- No category
Polymeric Membrane Characterization: Microscopy & Spectroscopy
advertisement
membranes Review Microscopy and Spectroscopy Techniques for Characterization of Polymeric Membranes Yousef Alqaheem * and Abdulaziz A. Alomair Petroleum Research Center, Kuwait Institute for Scientific Research, Safat 13109, Kuwait; aomair@kisr.edu.kw * Correspondence: yqhaeem@kisr.edu.kw; Tel.: +965-2495-6929; Fax: +965-239-0445 Received: 19 December 2019; Accepted: 24 January 2020; Published: 24 February 2020 Abstract: Polymeric membrane is a proven technology for water purification and wastewater treatment. The membrane is also commercialized for gas separation, mainly for carbon dioxide removal and hydrogen recovery. Characterization techniques are excellent tools for exploring the membrane structure and the chemical properties. This information can be then optimized to improve the membrane for better performance. In this paper, characterization techniques for studying the physical structure such as scanning electron microscopy (SEM), transmission electron microscopy (TEM), and atomic force microscopy (AFM) are discussed. Techniques for investigating the crystal structure such as X-ray diffraction (XRD), small-angle X-ray scattering (SAXS), and wide-angle X-ray scattering (WAXS) are also considered. Other tools for determining the functional groups such Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, and nuclear magnetic resonance (NMR) are reviewed. Methods for determining the elemental composition such as energy-dispersion X-ray spectroscopy (EDS), X-ray fluorescent (XRF), and X-ray photoelectron spectroscopy (XPS) are explored. The paper also gives general guidelines for sample preparation and data interpretation for each characterization technique. Keywords: gas-separation membrane; membrane characterization; surface analysis; crystal structure; functional groups; elemental composition 1. Introduction Characterization is an important field in material science. It refers to determining the physical and chemical properties of the material for a better understanding. This high level of knowledge can be then engineered to optimize the material performance. In membrane science, characterization techniques are widely used to confirm the quality and the purity of the prepared membranes. Furthermore, characterization techniques are powerful tools for interpreting the membrane performance and studying membrane degradation. Moreover, intensive research is carried out on preparing membranes from different materials to overcome the limitation in polymeric membranes. This limitation is due to the tradeoff between permeability and selectivity. This means that the produced gas can either have high flowrate or high purity [1]. This limitation was overcome by copolymerization, functionalization, or adding fillers [2]. Characterization techniques give valuable information on the interaction between the polymer and the fillers. This paper reviews common spectroscopy techniques for the characterization of polymeric membranes. The techniques usually study the morphology data, crystal structure, functional groups, and chemical composition. For membrane morphology, scanning electron microscopy (SEM), transmission electron microscopy (TEM), and atomic force microscopy (AFM) are widely implemented. For studying the crystal structure and its shape and size, X-ray diffraction (XRD) is classically applied. Techniques such as small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) can provide crystallography data with added information about the particle size and pore size distribution. Membranes 2020, 10, 33; doi:10.3390/membranes10020033 www.mdpi.com/journal/membranes Membranes 2020, 10, 33 Membranes 2020, 10, 33 2 of 37 2 of 36 and pore size distribution. Fourier‐transform infrared (FTIR) spectroscopy, Raman spectroscopy, and nuclear magnetic resonance spectroscopy (NMR) are generally utilized for determining the Fourier-transform infrared (FTIR) spectroscopy, spectroscopy, and nuclear magnetic resonance functional groups. For measuring the chemical Raman composition, energy‐dispersion X‐ray (EDS), X‐ray spectroscopy (NMR) are generally utilized for determining the functional groups. For measuring fluorescent (XRF), and X‐ray photoelectron spectroscopy (XPS) are commonly employed. In the this chemical X-rayof(EDS), X-ray sample fluorescent (XRF), andand X-ray photoelectron paper, eachcomposition, technology energy-dispersion is discussed in terms operation, preparation, limitations. The spectroscopy are guidelines commonly for employed. In this paper, technology is discussed in terms of paper also gives(XPS) general interpreting the data each for polymeric membranes. operation, sample preparation, and limitations. The paper also gives general guidelines for interpreting the data for polymeric 2. Morphology Analysismembranes. Morphology Analysis 2.1.2.Scanning Electron Microscopy 2.1.Scanning Scanning electron Electron Microscopy microscopy (SEM) is one of the fundamental techniques for membrane characterization as it gives the morphology topography data of thetechniques prepared membranes [3]. Scanning electron microscopy (SEM) and is one of the fundamental for membrane Furthermore, SEM as canit be used determine the pore size in thedata caseofofthe a porous membrane [4]. [3]. For characterization gives thetomorphology and topography prepared membranes a dense membrane, SEM is used to measure the thickness of the selective layer for calculation of Furthermore, SEM can be used to determine the pore size in the case of a porous membrane [4]. Forthe a permeability in Barrer [5].is used to measure the thickness of the selective layer for calculation of the dense membrane, SEM SEM worksinsimilarly permeability Barrer [5].to an optical microscope but in SEM, electrons are used instead of light [6]. A beamSEM of concentrated electrons will bemicroscope sent to the sample and electrons this will cause a release works similarly to an optical but in SEM, are used insteadofofsecondary light [6]. electrons [7]. After that, the electrons are collected by a detector and then analyzed to form an image A beam of concentrated electrons will be sent to the sample and this will cause a release of secondary as electrons demonstrated in Figure 1. Compared to collected a light microscope, which a maximum magnification [7]. After that, the electrons are by a detector and gives then analyzed to form an image of as 1500, the magnification can reach to 1microscope, million [8].which gives a maximum magnification demonstrated in Figurein1.SEM Compared to aup light of 1500, the magnification in SEM can reach up to 1 million [8]. 2.1.1. Sample Preparation 2.1.1. Sample Preparation Most of the membrane samples require preparation before using SEM. First, the samples should theexceed membrane samples before using SEM.cutting First, the samples should be be solidMost and of not an area of 10 require by 4 cmpreparation [9]. If the sample is larger, it into smaller pieces solid required. and not exceed an area of 10 by 4should cm [9]. be If the sample isconductive larger, cutting into smaller then is then Second, the samples electrically in it order to havepieces good is images required. Second, the samples should be electrically conductive in order to have good images [10]. [10]. Due to the weak conductivity in most polymers, the membrane surface needs to be coated Due with to the weak conductivity in most polymers, the membrane surface needs to be coated with a conductive a conductive material such as gold or palladium [11]. Usually, the sample will be coated with a material such as gold or palladium [11]. Usually, the sample will be coated with a sputter-coater to sputter‐coater to form a thin film of gold of 10 nm [12]. In addition to improving the quality of the form a thin film of gold of 10 nm [12]. In addition to improving the quality of the analyzed images, the analyzed images, the coating also reduces the thermal damage on the sample due to charge build‐up coating also reduces the thermal damage on the sample due to charge build-up [13]. [13]. FigureFigure 1. Components of SEM device magnification membranes 1. Components of SEM devicefor forsurface surface magnification of of membranes [14]. [14]. Membranes 2020, 10, 33 3 of 37 SEM can measure the thickness of the dense layer too by cutting the membrane and analyzing the cross‐section area. Before coating the sample, it is advised to insert the sample in liquid nitrogen Membranes 2020, 10, 33 3 of 36 for a few minutes then cut it by a sharp blade. This will result in a better image and eliminates surface bend [15]. SEM can measure the thickness of the dense layer too by cutting the membrane and analyzing the cross-section area. Before coating the sample, it is advised to insert the sample in liquid nitrogen for 2.1.2. Data Interpretation a few minutes then cut it by a sharp blade. This will result in a better image and eliminates surface Data[15]. in SEM is given in the form of a surface image. The membrane surface is expected to be bend free from defects such as large holes and cracks. For a porous membrane, the surface is expected to Data Interpretation have2.1.2. a uniform structure of small holes. SEM can be used to calculate the average pore size by processing the a software such MATLAB function “regionprops” shown Data in image SEM is by given in the form of a as surface image.and The the membrane surface is expected as to be free in Figure 2.defects such as large holes and cracks. For a porous membrane, the surface is expected to have a from SEM is structure an excellent toolholes. for analyzing composite membranes made from twobyorprocessing more polymers. uniform of small SEM can be used to calculate the average pore size the imagea by a software suchselective as MATLAB and the function “regionprops” assupport shown infor Figure 2. mechanical Usually, thin layer of the polymer is deposited over a porous better SEM is an excellent tool analyzing composite membranes made fromabout two orthe more polymers. properties [16]. Examining thefor cross‐section surface can give information structure of the a thin layer of the selective is deposited over of a porous support membrane for better mechanical twoUsually, polymers. For example, Figurepolymer 3 shows SEM images a composite made from properties [16]. Examining the cross-section surface can give information about (MFFK‐1). the structureSEM of thewas membrane poly(1‐trimethylsilyl‐1‐propyne) (PTMSP) over microfiltration two polymers. For example, Figure 3 shows SEM images of a composite membrane made from furtherly used to calculate the thickness of the PTMSP layer, which has an average value of 1.75 m poly(1-trimethylsilyl-1-propyne) (PTMSP) over microfiltration membrane (MFFK-1). SEM was furtherly [17]. used to calculate the thickness of the PTMSP layer, which has an average value of 1.75 µm [17]. For polymeric gas separation membranes, the transport mechanism is mainly based on the For polymeric gas separation membranes, the transport mechanism is mainly based on the solution‐diffusion model, which states that the gas dissolves through the polymer and then diffuses. solution-diffusion model, which states that the gas dissolves through the polymer and then diffuses. In these membranes, the dense and andfree freefrom fromfully fully penetrating holes. In these membranes, thesurface surfaceshould should be be fully fully dense penetrating holes. However, bybyanalyzing cross‐section surface, layers of and dense andstructures porous structures However, analyzing the the cross-section surface, twotwo layers of dense porous are usually are usually observed [5].isThis is because the membrane preparation, solvent used and then observed [5]. This because during during the membrane preparation, a solvent isaused and is then removed removed causing the formation ofstructure a porous as shown Figurethe 4. gas To permeability calculate the causing the formation of a porous as structure shown in Figure 4. To in calculate in gas Barrer, the in thickness thethickness dense layer needed and this is can be measured by SEM as demonstrated permeability Barrer,ofthe ofisthe dense layer needed and this can be measured byinSEM Figure 4. as demonstrated in Figure 4. Figure 2.2. Image processing of SEMofdata to calculate pore sizethe profile of asize silicon film using [18]. Figure Image processing SEM data to the calculate pore profile of a MATLAB silicon film using MATLAB [18]. Membranes2020, 2020,10, 10,33 33 Membranes 4 4ofof37 36 Membranes 2020, 10, 33 4 of 37 Figure3. 3. SEM image of: (a) Non‐modified microfiltration membrane (MFFK‐1), (b) Figure SEM image of: (a)of: Non-modified microfiltration membranemembrane (MFFK-1), (b) composite poly(1Figure 3. SEM image (a) Non‐modified microfiltration (MFFK‐1), (b) composite poly(1‐trimethylsilyl‐1‐propyne) (PTMSPM) with MFFK‐1 support [17]. trimethylsilyl-1-propyne) (PTMSPM) with MFFK-1 supportwith [17]. composite poly(1‐trimethylsilyl‐1‐propyne) (PTMSPM) MFFK‐1 support [17]. Figure 4. SEM image of the cross‐section polyetherimide membrane showing porous and Figure4.4.SEM SEMimage imageofofthe thecross-section cross‐section polyetherimide membrane showing porous and Figure polyetherimide membrane showing porous and dense dense structures (left) and of thethe dense layer (right)[19]. dense structures andmeasurement measurement dense layer (right)[19]. structures (left) and(left) measurement of the denseoflayer (right) [19]. Copolymerization produces aapolymer made from two or more monomers of different materials. Copolymerization or more more monomers ofdifferent different materials. Copolymerizationproduces produces a polymer polymer made from two or monomers of materials. This can be beneficial in improving the polymer characteristics such as mechanical properties, This beneficial in improving the polymer characteristics such as mechanical properties, thermal Thiscan canbebe beneficial in improving the polymer characteristics such as mechanical properties, thermal stability, or gas transport [20]. Porous particles made from polystyrene (PS) and polyacrylic stability, gas transport Porous particles frommade polystyrene (PS) and polyacrylic acid (PAA) thermal or stability, or gas [20]. transport [20]. Porousmade particles from polystyrene (PS) and polyacrylic acid (PAA) were analyzed by SEM to measure the particle size [21]. Figure 5 shows the particles with were by analyzed SEM to measure particle size Figure shows the particles with size ranging acid analyzed (PAA) were by SEMthe to measure the [21]. particle size 5[21]. Figure 5 shows the particles with size ranging from 1 to 7 m. The above reported studies on characterization of polymeric membranes from 1 to 7 µm. The above studies on characterization of polymeric using SEM size ranging from 1 to 7 m.reported The above reported studies on characterization ofmembranes polymeric membranes using SEM are summarized in Table 1. are summarized in33Table 1. in Table 1. using SEM2020, are 10, summarized Membranes 5 of 37 2.1.3. Limitations 2.1.3. Limitations In SEM, the sample should be in the solid form and free from moistures [22]. This is because SEM, the sample should be in solid form evaporation. and free from moistures the [22]. This isneed because SEM In operates under vacuum and this willthe cause sample Furthermore, samples SEM under vacuumand andifthis causeissample evaporation. to be operates electrically conductive not,will coating necessary. It should Furthermore, be noted that the for samples pore‐sizeneed to be electrically conductive if not,having coating is necessary. be noted that forforpore‐size measurement, SEM can be usedand for pores a diameter in tensItofshould micrometers. However, gas measurement, SEM canSEM be used forsuitable pores having a diameter in tens of micrometers. However, separation membranes, is not for pore size measurements because the pore size is for in gas the fractionsmembranes, of nanometer or units Angstrom measurements thin films of is in separation SEM is notofsuitable for [23]. poreFurthermore, size measurements becauseofthe pore size thickness less of than 10 nm is not recommended due to the Furthermore, lower resolution of SEM [24]. In the fractions nanometer or units of Angstrom [23]. measurements of this thincase, films of high‐resolution techniques transmission due electron are required, will becase, thickness less than 10 nm issuch not as recommended to themicroscopy lower resolution of SEMwhich [24]. In this discussed in the following section. high‐resolution techniques such as transmission electron microscopy are required, which will be discussed in the following section. Figure 5. Particle measurements of polysulfone (PS) and polyacrylic acidco-polymer (PAA) co‐ by Figure 5. Particle size size measurements of polysulfone (PS) and polyacrylic acid (PAA) polymer by SEM [21]. SEM [21]. Table 1. Performed studies using SEM for characterization of polymeric membranes. Study Membrane Dense surface Polyetherimide Pore size Methodology Cut samples in liquid nitrogen. Gold coat samples. Cut samples in liquid nitrogen. Conclusion No pores thus a dense membrane. Average pore size of 30 Ref. [15] Membranes 2020, 10, 33 5 of 36 2.1.3. Limitations In SEM, the sample should be in the solid form and free from moistures [22]. This is because SEM operates under vacuum and this will cause sample evaporation. Furthermore, the samples need to be electrically conductive and if not, coating is necessary. It should be noted that for pore-size measurement, SEM can be used for pores having a diameter in tens of micrometers. However, for gas separation membranes, SEM is not suitable for pore size measurements because the pore size is in the fractions of nanometer or units of Angstrom [23]. Furthermore, measurements of thin films of thickness less than 10 nm is not recommended due to the lower resolution of SEM [24]. In this case, high-resolution techniques such as transmission electron microscopy are required, which will be discussed in the following section. Table 1. Performed studies using SEM for characterization of polymeric membranes. Study Membrane Methodology Dense surface Polyetherimide Cut samples in liquid nitrogen. Gold coat samples. Pore size measurements Polyethersulfone Cut samples in liquid nitrogen. Used an imaging software to determine the pore size. Membrane thickness poly(1-trimethylsilyl-1-propyne) (PTMSP) Particle size measurements Copolymer of polysulfone (PS) and polyacrylic acid (PAA) Immersed samples in isopropanol. Cut samples in liquid nitrogen. Coated samples with gold. Freeze-dried samples. Sputter-coated samples with platinum. Conclusion Ref. No pores thus a dense membrane. [15] Average pore size of 30 nm. [25] Average PTMSP thickness of 1.5 µm. [17] Particle size ranging from 1 to 7 µm. [21] 2.2. Transmission Electron Microscopy Transmission electron microscopy (TEM) is another technique for observing the membrane surface. Compared to SEM, the magnification in TEM can reach 50 million, making it more suitable for measurements in the nanometer scale [26]. In TEM, an electron gun is used, as a source of electrons, and pointed to the sample. The electrons pass through the sample and collected thereafter. These transmitted electrons will form an image in a fluorescent screen giving more details about the internal structure of the membrane [27]. The components of the TEM instrument are shown in Figure 6. 