pdf2
advertisement

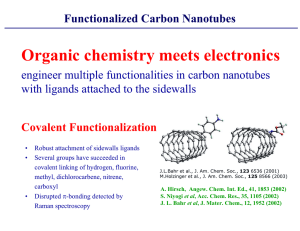
Multireference Configuration
Interaction:
Methodological Aspects and
Applications
Hans Lischka
University of Vienna
The concept of a reference wave
function
A simple example: H2 (1Σg+)
In a minimal basis we have two orbitals - σg
and σu
Ground state MO scheme
σu
σg
I
R→∞
σu
σg
II
At R ≈ Rmin configuration I will be a
qualitatively good representation of the exact
wave function. However, for R→∞ the
second orbital, σu, will become degenerate or
quasi-degenerate and configuration I will not
describe the system well. Within the given
symmetry a second configuration σu2 has to
be taken into account.
2
Generalization to more electrons
Single reference (SR) case (closed shell)
Reference wave function Ψ0:
ΨHF = Φ1Φ1 K Φ n Φ n
virtual
orbitals
M doubly occupied
Orbitals
M=n
3
Construction of the SR wave
function
excitation (substitution) of occupied orbitals
by virtual ones
Φ1Φ1 K Φ i Φi K Φ n Φ n
Φi → Φa
Φ1Φ1 K Φ a Φi K Φ n Φ n
Single-, double-, triple- … m-tuple excitations
Ψia , Ψijab , Ψijkabc K
Method of configuration interaction (CI):
ΨSR −CI = c0 Ψ0 + ∑ cia Ψia + ∑ cijab Ψijab + K
i ,a
i , j ,a ,b
Variation principle (Ritz)
Goal: Full CI – inclusion of all possible
configurations
4
Multireference (MR) case
virtual
orbitals
M occupied
orbitals, 2n El.
M>n
Reference wave function Ψ0: Ψ1 K ΨN
m-tuple excitations from Ψi , i = 1K N ref
into the virtual orbitals creates a set of
configurations {ΨI}
ΨMR −CI = ∑ cI ΨI
ref
Application of the Ritz variation principle
leads to the MR-CI approach
Select reference wave functions in order to
include all quasi-degeneracies
5
MR-CI approach – general
considerations
Stationary, nonrelativistic, clamped-nuclei,
electronic Schrödinger equation
HΨ = EΨ
H = ∑ hi + ∑ g ij + Vnn
i< j
i
hi = −1 2∇ i2 − ∑ Z A RAi
A
g ij = 1 rij
Vnn = ∑ Z A Z B RAB
A< B
Variational principle
E [Ψ ] = Ψ H Ψ
ΨΨ
The linear ansatz for the trial function Ψ
(Ritz variation principle)
n
Ψ = ∑ cs Φ s
s =1
6
leads to the following matrix
eigenvalue problem
with
Hc i = Ei c i , i = 1K n
H st = Φ s H Φ t
and assuming orthonormality of the Φ’s, i.e.
Φ s Φ t = δ st
Energies are ordered as
E1 ≤ E2 ≤ K ≤ En
Each eigenvalue Ep is an upper bound to the
corresponding exact eigenvalue. As
additional terms are added to the expansion
each eigenvalue Epn+1 of the (n+1)-term
expansion satisfies the inequalities
E pn −1 ≤ E pn +1 ≤ E pn
General references:
Shavitt 1977, Szabo 1989, Helgaker 2002
7
For practical calculations we have to make
several decisions concerning
• the representation of the many-electron basis
functions Φs
• Slater determinants
• Configuration state functions (CSFs) –
eigenfunctions of S2
• How to calculate the Hamiltonian matrix H
(calculate it at all?). In most applications the
dimension of H will be too large for explicit
storage.
Davidson subspace method for
diagonalization
Davisdon 1975, 1993
8
Algorithm:
A. If the kth eigenvalue is wanted, select a zeroth-order
orthonormal subspace v1, v2,…vl (l≥k) spanning
the dominant components of the first k eigenvectors.
~
Form Hv1 … Hvl and (vi,Hvj) = H ij , 1 ≤ i ≤ j ≤ l.
~
Diagonalize H using a standard method, select the
kth eigenvalue λk(l) and the eigenvector αk(l).
B. Form the residual vector
qM =
M
∑α
M
(M )
i ,k
i =1
(Hv i ) − ∑α i(,Mk ) λ(kM ) v i
i =1
~
M is the dimension of H .
C. Form ||qM || and check for convergence.
D. Form d I ,( M +1) = (λ
(M )
k
− H II ) q I , M , I = 1K N
−1
(and orthogonalize to the vi, i = 1…M).
