Chronic NG-Nitro-L-Arginine Methyl
Ester–Induced Hypertension
Novel Molecular Adaptation to Systolic Load in Absence of Hypertrophy
Jozef Bartunek, MD, PhD; Ellen O. Weinberg, PhD; Minori Tajima, MD, PhD;
Susanne Rohrbach, BA; Sarah E. Katz, BA; Pamela S. Douglas, MD; Beverly H. Lorell, MD
Downloaded from http://circ.ahajournals.org/ by guest on October 2, 2016
Background—Chronic NG-nitro-L-arginine methyl ester (L-NAME), which inhibits nitric oxide synthesis, causes
hypertension and would therefore be expected to induce robust cardiac hypertrophy. However, L-NAME has negative
metabolic effects on protein synthesis that suppress the increase in left ventricular (LV) mass in response to sustained
pressure overload. In the present study, we used L-NAME–induced hypertension to test the hypothesis that adaptation
to pressure overload occurs even when hypertrophy is suppressed.
Methods and Results—Male rats received L-NAME (50 mg 䡠 kg⫺1 䡠 d⫺1) or no drug for 6 weeks. Rats with
L-NAME–induced hypertension had levels of systolic wall stress similar to those of rats with aortic stenosis (85⫾19
versus 92⫾16 kdyne/cm). Rats with aortic stenosis developed a nearly 2-fold increase in LV mass compared with
controls. In contrast, in the L-NAME rats, no increase in LV mass (1.00⫾0.03 versus 1.04⫾0.04 g) or hypertrophy of
isolated myocytes occurred (3586⫾129 versus 3756⫾135 m2) compared with controls. Nevertheless, chronic pressure
overload was not accompanied by the development of heart failure. LV systolic performance was maintained by
mechanisms of concentric remodeling (decrease of in vivo LV chamber dimension relative to wall thickness) and
augmented myocardial calcium– dependent contractile reserve associated with preserved expression of ␣- and -myosin
heavy chain isoforms and sarcoplasmic reticulum Ca2⫹ ATPase (SERCA-2).
Conclusions—When the expected compensatory hypertrophic response is suppressed during L-NAME–induced hypertension,
severe chronic pressure overload is associated with a successful adaptation to maintain systolic performance; this adaptation
depends on both LV remodeling and enhanced contractility in response to calcium. (Circulation. 2000;101:423-429.)
Key Words: nitric oxide 䡲 calcium 䡲 NG-nitroarginine methyl ester 䡲 hypertrophy 䡲 remodeling
N
itric oxide (NO) and its donors increase cyclic GMP and
cause vasorelaxation, whereas a withdrawal of constitutive NO induces vasoconstriction and causes severe hypertension.1 This would be expected to induce cardiac hypertrophy as the fundamental compensatory response that maintains
left ventricular (LV) systolic performance in the presence of
chronic systolic pressure overload and prevents development
of heart failure.2 In addition, the NO-cyclic GMP pathway
inhibits cell growth in in vitro systems.3,4 Thus, it would be
expected that chronic systemic NO inhibition will induce
exuberant cardiac hypertrophy; however, L-N-nitro-Larginine methyl ester (L-NAME) treatment appears to suppress the expected increase in LV mass.5–12 This recognized
inhibitory effect on growth is independent of effects on tissue
NO synthesis13–16 and mediated by effects on amino acid
delivery and utilization by competing with amino acid transporters and by altering ornithine metabolism.13–18 Taken
together, this suggests that chronic L-NAME treatment provides a powerful tool to experimentally increase systolic load
and to simultaneously suppress compensatory hypertrophy.
It is questionable whether the suppression of LV hypertrophy
is beneficial or deleterious in pathologic pressure overload
because suppression of hypertrophy might be expected to cause
heart failure. Paradoxically, none of the previous studies which
used L-NAME to cause hypertension reported the development
of heart failure. Therefore, in the present study we used
L-NAME–induced hypertension to test the hypothesis that
successful molecular adaptation to chronic severe pressure
overload occurs even when hypertrophy is suppressed.
Methods
Preparation of Animals
Male Wistar rats (⬇300 g, Charles River Breeding Laboratories,
Wilmington, Mass) received no drug (controls, n⫽22) or L-NAME
Received March 5, 1999; revision received July 28, 1999; accepted August 11, 1999.
From the Charles A. Dana Research Institute and the Harvard-Thorndike Laboratory, Beth Israel Deaconess Medical Center, and Department of
Medicine, Cardiovascular Division, Harvard Medical School, Boston, Mass.
Current affiliation of J.B. is Cardiologisch Centrum, Aalst, Belgium; current affiliation of M.T. is Tohoku University, Sendai, Japan.
Correspondence to Beverly H. Lorell, MD, Cardiovascular Division, Beth Israel Deaconess Medical Center, 330 Brookline Ave, Boston, MA 02215.
E-mail blorell@caregroup.harvard.edu
© 2000 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org
423
424
Circulation
February 1, 2000
(Sigma Chemicals, St. Louis, Mo) at a dose of 50 mg 䡠 kg⫺1 䡠 d⫺1
(n⫽26) in drinking water for 6 weeks.6 –9 An additional group of rats
with 6 weeks ascending aortic stenosis was created19 –23 to compare
the levels of systolic wall stress and extent of LV remodeling for the
same duration of pressure overload.
