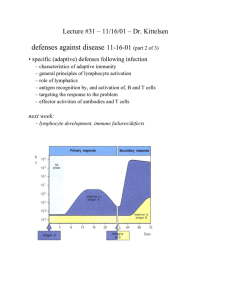
Note: Innate response takes 0-4 hours, and if needed up to 4 days. Adaptive response starts on day 4 Viral infected cell: Ub-tags it so proteosome can degrade it into linear peptide antigens (vs in APCs degradation happens in low acid phagosomes) TAP immediately sequesters these from proteosome into RER where MHC I is being newly made (vs in APCs the MHC II w/invariant chain is sent from RER to the endocytic vesicle) the two bind in RER w/chaperone of tapasin trafficked to cell membrane. o infected cell also releases IFN-a and IFN-B to (1) activate NK cells (2) stop viral replication, and (3) induce other body cells to express MHC I (virus tries to hide MHC I to evade CD8 Tcells) Tissue resident DCs (MHC low/phagocytic high) or macrophages also detects this viral pathogen (PAMP-- mannose receptor or antigen on cells’ MHC I) and either can activate NK cells via INF-a, INF-B, TNF-a, and IL-12. Often macrophages are doing it. o NK that is cytokine releasing/maintaining inflamm state (CD56hiCD16- ); i.e. IFN-y (send DC to LN and activate macrophages), TNF-a (inflam), IL-6 (acute phase proteins), IL-10 Note that the INF-y increases macrophage cytotoxicity and sends DC off to paracortex when its outnumbered by DC o NK that’s cytotoxic/releasing perforin/granzyme (CD56loCD16+). This effector NK will start killing cells in three possible ways (1) MHC I removed ----NKG2A co-receptor KIR has its ITAM activ tyrosine kinase signaling release granules (2) MIC ---- NKG2D co-receptor KIR has its ITAM activ tyrosine kinase signaling release granules (3) A few days later, it will kill cells antibodies have clustered around: Fc portion of Ab---- FcyR of NK cell Note that KIR binds only the MHC I (not the MHC I and Ag complex, as TCR does) and NK cell education involves its inhibitory KIR receptor being bound to MHC I during dev so it knows what its absence looks like Once in paracortex, the DC becomes a mature MHC/ B7.1 B7.2 expressing/ nonphagocytic DC. It will present its MHC I/Ag to CD8 naïve T-cells o 1st signal (NFAT, NFKKB, AP-1 making IL-2 mRNA) o 2nd signal (B7—CD28 stabilizing IL-2 and IL-2R mRNA so more secreted) o 3rd signal (binding of IL-2---y chain of IL-2R- STAT transcription factor make cytotoxic granules and cytokines like INF-y, TNFa and TNFB) Note that CD40 (will give Mo license to kill) and extravasion proteins also produced Of course, the DC will also present this to a naïve CD4 T-cell, turning it into TH1 cell (same process as above, signal #1, 2, and 3). This TH1 cell can provide the two other ways of activating a naïve CD8 cell: It can release some of its IL-2 onto the CD8 T-cell bound to APC so that 3rd signal can occur It can directly pair with that APC to induce it to express B7 so that 2nd signal can occur However of the three ways its made, the activated CD8 T cell (CTL) now leaves through efferent lymphatics: CXCR3, E and P selectin (vs CD26L-selectin), and integrin VFA-1 (vs LFA-1) that can bind VCAM on endothelial cells AT TISSUES o o o CTL’s TCR/CD8 ---MHC I/Ag of infected cell release of perforin/granzyme; and then moves on to the next cell (note it doesn’t need co-stimulation to kill) as it takes over NK’s job CTL can also talk to tissue resident DC (MHC I-Ag --TCR/CD8) CTL can also activate macrophages: CD40 “licenses” macrophages to kill, and the INF-y helps incr macrophage’s phagocytic abilities. CTL also releases TNFa and TNF-B (inflamm) in addition to INF-y, just like Th1! Th1 cells have a role here too Release IFN-y to have macrophages do microbial clean up In LN Th1 cells helped activate CTL by releasing IL-12 and INF-y, as well as bypassing the action of DC if needed (release IL-2, induce B7 expression on DC). Release IFN-y onto B-cells to promote secretion of IgG for ADCC killing by NK cells (NK cells has FyIIIR----Fc portion of IgG on pathogen *T effector memory cells will stay resident in the tissue (effector) and central memory cells in secondary lymphoid organs Macrophages TLR detects PAMP on bacteria o release of PG (“DIE”) for vasodilation, vasopermeation, and fever/pain (PGE2 does it), as well as LT (B,C,D,E) for vasopermeation and neutrophil cx (LTB4 does it) and TXs o release IL-1. TNF-a, IL-6, IL-8, CXCL8, IL-12 IL-1, TNF-a, IL,6 = “Inflammatory Group” “1 year olds start it