E. v(M+1) = d(M+1)/||d(M+1)||.
F. Form Hb(M+1)
~
G. Form H i ,M +1 = (v i , Hv ( M +1) ), i = 1K M + 1.
~
H. Diagonalize H and return to step B with
α (kM +1) and λ(kM +1) .
9
The main computational work is the matrixvector multiplication w = Hv.
Options:
• Compute H, store it on disk, and compute
Hvi afterwards – conventional CI
• We do not need to construct H, we only
need wi = Hvi! This is the basis of the
“direct CI” (Roos 1972, Roos 1977)
Matrix elements
Φ s H Φt =
∑
aijst hij +
ij
∑
st
bijkl
g ijkl
ijkl
Orbital integrals hij and gijkl are defined by
hij = (ϕ i h1 ϕ j ) and
g ijkl = (ϕi ϕ j g12 ϕ k ϕl )
ws =
∑∑
t
ij
aijst hij vt +
∑∑
t
st
bijkl
g ijkl vt
ijkl
10
The coupling elements are simple (-1 or +1)
in case of single determinants (Slater-Condon
rules). For CSFs several possibilities exist
(Shavitt 1977):
• Spin projection
• Symmetric group
• Unitary group
(Graphical) Unitary Group Approach
(G)UGA
We are using Gelfand states
Hamiltonian: H = ∑ hij Eij + ∑ (ij kl ) eij , kl
+
iσ
ij
ijkl
where X and X jσ are spinorbital creation
and annihilation operators, σ = α, β ,
and the sum Eij = ∑ X i+σ X jσ is over the spin.
σ
eijkl = Eij E kl − δ kj Eil
11
A matrix element of H between two Gelfand
states m′ and m is given as
H m , m′ = m′ H m
=
∑h
ij
ij
m′ Eij m +
∑ (ij kl ) m′ e
ij , kl
m
ijkl
Thus, the matrix elements of the one-body
and two-body unitary group operators are the
coupling coefficients of the corresponding
one- and two-electron integrals, respectively.
12
GUGA
•
•
•
•
•
This presentation describes the distinct row table
(DRT) and its graphical representation in the
graphical unitary group approach (GUGA). Further
details can be found, e.g., in:
I. Shavitt, in The Unitary Group (Lecture Notes in
Chemistry No. 22), J. Hinze, ed. (Springer, Berlin,
1981), pp. 51–99.
I. Shavitt, in Mathematical Frontiers in
Computational Chemical Physics, D. G. Truhlar,
ed. (Springer, New York, 1988), pp. 300–349,
and references therein.
GUGA slides taken from the contribution of I.
Shavitt presented at the COLUMBUS
Programmer's Workshop
(http://www.univie.ac.at/columbus/workshops/argo
nne2005/), Argonne National Laboratory, Chicago,
USA, August 15th-19th 2005,
13
Graphical representation of a DRT
6 orbitals
5 electrons
S = 1/2
The
circles
represent
the nodes
of the
graph
The directed walks through the DRT
represent all the configuration state functions
14
What possibilities do we have to connect
one node with another one above or
below?
There are only 4 possibilities!
Different step numbers di are
represented by arcs of different slope:
0
1
2
3
15
head
tail
Characterization of a configuration by a
step vector: d1d2d3d4d5d6
e.g.: 1 0 3 1 0 2
The step vector characterizes the orbital
16
occupation
Step Vector
di = 0 ⇒ orbital i is unoccupied (uo)
di = 1,2 ⇒ orbital i is singly occupied (so)
di = 3 ⇒ orbital i is doubly occupied (do)
Example:
Orbital 1 2 3 4 5 6
1 0 3 1 0 2
so uo do so uo so
17
general n electron case
Multireference CI singles and doubles
X
Z
Y
two electrons
W
Reference configurations in red
Inactive (reference doubly occupied)
Active (variable occupation in the reference)
External (virtual) orbitals: only singles and18
doubles
• The internal (active + closed shell) part
of the graph is complicated, but
relatively small in comparison to the
virtual (external) space.
• The graph for the external is space is
very simple due to the fact that we allow
only double excitations. Moreover, its
structure is independent of the internal
part. Respective loops (coupling
elements) can be computed once and
for all.
• The interface between internal and
external space is given by the vertices Z
(0-excitations), Y (single-excitations), X
(double excitations, triplet coupling) and
W (double excitations, singlet coupling)
19
• The loops are split into an internal part
and into an external one. The internal
part is computed explicitly and either
stored on a file (formula file) or
recomputed every Davidson iteration.
