Titrimetry Overview: Principles & Techniques
advertisement

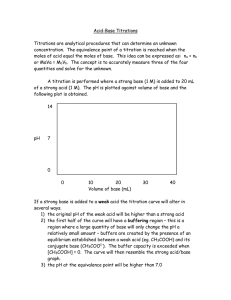
TITRIMETRY / Overview 105 Certified Reference Materials In order to validate speciation methods more effectively, a number of certified reference materials (CRMs) have been produced to allow laboratories to measure the accuracy of their techniques. A range of sediment and biota materials are available such as the sediment materials PACS-2 from the National Research Council of Canada, NIES-12 from the National Institute for Environmental Studies, Japan and the CRM-462 from Community Bureau of Reference (BCR), EU. These materials have been rigorously homogeneity and stability tested and the levels of organotins have been certified by a range of techniques utilizing either ‘definitive’ methods or multiple independent methods. See also: Atomic Absorption Spectrometry: Interferences and Background Correction. Atomic Emission Spectrometry: Principles and Instrumentation; Interferences and Background Correction; Flame Photometry; Inductively Coupled Plasma; Microwave-Induced Plasma. Atomic Mass Spectrometry: Inductively Coupled Plasma; Laser Microprobe. Countercurrent Chromatography: Solvent Extraction with a Helical Column. Derivatization of Analytes. Elemental Speciation: Overview; Practicalities and Instrumentation. Extraction: Solvent Extraction Principles; Solvent Extraction: Multistage Countercurrent Distribution; Microwave-Assisted Solvent Extraction; Pressurized Fluid Extraction; Solid-Phase Extraction; Solid-Phase Microextraction. Gas Chromatography: Overview. Isotope Dilution Analysis. Liquid Chromatography: Overview. Further Reading Abalos M, Bayona J-M, Comañó R, et al. (1997) Analytical procedures for the determination of organotin compounds in sediment and biota: A critical review. Journal of Chromatography A 788: 1–49. Alonso JIG, Encinar JR, Rodrı́guez-González P, and Sanz-Medel A (2002) Determination of butyltin compounds in environmental samples by isotope-dilution GC–ICP-MS. Analytical and Bioanalytical Chemistry 373: 432–440. Davies AG (1997) Organotin Chemistry. Weinheim: VCH. Ebdon L, Hill SJ, and Rivas C (1998) Organotin compounds in solid waste: A review of their properties and determination using high-performance liquid chromatography. Trends in Analytical Chemistry 17: 277–288. Ebdon L, Pitts L, Cornelis R et al. (eds.) (2001). Trace Element Speciation for Environment, Food and Health. Cambridge: Royal Society of Chemistry. Leroy MJF, Quevauviller P, Donard OFX, and Astruc M (1998) Determination of tin species in environmental samples. Pure and Applied Chemistry 70: 2051–2064. Rajendran RB, Tao H, Nakazato T, and Miyazaki A (2000) A quantitative extraction method for the determination of trace amounts of both butyl- and phenyltin compounds in sediments by gas chromatography-inductively coupled plasma mass spectrometry. The Analyst 125: 1757–1763. Rodrı́guez-González P, Encinar JR, Alonso JIG, and SanzMedel A (2003) Isotope dilution analysis as a definitive tool for the speciation of organotin compounds. The Analyst 128: 447–452. Smedes F, de Jong AS, and Davies IM (2000) Determination of (mono-, di- and) tributyltin in sediments. Analytical methods. Journal of Environmental Monitoring 2: 541–549. Suzuki T, Kondo K, Uchiyama M, and Murayama M (1999) Chemical species of organotin compounds in sediment at a marina. Journal of Agricultural and Food Chemistry 47: 3886–3894. TITRIMETRY Contents Overview Potentiometric Photometric Overview A Townshend, University of Hull, Hull, UK & 2005, Elsevier Ltd. All Rights Reserved. Introduction A titration is defined as ‘the process of determining the quantity of a substance A by adding measured increments of substance B, the titrant, with which it reacts until exact chemical equivalence is achieved 106 TITRIMETRY / Overview (the equivalence point)’. The titrant is usually added as a standardized solution, but electrolytic generation, as in coulometric titrations, is also possible. The achievement of the equivalence point is indicated in one of two