In Vivo Measurements
In vivo tail-cuff systemic blood pressure was measured weekly by a
single animal handler.19,20 At the end of the treatment period, rats
from each group were randomly selected for echocardiographic
measurements of LV dimensions, LV posterior wall thickness, and
relative wall thickness (ratio of 2⫻posterior wall thickness/LV
diastolic diameter).19,20 In vivo LV pressure measurements were
performed before euthanasia, and LV meridional systolic wall stress
(kdyn/cm2) was estimated.19,21
Calcium-Dependent Contractile Reserve
Downloaded from http://circ.ahajournals.org/ by guest on October 2, 2016
The contractile reserve in isolated hearts from control (n⫽7) and
L-NAME–treated rats (n⫽7) was evaluated using the isovolumic
buffer-perfused rat heart preparation with constant coronary
flow.19,21 To assess calcium-dependent contractile reserve, LV systolic pressure development was studied at 3 different perfusate
calcium concentrations (0.6, 1.2, and 3.0 mmol/L) as previously
described.20,21
To investigate the contractile reserve in isolated myocytes, LV
myocytes were prepared from additional control and L-NAME–
treated rats as previously described (n⫽7 to 8 per group).22,23 In
isolated myocytes, [Ca2⫹]i was measured with the Ca2⫹-sensitive
fluorescence indicator Fluo-3 as previously described.24 –26 Myocytes
were studied at 37°C and paced at 0.5 Hz. Simultaneous measurements of cell shortening and [Ca2⫹]i were measured after 5 minutes
of perfusion with 1.2 and 3.5⫻10 mmol/L Ca2⫹. In addition, the long
axis myocyte area was quantified in quiescent isolated myocytes
from control and L-NAME–treated rats using the NIH Image
software.
Measurements of Tissue Cyclic GMP
The LV and aortic cyclic GMP were determined by enzymatic assay
as previously described.25,27
Left Ventricular RNA Measurements
Northern blot analyses were performed using 20 g total RNA as
previously described.19,21 Probes used were the cDNA fragment
encoding the SR Ca2⫹ ATP-ase (SERCA-2, provided by D.H.
MacLennan, University of Toronto), the cDNA fragment encoding
the rat GAPDH, an 84-bp synthetic oligonucleotide complementary
to the coding region of rat ANF, a 20-bp synthetic oligonucleotide
complementary to the rat -myosin heavy chain (MHC) gene, and a
24-bp oligonucleotide fragment encoding the rat skeletal ␣-actin.
Ribonuclease Protection Assay of
angiotensin-converting enzyme (ACE) mRNA
LV ACE mRNA levels were quantified as previously described.21
The rat ACE probe was derived from clone pRace622 (provided by
Dr M.A. Lee, Harvard Medical School, Boston, MA) which after
linearization with Ava II yielded a 250-bp fragment. The rat -actin
probe was derived from clone pSKrBac and yielded a 150-bp
fragment after linearization with XhoI.
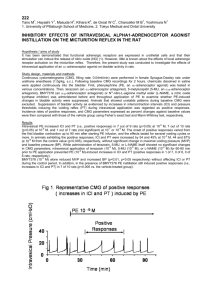
Figure 1. Tail cuff systolic arterial blood pressure in control and
L-NAME–treated rats. L-NAME administration induced sustained
severe systolic pressure overload over the entire treatment
period.
end and hybridized with 20 g of total RNA in molar excess. S1
digestion was performed using Multi-NPA kit (Ambion, Austin,
Tex) and was followed by separation of the protected fragments on
a 5% polyacrylamide gel.
Western Analysis of SERCA-2 Protein Level
SERCA-2 protein levels were analyzed by Western analysis as
previously described.26
Statistical Analysis
All data are expressed as mean⫾SEM. Student’s unpaired t test was
used where appropriate. Comparison between groups was performed
by ANOVA comparison or ANOVA for repeated measures, where
appropriate, followed by Fisher’s protected least significance test for
post hoc analyses. P⬍0.05 was considered significant.
Results
Effects of L-NAME on Blood Pressure and
LV Mass
Chronic L-NAME treatment caused persistent severe hypertension (Figure 1). L-NAME treatment was associated with a
lower body weight and no effect on tibial length, an index of
body growth independent of the body mass (Table 1). LV
weight and LV/body weight ratio were similar between
L-NAME rats and controls. In addition, the long-axis myoTABLE 1.
