Evaluation of Procedures for Amplification of Small
advertisement

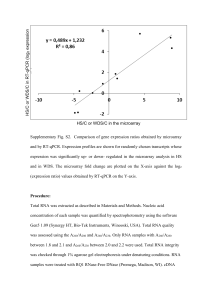
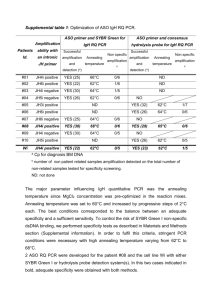
26-30 Microarray March17_RZ 18.03.2004 12:21 Uhr Seite 26 Evaluation of Procedures for Amplification of Small-Size Samples for Hybridization on Microarrays Sandra Klur1, Karen Toy2, Mickey P. Williams2, and Ulrich Certa1,* 1Roche Center for Medical Genomics, F. Hoffmann–La Roche, Ltd. Basel, Switzerland 2Department of Molecular Biology, Genentech, Inc., San Francisco, USA *Corresponding author: ulrich.certa@roche.com Microarrays Various approaches have been developed for the preparation of samples for gene expression monitoring. For Affymetrix chips, a standard protocol is widely used; however, this is inefficient for small samples such as laser capture microdissections. Several amplification procedures for such samples Sandra Klur already exist, and our goal was to test two of them: the first is based on random PCR amplification, and the second, linear amplification, involves performing the standard protocol twice. We analyzed a dilution of a commercially available mouse brain total RNA preparation and microdissections from mouse hippocampus and striatum. We evaluated the quality of microarray data by analyzing several chip parameters and performing multiple comparisons. At the biological level, brain microdissections prepared with either method gave similar expression results. At the technical level, analysis of the commercial sample showed that random PCR amplification is more reproducible, requires smaller RNA input, and generates cRNA of higher quality than linear amplification. Reprinted as a shortened version from Genomics 83 (2004), Klur S et al. ”Evaluation of procedures for amplification of small-size samples for hybridization on microarrays”, p. 508– 517 with permission from Elsevier. [...] designates where text has been shortened. [...] DNA microarrays are widely used in biology [3–5] and have applications in various fields such as tumor profiling [6, 7], analysis of complex diseases [8], or determination of genes involved in development [9]. Thanks to the development of techniques such as laser capture microdissection [10–12], cell flow sorting, or needle biopsies, one is now able to analyze complex organic structures more accurately. For example, microdissection is revolutionizing neurobiology: brain, one of the most complex biological structures, is made of around 500 different RNA isolation 5 g total RNA 10–50 ng total RNA Standard procedure PCR amplification Linear amplification cDNA synthesis cDNA synthesis cDNA synthesis In vitro transcription Random PCR amplification cDNA synthesis In vitro transcription Hybridization on array Figure 1: Strategies used to generate biotinylated targets for gene expression profiling 26 cell types, with a total of 1012 cells [13,14]. Laser microdissection allows for the excision of very precise small tissue pieces or even single cells for the analysis of site-specific functions (i.e., spatial learning in the hippocampus) or study of diseases (i.e., tumor cells disseminated in a network of normal cells). As standard protocols for sample preparation are inefficient with total RNA amounts smaller than 5 µg, it is evident that an amplification step is necessary for analyzing minute amounts of total RNA. Several strategies have been developed for target preparation and amplification in combination with Affymetrix chips. In this report, we will focus on procedures in which a doublestranded cDNA (ds cDNA) is produced from total RNA and then used as template to generate a labeled cRNA during an in vitro transcription reaction [15,16]. We compared microarray data from samples amplified through two different processes derived from this standard protocol. The first technique, random PCR amplification, includes a PCR amplification step at the ds cDNA level (Microarray Target Amplification Kit, Roche Applied Science). The second method, known as linear amplification, consists of two cycles of cDNA synthesis followed by in vitro transcription (GeneChip Eukaryotic Small Sample Target Labeling, Affymetrix) and is an adaptation of the method first described by Eberwine [15]. [...] To evaluate the performance of both amplification methods and to compare them to the standard protocol, we studied samples from two sources. [...] BIOCHEMICA · NO. 2 · 2004 26-30 Microarray March17_RZ 18.03.2004 12:22 Uhr Seite 27 Table 1: Samples analyzed and hybridization result Input Method R1 Commercial Mouse brain total RNA Mouse brain total RNA Mouse brain total RNA Mouse brain total RNA Mouse brain total RNA Microdissections Hippocampus—indpdt MD Hippocampus—pooled MD Striatum Hippocampus—indpdt MD