ways. The first is visually, for example, by the addition of an indicator, which changes color, or by fluorescence or a similar property at or close to the equivalence point. The change indicates the endpoint of the titration, and the indicator should be chosen so that the endpoint is as close as possible to the equivalence point. The second method is to measure a physical property of the solution being titrated (e.g., conductivity, pH, absorbance) and identify the equivalence point by processing the signals obtained. The latter approach is particularly useful when titrations are automated. The amount of titrant added is usually measured by volume (by dispensing the solution from a burette), and in this case, titrimetry is an example of volumetric analysis. Occasionally, the titrant is measured by weight (especially if greater accuracy is required) or by amount of electricity (as in coulometric titrations). Titrimetry is one of the oldest analytical techniques, originating in the middle of the eighteenth century as a rapid means of quality control of industrial processes, such as acid manufacture. Since that time, the equipment has been refined, the procedures have been automated, and the number of chemical reactions utilized greatly increased, but the basic principles are unchanged. Its continued popularity stems from the simplicity of equipment and execution, wide applicability, and high accuracy and precision (greater than most instrumental techniques), all of which make it particularly applicable to the determination of major and minor components of samples. Skilled titrimetric analysis should give results with a precision of o0.2% at the 1 10 2 mol l 1 level. Titrimetry may be classified with respect to the types of reaction that are involved. The major reactions are acid–base reactions (hence acid–base titrimetry), redox reactions (redox titrimetry), complexing reactions (compleximetric titrimetry), and precipitation reactions (precipitation titrimetry), which will be discussed in more detail below. Titrimetry may also be classified by the nature of the endpoint measurement. The use of electrical measurements gives rise to potentiometric, amperometric, and coulometric titrations. Measurement of heat changes is used in thermometric titrimetry, and of absorbance in photometric and turbidimetric titrations. Radiometric titrations measure changes in radioactivity during the titration. All of these techniques are dealt with in other articles in this Encyclopedia. This article discusses only those titrations that use visual indicators. General Manual Titrimetric Technique The simple equipment used for a typical titration is shown in Figure 1. A sample is measured into the flask from a pipette, or by weighing. The accuracy and precision of manual titrations using visual indicators is critically dependent on the use of correct experimental technique by the analyst. As in most analytical procedures, precise measurement of the amount of sample (or sample aliquot) is necessary, but it is most important in titrimetry if an accuracy of o0.2% is to be achieved. Thus, proper use of the pipette, burette, and balance, and a careful sample preparation procedure is crucial. Measurement devices of high quality, such as class A pipettes and burettes, should be used. When carrying out a titration the burette, holding the titrant solution, is clamped vertically just above the flask. Generally, the flask containing the solution being titrated should be placed on a white tile. Where the color change is somewhat difficult to detect, a reference solution should be used for comparison (this is a solution held in a flask similar to that being used for the titration, has the same volume, contains the same indicator, and has been adjusted to the endpoint). Titrant is added from the burette to the solution in the flask, which is then swirled by hand. As the endpoint approaches (which will be signaled by a transient color change in the portion of the solution where the titrant is added), the titrant should be added dropwise. When very