2.2.1. Sample Preparation Unlike SEM, TEM needs more preparation as the sample needs to be thin and small; usually less than 2.5 mm in diameter with thickness not exceeding 150 µm [27,28]. Before cutting the sample, most of the polymers are soft and it is suggested to harden them by immersing them in liquid nitrogen. To facilitate ion exchange and improve image contrast, the membranes are stained using a liquid containing positive stains that deposit electrons on the membrane surface such as sodium oxide and uranyl acetate [29]. After that, the sample is washed with deionized water and then dried. The sample is thereafter mounted on a TEM grid made of a metallic mesh and ready for analysis. 2.2.2. Data Interpretation TEM is widely used over SEM especially for analyzing mixed-matrix membranes made by adding nanoparticles to the polymer. The nanoparticles are expected to be uniformly distributed through the membrane [2]. However, agglomeration of the particles may occur due to molecular electrostatic forces [30]. TEM is an excellent instrument for studying the agglomeration of the particles. For example, Figure 7 shows the nanoparticles of titanium oxides deposited on polybenzimidazole (PBI) membrane for water vapor/gas separation. TEM revealed that some of the nanoparticles agglomerated but it is within the acceptable range [31]. Membranes 2020, 10, 33 6 of 37 Membranes 2020, 10, 33 6 of 37 particles. For example, Figure 7 shows the nanoparticles of titanium oxides deposited on polybenzimidazole (PBI) membrane for water vapor/gas separation. TEM revealed that some of the Membranes 33 particles.2020, For10, example, Figure 7 shows the nanoparticles of titanium oxides deposited6 ofon36 nanoparticles agglomerated but it is within the vapor/gas acceptable range [31]. polybenzimidazole (PBI) membrane for water separation. TEM revealed that some of the nanoparticles agglomerated but it is within the acceptable range [31]. Figure 6. Components of transmission electron microscopy (TEM) instrument [32]. 6. Components of transmissionelectron electron microscopy (TEM) instrument [32]. Figure 6.Figure Components of transmission microscopy (TEM) instrument [32]. Figure7.7.TEM TEM images of 2TiO 2 nanoparticles on polybenzimidazole (PBI) membrane with Figure images of TiO nanoparticles on polybenzimidazole (PBI) membrane with the addition the addition of TiO 2 nanoparticles: (a) At a magnification of 500 nm, (b) at a magnification of TiO nanoparticles: (a) At a magnification of 500 nm, (b) at a magnification of 50 nm [31]. 2 of 50 nm [31]. Thin-film composite (TFC) membranes are made mainly by depositing a selective layer over a support. Thecomposite film is usually nanometers TEM can be to measure the film thickness. Thin‐film (TFC)inmembranes areand made mainly byused depositing a selective layer over a Figure 8 shows aimages film of polyacrylic acid (PAA) deposited on polysulfone Based on TEM, the Figure 7. TEM of in TiO 2 nanoparticles on polybenzimidazole (PBI) membrane with support. The film is usually nanometers and TEM can be used to measure (PSf). the film thickness. Figure film thickness was about 20 nm [29]. The discussed characterization studies are summarized in Table 8 shows a film of polyacrylic acid (PAA) deposited on polysulfone (PSf). the film2. the addition of TiO 2 nanoparticles: (a) At a magnification of 500 nm,Based (b) atona TEM, magnification was about 20 nm [29]. The discussed characterization studies are summarized in Table 2. ofthickness 50 nm [31]. Thin‐film composite (TFC) membranes are made mainly by depositing a selective layer over a support. The film is usually in nanometers and TEM can be used to measure the film thickness. Figure 8 shows a film of polyacrylic acid (PAA) deposited on polysulfone (PSf). Based on TEM, the film thickness was about 20 nm [29]. The discussed characterization studies are summarized in Table 2. Membranes 2020, 10, 33 7 of 36 Membranes 2020, 10, 33 7 of 37 Figure 8.Figure TEM8.image of a nano‐film of of grafted polyacrylic acid (PAA) over polysulfone TEM image of a nano-film grafted on on polyacrylic acid (PAA) over polysulfone (PSf) [29].(PSf) [29]. Table 2. Different studies performed by TEM for membrane characterization. Table Study 2. Different studies performed by TEM for membrane characterization. Membrane Methodology Conclusion Ref. Agglomeration of Study particles in membranes of Agglomeration Polybenzimidazole MembraneCut samples in liquid nitrogen. Methodology (PBI) with titanium Gold coated samples. Polybenzimidazole (PBI) oxides particles Cut samples in liquid nitrogen. Low agglomeration Conclusion Ref. [31] effectLow particles in membranes Stained samples sodium hydroxide. Goldbycoated samples. Polyacrylicparticles acid Immersed samples in uranyl nitrate for 15 min. (PAA)/polysulfone Washed samples withsamples distilledby water. Stained sodium (PSf) Cut sampleshydroxide. to 60–100 nm in thickness by ultramicrotome. PAA film of 20 nm. Film thickness with titanium oxides agglomeration effect [31] [29] Immersed samples in uranyl nitrate PAA film of for 15 min. [29] 20 nm. Washed samples with distilled water. 2.2.3. Limitations Cut samples to 60‐100 nm in thickness Unlike SEM, TEM requires special preparation. The samples need to be generally no more than by ultramicrotome. Film thickness Polyacrylic acid (PAA)/polysulfone (PSf) 2.5 mm in diameter. Also, the thickness should be no more than 100 µm, otherwise reduction in thickness will be required. Furthermore, TEM gives the image in 2D format and this does not show the 2.2.3. Limitations topography data. Moreover, wet or volatile samples cannot be used in TEM due to the high vacuum, Unlike SEM, TEM evaporation requires special preparation. The samples need to be generally no more than which causes sample and this may damage the instrument. 2.5 mm in diameter. Also, the thickness should be no more than 100 m, otherwise reduction in 2.3. Atomic Microscopy thickness will Force be required. Furthermore, TEM gives the image in 2D format and this does not show the topography data. Moreover, wetisor volatile samples cannottechnique be used for in surface TEM due to the high Atomic force microscopy (AFM) a high-resolution microscopy examination. Figurewhich 9 shows the scale difference between SEM, and AFM, it is clear that AFM has a vacuum, causes sample evaporation and thisTEM, may damage theand instrument. similar magnification power compared to TEM. Yet, AFM can provide 3D images of the surface that provide surface topography along with surface roughness [33]. 2.3. can Atomic Force Microscopy Unlike SEM and TEM, AFM works by measuring the force between a probe and the sample. Atomic force microscopy (AFM) is a high‐resolution microscopy technique for surface The probe, which consists of a cantilever with a sharp tip, will try to touch the sample and this will cause examination. shows scale difference between SEM,and TEM, and AFM, it is clear the probe toFigure deflect9due to thethe attractive forces between the probe the surface [34]. and In reality, there that AFM similarformagnification power compared tosemi-contact TEM. Yet, AFM provide 3D images of the arehas twoamodes AFM operation: Contact mode and mode.can In contact mode, the probe surface that can provide surface topography along with surface roughness [33]. will constantly touch the sample surface by adjusting the applied force, while in semi-contact mode, the Unlike and TEM, AFM by[35]. measuring the force between probethe and the sample. probe willSEM periodically touch theworks sample A laser beam will be used toadetect deflection and The the reflected laser will through with a photo-detector form Because and a probe used, probe, which consists of pass a cantilever a sharp tip,towill trythe toimage touch[36]. the sample thisiswill cause properties as stiffness canforces also bebetween measuredthe by probe AFM based indentation [37]. the mechanical probe to deflect duesuch to the attractive and on thethe surface [34]. In reality, there are two modes for AFM operation: Contact mode and semi‐contact mode. In contact mode, the probe will constantly touch the sample surface by adjusting the applied force, while in semi‐contact mode, the probe will periodically touch the sample [35]. A laser beam will be used to detect the deflection and the reflected laser will pass through a photo‐detector to form the image [36]. Because a probe is used, mechanical properties such as stiffness can also be measured by AFM based on the Membranes 2020, 10, 33 8 of 36 2.3.1. Sample Preparation In AFM,2020, liquid Membranes 10, 33 samples, in addition to solids, can be analyzed due to the absence 8 of of 37 vacuum. Compared to SEM and TEM, AFM requires less or no preparation steps. For most polymers, coating Sample Preparation with a2.3.1. conductive material or staining is not needed. It is advised however to cut the samples no larger than 10 byIn10 mmliquid with thickness to 2 mm [38]. The sample is then a substrate made from AFM, samples, inup addition to solids, can be analyzed due fixed to the over absence of vacuum. to SEM and TEM, requires a flat Compared mica (silicate minerals) orAFM a glass [39].less or no preparation steps. For most polymers, coating with a conductive material or staining is not needed. It is advised however to cut the samples no 10 by 10 mm with thickness up to 2 mm [38]. The sample is then fixed over a substrate 2.3.2. larger Data than Interpretation made from a flat mica (silicate minerals) or a glass [39]. AFM is capable of displaying the topography data of the membrane surface in the form of a 3D 2.3.2. Data Interpretationcan be useful in studying the change in the membrane roughness due to image. This information fouling. With the rejected molecules will accumulate onmembrane the membrane causing AFMtime, is capable of displaying the topography data of the surface surface in the form of a 3Da swelling, image. This the information can roughness be useful in [40]. studying the 10 change in the roughness to thus alternating membrane Figure shows the membrane topography data of due polyvinylidene fouling. With time, the rejected molecules will accumulate on the membrane surface causing a Figure 9 fluoride (PVDF)/polyvinylalcohol (PA) membrane before and after the operation. The data in swelling, thus alternating the membrane roughness [40]. Figure 10 shows the topography data of are presented by two parameters, Ra and Rq. Ra mainly represents the roughness and it is the arithmetic polyvinylidene fluoride (PVDF)/polyvinylalcohol (PA) membrane before and after the operation. The average of the absolute values of the surface height deviations [41]. The higher the value of Ra, the data in Figure 9 are presented by two parameters, Ra and Rq. Ra mainly represents the roughness rougher On the otherofhand, Rq represents thesurface root mean square average of height andthe it issurface. the arithmetic average the absolute values of the height deviations [41]. The higherdeviation takenthe from theofplane. value Ra, the rougher the surface. On the other hand, Rq represents the root mean square average height deviation taken from the the plane. AFM is of also capable of determining pore size distribution especially if the pore size of the AFM is also capable of determining the pore distributionfor especially if the pore size of waste-water the membrane is larger than 2 nm [42]. Ultrafiltrationsize membranes water purification and membrane is larger than 2 nm [42]. Ultrafiltration membranes for water purification and waste‐water treatments usually have pores with a size of 10 nm making AFM a suitable technique for pore size treatments usually have pores with a size of 10 nm making AFM a suitable technique for pore size measurement [43]. Similar to SEM, pore size distribution can be determined by measuring the pore measurement [43]. Similar to SEM, pore size distribution can be determined by measuring the pore area of the images inFigure Figure [44]. However, provides better accuracy area of generated the generated imagesas asshown shown in 1111 [44]. However, AFM AFM provides better accuracy compared to SEM especially for mesoporous structure with pore size ranging from 2 to compared to SEM especially for mesoporous structure with pore size ranging from 2 to 50 nm. 50 nm. Figure 9. Differences between optical microscopy, SEM, TEM, and Figure 9. Differences between optical microscopy, SEM, TEM, andatomic atomicforce forcemicroscopy microscopy (AFM) in resolution [45]. Membranes 2020, 10, 33 9 of 37 (AFM) in resolution [45]. Figure 10. 3D AFM data of polyvinylidene fluoride (PVDF)/polyvinylalcohol (PA) membrane: (a) Figure 10. 3D AFM data of polyvinylidene fluoride (PVDF)/polyvinylalcohol (PA) Originalmembrane: surface, (b) after fouling to theafter exposure natural organic matter (pH3) [46]. (a)surface Original surface, (b)due surface foulingtodue to the exposure to natural organic matter (pH3) [46]. Because AFM works based on a moving probe, some mechanical properties can be measured by studyingBecause the indentation the on probe on the membrane surface.properties A graphcan ofbe the force versus the AFM worksof based a moving probe, some mechanical measured by studying the indentation of the probe on the membrane surface. A graph of the force versus the displacement can be plotted and then converted to a plot of the stress versus the strain to calculate the stiffness of the membrane (Youngʹs modulus) as shown in Figure 12. In Figure 12b, the linear relationship between force and displacement indicates an elastic deformation. Young’s modulus values in Figure 12c were calculated using the Derjaguin–Muller–Toporov (DMT) method and the Figure 10. 3D AFM data of polyvinylidene fluoride (PVDF)/polyvinylalcohol (PA) membrane: (a) Original surface, (b) surface after fouling due to the exposure to natural Membranes 2020, 10, 33matter (pH3) [46]. organic 9 of 36 Because AFM works based on a moving probe, some mechanical properties can be measured by studying can the indentation of thethen probe on the membrane A graph of thethe force versus displacement be plotted and converted to a plotsurface. of the stress versus strain to the calculate displacement be plotted(Young’s and then converted a plot of the versus calculate the stiffness of thecan membrane modulus)toas shown instress Figure 12. the In strain Figureto12b, the linear the stiffness of the membrane (Youngʹs modulus) as an shown in Figure 12. In Figure 12b,modulus the linearvalues relationship between force and displacement indicates elastic deformation. Young’s relationship between force and displacement indicates an elastic deformation. Young’s modulus in Figure 12c were calculated using the Derjaguin–Muller–Toporov (DMT) method and the first value values in Figure 12c were calculated using the Derjaguin–Muller–Toporov (DMT) method and the of 0.71first GPavalue at the an elastic deformation due to thedue elastic inner chains the polymer. of surface 0.71 GPashowed at the surface showed an elastic deformation to the elastic inner of chains of The change of Young’s modulus value to 0.39 GPa indicates lessindicates elasticity along withalong the membrane the polymer. The change of Youngʹs modulus value to 0.39 GPa less elasticity with depth.the It was also found later thatalso the found stiffer the the the lower the gas permeability membrane depth. It was latermembrane, that the stiffer membrane, the lower the due gas to the permeability due to the reduction in free volumes [47]. These volumes provide the path for gas reduction in free volumes [47]. These volumes provide the path for gas transport through the dense transport the studies dense membrane. The discussed studiescharacterization on using AFM are for given membrane membrane. Thethrough discussed on using AFM for membrane in Table 3. characterization are given in Table 3. 