all, need tissues, six of them!” IL-1 and TNF-a incr VP (loosen tight junction and up-reg E- P- selectins and ICAM’s) and in paracortex induce TH17 cells (extracell pathogen) IL-1 induces IL-6 production. IL-6 causes liver to secrete MLB (lecthin), Creactive protein (classical pathway even before IgG), and fibrinogen All three also induce anabolism in fat and muscle cells, both to provide energy and calor (heat). TNF-a can continue to do this systematically sepsis and cachexia (wasting syndrome) IL-8 and CXCL= attract and activate neutrophil! “8 year olds are neutral” IL-12= attract and activate NK cells “12 year olds just want to kill” complement system: C3b opsonization; C5b memb-attack complex, C3a/C4a/C5a for VP, C3a cx mast cell, and C5a cx neutrophils o 1-4 hrs: alternative pathway where C3Bb (sol C3 convertase) C3bBb (C3 convertase) C3bBbC3b (C5 convertase) o 4-96 hrs IL-6 drives liver to secrete CRP, fibrinogen and MBL (lecthin pathway; C4bC2a is C3 convertase C3bBbC3b C5 convertase) Lecthin good for capsulated pathogens cared for by spleen: P-Hi-N-K y and fragile (strp pneumo and pyogenes, H influenza, N. meningies, Klebsiella, bact fragilis) o 96+ hours: IgG and IgM surrounding pathogen activates C1 complex (classical pathway; C4bC2a is C3 convertase C3bBbC3b C5 convertase) Note that C3b can do 5 things here: opson, make C5 conv, make more C3 conv, or have Factor I turn it into C3dg that binds CR2 of B-Cell to incr Ig prod). barydkinin also released by HMW Kininogen for vasodil, permeation, and pain RUBOR (Redness/VD): NO, histamine, PG CALOR (fever): TNF, IL-1, PGE2 TUMOR (swell;VP): histamine PG, LT, C3a C5a, bradykinin DOLOR (pain): PGE2 and bradykinin neutrophils enter (sialyl-lewis and integrins to attach endothelial cells) and follow road made by LTB4, C5a, bacterial pathogens release defensins on opsonized bacteria (IgG and classical complement helps w/this later on) o macrophages later come and clean up dead neutrophils (pus) o granule release is constitutive in neutrophils o how neutrophil enters from bone marrow other cells that may enter: basophils/mast cells (histamine, heparin via IgE degran) or eosinophils (major basic protein) Macrophages and DC also travel to lymphoid to activate Th cells (MHC II-Ag ---TCR/CD4) and B-cells (MHC II-Ag ---BCR) (though its FDC for B cells) B-cell activation IL 7/CLP early then late Pro-B cell (V/D/J HC check w/surr LC) Pre-B cell (V/J lC made by tyring � loci 1st then � if needed) Immature B cell (IgM) enters peripheral circ to get to primary lymphoid follicle in bone marrow, throughout which time tested for negative selection (self ag = death, sol ag = anergy, multiv ag =try again) Mature B cell (IgM, IgD RNA pol kept going after first poly-A tail of miu Constant region) leave bone marrow venous to aorta afferent nodal artery to HEV settle in cortex Note that positive selection for B-cells occurs during the pre-B cell receptor checkpoint (vs for T cells its weak reactivity to self-MHC in thymus) Homing into HEV: B-cell’s L-selectin CD26L binds Gly-CAM and CD34 on HEV….causing its CXCR5 to bind CXCL-13…… activate B-cell’s integrin LFA-1 to bind ICAM-1……..enter! If no contact with pathogen, leave LN via efferent lymphatic and keep testing LNs until venous angle vena cava aortic blood circulation and again enter HEV of LN (same for T cell) PRIMARY FOLLICE : If pathogen/antigen present, the follicular DC will bring over the pathogen (early on pathogen dotted with C3b and binds comp recep and later on Ig has dotted it and DC has FcRs for) and present the pathogen in native conformation to a B-cell, who then takes up pathogen, processes it, and presents it on its MHC II………..OR….If that B-cell has a particular BCR (Ig) that can bind that particular pathogen in native conformation then it will do so process pathogen in low acid phagosomes present MHC II-Ag on membrane : B-cell moves towards paracortex by showing CCR7 (CCL19 and CCL21 there) and at the same CD4 Tcell moves towards cortex by showing CXCR5 until they meet at the border and form a conjugate pair: CD40L and TCR of TFH cell --- CD40 and MHC II-Ag of B cell o together they move to the cortex to form special follicle called… GERMINAL CENTER (SECONDARY FOLLICLE) o **CD40-CD40L interaction (co-stimulatory to BCR) activates cross-linking of BCRs - phosphorylation of ITAMs at the ends of Iga and IgB(CD79) by Syk tyrosine kinase signal transduction to enhance transcription factor needed for prolif and isotype switching o Cognate pair (CD40L---CD40 co-stim) will remain here will B-Cell is being re-presented with antigen by FDC o o TI-1 antigen (LPS) does BCR cross-linking w/o this CD40 interaction (so skip germinal center) and allows for fast response to endotoxin, but downsides are that you only get IgM secreted that does not have affinity maturation and no long-lived plasma cells TI-2 antigen (repeated epitotes ~ polysacch capsules) can also crosslink BCRs and then get second signal via BAFF cytokine from activated dendritic cells to do some small isotype switching to IgG (again, w/o TFH help). Isotype switching (IL-4) IFN-y, TGF-B, IL-4, IL-6 from TFH cell will show AICD which switch regions to make mutations at (so cuts can be made at those regions and bring those more distant Cgamma and Cepsilon dna segments next to the VDJ dna segment) CD40-CD40L interaction also needed and defect in this interaction causes hyper IgM Syndrome IL-4: IgG1, IgE TGF-B: IgG, IgA IFN-y: IgG 2a and 3 induced Affinity maturation (IL-4): Somatic hypermutation in HV region: AICD makes mutations in VDJ exon, forming CDR3 region within HV region B-cells who’s new CDR3 regions bind antigen better (stimulated) survive on to differentiate into plasma cells vs others that die Competition increases amongst B-cells as less antigen is available bc more antigen is being cleared. B-cells that don’t make the cut undergo Fas-FasL mediated apoptosis o Diff into plasma cell (Ab secretion) IL-6: RNA polymerase stops at the SC loci instead of continuing onto transmembrane loci Now have a cell that has no mIgs or CD40, its only job is to secrete Igs Ig will surround pathogen to (a) neutralize/block its entry into cell (IgG, IgA), (b) as opsonins, form immune complexes around pathogen to phagocytes, neutrophils (all have FcyRs) clear the complexes (note: medium complexes cleared best) o Note: at first IgM is covering pathogens, but that’s replaced with IgG so that phagocytes can clear the complex (c) activate complement so that C3b opsonins (classical pathway) can help clear the immune complexes RBC w/CR1 receptors for C3b can take it the complex the spleen! Also helps phagocytes (b) allow ADCC by NK/Tc (d) degranulation of mast/basophil via cross-linked IgE Two types of plasma cells made: Memory B-cells that stay in LN/blood circulation to monitor for 2nd infection, and long lived plasma cells that reside in bone marrow and tissues to secrete antibodies for years o Memory B-cells often have IgG and IgA (instead of D and M) and their Ig’s have higher affinity; and alterations in signal transduction to make them respond FASTER T-cell activation IL-7/CLP (CD34+) that leaves bone marrownotch ligand of thymus epithelial cell binding notch recep on CD34 = start T cell dev Pro-T (double neg) that makes V/D/J β chain is pass checkpoint #1 w/Tα to form pre-TCR a:B CD3 complex then becomes Pre-T cell (dble positive CD4,CD8, CD3) that makes an α chain (V/J) immature T cell (CD4, CD8, CD3, a:B TCR) undergoes positive selection (weak resp to MHC I-self peptide= CD8; weak resp to MHC II-self peptide= CD4) and neg selection (strong resp=death) leave mature and enter vena cava to aorta afferent artery to HEV of LN to settle in paracortex Note: A TCR in general can bind both MHC I and II, but it’s the expression of CD4 or CD8 that controls which is MOST stably bound; CD3 is needed to anchor TCR and to do signal transd Homing into HEV: CD4/CD8 T-cell’s L-selectin CD26L binds Gly-CAM and CD34….then its CCR7 binds CCL-21……this activates integrin LFA-1 to bind ICAM-1……..enter! Mature CD4 and CD8 T cells samples through whats presented to them by APC like mature DCs and macrophages EFFECTOR T-Cell formation if antigen present on APC ~ mature FDC…. CD4 T-cells… o 1st signal (activation) is MHC II-Ag---TCR/CD4 LFA-1---ICAM bond strengthened IL-2R expressed on cell surface (enters G1 phase) ……Even before 2nd signal, IL-2 is begun Ag binding TCR TCR clustering w/its CD4 or CD8 Lck at end of CD4 or CD8 phosphorylates ITAMs at ends of CD3 creates a scaffolding of phosphotyrosines onto which ZAP-70 can bind ZAP-70 phosphorylates LAT, created another scaffold that three