.Hemorrhage represents the most serious complication 2.Mortality rate from hemorrhage is approximately 1% in children and 5% in adult 3.Increase risk of severe bleeding in adult ITP 4.Spontaneous remission : occure in more than 80 % in children : uncommon in adults Treatment & Prognosis Acute ITP 1.Self remission 80 % 2.Corticosteroid therapy within 3-4 weeks • stand. dose - 1-2 mg / kg / day for 21 days • high doses (parenteral) - 4-8 mg / kg /day • Pulse therapy - i/v metypred 10-30 mg / kg per day for 3-5 days 3. IVIG (course dose) -1000mg/kg Treatment of ITP • Anti-D immunoglobulins • Splenectomy (rare in children under 5 years due to the risk of bacterial infections) Case 1. A newborn boy was discharged from the maternity home on the 9th day. Anamnesis vitae: the mother is 21 years old and the father is 26 years, both are healthy. Family history is not fully known. He was discharged later than usual from the maternity home due to cephalohematoma which appeared immediately after birth and has been progressing for 2 days. The umbilical cord fell off on the 9th day with scanty hemorrhagic discharge from the umbilical wound. The discharge continued till present and is the reason for the present hospitalization. He is not vaccinated. On examination: at the age of 11 days, the weight - 3000g (birth weight 3100 g), breastfeeding, sucking actively but inadequately, frequent vomiting, occasionally tremor of the chin, spontaneous Moro reflex. HR - 132 bpm, respiratory rate - 36 per minute. Cardiac tones are sonorous, rhythmic. In the lungs – puerile breathing, no rales. Liver + 1.5 cm beyond the costal margin, the edge of the spleen is palpable. Clinical blood count: Hb - 124 g/l; erythrocytes - 3,4x10x12/l; platelets – 300x10x9/l; leucocytes - 10,0x10x9/l, band neutrophils - 40%, lymphocytes - 51%, monocytes - 9%; ESR - 9 mm/hr. Coagulation: bleeding time - 2.2 minutes (normal up to 4 minutes), prothrombin time - 9,8 s (normal - 9,2-12,2 s), activated partial thromboplastin time – 76s (norm - 28-34s ), fibrinogen - 2.2 g/l (normal –1,25-3 g /l). The level and degree of activity of the Von Willebrand factor is normal. Questions: 1. Which symptoms of hemorrhagic syndrome are observable in this child? 2. What could be the reason for the cephalohematoma? 3. What does the prolonged discharge from the umbilical wound suggest? 4. Is there an indication for conducting neurosonography in this patient? 5. Evaluate the values of the blood coagulation system of the child. 6. What is the presumable diagnosis? 7. What tests can confirm your presumed diagnosis? 8. What is the cause of the disease? 9. What treatment should be prescribed? 10. What is the prognosis of the disease in this child? Case 3. A mother took her 8 years old girl to the doctor due to repeated profuse nosebleeds and frequent occurrence of ecchymosis on the limbs with no obvious causes. These complaints have been observed for the last 2-3 years. A day before, prolonged bleeding reoccurred after the removal of a tooth. Family history: The mother suffers from profuse menorrhagia, prone to ecchymosis. No hemorrhagic manifestation among the male relatives. On examination, the child revealed a moderate amount of ecchymosis of various limitations on the skin of the hands, the feet (mainly) and trunk. Skin is pale, pink. Lymph nodes are not enlarged. Heart sounds loud, rhythmic, heart rate 84 bpm. In the lungs- vesicular breathing, no rales, respiratory rate - 18 per minute. Liver 0.5 cm beyond the costal margin, the spleen is not palpable. Stool, urine output are normal. Clinical blood count: Hb - 110 g/l; erythrocytes - 4,0x10x12/l; platelets – 170x10x9/l; leucocytes - 6,5x10x9/l, neutrophils - 66%, lymphocytes - 24%, eosinophils - 1%, Monocytes - 9%; ESR - 8 mm/hr. Coagulation: activated partial thromboplastin time – 34s (norm - 29-34 s), prothrombin time - 10.1 s (norm - 9,2-12,2 s), fibrinogen - 2.8 g /l (normal 4.2 g /l), bleeding time - 5 minutes 10 seconds (normal–up to 4 min). Von Willebrand factor antigen concentration: nil. 1. Which symptoms of hemorrhagic syndrome are observable in this girl? 2. What in the family history may indicate an hereditary hemorrhagic disease and suggests the character? 3. Evaluate the conducted laboratory investigations. 4. What disease can be suspected? Justify your answer. 5. What forms of hemorrhagic disease are known to you? What form is the most common? 6. What treatment should be assigned to the child after confirmation of the diagnosis? 7. What other methods of treatment of this disease are known to you? Case 4. A boy of 2 years was admitted to the department with complaints of the appearance of hemorrhagic elements on the skin of the limbs, torso. From history, he had ARVI 14 days ago. 3 weeks before then, he was vaccinated (measles, rubella, mumps). On examination: body temperature is normal, no catarrhal phenomena, petechiae on the skin along with subcutaneous hemorrhages of various sizes (ecchymosis); simultaneous hemorrhages of different colors - from bluish to reddish-green and yellow; hemorrhagic elements are asymmetrical. There are singular hemorrhagic elements on the face. New elements appear more often in the morning, not related to trauma. Lymph nodes were not enlarged. The liver palpable 2 cm beyond the costal margin, the spleen at the edge of the costal margin. Clinical blood count: Hb - 122 g/l; erythrocytes - 3,8x10x12/l; platelets - units in the preparation; leucocytes - 7,4x109/l, neutrophils - 46%, lymphocytes - 38%, of monocytes - 14%; ESR - 9 mm/hr. Clinical urine analysis: without pathology. In blood serum and on the surface of platelet - antiplatelet antibodies identified. 