Photophosphorylation as Function of ADP Concentration at Varying
advertisement

Photophosphorylation as Function of A D P Concentration
at Varying Transmembrane Proton Gradients
Georg H einen and Heinrich Strotmann
In stitu t für B iochem ie d e r P flanzen, H ein rich -H ein e-U n iv ersität D ü sseld o rf, U n iv e rsitä tsstraß e 1,
D -4000 D ü sse ld o rf, B u n d esrep u b lik D eutschland
Z . N a tu rfo rsch .
44c, 473—479 (1989); received Ja n u a ry 16, 1989
D edicated to P rofessor A c h im Trebst on the occasion o f his 60th birthday
Iso la te d S pinach C h lo ro p lasts, P ho to p h o sp h o ry latio n , P roton-M otive F o rce,
E nzym e K inetic C o n sta n ts
R a te s o f p h o to p h o sp h o ry latio n w ere m easured at co n stan t sa tu ratin g p h o sp h a te c o n c e n tra tio n ,
varying A D P c o n c e n tra tio n , and varying light intensity. A s the tran sm e m b ra n e p ro to n g ra d ie n t is
d e cre ased by p h o sp h o ry la tio n to d ifferent extents dep en d in g on the c o n ce n tra tio n o f A D P . rates
o f A T P fo rm atio n o b tain e d at the d ifferent A D P c o n cen tratio n s w ere p lo tte d versus th e actual
stead y sta te A p H (in the absence o f Ai|>) during the course o f the re ac tio n . A pH w as m o n ito re d by
th e c alib ra ted 9-am inoacridine fluorescence technique. In secondary plots p h o sp h o ry la tio n as
function o f A D P c o n ce n tra tio n at d ifferent constant A pH values w ere o b tain e d . T he results
indicate M ichaelis-M enten kinetics. T he tru e K m for A D P is virtually u n affected by A p H w h ereas
(a t A D P sa tu ra tio n ) strongly dep en d s on A pH . T he results are discussed in the fram ew o rk of
a sim ple enzym e kinetic m odel which considers the in trath y lak o id al p ro to n (at c o n stan t ex tern al
p H ) as a th ird su b stra te for A T P form ation. T he m odel is capable o f explaining th e re p o rte d
resu lts as well as a variety o f im p o rta n t results from the lite ratu re.
Introduction
Photophosphorylation is catalyzed by a protontranslocating ATPase of the F0F] type which is a con­
stituent of the thylakoid membrane of the chloro­
plast. The mechanism of A|iH+-coupled phosphoryl
transfer to A D P is a subject of intensive investiga­
tion. As the catalytic site is located on ß subunit or at
the interface between a and ß [for review see 1—3],
three catalytic entities must be assumed to be present
per one A TPase molecule because the num ber of a
and ß copies per enzyme is three [4—5]. Nevertheless
Michaelis-M enten kinetics are usually observed
when phosphorylation is measured as function of
A D P or phosphate concentration.
This fact may be explained within the framework
of the three-site binding change mechanism propos­
ed by Boyer and his coworkers [6]. This hypothesis
assumes that the three catalytic sites change their
properties in a sequential mode, so that at a given
mom ent only one of them is in a condition to take up
the substrates. Substrate binding to one site faciliA b b revia tio n s: C hi, c h lorophyll; D C M U , 3-(3 ',4 '-d ich lo ro p h e n y l)-l,l-d im e th y lu re a ; PM S, phenazine m ethosulfate;
T ric in e, N -[tris(hydroxym ethyl)m ethyl]glycine.
R e p rin t re q u ests to P rof. D r. H . S trotm ann.
V erlag d e r Z eitsc h rift fü r N a turforschung, D-7400 T übingen
0341 -038 2 /8 9 /0 5 0 0 - 0473 $ 0 1 .3 0 /0
tates product release from another site. A t very low
A D P concentration (< 1 ^im), however, a deviation
from monophasic Michaelis-Menten behaviour was
observed [7], a result which was interpreted to indi­
cate the operation of a single site in this concentra­
tion range of A D P. Although im portant for the elu­
cidation of the catalytic mechanism, single site
catalysis can be disregarded in a physiological sub­
strate concentration range.