Hippocampus—pooled MD Striatum Yield (µg) R2 R3 R4 3′/5′ ratio Mean STDEV % present calls Mean STDEV 5 µg 50 ng 10 ng 50 ng 10 ng Standard PCR PCR 2x Amp 2x Amp 73.6 52.6 63.2 42.2 51.6 56.0 57.6 59.2 38.2 53.4 58.9 58.0 63.7 55.0 45.6 76.2 56.2 65.7 57.3 45.1 1.1 1.0 1.0 3.9 5.7 0.1 0.1 0.1 0.1 0.8 50.2 51.3 50.8 40.7 33.7 2.9 1.4 1.4 5.0 7.0 50 ng 50 ng 50 ng 50 ng 50 ng 50 ng PCR PCR PCR 2x Amp 2x Amp 2x Amp 98.8 82.8 88.4 48.8 71.1 75.5 77.6 73.6 72.0 76.4 74.0 60.8 64.0 73.6 82.4 83.7 73.4 58.8 70.0 72.0 75.2 81.2 80.6 74.2 1.1 1.1 1.2 4.7 5.8 7.7 0.2 0.0 0.1 0.9 0.8 2.0 38.8 37.2 35.7 37.9 37.8 33.7 3.5 3.3 2.3 3.7 1.0 3.1 Source, source of RNA; Input, amount of total RNA used for target synthesis, the amount indicated for microdissections is an evaluation; Method, method used for generation of labeled cRNA; Yield, yield of cRNA from the four replicates of each condition; 3′/5′ ratio, ratio of hybridization signals obtained from GeneChip probe pairs directed at the 3′ or 5′ end of GAPDH and β-actin transcripts; % present calls, percentage of genes detected as present by MAS 5.0; Mean and STDEV, mean and corresponding standard deviation of ratio and present calls of the four replicates of each condition. Results Amplification strategy The aim of our study was to evaluate the sensitivity, reproducibility, and reliability of two amplification protocols for gene expression analysis of small-size samples. Both amplification techniques are derived from a standard protocol [16]. Figure 1 shows the different sample preparation strategies we have tested in our analysis. The first step of classical sample preparation consists of reverse transcription of total RNA with an oligo(dT) primer containing a T7 promoter for generation of a ds cDNA. Then, the second strand of cDNA is synthesized through RNA nicking and priming. Finally, in vitro transcription (IVT) of ds cDNA with a T7 RNA polymerase and biotinylated nucleotides produces a labeled cRNA, which is hybridized to the chip after fragmentation. The random PCR protocol relies on amplification of the cDNA before IVT. For this purpose, the primers used for cDNA synthesis are slightly modified: they contain an additional sequence (target amplification sequence, TAS) which is used for PCR amplification. The linear amplification protocol consists of two cycles of cDNA synthesis followed by IVT: the first cycle provides initial amplification of total RNA, resulting in unlabeled cRNA; the second cycle achieves further amplification and incorporation of biotinylated nucleotides for labeling. Samples analyzed To evaluate the performance of both amplification protocols, we analyzed a commercial mouse brain total RNA solution: we amplified 50- and 10-ng aliquots through random PCR or by linear amplification (four replicates for each quantity and each condition) and we compared the chip results to a ‘‘gold standard’’ reference comprising a 5-µg aliquot processed with the standard protocol (four replicates). To see if we could also obtain reliable data from biological samples, we included laser microdissections from different mouse brain regions in our study: two groups of samples that were constituted of independent microdissections from either hippocampus or striatum and a third group, which was a pool of hippocampus microdissections. The reason for preparing a pool from microdissected samples was to test the effect of amplification on homogeneous biological samples, and the independent microdissections served to pinpoint differences resulting from biological variability. Microarrays Source Gene expression profiles were obtained from all these samples using the murine genome U74Av2 microarray from Affymetrix, allowing analysis of ≈ 6000 functionally characterized Mouse Unigene Sequences (Build 74) and ≈ 6000 EST clusters. For each condition, we prepared four samples in parallel that were independently hybridized on chips. Resulting chip data were normalized before multiple comparison was performed. [...] Amplification method performance The yield of labeled cRNA is a critical issue because sufficient material has to be prepared for hybridization onto a microarray. Generally, a minimum of 10 µg of labeled cRNA is loaded on a chip. The total RNA amount contained in the microdissections has been deduced from the number of PCR cycles necessary for amplification of 10- and 50-ng mouse brain total RNA aliquots and of 50 ng of control total RNA (provided in the RAS Microarray Target Amplification Kit). We carried out 25 PCR cycles for all samples and for the positive control. From this observation, we deduced that there was around 10 to 50 ng of total RNA in a microdissected sample (≈ 1000 cells). The quantity of labeled cRNA obtained