The external part is added “on-the-fly”
when the total coupling elements are
computed.
• The two-electron integrals are sorted
according to the number of internal
indices: all (four)-internal, three-internal,
two-internal, one internal and allexternal.
20
Algorithm for w=Hv
ws =
∑∑ a
t
ij
hv +
st
ij ij t
∑∑ b
st
ijkl
t
g ijkl vt
ijkl
In this approach (COLUMBUS program,
Lischka 1981, Shepard 1988, Lischka 2001,
2006) the computational scheme is driven by
the integral indices
Loop over types of two-electron integrals
For each set
Loop over the quadruple of indices
Get coupling elements, integrals and v
Compute the respective contribution to w
Update w
End loop indices
End loop integral types
The remaining steps of the Davidson iteration
are simple.
21
The update of w can be performed via matrix
and vector operations if the whole virtual
space if the same virtual orbital space is used
for excitations from all reference
configurations. In this case selection schemes
can only be applied at the level of the
reference configurations.
Individual selection schemes specific for each
reference configurations (e.g. multireference
double excitation CI (MRD-CI) Buenker
1968, 1974, 1975, Hanrath 1997) do not have
this restriction.
22
Overall CI scheme
• AO integrals
• Determination of MOs by means of a SCF
or MCSCF calculation
• Transformation of the two-electron
integrals into the MO basis
• Sorting of the integrals
• Davidson diagonalization
• Properties
• Energy gradient and nonadiabatic
couplings
• Geometry optimization routines
23
Overview of different CI approaches
• Selection at the reference configuration
level
– Uncontracted CI: single- and double
excitations are constructed from each
reference CSF and the unique set of
configurations (ΨI, I = 1…NCI) is taken.
The CI coefficient of each CSF is
included in the variational procedure.
– Contracted MRCI (IC-MRCI) (Meyer
1977, Werner 1988):
Ψijpab = 1 2(Eai ,bj + pEbi ,aj ) Ψ0
(p = ±1 for external singlet and triplet
pairs)
Ψ0 =
∑
a Rμ ΨRμ
Rμ
ab
Ψ
The configurations ijp span exactly
the first-order interacting space
24
Uncontracted vs. contracted CI
Uncontracted CI
Advantages:
• Larger flexibility of
the wave function,
full variation,
Excited states!
• Computation of
analytic gradient
relatively easy
(available in
COLUMBUS)
Disadvantage:
Much larger
computational effort
Contracted CI
Advantages:
• High
computational
efficiency
Disadvantages:
• Relaxation effects
for Ψ0
• Potential energy
surfaces for
excited states,
avoided
crossings,…
• Analytic gradient
difficult (not
available)
25
Overview of MRD-CI
Original work: Buenker 1968, 1974, 1975
• Selection scheme based on perturbation
theory – energy threshold T
• Extrapolation scheme T → 0
Conventional CI program
• Hanrath 1997:
DIESEL-MR-CI (direct internal external
separated individually selecting MR-CI):
direct, division of internal/external space
Advantage of MRD-CI: great flexibility,
relatively large molecules
Disadvantage: Extrapolation, gradient for
extrapolated energy
26
When do we use which method?
• MR-CI, MR-CI+Q, MR-AQCC (Szalay
1993, 1995) in multireference cases,
otherwise use single reference method
• When geometry optimization at the CI
level is not required or when pointwise
optimization is possible – IC-MRCI or
MRD-CI
• Difficult cases: robust method is required
for description of large portions of the
energy surfaces – uncontracted MR-CI or
MR-AQCC
Special feature: analytic energy gradients
and nonadiabatic couplings (conical
intersections)
27
Characterization of Internal Orbital
spaces
auxiliary orbitals
active orbitals
Reference doubly
occupied
frozen core
Active orbitals: usually as CAS
Auxiliary orbitals: for the description of
Rydberg states (single excitations or
individual configurations)
28
Selection of CSF space
Three options:
1. a) The reference configurations are
chosen to have the same symmetry as the
state symmetry
b) Only those singles and doubles are
constructed, which have a nonzero matrix
element with one of the references
(interacting space restriction (Bunge
1970)
2. Restriction 1b is lifted
3. All possible reference configurations are
constructed within the specified orbitaloccupation restrictions. Singles and
doubles from reference with ”wrong”
symmetry can have the correct symmetry.
Condition 3 allows consistent calculations
using different subgroups of the actual
molecular symmetry.
29
COLUMBUS project
•
•
•
•
•
•
•
Set of programs for high-level ab initio calculations
Methods: MCSCF, MR-CISD, MR-ACPF/AQCC,
Spin-orbit CI
Focus: multireference calculations on ground and
excited states
Recent achievements: analytic MR-CI gradient,
nonadiabatic couplings, parallel CI
Authors: H. Lischka, R. Shepard, I. Shavitt, R. M.