Manyfold determ ination of K m for A D P are re­
ported in the literature with rather diverging results.
A systematic investigation showed that K m increases
with increasing light intensity [8—10], decreases with
increasing concentration of electron transport in­
hibitors like DCM U [8], increases or decreases in the
presence of an uncoupler depending on the concen­
tration employed [9, 12]. Different conclusions were
drawn from these experim ental findings. D avenport
and McCarty [11] were right in criticizing that all
these m easurem ents suffered from uncontrolled ex­
perim ental conditions with regard to A|iH+ during the
phosphorylation reaction. Ap,H* is a function of the
rate of electron transport, as well as the rates of pas­
sive and productive proton efflux. With increasing
substrate concentration we can expect a progressive
decrease of the electrochemical proton gradient even
at constant light intensity, since the rate of phos­
phorylation is dependent on the concentrations of
Unauthenticated
Download Date | 10/2/16 3:48 PM
474
G . H ein en and H . S tro tm an n • P hoto p h o sp h o ry latio n as Function of A D P C o n c en tratio n
the substrates. As electron transport is controlled by
intrathylakoidal pH [12, 13] varying extents of accel­
eration of electron transport must be taken into ac­
count, and the rate of passive proton leakage which
is a function of the internal proton concentration will
be changed, too. Analysis of the effect of A D P [10]
and phosphate concentrations [14] on the magnitude
of steady state ApH indeed verified these predic­
tions. The goal of this study is the determ ination of
the kinetic constants of the H*-ATPase at controlled
ApH (in the absence of Aty) and to give an answer to
the question w hether the true K m is constant or
variable with ApH. This decision has an im portant
implication for the understanding of the enzymatic
mechanism.
The experim ental approac!' includes m easure­
ments of photophosphorylation ai varying light in­
tensity or uncoupler concentration, respectively, and
varying A D P concentration. By simultaneous regis­
tration of 9-aminoacridine fluorescence which was
calibrated for the employed experimental conditions
[15, 16], the rates of ATP form ation could be related
to the actual ApH. K m and Vmax at constant actual
ApH were ascertained by secondary plots.
M ethods
Thylakoids were isolated from spinach leaves as in
ref. [17].
All m easurem ents were conducted in a cylindrical
glass cuvette which was placed in a self-constructed
fluorom eter [16]. The experim ental set-up perm itted
easy addition of substrates and taking of aliquots for
analysis. During the whole experim ent the fluores­
cence of 9-aminoacridine was recorded as a measure
of ApH.
The final cuvette volume was 2.5 ml, the tem pera­
ture 20 °C. The medium consisted of 25 mM Tricine
buffer, pH 8.0, 50 mM KC1, 5 mM MgCl, 10 mM dithiothreitol, 50 piM PMS, 50 nM valinomycin, 10 mM glu­
cose and 30 units/ml hexokinase (salt-free, Sigma).
A fter addition of thylakoids corresponding to 25 ^ig
chlorophyll/ml, 5 ^im 9-aminoacridine was injected.
Then full light was given for 2 min to bring the A T P ­
ase in its thiol-m odulated state.
A fter dark relaxation of the fluorescence signal,
the indicated concentrations of A D P were added.
Since A D P caused some instantaneous artificial
fluorescence quenching [15], the fluorescence emis­
sion obtained after A D P addition minus basal fluo­
rescence (absence of 9-aminoacridine) was used as
standard (<J>0) for calculation of ApH.
Subsequently, light of varying intensity was turned
on to produce different initial ApH values. 2 min
later, after reaching a steady fluorescence, photo­
phosphorylation was started by the addition of 32Plabeled phosphate. Initiation of ATP synthesis
causes a decrease of ApH. The steady state fluores­
cence during phosphorylation (<I>) was used for cal­
culation of the actual ApH. For analysis of the
formed 32P-labeled glucose-6-phosphate 0.15 ml
samples were taken 15, 30, 45 and 60 s after addition
of [3: P]Pj and deproteinized by H C104 (0.5 m final
concentration). Organic 32P was separated from in­
organic 32P by precipitation [18] and m easured in a
scintillation counter.