LV Hypertrophy and Blood Pressure
Control
(n⫽9)
L-NAME
(n⫽15)
Blood pressure, mm Hg
110⫾5
183⫾3*
Body weight, g
491⫾10
Tibial Length, mm
MHC iso-mRNA Analysis by Nuclease S1
Protection Assay
S1 nuclease protection assay of the myosin heavy chain (MHC-iso)
mRNA was performed as described by Waspe et al.28 The probe was
a 61-base synthetic oligonucleotide that was designed to be complementary to a 41-nucleotide common coding sequence at the carboxyl
end of both ␣- and -MHC iso-mRNAs28 and complementary to the
final 15 nucleotides of -MHC iso-mRNA that significantly differs
from those of ␣-MHC iso-mRNA.28 The probe was labeled with
[␣-32P dideoxyl] ATP (Amersham Corp, Arlington Heights, Ill) at 3⬘
438⫾8*
42⫾0.6
41.8⫾0.5
LV weight, g
0.95⫾0.04
0.98⫾0.02
LV/BW, g/kg
1.93⫾0.09
2.04⫾0.04
LV/Tibia, g/mm
23.01⫾1.57
22.78⫾0.96
Myocyte area, m2
3756⫾135
3586⫾129
RV Weight, g
0.21⫾0.02
0.19⫾0.01
RV/BW, g/kg
0.43⫾0.04
0.40⫾0.01
*P⬍0.05 vs controls.
BW indicates body weight; RV, right ventricular.
Bartunek et al
TABLE 2. In Vivo Hemodynamic and
Echocardiographic Measurements
Control
L-NAME
AS
Heart rate, bpm
264⫾3
247⫾6
260⫾15
LV Dd, mm
8.94⫾0.14
7.72⫾0.26*
8.77⫾0.55*†
LV Ds, mm
5.83⫾0.24
5.38⫾0.24
5.89⫾0.61
LV Dd/BW, mm/g
1.87⫾0.01
1.71⫾0.01*
2.18⫾0.02†
LV Ds/BW, mm/g
1.20⫾0.01
1.17⫾0.01
1.38⫾0.01†
35⫾1.0
31⫾1.0
36⫾1.0†
LV mass, g
1.00⫾0.03
1.04⫾0.04
1.75⫾0.24*†
LV mass/BW, g/kg
0.21⫾0.01
0.23⫾0.01
0.43⫾0.08*†
FS, %
Wall stress, kdyne/cm
37⫾4
85⫾19*
92⫾16*
FS/Wall stress, %/kdyne/cm
3.7⫾0.4
4.4⫾0.8
3.6⫾0.5
RW T, mm/mm
36.5⫾1.5
51.1⫾3.9*
51.0⫾3.4*
LV SP, mm Hg
107⫾6
165⫾5*
198⫾8*†
LV devP/g, mm Hg/g
Downloaded from http://circ.ahajournals.org/ by guest on October 2, 2016
95⫾6
143⫾8*
113⫾7*†
LV dP/dt max, mm Hg/s
8716⫾629
11840⫾630*
8107⫾519†
LV dP/dt min, mm Hg/s
6348⫾375
10242⫾527*
8396⫾261†
LVEDP, mm Hg
4.5⫾0.5
5.2⫾0.7
26.9⫾3.9*†
AS indicates aortic stenosis; LV Dd, left ventricular diastolic diameter; LV Ds,
left ventricular systolic diameter; FS, fractional shortening; RW T, relative wall
thickness calculated as the ratio of 2⫻ posterior wall thickness and LVDd; LV
SP, left ventricular systolic pressure; LV devP/g, left ventricular developed
pressure per g LV; LVEDP, left ventricular end diastolic pressure. Hemodynamic
measurements were obtained in vivo from 19 control and 17 L-NAME rats and
7 AS rats. Echocardiographic measurements were obtained from 10 control, 13
L-NAME and 6 AS rats. Other abbreviations as in Table 1.
*P⬍0.05 vs controls; †P⬍0.05 vs L-NAME 50 mg 䡠 kg⫺1 䡠 d⫺1.
cyte area (⬎50 myocytes per rat from 3 to 4 rats per group)
was similar between L-NAME rats and controls.
In Vivo Measurements
No L-NAME rat showed clinical signs of failure (tachypnea
or edema). To investigate in vivo LV systolic function, we
L-NAME and Cardiac Remodeling
425
performed echocardiographic measurements in control and
L-NAME rats before sacrifice (Table 2). In addition, we
compared echocardiographic and hemodynamic indices of
the L-NAME rats with the cohort of 6 weeks ascending aortic
stenosis rats (Figure 2).19 –23
In L-NAME rats, both LV systolic and developed pressure
per gram were significantly higher as compared with controls.
Midwall fractional shortening, which is relatively independent of loading conditions,29,30 was also preserved as compared with controls (0.25⫾0.01% versus 0.23⫾0.01%,
P⫽NS). In addition, the LV end-diastolic pressure (LVEDP)
was not elevated as compared with controls. Thus, in spite of
the absence of an increase in LV mass, L-NAME–induced
hypertension was not associated with the depression of LV
systolic pressure or elevation of LVEDP. Second, the level of
LV systolic wall stress was elevated and similar in L-NAME
and aortic stenosis rats; however, there was a marked difference in LV mass (Table 2). The relative wall thickness2,29,30
was similar between aortic stenosis rats and L-NAME rats.
However, the increase in relative wall thickness in aortic
stenosis rats reflects a marked increase in the wall thickness
associated with an increase in LV mass; in contrast, in
L-NAME rats, it reflects chiefly a decrease in internal LV
dimension.