downstream pathways use as a starting point PLCy hydrolyzes inositol phospholipid PIP2 into IP3and DAG (diacylglycerol) o IP3 release of intracellular vesicles Ca2+ and influx of extracellular Ca2+ calcineurin (phosphatase) NFAT allowed to act (transc factor) Cyclosporin and Tacrolimus (FK506) stop this activ of calcineurin DAG Protein Kinase C I𝛋B inhibited disinhibition of NFK𝛋B (trans factor) Ras/Rac pathway o GTP-bound and thus activated Ras and Rac MAP kinases activate Fos and Jun Fos-Jun complex AP-1 (transcrip factor) NOTE: other IL’s are made this way! Just showing how its done for IL-2 2nd signal (continue activation) is B7---CD28 of Th cell IL-2 released by T-cell autocrine activation of rest of cell cycle So B7--CD28 increases the of IL-2R and IL-2 by stabilizing these mRNA products CD40L expressed, as are P- and E-selectin and VLA-4 for extravasion into tissue; CTLA-4 (for when its time to die, binds to B7 x20 better than CD28) 3rd signal (start proliferation) is the cytokines the APC releases (so in addition to IL-2 naïve T-cell releases) that activates certain STAT transcription factors that produce the proteins determining what subtype of CD4 cell it becomes. Note: IL receptors all have a CD3 gamma chain (defect here=SCID) that are involved in signal transduction leading to the transcription of proteins needed for T-cell diff: dimerization of the two arms of a cytokine receptor activates JAK to phosphorylate (and thus dimerize) the STAT proteins recruited to the gamma chain- translocation of STAT dimer to nucleus to act as transcrip factor o o o CD4 T-cell effectors o TFH (TGF-B) CD40L/CD40 interaction conjugate pair w/B-cell to oversee B-cell differentiation Secretes IL-4, IL-6, INF-y, TGF IL-4: IgG1, IgE TGF-B: IgG, IgA IFN-y: IgG 2a and 3 induced Note: TH1 and TH2 cells also are involved in promoting isotype switching o o o o TH1 (IL-12, INF-y) Macrophages, intracellular pathogens (Chlamydia trachomatis/pneumo, viruses IL-2, IL-12 (activates NK, Tc cells) INF-y (enhance Macrophage activity), GM-CSF, TNF-a (keep inflam resp going) o All of these activate macrophages! And are immunoactivating! o INF-y also inhibits Th2 TH2 (IL-4, IL-25, IL-33) Eosinophils/basophils; helmith worm and allergens IL-4 (affinity mat, switching to IgE (G-E, for mE?), IL-5 (eosinophil prog/surv/cx), IL-6 (Ab secretion, acute phase proteins), IL-13 (IgE switch, goblet cells), TGF-β, IL-10 o All of these promote activation of B-cells! Which secrete IgE needed for crosslinking so granules of histamine and major basic protein can be secreted o TGF-B inhibits Th1 and IL-10 inhibits Macrophage so it wont secrete IL-12 TH17 (IL-6, TNF-a) Neutrophils, extracellular bacteria and fungi IL-17, IL-21 Treg (TGF-B) Prevent abnormal proliferation and differentation IL-10, TGF-B CD8 T-cell effectors= CTL o 3 ways to be activated 1) APC’s MHC I/Ag---TCR (1st signal, IL-2R mRNA made via NFAT, NFKKB, and AP-1)…..B7— CD28 (2nd signal, IL-2 secreted)…..(3rd signal is STAT pathways from gamma chain of IL-2R being bound) Only difference from B-cell activation is that MHC I is presenting the processed linear peptide (often of inner folded portion) via proteosome/TAP/tapasin 2) Activated CD4 T-cell that is releasing IL-2 also releases it onto the CD8 T-cell that’s paired with the same APC Or IT notices the APC paired with CD8 T-cell is lacking B7 so it binds to the APC via CD40L/CD40 to induce its B7 expression (GIVE IT LICENSE TO BE AN APC) o CTL cell leaves via efferent lymphatics does so by showing CXCR3 (3rds strike and Im out!) , E and P selectin (instead of CD26L-selectin), and integrin VFA-1 (instead of LFA-1) that can bind VCAM on endothelial cells Cytokines released by CTL is INF-y (activate macrophages), TNFa and TNF-B (inflamm) just like Th1! CTLs are “serial killers” that don’t need co-stimulation to kill. Memory T cell o Exist either in secondary lymphoid organs (central memory) or in tissue (effector memory). Central memory – proliferate/differentiate more rapidly than naïve cells Effector memory - Already differentiated; rapidly produce effector molecules o Basis for type IV hypersensitivities. o Faster Intracellular signaling events, but they also display altered levels of adhesion molecules, which allow them to home to certain tissues as well as peripheral lymphoid organs.