2.3.3. Limitations 2.3.3. Limitations AFM has some benefits over SEM and TEM in terms of sample preparation. However, AFM AFM has some benefits over SEM and TEM in terms of sample preparation. However, AFM analysis requires more processing time. Also, AFM has a lower depth of field compared to SEM [48]. analysis requires more processing time. Also, AFM has a lower depth of field compared to SEM [48]. Nevertheless, the contrast in AFM images better[49]. [49]. Nevertheless, the contrast in AFM imagesisisgenerally generally better 11. Determination pore size distribution of polyvinylidene (PVDF) FigureFigure 11. Determination of poreofsize distribution of polyvinylidene fluoride fluoride (PVDF) membrane by membrane by analyzing the 3D image generated by AFM [50]. analyzing the 3D image generated by AFM [50]. Membranes 2020, 10, 33 10 of 37 12.ofUse of for AFM the measurements of the stiffness of polymer of intrinsic FigureFigure 12. Use AFM thefor measurements of the stiffness of polymer of intrinsic microporosity microporosity by plotting the force versus displacement and was (PIM-1) membrane by(PIM‐1) plottingmembrane the force versus displacement and indentation. Young’s modulus indentation. modulus was calculated(DMT) based method on the Derjaguin–Muller–Toporov calculated based onYoung’s the Derjaguin–Muller–Toporov [51]. (DMT) method [51]. Table 3. Studies performed by AFM for membrane characterization. Table 3. Studies performed by AFM for membrane characterization. Study Membrane Methodology Conclusion Study Membrane Methodology Conclusion Ref. Roughness decreased Polyvinylidene fluoride Samples in contact mode with silicon nitride probe. Roughness Samples in contact mode with silicon (PVDF)/polyvinylalcohol Scan area of 5 µm by 5 µm. from 54 to 37 nm Polyvinylidene fluoride Roughness decreased measurements Roughness nitride probe. (PA) /polyvinylalcohol Five areas were analyzed. (PVDF) from indicating 54 to 37 nma fouling. [46] measurements Scan area of 5 m by 5 m. ◦ C overnight. (PA) indicating a fouling. Dried samples at 40 Five areas were analyzed. Pore size Polyvinylidene fluoride Average pore Semi-contact mode. Dried samples at 40C overnight. distribution (PVDF) size of 0.3 µm. Image processing by NT-MDT software. Pore size Polyvinylidene fluoride Semi‐contact mode. Average pore size of [50] Contact-mode analysis. by NT‐MDT distribution (PVDF) Image processing 0.3 m. Membrane Polymer of intrinsic Probe radius of 8 nm. Elasticity decrease with software. stiffness microporosity (PIM-1) Derjaguin–Muller–Toporov (DMT) method to membrane depth. Contact‐mode analysis. Young’s modulus. Membrane Polymer of intrinsiccalculate Probe radius of 8 nm. Elasticity decrease [51] stiffness microporosity (PIM‐1) Derjaguin–Muller–Toporov (DMT) with membrane depth. method to calculate Young’s modulus. 3. Crystal Structure Analysis 3.1. X‐ray Diffraction X‐ray diffraction (XRD) is a technique for studying the crystal structure of the membrane. The Ref. [46] [50] [51] Membranes 2020, 10, 33 10 of 36 3. Crystal Structure Analysis 3.1. X-ray Diffraction X-ray diffraction (XRD) is a technique for studying the crystal structure of the membrane. The technology is unique in determining the crystal structure type and the distance between the polymeric chains [52]. Moreover, the polymer structure, either glassy or amorphous, can be identified by XRD. Glassy polymers feature ordered, more crystallized structure with better mechanical properties [53–55]. They are widely used for gas separation due to the high selectivity [56]. This means that they separate gases with a higher product purity. The high selectivity in glassy polymers is related to the high gas solubility due to the excess of free volumes [57]. On the other hand, amorphous (rubbery) polymers have a random structure but with lower mechanical properties [58]. However, high gas permeability is achieved by amorphous membranes due to the absence of gas diffusion limitation [59]. In addition to crystal structure studies, the chemical compounds presented in the sample can be identified by XRD. This information is valuable for confirming sample purity. The formation of secondary phases due to membrane degradation can also be spotted by XRD. In XRD, a filament such as tungsten is used to generate an X-ray beam [60]. This beam is directed to the sample and the scattered X-ray is detected and analyzed. Figure 13 shows how XRD analysis is performed. Some X-rays will be reflected upon hitting the surface while some will enter the surface and then reflect as shown in Figure 14. This variation is called diffraction and Bragg’s law can be applied to calculate the diffraction angle (2θ) [61]: nλ = 2dsin θ Membranes 2020, 2020,10, 10, 33 33 Membranes (1) 11 of of 37 37 11 where nwhere is the order of reflection (usually unity), λ is is the thewavelength wavelength andis it is determined from the is the the order order of of reflection reflection (usually (usually unity), unity), is and itit is determined from from the the where nn is the wavelength and determined source source of radiation usedused (commonly copper anddddrepresents represents plane distance between source of radiation radiation used (commonly copperK-α), K‐α), and and represents thethe plane distance between set a set of (commonly copper K‐α), the plane distance between aa set of atoms. of atoms. of atoms. Figure 13. 13. XRD XRD instrumentation instrumentation diagram diagram [62]. [62]. Figure Figure 13. XRD instrumentation diagram [62]. Figure 14. 14. Diffraction Diffraction of of an an X‐ray X‐ray beam beam from from aa sample sample [63]. [63]. Figure Figure 14. Diffraction of an X-ray beam from a sample [63]. 3.1.1. Sample Sample Preparation Preparation 3.1.1. XRD is is aa non‐destructive non‐destructive test test with with minimum minimum preparation. preparation. ItIt can can be be used used to to analyze analyze solids, solids, as as XRD well as as powders. powders. For For solid solid membranes, membranes, the the samples samples are are usually usually cut cut into into small small sections sections of of 20 20 by by 20 20 well mm. The The sample sample is is then then mounted mounted on on the the XRD XRD chamber. chamber. For For polymer polymer powders, powders, generally, generally, 100 100 to to 200 200 mm. mg of of powder powder is is needed needed and and spread spread over over aa carbon carbon tape. tape. This This tape tape has has aa very very low low XRD XRD intensity intensity and and mg Membranes 2020, 10, 33 11 of 36 3.1.1. Sample Preparation XRD is a non-destructive test with minimum preparation. It can be used to analyze solids, as well as powders. For solid membranes, the samples are usually cut into small sections of 20 by 20 mm. The sample is then mounted on the XRD chamber. For polymer powders, generally, 100 to 200 mg of powder is needed and spread over a carbon tape. This tape has a very low XRD intensity and will not affect the analysis [64]. 3.1.2. Data Interpretation The data in XRD are represented by intensity (counts) versus diffraction peak angle (2θ). Usually, Membranes 2020, 10, 33 12 of 37 the angle starts from 10 to 80 and each compound has a defined set of peaks. For example, polyetherimide is known to have sharp peaks at the 21.4mechanical and 23.8 properties (2θ) [15]. (Figure If additional peaks were detected for a pure crystallized, which enhanced 15). Some researchers also observed the relationship between themay intensity and gas permeation they concluded that the higher the polyetherimide membrane, this indicate impurities or and membrane degradation. themembranes, lower the permeation This was interpreted by the limitation in gas diffusion In intensity, polymeric there is[67]. a tradeoff between permeability and selectivity due to the through the membrane as the structure becomes more crystallized [68]. nature of the polymer [1]. Mixed-matrix membranes can overcome this limitation by adding fillers of Amorphous and glassy structures can be distinguished in XRD by observing the peak shape another[62]. material to the polymer during the membrane preparation [65]. XRD can be beneficial for Sharp peaks refer to a more crystallized structure while broad peaks refer to an amorphous studying the effect of adding the polymer The fillersparameter are supposed to be detected structure as demonstrated in fillers Figure to 16.the d‐space (d) is matrix. another important for membrane in the XRD peak profile along with polymer. example, a mixed-matrix membrane was made characterization. In Equation (1), dthe represents the For distance between two planes of atoms. In polymeric membranes, d‐space can representtothe distance between the polymer chains and it is calculated based by adding aluminosilicate particles polyethersulfone with different concentrations [66]. XRD data on the highest peak of the XRD pattern. Long‐chain polymers are presumed to have a higher d‐space revealed that the intensity of polyethersulfone peaks increased making the structure more crystallized, value. In polymer science, it is observed that the longer the chain, the less the structure crystallinity which enhanced the mechanical properties (Figure 15). Some researchers also observed the relationship due to the transformation from a glassy to a rubbery structure [52]. In terms of gas permeability, it is betweeneasier the intensity and gas permeation and they concluded that the higher the intensity, the lower for the gas to pass through an amorphous structure due to the absence of diffusion limitation. the permeation [67]. This was by interpreted the limitation in gas diffusion through the membrane as This was also confirmed noticing anbyincrease in the membrane permeability for longer chain the structure becomes more crystallized polymers [69]. Table 4 summarizes the[68]. mentioned studies for membrane analysis by XRD. Figure XRD of a mixed‐matrix membrane of polyethersulfone membrane with Figure 15. XRD15. data of adata mixed-matrix membrane of polyethersulfone membrane with aluminosilicate aluminosilicate particles [66]. particles [66]. Amorphous and glassy structures can be distinguished in XRD by observing the peak shape [62]. Sharp peaks refer to a more crystallized structure while broad peaks refer to an amorphous structure as demonstrated in Figure 16. d-space (d) is another important parameter for membrane characterization. In Equation (1), d represents the distance between two planes of atoms. In polymeric membranes, d-space can represent the distance between the polymer chains and it is calculated based on the highest peak of the XRD pattern. Long-chain polymers are presumed to have a higher d-space value. In polymer science, it is observed that the longer the chain, the less the structure crystallinity due to the transformation from a glassy to a rubbery structure [52]. In terms of gas permeability, it is easier for Figure 16. Differentiation of XRD peaks for determining the glassy and amorphous structures [62]. Membranes 2020, 10, 33 12 of 36 the gas to pass through an amorphous structure due to the absence of diffusion limitation. This was alsoFigure confirmed by noticing in the membrane for longer membrane chain polymers 15. XRD data ofanaincrease mixed‐matrix membranepermeability of polyethersulfone with[69]. Table 4 summarizesparticles the mentioned aluminosilicate [66]. studies for membrane analysis by XRD. Figure of XRD for determining theamorphous glassy and amorphous Figure16. 16. Differentiation Differentiation of XRD peaks peaks for determining the glassy and structures [62]. structures [62]. Table 4. XRD studies for characterization of polymeric membrane. Study Membrane Methodology Conclusion Ref. Membrane purity Polyetherimide Samples fractured in liquid nitrogen. Carbon tape to hold the samples. Only polyetherimide peaks were detected indicating a pure sample. [15] Polymer Crystallinity Polyethersulfone with aluminosilicate particles Intensity increased indicating a more crystallized structure with better mechanical properties. [66] Polymer chain distance (d-space) 6H,12H-5,11methanodibenzo[b,f][1,5]diazocine [PIM-EA(Me2 )-TB] Increase in d-space indicated transformation from glassy to rubbery phase. [69] Not reported. Not reported. 3.1.3. Limitations X-ray beam does not interact strongly with lighter elements and this may limit the elements detection in XRD [70]. Also, XRD is more accurate for measuring large crystal structures rather than small ones [71]. Furthermore, if more compounds are presented in the sample, peak overlap may complicate the analysis. 3.2. X-ray Scattering XRD is performed widely for materials with well-ordered (crystalline) structure. However, for semi-crystalline or non-crystalline (amorphous) materials, X-ray scattering is preferred [72]. In X-ray scattering, a monochromatic beam of X-rays is sent to the sample in which some of the X-rays will be scattered. These scattered X-rays are then detected and analyzed. There are two types of X-ray scattering: Small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS). In SAXS, the scattering angle (2θ) is less than 5◦ and the technique focuses on studying the particles in a system. For instance, SAXS can determine the nanoparticle size distribution and pore size [73,74]. These properties can still be measured by microscopic techniques, however SAXS is better in giving the average values for larger areas [75]. On the other hand, WAXS scans at a scattering angle of over 5◦ with the ability to study the degree of crystallinity and the chemical composition [76]. The SAXS and WAXS are usually combined in one instrument and its components are given in Figure 17. 3.2.1. Sample Preparation SAXS and WAXS barely need sample preparation for polymeric membranes similar to XRD. Membranes 2020, 10, 33 13 of 36 3.2.2. Data Interpretation The data in SAXS/WAXS are presented by the intensity and the scattering vector (q) and the latter is calculated by: 4π θ (2) q= sin λ 2 where λ is the wavelength of the incident X-rays and θ is the scattering angle. SAXS was used to study the preparation method of sulfonated poly(aryl ether ketone) (SPEEK) membranes. The absence of the ionomer peak (–SO3 H) indicated no nano-phase separation between the hydrophilic segments and the polymer matrix (Figure 18). Addition of inorganic filler resulted in a significant increase in intensity. The high intensity revealed that the filler was merged into the mass-fractal structure meaning that PMoA was successfully crosslinked with the polymer [77]. SAXS was also used to calculate the particle radius (Lm ) in the solution using Schimdt equation: Lm = Membranes 2020, 10, 33 π 14 of 37 (3) qmin where 𝑞 is the lower limit of the scattering vector (q) in SAXS graph. PMoA particles were found where qmin is the lower limit of the scattering vector (q) in SAXS graph. PMoA particles were found to to have an average radius of 524Å. In addition to the above, WAXS was implemented to calculate the have an average radius of 524Å. In addition to the above, WAXS was implemented to calculate the pore pore size distribution of PIM membranes. Equation (3) was used to calculate the pore size but with size distribution of PIM membranes. Equation (3) was used to calculate the pore size but with the lower the lower and upper limit of scattering vector (Figure 19). The intensity is proportional to the amounts and upper limit of scattering vector (Figure 19). The intensity is proportional to the amounts of pores of pores having that pore size. The reported pore size for amidoxime‐grafted PIM membranes ranged having pore The reported pore sizethe forstudies amidoxime-grafted membranes ranged and 3.9 fromthat 3.9 to 5.9 size. Å [78]. Table 5 summarizes for membranePIM characterization by SAXSfrom to 5.9 Å [78]. Table 5 summarizes the studies for membrane characterization by SAXS and WAXS. WAXS. Figure 17. Small‐angle scattering (SAXS)/wide‐angle X‐ray (WAXS) scattering (WAXS) for Figure 17. Small-angle X-ray X‐ray scattering (SAXS)/wide-angle X-ray scattering instrument instrument for crystal and molecular analysis: (1) Rotation unit, (2) arm, (3) counterweight, crystal and molecular analysis: (1) Rotation unit, (2) arm, (3) counterweight, and (4) hot stage [79]. and (4) hot stage [79]. Table 5. SAXS/WAXS studies for characterization of polymeric membrane. Study Membrane Methodology Conclusion Ref. Polymer-filler interaction and particle size measurements Sulfonated poly(aryl ether ketone) (SPEEK) with phosphomolybdic acid (PMoA) filler Fixed wavelength of 1.5Å. Vacuum operation at room temperature. Data calibrated using positron-emission tomography (PET). No detection of –SO3 H group indicated no nano-phase separation. Broad peak showed amorphous SPEEK structure. Particle radius of 524 Å. [80] Pore size measurements Polymer of intrinsic microporosity (PIM) with amidoxime groups Not reported. Pore size distribution from 3.9 to 5.9 Å. [78] Figure 17. Small‐angle X‐ray scattering (SAXS)/wide‐angle X‐ray scattering (WAXS) instrument for crystal and molecular analysis: (1) Rotation unit, (2) arm, (3) counterweight, Membranes 2020, and10, (4)33 hot stage [79]. 14 of 36 FigureFigure 18. SAXS analysis of sulfonated poly(aryl ether ether ketone) (SPEEK)/phosphomolybdic acid (PMoA) 18.10,SAXS analysis of sulfonated poly(aryl ketone) (SPEEK)/phosphomolybdic Membranes 2020, 33 15 of 37 membranes (a), and pristine SPEEK membranes (b) [80]. acid (PMoA) membranes (a), and pristine SPEEK membranes (b) [80]. Figure 19. WAXS analysis revealed an amorphous structure of SPEEK due to the peak [80]. Figure 19. WAXS analysis revealed an amorphous structure of SPEEK due tobroad the broad peak [80]. 