Phosphorylation and Acetylation of Histone H3 and
Autoregulation by Early Growth Response 1 Mediate
Interleukin 1 Induction of Early Growth Response
1 Transcription
Bo Wang, Jinbiao Chen, Fernando S. Santiago, Mary Janes, Mary M. Kavurma, Beng H. Chong,
John E. Pimanda, Levon M. Khachigian
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
Objective—The transcription factor early growth response (EGR)-1 has been implicated as a key vascular phenotypic
switch through its control of inducible transcription. EGR-1 autoregulation, and histone modification in the EGR-1
promoter, represent key mechanisms in EGR-1 control, but have not been explored.
Methods and Results—We demonstrate that EGR-1 regulates its own transcription and that this involves histone H3
phosphorylation and acetylation. EGR-1 transactivates its promoter in smooth muscle cells exposed to interleukin (IL)
1 through a novel cis-acting element (⫺211/⫺203). PD98059, which inhibits mitogen-activated protein kinase
kinase/extracellular regulated kinase (MEK/ERK) attenuates IL-1–inducible phosphorylation of extracellular signal–
regulated kinase 1/2 and mitogen and stress–activated protein kinases 1/2; and reduces levels of phosphorylated and
acetylated histone H3. Histone deacetylase inhibition enhances EGR-1 transcription in response to cytokine. Conversely,
suppression of histone modification with mitogen and stress–activated protein kinase 1/2 short interfering RNA, or the
histone H3 acetyltransferase inhibitor Garcinol, inhibits IL-1–inducible EGR-1 transcription. EGR-1 interacts with the
acetyltransferase p300. Acetylated H3 and phosphorylated H3 are enriched at the promoter of EGR-1; and EGR-1 is
enriched at the promoters of tissue factor and plasminogen activator inhibitor 1 in response to IL-1, and attenuated by
PD98059, Garcinol, and mitogen and stress–activated protein kinase 1/2 short interfering RNA.
Conclusion—IL-1 induction of EGR-1 transcription involves histone H3 phosphorylation, acetylation, and autoregulation
by EGR-1. (Arterioscler Thromb Vasc Biol. 2010;30:536-545.)
Key Words: histone modification 䡲 immediate-early gene 䡲 transcription
E
arly growth response 1 (human EGR-1 or mouse EGR-1,
also known as NGFI-A, Zif268, Krox24, and Tis8) is an
inducible zinc finger transcription factor that plays a critical
role in controlling cell growth, proliferation, differentiation,
and apoptosis.1–5 EGR-1 belongs to a family of zinc finger
transcription factors that also includes EGR-2, EGR-3,
EGR-4, EGR-␣, and the Wilm tumor gene product.6 EGR-1
contains a highly conserved DNA-binding domain (three zinc
finger domains of the C2H2 subtype), an activation domain,
a repression domain, and a nuclear localization signal.7,8
Carman and Monroe9 identified an activation domain between amino acids 174 and 270, a serine/threonine/prolinerich region that is critical for its activity. Mutants lacking this
domain perturb wild-type EGR-1 function.9 EGR-1 regulates
the transcription of target genes by binding to GC-rich
consensus DNA elements present in the regulatory regions in
vascular pathophysiologically relevant genes, such as insu-
linlike growth factor II, platelet-derived growth factor A and
B chains, tissue factor, fibroblast growth factor 2, transforming growth factor 1, urokinase plasminogen activator, and
plasminogen activator inhibitor (PAI) 1.5,7,10 –13 EGR-1 also
regulates the expression of its corepressor nerve growth
factor I-A binding protein-2.11 Functional cis-acting elements
within the human EGR-1 promoter have been identified in
different cell types and include serum-response elements
(SREs) and sites for Elk1, Ets, AP-1, and nuclear factor
(NF)-B binding sites.14,15
EGR-1 is induced by a wide range of stimuli, including
growth factors, cytokines, ionizing radiation, UV light, and
mechanical injury.16 –19 Increasing evidence indicates that
EGR-1 activation may function as a key switch in many
pathological processes, including cancers and cardiovascular
diseases. EGR-1 has been indicated in atherosclerosis, intimal
thickening after acute vascular injury, ischemia, angiogene-
Received July 2, 2009; revision accepted November 23, 2009.
From the Centre for Vascular Research, University of New South Wales, Sydney, Australia (B.W., J.C., F.S.S., M.M.K., B.H.C., and L.M.K.); Qiqihar
Medical University, Qiqihar, People’s Republic of China (B.W.); and the Cancer Research Centre and Prince of Wales Clinical School, Faculty of
Medicine, University of New South Wales, Sydney, Australia (M.J. and J.E.P.).
Correspondence to Levon M. Khachigian, PhD, DSc, Centre for Vascular Research, University of New South Wales, Sydney NSW 2052, Australia.
E-mail L.Khachigian@unsw.edu.au
© 2010 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol is available at http://atvb.ahajournals.org
536
DOI: 10.1161/ATVBAHA.109.193821
Wang et al
EGR-1 Autoregulation and Histone Modification
537
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
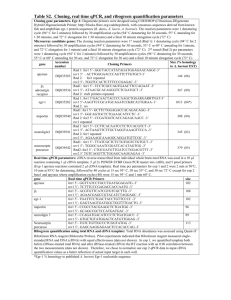
Figure 1. Early growth response (EGR) 1 transactivates its own transcription via a novel functional EGR-1 element within its promoter.
A, The EGR-1 promoter sequence was retrieved from the University of Calif., Santa Cruz genome browser. Consensus elements are as
follows: nuclear factor–B, GGGRNNYYY; Elk1, MMGGAW; serum-response element, CCWWWWWWGG; EGR-1, WTGCGTGGGCGK.
B, Human aortic smooth muscle cells (HASMCs) grown to 70% to 80% confluence were transfected with EGR-1 expression vector
(pCB6–EGR-1) or empty vector (pCB6), and EGR-1 promoter-reporter construct containing wild type or mutant putative EGR-1 binding
site. The cells were incubated for a further 24 hours in serum-free medium before determination of relative luciferase activity, as described in
the “Methods” section. Western blotting 24 hours after transfection confirmed that pCB6 –EGR-1, but not the empty vector (pCB6⫹),
increased EGR-1 levels in the HASMCs. Increased EGR-1 immunoreactivity represents total EGR-1, which includes both endogenous
and exogenous proteins. C, Rat aortic smooth muscle cells (RASMCs) were transfected with empty plasmid or wild-type EGR-1
538
Arterioscler Thromb Vasc Biol
March 2010
Figure 1 (Continued). reporter or a series of
mutant EGR-1 promoter-reporter plasmids for 24
hours. The cells were serum starved for another 24
hours and stimulated with 10 ng/mL of interleukin
(IL) 1 for 30 minutes before the assessment of
relative luciferase activity. D, An electrophoretic
mobility-shift assay (EMSA) was performed with 32
phosphorylated oligo EGR-1-225/-187 or messenger
EGR-1-225/-187 units, and increasing amounts of
EGR-1– glutathione S-transferase protein, 4 and 8
g. The antibody used (sc-110; Santa Cruz Biotechnology) targets native EGR-1. E, Twenty-four
hours after starvation, HASMCs were exposed to
10 ng/mL of IL-1 for 30 minutes. Conventional
chromatin immunoprecipitation (ChIP) (right) and
real-time ChIP (lower) were performed with EGR-1
antibodies or IgG, as described in the “Methods”
section. Quantitation of EGR-1 enriched at its promoter incorporates an IgG control, which was subtracted from the EGR-1 value before quantitative
comparison. The asterisk indicates P⬍0.05.