close to the endpoint, fractions of drops can be added by touching the tip of the burette with a partly formed drop on the inside of the flask, and washing down with water from a wash bottle. The detection of the endpoint, when using a 50 or 10 cm3 burette, should be possible to within 0.02 cm3. The volume of titrant run out of the burette is used to calculate the concentration of analyte in the titrated solution. Standards Titrimetric analysis depends upon the availability of solutions of accurately known concentration for use as titrants. Such standard solutions may be prepared and themselves standardized by titration with solutions prepared from materials of guaranteed purity and composition, preferably solutions of chemicals known as primary standards. These materials have the following properties, as listed by Dodge: 1. They are easily obtained in an analytically pure state. TITRIMETRY / Overview 107 D 20°C ml Maker (here: Brand) 0 Country of manufacture 1 Trademark of Brand 2 Rated volume 49 BRAND Class: all instruments having the international symbol 'A' are suitable for official certification. 'S' stands for rapid delivery 50 25 25 ml D 20°C ml AS 0.03 ml Ex=15s 20°C Tolerance Calibration (here Ex = deliver) and waiting time (here 15 s) Calibration temperature (A) (B) (C) 0 1 2 3 4 (D) (E) Figure 1 Equipment used for a manual titration. (A) Transfer pipette; (B) burette; (C) information provided on volumetric glassware; (D) conical or Erlenmeyer flask (normally 250 or 100 cm3); (E) burette reader. (Reproduced with permission from Belcher et al. (1970) and Rudolf Brand and Co. Working with Volumetric Instruments. Wertheim: Brand.) 2. They should be unalterable in air at ambient or moderately high temperatures. 3. They should have a high equivalent weight, thus decreasing the effect of small weighing errors. 4. They should be readily soluble under the conditions of the analysis, thus allowing immediate titration in the cold. 5. On titration, no interfering product should be present. 108 TITRIMETRY / Overview Table 1 Some primary titrimetric standards Compound Name Formula Anhydrous sodium carbonate Sodium borate (borax) (recrystallized) Sulfamic acid Potassium hydrogenphthalate Potassium hydrogenbiiodate Silver nitrate Sodium oxalate Arsenic(III) oxide Potassium dichromate Ammonium hexanitrocerate(IV) Potassium iodate Potassium bromide Calcium carbonate Zinc oxide Ni, Zn, Cu metals Anhydrous disodium EDTA Na2CO3 Na2B4O7.10H2O NH2SO3H KHC8H4O4 KH(IO3)2 AgNO3 Na2C2O4 As2O3 K2Cr2O7 (NH4)2Ce(NO3)6 KIO3 KBrO3 CaCO3 ZnO Ni, Zn, Cu C10HI4N2O8Na2 Type Type of titration Weak base Weak base Strong acid Weak acid Strong acid Acid–base Acid–base Acid–base Acid–base Acid–base Argentimetric Redox Redox Redox Redox Redox Redox Compleximetric Compleximetric Compleximetric Compleximetric Reductant Reductant Oxidant Oxidant Oxidant Oxidant Source of calcium ions Source of zinc ions Source of metal ions Complexing agent 6. They should be colorless, before and after titration, to avoid interference with indicators. Equivalence point Primary standards for particular titrations are given in Table 1. There are relatively few compounds that satisfy all these conditions. It is also possible to buy concentrated standard solutions, which, after accurate dilution, can be used for titrimetry. A −1 12 A = 0.1 mol 10 B = 0.01 mol −1 C = 0.001 mol B −1 C pH 8 6 Acid–Base Titrations In acid–base titrations, an acid is determined by titration with a base, or vice versa. The essential reaction is between H þ and OH , giving water. The pH at the endpoint depends on the dissociation constants of the reactants and products. Thus, the titration of a strong, i.e., completely dissociated, acid with a strong, almost completely dissociated, base reaches equivalence at pH 7.0. A typical example is the titration of hydrochloric acid with sodium hydroxide solution. If a weaker, i.e., less dissociated base, such as ammonia, is used, the equivalence pH is o7.0; the weaker the base, the lower the equivalence pH. Likewise, if a weak, i.e., less dissociated, acid is titrated with a strong base, the equivalence pH is 47.0, the pH increasing with increasing weakness of the acid. For the titration of a weak acid with a weak base, the equivalence pH depends on the relative dissociation constants of the acid and base, but the limited pH change means that the equivalence point is not as sharp as in a strong acid or strong base titration. The change of pH during the course of such titrations is illustrated in Figures 2 and 3. 