3.2.3. Limitations Table 5. SAXS/WAXS studies for characterization of polymeric membrane. Sample homogeneity is critical for SAXS analysis as sample aggregation or degradation will Study Ref. result in incorrect data [81].Membrane Sample damage by Methodology radiation is also possibleConclusion and can distort the data [82]. No detection of –SO3H group Fixed wavelength of 1.5Å. Furthermore, the scattered intensity can be weak for some systems [83]. no nano‐phase indicated Polymer‐filler Sulfonated poly(aryl 4. interaction ether ketone) (SPEEK) and particle size with phosphomolybdic Functional Groups Analysis measurements acid (PMoA) filler Vacuum operation at room temperature. Data calibrated using positron‐ emission tomography (PET). 4.1. Fourier-Transform Infrared PolymerSpectroscopy of intrinsic Pore size microporosity (PIM) separation. Broad peak showed amorphous SPEEK structure. Particle radius of 524 Å. [80] Pore size distribution from Not reported. [78] Fourier-transform infrared spectroscopy (FTIR) is a technique3.9toto determine the functional groups. measurements with amidoxime 5.9 Å. groups Usually, the functional groups give the chemical reaction properties of the compound. Some examples of the functional groups are hydroxyl (OH–) and carbonyl (C=O). If the functional groups are known, the 3.2.3. Limitations Sample homogeneity is critical for SAXS analysis as sample aggregation or degradation will result in incorrect data [81]. Sample damage by radiation is also possible and can distort the data [82]. Furthermore, the scattered intensity can be weak for some systems [83]. 4. Functional Groups Analysis made mainly from diamond [86]. This will cause creation of evanescent waves. The reflected beam is then detected and analyzed to generate the FTIR data. 4.1.1. Sample Preparation Membranes 2020, 10, 33 15 of 36 FTIR spectroscopy usually requires sample preparation before analysis. Generally, the membrane will be crushed into a powder of 2 to 5 mg then mixed with a solvent commonly potassium compound class can be determined. For example, the hydroxyl group (OH–) usually represents bromide with a mixing ratio of 1 to 100 [87, 88]. Then, the mixed powder is pressed in a diealcohols at a load carboxyl acids and FTIR easily those compounds discussed of 13can mm [89].distinguish The pellet between is then inserted in the FTIRaschamber forlater. analysis. ofand 10 tons to form a pellet In FTIR spectroscopy, thepotassium analysis canbromide be performed on gases, liquids, and solids.material It is a quantitative It should be mentioned that is usually used as a reference because it −1 [90]. Also, technique as wellin[84]. works window based onfrom sending radiation to the itsample the use does not interfere the It infrared 400 infrared to 4000 cm acts aswith a carrier forof the a reference solvent [85]. Some of the radiation will be absorbed by the sample and others will pass infrared spectrum. Furthermore, the reference material of potassium bromide will be used to plot the through or will bein transmitted. A mathematical modeltechniques (Fourier transform will be used transmittance axis the FTIR graph. Modern FTIR such as function) ATR requires little ortono covert the data into the spectrum. Figure 20 shows the components of the FTIR spectrometer. sample preparation with the ability to analyze solid samples [91]. Figure 20. Components of of FTIR fordetection detection functional groups Figure 20. Components FTIRspectroscopy spectroscopy for of of functional groups [32]. [32]. Attenuated total reflection (ATR) is another method for generating FTIR spectrum without the 4.1.2. Data Interpretation use of a reference material. In ATR, a beam of infrared light is sent to the sample through ATR crystal The data in FITR spectroscopy is given by the transmittance versus the wavelength. Zero made mainly from diamond [86]. This will cause creation of evanescent waves. The reflected beam is transmittance means that the sample absorbed all the radiation while 100% transmittance means that then detected and analyzed to generate the FTIR data. the sample absorbed the same amounts of radiation as the reference. The generated graph is unique for eachSample molecule and this can be used in identifying the functional groups. For instance, the hydroxyl 4.1.1. Preparation group (OH–) represents both alcohols and carboxylic acids but FTIR can differentiate between them FTIR spectroscopy usually requires sample preparation before analysis. Generally, the membrane based on the wavelength. For instance, alcohol’s peak is detected at a wavelength of 3200 to 3600 cm−1 will be crushed into a powder of 2 to 5 mg then mixed with a solvent commonly potassium bromide while carboxylic acids peak is detected at 2500 to 3300 cm−1 [92]. Table 6 shows the wavelength of with a mixing ratio of 1 to 100 [87,88]. Then, the mixed powder is pressed in a die at a load of 10 tons to different functional groups FTIR analysis. form a pellet of 13 mm [89].for The pellet is then inserted in the FTIR chamber for analysis. It should be FTIR spectroscopy can be very useful determining thematerial compatibility theinterfere polymer mentioned that potassium bromide is usuallyfor used as a reference becausebetween it does not and theinfrared solvent.window The membrane preparation involves useasofa carrier solventforfor solvent in the from 400 to 4000 cm−1 [90]. Also,the it acts thecasting. infraredThe spectrum. should not react with the polymer to avoid changing the physical and chemical properties of in the Furthermore, the reference material of potassium bromide will be used to plot the transmittance axis polymer A Modern study was by preparing a PSf membrane different solventswith such the FTIR[93]. graph. FTIRmade techniques such as ATR requires little orwith no sample preparation theas diethylene glycol (DEG), dimethylacetamide (DMAc), dimethylformamide (DMF), and n‐ ability to analyze solid samples [91]. methylpyrrolidone (NMP). FTIR data is given in Figure 21 and none of the solvents altered the FTIR 4.1.2. Data Interpretation spectrum indicating a pure PSf membrane. In mixed matrix membranes, FTIR analysis is widely used to determine thein interaction between the fillers for better compatibility [94]. This can The data FITR spectroscopy is polymer given byand thethe transmittance versus the wavelength. Zero transmittance means that the sample absorbed all the radiation while 100% transmittance means that the sample absorbed the same amounts of radiation as the reference. The generated graph is unique for each molecule and this can be used in identifying the functional groups. For instance, the hydroxyl group (OH–) represents both alcohols and carboxylic acids but FTIR can differentiate between them based on the wavelength. For instance, alcohol’s peak is detected at a wavelength of 3200 to 3600 cm−1 while carboxylic acids peak is detected at 2500 to 3300 cm−1 [92]. Table 6 shows the wavelength of different functional groups for FTIR analysis. FTIR spectroscopy can be very useful for determining the compatibility between the polymer and the solvent. The membrane preparation involves the use of solvent for casting. The solvent should not react with the polymer to avoid changing the physical and chemical properties of the polymer [93]. A study was made by preparing a PSf membrane with different solvents such as diethylene glycol such as oxygen and nitrogen as they do not absorb infrared radiation [96]. Table 6. Identification of chemicals based on the wavelength of the functional groups by FTIR spectrometer [97]. Membranes 2020, 10, 33 16 of 36 Wavelength (cm−1) Functional Group Chemical Class 3200–3500 OH– Alcohols 2500–3300 OH– Carboxylic acids (DEG), dimethylacetamide2800–3000 (DMAc), dimethylformamide (DMF), and n-methylpyrrolidone (NMP). N–H Amine salts FTIR data is given in Figure 21 and none of the solvents altered the FTIR spectrum indicating a pure 3267–3333 C–H alkynes PSf membrane. In mixed matrix membranes, FTIRC–H analysis is widelyalkenes used to determine the interaction 3000–3100 between the polymer and the fillers for better compatibility [94]. This can be studied by detecting the 2840–3000 C–H Alkanes functional group of the fillers2349 within the FTIR spectrum of the mixed matrix O=C=O carbon dioxidemembrane. Table 7 sum up the discussed membrane characterization studies 1380–1415 S=Oby FTIR. sulfates Figure FTIR analysis indicating significant change preparation polysulfone Figure 21.21. FTIR analysis indicating nono significant change in in thethe preparation of of polysulfone (PSF) (PSF) membrane using diethylene glycol (DEG), (DMAc), dimethylacetamide (DMAc), membrane using diethylene glycol (DEG), dimethylacetamide dimethylformamide (DMF), dimethylformamide n‐methylpyrrolidone (NMP) as solvents [93]. and n-methylpyrrolidone(DMF), (NMP)and as solvents [93]. 4.1.3. Limitations Table 7. FTIR studies for characterization of polymeric membrane. Membrane Ref. AsStudy discussed before, FTIR may requireMethodology sample preparation but Conclusion for modern FTIR techniques, Functional groups of only aryl almost no preparation is needed. Aqueous solutions are difficult to analyze by FTIR due to the strong Polymer‐ ethers, aryl sulfones and methyl infraredsolvent absorption of water [95]. Furthermore, FTIR cannot detect of twonoidentical Polysulfone Not reported. weremolecules detected indicating [93]atoms such compatibility as oxygen and nitrogen as they do not absorb infrared radiation [96].with the solvent interaction (solvent is compatible). Peaks of (C‐N), (Si‐O), (CO2–), and Table 6. Identification of chemicals based Polyetherimide with on the wavelength of the functional groups by FTIR spectrometer [97]. Polymer‐filler metal‐organic framework interaction Wavelength (cm−1 ) filler (MIL‐53) 3200–3500 2500–3300 2800–3000 3267–3333 3000–3100 2840–3000 2349 1380–1415 FTIR Spectrum recorded at 4000–500 cm‐1Group . Functional OH– OH– N–H C–H C–H C–H O=C=O S=O (Al‐O) indicated that MIL‐53 was successfully incorporated Chemical Classin the polymer matrix. [94] Alcohols Carboxylic acids Amine salts alkynes alkenes Alkanes carbon dioxide sulfates Table 7. FTIR studies for characterization of polymeric membrane. Study Membrane Polymer-solvent compatibility Polysulfone Polymer-filler interaction Polyetherimide with metal-organic framework filler (MIL-53) Methodology Conclusion Ref. Not reported. Functional groups of only aryl ethers, aryl sulfones and methyl were detected indicating no interaction with the solvent (solvent is compatible). [93] FTIR Spectrum recorded at 4000–500 cm−1 . Peaks of (C-N), (Si-O), (CO2 –), and (Al-O) indicated that MIL-53 was successfully incorporated in the polymer matrix. [94] Membranes 2020, 10, 33 Membranes 2020, 10, 33 18 of 37 17 of 36 4.2. Raman Spectroscopy 4.2. Raman Spectroscopy Raman spectroscopy studies the vibration and rotation modes of molecules [98]. Each Raman studies the vibration and be rotation modes of molecules compound compound hasspectroscopy its own unique spectrum that can cross‐referenced with a [98]. set ofEach known Raman has its own unique spectrum can be with a set of cause known Raman spectrum. spectrum. Basically, laser light isthat focused on cross-referenced the sample and this light will molecular vibration. Basically, light iswill focused the sample andthat thisresults light will cause molecular vibration. Thisphoton form This form oflaser excitation causeonlight scattering in shifting up or down the laser of excitation will cause light scattering that results shifting up or down energy [99]. Components of Raman instrument areingiven in Figure 22. the laser photon energy [99]. Components of Raman instrument are given Figure 22. technique that gives information about the Raman spectroscopy is a quantitative andinqualitative Raman spectroscopy is a quantitative andtoqualitative technique that gives information about functional groups in the polymer. Compared FTIR, Raman is more sensitive to the functional the functional groups in the polymer. Compared to FTIR, Raman is more sensitive to the functional groups giving sharper peaks [100]. Furthermore, diatomic molecules such as oxygen and nitrogen giving sharper peaks [100]. Furthermore, diatomic molecules such oxygen tool and nitrogen can cangroups be detected in Raman spectroscopy. Also, Raman spectrometer is an as excellent for studying detectedin in Raman spectroscopy. Raman spectrometer an the excellent tool for the thebechanges the polymer crystal Also, structure due to changesisin chemical andstudying mechanical changes in the polymer crystal structure due to changes in the chemical and mechanical properties [101]. properties [101]. Furthermore, polymer chain orientation and interfacial surface properties can be Furthermore, polymer chain orientation and interfacial surface properties can be obtained by Raman obtained by Raman spectrum [32]. spectrum [32]. 4.2.1. Sample Preparation 4.2.1. Sample Preparation Raman is is considered asas a non‐destructive test and thethe preparation is is much easier compared Raman considered a non-destructive test and preparation much easier comparedto other methods such as FTIR where dissolving the sample with a proper solvent may be needed. The to other methods such as FTIR where dissolving the sample with a proper solvent may be needed. 2 with 2thickness less than 2.5 cm placed sample area for Raman spectrometer is usually less than 10 cm The sample area for Raman spectrometer is usually less than 10 cm with thickness less than 2.5 cm over a glass orasilicon substrate [102]. [102]. placed over glass or silicon substrate Figure 22. Components Ramanspectroscopy spectroscopy instrumentation [103]. Figure 22. Components of of Raman instrumentation [103]. 4.2.2. Data Interpretation 4.2.2. Data Interpretation The data in Raman spectroscopy is given in the form of intensity over wavelength of the scattered The data in Raman spectroscopy is given in the form of intensity over wavelength of the photons (Raman shift). The technique can be used to determine the form of the crystal structure by scattered photons (Raman shift). technique can in beintensity used toofdetermine the indicates form of athe crystal observing the intensity peaks. For The example, the change the same peak variation structure by observing the intensity peaks. For example, the change in intensity of the same peak in the structure asymmetry. For instance, PVDF membrane is known to have three different structures; indicates a variation in the structure asymmetry.sequence For instance, PVDFand membrane is known have form I (all trans structure), form II (trans-Gauche structure), form III (three transto bonds three different structures; form I (all trans structure), form II (trans‐Gauche sequence structure), and separated by Gauche structure) [101]. Raman spectroscopy distinguished between these structures −1 form (threethe trans bonds separated by Gauche structure) [101]. spectroscopy distinguished by III noticing peak shape and intensity at a wavelength of 793Raman cm . The use of various solvents −1. The between these structures by noticing the peak shape and intensity at a wavelength of 793 cm such as DMF, NMP, and triethyl phosphate (TEP) for PVDF membrane preparation resulted in different usestructures of various solvents such as NMP, and oftriethyl phosphate (TEP) for additional PVDF membrane as shown in Figure 23.DMF, Form II structure PVDF was recognized by the peak at preparation resulted in different structures as shown in Figure 23. Form II structure of PVDF was 795 cm−1 (Figure 23c). recognized by the additional peak at 795 cm−1 (Figure 23c). Raman peaks can also be used to determine the functional groups of the polymer. For example, ethylene compounds with functional group of C=C can be recognized at a wavelength of 1623 cm‐1. Membranes 2020, 10, 33 18 of 36 Raman peaks can also be used to determine the functional groups of the polymer. For example, ethylene compounds with functional group of C=C can be recognized at a wavelength of 1623 cm−1 . Membranes 2020, 10, 33 19 of 37 Some chemicals have the same functional group such as ketones and aldehydes in which they share the carbonyl (C=O) group. spectroscopy can differentiate between ketones (RCOR’) aldehydes Some chemicals have Raman the same functional group such as ketones and aldehydes in whichand they share −1 , respectively [104,105]. Figure 24 demonstrates the (RCHO) at the wavelength of 1659 and 1725 cm the carbonyl (C=O) group. Raman spectroscopy can differentiate between ketones (RCOR’) and peaks and wavelengths different functional groups. aldehydes (RCHO) atofthe wavelength of 1659 and 1725 cm−1, respectively [104, 105]. Figure 24 In addition to the above, Raman spectroscopy be utilized to study the aging effect of the demonstrates the peaks and wavelengths of differentcan functional groups. membrane due to to operation. example, the presence new functional groups In addition the above,For Raman spectroscopy can beof utilized to study the aging within effect ofRaman the spectrum maydue indicate degradation. Another way to detect of membrane degradation by observing membrane to operation. For example, the presence new functional groupsis within Ramanthe spectrum may degradation. Another way to detect membrane degradation is by observing transformation ofindicate the main backbone into shorter chains [106]. theRaman transformation of the main backbone into shorter chains spectroscopy is a quantitative technique as well [106]. and for some mixtures, the intensity can Raman spectroscopy a quantitative as wellTo and for somethe mixtures, theofintensity canin a be related to the amounts ofisthe compoundtechnique in the sample. illustrate, volume methanol −1 volume in athe be related to thelinearly amountswith of the compound sample.peak To illustrate, the methanol sample increased the decreasein ofthe intensity at 729 cm and thisofwas used for −1 and this was used for the sample increased with the decrease of Raman intensityspectroscopy peak at 729 cm quantification [107].linearly The mentioned studies by for membrane characterization arequantification given in Table[107]. 