The calculation of ApH was based on the calibra­
tion perform ed in ref. [16] under the same experi­
mental conditions. Because of the observed linear
relationship between log («J^- ^ ) / ^ and ApH in the
relevant ApH range between 2.5 and 3.5 [15, 16], the
equation by Schuldiner et al. [13] could be employed.
From the calibration curve [16] an apparent internal
thylakoid volume of 30 ^il/mg chlorophyll was esti­
m ated. The empiric formula for calculation of ApH
under the employed experimental conditions was
ApH = log (<E>0—<t»)/<I> + 3.12.
Results
Fig. 1 shows rates of photophosphorylation as a
function of the actual ApH at 6 different A D P con­
centrations ranging from 10 to 152 ^im. The data are
taken from a large num ber of single independent ex­
periments; in every individual experiment up to 20
points were gained. As the activity of phosphoryla­
tion varied between the experiments, the rates were
normalized by relating them to mean maximal rates.
For every ADP concentration in every experim ent
Vrnax values at saturating ApH were estim ated by
extrapolation. For this purpose reciprocal rates (1/v)
were plotted versus reciprocal internal proton con­
centration to the power n ([H +in]"). The exponent n
was chosen, so that the coefficient of determ ination
(r2) of a linear regression attained its maximum. Usu­
ally n was between 2.5 and 3. In order to ensure
comparability of the ApH curves at different ADP
concentrations, ApH was varied in one experim ent
with not less than two ADP concentrations in an
overlapping mode.
Unauthenticated
Download Date | 10/2/16 3:48 PM
G . H ein en and H . S tro tm an n • P h o to p h o sp h o ry la tio n as Function o f A D P C oncentration
475
Fig. 1. R a te o f ph o to p h o sp h o ry latio n at 6 dif­
ferent A D P c o n cen tratio n s as function o f the
actual steady state A pH m easu red during the
course of the reaction.
ApH was also changed by addition of varying con­
centrations of the uncoupler nigericin at constant
(maximal) light intensity. The results (not shown)
indicate that these data fit into the curves of Fig. 1.
This finding which could be expected on the basis of
the chemiosmotic theory, dem onstrates that solely
the magnitude of ApH is critical, not the m ethod of
its manipulation.
Rates of ATP form ation as a function of ADP
concentration at constant ApH values are shown in
Fig. 2. The points were either taken directly as single
or mean values from Fig. 1 or gained by linear inter­
polation between vicinal experimental points. Be­
cause of the low rates, a fair evaluation of data below
pH = 2.6 is impossible. In a reasonable ApH range
the plots of phosphorylation rate versus A D P con­
centration
are essentially hyperbolic; hence
Michaelis-M enten kinetics may be presupposed. In
order to determ ine Vmax and K m the graphs were
linearized according to Lineweaver-Burk (1/v versus
1/[ADP]), Eadie-Hoffstee (v versus v/[ADP]) and
Hanes-W oolf ([ADP]/v versus [ADP]). The three
modes of evaluation give different emphasis to the
single experim ental points, depending on the ADP
concentration and the obtained rates. Therefore ex­
perim ental scattering affects the determ ination of the
kinetic param eters by the three methods in different
ways. As the confidence of the estimates is restricted
by the relatively low num ber of A D P concentrations
employed, the evaluation by different plots may give
o
U)
E
(X
5
HM ADP
Fig. 2. R ate of p h o to p h o sp h o ry latio n at different actual
steady state A pH values as function of A D P c o n cen tratio n .
T h e experim ental points w ere tak e n from Fig. 1. T he
curves w ere com puted by the m odel p re sen te d in the D is­
cussion.
additional certainty. To exclude any subjectivity in
preparation of the plots, the graphs were computed
from the experimental data by linear regression. The
results are summarized in Table I. As expected, Vmax
increases progressively with rising ApH. Between
ApH 2.6 and 3.0 maximal velocity increases by a
factor of about 7. On the other hand, K m is virtually
unaffected by the magnitude of ApH in this range.
