Methods: X-ray Crystallography Biochemistry 4000 Dr. Ute Kothe
advertisement

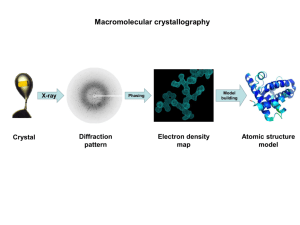
Methods: X-ray Crystallography Biochemistry 4000 Dr. Ute Kothe Why X-rays? Optics: Limit of resolution ~ l/2 Distance between atoms: 0.1 – 0.3 nm Wavelenght of X-rays: 0.1 – 10 nm Allows structure determination at atomic resolution Problem: No lenses available for X-rays (as for visible light in microscope) Remember: Wavelength, Amplitude, Phase Overview of steps Growing a Crystal Collecting X-ray diffraction pattern Solving of Phase Problem Calculate Electron Density Map Constructing a Structural Model Refining the Structural Model Growth of Crystals - Background Why? Diffraction pattern of single molecule too weak for detection => sum of several diffraction patterns of identically oriented molecules with identical conformation Porcine Elastase Supersaturated Solution: Concentration higher than intrinsic solubility S0 Growth of Crystals - Approaches Hanging Drop Method: Empirical Approach: Test hundreds of conditions Parameters: • pH • salt type & concentration • precipitant (ammonium sulfate, polyethylene glycol, 2-methyl-2,4-dimethyl-pentane diol (MPD), propanol) • temperature (2 – 30°C) • protein concentration (1 – 100 mg/ml) • additional substances (substrates, inhibitors, cofactors etc.) Alternative: Sitting Drop Microdialysis Description of Crystals a Unit cell: γ characterized by edge length and angles b can contain several molecules is repeated multiple times along the axis of the crystal Assymetric Unit: minimal part of the unit cell which is related to other parts by defined symmetry operations Space Groups Crystal can be described by shape of unit cells (called lattice type, see figure) and symmetry type of asymmetric unit 65 different space groups possible for natural biomolecules Example: P212121 Diffraction and Interference Huygen’s principle of diffraction: Each point in front of a wavefront acts as a point of progapation for a new wavelet. b) Constructive interference of scattered waves => Amplitude = 2E c) Destructive interference of scattered waves => Amplitude = 0 (wave 180° out of phase) Bragg’s Law Requirement for constructive interference: 2 d sin θ = n l n: integer values (0, 1, 2, ...) If angle θ and wavelenght l are known, the distance d between planes can be determined. Simplification: reflection at planes (one dimension: distance d of planes)! Diffraction in 3D: Miller Indices Van Laue Conditions replace Bragg’s Law (too much to explain in Bchm 4000) Instead of “n”, three Miller Indices (h, k, l) define the integer number of wavelenght that result in an observed reflection froma 3D crystal. Example: 2 points in space 1D reflection 2D reflection More Diffraction Patterns 2D example: Each diffraction spot corresponds to one set of Miller indices (h, k, l). Friedel’s law: I(h,k,l) = I(-h,-k,-l) http://www.esc.cam.ac.uk/teaching/teaching.html Diffraction Photograph Each spot contains information from all atoms! “Pattern of spots” (diffraction pattern) allows determination of crystal parameters (size of unit cell, symmetry within unit cell). Structural information has to de derived (see below) from difference in spot intensities. X-ray diffraction photograph of Sperm Whale Myoglobin crystal To obtain enough information, diffraction photographs are taken at various different angles of the crystal in the X-ray beam. The Structure Factor: F(S) How to get to an electron density map from the intensities of reflections? • Intrinsic amplitude of scattered X-ray from each type of atom depends on its electron density r(xyz) (described by atomic scattering factor f) • f is used to describe scattering vector for each atom for one reflection (with Miller indices h,k,l) • Sum of atomic scattering vectors is molecular scattering factor F, also called structure factor (can be calculated for a given structure): F(hkl) = ∫r(xyz) e-2pi(hx+ky+lz) Fourier Transformation The Phase Problem Continued: How to get to an electron density map from the intensities of reflections? Fourier Transformation (other way around) r(xyz) = ∫F(hkl) e2pi(hx+ky+lz) Magnitude of F is proportional to square root of intensity: |F(hkl)| ~ √ I(hkl) Thus: r(xyz) = 1/NV ∑ ∑ ∑ √I(hkl) e-i(hkl) e2pi(hx+ky+lz) h k Electron Density l Intensity Phase Missing Information to get electron density! Molecular Replacement 1. Method to obtain Phase information • Only possible if structure of a very similar molecule is known (native vs. Mutant, close homolog), at least 25% sequence identity • Calculate structure factors Fcalc for known molecule • Compare with data set to estimate phase for known structure (acalc) • Use this Phase information together with observed structure factors (Fobs) to calculate first electron density map • Refine Structure (see below) Multiple Isomorphous Replacement 2. Method to obtain Phase information • Use crystals containing heavy atoms at specific positions (isomorphous crystals) as well as crystal of native protein • Heavy atoms strongly diffract • Difference data set of heavy atom derivative and native crystals gives structure factors of heavy atoms only = limited data set • Can be used to determine position and phase of heavy atoms (as for small molecule crystals, Patterson function) • Needs to be done for at least 2 heavy atom derivatives to obtain enough information to estimate phase of native data set and to solve the structure Multiple Anomalous Dispersion 3. Method to obtain Phase information • Use one crystal containing heavy atoms at specific positions as well as crystal of native protein • Heavy atoms not only diffract X-rays, but also absorbs X-rays of certain wavelength • Using synchroton X-ray source, diffraction data are generated at different wavelenghts of the heavy-atom crystal • Different data sets can be used to obtain phase information similar to multiple isomorphous replacement Electron Density Map Computed electron density map can be represented by “chicken wire” on computer. Knowing the primary sequence of a protein (or nucleic acid) the atoms can be placed by hand into the electron density map. Rough, inaccurate model of protein structure What about hydrogen atoms? Structural Refinement Iterative Process: • model is subtly changed • Structure Factors are calculated (Fcalc) and compared by Rcrys factor with measured Fobs to see how well a model fits the data: • differences are minimized (minimal R) • good structure: Rcrys < 20% (0.2) Problem: • R factor can be artifically lowered by adding more solvent • better Rfree: as Rcrys, but calculated with 10% reflections which have not been used for refinement (unbiased data set) Resolution Limited by: 1. Quality of crystal (Conformational inhomogenities, Mobility of individual atoms) 2. Number of unique reflections used for structure determination (at least four times more than atoms required) 3. Typically not by wavelength 5Å 3Å 2Å - a helices roughly visible - peptide chain visible, side chains if sequence known - conformations of side chains visible Temperature Factor (B factor) • Describes local imprecision in electron density • Calculated during structure refinement • Measured in Ų The higher the B factor, the more imprecise the atom position. Good protein structure: B < 20 - 30 Ų Reasons for higher B factor: 1. Thermal motion of atom 2. Different conformations of side chain, i.e. lower occupancy at specific position 3. Protein disordered, e.g. In surface exposed loops Other Important Parameters • Protein backbone should have only allowed conformations (as described in Ramachandran plot) • Completenes of structure (should be >90%) Parameters in Publication Rasmussen et al., Science 2007 Protein Data Base • structure data (x, y, z coordinates of each atom) are submitted to Protein Data Base • freely accessible, structure can be downloaded and visualized with free programs such as RasMol or Swiss PDB Viewer Native Conformation? Is protein structure determined from crystal the same as in vivo solution structure? Most likely: 1. Crystals contain 40 – 60 % solvent, i.e. the protein is in simialr environment as in cell 2. Often proteins crystallize in different space groups with different crystal contacts between proteins, but the determined structure is the same, i.e. It is independent of crystal packing. 3. Often X-ray structures and solution NMR structures superimpose well. 4. Many enzymes are active in crystalline state BUT: You never know! Exceptions are possible!