4 C B A 2 0 5 Titrant (ml) 10 Figure 2 Titration of 10 ml of hydrochloric acid of various concentrations with sodium hydroxide solution of the same concentration. (Redrawn from Belcher et al. 1970.) The extent of the pH change also depends on the concentration of the analyte and titrant. Figure 2 shows how the pH change decreases with decreasing concentration of HCl and NaOH. The indicators for such titrations are chosen to change color very close to the pH at equivalence, and are described in detail in a separate article. Some acids are polybasic, i.e., they give rise to more than one hydrogen ion. Phosphoric acid, for TITRIMETRY / Overview 109 a 12 12 10 10 b 8 pH pH 8 6 6 4 4 2 2 0 5 Titrant (ml) 0 10 25 50 75 NaOH (ml) Figure 3 Titration of 10 ml of 0.1 mol l 1 acetic acid with (A) 0.1 mol l 1 sodium hydroxide; (B) 0.1 mol l 1 ammonia. (Redrawn from Belcher et al. 1970.) Figure 4 Titration of 25 ml of 0.1 mol l 1 H3PO4 with 0.1 mol l 1 NaOH. (Redrawn from Belcher et al. 1970.) Table 2 Examples of acid–base titrimetric analyses Analyte Titrant Indicator Conditions CaCO3 H3PO4 HCl NaOH NH4þ Boric acid HCl NaOH Methyl red or phenolphthalein Methyl orange Thymolphthalein Methyl red Phenolphthalein – First endpoint Second endpoint Distillation from NaOH solution Binding with mannitol or sorbitol to increase acidity example, produces three hydrogen ions: þ H3 PO4 "H2 PO 4 þH ½I 2 þ H2 PO 4 "HPO4 þ H ½II 3 þ HPO2 4 "PO4 þ H ½III The change in pH on titration with NaOH is shown in Figure 4. The first dissociation (reaction [I]) occurs most easily, and titration with NaOH gives an equivalence point at pH 4. Further titration, of the second, more strongly bound hydrogen ion (reaction [II]), also gives rise to an equivalence point, at pH 9. The third hydrogen ion does not give rise to a sharp endpoint. Thus, phosphoric acid may be determined by titration to the first or second endpoints. Some examples of acid–base titrimetric analyses are given in Table 2. Precipitation Titrations These are titrations in which the analyte and titrant react to form a precipitate. The only common titrant used is silver nitrate (argentimetric titrations), and its use is mainly restricted to the determination of chloride, bromide, iodide, cyanide, and thiocyanate, although in principle any species that is precipitated by silver ions could be determined. Direct titrations involve the use of potassium chromate (Mohr’s method) or fluorescein derivatives as indicator. An indirect (back-titration) procedure is also popular, in which excess of precipitant (Ag þ ) is added to the sample, and the unreacted Ag þ titrated with thiocyanate ions (Volhard’s method), using iron(III) as indicator. The mechanisms of the indicator reactions are described in another article. Redox Titrations In these titrations, a reducing agent is titrated with an oxidizing agent, or vice versa. The common oxidizing titrants are potassium permanganate (KMnO4), potassium dichromate (K2Cr2O7), cerium(IV) sulfate (Ce(SO4)2), iodine (I2), potassium iodate (KIO3), and potassium bromate (KBrO3), all of which are solids, and sodium hypochlorite (NaClO), which is available as a solution. The most important reducing titrants are iron(II) salts, often ammonium iron(II) sulfate ((NH4)2Fe(SO4)2 6H2O), sodium thiosulfate (Na2S2O3 H2O), and arsenic(III) oxide (As2O3). 