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
sis, allograft rejection, and cardiac hypertrophy.17 EGR-1 is
overexpressed in esophageal cancer and plays a key role in
mediating and maintaining growth-related oncogene/CXC
chemokine receptor 2 proliferative signaling.20 Primary human
prostate carcinomas express high levels of EGR-1,21–23 and
several EGR-1 target genes (eg, insulinlike growth factor II,
transforming growth factor 1, and platelet-derived growth
factor A-chain) have been implicated in prostate tumorigenesis.22 EGR-1 knockdown inhibits prostate cancer cell
proliferation and tumor development in transgenic adenocarcinoma mouse prostate mice.24 Furthermore, an indispensable role for EGR-1 has been demonstrated in breast
cancer cell proliferation, migration, and invasion in nude
mice using DNAzymes.25–28 This is due, in part, to EGR-1
regulation of angiogenesis.26
Although EGR-1 plays a key role in many pathological
processes, it is unknown whether EGR-1 regulates its own
transcription. Moreover, information on the role of chromatin
remodeling and histone modification in the control of EGR-1
transcription is lacking. Herein, we provide novel insights on
the role of EGR-1 and histone H3 modifications in the
transcriptional regulation of the human EGR-1 gene.
Methods
Primary human aortic smooth muscle cells (HASMCs) and rat aortic
smooth muscle cells were purchased from the American Type
Culture Collection and cultured at 37°C in a humidified atmosphere
of 5% carbon oxide and 95% air in Waymouth medium containing
10% fetal bovine serum. SMCs grown to 90% confluence were
starved of serum for 24 hours and then treated with 10 ng/mL of
interleukin (IL) 1 for various time intervals; where indicated, they
were treated, after 1 hour of incubation, with inhibitors such as
PD98059, SB202190, BAY 11 to BAY 7085, or Oridonin. Cells
were used between passages 5 and 8.
Details on the generation of promoter reporter constructs, sitedirected mutagenesis, transient transfection, luciferase assays, realtime quantitative reverse transcription–polymerase chain reaction
(RT-PCR), immunocytofluorescence, Western blot analysis, phosphorylation and acetylation of histone H3, electrophoretic mobilityshift assay, immunoprecipitation experiments, chromatin immunopre-
cipitation (ChIP), and statistical analysis are found in the Supplemental
Materials section, available online at http://atvb.ahajournals.org.
Results
EGR-1 Regulates Transcription of Its Own Gene
EGR-1 is implicated as a key regulatory switch in a wide
variety of vascular disorders through its control of inducible
transcription.5,17 Although the EGR-1 promoter has previously been studied,11,15 it is unknown whether EGR-1 can
control its own transcription. Moreover, the critical role of
histone modification in EGR-1 transcription is completely
unknown. Bioinformatic analysis of the proximal region of
the EGR-1 promoter shows the existence of numerous conserved putative cis-acting elements, including SREs, and
motifs for Ets, Elk1, and NF-B. Interestingly, the EGR-1
promoter contains a putative atypical binding motif (5⬘GCGCGGGCG-3⬘) for EGR-1 located at ⫺211/⫺203 (Figure 1A). We used the proinflammatory cytokine IL-1 as a
model agonist of EGR-1 to gain valuable insights into the
EGR-1 transcription in HASMCs. Quantitative real-time
RT-PCR showed both a time- and dose-dependent induction
in EGR-1 messenger RNA (mRNA) expression in HASMCs
exposed to the IL-1 (Supplemental Figure IA and B).
EGR-1 was induced by as little as 2.5 ng/mL of IL-I
(Supplemental Figure IA) and peaked at 30 minutes in cells
exposed to 10 ng/mL of IL-1 (Supplemental Figure IB).
Immunofluorescence staining revealed that IL-1 triggered
the rapid accumulation of EGR-1 in HASMC nuclei within
0.5 to 1.0 hours (Supplemental Figure IC), which correlates
with the time course of IL-1–inducible EGR-1 expression
(Supplemental Figure IB).
Overexpression of EGR-1 in HASMCs transfected with a
luciferase reporter construct driven by 450 base pairs of the
human EGR-1 promoter spanning the putative EGR-1 binding motif significantly increased luciferase activity (Figure
1B, upper). When the –208CGG–206 motif in the promoter was
mutated to –208AAA–206, induction by EGR-1 of the EGR-1
promoter was abolished (Figure 1B, lower). Western blotting
Wang et al
EGR-1 Autoregulation and Histone Modification
539
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
Figure 2. Pathways mediating interleukin
(IL) 1– dependent early growth
response (EGR) 1 expression in human
aortic smooth muscle cells (HASMCs). A,
HASMCs were starved for 24 hours and
stimulated with 10 ng/mL of IL-1 for 30
minutes or treated with different concentrations of PD98059, SB202190, BAY 11
to 7085, or Oridonin for 1 hour before
IL-1 treatment. Total RNA was isolated
and subjected to real-time reverse transcription–polymerase chain reaction
using EGR-1 primers, as described in
the “Methods” section. B, Western blot
analysis was performed using antibodies
to phosphorylated extracellular signal–
regulated kinase (ERK) 1/2 or ERK1/2,
phosphorylated p38 or p38, and EGR-1
or -actin. The asterisk indicates
P⬍0.05.
confirmed that pCB6–EGR-1, but not the empty vector
(pCB6⫹), increased EGR-1 levels in the HASMCs (Figure
1B, lower, inset). Luciferase activity was induced by IL-1 in
SMCs transfected with wild-type pGL3– 450EGR-1-luc (Figure 1C). IL-1 responsiveness of the EGR-1 promoter was
attenuated in cells transfected with the EGR-1 mutant
promoter-reporter construct (Figure 1C). Mutation of SREs in
the promoter also abolished IL-1 inducibility (Figure 1C). In
contrast, mutation of the NF-B element had no effect on
both basal expression and inducibility of EGR-1. Electrophoretic mobility shift analysis using a double-stranded
oligonucleotide spanning the putative EGR-1 binding site
(Oligo EGR-1-225/-187) and recombinant EGR-1 protein revealed the formation of an EGR-1/DNA complex, which was
supershifted by EGR-1 antibodies (Figure 1D). In contrast,
the –208CGG–206 to –208AAA–206 mutation in Oligo EGR-1-225/-187
failed to support the complex formation with recombinant
EGR-1 protein (Figure 1D). ChIP analysis, using conventional PCR, confirmed that IL-1 induced the interaction
of EGR-1 with its own promoter (Figure 1E, top right).
Moreover, real-time ChIP revealed the enrichment of
EGR-1 protein at the EGR-1 promoter in HASMCs exposed to IL-1 (Figure 1E, lower). Therefore, by using a
variety of approaches, these results demonstrate that IL1–inducible EGR-1 binds to, and transactivates, its own
promoter.
IL-1 Stimulates EGR-1 Expression in HASMCs
via the MEK-ERK1/2 Pathway
To identify the signaling pathway(s) mediating IL-1 induction of EGR-1, we first evaluated the influence of IL-1 on
levels of phosphorylated extracellular signal–regulated kinase
(ERK) 1/2, phosphorylated p38, and phosphorylated c-Jun
NH2-terminal kinases 1/2 (JNK1/2) in this cell type. IL-1
activated ERK1/2 within 15 minutes (Supplemental Figure
IIA and B). The cytokine increased levels of phosphorylated
p38, albeit less impressively (Supplemental Figure IIA), and
had no effect on levels of phosphorylated JNK1/2 or phosphorylated c-Jun (Supplemental Figure IIA). Levels of phosphorylated c-Jun were also unaffected (Supplemental Figure
IIA). Because Elk1 is immediately downstream of ERK1/2,
we assessed the effect of IL-1 on Elk1 phosphorylation in
the HASMCs. Levels of phosphorylated Elk1 in nuclear
extracts increased within 15 minutes (Supplemental Figure
IIC). As expected, IL-1 also increased p65 levels in the
nucleus (Supplemental Figure IIC). These results indicate that
ERK1/2-Elk1, p38, and NF-B pathways are activated in
HASMCs in response to IL-1.