after amplification was measured by BIOCHEMICA · NO. 2 · 2004 27 18.03.2004 12:22 Uhr Seite 28 absorbance at 260 nm. The values are reported in Table 1. Both amplification methods afforded the production of enough material for hybridization to microarray: for all samples we obtained at least 40 µg cRNA. This is comparable to the values obtained for a 5-µg sample prepared with the classical protocol. Slightly more material was yielded from random PCR amplification. We first examined the present call percentage. This value corresponds to the percentage of probe sets that have a detectable and specific hybridization signal, thus reflecting directly the percentage of genes that are expressed in a sample. At first sight, present call percentage is higher for mouse brain samples than for microdissections (Table 1). As this parameter is related to sample complexity, this result is not surprising: in a mouse brain total RNA extract (whole organ), much more genes are expressed than in a brain microdissection containing only around 1000 cells (small piece of organ). For mouse brain total RNA, the highest value, around 50%, is obtained when preparing 5 µg aliquots with the standard protocol or amplifying 50and 10-ng aliquots through random PCR. For linear amplification of 50- and 10-ng aliquots, only 40% present calls or even less was detected, indicating a clear loss of information (10–15% of present calls are not detected) with this amplification method. With regard to microdissections, all samples had similar present call percentages, around 28 a An additional important issue is the degree of reproducibility within individual replicates and different conditions. We thus compared the methods using scatter plots of data from mouse brain total RNA (Figure 2). For the standard protocol (Figure 2A), the point distribution between two chips looks like the classical scatter-plot shape typical for Affymetrix chips. When scatter plots of chips prepared from PCR- or linear-amplified samples (2B and 2C) are examined, the data display also looks very similar to the classical pattern. However, comparing amplified to nonamplified samples, the point distribution is more widely spread. For random PCR amplification, we observe a moderate bias for all signal intensities, whereas for linear amplification, the scatter plot has a banana shape: those genes expressed at either a low (intensity smaller than 50) or high (intensity higher than 5000) level are amplified to a greater extent than those expressed at a medium level. Furthermore, we examined the correlation factors (r2) between chips, and the values reflected the results of scatter plots (Table 2). Within conditions, the correlation was very good, at around 0.98 for nonamplified samples and around 0.97 for random PCR-amplified samples. For linear-amplified samples, correlation factors were not as good, but still acceptable. As expected, comparing amplified samples to nonamplified ones, the correlation factors drastically decreased. Actually, linear-amplified samples showed a better correlation to nonamplified samples c 10000 1000 100 10 1 1 10 100 1000 10000 100000 Signal intensity - PCR amplified b 100000 Signal intensity - non amplified 100000 10000 1000 100 10 1 1 Signal intensity - non amplified 10 100 1000 10000 100000 Signal intensity - PCR amplified e d 100000 10000 1000 100 10 1 1 10 100 1000 10000 100000 Signal intensity - non amplified Signal intensity - linear amplified Figure 2: Scatter plots: signal intensities in a log10 scale. Samples are mouse brain total RNA. (a) Standard protocol: 5 µg against 5 µg. (b) PCR amplification: 50 ng against 50 ng. (c) Linear amplification: 50 ng against 50 ng. (d) PCR amplification on 50 ng against standard protocol on 5 µg. (e) Linear amplification on 50 ng against standard protocol on 5 µg. (a to c) Signal intensities of one chip were plotted against signal intensities of another chip from the same condition. (d and e) Mean of signal intensities of the four chips from one condition plotted against mean of signal intensities of the four chips from another condition. Signal intensity - PCR amplified Microarrays Array quality parameters To evaluate whether the two amplification protocols allow for preparation of high-quality samples for microarray analysis, we examined various chip quality parameters calculated after analysis with the RACE-A software, and the data resulting from the different experimental conditions were compared. 37%, whether they were amplified through random PCR or with the linear protocol. In that case, no clear difference between PCR- and linear-amplified samples was observed. Nevertheless, the data from mouse brain total RNA indicate a better sensitivity of PCR amplification (10 ng) compared to linear amplification (50 ng). 