Isolated Heart and Myocytes Studies:
Contractile Reserve
To study whether an increase in contractility also contributes
to preserved in vivo LV systolic function in the L-NAME
rats, we performed in vitro studies of the LV pressurecalcium relationship19,21 in isolated hearts (n⫽7 per group)
and the shortening calcium relationship in myocytes22,26 (n⫽7
to 8 per group). At the identical LV balloon volume,
comparable levels of LVEDP (⬇10 mm Hg), heart rate and
coronary flow per gram (data not shown), LV systolic
pressure was similar at the low baseline calcium concentration of 0.6 mmol/L (Figure 3). However, at higher calcium
Figure 2. Representative examples of in
vivo M-mode (top) and LV pressure (bottom) measurements in control (left),
L-NAME–treated (center) and aortic stenosis (right) rats. Both L-NAME rats and
aortic stenosis rat show similar increase
in LV systolic pressure and preserved LV
fractional shortening as compared with
control rat. At this stage of compensatory LV hypertrophy in the aortic stenosis
rat, LV diastolic pressure is already elevated. Remarkably, severe hypertrophy is
present in the aortic stenosis rat,
whereas no increase in the wall thickness or LV mass is observed in the
L-NAME rat.
426
Circulation
February 1, 2000
TABLE 3.
LV Gene Expression
Control
(n⫽6)
L-NAME
(n⫽8)
Atrial natruiretic factor
2.66⫾1.07
9.23⫾1.55*
␣-skeletal actin
0.36⫾0.07
1.96⫾0.32*
-MHC
1.49⫾0.18
1.82⫾0.19
ACE
1.81⫾0.43
1.61⫾0.22
SERCA-2
0.86⫾0.18
2.16⫾0.43*
Data are expressed as densitometric units normalized to message levels of
LV GAPDH.
*P⬍0.05 vs controls. ACE indicates angiotensin converting enzyme normalized to -actin.
Downloaded from http://circ.ahajournals.org/ by guest on October 2, 2016
Figure 3. The LV pressure-calcium relationship in isolated
buffer-perfused hearts from control and L-NAME–treated rats. At
baseline calcium level (0.6 mmol/L), LV systolic pressure (LVSP)
was similar between hearts from control and L-NAME–treated
rats. In response to high calcium concentrations, L-NAME rats
showed an upward shift in pressure-calcium relation as compared with controls.
concentrations of 1.2 and 3.0 mmol/L, the relationship
between LV systolic pressure and calcium was shifted upward in L-NAME–treated rats compared with controls.
To further examine calcium-dependent contractile function, LV-isolated myocytes from control and L-NAME–
treated rats were paced at 0.5 Hz, and myocyte fractional
shortening and [Ca2⫹]i was measured in response to 1.2 and
3.5 mmol/L CaCl2 at 37°C. Figure 4 shows the relationship
between fractional myocyte shortening and peak systolic
[Ca2⫹]i in response to elevated perfusate calcium. There was
no difference in baseline peak systolic [Ca2⫹]i or fractional
shortening between myocytes from L-NAME–treated rats
and controls. Similar to the response of the isolated hearts,
there was an upward shift in the relationship between myocyte shortening and peak systolic [Ca2⫹]i at high perfusate
calcium in myocytes from L-NAME–treated rats compared
with control myocytes (P⫽0.09).
Figure 4. Relationship between myocyte shortening and peak
systolic [Ca2⫹]i at baseline (1.2 mmol/L) and high (3.5 mmol/L)
perfusate calcium concentrations in myocytes from control and
L-NAME–treated rats. Peak systolic [Ca2⫹]i was similar between
both groups at baseline and high perfusate calcium. There was
a trend for enhanced myocyte shortening in response to calcium related to an upward shift in the cell-shortening peak systolic [Ca2⫹]i relationship consistent with an enhanced myofilament responsiveness.
Tissue Cyclic GMP Levels
Tissue cyclic GMP content was determined in aortic and LV
tissues. As expected, L-NAME treatment caused a decrease
in aortic cyclic GMP content (253⫾83 versus 746⫾103
fmol/mg, P⬍0.01). In contrast, LV cyclic GMP was unchanged in L-NAME rats compared with controls (87⫾10
versus 73⫾9 fmol/mg, P⫽NS).
Effects of L-NAME on LV Gene Expression
We then examined whether L-NAME–induced hypertension
is associated with the load-induced changes in LV gene
expression despite the absence of LV hypertrophy (Table 3).
L-NAME treatment was associated with a 4-fold increase of
LV ANF mRNA levels and a 5.5-fold increase in ␣-skeletal
actin LV mRNA levels. Unexpectedly, LV mRNA levels of
-MHC and LV ACE mRNA were unchanged in L-NAME–
treated rats. This suggests a dissociation between gene
induction associated with pressure overload and hypertrophic
growth per se. In addition, LV mRNA levels of SERCA-2
were increased; however, this did not translate into an
increase in LV SERCA-2 proteins relative to controls
(127⫾14% versus 99.9⫾6%, P⫽NS).