Next, HASMCs were treated with 10 ng/mL of IL-1 in
the absence or presence of various concentrations of
PD98059 (MEK-ERK1/2 inhibitor), BAY 11 to 7085, and
Oridonin (2 NF-B inhibitors) or SB202190 (a p38 inhibitor);
then, the EGR-1 mRNA levels were examined. Quantitative
real-time RT-PCR analysis revealed that PD98059, 5 to
540
Arterioscler Thromb Vasc Biol
March 2010
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
Figure 3. Histone H3 modification is associated with inducible early growth response (EGR) 1 transcription. A, Human aortic smooth
muscle cells (HASMCs) were starved for 24 hours and treated with 10 ng/mL of interleukin (IL) 1 for 30 minutes or treated with 5 and
10 mol/L of PD98059 1 hour before IL-1 treatment. Whole cell lysates were prepared in RIPA buffer and separated on 10% sodium
dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE). Western blot analysis was performed using antibodies specific to
phosphorylated mitogen and stress–activated protein kinase (MSK) 1/2, phosphorylated H3, acetyl-H3, total H3, phosphorylated ribosomal S6 kinase (RSK), or -actin, as described in the “Methods” section. B, HASMCs were starved for 24 hours and treated with
2.5 mol/L of suberoylanilide hydroxamic acid (SAHA) for 4 hours before 30-minute incubation with IL-1, 10 ng/mL. Whole cell lysates
were prepared in RIPA buffer and separated on 10% SDS-PAGE. Western blot analysis was performed as described in the “Methods”
section. Total RNA was isolated and subjected to real-time reverse transcription–polymerase chain reaction (RT-PCR) with EGR-1 primers, as described in the “Methods” section. The asterisk indicates P⬍0.05.
10 mol/L, inhibits IL-1–inducible EGR-1 mRNA expression in a dose-dependent manner (Figure 2A, column 2 versus
column 3; and Figure 4), whereas SB202190, 5 to 10 mol/L,
stimulates EGR-1 mRNA expression (Figure 2A, column 2
versus columns 9 and 10). In contrast, BAY 11 to 7085 and
Oridonin had no effect on IL-1–inducible EGR-1 expression
(Figure 2A, column 2 versus columns 5 and 6 or 7 and 8).
PD98059 completely inhibits IL-1–inducible EGR-1 protein
expression (Figure 2B, left), whereas SB202190 stimulates
EGR-1 protein expression (Figure 2B, right). This demonstrates the negative regulatory role of p38 and the positive
regulatory role of ERK1/2 in IL-1–inducible EGR-1 expression. These results indicate that, in HASMCs, IL-1 activates
EGR-1 via the MEK-ERK1/2-Elk1 pathway.
Influence of IL-1–Inducible Histone H3
Modifications on EGR-1 Transcription
Although chromatin remodeling is a key process in transcriptional regulation,29 surprisingly there is nothing known about
the role of histone modifications in the control of EGR-1
transcription. Mitogen and stress–activated protein kinases
(MSK1/2) mediate the phosphorylation of histone H3.30,31
Having demonstrated that PD98059 inhibits IL-1–inducible
ERK1/2 activation (Figure 2B, left) and EGR-1 expression in
HASMCs (Figure 2A and B), we next investigated the effect
of MEK-ERK1/2 inhibition on levels of phosphorylated
MSK1 and MSK2 in HASMCs treated with IL-1. MSK1
and MSK2 were phosphorylated within 30 minutes of exposure to IL-1 (Figure 3A). PD98059 suppressed MSK1 and
MSK2 phosphorylation in a dose-dependent manner (Figure
3A). H3 phosphorylation and acetylation also increased in
response to IL-1, as did ribosomal S6 kinase phosphorylation32; each was reduced by MEK-ERK1/2 inhibition (Figure
3A), demonstrating the importance of ERK1/2-MSK1/2 signaling in IL-1–mediated histone H3 phosphorylation and
acetylation. To establish a causal relationship between histone H3 modification and EGR-1 transcription, we then
determined the levels of phosphorylated, acetylated, and
methylated H3 in HASMCs treated with IL-1 over time.
Western blot analysis indicated a rapid and transient induction in both phosphorylated and acetylated H3 (Supplemental
Figure 3A and B) but not methylated H3 (Supplemental
Figure IIIA). We further explored the role of H3 acetylation
in IL-1–mediated EGR-1 transcription in HASMCs using
Wang et al
EGR-1 Autoregulation and Histone Modification
541
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
Figure 4. Inhibition of histone H3 modification by either short interfering RNA
(siRNA) or inhibitor attenuates interleukin
(IL) 1–induced early growth response
(EGR) 1 transcription. Human aortic
smooth muscle cells (HASMCs) were
transfected with 0.05 mol/L of mitogen
and stress–activated protein kinase
(MSK) 1 siRNA, MSK2 siRNA, or control
siRNA for 24 hours. Alternatively, 18
hours after starvation, the cells were
treated with Garcinol for 6 hours. The
cells were then incubated with IL-1, 10
ng/mL, for 30 minutes. A, Western blot
analysis of whole-cell lysates for phosphorylated H3, acetyl H3, total H3, or
-actin (left). The effect of MSK1 and
MSK2 siRNA on levels of MSK1 and
MSK2 is shown (right). B, Real-time
reverse transcription–polymerase chain
reaction (RT-PCR) with total RNA and
EGR-1 primers. C, EGR-1 and p300
interact in cells exposed to IL-1.
Extracts from untreated or IL-1–treated
HASMCs were incubated with EGR-1
antibodies, pulled down with protein A/G
beads, and immunoblotted for p300 or
EGR-1. An immunoblot for p300 is also
shown; this serves as a loading control
for immunoprecipitated p300 (upper).
Alternatively, recombinant p300,
0.6 g, was incubated with glutathione
S-transferase–EGR-1, 1 or 2 g, pulled
down with glutathione-agarose beads,
then blotted with p300 antibodies (middle), or pulled down with p300 antibodies
and protein A/G, and blotted with glutathione S-transferase antibodies (lower).
D, Real-time chromatin immunoprecipitation (ChIP) with p300 antibodies and
amplification of the EGR-1 promoter. E,
Transient transfection analysis in rat aortic smooth muscle cells (RASMCs) transfected with pCB6 –EGR-1 or pCMVp300. Luciferase assays were performed
as described in the “Methods” section. F,
Enrichment of phosphorylated and acetylated H3 at the EGR-1 promoter in
response to IL-1. Chromatin from
HASMCs stimulated with 10 ng/mL of
IL-1 was sonicated and immunoprecipitated using ChIP-grade antibodies to acetyl-H3, phosphorylated H3, total H3, and EGR-1, and
DNA was amplified by real-time ChIP. Enrichment was normalized to that obtained with control rabbit antibody and expressed as fold
increase over that measured at a control region in the LMO2 proximal promoter. The asterisk indicates P⬍0.05.
the histone deacetylase inhibitor, suberoylanilide hydroxamic
acid (SAHA).33 SAHA caused a dramatic increase in the
levels of acetylated H3 (Figure 3B). Interestingly, it also
increased the levels of phosphorylated H3 (Figure 3B).
Real-time RT-PCR revealed that EGR-1 expression induced by IL-1 was further increased by SAHA (Figure
3B). This indicates the permissive role of H3 acetylation in
IL-1–inducible EGR-1 expression.