110 TITRIMETRY / Overview The stoichiometry of the reaction between one of these titrants and a particular analyte is established by combining the appropriate half-reactions. For the titrants above, the half-reactions are as follows: given by the Nernst equation: Oxidants where n is the number of electrons involved in the above half-reaction, and [ox] and [red] are the concentrations (or better, activities) of the oxidized and reduced forms of the species, respectively. E0 is a constant known as the standard potential, which is the idealized potential when [ox] ¼ [red]. A similar parameter, but measured under actual experimental conditions, is known as the formal potential (EF), and is more immediately useful for application in redox titrimetry. Some potentials for the above half-reactions are given in parentheses in the above equations. The change in oxidation potential during the titration of a reductant (iron(II)) by an oxidant (cerium(IV)) is shown in Figure 5. The final oxidation potential increases with the strength of the oxidant used. Sometimes, it is possible to make a simple calculation of the equivalence potential (EEP) as follows. For the reaction that can be written as in [XVII]: þ 2þ MnO þ 4H2 O 4 þ 8H þ 5e -Mn ðacidic conditions; E0 ¼ 1:50 VÞ ½IV þ MnO 4 þ 4H þ 3e -MnO2 þ 2H2 O ðneutral conditionsÞ ½V 2 MnO 4 þ e -MnO4 ðalkaline conditionsÞ ½VI 3 þ Cr2 O2 7 þ 14H þ 6e -2Cr þ 7H2 O ðEF ¼ 1:0 V; 1 mol l1 HClÞ ½VII Ce4þ þe -Ce3þ ðEF ¼ 1:44 V; 1 mol l1 H2 SO4 Þ I2 þ 2e -2I ðE0 ¼ 0:54 VÞ þ IO 3 þ 6H þ 6e -I þ 3H2 O ½VIII E ¼ E0 þ 0:059 ½ox log n ½red ½1 Fe2þ þ Ce4þ "Fe3þ þ Ce3þ ½IX ½XVII then ½X EF ðCe4þ =Ce3þ Þ þ EF ðFe3þ =Fe2þ Þ 2 1:44 þ 0:68 ¼ 2 ¼ 1:06 V EEP ¼ þ BrO 3 þ 6H þ 6e -Br þ 3H2 O ðE0 ¼ 1:51 VÞ ClO þ 2Hþ þ 2e -Cl þ H2 O ½XI ½XII Reductants ½2 1.50 Fe2þ -Fe3þ þ e ðEF ¼ 0:68 VÞ Excess Ce(IV) 1.40 ½XIII 1.30 AsðIIIÞ-AsðVÞ þ 2e ðE0 ¼ 0:09 VÞ ½XIV ðEF ¼ 0:58 V; 1 mol l1 HClÞ 1.10 1.00 ½XV 0.90 For example, iron(II) can be determined by titration with dichromate, so combination of the appropriate half-reactions [VII] and [XIII], so as to achieve a charge and mass balance, gives the overall reaction [XVI]: 2þ Cr2 O2 þ 14Hþ -2Cr3 þ 6Fe3þ þ 7H2 O 7 þ 6Fe 1.20 E (V ) 2 2S2 O2 3 -S4 O6 þ 2e ½XVI The driving force for each half-reaction is measured by its oxidation potential, E, measured in V, which is 0.80 Excess Fe(II) 0.70 0 25 Titrant (ml) 50 Figure 5 Titration of 25 ml of 0.1 mol l 1 iron(II) with 0.1 mol l 1 cerium(IV). (Redrawn from Vogel’s Textbook of Quantitative Inorganic Analysis, 4th edn (1978). London: Longman.) TITRIMETRY / Overview 111 For more complex systems, however, especially those involving oxoanions, such simple calculations are not valid. Indicators for redox titrations will be chosen to change color reversibly by oxidation or reduction at a potential as close as possible to the equivalence potential (starch indication for iodine is an exception). This aspect is described in detail in another article. Some analytes may be determined by titration with an oxidant, after their reduction. There are several ways of carrying out such reductions. One commonly used reductant is tin(II) chloride, in which the excess of tin(II) is destroyed by addition of mercury(II) chloride: SnðIIÞ þ 2HgðIIÞ þ 2Cl -SnðIVÞ þ Hg2 Cl2 Redox titrations are still widely used. Table 4 summarizes some applications of redox titrations. Compleximetric Titrations Compleximetric titrations are used mainly to determine metal ions by use of complex-forming reactions. Although in theory many complexing agents (cyanide, thiocyanate, fluoride, 1,2-diaminoethane, etc.) could be used for this purpose, in practice the titrants are almost always compounds having the iminodiacetic acid functional group: CH2COOH N ½XVIII The mercury(I) chloride is unaffected by oxidants during the subsequent titration. Sulfite (or SO2) and hydrogen sulfide are alternative reductants. Metals may also be used. Small pieces of metal (zinc – a Jones reductor, silver – a Walden reductor) are used to fill a column, through which the analyte solution is passed. The effluent is titrated with oxidant. A comparison of the reduction products of the two reductor columns is given in Table 3. Table 3 Comparison of the Jones and Walden reductors Walden (HCl) Jones (H2SO4) Fe(III)-Fe(II) Ti(IV) not