To investigate whether the preserved LV function in vivo
in the presence of L-NAME–induced pressure overload is
related to relative changes of steady state ␣- and -MHC
iso-mRNA levels, we performed quantitative S1 endonuclease assay in LV tissue of control, L-NAME rats, and aortic
stenosis rats (Figure 5). Consistent with previous studies,27,28
control hearts expressed predominantly ␣-MHC iso-mRNA
(69⫾5% of total MHC). In LV tissue from aortic stenosis
rats, there was a relative reduction in ␣-MHC iso-mRNA and
an increase in -MHC iso-mRNA (relative amount of
␣-MHC iso-mRNA 24⫾9% of total MHC, P⬍0.05 versus
controls). In contrast, in LV tissue from L-NAME rats, there
was no significant change in the relative ␣- and -MHC
expression (relative amount of ␣-MHC 60⫾4% of total
MHC, P⫽NS versus controls). Thus, L-NAME–induced
pressure overload is not associated with a switch in MHC
isoform expression.
Discussion
The present study used chronic treatment with L-NAME as a
potent tool to simultaneously induce hypertension and suppress adaptive LV hypertrophic growth to test the hypothesis
that adaptation to pressure overload occurs even when hyper-
Bartunek et al
L-NAME and Cardiac Remodeling
427
Figure 5. Representative blots of S1
endonuclease assay of MHC iso-mRNA
expression. ␣-MHC iso-mRNA is predominantly expressed in adult control
LV, whereas in the AS rat, there is an
upregulation of -isoform and decrease
of ␣-isoform. In L-NAME rats, there was
a slight increase in the expression of
-MHC iso-mRNA and no change in
␣-MHC-iso-mRNA. AS indicates aortic
stenosis.
Downloaded from http://circ.ahajournals.org/ by guest on October 2, 2016
trophy is absent. L-NAME–induced pressure overload is
associated with a distinct pattern of LV remodeling characterized by a decrease in LV chamber size relative to wall
thickness in the absence of an increase in LV mass. Despite
the lack of compensatory hypertrophy, L-NAME hypertension for 6 weeks is associated with the absence of heart failure
and preserved in vivo LV function related to an enhanced
pressure development and cardiomyocyte shortening in response to calcium.
Dissociation Between LV Hypertrophy and
Induction of Fetal Genes
A novel feature of the present study was the comparison of
hypertrophic growth in response to pressure overload in
L-NAME and aortic stenosis animals. It is striking that
hypertrophy did not develop in 6-week L-NAME–treated
animals despite an elevated and similar LV systolic wall
stress sufficient to cause an ⬇2-fold increase in LV mass in
6-week aortic stenosis animals. Our comparison of the change
in LV mass for a similar increase in LV pressure overload in
6-week L-NAME versus aortic stenosis rats supports the
interpretation of earlier studies that the LV growth response is
inappropriately low in L-NAME rats.5– 8,11,12 The present
study does not exclude the possibility of changes in LV mass
in response to more prolonged L-NAME–induced hypertension. Some previous studies with higher doses or more
prolonged treatment have observed a relative increase in LV
mass ranging from 9% to 30%.9,10,31,32 Taken together, our
study and these previous studies show that changes in LV
mass are absent or modest in the face of severe sustained
hypertension.
Several mechanisms are likely to account for blunted
hypertrophic response. First, in addition to NO inhibition,
L-NAME modulates amino acid delivery and polyamino acid
synthesis.13–18 Second, in contrast to expected inhibitory
effects of L-NAME on tissue cyclic GMP content,1,6,7,33,34 LV
cyclic GMP content in response to chronic L-NAME treatment remained unchanged.6 Third, we did not observe
changes in LV ACE mRNA expression that could result in
increased angiotensin II production and modulation of the
hypertrophic growth response.7,10,19,21 This contrasts with the
observation of Takemoto et al,10 who (using higher doses of
L-NAME) observed an upregulated systemic and local angiotensin system and a relative increase in LV mass. Fourth,
the absence of hypertrophic growth may be related to
L-NAME–induced vasoconstriction with myocardial ischemia;9,35 however, the absence of depressed LV systolic
function in vivo or in the isolated heart preparation strongly
argues against chronic ischemia.
Adaptation of the Adult Heart to Pressure
Overload in Absence of Hypertrophy
The absence of compensatory hypertrophy in the presence of
severe pressure overload would be expected to promote early
cardiac dilatation and heart failure.2 Our study indicates
several mechanisms by which the heart can adapt to high
systolic load without a pathologic increase in LV mass. The
first mechanism is concentric geometric remodeling with a
reduction of the LV chamber size relative to wall thickness
that increases relative wall thickness (the ratio of posterior
wall to the LV diastolic diameter), an adaptation which
preserves LV pump function.29,30,36 Our observations are
consistent with a recent study of Matsubara et al,12 who also
observed a decrease in LV volume, absence of LV hypertrophy, and preserved LV function in response to L-NAME–
induced hypertension. The mechanisms which underlie this
geometric remodeling are unclear because the decrease in LV
dimension did not appear to be related to a change in myocyte
size. It is a possibility that the decrease in LV dimension is
related in part to the slight reduction in body mass or change
in venodilation in L-NAME rats. We also did not address the
possibility of differences in myocyte size in subendocardium
versus subepicardium.