To further explore the role of histone acetylation and
phosphorylation in IL-1–inducible EGR-1 transcription, we
treated HASMCs with Garcinol, an inhibitor of H3 acetyltransferases,34 or transfected the cells with short interfering
RNA (siRNA) targeting MSK1 and MSK2. H3 phosphorylation and acetylation induced by IL-1 were reduced by
MSK1 and MSK2 siRNA (Figure 4A). H3 acetylation was
inhibited by Garcinol, with less effect on H3 phosphorylation
(Figure 4A). Real-time RT-PCR revealed that IL-1–inducible EGR-1 expression is indeed reliant on H3 acetylation and
phosphorylation (Figure 4B). Co-IP analysis revealed the
formation of an IL-1–inducible complex between EGR-1
and the histone acetyltransferase p300 in HASMC extracts
(Figure 4C, upper). EGR-1 and p300 also interact as recombinant proteins (Figure 4C, lower). Real-time ChIP showed
the enrichment of p300 at the EGR-1 promoter in response to
IL-1 (Figure 4D), whereas transient transfection analysis
demonstrated that p300 stimulates EGR-1 promoter activity
through the EGR-1 binding site (Figure 4E). Moreover, both
phosphorylated and acetylated H3 are enriched, together with
EGR-1 itself, at the EGR-1 promoter in response to IL-1
stimulation (Figure 4F). Figure 3A and Supplemental Figure
IIIA and Supplemental Figure IVA indicate that although
IL-1 affects change in H3 acetylation and phosphorylation,
542
Arterioscler Thromb Vasc Biol
March 2010
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
Figure 5. Early growth response (EGR) 1 is enriched at the promoters of EGR-1–dependent genes in a mitogen and stress–activated
protein kinase (MSK)– and histone H3 acetyltransferase–dependent
manner. Twenty-four hours after starvation, human aortic smooth
muscle cells (HASMCs) were exposed to 10 ng/mL of interleukin (IL)
1 for 30 minutes. Chromatin immunoprecipitation (ChIP) and realtime ChIP were performed with EGR-1 antibodies or control IgG,
amplifying the plasminogen activator inhibitor (PAI) 1 promoter (A) or
the TF promoter (B), as described in the “Methods”. C, Twenty-four
hours after starvation, HASMCs were exposed to 10 mol/L of
PD98059 for 1 hour and 20 mol/L of Garcinol for 4 hours, before
treatment with 10 ng/mL of IL-1 for 30 minutes. Real-time ChIP was
performed with EGR-1 antibodies or IgG, as described in the “Methods” section. D, EGR-1 short interfering RNA (siRNA) inhibits IL-1–
induced EGR-1, PAI-1, and tissue factor expression. Serum-arrested
HASMCs were transfected with 0.1 mol/L of siRNA using FuGENE6
and harvested 30 minutes or 2 hours after the addition of 10 ng/mL
of IL-1; Western blotting was then performed. The asterisk indicates
P⬍0.05.
there is no change in total H3 levels in cells exposed to the
cytokine. We used an antibody against total H3 (nonacetylated and nonphosphorylated forms) as a control in the ChIP
assay to show that H3 acetylation and phosphorylation are
increased at the EGR-1 promoter in response to IL-1. Figure
4F demonstrates that total H3 occupancy of the EGR-1
promoter does not differ between untreated and IL-1–
treated samples. Taken together, these data indicate that H3
Wang et al
EGR-1 Autoregulation and Histone Modification
543
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
Figure 6. Model of interleukin (IL) 1–inducible early growth response (EGR) 1
transcription. The proinflammatory cytokine, IL-1, binds to its cell surface
receptor and activates the mitogen and
stress–activated protein kinase (MEK)–
extracellular signal–regulated kinase
(ERK) 1/2 pathway. Mitogen and stress–
activated protein kinase (MSK) 1/2 lies
downstream of ERK1/2. MSK1/2 phosphorylation causes the enrichment of
phosphorylated H3 at the EGR-1 promoter, and the phosphorylation of ternary complex factors, such as Elk1,
which interact with serum response factor (SRF) at serum response elements in
the EGR-1 promoter. MSK1/2 is also
required in the recruitment of H3 acetyltransferases, such as p300, to the
EGR-1 promoter. This increases EGR-1
transcription and protein synthesis,
allowing EGR-1 to enter the nucleus,
bind its own promoter and associated
factors, such as p300, and transactivate
itself and other genes, such as TF and
plasminogen activator inhibitor (PAI) 1.
acetylation and phosphorylation, and EGR-1, are increased at
the EGR-1 promoter in response to IL-1.
EGR-1 Is Enriched at the Promoters of
EGR-1–Dependent Genes in an MSK- and Histone
H3 Acetyltransferase–Dependent Manner
To determine whether IL-1–inducible EGR-1 functionally
regulates transcription of inflammatory mediators, we treated
HASMCs with a course of IL-1, and the mRNA levels of
tissue factor and PAI-1 were examined. Quantitative realtime RT-PCR showed a time-dependent induction in both TF
and PAI-1 in HASMCs exposed to IL-1 (Supplemental
Figure IV), consistent with the time-course induction of
EGR-1 expression (Supplemental Figure IA). Both conventional
and real-time ChIP revealed the enrichment of EGR-1 at the
promoter region of TF and PAI-1 in response to IL-1 (Figure
5A and B), which is suppressed by PD98059, Garcinol, and
MSK1/2 siRNA (Figure 5C). EGR-1 siRNA blocked IL-1–
inducible EGR-1, PAI-1, and tissue factor expression (Figure
5D). Thus, EGR-1 induced by IL-1 binds to and induces TF
and PAI-1 expression, in an MEK/ERK, MSK, and H3 acetyltransferase– dependent manner (Figure 6).
Discussion
The proinflammatory cytokine IL-1 has long been implicated in the pathogenesis of cardiovascular disease. IL-1
and its receptor are expressed in atheromatous tissue; IL-1
activates macrophages, stimulates SMC proliferation, and
upregulates endothelial adhesion molecule expression.35 The
zinc finger transcription factor EGR-1 is a “master regulator”
in that it controls the expression of many pathophysiologically relevant genes.5,17 EGR-1– dependent genes have been
strongly implicated in the initiation and progression of
atherosclerosis and postangioplasty restenosis.36 –38 Understanding the mechanisms regulating the inducible expression
of EGR-1 in vascular cells by IL-1 would provide an
important backdrop for future interventional approaches targeting this transcription factor.
To our knowledge, this study has revealed, for the first
time, that EGR-1 regulates its own transcription and provides
important clues on the role of chromatin remodeling and
histone modification in EGR-1 transcription. We show that
IL-1 controls EGR-1 expression via the MEK-ERK1/2 and
MSK pathway. EGR-1 binds to and transactivates its own
gene, and IL-1 induction of EGR-1 transcription involves
histone H3 phosphorylation and acetylation. In contrast, p38
is a negative regulator of EGR-1, because SB202190, a
selective p38 inhibitor, dose dependently enhances IL-1–
inducible EGR-1 transcription in HASMCs. The opposing
influence of different mitogen-activated protein kinases on
EGR-1 transcription demonstrates the tight control of this
transcription factor at the level of signaling and transcription.
Chromatin structure and remodeling can have a profound
influence on gene transcription; however, this influence has
been unexplored because of the EGR-1 promoter until the
present study. DNA methylation and histone modifications
can lead to recruitment of protein complexes that regulate
transcription. Our findings demonstrate that histone H3 modifications, acetylation, and phosphorylation, in particular, are
associated with IL-1–induced EGR-1 transcription in
HASMCs. This study investigated the role of histone modification in the EGR-1 promoter, with a focus on H3 (rather
than H4), given that H3 is the most extensively modified of
the 5 known histones (H1-H5) and that it builds on prior
work39 showing that EGR-1 transcription can be repressed by
histone demethylase lysine demethylase 5b, which demethylates lysine 4 of H3. IL-1–inducible phosphorylation and
acetylation of histone H3 are attenuated by PD98059, consistent with the stimulatory effect of IL-1 on phosphorylated
ERK1/2, phosphorylated MSK1/2, and EGR-1 mRNA. IL-1
544
Arterioscler Thromb Vasc Biol
March 2010
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
causes a rapid and transient induction in acetylated and
phosphorylated histone H3, in line with the time course of
IL-1–induced EGR-1 mRNA expression. Furthermore, histone deacetylase inhibition enhances EGR-1 transcription in
HASMCs treated with IL-1, clearly indicating a role for
histone acetylation in EGR-1 expression. Conversely, suppression of histone modification reduced cytokine-inducible
EGR-1 transcription. Although IL-1–inducible EGR-1 expression involves H3 acetylation and phosphorylation, Figure
3B and Figures 4A and B do not suggest absolute specificity
of H3 acetylation on the EGR-1 gene. SAHA, in the absence
of cytokine, caused a dramatic increase in acetylated H3 and
EGR-1 mRNA levels (Figure 3B). SAHA potentiated IL-1–
inducible EGR-1 expression (Figure 3B, lower); however,
SAHA caused little or no change in the total levels of H3
acetylation (Figure 3B, upper). There are several possible
explanations for this. First, it is possible that acetylated H3
was totally recovered from many gene loci, not just those of
EGR-1. Second, there may be the involvement of transaction
effects, additional to cis-action, induced by H3 acetylation.
Alternatively, saturating acetylated H3 levels induced by
SAHA would render further increases difficult to detect.