reduced Cr(III) not reduced V(V)-V(IV) Mo(VI)-Mo(V) Cu2 þ -Cu(I) Fe(III)-Fe(II) Ti(IV)-Ti(III) Cr(III)-Cr(II) V(V)-V(II) Mo(VI)-Mo(III) Cu2 þ -Cu(0) CH2COOH and by far the most popular of these is ethylenediaminetetraacetic acid (EDTA): CH2COOH HOOCCH2 N CH2 CH2 N HOOCCH2 CH2COOH (H4L) This ligand fulfills many of the qualities of a good compleximetric titrant. It forms complexes with most metal ions (those with the alkali metals are too weak to be useful); all the complexes have exact 1:1 stoichiometry, because the ligand is hexadentate, and can therefore occupy up to six coordination positions on the metal ions, as shown in Figure 6. The reaction with most metal ions is rapid (Cr3 þ is a well-known exception), and the complexes are water Table 4 Examples of redox titrations Analyte Titrant Indicator Iron(II) Iron(III) H2O2 Iron Ethanol Oxalate Nitrite Copper(II) Acids Available chlorine Antimony(III) Magnesium(II) KMnO4 KMnO4 KMnO4 K2Cr2O7 K2Cr2O7 Ce(SO4)2 Ce(SO4)2 Na2S2O3 Na2S2O3 Na2S2O3 I2 Na2S2O3 Self-indicating Self-indicating Self-indicating Diphenylamine sulfonic acid N-Phenylanthranilic acid Nitroferroin Ferroin Starch Starch Starch Starch Starch Ascorbic acid Ammonia KIO3 NaClO Self-indicating (I2/CCl4) Bromothymol blue Condition SnCl2 reduction In iron ore, SnCl2 reduction Add excess oxidant, heat, back-titrate with iron(II) Add excess oxidant, heat, back-titrate with iron(II) Add excess oxidant, back-titrate with iron(II) Iodide oxidized to iodine þ 5I þ IO 3 þ 6H -3I2 þ 3H2 O Oxidation of I to I2 Precipitate Mg2 þ with 8-quinolinol, add excess KBrO3/KBr to brominate precipitate, determine excess KBrO3 by oxidation of I -I2 Add Br and excess NaClO 112 TITRIMETRY / Overview Table 5 Stability constants of some EDTA complexes and optimum pH for titration of the metal ions with EDTA 2− CO Metal ion O CH2 CO 2þ Mg Ca2 þ Ba2 þ Mn2 þ Zn2 þ Ni2 þ Co2 þ Fe2 þ Fe3 þ Cu2 þ Hg2 þ Pb2 þ Bi3 þ CH2 O N CH2 M CH2 N O CO CH2 CH2 O log k1 Optimum pH for titration 8.7 10.7 7.8 13.8 16.5 18.6 16.3 14.3 25.1 18.8 21.8 18.0 27.9 10 7.5 12–13 5.5 4 3 4 5 1 3 5.0–5.5a 4 1–3 The behavior of Hg2 þ is somewhat anomalous because of hydroxocomplex formation. Values obtained from Pribil (1982). a CO Figure 6 Structure of chelate of EDTA anion (L4 ) with a metal ion M2 þ . (Redrawn from Vogel’s Textbook of Quantitative Inorganic Analysis, 4th edn (1978). London: Longman.) CH2COOH N CH2COOH soluble and colorless (unless the metal ion itself is colored). The reaction between a typical metal ion and EDTA (H4L) can be written as nþ M þ H4 L"ML ð4nÞ þ þ 4H ½XIX that is, as a competition between the metal ion and hydrogen ions for binding with L2 . The stability of binding of Mnþ with L2 is measured by its stability constant k1, which is the equilibrium constant for the reaction: Mnþ þ L4 "MLð4nÞ k1 ¼ ½MLð4nÞ ½Mnþ ½L4 ½XX ½3 where [ ] denotes concentrations (better, activities). Some typical stability constants are given in Table 5. Metals forming weaker complexes, therefore, require less acidic (i.e., higher pH) conditions for complex formation. The optimum pH values for the titration of a number of metal ions are included in Table 5. Metals forming stronger complexes can be titrated at lower pH values, at which the weaker complexing metals do not react, thus selective titration of, for example, bismuth can be carried out in the presence of lead at pH 1–2. The reaction of metal ions with EDTA (reaction (XIX)) generates H þ . Thus, to prevent a pH change during the titration, the solution must be adequately buffered. N(CH2COOH)3 CH2COOH N CH2COOH DCTA NTA Figure 7 Two alternative titrants to EDTA. Adjustment of pH often will not give sufficient selectivity and, of course, is inappropriate if a weakercomplexing metal (MW) has to be titrated in the presence of a more strongly complexing metal (MS). In such circumstances it is possible to ‘mask’ MS by adding another complexing agent that complexes much more strongly with MS than with MW, so that MW but not MS will react with EDTA. The use of such masking agents is widespread in