A second compensatory mechanism is an enhanced LV
contractile reserve in response to calcium, which we observed
in isolated hearts. Although we did not examine histological
changes in matrix composition, other studies have demonstrated an increase in collagen in this model.9,10 Because
changes in collagen deposition and matrix can alter the
contractile response in the intact heart in vivo or in isolated
hearts, we also examined the contractile response at the level
of isolated myocytes. The enhanced pressure development in
isolated hearts and enhanced myocyte shortening in response
to calcium implicate an increased myocardial responsiveness
to calcium. We23 and others37,38 have shown that the acute
depression of contractility by NO in rat myocytes is predominantly related to the depression of myofilament calcium
responsiveness. Thus, we postulate that the augmentation of
contractility in L-NAME–treated rats is related in part to the
withdrawal of these mild depressant effects of constitutive
NO on contractility and myofilament calcium sensitivity. In
addition, in the present study, severe hypertension was not
associated with an isoform switch of ␣- and -MHC isomRNAs, typical for sustained mechanical pressure over-
428
Circulation
February 1, 2000
Downloaded from http://circ.ahajournals.org/ by guest on October 2, 2016
load.39,40 The absence of such isoform switch could also
contribute to preserved contractile function in vivo in the
presence of pressure overload.41 Of interest, studies in transgenic animals with increased SERCA-2 expression have
reported enhanced calcium transients and myocardial contractility.42 However, alterations in SERCA-2 expression do
not appear to play an adaptive role in the present study
because protein levels of SERCA-2 and calcium transients
were unchanged by L-NAME treatment.
These observations in L-NAME hypertensive rats contrast
with the molecular adaptation of aortic stenosis animals to
pressure overload, which is characterized by a relative increase in -MHC expression and reduction in ␣-MHC expression, as well as reduction in SERCA-2 expression. In
contrast to L-NAME hypertensive rats, we22 and others43
have previously shown that aortic banded rats at the stage of
early concentric hypertrophy do not exhibit an enhanced
myocyte contractile response to calcium, whereas this relationship is depressed during progression to failure.
Limitations and Conclusions
This study does not determine whether this early adaptation
after 6 weeks of L-NAME–induced pressure overload will be
successful in preventing the progression to heart failure
during a longer period of L-NAME–induced hypertension.
Second, determination of beneficial or adverse effects of
chronic in vivo NO inhibition with L-NAME on contractile
performance and hypertrophic growth in the conditions associated with excessive NO production, such as advanced heart
failure, will require further investigation. Third, the potential
use of L-NAME to suppress pathologic hypertrophy is
limited by confounding vasoconstriction. Nonetheless, the
present study supports the possibility that novel pharmacologic measures that suppress hypertrophic growth may be
associated with beneficial geometric and molecular adaptations in pathologic pressure overload which suppress the
progression to heart failure.
Acknowledgments
This study was supported by a grant from National Heart, Lung, and
Blood Institute Grant HL-38189 (to B.H.L. and E.O.W.), an award
from NASA (to B.H.L.), and by the US Fogarty International
Fellowship Award (NIH, F05 TW05261-01 [to J.B.]). We thank Lois
Wiltberger for assistance in preparing this manuscript.
References
1. Baylis C, Mitruka B, Deng A. Chronic blockade of nitric oxide synthesis
in the rat produces systemic hypertension and glomerular damage. J Clin
Invest. 1992;90:278 –281.
2. Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56:56 – 64.
3. Carg UC, Hassid A. Nitric oxide vasodilators and 8-bromo-cyclic
guanosine monophosphate inhibit mitogenesis and proliferation of
cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83:
1774 –1777.
4. Kolpakov V, Gordon D, Kulik TJ. Nitric-oxide generating compounds
inhibit total protein and collagen synthesis in cultured vascular smooth
muscle cells. Circ Res. 1995;76:305–309.
5. Ribeiro MO, Antunes E, de Nucci G, Lovisolos SM, Zatz R. Chronic
inhibition of nitric oxide synthesis: a new model of arterial hypertension.
Hypertension. 1992;20:298 –303.
6. Arnal JF, Warin L, Michel JP. Determinants of aortic cyclic guanosine
monophosphate in hypertension induced by chronic inhibition of nitric
oxide synthase. J Clin Invest. 1992;90:647– 652.
7. Arnal JF, Amrani AI, Chatellier G, Menard J, Michel JP. Cardiac weight
in hypertension induced by nitric oxide synthase blockade. Hypertension.
1993;22:380 –387.
8. Rhaleb NE, Yang XP, Scicli AG, Carretero OA. Role of kinins and nitric
oxide in the antihypertrophic effect of ramipril. Hypertension. 1994;
23(part 2):865– 868.
9. Moreno H Jr, Metze K, Bento AC, Antunes E, Zatz R, de Nucci G.
Chronic nitric oxide inhibition as a model of hypertensive heart muscle
disease. Basic Res Cardiol. 1996;91:248 –255.