Unauthenticated
Download Date | 10/2/16 3:48 PM
476
G. H ein en and H . S tro tm an n • P h o to p h o sp h o ry la tio n as F unction o f A D P C o n c en tratio n
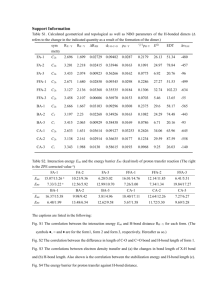
T ab le I. D e term in a tio n o f Vmax and ATm(A D P ) for p h o to p h o s­
p h o ry latio n at different actual A pH values. T he d ata w ere taken
from Fig. 1 and p lo tted according to L inew eaver-B urk (A ). E adieH o ffste e (B ), and H an es-W o o lf (C ). T he kinetic p aram eters w ere
gained from lin ear regression analysis of the experim ental points.
yy max [ixmol A T P /m g Chi p e r h] K m(A D P ) [[am]
A ctu al
A pH A
B
C
m ean
A
B
C
m ean
2.60
2.65
2.70
2.75
2.80
2.85
2.90
2.95
3.00
55.6
66.5
60.3
63.5
61.8
70.8
55.5
56.1
50.9
44
37 46 42
62
63
71
66
67
76
88 76
94
96 102 97
131 123 169 141
209 184 217 203
196 205 238 213
270 247 303 274
265 277 351 298
58.3
60.9
48.1
49.3
54.4
72.9
47.9
55.4
42.1
43.6
62.5
56.9
51.3
46.9
59.6
50.9
45.7
43.1
65.0
76.1
75.2
56.8
84.1
79.8
67.6
67.3
67.4
The single estimates of K m obtained by the three
different plots vary by a factor of about 2. However,
they reveal no clear-cut increasing or decreasing
tendency in dependence of ApH. Hence we conclude
that ApH in the indicated range from 2.6 to 3 does
not affect the Michaelis constant significantly.
The reciprocal
values of Table I were plotted
versus the reciprocal internal H + concentrations to
the pow er n, [H +jn]". The value of n was chosen so
that the plot gave a linear approximation. Linear re­
lationships were found at values of n between 2.5 and
3, Fig. 3 shows a fit perform ed with n = 2.6. n is
identical with the Hill coefficient for H +in which can
be classified as the third substrate of photophos­
phorylation according to
A D P + Pi + n H +in -> ATP + H 20 + n H +out.
The reaction can be written as a unidirectional
process, because A TP is recycled to ADP by the
subsequent hexokinase reaction. The maximal veloc­
ity obtained at saturation of A D P and Pj as function
of [H +in] can be expressed by the Michaelis-Menten
equation as
1 / (M t-Tjn)2'6
Fig. 3. L in ew eav er-B u rk plot o f Vmax (sa tu ratin g A D P co n ­
c en tratio n ) versus in te rn al H + co n ce n tra tio n . T he p ro to n
c o n ce n tra tio n is e xpressed in M 26 (see tex t). T he results
w ere taken from T ab le I. T he points are m ean values of
the bars indicate the lim its o f variation.
V'max = [Et] •kc ■[H +in]”/([H +in]" + K (H +in)),
where [Et] is the total enzyme concentration, kc is the
rate-lim iting velocity constant and K (H +in) means an
equilibrium constant for the internal proton with the
dimension (mol/1)". Theoretically n is equal to the
stoichiom etry of H + translocated through the A TP­
ase complex per ATP formed. The approximation
towards 3 is in agreem ent with determ inations of H +/
ATP stoichiometry obtained by different experim en­
tal approaches [15, 19-22], The plot 1/Vmax versus
l/[H +in]" permits the estimation of the maximal rate
of ATP form ation at saturation of all involved sub­
strates. This param eter (V abs) was determ ined as
1000 to 2000 nmol/mg chlorophyll per h, depending
on the chosen value for n , which agrees well with the
reported turnover num ber of the enzyme [23], In
Unauthenticated
Download Date | 10/2/16 3:48 PM
477
G . H ein en and H . S tro tm an n • P h o to p h o sp h o ry la tio n as Function of A D P C o n cen tratio n
Fig. 3, Kabs was extrapolated to 1250 [imol/mg
Chlorophyll per h. The received value of K (H Tln) per­
mits the calculation of a half-maximal ApH of about
3.1.