compleximetric titrimetry. A typical example is the use of triethanolamine to mask iron(III) when calcium is titrated with EDTA in alkaline solution. A wide range of visual indicators is available for compleximetric titrations. These generally function by forming a colored complex with the metal ion being titrated, which causes a color change when the metal ion is removed from the complex by reaction with EDTA and releases the free ligand. These indicators are described in detail in another article. Other EDTA-type compounds are sometimes used as titrants, including 1,2-diaminocyclohexaneN,N,N0 ,N0 -tetraacetic acid, which generally forms stronger complexes than EDTA, and nitrilotriacetic acid, which generally forms weaker complexes than EDTA (Figure 7). TITRIMETRY / Overview 113 Nonaqueous Titrimetry Most titrations are carried out in aqueous solution, including all those described above. In some circumstances, however, it is advantageous to use other solvents, especially organic solvents. Such nonaqueous titrations are normally used for acid–base reactions, but redox reactions may also be applicable. The Karl–Fischer titration of water, in particular, is based upon redox reactions in a nonaqueous medium. The ionization of a molecule HB in a solvent S is influenced by the solvation of the ions: HB þ nS"HSþ þ BS ½XXI The ease of dissociation to form HS þ (solvated H þ ) increases with increasing basicity of the solvent, i.e., N2 N2 with increasing binding strength between H þ and the solvent. Thus, an acid that is very weak in aqueous solution will be stronger in a more basic solvent such as pyridine or dimethylformamide, and will give a bigger ‘pH’ change on titration. Phenols, for example, which are too weak acids to be titrated in aqueous solution, can be titrated in pyridine solution with tetrabutylammonium hydroxide in benzene– methanol (9:1, v/v) as titrant, and thymolphthalein in methanol as indicator. Similarly, bases that are very weak in aqueous solution (e.g., amines) show increased basicity in solvents of greater acidity, such as anhydrous acetic acid. Perchloric acid in acetic acid may be used as the titrant, with crystal violet in acetic acid as indicator. Because many of the solvents used are aggressive, volatile, and obnoxious, nonaqueous titrations are normally carried out in a closed environment, which also minimizes the ingress of moisture (Figure 8). It is essential to ensure that all apparatus used is dry and, especially for titrations in basic solvents, a stream of nitrogen is used to prevent access of carbon dioxide to the solution being titrated. The titrant is stored in a reservoir connected directly to the burette. Compounds that may be determined by nonaqueous titrimetry include amines, amino acids, phenols, and Schiff’s bases. Carbonyl compounds (by oxidation and titration of the released H þ ) can also be determined. Such titrations are especially useful in the pharmaceutical industry. See also: Indicators: Acid–Base; Redox; Complexometric, Adsorption, and Luminescence Indicators. pH. Quality Assurance: Internal Standards. Water Determination. Further Reading Figure 8 Apparatus for visual titration with nonaqueous solvents. Automatic burette protected with guard tubes; a guard tube and blow-bulb are attached to the bottom outlet of the burette. The nitrogen flow can be omitted for titrations in acidic solvents. (Redrawn from Belcher et al. 1970.) Belcher R, Nutten AJ, and Macdonald AMG (1970) Quantitative Inorganic Analysis, 3rd edn. London: Butterworths. Kolthoff IM and Belcher R (1957) Volumetric Analysis (Redox Titrations), vol. III. New York: Interscience. Kolthoff IM and Stenger VA (1947) Volumetric Analysis, vols. I and II. New York: Interscience. Kucharsky J and Safarik L (1965) Titrations in Non-Aqueous Solvents. Amsterdam: Elsevier. Pribil R (1982) Applied Complexometry. Oxford: Pergamon. Schwarzenbach G and Flaschka H (1969) Complexometric Titrations, 2nd English edition. London: Methuen. West TS (1969) Complexometry with EDTA and Related Reagents. Poole: BDH Chemicals.