10. Takemoto M, Egashira K, Usui M, Numaguchi K, Tomita H, Tsutsui H,
Shimokawa H, Sueishi H, Takeshita T. Important role of tissue angiotensin-converting enzyme activity in the pathogenesis of coronary
vascular and myocardial structural changes induced by long-term
blockade of nitric oxide synthesis in rats. J Clin Invest. 1997;99:278 –287.
11. Hu L, Manning D Jr, Brands MW. Long-term cardiovascular role of nitric
oxide in conscious rats. Hypertension. 1994;23:185–194.
12. Matsubara BB, Matsubara LS, Zornoff LA, Franco M, Janicki JS. Left
ventricular adaptation to chronic pressure overload induced by inhibition
of nitric oxide synthase in rats. Basic Res Cardiol. 1998;93:173–181.
13. Baydoun AR, Mann GE. Selective targeting of nitric oxide synthase
inhibitors to system y⫹ in activated macrophages. Biochem Biophys Res
Commun. 1994;200:726 –731.
14. Bogle RG, Moncada S, Pearson JD, Mann GE. Identification of inhibitors
of nitric oxide synthase that do not interact with the endothelial cell
L-arginine transporter. Br J Pharmacol. 1992;105:768 –770.
15. Morgan DM. Polyamines, arginine, and nitric oxide. Biochem Soc Trans.
1994;22:879 – 883.
16. Kerwin JF Jr, Lancaster JR Jr, Feldman PF. Nitric oxide: a new paradigm
for second messengers. J Med Chem. 1995;38:4343– 4362.
17. Greenberg SS, Lancaster JR, Xie J, Sarphie TG, Zhao X, Hua L, Freeman
T, Kapusta DR, Giles TD, Powers DR. Effects of NO synthase inhibitors,
arginine-deficient diet and amiloride in pregnant rats. Am J Physiol.
1997;273:R1031–R1045.
18. Young M, Prenton M. Maternal and fetal plasma concentrations of amino
acids during gestation and in retarded fetal growth. Br J Obstet Gynaecol.
1989;76:333–341.
19. Weinberg EO, Schoen FJ, George D, Kagaya Y, Douglas PS, Litwin SE,
Schunkert H, Benedict CR, Lorell BH. Angiotensin-converting enzyme
inhibition prolongs survival and modifies the transition to heart failure in
rats with pressure overload hypertrophy due to ascending aortic stenosis.
Circulation. 1994;90:1410 –1422.
20. Litwin SE, Katz SA, Weinberg EO, Lorell BH, Aurigemma GP, Douglas
PS. Serial echocardiographic assessment of left ventricular geometry and
function in rats with pressure overload hypertrophy: chronic angiotensinconverting enzyme inhibition attenuates the transition to heart failure.
Circulation. 1995;91:2642–2654.
21. Weinberg EO, Lee MA, Weigner M, Lindpaintner K, Bishop SP,
Benedict CR, Ho KKL, Douglas PS, Chafizadeh E, Lorell BH. Angiotensin AT1 receptor inhibition: effects on hypertrophic remodeling and
ACE mRNA expression in rats with pressure overload hypertrophy due to
ascending aortic stenosis. Circulation. 1997;95:1592–1600.
22. Kagaya Y, Hajjar RJ, Gwathmey JK, Barry WH, Lorell BH. Long-term
angiotensin-converting enzyme inhibition with fosinopril improves
depressed responsiveness to Ca2⫹ in myocytes from aortic-banded rats.
Circulation. 1996;94:2915–2922.
23. Ito N, Bartunek J, Spitzer KW, Lorell BH. Effects of nitric oxide donor
sodium nitroprusside on intracellular pH and contraction in hypertrophied
myocytes. Circulation. 1997;94:2303–2311.
24. Yao A, Spitzer KW, Bridge JH, Barry WH. Sarcoplasmic reticulum and
Na⫹/Ca2⫹ exchanger function during early and late relaxation in ventricular myocytes. Am J Physiol. 1997;274:H2765–H2773.
25. Tajima M, Bartunek J, Weinberg EO, Lorell BH. Atrial natriuretic
peptide has different effects on contractility and intracellular pH in
normal and hypertrophied myocytes from pressure-overloaded hearts.
Circulation. 1998;98:2760 –2764.
26. Tajima M, Weinberg EO, Bartunek J, Jin H, Yan R, Paoni NF, Lorell BH.
Treatment with growth hormone enhances contractile reserve and intracellular calcium in myocytes from rats with post-infarction heart failure.
Circulation. 1999;99:127–134.
27. Bartunek J, Dempsey S, Weinberg EO, Ito N, Tajima M, Rohrbach S,
Lorell BH. Chronic L-arginine treatment increases cardiac cyclic
guanosine 5⬘-monophosphate in rats with aortic stenosis: effects on left
ventricular mass and beta-adrenergic contractile reserve. J Am Coll
Cardiol. 1998;32:528 –535.
Bartunek et al
Downloaded from http://circ.ahajournals.org/ by guest on October 2, 2016
28. Waspe LE, Ordahl CP, Simpson PC. The cardiac -myosin heavy chain
isogene is induced selectively in ␣1-adrenergic receptor-stimulated hypertrophy of cultured rat heart myocytes. J Clin Invest. 1990;85:1206–1214.