Discussion
The here reported results show that the Michaelis
constant for A D P is largely independent of ApH in a
range from 2.6 to 3.0 when m easured at phosphate
saturation and controlled thylakoid energization, i.e.
at a constant actual pH gradient during variation of
the A D P concentration. The evaluation proposes
Michaelis-Menten type kinetics, an assumption
which is based on the hyperbolic dependence of ve­
locity on A D P concentration in the employed range.
We have to admit considerable experimental error
due to scattering in ApH determ ination and the utili­
zation of results from diverse independent experi­
ments. Nevertheless, the large num ber of data and
their treatm ent by different secondary plots confers
some confidence on the determ ination of the kinetic
param eters. The evaluated ApH range from 2.6 to
3.0 appears rather small; however, in this range the
activity of phosphorylation increases by almost one
order of m agnitude, which should be sufficient to
ensure possible substantial changes in K m. Since no
clear-cut tendency of an increase or decrease of
could be detected in dependence of ApH, it appears
highly probable that the true K m is ApH-independent. The same presumption was made by Quick and
Mills [10] in explaining the results on apparent K m
(A D P) at uncontrolled ApH on the basis of a reason­
able theoretical model. In variance to their assump­
tion (K m = 12 |j ,m ) , however, our data indicate a true
K m which is 5 times higher (59 |j .m ) .
In a formal enzyme kinetic model the internal H +
can be considered as a kind of substrate in phos­
phorylation when the external H + concentration is
kept constant as in the here conducted experiments.
Invariability of ATm(A D P) at F max being increased by
raising [H +in] may be interpreted by conceiving H +jn
as an “inverse” non-com petitive inhibitor. In analogy
to the formalism of non-competitive inhibition, H +in
would act on a deenergized free enzyme to create an
energized enzyme which is ready to bind substrate
A D P, as well as on a deenergized form of the en­
zyme with bound A D P to create the energized enzyme-ADP complex. The active enzyme-ADP com­
plex interacts with phosphate in a reversible reaction
to form the enzyme-product complex. In the model
shown below, it is assumed that the active enzymeATP complex can also undergo transfer to a deener­
gized complex and that the reverse reaction depends
on [H+in], too. The final reaction, the dissociation
into the free enzyme and free A TP, can be written as
an irreversible step since the product ATP is trapped
by hexokinase. As a consequence of product release,
the energized state of the enzyme is discharged. The
model is a different view of a conformational cou­
pling hypothesis which is basically not far from the
mechanism proposed by Boyer and his colleagues
[ 6],
E9 .
nH+h/fi
ADP k2
P, k3
...- Ee ADP ^-----flHV,
nH+*k,
Ed-ADP
E.-ATP
/r_t
E„+ATP
/f_i
Ed-ATP
The respective energized and deenergized forms
are designated by the indices e and d, the num ber of
protons reacting from the inner phase of the thy­
lakoid is n. The value of n is assumed to be 3. Com­
putation of the model yields for the param eters Vmax
and ATm(ADP):
Kmax = E, • [H +in]" • k m k y k j
( [ H + in]'' • k\ + f c - O t t P i f o + * - 3 + * 4 )
and
Km(A D P) = ( k . 2k - 3 + k^_k4 + [P;]*3*4)/
(£2[Pj]^3 + kik-T, + k2k4).
The model which is similar but not identical to a
scheme proposed by Quick and Mills [10, 14] is cer­
tainly incomplete. So, it does not include site
cooperativity and explains ATP synthesis but not
hydrolysis without making additional assumptions.
Nevertheless, the model is capable of explaining a
variety of im portant results reported in the literature
as well as the findings of the present study.
(1) The independence of the true /Cm(A D P) as well
as the dependence of F max on the actual ApH or
[H +in], respectively, is fulfilled by the model. As in
the scheme of Quick and Mills [10], variability of the
apparent K m obtained at different light intensities or
uncoupler concentrations can be explained, too.