8. The mentioned studies by Raman spectroscopy for membrane characterization are given in Table 8. 4.2.3. Limitation 4.2.3. Limitation Unlike the FTIR spectrometer, Raman spectroscopy requires a longer time for the analysis. Due to Unlike the FTIR spectrometer, Raman spectroscopy requires a longer time for the analysis. Due the use of laser in Raman spectroscopy, some samples may release fluorescent light that interferes with to the use of laser in Raman spectroscopy, some samples may release fluorescent light that interferes the spectrum causing an increase in the background noise [108]. Moreover, polar molecules often have with the spectrum causing an increase in the background noise [108]. Moreover, polar molecules weak Raman signals as the atoms hold electrons so closely [109]. often have weak Raman signals as the atoms hold electrons so closely [109]. Figure Raman spectroscopy of PVDF membrane different solvents: (a) DMF; (b) (c) Figure 23. 23. Raman spectroscopy of PVDF membrane usingusing different solvents: (a) DMF; (b) NMP; NMP; (c) triethyl phosphate (TEP) [101]. triethyl phosphate (TEP) [101]. Membranes 2020, 10, 33 19 of 36 Membranes 2020, 10, 33 20 of 37 Figure 24. Functional identified byspectroscopy Raman spectroscopy and the corresponding Figure 24. Functional groups groups identified by Raman and the corresponding wavelength [110]. wavelength [110]. Table 8. Raman studies for characterization of polymeric membrane. Study Table 8. Raman studies for characterization of polymericConclusion membrane. Membrane Methodology Study Membrane Methodology Conclusion Membrane layers were Additional peak at 795 cm−1 Membrane in layers were Polyvinylidene superimposed a solid sampler. Additional peak at 795 indicated formation of cm−1 fluoride (PVDF) Spectrum recordedinata solid sampler. Polyvinylidene superimposed trans-Gauche sequence structure. Crystal structure indicated formation of trans‐ −1 resolution. fluoride (PVDF) 2 cmSpectrum recorded at 2 cm−1 Crystal structure Membrane Membrane degradation degradation resolution. Samples mounted vertically. Samples mounted vertically. Perfluorinated He-Ne laser and Peltier-cooled He‐Ne laser and Peltier‐cooled sulfonic-acid Perfluorinated charge coupled device (CCD) to charge coupled device (CCD) to (PFSA) (PFSA)detect Raman spectrum. sulfonic‐acid detect Raman Resolution of < 2 spectrum. cm−1 . Resolution of < 2 cm−1. Ref. Ref. [101] [101] Gauche sequence structure. Decrease in intensity of C–O–C, Decrease in intensity of C–O– C–S andC, S–O indicated C–Sbonds and S–O bonds membrane degradation. indicated membrane [111] [111] degradation. 4.3.4.3. Nuclear Magnetic Resonance Nuclear Magnetic ResonanceSpectroscopy Spectroscopy Nuclear magnetic resonance (NMR) spectroscopy is another technique for analyzing the molecular Nuclear magnetic resonance (NMR) spectroscopy is another technique for analyzing the structure and identifying the functional groups. It is also used to study polymer blend miscibility molecular structure and identifying the functional groups. It is also used to study polymer blendand polymer degradation [112,113]. NMR spectroscopy is based on placing the sample between two poles miscibility and polymer degradation [112,113]. NMR spectroscopy is based on placing the sample The radio sample will spin andbroadcast radio waves will be two poles of a powerful magnet of abetween powerful magnet [114]. The sample will[114]. spin and waves will be to the sample. broadcast to excitation the sample. will cause excitation of the nuclei and this will generate a resonance This will cause of This the nuclei and this will generate a resonance frequency detected by a radio frequency detected by a radio receiver as to shown in Figure 25. Compared to Raman, NMR spectrum receiver as shown in Figure 25. Compared Raman, NMR spectrum has less background interference has less background interference and it is capable of detecting polar compounds [115]. and it is capable of detecting polar compounds [115]. 4.3.1. Sample Preparation 4.3.1. Sample Preparation Similar FTIR spectroscopy,the thesample sample in in NMR NMR spectroscopy toto bebe in in thethe liquid form. Similar to to FTIR spectroscopy, spectroscopyneeds needs liquid form. Generally, the membrane is cut into small pieces having a mass of 20 to 50 mg [116]. The sample Generally, the membrane is cut into small pieces having a mass of 20 to 50 mg [116]. The sample is is then then dissolved in a strong solvent such as deuterated chloroform [117]. The concentration of the dissolved in a strong solvent such as deuterated chloroform [117]. The concentration of the sample sample in the mixture should be about 1 wt% [118]. The liquid is then placed in a 5 mm tube and in the mixture should be about 1 wt% [118]. The liquid is then placed in a 5 mm tube and ready for ready for analysis [119]. analysis [119]. 4.3.2. Data Interpretation 4.3.2. Data Interpretation The data in NMR spectroscopy is presented by the chemical shift () in parts per million (ppm). The data in NMR spectroscopy is presented by the chemical shift (δ) in parts per million (ppm). This property is calculated from the resonance frequency of the sample (v) with a reference: This property is calculated from the resonance frequency of the sample (v) with a reference: 𝛿 δ= 𝑣 𝑣 vsample 𝑣 − vref vref (4) (4) Membranes 2020, 10, 33 20 of 36 Membranes 2020, 10, 33 21 of 37 Routinely, tetramethylsilane (TMS) is used as a reference and it has a chemical formula of Si(CH3 )4 . TMS hasRoutinely, a sharp resonance line with(TMS) its 12 protons Furthermore, TMS with most samples. tetramethylsilane is used [120]. as a reference and it hasisainert chemical formula of TheSi(CH chemical shifthas is aunique each functional as given[120]. in Figure 26. 3)4. TMS sharp for resonance line with group its 12 protons Furthermore, TMS is inert with NMR spectroscopy was used tounique determine the functional functionalgroup groups a polyetherimide most samples. The chemical shift is for each as of given in Figure 26. thin-film 0 -oxydianiline (ODA) and trimesoyl chloride [19]. The technique composite membrane made from 4,4 NMR spectroscopy was used to determine the functional groups of a polyetherimide thin‐film wascomposite useful in membrane determining the molecular structure of(ODA) the composite membrane by detecting compounds made from 4,4′‐oxydianiline and trimesoyl chloride [19]. The technique was in determining the molecular structure of with the composite by an detecting such as useful carboxylic acid at 172 ppm, phenyl rings attached oxygen at membrane 155 ppm, and aromatic compounds such as carboxylic acid at 172 ppm, phenyl rings attached with oxygen at 155 ppm, and ring attached to amine at 146.5 ppm (Figure 27). anFunctionalization aromatic ring attached to amine at 146.5 ppm (Figure 27). refers to adding a chemical group into the polymer molecules. It can enhance Functionalization refers to adding chemical group into theFor polymer molecules. It can enhance the polymer physical, mechanical, anda chemical properties. instance, amino-functionalized the polymer physical, mechanical, and chemical properties. For instance, amino‐functionalized carbon nanotubes were added to polysulfone (PSf) during the membrane preparation to improve the carbonreactivity nanotubes[121]. were added to polysulfone (PSf) during the membrane to improve the polymer It is known that the functionalization reactionpreparation of amino-carbon nanotubes polymer reactivity [121]. It is known that the functionalization reaction of amino‐carbon nanotubes with the polymer will result in formation of new functional groups. NMR was used to analyze with the polymer will result in formation of new functional groups. NMR was used to analyze the the mixed-matrix membrane and functional group such as NH2 protons was detected at 1.845 ppm. mixed‐matrix membrane and functional group such as NH2 protons was detected at 1.845 ppm. The The amino-benzo-crown-ether group was measured at 2.975 ppm, which was attributed to the –NH– amino‐benzo‐crown‐ether group was measured at 2.975 ppm, which was attributed to the –NH– bond confirming the functionalization reaction (Figure 28). bond confirming the functionalization reaction (Figure 28). Figure instrument[122]. [122]. Figure25. 25.Components Components of of NMR NMR instrument addition studyingthe thechemical chemical structure, structure, NMR the polymer blend In In addition totostudying NMRcan canbe beused usedtotostudy study the polymer blend miscibility. Two or more polymers can be blended to form a single membrane. NMP can be miscibility. Two or more polymers can be blended to form a single membrane. NMP can be implemented implemented to study between the interaction between the observing in the in intensity to study the interaction the polymers by polymers observingbythe changesthe in changes the intensity chemical in chemical shift. For instance, the miscibility between polysulfone and polyvinyl methyl ether shift. For instance, the miscibility between polysulfone and polyvinyl methyl ether (PVME) were (PVME) were studied at a weight percentage of 65 wt% and 35 wt%, respectively. NMR was studied at a weight percentage of 65 wt% and 35 wt%, respectively. NMR was performed for pure performed for pure polysulfone and PVME for comparison. The significant increase in polysulfone polysulfone and PVME for comparison. The significant increase in polysulfone intensity in the polymer intensity in the polymer mixture indicated a good miscibility [123]. mixture indicated a good miscibility [123]. NMR spectroscopy is an excellent tool for monitoring the membrane degradation. With time, NMR spectroscopy is an excellent monitoring the time, the membrane performance reduces tool due for to accumulation of membrane the rejecteddegradation. molecules orWith due to thethe membrane due[124]. to accumulation of the rejected or due toaccelerate the reaction reaction performance with the feed reduces impurities It is known that some acidicmolecules environments can with the feed impurities [124]. It is known that some acidic environments can accelerate membrane membrane degradation and this can be studied by detecting the free radical peaks by NMR [125]. degradation andthe this can be studied by detecting radical peaks by NMRdegradation [125]. Furthermore, shiftfree peaks indicates membrane [126]. Furthermore, disappearance of some chemicalthe theFor disappearance of some chemical shiftmembrane peaks indicates membrane [126]. For instance, a instance, a perfluorinated ionomer (Nafion® 117) has degradation been examined for degradation ® 117) has been examined for degradation due to fuel cell perfluorinated ionomer membrane (Nafion due to fuel cell operation. It was found that the C–F bond was decomposed to fluoride and sulfate ions indicating a membrane detection of hydroxyl and ions hydroperoxyl operation. It was found that thedecomposition. C–F bond wasMoreover, decomposed to fluoride and sulfate indicating a radicals also confirmed Nafion® degradation Table 9 summarizes the mentioned in this membrane decomposition. Moreover, detection[125]. of hydroxyl and hydroperoxyl radicalsstudies also confirmed ® for paper membrane[125]. analysis by NMR. Nafion degradation Table 9 summarizes the mentioned studies in this paper for membrane analysis by NMR. Membranes 2020, 10, 33 21 of 36 Table 9. Characterization of polymeric membranes by NMR. Study Membrane Methodology Conclusion Ref. Only peaks of carboxylic acid, carboxylic-amide-carbon, phenyl-carbon-oxygen, and carbon-aromatic ring-amine were detected indicating a pure polyetherimide. [19] Peaks of NH2 protons and amino-benzo-crown ether demonstrated the functionalization of carbon nanotubes in the polymer. [121] Increase in polymer intensity in the mixture indicated a good mixing. [123] Detection of F− , SO4 −2 , and OH− indicated membrane decomposition [125] Membrane purity Polyetherimide Spinning carbon (13 C) to generate the magnetic field. Polymer-filler interaction Polysulfone with functionalized carbon nanotubes Dissolved samples in deuterated chloroform. Polymer miscibility Polysulfone and polyvinyl methyl ether(PVME) Membrane degradation Perfluorinated ionomer (Nafion® 117) Membranes 2020, 10, 33 Membranes 2020, 10, 33 Not reported Solid-state 19 F NMR. Inserted samples in rotors filled with alcohol or water. 22 of 37 22 of 37 Figure 26. NMR chemical shift for various classes and functional groups [127]. Figure 26. NMR chemical shift for various classes and functional groups [127]. Figure 26. NMR chemical shift for various classes and functional groups [127]. Figure 27. Use of NMR for NMR study the structure of polyetherimide film composite membrane [19]. Figure 27. Use of formolecular study the molecular structure thin of polyetherimide thin film Figure 27. membrane Use of NMR composite [19].for study the molecular structure of polyetherimide thin film composite membrane [19]. Membranes 2020, 10, 33 Use of NMR for study the molecular structure of polyetherimide thin film 22 of 36 Figure 27. composite membrane [19]. Figure NMR analysis a mixed matrix membrane made polysulfone Figure 28. 28. NMR analysis of aofmixed matrix membrane made fromfrom polysulfone (PSf)(PSf) and and carbon carbon nanotubes showing the detection of amino‐benzo‐crown‐ether group at 2.975 ppm nanotubes showing the detection of amino-benzo-crown-ether group at 2.975 ppm [121]. [121]. 4.3.3. Limitations Paramagnetic elements such as oxygen and sodium have less NMR signals [128]. This is because those elements lose their magnetic properties after the magnet is removed. Furthermore, the quantitative analysis of heavy elements may take hours to complete [115]. 5. Elemental Composition Analysis 5.1. Energy-Dispersion X-Ray Spectroscopy Energy-dispersion X-ray spectroscopy (EDS) is a method for detecting the elements of a sample. EDS also gives the mass fraction of each element. EDS is considered as a “bulk” analysis technique that covers a larger area with a higher depth of generally of 1 µm [129]. The analysis processing time is also fast and data are displayed within seconds. EDS is commonly integrated with the SEM because both EDS and SEM share the same electron beam. This beam will excite the atom and will cause the release of electrons. The electrons from a higher state will move to fill in the vacancies and this will emit X-rays to balance the energy difference between the electrons (Figure 29). When the X-rays hit the EDS detector, pulses are created and then converted to a voltage by an analyzer in which the voltage is proportional to the energy of the X-rays [130]. 5.1.1. Sample Preparation The prepared samples for SEM can be directly analyzed by EDS. However, if the sample was coated due to poor conductivity, the coating material will be detected in the EDS spectrum. Therefore, the coating material should be carefully selected so it cannot overlap with the elements found in the sample. 