100000 10000 1000 100 10 1 1 10 100 1000 10000 100000 Signal intensity - non amplified BIOCHEMICA · NO. 2 · 2004 Signal intensity - linear amplified 26-30 Microarray March17_RZ 100000 10000 1000 100 10 1 1 10 100 1000 10000 100000 Signal intensity - linear amplified 26-30 Microarray March17_RZ 18.03.2004 12:22 Uhr Seite 29 Table 2: Chip correlation factors (r2) None, 5 µg PCR, 50 ng PCR, 10 ng Linear, 50 ng Linear, 10 ng (B) Amplification Standard 5 µg Random PCR 50 ng 10 ng 0.977 — — — — 0.593 0.974 — — — Random PCR Independent Pooled hippocampus hippocampus Linear amplification 50 ng 0.591 0.959 0.968 — — Striatum 0.744 0.499 0.495 0.955 — 10 ng 0.570 0.386 0.385 0.875 0.925 Linear amplification Independent Pooled Striatum hippocampus hippocampus PCR Independent hippocampus Pooled hippocampus Striatum 0.919 — — 0.945 0.980 — 0.811 0.840 0.881 0.465 0.513 0.459 0.504 0.524 0.465 0.422 0.444 0.472 Linear Independent hippocampus Pooled hippocampus Striatum — — — — — — — — — 0.923 — — 0.951 0.983 — 0.868 0.887 0.944 Correlation factors were calculated with RACE-A for all possible pair-wise comparisons of chips analyzed in our study. These factors were then merged within each condition and reported in (A) for mouse brain total RNA and (B) for mouse brain microdissections. (r2 = 0.74) than to PCR-amplified (r2 = 0.6), probably because the same enzymatic reactions are applied in both methods. We also asked whether it was possible to detect differences in correlation factors within and between the three groups of microdissections. The results were very interesting: correlation factors for pooled hippocampus replicates were better than those for independent hippocampus, and striatum microdissections correlated better with other striatum microdissections than with hippocampus microdissections (pooled or not). Next, we wanted to examine the quality of amplified cRNA and therefore we looked at the 3′/5′ ratio. This quality parameter is automatically calculated by the analysis software, after scanning and data normalization. The value corresponds to the hybridization signal intensity of probe sets specific for the 3′ and 5′ ends of certain transcripts. The optimal value is 1 and indicates that the mRNA sequences are full length, neither degraded during total RNA extraction nor shortened during cDNA synthesis. In our experiment, ratios for nonamplified mouse brain total RNA were very close to 1 (Table 1). All PCR-amplified samples (mouse brain total RNA and microdissections) also had ratios of around 1. For linearamplified samples the ratios were higher, between 3.9 and 5.7 for mouse brain total RNA, and microdissections also showed very high ratios. This reflects the fact that linear amplification yields truncated transcripts without 5′ ends. This was somehow expected because random primers are used for the second cDNA synthesis cycle. Since most of the probe sets present on the microarray represent the 3′ end of each transcript, this shortening should not have a major impact on data quality. Microarrays (A) Amplification We concluded from all these technical parameters that both methods can be used for amplification of samples for microarray analysis, with a small advantage for PCR amplification: samples amplified through PCR are of higher quality and the method offers increased sensitivity. PCR amplification has also some technical advantages: the protocol is easier to set up and less time consuming. Finally, an important observation is that the same preparation protocol should be used for all samples within a set of experiments; if one uses different protocols in a given study, there is a risk of adding variability due to technical manipulation and therefore of disturbing overall gene expression level within replicates of a condition and ultimately, the results will not be as accurate. However, this can be a drawback if it is desired to compare results from amplified samples to previous results from nonamplified samples. Detection of genes differentially expressed in two brain regions We also wanted to assess if it was possible to detect genes differentially expressed in different brain areas and if the same genes were identified by each amplification method. For this purpose, we compared the microarray data from hippocampus and striatum microdissections (Figure 3). We first filtered for genes specific for hippocampus and having a change factor higher than 2: we identified 50 genes by PCR amplification and 41 by linear amplification. We then checked if genes that were detected as ‘‘upregulated’’ by one method were similarly detected by the other. No expression contradiction between the two BIOCHEMICA · NO. 2 · 2004 29 