(2) Energy of the electrochemical gradient is not
invested directly in the chemical reaction at the
Unauthenticated
Download Date | 10/2/16 3:48 PM
478
G . H ein en and H . S trotm ann • P h o to phosphorylation as F un ctio n o f A D P C o n c en tratio n
catalytic site [6]. Here the transformation of
“deenergized” into “energized” enzyme forms is the
only site of energy input. This assumption was also
made by Schumann [24] in a comprehensive hypo­
thesis of the H +-coupled chloroplast ATPase. The
deenergized species may be conceived as enzyme
molecules characterized by “closed” i.e. unavailable
catalytic sites, whereas the energized forms have
“o pen” catalytic sites which are accessible to the sub­
strates of the medium. If a closed site is containing a
nucleotide molecule, this ligand therefore appears
“tightly bound” . In variance with the “energy-linked
binding change mechanism” [6] the formation of
tightly bound A D P or ATP would be a side reaction
rather than an obligate interm ediate step of the
catalytic sequence. The ratio of ATPase molecules
with exchangeable nucleotides to those with tightly
bound nucleotides depends on the energy state of the
m em brane [8, 25] and the level of tightly bound nu­
cleotides changes inversely with the activity of photo­
phosphorylation as function of energy input [26].
W hen light is turned off, the level of tightly bound
nucleotides increases to a maximum of one per A TP­
ase when A D P or ATP is present in the medium [27],
Tight binding of A D P is related with inactivation of
the enzyme [28]. On the other hand, the tightly
bound nucleotides are released or exchanged upon
reillum ination, a reaction which is related with en­
zyme reactivation [28]. U nder certain conditions the
initial rate of nucleotide release matches the rate of
A TP form ation [29, 30]. All these results can be easi­
ly explained by the model. H ence, the reactions re­
lated with enzyme activation are basically identical
with the energy transfer reactions involved in the
catalytic process and deactivation is a compulsory
consequence of relaxation of the proton gradient
leading to closure of the catalytic sites. From the
standpoint of this model it is unnecessary to assume
extra nucleotide binding sites for nucleotide-depend­
ent regulatory processes. This view is in line with the
recent finding that tightly bound A D P and ATP in
darkened thylakoids are on catalytic sites, a result
which was obtained by the photoreactive nucleotide
analogue 2-azido-ADP [32], The general structural
identity of the sites containing tightly bound nucleo­
tides with those performing catalysis had been sug­
gested before on the basis of photolabeling experi­
ments with the same analogue [31]. Although up to 6
nucleotide-binding sites were identified in isolated
CF! [33], the participation of all of them in catalysis
and control of the m em brane-bound enzyme has
never been proven.
(3) It was shown by lsO exchange that decrease of
substrate concentration increased the num ber of re­
versible cycles of ATP form ation at the catalytic site
before the product ATP is released [34], This experi­
mental fact was concluded to indicate catalytic site
cooperativity, i.e. release of the product is facilitated
by binding of substrates to a second catalytic site. For
the reason of simplicity, site cooperativity was om it­
ted, but could be integrated in the scheme without
changing the general features significantly. The same
effect on lfsO exchange was reported to occur when
light intensity was reduced [35] and this result was
taken as evidence for A|IH+ dependence of product
release. In contrast, our model proposes that release
of ATP from the energized enzyme is a spontaneous
reaction. This conclusion which is derived from the
formal enzyme kinetic interpretation of the reported
results, is in line with the earlier finding that the
affinity of the energized enzyme for ATP is by one to
two orders of magnitude lower than for A D P [36].
On the energized enzyme, A TP acts as a competitive
inhibitor of phosphorylation with a K\ as high as
4 m M [36], indicating that the dissociation equilib­
rium is far on the side of free A TP. In the framework
of the model, the effect of low light intensity on 1kO
exchange may be interpreted by assuming that the
cycles of reversible ATP hydrolysis occur in the state
of the enzyme where A TP is tightly bound (EdATP). Actually the exchange of [IS0 ] H 20 with tight­
ly bound ATP was dem onstrated experimentally
[37], If k -\ > &4, the equilibrium may be on the side
of E d-ATP rather than free A TP at low ApH, thus
permitting repetitive lsO exchange with medium
[180 ] H 20 .
(4) The “kinetic com petence” of tightly bound
ATP is an unsolved problem connected with the
energy-linked binding change mechanism. In this re­
spect conflicting results were reported [38, 39]. It is
quite certain that the exchange of at least part of the
tightly bound ATP is too slow to be a step in the
catalytic process [37]. In our model the exchange of
tightly bound nucleotides is not necessarily in pace
with the catalytic cycle. In this context it should be
noted that for sake of simplicity the rate constants k\
and k_\ were assumed to be identical for the inter­
conversion of all deenergized to energized enzyme
forms. They could well be different for the reversible
transitions of E d, E d-ADP and E d-ATP, respectively.