5.1.2. Data Interpretation Data of EDS analysis are presented by the intensity in counts and X-ray energy in keV. Each element has a peak at specific X-ray energy and this information can be utilized as a reference to pinpoint the element. EDS is a quantitative technique too and the composition can be calculated based on the intensity. This is useful as well for determining the chemical formula of the sample. Actually, Membranes 2020, 10, 33 23 of 36 EDS gives the data in atomic composition then the data are used to calculate the chemical formula. For example, to calculate the formula of a compound containing two elements (A and B), the following equation can be used: Ax B y Membranes 2020, 10, 33 24 of 37 (5) where x and y are the atomic percentages determined by EDS. Now, the formula should be normalized 𝐴 𝐵 (4) by the lowest number (assuming x is the lower number): where x and y are the atomic percentages determined by EDS. Now, the formula should be AB y/x (6) normalized by the lowest number (assuming x is the lower number): 𝐴𝐵 / (5) The mass percentage can be then calculated using the atomic weight and the atomic number of The mass percentage can be then calculated using the atomic weight and the atomic number of thethe elements: elements: x(at%) × m x(wt%) = 𝑥 𝑤𝑡% x 𝑎𝑡% x(at%) 𝑥× m at%) × m y x + y(𝑚 𝑥 𝑎𝑡% 𝑚 𝑦 𝑎𝑡% 𝑚 (7) (5) where mx and my are the atomic masses of element x and y, respectively. EDS can also be used to whereany mx and my are masses of elementdue x and y, respectively. EDS can also be used to monitor changes in the the atomic membrane composition to operation. For example, the sulfur content monitor any changes in the membrane composition due to operation. For example, the sulfur content in polypiperazine-amide membrane was measured to study the membrane fouling due to coal mine in polypiperazine‐amide membrane was measured study the due to mine drainage [131]. EDS assisted the chemical cleaningtomethod to membrane restore thefouling membrane bycoal measuring the chemical cleaning method to restore the membrane by measuring drainage [131]. EDS assisted the sulfur after the treatment. Furthermore, EDS was used to determine the chemical formula of the sulfur after the treatment. Furthermore, EDS was used to determine the chemical formula of nanoparticles inserted in a poly(vinyl alcohol) membrane [132]. EDS matched the added filler of nanoparticles inserted in a poly(vinyl alcohol) membrane [132]. EDS matched the added filler of bismuth(III) oxide with the formula of Bi2 O3 as given in Figure 30. EDS studies for characterization of bismuth(III) oxide with the formula of Bi2O3 as given in Figure 30. EDS studies for characterization polymeric membranes are given in Table 10. of polymeric membranes are given in Table 10. Figure 29.29. Atom excitation of -rays ‐raysfor forEDS EDSanalysis analysis[133]. [133]. Figure Atom excitationby byelectron electronand and the the release release of 4.1.3. Limitations Table 10. Characterization of polymeric membranes by EDS. Study Membrane Methodology Conclusion Ref. Unfortunately, EDS cannot give the full analysis of polymers due to the inability to detect No additional elements to hydrogen. This is because hydrogen has no electrons ininthe k‐shell [134]. However, EDS can still be Cut samples liquid nitrogen. Membrane purity Polyetherimide polyetherimide were detected [15] Coated samples gold. useful to detect elements that are usually presented in by polymers such indicating as carbon, and a purenitrogen, sample. oxygen. Nevertheless, the quantitative analysis of EDS is more effective for elements with highdue atomic Reduction in sulfur content Restoration of a fouled Polypiperazine-amide Not reported to chemical cleaning showed [131] membrane by chemicalfrom cleaning20 and above [135]. Because polymers are organic materials with mainly mass starting membrane restoration. hydrogen, carbon, nitrogen, and oxygen, it is not suggested to use EDS to quantify the polymer. Poly(vinyl alcohol) and The calculated formula matched This Filler chemical formula Not reported [132] oxide fillers bismuth(III) oxide. is because the low atomic bismuth(III) mass elements produce weak X‐ray signals and therefore the composition data might be inaccurate. The typical detection limit of EDS is 0.1 wt% or 1000 ppm. This means that 5.1.3. EDSLimitations cannot detect elements below that range. The general error expected from EDS is between ‐2 to 2 wt%. Unfortunately, EDS cannot give the full analysis of polymers due to the inability to detect hydrogen. This is because hydrogen has no electrons in the k-shell [134]. However, EDS can still be useful to detect elements that are usually presented in polymers such as carbon, nitrogen, and oxygen. Nevertheless, Membranes 2020, 10, 33 24 of 36 the quantitative analysis of EDS is more effective for elements with high atomic mass starting from 20 and above [135]. Because polymers are organic materials with mainly hydrogen, carbon, nitrogen, and oxygen, it is not suggested to use EDS to quantify the polymer. This is because the low atomic mass elements produce weak X-ray signals and therefore the composition data might be inaccurate. The typical detection limit of EDS is 0.1 wt% or 1000 ppm. This means that EDS cannot detect elements below that range. Membranes 2020, 10,The 33 general error expected from EDS is between -2 to 2 wt%. 25 of 37 3 particles deposited poly(vinyl alcohol) membrane, Figure 30. SEM (a) SEM image of2 O Bi32O Figure 30. (a) image of Bi particles deposited on on poly(vinyl alcohol) membrane, (b) (b) EDS EDS analysis of Bi 2 O 3 , (c) membrane image [132]. analysis of Bi2 O3 , (c) membrane image [132]. 5.2. X-Ray Fluorescence Table 10. Characterization of polymeric membranes by EDS. Study Membrane Methodology the elementalConclusion X-ray fluorescence (XRF) is a method for determining composition of a Ref. sample additional elementsto to the sample. in wt%. Compared to EDS, XRF is a non-destructive test and can beNo applied directly Membrane Cut samples in liquid nitrogen. polyetherimide were Polyetherimide [15] Furthermore, thesamples need by forgold. a vacuum, detected liquid samples purity because XRF operates without Coated indicating acan purebe analyzed. In XRF, when an X-ray beam is concentrated on the sample, secondary (orsample. fluorescent) X-rays will a emit Restoration from the ofsample due to atom excitation as demonstrated. TheReduction high-energy in sulfurX-ray content source will fouled due to chemical cleaning cause membrane dislodging of an electron from the inner orbital shell of an atom [136]. To fill in this vacancy, an [131] by Polypiperazine‐amide Not reported showed membrane electronchemical will drop from another atom with a high-energy orbital shell [137]. This drop will cause the restoration. cleaning release of secondary or fluorescent X-ray. This emitted X-ray is unique for each element and this will Filler chemical Poly(vinyl alcohol) and The calculated formula reportedare available and they are intensively used [132] be used formula for identification. XRF handheldNot devices in the bismuth(III) oxide fillers matched bismuth(III) oxide. analysis of metals, soils, and food [138]. Figure 31 shows the components of a portable XRF device and electron excitation. 5.2. X‐Ray Fluorescence X‐ray Preparation fluorescence (XRF) is a method for determining the elemental composition of a sample in 5.2.1. Sample wt%. Compared to EDS, XRF is a non‐destructive test and can be applied directly to the sample. XRF is a fastbecause and non-destructive methodthe forneed identifying and quantifying elements. The results Furthermore, XRF operates without for a vacuum, liquid samples can be analyzed. are usually Most of the samples require preparation. In XRF, given when in an seconds. X‐ray beam is concentrated on do thenot sample, secondary (or fluorescent) X‐rays will emit from the sample due to atom excitation as demonstrated. The high‐energy X‐ray source will 5.2.2. Datadislodging Interpretation cause of an electron from the inner orbital shell of an atom [136]. To fill in this vacancy, an electron willpresented drop from with a high‐energy orbital shell(keV). [137].Each This element drop willwill cause XRF data are byanother plottingatom the intensity (counts) with energy have the release of secondary or fluorescent X‐ray. This emitted X‐ray is unique for each element and this a unique energy peak for easier identification. Figure 32 shows some elements and their corresponding willusing be used for identification. XRF handheld are available and they are intensively energy cadmium as an excitation source. devices In addition, the relative amplitude of each used peak in can the analysis of metals, soils, and food [138]. Figure 26 shows the components of a portable XRF be used to calculate the mass percentage of the elements [139]. XRF was used to analyze adevice doped and electron excitation. membrane made from polycarbonate and iron(III) chloride. The technique detected iron and chloride with5.2.1. the chemical formula of FeCl3 as given in Figure 33. Furthermore, a study was performed by Sample Preparation XRF to determine the capability of trioctylmethylammonium thiosalicylate (TOMATS) membrane in XRF is a fastfrom and non‐destructive for identifying and quantifying elements. The results extracting mercury natural waters.method The membranes were immersed inside the contaminated are usually given in seconds. Most of the samples do not require preparation. 5.2.2. Data Interpretation XRF data are presented by plotting the intensity (counts) with energy (keV). Each element will have a unique energy peak for easier identification. Figure 32 shows some elements and their Membranes Membranes 2020, 2020, 10, 10, 33 33 26 26 of of 37 37 each peak can be used to calculate the mass percentage of the elements [139]. XRF was used to analyze a membrane made from polycarbonate and iron(III) chloride. The technique detected iron and 25 of 36 Membranesdoped 2020, 10, 33 chloride with the chemical formula of FeCl33 as given in Figure 33. Furthermore, a study was performed by XRF to determine the capability of trioctylmethylammonium thiosalicylate (TOMATS) membrane in extracting mercuryusing from natural Theamounts membranes immersed inside the with water and mercury was measured in-situ waters. XRF. The of were mercury was monitored contaminated water and mercury was measured using in‐situ XRF. The amounts of mercury was time to determine the removal efficiency [140]. Table 11 summarized the above studies for the use of monitored with time to determine the removal efficiency [140]. Table 11 summarized the above XRF forstudies membrane analysis. for the use of XRF for membrane analysis. Figure 31. Components a handheld XRF device measuring the elementalcomposition composition[141]. Figure 31. Components of a of handheld XRF device forfor measuring the elemental [141]. Figure 32. Identification of compounds using XRF by radioactive cadmium [139]. Figure 32. Identification of compounds using XRF by radioactive cadmium [139]. Table 11. XRF studies for membrane characterization. Study Chemical formula of filler Mercury extraction from natural waters Membrane Polycarbonate and iron chloride filler Trioctylmethylammonium thiosalicylate (TOMATS) Methodology Conclusion Ref. Not reported Elemental composition of iron and chloride gives chemical formula of FeCl3 that matched the added filler. [139] Cut sample to disks of 1 to 3 cm in diameter. Palladium target X-ray tube with beryllium window. SPECTRA EDX software for intensity measurements. Mercury extraction by the membrane was monitored by detecting the amounts of mercury in the polymer. [140] Membranes 2020, 10, 33 26 of 36 Membranes 2020, 10, 33 27 of 37 Figure Detection FeCl a polycarbonatemembrane membranefor for hydrogen hydrogen separation 3 in a polycarbonate separation[142]. [142]. Figure 33.33. Detection ofof FeCl 3 in 5.2.3. Limitations Table 11. XRF studies for membrane characterization. XRF is a very useful tool for detecting the Methodology elements and their weightConclusion percentages. However, the Study Membrane Ref. Elemental composition of quantification data will be given for the total element, not the ions. For example, if iron is measured, iron and chloride gives Chemical Polycarbonate ironiron, not the mass percentage of each ion such as Fe+2 and Fe+3 . XRF will give the data for the and total Not reported chemical formula of FeCl3 [139] of filler the difference chloride filler Thisformula is because in characteristic energy of ions is so small that thetheXRD detector cannot that matched added filler. distinguish [143]. Moreover, some elements cannot be detected by XRF due to its weak fluorescent Cut sample to disks of 1 to 3 X-ray [142]. Some examples are hydrogen, carbon, and oxygen. However, polymers are organic cm in diameter. Mercury extraction by the Mercury containing mainly hydrocarbons and therefore, and XRF cannot be used to analyze the compounds Trioctylmethylammonium Palladium target X‐ray tube membrane was monitored extraction from [140] thiosalicylate the (TOMATS) withpenetrates beryllium window. detecting the for amounts of mm making purenatural polymer. Furthermore, XRF beam deeply insidebythe sample a few waters SPECTRA EDX software for mercury in the polymer. it unsuitable for measurements of thin films of micrometers. intensity measurements. 5.3. X-ray Photoelectron Spectroscopy 5.2.3. Limitations X-ray photoelectron spectroscopy (XPS) is a tool for identifying and quantifying the elements. XRF is a very useful tool for detecting the elements and their weight percentages. However, the XPS is considered a be sensitive surface-characterization as it gives theis profile of the quantification dataas will given for the total element, not thetechnique ions. For example, if iron measured, elemental with theiron, depth 1 tomass 10 nm [48]. Thisofiseach favorable for as better XRF willcomposition give the dataalong for the total notofthe percentage ion such Fe+2 and and accurate Fe+3. measurements of the surface especially for thin-film membranes [144]. Furthermore, XPS can easily This is because the difference in characteristic energy of ions is so small that the XRD detector cannot detect elements thatMoreover, EDS and some XRF cannot detect. distinguish [143]. elements cannot be detected by XRF due to its weak fluorescent XPS[142]. works by sending a beam of X-ray to the sample and thisHowever, will cause the release electrons X‐ray Some examples are hydrogen, carbon, and oxygen. polymers are of organic along with kinetic energymainly [145]. A detector is used to measure theXRF number of be electrons the the kinetic compounds containing hydrocarbons and therefore, and cannot used towith analyze pure polymer. Furthermore, the XRF beamelement penetrates inside the sample forand a few energy as demonstrated in Figure 34. Each hasdeeply a specific binding energy thismm canmaking be used to it unsuitable for measurements of thin for films of micrometers. identify the element. XPS is also useful determining the oxidation state of the element. For instance, sulfur compounds can have different forms such as sulfide (S−2 ), sulfite (SO3)−2 , and sulfate (SO4)−2 and 5.3.can X‐ray Photoelectron Spectroscopy XPS distinguish between these forms due to the difference in binding energy [146]. Furthermore, the detection limit in XPS isspectroscopy similar to EDS of 1000 ppm X‐ray photoelectron (XPS) is a tool for[147]. identifying and quantifying the elements. XPS is considered as a sensitive surface‐characterization technique as it gives the profile of the 5.3.1. Samplecomposition Preparationalong with the depth of 1 to 10 nm [48]. This is favorable for better and elemental accurate of thetest surface for thin‐film membranes Furthermore, XPS XPS ismeasurements a non-destructive andespecially usually does not require special [144]. preparation for polymeric can easily detect elements that EDS and XRF cannot detect. 2 membranes. Generally, the membrane is cut into small pieces with an area of 5 to 10 mm with sample XPS works by sending a beam of X‐ray to the sample and this will cause the release of electrons depth not exceeding 4 mm to fit inside the chamber. along with kinetic energy [145]. A detector is used to measure the number of electrons with the kinetic (SO4)‐2 and XPS can distinguish between these forms due to the difference in binding energy [146]. Furthermore, the detection limit in XPS is similar to EDS of 1000 ppm [147]. 5.3.1. Sample Preparation XPS is10, a 33 non‐destructive test and usually does not require special preparation for polymeric Membranes 2020, 27 of 36 membranes. Generally, the membrane is cut into small pieces with an area of 5 to 10 mm2 with sample depth not exceeding 4 mm to fit inside the chamber. 5.3.2. Data Interpretation 5.3.2. Data Interpretation The data in XPS are presented by the intensity (counts) and the binding energy (eV). First, a Theisdata in XPS are by the intensity thethat, binding energy (eV).will First, wide wide scan performed topresented find the elements in the(counts) sample.and After each element beascanned scan is performed to find the elements in the sample. After that, each element will be scanned separately to determine the functional groups and the oxidation state of the element. For instance, separately to polysulfone determine themembrane functional was groups and theby oxidation state of the voltage element.range For instance, polyol-grafted analyzed XPS with a wide from 0 to polyol‐grafted polysulfone membrane was analyzed by XPS with a wide voltage range from 0 to 700 700 eV and five elements were detected such as oxygen, nitrogen, carbon, sulfur, and chlorine [148]. eV and five elements were detected such as oxygen, nitrogen, carbon, sulfur, and chlorine [148]. The The carbon was again analyzed from 280 to 296 eV and a large single peak with asymmetrical shape carbon was again analyzed from 280 to 296 eV and a large single peak with asymmetrical shape was was detected. This peak is formed due to the combination of other smaller peaks as shown in Figure 30. detected. This peak is formed due to the combination of other smaller peaks as shown in Figure 30. Different chemical classes were found such as ketones (C–O), alkanes (C–H), and amines (C–N) as Different chemical classes were found such as ketones (C–O), alkanes (C–H), and amines (C–N) as demonstrated in Figure 35. Furthermore, the successful grafting of polyol on polysulfone was observed demonstrated in Figure 35. Furthermore, the successful grafting of polyol on polysulfone was by observed the increase in O1s intensity and the reduction in C1s intensity [148]. [148]. by the increase in O1s intensity and the reduction in C1s intensity Membranes 2020, 10, 33 29 of 37 hydrogen and helium still cannot be recognized in XPS due to the absence of core electrons [153]. In addition, functional groups of analysis byphotoelectron XPS couldspectroscopy be spectroscopy difficult due to peaks Figure 34. photoelectron (XPS) for elemental analysis [149]. Figure 34. Components Components ofX-ray X‐ray (XPS) foroverlap. elemental analysis [149]. XPS is also a very useful tool for studying the membrane degradation. During fuel cell operation, hydrogen peroxide is usually formed due to diffusion of oxygen across the membrane and its reaction with hydrogen [150]. Degradation of Nafion® membrane was noticed by the decomposition of (CF2)n backbone to fluoride and sulfate ions [151]. Quantification of elements is generally performed in XPS based on the intensity peak [152]. The area under the curve is calculated then used to estimate the weight fraction of each element. This will also help in determining the chemical formula of the sample similar to EDS. Table 12 summarizes the discussed studies for membrane characterization by XPS. 5.3.3. Limitations XPS is an excellent technique for determining the oxidation state of the elements in a sample. Nevertheless, the analysis is only limited to solids due to the requirement of a high vacuum. XPS analysis is also