Nevertheless, acetylated H3 and phosphorylated H3 were
markedly enriched at the EGR-1 promoter by real-time ChIP,
further confirming the role of histone modifications in EGR-1
transcription. Histone H3 acetyltransferase and MSK1/2, as
well as MEK-ERK1/2, are needed for EGR-1 occupancy of
the PAI-1 and TF promoters. EGR-1 physically and functionally interacts with p300 and interacts with its own promoter.
Thus, these findings demonstrate that histone H3 modifications are crucial for the transcriptional activation of EGR-1 in
cells exposed to IL-1, and that EGR-1 controls transcription
of its own gene. EGR-1 control of EGR-1 transcription
suggests the existence of a “self-amplification” cascade in
injured arteries, involving IL-1– driven chromatin remodeling, EGR-1 and EGR-1– dependent gene expression, a shift to
a synthetic cellular and proinflammatory phenotype, and
clinical manifestation of vascular disease.
Acknowledgments
We thank Sudha Rao, PhD (John Curtin School of Medical Research,
Australia National University, Canberra) for her helpful advice.
Sources of Funding
This study was supported by grants from the National Health and
Medical Research Council and the National Heart Foundation of
Australia. Dr Khachigian is an Australia Fellow of the NHMRC.
Disclosures
None.
References
1. Muthukkumar S, Nair P, Sells SF, Maddiwar NG, Jacob RJ, Rangnekar
VM. Role of EGR-1 in thapsigargin-inducible apoptosis in the melanoma
cell line A375–C6. Mol Cell Biol. 1995;15:6262– 6272.
2. Nguyen HQ, Hoffman-Liebermann B, Liebermann DA. The zinc finger
transcription factor Egr-1 is essential for and restricts differentiation
along the macrophage lineage. Cell. 1993;72:197–209.
3. Santiago FS, Atkins DA, Khachigian LM. Vascular smooth muscle cell
proliferation and regrowth after injury in vitro is dependent upon NGFIA/Egr-1. Am J Pathol. 1999;155:897–905.
4. Sukhatme VP, Cao X, Chang LL, Tsai-Morris C-H, Stamenkovich D,
Ferreira PCP, Cohen DR, Edwards SA, Shows TB, Curran T, Le Beau
MM, Adamson ED. A zinc-finger encoding gene coregulated with c-Fos
during growth and differentiation and after depolarization. Cell. 1988;53:
37– 43.
5. Khachigian LM, Lindner V, Williams AJ, Collins T. Egr-1-induced
endothelial gene expression: a common theme in vascular injury. Science.
1996;271:1427–1431.
6. Ahmed MM. Regulation of radiation-induced apoptosis by early growth
response-1 gene in solid tumors. Curr Cancer Drug Targets. 2004;4:
43–52.
7. Gashler A, Sukhatme V. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucl Acid
Res. 1995;50:191–224.
8. Gashler AL, Swaminathan S, Sukhatme VP. A novel repression module,
an extensive activation domain, and a bipartite nuclear localization signal
defined in the immediate-early transcription factor Egr-1. Mol Cell Biol.
1993;13:4556 – 4571.
9. Carman JA, Monroe JG. The EGR1 protein contains a discrete transcriptional regulatory domain whose deletion results in a truncated protein that
blocks EGR1-induced transcription. DNA Cell Biol. 1995;14:581–589.
10. Khachigian LM, Williams AJ, Collins T. Interplay of Sp1 and Egr-1 in
the proximal PDGF-A promoter in cultured vascular endothelial cells.
J Biol Chem. 1995;270:27679 –27686.
11. Hasan RN, Schafer AI. Hemin upregulates Egr-1 expression in vascular
smooth muscle cells via reactive oxygen species ERK-1/2-Elk-1 and
NF-kappaB. Circ Res. 2008;102:42–50.
12. Rafty LA, Khachigian LM. Zinc finger transcription factors mediate high
constitutive PDGF-B expression in smooth muscle cells derived from
aortae of newborn rats. J Biol Chem. 1998;273:5758 –5764.
13. Sumpio BE, Du W, Galagher G, Khachigian LM, Collins T, Gimbrone
MA Jr, Resnick N. Regulation of PDGF-B in endothelial cells exposed to
cyclic strain. Arterioscler Thromb Vasc Biol. 1998;18:349 –355.
14. Aicher WK, Sakamoto KM, Hack A, Eibel H. Analysis of functional
elements in the human Egr-1 gene promoter. Rheumatol Int. 1999;18:
207–214.
15. Hoffmann E, Ashouri J, Wolter S, Doerrie A, Dittrich-Breiholz O,
Schneider H, Wagner EF, Troppmair J, Mackman N, Kracht M. Transcriptional regulation of EGR-1 by the interleukin-1-JNK-MKK7-c-Jun
pathway. J Biol Chem. 2008;283:12120 –12128.
16. Khachigian LM, Collins T. Early growth response factor 1: a pleiotropic
mediator of inducible gene expression. J Mol Med. 1998;76:613– 616.
17. Khachigian LM. Early growth response-1 in cardiovascular pathobiology.
Circ Res. 2006;98:186 –191.
18. Santiago FS, Lowe HC, Day FL, Chesterman CN, Khachigian LM. Early
growth response factor-1 induction by injury is triggered by release and
paracrine activation by fibroblast growth factor-2. Am J Pathol. 1999;
154:937–944.
19. Delbridge GJ, Khachigian LM. FGF-1-induced PDGF A-chain gene
expression in vascular endothelial cells involves transcriptional activation
by Egr-1. Circ Res. 1997;81:282–288.
20. Wang B, Khachigian LM, Esau L, Birrer MJ, Zhao X, Parker MI,
Hendricks DT. A key role for early growth response-1 and nuclear
factor-kappaB in mediating and maintaining GRO/CXCR2 proliferative
signaling in esophageal cancer. Mol Cancer Res. 2009;7:755–764.
21. Eid MA, Kumar MV, Iczkowski KA, Bostwick DG, Tindall DJ.
Expression of early growth response genes in human prostate cancer.
Cancer Res. 1998;58:2461–2468.
22. Svaren J, Ehrig T, Abdulkadir SA, Ehrengruber MU, Watson MA, Milbrandt J. EGR1 target genes in prostate carcinoma cells identified by
microarray analysis. J Biol Chem. 2000;275:38524 –38531.
23. Thigpen AE, Cala KM, Guileyardo JM, Molberg KH, McConnell JD,
Russell DW. Increased expression of early growth response-1 messenger
ribonucleic acid in prostatic adenocarcinoma. J Urol. 1996;155:975–981.
24. Baron V, Duss S, Rhim J, Mercola D. Antisense to the early growth
response-1 gene (Egr-1) inhibits prostate tumor development in TRAMP
mice. Ann N Y Acad Sci. 2003;1002:197–216.
25. Khachigian LM. Catalytic DNA as therapeutic agents and molecular tools
to dissect biological function. J Clin Invest. 2000;106:1189 –1195.
26. Fahmy RG, Dass CR, Sun LQ, Chesterman CN, Khachigian LM. Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nature Med. 2003;9:1026 –1032.
27. Mitchell A, Dass CR, Sun L-Q, Khachigian LM. Inhibition of human
breast carcinoma proliferation, migration, chemoinvasion and solid tumor
Wang et al
28.
29.
30.
31.
32.
33.
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
growth by DNAzymes targeting the zinc finger transcription factor
EGR-1. Nucleic Acids Res. 2004;32:3065–3069.
Fahmy R, Waldman A, Zhang G, Mitchell A, Tedla N, Cai H, Chesterman CN, Geczy CR, Perry MA, Khachigian LM. Suppression of
vascular permeability and inflammation by targeting of the transcription
factor c-Jun. Nature Biotech. 2006;24:856 – 863.
Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407– 412.
Yu M, Blomstrand E, Chibalin AV, Krook A, Zierath JR. Marathon
running increases ERK1/2 and p38 MAP kinase signalling to downstream
targets in human skeletal muscle. J Physiol. 2001;536:273–282.
Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH,
Hazzalin CA, Mahadevan LC, Arthur JS. MSK2 and MSK1 mediate the
mitogen- and stress-induced phosphorylation of histone H3 and HMG-14.
EMBO J. 2003;22:2788 –2797.
Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S,
Hanauer A, Allis CD. Requirement of Rsk-2 for epidermal growth factoractivated phosphorylation of histone H3. Science. 1999;285:886 – 891.
Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E,
Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh
JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide
hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor
deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci
U S A. 2003;100:2041–2046.
EGR-1 Autoregulation and Histone Modification
545
34. Sailhamer EA, Li Y, Smith EJ, Shuja F, Shults C, Liu B, Soupir C,
deMoya M, Velmahos G, Alam HB. Acetylation: a novel method for
modulation of the immune response following trauma/hemorrhage and
inflammatory second hit in animals and humans. Surgery. 2008;144:
204 –216.
35. Apostolakis S, Vogiatzi K, Krambovitis E, Spandidos DA. IL-1 cytokines in cardiovascular disease: diagnostic, prognostic and therapeutic implications. Cardiovasc Hematol Agents Med Chem. 2008;6:
150 –158.
36. McCaffrey TA, Fu C, Du C, Eskinar S, Kent KC, Bush H Jr, Kreiger K,
Rosengart T, Cybulsky MI, Silverman ES, Collins T. High-level
expression of Egr-1 and Egr-1-inducible genes in mouse and human
atherosclerosis. J Clin Invest. 2000;105:653– 662.
37. Lowe HC, Fahmy RG, Kavurma MM, Baker A, Chesterman CN,
Khachigian LM. Catalytic oligodeoxynucleotides define a key regulatory
role for early growth response factor-1 in the porcine model of coronary
in-stent restenosis. Circ Res. 2001;89:670 – 677.
38. Lowe HC, Chesterman CN, Khachigian LM. Catalytic antisense DNA
molecules targeting Egr-1 inhibit neointima formation following permanent ligation of rat common carotid arteries. Thromb Haemost. 2002;
87:134 –140.
39. Dey BK, Stalker L, Schnerch A, Bhatia M, Taylor-Papidimitriou J,
Wynder C. The histone demethylase KDM5b/JARID1b plays a role in
cell fate decisions by blocking terminal differentiation. Mol Cell Biol.
2008;28:5312–5327.
Downloaded from http://atvb.ahajournals.org/ by guest on October 1, 2016
Phosphorylation and Acetylation of Histone H3 and Autoregulation by Early Growth
Response 1 Mediate Interleukin 1 β Induction of Early Growth Response 1 Transcription
Bo Wang, Jinbiao Chen, Fernando S. Santiago, Mary Janes, Mary M. Kavurma, Beng H. Chong,
John E. Pimanda and Levon M. Khachigian
Arterioscler Thromb Vasc Biol. 2010;30:536-545; originally published online December 17,
2009;
doi: 10.1161/ATVBAHA.109.193821
Arteriosclerosis, Thrombosis, and Vascular Biology is published by the American Heart Association, 7272
Greenville Avenue, Dallas, TX 75231
Copyright © 2009 American Heart Association, Inc. All rights reserved.
Print ISSN: 1079-5642. Online ISSN: 1524-4636
The online version of this article, along with updated information and services, is located on the
World Wide Web at:
http://atvb.ahajournals.org/content/30/3/536
Data Supplement (unedited) at:
http://atvb.ahajournals.org/content/suppl/2010/02/18/ATVBAHA.109.193821.DC1.html
Permissions: Requests for permissions to reproduce figures, tables, or portions of articles originally published
in Arteriosclerosis, Thrombosis, and Vascular Biology can be obtained via RightsLink, a service of the
Copyright Clearance Center, not the Editorial Office. Once the online version of the published article for
which permission is being requested is located, click Request Permissions in the middle column of the Web
page under Services. Further information about this process is available in the Permissions and Rights
Question and Answer document.
Reprints: Information about reprints can be found online at:
http://www.lww.com/reprints
Subscriptions: Information about subscribing to Arteriosclerosis, Thrombosis, and Vascular Biology is online
at:
http://atvb.ahajournals.org//subscriptions/
Supplementary Material; ATVB/2009/193821/R3, page 1
Phosphorylation and Acetylation of Histone H3 and Autoregulation by EGR-1 Mediate IL-1beta
Induction of EGR-1 Transcription
Bo Wang, Jinbiao Chen, Fernando S. Santiago, Mary Janes, Mary M. Kavurma, Beng H. Chong, John
E. Pimanda, Levon M. Khachigian
Materials and Methods
Generation of promoter reporter constructs. To evaluate the contribution of transcription factor
binding sites in the human EGR-1 promoter, the 506 bp Not I-Xho I fragment of the human EGR-1
promoter was amplified by PCR and cloned in pGL3 Basic plasmid (Promega) using the following
primers, -450 forward: 5’-ATA TAA GGT ACC CAG CTG CGA C-3’, +56 reverse: 5’-ATA TAA
CTC GAG GAA GCT GGC TG-3’. The murine Egr-1 expression vector (pCB6-Egr-1) was a gift from
Dr Vikas Sukhatme (Beth Israel Deaconess Hospital, Boston). Sequence identity of constructs was
confirmed by automated DNA sequencing. pRL-null was purchased from Promega.
Site-directed mutagenesis. Site-directed mutation of putative transcription factor binding sites was
carried out using QuickChange II Site-Directed Mutagenesis Kit (Stratagene) as the manufacturer’s
instruction. The following primers were utilized to generate site-directed mutants, SRE1mt forward: 5’GCT TCC TGC TTC CCA TGG GTG GCC ATG TAC GTC ACG-3’, SRE1mt reverse: 5’-CGT GAC
GTA CAT GGC CAC CCA TGG GAA GCA GGA AGC-3’; SRE2mt forward: 5’-GGC CGG TCC
TGC CAT GGG AGG GCT TCC TGC-3’, SRE2mt reverse: 5’-GCA GGA AGC CCT CCC ATG
GCA GGA CCG GCC-3’; EGR-1mt forward: 5’-CCT GGG ATG CGG GCG AAA GCG CGG GCC
CTA GG-3’, EGR-1mt reverse: 5’-CCT AGG GCC CGC GCT TTC GCC CGC ATC CCA GG-3’;
Supplementary Material; ATVB/2009/193821/R3, page 2
NF-kappaBmt forward: 5’-GGG GCA ACG CGG GCC TTC CGG AGC TGC GC-3’, NF-kappaBmt
reverse: 5’-GCG CAG CTC CGG AAG GCC CGC GTT GCC CC-3’. All mutants were confirmed by
automated DNA sequencing.
Transient transfection and luciferase assays. Human and rat aortic SMCs grown to 70-80%
confluence in 6-well plate in Waymouth medium containing 10% FBS were transiently transfected
with 0.5 μg reporter plasmid, 1 μg pCB6-Egr-1 plasmid, 1 μg pCMVβ-p300 and 0.1 μg pRL-null
plasmid using FuGENE6 (Roche) as manufacturer’s instruction (ratio of DNA:FuGENE6 is 1:3). The
total amount of transfected DNA was kept constant using the corresponding empty vector. 24 h after
co-transfection, cells were incubated in serum-free medium for another 24 h, and/or treated with 10
ng/mL IL-1beta for the indicated times, the relative luciferase activity was measured by DualLuciferase Reporter Assay system (Promega) using a TD-20/20 luminometer (Turner Designs) and
with Firefly luciferase data normalized to Renilla.