1. Describe the hemorrhagic syndrome and its features in this given patient. 2. What disease can be suspected based on the clinical, history and laboratory data? 3. What can the progression of the disease be connected to? 4. What forms of the disease are known to you, depending on the mechanism, the clinical manifestations and the duration? Which form of the disease occurred in this child? 5. Conduct a differential diagnosis. What diseases is it necessary to carry out the differential diagnosis in this case, considering the patient's age? 6. What further investigations do the child need? 7. What treatment should be prescribed urgently? 8. What modern methods of treatment of this disease do you know? 9. What is the prognosis of this form of the disease? Case 6. A boy of 5 years was admitted to the department with complaints of severe pain in the elbow joints, ankle joints and a febrile fever. From his history, we know that these complaints started a week ago, when the mother first noticed multiple bruises on the skin of the feet, the trunk. At first, there was moderate nasal bleeding which stopped quickly. A month prior to that, he became less active, pale, tired during sport periods, refused to go to the pool. On examination: pale skin, hemorrhagic elements on the limbs, torso: petechiae, ecchymosis of varying degrees, not abundant. Capricious, sluggish, muscle weakness, body temperature - 37,9 ºC; palpable cervical, occipital, axillary submandibular, axillary, inguinal lymph nodes up to 1x1,5 cm, painless, mobile. In the lung- no rales. Tachycardia, heart rate – 112 bpm, heart sounds are muffled, soft systolic murmur at the apex. Palpation-Liver 4 cm, spleen 2 cm beyond the costal margin. Elbow and ankle joints without apparent changes, skin temperature is normal, painful on palpation, expressed pain syndrome with passive movements. Clinical blood count: Hb - 80 g/l; erythrocytes - 2,1x1012/l; platelets – 28x109/l; leucocytes – 25x109/l, band cells- 18%, segmented cells - 7%, lymphocytes 65%, monocytes - 10%, blasts; ESR - 70 mm/hr. 1. Describe the hemorrhagic syndrome in this child. 2. What other clinical syndromes are observable in this child? Give their clinical characteristics. 3. Evaluate the values of the clinical analysis of blood. 4. What disease is the child most likely to have? 5. What examination and treatment should be carried out urgently? 6. What causes thrombocytopenia in this disease? Case 10. A girl of 6 years was admitted to the department with complaints of severe, cramping abdominal pain which started at night, diarrhea, body temperature rise to 37,5 ºC. From history, we know that 10 days ago, she suffered from ARVI. On examination: state of moderate severity, negative attitude, concerned about pain in the abdomen. The skin is pale pink, on the legs, mainly the extensor surfaces around the ankle and knee, there are multiple symmetrical exudative hemorrhagic reddish-purple color elements of different sizes from 1-2 mm to 3-4 mm, the individual elements with signs of necrosis. Swelling of the dorsum of the foot with a cyanotic shade. In the lungs – no rales, heart sounds loud, rhythmic, heart rate - 90 bpm. The abdomen is soft, painful on palpation along the intestine, around the navel. Liver can be palpated at the edge of the costal margin, the spleen is not palpable. She was examined by the emergency room surgeon: no sign of acute surgical pathology. Clinical blood count: Hb - 110 g/l; platelets – 400x109/l; leucocytes – 14x109/l, neutrophils band cells5%, segmented cells - 67%, eosinophils - 3%, lymphocytes - 18%, monocytes - 7%; ESR - 35 mm/hr. Clinical urine analysis: leucocytes - 2-4, erythrocytes - 25-30 in the field of vision. Daily urine: protein - 0.126 g/l. Biochemical blood test: total protein - 50 g/l (norm - 40-60 g/l), albumin - 49% (norm - 40-60%), globulin: α1 - 6% (norm - 2-5%), α2 - 14% (norm - 7-13%),γ - 24% (normal: 12-22%). C-reactive protein - 0.05 g/l (normal - 0.003 g/l). Coagulation: bleeding time - 30 seconds, activated partial thromboplastin time – 24s (norm - 29-34 s), prothrombin time - 9.2 sec (norm - 9,2-12,2 s), fibrinogen - 5.2 g/l (normal - 2.4 g/l) reduced concentration of antithrombin-III, increased activity of Von Willebrand factor. Questions: 1. Describe the hemorrhagic syndrome in this child. What group of bleeding does it belong to? 2. What other clinical syndromes are present in the clinical picture of the disease in this child, beside hemorrhagic? 3. Evaluate the laboratory values. 4. What is the most likely diagnosis? Explain the diagnosis on the basis of existing patient diagnostic criteria for the disease, give specifics in accordance with the classification. 5. Explain the pathogenesis and pathmorphology of the disease? 6. What was the most likely trigger of this disease? What confirmations in history should be done? 7. What treatment should be assigned to the child?