Pitzer, M. Dallos, Th. Müller, P. G. Szalay, F. B.
Brown, R. Ahlrichs, H. J. Böhm, A. Chang, D. C.
Comeau, R. Gdanitz, H. Dachsel, C. Ehrhardt, M.
Ernzerhof, P. Höchtl, S. Irle, G. Kedziora, T. Kovar,
V. Parasuk, M. J. M. Pepper, P. Scharf, H. Schiffer,
M. Schindler, M. Schüler, M. Seth, E. A. Stahlberg,
J.-G. Zhao, S. Yabushita, Z. Zhang, M. Barbatti, S.
Matsika, M. Schuurmann, D. R. Yarkony, S. R.
Brozell, E. V. Beck, and J.-P. Blaudeau,
COLUMBUS, an ab initio electronic structure
program, release 5.9.1 (2006).
Web page: http://www.univie.ac.at/columbus/
30
COLUMBUS program package
Recent review: H. Lischka, R. Shepard, R. M.
Pitzer, I. Shavitt, M. Dallos, Th. Müller, P.
G. Szalay, M. Seth, G. S. Kedziora, S.
Yabushita, and Z. Zhang 2001, Phys.
Chem. Chem. Phys. 3, 664
• User-friendly interface
• Public domain – free of charge
• Distribution of source code
31
References
R. J. Buenker and S. D. Peyerimhoff 1968, Theor. Chim. Acta 12, 183.
R. J. Buenker and S. D. Peyerimhoff 1974, Theor. Chim. Acta 35, 33.
R. J. Buenker and S. D. Peyerimhoff 1975, Theor. Chim. Acta 39, 217.
E. R. Davidson 1975, J. Comp. Physics 17, 87.
E. R. Davidson 1993, Computers in Physics 7, 519.
M. Hanrath and B. Engels 1997, Chem. Phys. 225 (1997)
T. Helgaker, P. Jørgensen and J. Olsen 2002, Molecular Electronic-Structure
Theory, John Wiley.
H. Lischka, R. Shepard, F. B. Brown, and I. Shavitt 1981, Int. J. Quantum
Chem. Quantum Chem. Symp. 15, 91.
H. Lischka, R. Shepard, R. M. Pitzer, I. Shavitt, M. Dallos, Th. Müller, P. G.
Szalay, M. Seth, G. S. Kedziora, S. Yabushita, and Z. Zhang 2001, Phys.
Chem. Chem. Phys. 3, 664.
H. Lischka, R. Shepard, I. Shavitt, R. M. Pitzer, M. Dallos, Th. Müller, P. G.
Szalay, F. B. Brown, R. Ahlrichs, H. J. Böhm, A. Chang, D. C. Comeau,
R. Gdanitz, H. Dachsel, C. Ehrhardt, M. Ernzerhof, P. Höchtl, S. Irle, G.
Kedziora, T. Kovar, V. Parasuk, M. J. M. Pepper, P. Scharf, H. Schiffer,
M. Schindler, M. Schüler, M. Seth, E. A. Stahlberg, J.-G. Zhao, S.
Yabushita, Z. Zhang, M. Barbatti, S. Matsika, M. Schuurmann, D. R.
Yarkony, S. R. Brozell, E. V. Beck, and J.-P. Blaudeau, COLUMBUS, an
ab initio electronic structure program, release 5.9.1 (2006)
I. Shavitt 1977, in: Methods in Electronic Structure Theory, H. F. Schaefer III,
Ed., Plenum Press, p. 189.
B. O. Roos 1972, Chem. Phys. Lett. 15, 153.
B. O. Roos and P. E. M. Siegbahn, 1977, in Methods of Electronic Structure
Theory, H. F. Schaefer III, Ed., Plenum Press, p. 277.
R. Shepard, I. Shavitt, R. M. Pitzer, D. C. Comeau, M. Pepper, H. Lischka, P.
G. Szalay, R. Ahlrichs, F. B. Brown, J. Zhao 1988, Int. J. Quantum Chem.
Quantum Chem. Symp. 22, 149.
A. Szabo and N. S. Ostlund 1989, Modern Quantum Chemistry. Introduction
to Advanced Structure Theory, McGraw-Hill.
P.G. Szalay, R.J. Bartlett, 1993, Chem. Phys. Lett. 214, 481.
P.G. Szalay, R.J. Bartlett, 1995, J. Chem. Phys. 103, 3600.
32