26-30 Microarray March17_RZ 18.03.2004 a PCR amplification b PCR amplification 12:22 Uhr Seite 30 Genes specific for hippocampus 4 60 5 Linear amplification Genes specific for striatum 4 49 3 Linear amplification Microarrays Figure 3: Overview of the number of genes that are differentially expressed in hippocampus and striatum. Numbers within the circles indicate genes identified by the corresponding method and numbers within the intersections indicate number of genes commonly detected by both methods. amplification methods was observed: all genes detected as induced in hippocampus after PCR amplification were also detected as induced in hippocampus after linear amplification and few genes had no change in one method. Ultimately, 60 genes specific for hippocampus were successfully identified by both methods, with 4 genes being detected only by PCR amplification and 5 only by linear amplification. To complete the technical and statistical issues of this comparison, we then evaluated the biological meaning of our data. We briefly examined the function of genes induced in hippocampus compared to striatum. Most of the genes identified and for which enough data about function and tissue specificity were known were brain-specific. Moreover, we successfully detected hippocampus-specific genes like hippocalcin, transcription factor 4, and eph receptor a7. Genes described as specific for other tissues and overexpressed in hippocampus were not necessarily false positives; the data indicated only that expression in hippocampus was higher than in striatum, rather than implying that these genes have a biological function in hippocampus. We performed a similar analysis for striatum-specific genes. Forty-eight genes were identified through PCR amplification and 19 through linear amplification. As before, we did not notice any expression contradiction, and very few genes had no change in one amplification method. Finally, 49 genes specific for striatum were successfully detected by both methods, with 4 detected only by PCR amplification and 3 only by linear amplification. We did not identify genes known as being specific for striatum but most of the genes whose expression was higher in striatum than in hippocampus were brainspecific. Our microarray results demonstrated that random PCR amplification and linear amplification are both 30 reliable for sample analysis on oligonucleotide chips. Even if some genes have not been identified after both amplification processes, they represent only a minority (16 of 125 genes, around 13%). An important finding is that direction of change (induction or repression) was identical for the two protocols, and also change factor values were very similar. In addition, the data were biologically significant and moreover, they correlated with another expression study done using SAGE on whole hippocampi, without sample amplification [23]. This is clearly a further validation for the use of amplification in functional genomics studies. Discussion We have shown that two unrelated sample amplification protocols give good quality microarray data, with a small advantage for random PCR amplification, which provides the best 3′/5′ ratios and cRNA yield. Comparing expression profiles obtained by both methods, we found that the direction of change was essentially identical for both amplification techniques. Moreover, our results are biologically relevant and show that amplification can be used for gene expression analysis because no major distortion is introduced into the sample, thus allowing for reliable study of very small samples. Another important fact is that PCR, often described as introducing bias to samples [5,15], gave results comparable to those of other methods: when performing a reasonable number of PCR cycles (around 25), almost no bias was detected. Whatever tendency may be introduced is compensated for since all samples are prepared with the same protocol and so are submitted to the same bias. The relative expression level between samples is preserved, even though the absolute expression level may be altered. [...] Global gene expression analysis has a wide range of applications in biological research and has already provided interesting results. The combination of DNA microarray technology with microdissection and sample amplification is promising and allows access to analysis of discrete cell populations [24–26]. Please refer to the publisher for Materials and Methods and for References. Product Pack Size Cat. No. Microarray Target Amplification Kit 1 kit (10 reactions) 3 310 191 Microarray RNA Target Synthesis Kit (T7) 1 kit (25 reactions) 3 266 877 Microarray Target Purification Kit 1 kit 3 266 885 (50 purifications) BIOCHEMICA · NO. 2 · 2004