29. Gaasch WH. Left ventricular radius to wall thickness ratio. Am J Cardiol.
1979;43:1189 –1194.
30. Aurigemma GP, Silver KH, Priest MA, Gaasch WH. Geometric changes
allow normal ejection fraction despite depressed myocardial shortening in
hypertensive left ventricular hypertrophy. J Am Coll Cardiol. 1995;26:
195–202.
31. Pechanova O, Bernatova I, Pelouch V, Simko F. Protein remodeling of
the heart in NO-deficient hypertension: the effect of captopril. J Mol Cell
Cardiol. 1997;29:3365–3374.
32. Sladek T, Gerova M, Znojil V, Devat L. Morphometric characteristics of
cardiac hypertrophy induced by long-term inhibition of NO synthase.
Physiol Res. 1996;45:335–338.
33. Delacretez E, Hayoz D, Osterheld MC, Genton CY, Brunner HR, Waeber
B. Long-term nitric oxide synthase inhibition and distensibility of carotid
artery in intact rats. Hypertension. 1994;23(part 2):967–970.
34. Keaney JF, Hare JM, Balligand JL, Loscalzo J, Smith TW, Colucci WS.
Inhibition of nitric oxide synthase augments myocardial contractile
responses to  -adrenergic stimulation. Am J Physiol. 1996;271:
H2646 –H2652.
35. Numaguchi K, Egashira K, Takemoto M, Kadokami T, Shimokawa H,
Sueshi K, Takeshita A. Chronic inhibition of nitric oxide synthesis causes
coronary microvascular remodeling in rats. Hypertension. 1995;26(part
1):957–962.
36. Litwin SE, Raya TE, Anderson PG, Litwin CM, Breezier R, Goldman S.
Induction of myocardial hypertrophy following coronary ligation in rats
37.
38.
39.
40.
41.
42.
43.
L-NAME and Cardiac Remodeling
429
decreases left ventricular dilatation and improves systolic function. Circulation. 1991;84:1819 –1827.
Kelly RA, Balligand JL, Smith TW. Nitric oxide and cardiac function.
Circ Res. 1996;79:363–380.
Shah AM, Spurgeon HA, Sollot SJ, Talo A, Lakatta EG. 8-bromo-cGMP
reduces the myofilament response to Ca2⫹ in intact cardiac myocytes.
Circ Res. 1994;74:970 –978.
Izumo S, Nadal-Ginard B, Mahdavi V. Protooncogene induction and
reprogramming of cardiac gene expression produced by pressure
overload. Proc Natl Acad Sci U S A. 1988;85:339 –343.
Chang KC, Figueredo VM, Sreur JHM, Kariya K, Weiner MW, Simpson
PC, Camacho CA. Thyroid hormone improves function and Ca2⫹
handling in pressure overload hypertrophy. Association with increased
sarcoplasmic reticulum Ca2⫹-ATPase and ␣-myosin heavy chain in rat
hearts. J Clin Invest. 1997;100:1742–1749.
Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin
heavy chain gene expression in human heart failure. J Clin Invest. 1997;
100:2362–2370.
He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA,
McDonough PM, Bluhm WF, Meyer M, Sayen MR, Dillman WH. Overexpression of the rat sarcoplasmic reticulum Ca2⫹-ATPase gene in the
heart of transgenic mice accelerates calcium transients and cardiac
relaxation. J Clin Invest. 1997;100:380 –389.
McCall E, Ginsburg KS, Bassani RA, Shannon TR, Qi M, Samarel AM,
Bers DM. Ca2⫹ flux, contractility and excitation-contraction coupling in
hypertrophied rat ventricular myocytes. Am J Physiol. 1998;274:
H1348 –H1360.
Chronic NG-Nitro-l-Arginine Methyl Ester−Induced Hypertension : Novel Molecular
Adaptation to Systolic Load in Absence of Hypertrophy
Jozef Bartunek, Ellen O. Weinberg, Minori Tajima, Susanne Rohrbach, Sarah E. Katz, Pamela
S. Douglas and Beverly H. Lorell
Downloaded from http://circ.ahajournals.org/ by guest on October 2, 2016
Circulation. 2000;101:423-429
doi: 10.1161/01.CIR.101.4.423
Circulation is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231
Copyright © 2000 American Heart Association, Inc. All rights reserved.
Print ISSN: 0009-7322. Online ISSN: 1524-4539
The online version of this article, along with updated information and services, is located on the
World Wide Web at:
http://circ.ahajournals.org/content/101/4/423
Permissions: Requests for permissions to reproduce figures, tables, or portions of articles originally published
in Circulation can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial
Office. Once the online version of the published article for which permission is being requested is located,
click Request Permissions in the middle column of the Web page under Services. Further information about
this process is available in the Permissions and Rights Question and Answer document.
Reprints: Information about reprints can be found online at:
http://www.lww.com/reprints
Subscriptions: Information about subscribing to Circulation is online at:
http://circ.ahajournals.org//subscriptions/