time‐consuming as it can take hours to cover a large area of the sample. Unfortunately, Figure 35. XPS of: (a) Polyol-grafted polysulfone membranes, (a’) with the (a’) determination Figure 35. analysis XPS analysis of: (a) Polyol‐grafted polysulfone membranes, with the of carbon compounds [148]. determination of carbon compounds [148]. XPS is also a very useful for studying the membrane degradation. During fuel cell operation, Table tool 12. Studies for membrane characterization by XPS. hydrogen peroxide is usually formed due to diffusion of oxygen across the membrane and its reaction Study Membrane Methodology Conclusion Ref. with hydrogen [150]. Degradation of Nafion® membrane was noticed by the decomposition of (CF2 )n Monochromatic Al K X‐ray backbone to fluoride and sulfate ions [151]. source. O1s intensity increased while Surface Measurements at 45 take‐off C1s intensity Quantification of polyol‐grafted elements is generally performed in XPS based on thedecreased intensity peak [152]. chemical angle. confirming the grafting of [148] The area under the curvepolysulfone is calculated then used to estimate the weight fraction of each element. This composition Survey scan from 0 to 1000 eV hydroxyl groups on the will also help in determining the chemicalthen formula of thescans sample to EDS.surface. Table 12 summarizes high‐resolution of similar membrane C1s regions. the discussed studies for membrane characterization by XPS. Membrane degradation Nafion® 112 Monochromator with Al K source. Step of 0.025 eV with 100ms. Polymer backbone was decomposed due to detection of fluoride and sulfate ions. [151] 6. Conclusions Characterization techniques are powerful tools for studying the physical structure and chemical Membranes 2020, 10, 33 28 of 36 Table 12. Studies for membrane characterization by XPS. Study Membrane Methodology Conclusion Ref. [148] [151] Surface chemical composition polyol-grafted polysulfone Monochromatic Al Kα X-ray source. Measurements at 45◦ take-off angle. Survey scan from 0 to 1000 eV then high-resolution scans of C1s regions. O1s intensity increased while C1s intensity decreased confirming the grafting of hydroxyl groups on the membrane surface. Membrane degradation Nafion® 112 Monochromator with Al Kα source. Step of 0.025 eV with 100ms. Polymer backbone was decomposed due to detection of fluoride and sulfate ions. 5.3.3. Limitations XPS is an excellent technique for determining the oxidation state of the elements in a sample. Nevertheless, the analysis is only limited to solids due to the requirement of a high vacuum. XPS analysis is also time-consuming as it can take hours to cover a large area of the sample. Unfortunately, hydrogen and helium still cannot be recognized in XPS due to the absence of core electrons [153]. In addition, functional groups analysis by XPS could be difficult due to peaks overlap. 6. Conclusions Characterization techniques are powerful tools for studying the physical structure and chemical properties of the membranes. These data are used thereafter for membrane development to enhance the performance. The basic characterization technique is the SEM where the membrane surface is examined. TEM provides a higher resolution compared to SEM for better studies on nanomaterials. One the other hand, AFM has a similar resolution to TEM but it can also provide some mechanical properties such as roughness. XRD is another tool for studying the crystal structure with the ability to name the compounds. SAXS and WAXS are more suitable for semi- and non-crystalline polymers with more statistics on the pore and particle sizes. FTIR, on the other hand, focuses on determining the functional groups, which can be used to identify the chemicals. Nevertheless, Raman spectroscopy is more sensitive in detecting these functional groups and requires almost no preparation. NMR spectroscopy provides another route for identifying the functional groups with minimum background interference. For the elemental analysis, EDS provides a basic elemental scan with a depth of 10 µm. XRF is a less sensitive technique for elemental analysis as the scan can reach up to 3 mm of depth, however the analysis can be directly applied by a handheld device with no sample preparation. On the other hand, XPS is a more advanced method for determining the elements and it is more of a surface technique due to the scanning depth of 10 nm. XPS can also give the weight composition along with the oxidation state of the elements. It is clear that the selection of the characterization technique depends on the membrane nature and the required study. However, techniques such as SEM, XRD, and EDS are considered as fundamentals for membrane characterization. More advanced techniques can give additional information on the interfacial surface properties, molecular structure, and chemical properties. Comparison between the techniques in term of performed studies, advantages, and limitations is given Table 13. Membranes 2020, 10, 33 29 of 36 Table 13. Comparison of various characterization techniques for analysis of polymeric membranes. Technique SEM Performed studies Surface topography. Pore size. Particle size Membrane Thickness Advantages Magnification of up to 1 million. Samples needs to be conductive. Not accurate for measurements less 10 nm. Images does not show topography data. Staining the sample may be required. Thick samples (> 100µm) are difficult to be analyzed in TEM. TEM Nanoparticles. Nanofilms. Nanopores. Membrane thickness. Higher resolution than SEM. AFM Nano-profiling. Surface roughness. Pore size distribution. Membrane stiffness. No sample preparation. Similar resolution to TEM. Measurements of mechanical properties. Membrane purity. Compounds chemical formula. Crystal structure. Polymer chain distance. Limitations Requires more processing time. Lower depth of field. No sample preparation. Detection of wide range of crystalline compounds. Heavy elements are less sensitive to XRD. Less accuracy for small crystals. Peaks overlap for some compounds. Crystal structure. Polymer-filler interaction. Particle size distribution. Pore size measurements. No sample preparation. Suitable for semi- and non-crystalline materials. More accurate average measurements for particle size and pore size. Scattering intensity can be weak for some systems. Functional groups. Polymer-solvent compatibility. Polymer-filler interaction. Miscibility of polymer blends. Membrane degradation. Detection of variety of compounds. High sensitivity of parts per million (ppm). Fast analysis time (in seconds). Raman Functional groups. Crystal structure. Polymer chain orientation. Polymer blends. Membrane fouling. No sample preparation. More sensitive to functional groups with better intensity peaks. Release of fluorescent light of some samples may cause background noise. Polar molecules have lower Raman signal. NMR Functional groups. Polymer-blend miscibility. Membrane decomposition. Less background interference. Detection of polar molecules. Liquid samples. Paramagnetic elements have less NMR signal. EDS Elemental composition. Chemical formula of fillers. Membrane fouling. Fast analysis time. Samples needs to be conductive. Limitation in detecting light elements. Cannot quantify ions. No sample preparation. In-situ analysis. Very low sensitivity to hydrogen, carbon and oxygen. Cannot quantify ions. Unsuitable for thin film measurements. High sensitivity. Quantification of ions. Measurements of thin films of nm. Solid samples. Cannot detect hydrogen and helium. Peaks Overlap for some elements. Long processing time. XRD SAXS WAXS FTIR XRF XPS Elemental composition. Chemical formula of fillers. Membrane degradation. Elemental composition. Functional groups. Formula of chemical compounds. Membrane degradation. Cannot analyze aqueous samples. Cannot detect molecules of two identical atoms. Membranes 2020, 10, 33 30 of 36 Author Contributions: Conceptualization, data collection, and writing by Y.A.; data discussion and interpretation by A.A.A. All authors have read and agreed to the published version of the manuscript. Funding: This research received no external funding. Conflicts of Interest: The authors declare no conflict of interest. References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. Freeman, B. Basis of permeability/selectivity tradeoff relations in polymeric gas separation membranes. Macromolecules 1999, 32, 375–380. [CrossRef] Goh, P.; Ismail, A.; Sanip, S.; Ng, B.; Aziz, M. Recent advances of inorganic fillers in mixed matrix membrane for gas separation. Sep. Purif. Technol. 2011, 81, 243–264. [CrossRef] Czanderna, A.; Madey, T.; Powell, C. Beam Effects, Surface Topography, and Depth Profiling in Surface Analysis; Springer: New York, NY, USA, 1998. Ziel, R.; Haus, A.; Tulke, A. Quantification of the pore size distribution (porosity profiles) in microfiltration membranes by SEM, TEM and computer image analysis. J. Membr. Sci. 2008, 323, 241–246. [CrossRef] Mohamad, M.; Fong, Y. Preparation of defect—Free Polysulfone membrane: Optimization of fabrication method. J. Sci. Res. Dev. 2016, 3, 126–131. Ghorbanpour, M.; Wani, S. Advances in Phytonanotechnology; Academic Press: Cambridge, MA, USA, 2019; pp. 45–121. Hawkes, P.; Reimer, L. Scanning Electron Microscopy: Physics of Image Formation and Microanalysis; Springer: Berlin, Germany, 2013. Fails, A.; Magee, C. Anatomy and Physiology of Farm Animals; Wiley: Hoboken, NJ, USA, 2018. Klapetek, P. Quantitative Data Processing in Scanning Probe Microscopy: SPM Applications for Nanometrology; Elsevier Science: Amsterdam, The Netherlands, 2013. Aharinejad, S.; Lametschwandtner, A. Microvascular Corrosion Casting in Scanning Electron Microscopy: Techniques and Applications; Springer: Berlin, Germany, 2012. Sawyer, L.; Grubb, D.; Meyers, G. Polymer Microscopy; Springer: New York, NY, USA, 2008. Morozov, O.; Bulgakov, B.; Ivanchenko, A.; Shachneva, S.; Nechausov, S.; Bermeshev, M.; Kepman, A. Data on synthesis and characterization of sulfonated poly(phenylnorbornene) and polymer electrolyte membranes based on it. Data Brief 2019, 27, 104626. [CrossRef] Fleck, R.; Humbel, B. Biological Field Emission Scanning Electron Microscopy; Wiley: Hoboken, NJ, USA, 2019. Choudhary, O.; Choudhary, P. Scanning electron microscope: Advantages and disadvantages in imaging components. Int. J. Curr. Microbiol. Appl. Sci. 2017, 6, 1877–1882. [CrossRef] Alqaheem, Y.; Alomair, A.; Alhendi, A.; Alkandari, S.; Tanoli, N.; Alnajdi, N.; Quesada-Peréz, A. Preparation of polyetherimide membrane from non-toxic solvents for the separation of hydrogen from methane. Chem. Cent. J. 2018, 12, 80. [CrossRef] Ren, Y.; Zhu, J.; Cong, S.; Wang, J.; Van der Bruggen, B.; Liu, J.; Zhang, Y. High flux thin film nanocomposite membranes based on porous organic polymers for nanofiltration. J. Membr. Sci. 2019, 585, 19–28. [CrossRef] Bazhenov, S.; Borisov, I.; Bakhtin, D.; Rybakova, A.; Khotimskiy, V.; Molchanov, S.; Volkov, V. High-permeance crosslinked PTMSP thin-film composite membranes as supports for CO2 selective layer formation. Green Energy Environ. 2016, 1, 235–245. [CrossRef] Gaur, G. Quantum Dot Integrated Silicon Photonic Devices for Optical Sensor Applications. Ph.D. Thesis, Vanderbilt University, Nashville, TN, USA, 2015. Almijbilee, M.; Wu, X.; Zhou, A.; Zheng, X.; Cao, X.; Li, W. Polyetheramide organic solvent nanofiltration membrane prepared via an interfacial assembly and polymerization procedure. Sep. Purif. Technol. 2020, 234, 116033. [CrossRef] Diab, M.; El-Sonbati, A.; El-Bindary, A.; Abd El-Ghany, H. Thermal stability and degradation of poly (N-phenylpropionamide) homopolymer and copolymer of N-phenylpropionamide with methyl methacrylate. Arabian J. Chem. 2017, 10, S3732–S3739. [CrossRef] Alvarez, J.; Saudino, G.; Musteata, V.; Madhavan, P.; Genovese, A.; Behzad, A.; Sougrat, R.; Boi, C.; Peinemann, K.-V.; Nunes, S. 3D analysis of ordered porous polymeric particles using complementary electron microscopy methods. Sci. Rep. 2019, 9, 13987. [CrossRef] [PubMed] Membranes 2020, 10, 33 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 31 of 36 Taheri, S.; Ams, M.; Bustamante, H.; Vorreiter, L.; Withford, M.; Clark, S. A practical methodology to assess corrosion in concrete sewer pipes. MATEC Web Conf. 2018, 199, 6010. [CrossRef] Kaufmann, E. Characterization of Materials; Wiley: Hoboken, NJ, USA, 2012. Brunette, D.; Tengvall, P.; Textor, M.; Thomsen, P. Titanium in Medicine: Material Science, Surface Science, Engineering, Biological Responses, and Medical Applications; Springer: New York, NY, USA, 2001. Kellenberger, C.; Pfleiderer, F.; Raso, R.; Burri, C.; Schumacher, C.; Grass, R.; Stark, W. Limestone nanoparticles as nanopore templates in polymer membranes: Narrow pore size distribution and use as self-wetting dialysis membranes. RSC Adv. 2014, 4, 61420–61426. [CrossRef] Bhowmick, A. Current Topics in Elastomers Research; CRC Press: Boca Raton, FL, USA, 2008. Sharma, S.; Verma, D.; Khan, L.; Kumar, S.; Khan, S. Handbook of Materials Characterization; Springer International Publishing: New York, NY, USA, 2018. Li, Z. Industrial Applications of Electron Microscopy; CRC Press: Boca Raton, FL, USA, 2002. Freger, V.; Gilron, J.; Belfer, S. TFC polyamide membranes modified by grafting of hydrophilic polymers: An FT-IR/AFM/TEM study. J. Membr. Sci. 2002, 209, 283–292. [CrossRef] Knez, S.; Stražišar, J.; Golob, J.; Horvat, A. Agglomeration of zeolite in the fluidized bed. Acta Chimica Slovenica 2001, 48, 487–504. Akhtar, F.; Kumar, M.; Villalobos, L.; Shevate, R.; Vovusha, H.; Schwingenschlogl, U.; Peinemann, K.-V. Polybenzimidazole-based mixed membranes with exceptional high water vapor permeability and selectivity. J. Mater.l Chem. A 2017, 5, 21807–21819. [CrossRef] Hilal, N.; Ismail, A.; Matsuura, T.; Oatley-Radcliffe, D. Membrane Characterization; Elsevier Science: Amsterdam, The Netherlands, 2017. Erinosho, M.; Akinlabi, E.; Johnson, O. Characterization of surface roughness of laser deposited titanium alloy and copper using AFM. Appl. Surf. Sci. 2018, 435, 393–397. [CrossRef] Sharpe, W. Springer Handbook of Experimental Solid Mechanics; Springer: New York, NY, USA, 2008. Kargarzadeh, H.; Ahmad, I.; Thomas, S.; Dufresne, A. Handbook of Nanocellulose and Cellulose Nanocomposites; Wiley: Hoboken, NJ, USA, 2017. Giordano, N. College Physics: Reasoning and Relationships; Cengage Learning: Boston, MA, USA, 2009. Grant, C.; Twigg, P.; Bell, G.; Lu, J. AFM relative stiffness measurement of the plasticising effect of a non-ionic surfactant on plant leaf wax. J. Colloid Interface Sci. 2008, 321, 360–364. [CrossRef] Cahn, R.; Lifshitz, E. Concise Encyclopedia of Materials Characterization; Elsevier Science: Amsterdam, The Netherlands, 2016. Cohen, S.; Lightbody, M. Atomic Force Microscopy/Scanning Tunneling Microscopy 3; Springer US: New York, NY, USA, 2007. Mohammad, A.; Hilal, N.; Lim, Y.; Amin, I.; Raslan, R. Atomic force microscopy as a tool for asymmetric polymeric membrane characterization. Sains Malays. 2011, 40, 237–244. Shamohammadi, M.; Hormozi, E.; Moradinezhad, M.; Moradi, M.; Skini, M.; Rakhshan, V. Surface topography of plain nickel-titanium (NiTi), as-received aesthetic (coated) NiTi, and aesthetic NiTi archwires sterilized by autoclaving or glutaraldehyde immersion: A profilometry/SEM/AFM study. Int. Orthod. 2019, 17, 60–72. [CrossRef] [PubMed] Khulbe, K.; Feng, C.; Matsuura, T. Synthetic Polymeric Membranes: Characterization by Atomic Force Microscopy; Springer: Berlin, Germany, 2007. ElHadidy, A.; Peldszus, S.; Van Dyke, M. Development of a pore construction data analysis technique for investigating pore size distribution of ultrafiltration membranes by atomic force microscopy. J. Membr. Sci. 2013, 429, 373–383. [CrossRef] Ochoa, N.; Prádanos, P.; Palacio, L.; Pagliero, C.; Marchese, J.; Hernández, A. Pore size distributions based on AFM imaging and retention of multidisperse polymer solutes: Characterisation of polyethersulfone UF membranes with dopes containing different PVP. J. Membr. Sci. 2001, 187, 227–237. [CrossRef] Arita, M.; Sakaguchi, N. Electron Microscopy: Novel Microscopy Trends; IntechOpen: London, UK, 2018. Meng, X.; Tang, W.; Wang, L.; Wang, X.; Huang, D.; Chen, H.; Zhang, N. Mechanism analysis of membrane fouling behavior by humic acid using atomic force microscopy: Effect of solution pH and hydrophilicity of PVDF ultrafiltration membrane interface. J. Membr. Sci. 2015, 487, 180–188. [CrossRef] Najafi, M.; Sadeghi, M.; Bolverdi, A.; Pourafshari Chenar, M.; Pakizeh, M. Gas permeation properties of cellulose acetate/silica nanocomposite membrane. Adv. Polym. Technol. 2018, 37, 2043–2052. [CrossRef] Membranes 2020, 10, 33 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. 71. 72. 73. 74. 