Real-time quantitative reverse transcription-PCR. Total RNA was prepared from HASMC treated
with 10 ng/mL IL-1beta for the indicated times using TRIzol (Invitrogen) according to the
manufacturer’s instructions and quantified by UV absorbance at 260 nm in a GeneQuant Pro
spectrophotometer (Amersham Biosciences). cDNA was synthesized using 2 μg total RNA and 25
μg/mL oligo (dT)12-18 primer using the SuperScript II reverse transcriptase (Invitrogen). The target
primers for amplifying EGR-1 were forward primer: 5’-AGC AGC ACC TTC AAC CCT CA-3’ and
reverse primer: 5’-CAG CAC CTT CTC GTT GTT CAG A-3’1. For amplifying PAI-1, forward primer:
5’-GTG ATT CCA TCA ATG CAT GGG TTA-3’ and reverse primer: 5’-TTG GAA CCG TGA TTT
CCA CAG A-3’2. For amplifying TF, forward primer: 5’-TCC CCA GAG TTC ACA CCT TAC C-3’
and reverse primer: 5’-TGA CCA CAA ATA CCA CAG CTC C-3’3. Each 20 μL of reaction mixture
Supplementary Material; ATVB/2009/193821/R3, page 3
for real-time RT-PCR contained 1 mmol/L MgCl2, 0.15 μmol/L of each primer and 1 μL of SYBR
Green PCR Master Mix (Applied Biosystems). Real-time quantitative RT-PCR analysis was carried out
with Corbett Research RG-6000 (Corbett Life Science, Australia). Glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) was used as the internal control to standardize and to test the RNA integrity
with sequence for forward primer: 5’-GAA GGC TGG GGC TCA TTT-3’ and for reverse primer: 5’CAG GAG GCA TTG CTG ATG AT-3’4. All experiments for real-time RT-PCR were performed in
triplicate and data was analysed using the comparative Ct method5. Results are shown as fold induction
of mRNA.
Immunocytofluorescence. To detect the expression and subcellular distribution of EGR-1, phosphop44/42 MAPK, and phospho-H3 in HASMC treated with 10 ng/mL IL-1beta, the cells were cultured on
coverslips, fixed and permeabilized with methanol and 4% paraformaldehyde, and incubated with
1:400 dilution of rabbit monoclonal antibody to EGR-1 (Cell Signalling Technology) or 1:200 dilution
of rabbit monoclonal antibody to phospho-p44/42 MAPK, or 1:100 dilution of rabbit polyclonal
antibody to phospho-histone H3 (Cell Signalling Technology) in blocking buffer at 4°C overnight.
After incubation with primary antibody, cells were washed five times in PBS (pH 7.4) and incubated
with 1:100 dilution of FITC-conjugated goat anti-rabbit antibody (Zymed) for 2 h at room temperature.
After five-time washes in PBS, nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI; Sigma).
Fluorescence was observed under 600x on an inverted microscope (Olympus Digital DP70).
Western blot analysis. Cells were rinsed three times with ice-cold PBS and scraped off the plate in
RIPA buffer (150 mmol/L NaCl, 1% Triton X-100, 0.1% SDS, 25 mmol/L Tris-Cl (pH 7.5), 1%
sodium deoxycholate, 1mmol/L Na3VO4, 20 μg/mL pepstatin, 5 μg/mL aprotinin and 1 mmol/L
PMSF), pipetted up and down 8 times, and centrifuged for 10 min at 13,000g. Nuclear lysates were
Supplementary Material; ATVB/2009/193821/R3, page 4
prepared as previously described6. Protein concentrations of the lysates were determined using the
BCA Protein Assay Kit (Pierce). 20-40 μg protein per sample was electrophoresed on 10% SDS-PAGE
and electrophoretically transferred to PVDF membranes (Millipore) at 4°C for 1.5 h. Blots were
incubated for 1 h with 5% non-fat dry milk or 5% BSA to block non-specific binding sites and then
incubated with either 1:2,000 dilution of rabbit monoclonal antibody against Phospho-p44/42 MAPK
or 1:2,000 dilution of mouse monoclonal antibody to p42 MAP kinase or 1:1,000 dilution of rabbit
monoclonal antibody to p-p38 or 1:1,000 dilution of rabbit polyclonal antibody to p38 or 1:1,000
dilution of rabbit polyclonal antibody to phospho-MSK1 or 1:1000 dilution of mouse monoclonal
antibody to phospho-Elk1 or 1:1000 dilution of rabbit polyclonal antibody to Elk1 or 1:1,000 dilution
of rabbit monoclonal antibody to phospho-SAPK/JNK or 1:1,000 dilution of rabbit monoclonal
antibody to SAPK/JNK or 1:1,000 dilution of monoclonal antibody to EGR1 (Cell Signaling
Technology) or 0.5 μg/mL anti-phospho-MSK2/RSKB antibody (R&D Systems) or 1:2,000 dilution of
rabbit polyclonal antibody to MSK2/RSKB (Abcam) or 1:1,000 dilution of rabbit polyclonal antibody
to MSK1 or 1:500 dilution of mouse monoclonal antibody to p65 or 1:500 dilution of mouse
monoclonal antibody to p-c-Jun (sc-822, Santa Cruz Biotechnology) or 1:1,000 dilution of rabbit
monoclonal antibody to c-Jun (Abcam) or 1:2,000 dilution of rabbit monoclonal antibody to phosphohistone H3 (Ser10, Upstate) or 1:500 dilution of rabbit polyclonal antibody to acetyl-Histone H3
(Lys23, Cell Signaling) or 1:1,000 dilution of rabbit polyclonal antibody to di-Methyl-Histone H3
(Lys4, Cell Signaling Technology) or 1:1,000 dilution of rabbit polyclonal antibody to phospho-RSK
or RSK (Cell Signaling Technology) or 2 μg/mL monoclonal antibodies to p300 (Upstate Biolabs) at
4°C overnight. Immunoreactivity was detected using peroxidase-conjugated antibody and visualized by
enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, Pierce). Blots
were stripped and reprobing with antibody to beta-actin (Sigma).
Supplementary Material; ATVB/2009/193821/R3, page 5
siRNA. EGR-1 siRNA used in this study was SMARTpool L-006526-00-0010 (Dharmacon). MSK1
siRNA (s17691) and MSK2 siRNA (s17139) were obtained from Ambion. ON-TargetPlus Control
Pool D-001810-10-20 (Dharmacon) was used as control siRNA.
Phosphorylation and acetylation of histone H3. HASMC (at 80% confluence) were transfected with
50 nmol/L MSK1 or MSK2 siRNA using FuGENE6 (Roche) for 24 h, followed by starvation in serumfree medium for 24 h. Cells were then treated with 10 μmol/L or 20 μmol/L Garcinol (BIOMOL) for 6
h prior to IL-1beta exposure. After preparation of total cellular RNA and protein samples, the levels of
EGR-1 mRNA, phosphorylated-H3 and acetylated-H3 were determined by real-time RT-PCR and
Western blot analysis.
Electrophoretic mobility shift assay (EMSA). EMSA was carried out as described previously 7.
Briefly, 20 μL of reaction contained 1 or 2 µg of Egr-1-GST protein (mouse Egr-1 from pGEX-2Tmurine Egr-1 and purified in-house using glutathione-agarose beads (Sigma), 1 μg poly(dI/dC)poly(dI/dC) (Sigma), 1 μg salmon sperm DNA (Sigma), 10 mmol/L Tris-Cl, pH 7.5, 50 mmol/L NaCl,
1 mmol/L EDTA, 1 mmol/L dithiothreitol, 2.5% glycerol, 0.001% NP-40, 1 μg/μL BSA, and 150,000
cpm of 32P-labeled probe (EGR-1 EMSA1: 5’-CGC CTG GGA TGC GGG CGC GGG CGC GGG
CCC TAG GGT GCA-3’, and EGR-1 EMSA2: 5’-TGC ACC CTA GGG CCC GCG CCC GCG CCC
GCA TCC CAG GCG-3’; EGR-1 EMSA-mt1: 5’-CGC CTG GGA TGC GGG CGA AAG CGC GGG
CCC TAG GGT GCA-3’, AND EGR-1 EMSA-mt2: 5’- TGC ACC CTA GGG CCC GCG CTT TCG
CCC GCA TCC CAG GCG-3’). DNA/protein complexes were resolved electrophoretically on 6%
non-denaturing polyacrylamide gels. Gels were dried and exposed to X-ray film at -20°C.
Supplementary Material; ATVB/2009/193821/R3, page 6
Co-immunoprecipitation (Co-IP). Co-IP was performed with cell extracts as previously described8
using 50µg nuclear extract from IL-1beta-treated and untreated HASMC. To demonstrate an interaction
between recombinant Egr-1 and p300, a fixed amount (0.6 µg) of purified recombinant p300 (Active
Motif) was combined with 1 or 2 µg GST-Egr-1 and precipitated with glutathione-agarose beads
(Sigma) then immunoblotted with p300 antibodies (sc-585, Santa Cruz Biotechnology). Alternatively,
pull down was achieved with p300 antibodies and Protein A/G (GE Healthcare) prior to blotting with
GST antibodies (GE Healthcare). Samples were loaded into 6% SDS-PAGE gels and transferred into
PVDF membrane, and visualised by chemiluminescence.