Unauthenticated
Download Date | 10/2/16 3:48 PM
G . H einen and H . S tro tm a n n ■P h o to p h o sp h o ry la tio n as Function o f A D P C oncen tratio n
to the corresponding energized forms. Independence
of K m on ApH would be fulfilled if the ratios of the
forward and backward constants were the same.
The curves drawn in Fig. 2 were com puted with the
model by chosing the following constants: £ t -&4 =
Vabs = 1250 ^imol/mg chlorophyll per h (Fig. 3),
Km(A D P) = 59 [am (Table I), (*_3 + k4)/k3 = K m(P{) =
500 ^im [40], and k - X!k\ = 2000 |xm3. The latter value
was calculated from the ApH at half-maximal phos­
phorylation rate which was found to be 3.1 (Fig. 3):
k - x!kx = (10(31"8)-106)3 = 2000 ^m3.
[1] H . S tro tm a n n and S. B ick e l-S a n d k ö tter, A n n u . Rev.
P lant Physiol. 35, 9 7 - 1 2 0 (1984).
[2] S. M erch an t and B. R. S elm an, P hoto sy n th . R es. 6,
3 - 3 1 (1985).
[3] J. M. G alm ich e, G . G ira u lt, an d C. L em aire. Photochem . P h otobiol. 41, 7 0 7 -7 1 3 (1985).
[4] K. H . Süss and O . S chm idt, FE B S L ett. 144, 213—218
(1982).
[5] J. V. M o roney, L. L o p estri, B. F. M cE w en. R. E.
M cC arty, and G . G . H a m m e s, F E B S L ett. 158, 58—62
(1983).
[6] P. D. B oyer and W. E . K o h lb re n n er, in: E nergy
C oupling in P hoto sy n th esis (B. R. Selm an and S. Selm an -R e im e r, e d s.), pp. 231—240, E lsevier/N orth H o l­
land In c ., New Y o rk 1981.
[7] S. D . S tro o p and P. D . B o y er, B iochem istry 24,
2 3 0 4 -2 3 1 0 (1985).
[8] S. B ick e l-S a n d k ö tter and H . S tro tm a n n , F E B S L ett.
125, 1 8 8 -1 9 2 (1981).
[9] C. V in k ler, B iochem . B iophys. R es. C om m un. 99,
1 0 9 5 -1 1 0 0 (1981).
[10] W . P. Q uick and J. D . M ills, B iochim . B iophys. A cta
893, 1 9 7 -2 0 7 (1987).
[11] J. W. D a v en p o rt and R. E . M cC arty, Biochim . B io­
phys. A cta 851, 1 3 6 -1 4 5 (1986).
[12] U . Siggel, in: P roceedings o f the T h ird In tern atio n al
C ongress on P hoto sy n th esis (M . A v ro n , e d .). Vol. I,
pp. 645—654, E lsevier, A m ste rd am 1975.
[13] S. S chuldiner, H . R o tte n b e rg , and M . A v ro n , E u r. J.
B iochem . 25, 6 4 - 7 0 (1972).
[14] W. P. Q uick and J. D . M ills, B iochim . Biophys. A cta
932, 2 3 2 -2 3 9 (1988).
[15] H . S tro tm a n n and D . L ohse, F E B S L ett. 229,
3 0 8 -3 1 2 (1988).
[16] D . L o h se, R. T h e le n , and H . S tro tm a n n . Biochim .
B iophys. A c ta, su b m itte d .
[17] H . S tro tm an n an d S. B ick e l-S a n d k ö tter, Biochim .
B iophys. A cta 460, 1 2 6 -1 3 5 (1977).
[18] Y. Sugino and Y. M iyoshi, J. Biol. C hem . 239,
2 3 6 0 -2 3 6 4 (1964).
[19] M . R a th e n o w and B. R u m b erg , B er. B unsenges.
Phys. C hem . 84, 1 0 5 9 -1 0 6 2 (1980).
[20] W . Ju n g e , B. R u m b erg , and H . S chröder. E ur. J.