32 of 36 Ross, R. Microelectronics Failure Analysis Desk Reference, 6th ed.; ASM International: Geauga County, OH, USA, 2011. Mittal, V.; Matsko, N. Analytical Imaging Techniques for Soft Matter Characterization; Springer: Berlin, Germany, 2012. Mistry, R.; Saxena, M.; Ray, P.; Singh, P. Octadecyl-silica—PVDF membrane of superior MD desalination performance. J. Appl. Polym. Sci. 2018, 135, 46043. [CrossRef] Yang, Q.; Li, W.; Dong, C.; Ma, Y.; Yin, Y.; Wu, Q.; Xu, Z.; Ma, W.; Fan, C.; Sun, K. PIM-1 as an artificial solid electrolyte interphase for stable lithium metal anode in high-performance batteries. J. Energy Chem. 2020, 42, 83–90. [CrossRef] Chowdhury, G.; Kruczek, B.; Matsuura, T. Polyphenylene Oxide and Modified Polyphenylene Oxide Membranes: Gas, Vapor and Liquid Separation; Springer: New York, NY, USA, 2013. Li, B.; Wang, B.; Liu, Z.; Qing, G. Synthesis of nanoporous PVDF membranes by controllable crystallization for selective proton permeation. J. Membr. Sci. 2016, 517, 111–120. [CrossRef] Chen, Y.; Wang, B.; Zhao, L.; Dutta, P.; Winston Ho, W. New Pebax® /zeolite Y composite membranes for CO2 capture from flue gas. J. Membr. Sci. 2015, 495, 415–423. [CrossRef] Powell, C.; Qiao, G. Polymeric CO2 /N2 gas separation membranes for the capture of carbon dioxide from power plant flue gases. J. Membr. Sci. 2006, 279, 1–49. [CrossRef] Minelli, M.; Sarti, G. Elementary prediction of gas permeability in glassy polymers. J. Membr. Sci. 2017, 521, 73–83. [CrossRef] Saberi, M.; Rouhi, P.; Teimoori, M. Estimation of dual mode sorption parameters for CO2 in the glassy polymers using group contribution approach. J. Membr. Sci. 2020, 595, 117481. [CrossRef] Swallowe, G. Mechanical Properties and Testing of Polymers: An a–z Reference; Springer: Dordrecht, The Netherlands, 2013. Ismail, A.; Khulbe, K.; Matsuura, T. Gas Separation Membranes: Polymeric and Inorganic; Springer International Publishing: New York, NY, USA, 2015. Weller, M. Inorganic Materials Chemistry; Oxford University Press: Oxford, UK, 1995. Suryanarayana, C.; Norton, M. X-ray Diffraction: A Practical Approach; Springer: New York, NY, USA, 2013. Thomas, S.; Rouxel, D.; Ponnamma, D. Spectroscopy of Polymer Nanocomposites; William Andrew Publishing: Oxford, UK, 2016. Zakhariev, Z. Polycrystalline Materials: Theoretical and Practical Aspects; IntechOpen: London, UK, 2012. Shahien, M.; Yamada, M.; Yasui, T.; Fukumoto, M. Reactive atmospheric plasma spraying of AlN coatings: Influence of aluminum feedstock particle size. J. Therm. Spray Technol. 2011, 20, 580–589. [CrossRef] Shen, Q.; Cong, S.; He, R.; Wang, Z.; Jin, Y.; Li, H.; Cao, X.; Wang, J.; Van der Bruggen, B.; Zhang, Y. SIFSIX-3-Zn/PIM-1 mixed matrix membranes with enhanced permeability for propylene/propane separation. J. Membr. Sci. 2019, 588, 117201. [CrossRef] Velu, S.; Arthanareeswaran, G.; Lade, H. Removal of organic and inorganic substances from industry wastewaters using modified aluminosilicate-based polyethersulfone ultrafiltration membranes. Environ. Prog. Sustain. Energy 2017, 36, 1612–1620. [CrossRef] Sawada, H.; Takahashi, Y.; Miyata, S.; Kanehashi, S.; Sato, S.; Nagai, K. Gas transport properties and crystalline structures of poly(lactic acid) membranes. Trans. Mater. Res. Soc. Japan 2010, 35, 241–246. [CrossRef] Karimi, S.; Firouzfar, E.; Khoshchehreh, M. Assessment of gas separation properties and CO2 plasticization of polysulfone/polyethylene glycol membranes. J. Pet. Sci. Eng. 2019, 173, 13–19. [CrossRef] Lasseuguette, E.; Malpass-Evans, R.; Carta, M.; McKeown, N.; Ferrari, M.-C. Temperature and pressure dependence of gas permeation in a microporous Tröger’s base polymer. Membranes 2018, 8, 132. [CrossRef] Ali, A. Failure Analysis and Prevention; IntechOpen: London, UK, 2017. Fendler, J. Nanoparticles and Nanostructured Films: Preparation, Characterization, and Applications; Wiley: Hoboken, NJ, USA, 2008. Mishra, S. Fibre Structure; Woodhead Publishing: Cambridge, UK, 2016. Paradies, H. Particle size distribution and determination of characteristic properties of colloidal bismuth—silica compounds by small-angle x-ray scattering and inelastic light scattering. Colloids Surf. A Physicochem. Eng. Asp. 1993, 74, 57–69. [CrossRef] Radlinski, A.; Mastalerz, M.; Hinde, A.; Hainbuchner, M.; Rauch, H.; Baron, M.; Lin, J.; Fan, L.; Thiyagarajan, P. Application of SAXS and SANS in evaluation of porosity, pore size distribution and surface area of coal. Int. J. Coal Geol. 2004, 59, 245–271. [CrossRef] Membranes 2020, 10, 33 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. 33 of 36 Agbabiaka, A.; Wiltfong, M.; Park, C. Small angle X-Ray scattering technique for the particle size distribution of nonporous nanoparticles. J. Nanopartic. 2013, 2013, 11. [CrossRef] Schneider, K.; Schöne, A.; Jun, T.-S.; Korsunsky, A. Investigation of changes in crystalline and amorphous structure during deformation of nano-reinforced semi-crystalline polymers by space-resolved synchrotron SAXS and WAXS. Procedia Eng. 2009, 1, 159–162. [CrossRef] Niu, Q.; Pan, J.; Jin, Y.; Wang, H.; Li, M.; Ji, Z.; Wang, K.; Wang, Z. Fractal study of adsorption-pores in pulverized coals with various metamorphism degrees using N2 adsorption, X-ray scattering and image analysis methods. J. Pet. Sci. Eng. 2019, 176, 584–593. [CrossRef] Baran, M.; Braten, M.; Sahu, S.; Baskin, A.; Meckler, S.; Li, L.; Maserati, L.; Carrington, M.; Chiang, Y.-M.; Prendergast, D.; et al. Design rules for membranes from polymers of intrinsic microporosity for crossover-free aqueous electrochemical devices. Joule 2019, 3, 2968–2985. [CrossRef] Rueda, D.; García-Gutiérrez, M.; Nogales, A.; Capitán, M.; Ezquerra, T.; Labrador, A.; Fraga, E.; Beltrán, D.; Juanhuix, J.; Herranz, J.; et al. Versatile wide angle diffraction setup for simultaneous wide and small angle x-ray scattering measurements with synchrotron radiation. Rev. Sci. Instru. 2006, 77, 033904. [CrossRef] De Prado, S.A.L.; Ponce, M.; Funari, S.; Schulte, K.; Garamus, V.; Willumeit, R.; Nunes, S. SAXS/WAXS characterization of proton-conducting polymer membranes containing phosphomolybdic acid. J. Non-Cryst. Solids 2005, 351, 2194–2199. [CrossRef] Mrozowich, T.; McLennan, S.; Overduin, M.; Patel, T. Structural studies of macromolecules in solution using small angle x-ray scattering. JoVE 2018, 141, e58538. [CrossRef] Kikhney, A.; Svergun, D. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [CrossRef] Tant, M.; Mauritz, K.; Wilkes, G. Ionomers: Synthesis, Structure, Properties and Applications; Springer: Dordrecht, The Netherlands, 1997. Xiao, T.; Yuan, H.; Ma, Q.; Guo, X.; Wu, Y. An approach for in situ qualitative and quantitative analysis of moisture adsorption in nanogram-scaled lignin by using micro-FTIR spectroscopy and partial least squares regression. Int. J. Biol. Macromol. 2019, 132, 1106–1111. [CrossRef] Wypych, G. Handbook of Solvents; Noyes Publications: Park Ridge, NJ, USA, 2001. Sun, D. Modern Techniques for food Authentication; Elsevier Science: Amsterdam, The Netherlands, 2008. Bouis, P. Reagent Chemicals: Specifications and Procedures; American Chemical Society Specifications: Washington, DC, USA, 2006. Mishra, M. Encyclopedia of Polymer Applications; CRC Press: Boca Raton, FL, USA, 2018. Townshend, A. Encyclopedia of Analytical Science: Gast-Lip; Academic Press: Cambridge, MA, USA, 1995. Dalton, D. Foundations of Organic Chemistry: Unity and Diversity of Structures, Pathways, and Reactions; Wiley: Hoboken, NJ, USA, 2011. Bonilla, J.; Srivatsa, G. Handbook of Analysis of Oligonucleotides and Related Products; CRC Press: Boca Raton, FL, USA, 2011. Hansen, S.; Pedersen-Bjergaard, S.; Rasmussen, K. Introduction to Pharmaceutical Chemical Analysis; Wiley: Hoboken, NJ, USA, 2011. Adewole, J.; Ahmad, A.; Ismail, S.; Leo, C.; Sultan, A. Comparative studies on the effects of casting solvent on physico-chemical and gas transport properties of dense polysulfone membrane used for CO2 /CH4 separation. J. Appl. Polym. Sci. 2015, 132, 42205. [CrossRef] Zhu, H.; Wang, L.; Jie, X.; Liu, D.; Cao, Y. Improved interfacial affinity and CO2 separation performance of asymmetric mixed matrix membranes by incorporating postmodified MIL-53(Al). ACS Appl. Mater. Interfaces 2016, 8, 22696–22704. [CrossRef] [PubMed] Grdadolnik, J. ATR-FTIR spectroscopy: Its advantages and limitations. Acta Chimica Slovenica 2002, 49, 631–642. Smith, B. Fundamentals of Fourier Transform Infrared Spectroscopy; Taylor & Francis: Abingdon-on-Thames, UK, 1995. Winter, A. Organic Chemistry I for Dummies; Wiley: Hoboken, NJ, USA, 2014. Zhang, S. Raman Spectroscopy and its Application in Nanostructures; Wiley: Hoboken, NJ, USA, 2012. Korb, C. Differential Absorption Lidars for Remote Sensing of Atmospheric Pressure and Temperature Profiles; National Aeronautics and Space Administration: Washington, DC, USA, 1995. Membranes 2020, 10, 33 34 of 36 100. Correia, J.; Detrich, H. Biophysical Tools for Biologists: In Vivo Techniques; Elsevier Science: Amsterdam, The Netherlands, 2009. 101. Boccaccio, T.; Bottino, A.; Capannelli, G.; Piaggio, P. Characterization of PVDF membranes by vibrational spectroscopy. J. Membr. Sci. 2002, 210, 315–329. [CrossRef] 102. Andrews, D.; Nann, T.; Lipson, R. Comprehensive Nanoscience and Nanotechnology; Elsevier Science: Amsterdam, The Netherlands, 2019. 103. Amer, M. Raman Spectroscopy for Soft Matter Applications; Wiley: Hoboken, NJ, USA, 2009. 104. Katoh, T.; Imamura, G.; Obata, S.; Bhanuchandra, M.; Copley, G.; Yorimitsu, H.; Saiki, K. The influence of source molecule structure on the low temperature growth of nitrogen-doped graphene. Phys. Chem. Chem. Phys. 2015, 17, 14115–14121. [CrossRef] [PubMed] 105. Jacox, M. Vibrational and electronic energy levels of polyatomic transient molecules. Supplement B. J. Phys. Chem. Ref. Data 2003, 32, 1–441. [CrossRef] 106. Grumezescu, A. Nanostructures for the Engineering of Cells, Tissues and Organs: From Design to Applications; Elsevier Science: Amsterdam, The Netherlands, 2018. 107. Asundi, A.K.; Rao, V.V. Measurement of Methanol Proportion in Methanol-Gasoline Mixtures—An Application of Fiber Optic Raman Spectroscopy; SAE International: Warrendale, PA, USA, 2000. 108. Wild, D. The Immunoassay Handbook: Theory and Applications of Ligand Binding, ELISA and Related Techniques; Elsevier Science: Amsterdam, The Netherlands, 2013. 109. Pathak, Y.; Lokhande, J. Handbook of Metallonutraceuticals; Taylor & Francis: Abingdon, UK, 2014. 110. Schreiner, M.; Strlič, M.; Salimbeni, R. Handbook of the Use of Lasers in Conservation and Conservation Science; COST Office: Brussels, Belgium, 2008. 111. Holber, M.; Johansson, P.; Jacobsson, P. Raman spectroscopy of an aged low temperature polymer electrolyte fuel cell membrane. Fuel Cells 2011, 11, 459–464. [CrossRef] 112. Sundarapandiyan, S.; Renitha, T.; Sridevi, J.; Saravanan, P.; Chandrasekaran, B.; Raju, G. Photocatalytic degradation of highly refractive phenolic polymer—mechanistic insights as revealed by electron spin resonance (ESR) and solid-state 13 C NMR spectroscopy. Chem. Eng. J. 2017, 313, 1112–1121. [CrossRef] 113. Sangroniz, A.; Gonzalez, A.; Martin, L.; Irusta, L.; Iriarte, M.; Etxeberria, A. Miscibility and degradation of polymer blends based on biodegradable poly(butylene adipate-co-terephthalate). Polym. Degrad. Stab. 2018, 151, 25–35. [CrossRef] 114. Ebbing, D.; Gammon, S. General Chemistry, 10th ed.; Cengage Learning: Boston, MA, USA, 2012. 115. Emwas, A.; Roy, R.; McKay, R.; Tenori, L.; Saccenti, E.; Gowda, G.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR spectroscopy for metabolomics research. Metabolites 2019, 9, 7. [CrossRef] 116. Claridge, T.; Claridge, N. High-Resolution NMR Techniques in Organic Chemistry; Elsevier Science: Amsterdam, The Netherlands, 2009. 117. Pavia, D.; Kriz, G.; Lampman, G.; Engel, R. A Microscale Approach to Organic Laboratory Techniques; Cengage Learning: Boston, MA, USA, 2016. 118. Kobayashi, Y.; Nakamitsu, Y.; Zheng, Y.; Takashima, Y.; Yamaguchi, H.; Harada, A. Preparation of cyclodextrinbased porous polymeric membrane by bulk polymerization of ethyl acrylate in the presence of cyclodextrin. Polymer 2019, 177, 208–213. [CrossRef] 119. Mueller, R.; Zhang, S.; Zhang, C.; Lively, R.; Vasenkov, S. Relationship between long-range diffusion and diffusion in the ZIF-8 and polymer phases of a mixed-matrix membrane by high field NMR diffusometry. J. Membr. Sci. 2015, 477, 123–130. [CrossRef] 120. Becker, E. High Resolution NMR: Theory and Chemical Applications; Elsevier Science: Amsterdam, The Netherlands, 1999. 121. Nechifor, A.; Panait, V.; Naftanaila, L.; Batalu, D.; Voicu, Ş. Symmetrically polysulfone membranes obtained by solvent evaporation using carbon nanotubes as additives. Synthesis, characterization and applications. Dig. J. Nanomater. Biostruc. 2013, 8, 875–884. 122. Reusch, W. Nuclear Magnetic Resonance Spectroscopy; LibreTexts: Davis, CA, USA, 2019. 123. Parmer, J.; Dickinson, L.; Chien, J.; Porter, R. Polymer-polymer miscibility determination via CP-MAS NMR in blends of deuteriated and protonated polymers. Macromol. 1987, 20, 2308–2310. [CrossRef] 124. Zatoń, M.; Rozière, J.; Jones, D.J. Current understanding of chemical degradation mechanisms of perfluorosulfonic acid membranes and their mitigation strategies: A review. Sustain. Energy Fuels 2017, 1, 409–438. [CrossRef] Membranes 2020, 10, 33 35 of 36 125. Kinumoto, T.; Inaba, M.; Nakayama, Y.; Ogata, K.; Umebayashi, R.; Tasaka, A.; Iriyama, Y.; Abe, T.; Ogumi, Z. Durability of perfluorinated ionomer membrane against hydrogen peroxide. J. Power Sources 2006, 158, 1222–1228. [CrossRef] 126. Parrondo, J.; Arges, C.; Niedzwiecki, M.; Anderson, E.; Ayers, K.; Ramani, V. Degradation of anion exchange membranes used for hydrogen production by ultrapure water electrolysis. RSC Adv. 2014, 4, 9875–9879. [CrossRef] 127. McMurry, J. Organic Chemistry; Cengage Learning: Boston, MA, USA, 2011. 128. Kong, X.; Terskikh, V.; Khade, R.; Yang, L.; Rorick, A.; Zhang, Y.; He, P.; Huang, Y.; Wu, G. Solid-state 17 O NMR spectroscopy of paramagnetic coordination compounds. Angew. Chem. 2015, 54, 4753–4757. [CrossRef] 129. Moore, T. Characterization of Integrated Circuit Packaging Materials; Elsevier Science: Amsterdam, The Netherlands, 2013. 130. Xiong, Y.; Lu, X. Metallic Nanostructures: From Controlled Synthesis to Applications; Springer International Publishing: New York, NY, USA, 2014. 131. Wadekar, S.; Vidic, R. Comparison of ceramic and polymeric nanofiltration membranes for treatment of abandoned coal mine drainage. Desalination 2018, 440, 135–145. [CrossRef] 132. Ayesh, A.; Salah, B.; Al-Sulaiti, L. Production and characterization of flexible semiconducting polymernanoparticle composites for x-ray sensors. Radiat. Phys. Chem. 2019, 167, 108233. [CrossRef] 133. Thermo Fisher Scientific. EDX Analysis with SEM: How Does it Work? Available online: https://www. thermofisher.com/blog/microscopy/edx-analysis-with-sem-how-does-it-work/ (accessed on 1 November 2019). 134. Saxena, P. IIT Chemistry-I; Krishna Prakashan: Uttar Pradesh, India, 1995. 135. Russ, J.; Frs, M.; Kiessling, R.; Charles, J. Fundamentals of Energy Dispersive X-ray Analysis: Butterworths Monographs in Materials; Elsevier Science: Amsterdam, The Netherlands, 2013. 136. Taylor, P.; Yurdakul, E.; Ceylan, H. Concrete Pavement Mixtures Design and Analysis: The Application of Portable X-ray Fluorescence Technique to Assess Concrete Mix Proportions; National Concrete Pavement Technology Center: Ames, IA, USA, 2012. 137. Verma, H. Atomic and Nuclear Analytical Methods: XRF, Mössbauer, XPS, NAA and Ion-Beam Spectroscopic Techniques; Springer: Berlin, Germany, 2007. 138. Shugar, A.; Mass, J. Handheld XRF for Art and Archaeology; Leuven University Press: Leuven, Belgium, 2012. 139. Crain, E. Crain’s Petrophysical Handbook; Spectrum 2000 Mindware: Rocky Mountain House, AB, Canada, 2010. 140. Elias, G.; Marguí, E.; Díez, S.; Fontàs, C. Polymer inclusion membrane as an effective sorbent to facilitate mercury storage and detection by x-ray fluorescence in natural waters. Anal. Chem. 2018, 90, 4756–4763. [CrossRef] 141. Thermo Fisher Scientific. The Power Behind the Analyzer: X-ray Tubes. Available online: https://www. thermofisher.com/blog/metals/the-power-behind-the-analyzer-x-ray-tubes (accessed on 24 January 2020). 142. Acharya, N.; Kulshrestha, V.; Awasthi, K.; Jain, A.; Singh, M.; Vijay, Y. Hydrogen separation in doped and blend polymer membranes. Int. J. Hydrogen Energy 2008, 33, 327–331. [CrossRef] 143. Panchuk, V.; Rabdano, N.; Goidenko, A.; Grebenyuk, A.; Irkaev, S.; Semenov, V. Determination of the oxidation state of iron by x-ray fluorescence spectroscopy using chemometric approaches. J. Anal. Chem. 2017, 72, 662–670. [CrossRef] 144. Weidner, J. Separators and Membranes for Batteries, Capacitors, Fuel Cells, and Other Electrochemical Systems; Electrochemical Society: Pennington, NJ, USA, 2009. 145. Hüfner, S. Photoelectron Spectroscopy: Principles and Applications; Springer: New York, NY, USA, 2003. 146. Aulisa, E.; Gilliam, D. A Practical Guide to Geometric Regulation for Distributed Parameter Systems; CRC Press: Boca Raton, FL, USA, 2015. 147. Shard, A. Detection limits in XPS for more than 6000 binary systems using Al and Mg Kα X-rays. Surf. Interface Anal. 2014, 46, 175–185. [CrossRef] 148. Du, X.; Meng, J.; Xu, R.; Shi, Q.; Zhang, Y. Polyol-grafted polysulfone membranes for boron removal: Effects of the ligand structure. J. Membr. Sci. 2015, 476, 205–215. [CrossRef] 149. Van der Heide, P. X-ray Photoelectron Spectroscopy: An Introduction to Principles and Practices; Wiley: Hoboken, NJ, USA, 2011. 150. Futter, G.A.; Latz, A.; Jahnke, T. Physical modeling of chemical membrane degradation in polymer electrolyte membrane fuel cells: Influence of pressure, relative humidity and cell voltage. J. Power Sources 2019, 410–411, 78–90. [CrossRef] Membranes 2020, 10, 33 36 of 36 151. Chen, C.; Levitin, G.; Hess, D.; Fuller, T. XPS investigation of Nafion® membrane degradation. J. Power Sources 2007, 169, 288–295. [CrossRef] 152. Riviere, J.; Myhra, S. Handbook of Surface and Interface Analysis: Methods for Problem-Solving; CRC Press: Boca Raton, FL, USA, 2009. 153. Stojilovic, N. Why can’t we see hydrogen in x-ray photoelectron spectroscopy? J. Chem. Edu. 2012, 89, 1331–1332. [CrossRef] © 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
 0
0
advertisement


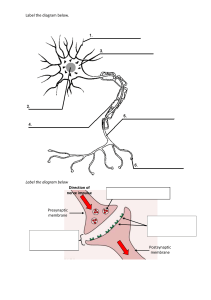
Download
advertisement
Add this document to collection(s)
You can add this document to your study collection(s)
Sign in Available only to authorized usersAdd this document to saved
You can add this document to your saved list
Sign in Available only to authorized users