ChIP. Quantitative ChIP assays were performed on primary human vascular SMCs as detailed
elsewhere9. Briefly cells were treated with 0.4% formaldehyde and the cross-linked chromatin
retrieved by nuclei isolation and lysis. The chromatin was sonicated to ~300 bp, pre-cleared with
rabbit serum and immunoprecipitated with anti–Egr-1 (sc-110) from Santa Cruz Biotechnology, antiacetyl H3 (06-599), anti-phospho H3 (05-817) and anti-H3 (06-755) antibodies from Upstate (Lake
Placid, NY). Enrichments were measured by real-time PCR using Sybr Green (Stratagene) as
previously described9. The levels of enrichment were normalized to that obtained with a control rabbit
antibody or total input and were calculated as a fold increase over that measured at a control region, the
LMO2 proximal promoter. The following primer pairs were used; EGR-1F: 5’-TTA TTT GGG CAG
CAC CTT ATT TG-3’; EGR-1R: 5’-CTC CCT CCG CCT TCT TCC-3’; LMO2F: 5’-CCC AGC CTC
CTT TCC AAG-3’, LMO2R: 5’-CAA TAC AAA CAG CAG CAT CAG-3’; PAI-1-330F: 5’-ACC
CGG CAT GGC AGA CAG TCA ACC-3’, PAI-1+22R: 5’-GGC CCT GCA GCC AAA CAC AGC-3’;
TF-205F: 5’-CAT CCC TTG CAG GGT CCC GGA GTT-3’, TF+2R: 5’-GGG GTG CGG GGA GCT
CGC AGT C-3’10.
Supplementary Material; ATVB/2009/193821/R3, page 7
Statistical analysis. Data was analysed by ANOVA, and significant differences (where the P
value<0.05) are indicated by asterisk. Experiments were performed in triplicate on at least two
independent occasions, and studies (such as transient transfection analysis) subject to quantitative
analysis were performed in triplicate.
Supplementary Figure Legends
Supplementary Fig. 1.
IL-1beta stimulates EGR-1 expression and nuclear accumulation in
HASMC. A, HASMC grown to 90% confluence were incubated in serum-free medium for 24 h then
exposed to different concentrations of IL-1beta for 30 min. Total RNA was isolated and subjected to
real-time RT-PCR using primers for EGR-1 as described in the Methods. B, HASMC grown to 90%
confluence were incubated in serum-free medium for 24 h and exposed to 10 ng/mL IL-1beta for the
indicated times. Total RNA was isolated and subjected to real-time RT-PCR. C, HASMC grown on
coverslips were starved for 24 h in serum-free medium and treated with 10 ng/mL IL-1beta for 30 min,
immunofluorescence staining was then performed at 0.5, 1, 2 and 4 h using EGR-1 antibodies as
described in the Methods. *indicates P<0.05.
Supplementary Fig. 2.
Pathways mediating IL-1beta-dependent EGR-1 expression in
HASMC. A, HASMC were starved for 24 h and stimulated with 10 ng/mL IL-1beta for the indicated
times. Whole cell lysates were prepared in RIPA buffer and separated by 10% SDS-PAGE. Western
blot analysis was carried out using antibodies specific to either phospho-ERK1/2 or ERK1/2, phosphop38 or p38, phospho-JNK1/2 or JNK1/2, phospho-c-Jun or c-Jun, as described in the Methods. B,
HASMC grown on coverslips were starved for 24 h in serum-free medium and treated with 10 ng/mL
IL-1beta for 30 min. Immunofluorescence staining was performed using phospho-ERK1/2 specific
antibodies to determine the p-ERK1/2 immunoreactivity and its subcellular localization. C, HASMC
Supplementary Material; ATVB/2009/193821/R3, page 8
were starved for 24 h and exposed to 10 ng/mL IL-1beta for the indicated times. Nuclear lysates were
prepared and separated by 10% SDS-PAGE. Western blot analysis was performed using antibodies
specific to p-Elk1 or Elk1, or p65 as described in the Methods. Quantitative densitometric analysis of
band intensity is also shown (right).
Supplementary Fig. 3.
IL-1beta mediated histone H3 modification. A, HASMC were starved
for 24 h and treated with 10 ng/mL IL-1beta for the indicated times. Whole cell lysates were prepared
in RIPA buffer and separated on 10% SDS-PAGE. Western blot analysis was performed for phosphoH3, acetyl-H3, dM-H3, total H3 or beta-actin. B, HASMC grown on coverslips were starved for 24 h in
serum-free medium and treated with 10 ng/mL IL-1beta for 30 min. Immunofluorescence staining was
performed using phospho-H3 specific antibodies to determine phospho-H3 immunoreactivity and its
subcellular localization.
Supplementary Fig. 4.
IL-1beta inducible TF and PAI-1 expression. HASMC were incubated
in serum-free medium for 24h and exposed to 10 ng/mL IL-1beta for the indicated times. Total RNA
was isolated and subjected to real-time RT-PCR using primers for TF (upper) and PAI-1 (lower).
References
1.
Ning W, Li CJ, Kaminski N, Feghali-Bostwick CA, Alber SM, Di YP, Otterbein SL, Song R,
Hayashi S, Zhou Z, Pinsky DJ, Watkins SC, Pilewski JM, Sciurba FC, Peters DG, Hogg JC,
Choi AM. Comprehensive gene expression profiles reveal pathways related to the pathogenesis
of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2004;101:14895-14900.
Supplementary Material; ATVB/2009/193821/R3, page 9
2.
Katono T, Kawato T, Tanabe N, Tanaka H, Suzuki N, Kitami S, Morita T, Motohashi M,
Maeno M. Effects of nicotine and lipopolysaccharide on the expression of matrix
metalloproteinases, plasminogen activators, and their inhibitors in human osteoblasts. Arch
Oral Biol. 2009;54:146-155.
3.
Camici GG, Steffel J, Akhmedov A, Schafer N, Baldinger J, Schulz U, Shojaati K, Matter CM,
Yang Z, Luscher TF, Tanner FC. Dimethyl sulfoxide inhibits tissue factor expression, thrombus
formation, and vascular smooth muscle cell activation: a potential treatment strategy for drugeluting stents. Circulation. 2006;114:1512-1521.
4.
Carraro G, Albertin G, Forneris M, Nussdorfer GG. Similar sequence-free amplification of
human glyceraldehyde-3-phosphate dehydrogenase for real time RT-PCR applications. Mol
Cell Probes. 2005;19:181-186.
5.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative
PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-408.
6.
Khachigian LM, Williams AJ, Collins T. Interplay of Sp1 and Egr-1 in the proximal PDGF-A
promoter in cultured vascular endothelial cells. J. Biol. Chem. 1995;270:27679-27686.
7.
Khachigian LM, Anderson KA, Halnon NJ, Resnick N, Gimbrone MA, Jr., Collins T. Egr-1 is
activated in endothelial cells exposed to fluid shear stress and interacts with a novel shear-stress
response element in the PDGF A-chain promoter. Arterioscl. Thromb. Vasc. Biol.
1997;17:2280-2286.
8.
Kavurma MM, Bobryshev Y, Khachigian LM. Ets-1 positively regulates Fas ligand
transcription via cooperative interactions with Sp1. J Biol Chem. 2002;277:36244-36252.
9.
Pimanda JE, Chan WY, Donaldson IJ, Bowen M, Green AR, Gottgens B. Endoglin expression
in the endothelium is regulated by Fli-1, Erg, and Elf-1 acting on the promoter and a -8-kb
enhancer. Blood. 2006;107:4737-4745.
Supplementary Material; ATVB/2009/193821/R3, page 10
10.
Hasan RN, Schafer AI. Hemin upregulates Egr-1 expression in vascular smooth muscle cells
via reactive oxygen species ERK-1/2-Elk-1 and NF-kappaB. Circ Res. 2008;102:42-50.