B iochem . 14, 5 7 5 -5 8 1 (1970).
[21] A . R. P ortis and R. E. M cC arty, J. Biol. C hem . 249,
6 2 5 0 -6 2 5 4 (1974).
479
It should be mentioned that this value of course
relies on the correct quantitative determ ination of
ApH in our experiments. The problems related with
calibration of the 9-aminoacridine fluorescence
method by the phosphate potential in energetic
equilibrium are discussed elsewhere [16].
Acknow ledgem ents
This work was supported by grants from Deutsche
Forschungsgemeinschaft (SFB 189) and Fonds der
Chemischen Industrie. The authors thank Mrs. Rita
Reidegeld for help in preparing the manuscript.
[22] J. W. D a v en p o rt and R. E . M cC arty. J. Biol. C hem .
256, 8 9 4 7 -8 9 5 4 (1981).
[23] P. G rä b e r, E . S chlodder, and H. T. W itt, Biochim .
B iophys. A cta 461, 426—440 (1977).
[24] J. S chum ann, Z . N a tu rfo rsc h ., subm itted.
[25] S. B ickel-S andkötter, Biochim . B iophys. A cta 723,
7 1 - 7 7 (1983).
[26] H . S tro tm a n n , S. B ick el-S an d k ö tter, U. F ra n e k , and
V. G e rk e , in: E nergy C oupling in P hotosynthesis
(B. R. Selm an and S. S elm an -R eim er. e d s.), pp.
187—196, E lsev ier/N o rth H ollan d In c ., N ew Y ork
1981.
[27] H . S tro tm a n n , S. B ick e l-S a n d k ö tter. an d V. S hoshan.
FE B S L ett. 101, 3 1 6 -3 2 0 (1979).
[28] J. S chum ann and H . S tro tm a n n , in: P hotosynthesis II.
E lectron T ran sp o rt and P h o to p h o sp h o ry la tio n (G .
A k o yunoglou. e d .), pp. 881—892, B alaban Int. Sei.
Service, P hiladelphia, Pa. 1981.
[29] E. S chlodder and H . T. W itt, Biochim . B iophys. A cta
635, 5 7 1 -5 8 4 (1981).
[30] H. S tro tm a n n , in: A dvances in P hotosynthesis R e ­
search (C. Sybesm a, e d .). Vol. II, pp. 477—484, M artinus N ijhoff/D r. W. Ju n k P ublishers, T he H ague
1984.
[31] J. J. C zarnecki, M . S. A b b o tt, and B. R . Selm an,
Proc. N atl. A cad. Sei. U .S .A . 79, 7 7 4 4 -7 7 4 8 (1982).
[32] J.-M . Z h o u , Z . X u e, Z . D u , T . M elese, and P. D.
B oyer, B iochem istry 27, 51 2 9 -5 1 3 5 (1988).
[33] Z . X ue, J.-M . Z h o u , T . M elese. R. L. C ross, and
P. D. B oyer. B iochem istry 26, 3749—3753 (1987).
[34] D . D. H ack n ey , G . R o se n , and P. D . B o y er, Proc.
N atl. A cad. Sei. U .S .A . 76, 3 6 4 6 -3 6 5 0 (1979).
[35] S. D. S tro o p and P. D . B oyer, B iochem istry 26,
1479 -1 4 8 4 (1987).
[36] U. F ran ek and H . S tro tm a n n , FE B S L ett. 126, 5 —8
(1981).
[37] L. T. Sm ith, G . R o se n , and P. D . B oyer, J. Biol.
C hem . 258, 10887-10894 (1982).
[38] D. J. Sm ith and P. D . B o y er, Proc. N atl. A cad. Sei.
U .S .A . 73, 4 3 1 4 -4 3 1 8 (1976).
[39] C. A flalo and N. S havit, E u r. J. B iochem . 126, 61—68
(1982).
[40] H. S tro tm a n n , S. N iggem eyer, and A .-R . M ansy, in:
Progress in P hotosynthesis R esearch (J. Biggins, e d .).
Vol. Ill, pp. 29—36, M artinus N ijhoff P ublishers,
D o rd re ch t 1987.
Unauthenticated
Download Date | 10/2/16 3:48 PM