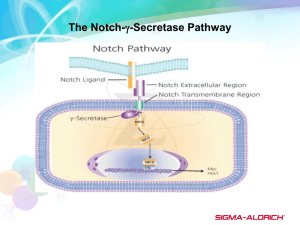
Biochemical Origins of Alzheimer's Disease With Treatment Techniques
advertisement

Biochemical Origins of Alzheimer’s Disease With Treatment Techniques Nikolai Wajda Senior Comprehensive Paper The Catholic University of America Spring 2007 Abstract: Alzheimer’s disease (AD) is a neurodegenerative disease caused by irregular protein formations in the brain leading to neuronal loss and ultimately affecting the patient’s cognitive ability and memory. AD affects nearly 4.5 million Americans, and this number is expected to continue to rise1. The pathological manifestations of AD occur in the neurons and are two-fold; the primary cause is the accumulation amyloid (Aprotein depositions, which aggregate into pathogenic plaques. The second is the accumulation of paired helical filaments that form into neurofibrillary tangles (NFTs). A plaques result from the sequential cleavage of the amyloid precursor protein (APP) by -secretase and secretase. NFTs result from the hyperphosphorylation of tau, a stabilizing component of microtubules. Based on current understanding of the Apathway, two major strategies will be discussed that aim at decreasing the deposition of Aplaques in the brain. In the first approach, non-streroidal anti-inflammatory drugs alter the APP cleavage site by -secretase to produce less amyoidogenic plaques. A second method aims at inhibiting -secretase activity on APP through allosteric inhibition of ATP binding. 2 Introduction: The first diagnosis of the disease that would eventually carry his name was done by Alois Alzheimer while treating a woman who showed early signs of dementia, which included progressive memory loss, delusions, and hallucinations2. Alzheimer, a German psychiatrist working at a mental institution in Frankfurt, Germany, made the critical breakthrough leading to the discovery of the pathological causes of this disease when he applied Max Bielschowsky’s silver staining technique to brain slides of the woman after her death. In the slides, Alzheimer identified two abnormal protein products accumulating in the neurons. While the composition was not known at that time, the components would later be identified as -amyloid plaques and neurofibrillary tangles (NFTs), the hallmark causes of AD. amyloid is an irregular protein product that accumulates into large plaques in extra-cellular spaces and inhibits intercellular communication, ultimately leading to neuronal death. NFTs result from the hyperphosphorylation of tau, which leads to the destabilization of microtubules in neurons. This allows the hyperphosphorylated tau to accumulate into tangled protein masses that blocks both intracellular communication and transportation of nutrients in the cell. Drugs are being developed that target these pathological causes. Many biochemical techniques have been utilized for the development of therapeutics that will inhibit the production of these pathological proteins. In this paper, nucleotide binding site inhibition and cleavage site alteration by nonsteroidal anti-inflammatory drugs (NSAIDs) will be discussed. 3 Production of Amyloid Plaques by Protease Activity Amyloid-plaques result from the cleavage of the amyloid precursor protein (APP) by a family of proteinases, termed secretases. The gene encoding APP is located on chromosome 21, which is consistent with the predominance of plaques developing in patients with Down’s Syndrome3. The normal function of APP is not well understood, but it is believed that APP could act as a cell-surface receptor, could participate in cellcell adhesions, or could promote neuritic growth. APP is a transmembrane protein consisting of 770 Amino acids; the protein has both intra- and extra- cellular segments. Three sites for secretase cleavage occur on APP, secretase cleaves APP at AA 687 in the extracellular region, secretase cleaves APP at AA 671 in the extracellular region, and -secretase cleavage occurs at various locations in the transmembrane region. cleavage precludes production of the amyloidogenic A proteins, however, sequential cleavage of APP by and secretases produces the amyloidogenic A proteins4. Amyloid-is a normal protein product, with an unknown function, that is formed by the cleavage of APP by secretases. Two routes exist for the sequential cleavage of APP, the first pathway produces the normally secreted product, p3, while the second results in the pathological amyloid plaques6. The transmembrane region of APP is between amino acids 700-723; the N-terminus, amino acids 1-699, is extracellular while the COOH terminus, amino acids 724-770, is in the cytoplasm of the cell. When APP is cleaved by -secretase after Lys687, the result is the release of a large soluble ectodomain fragment ( -APPs) leaving the 83 residue COOH-terminal fragment (APP-CTFor C83) with a transmembrane, extracellular, and an intracellular region. APP-CTF is subsequently cleaved by -secretase at either Val711 or Ala713, which results in p3, a non4 pathogenic peptide fragment consisting of either 26 or 24 amino acids; a second protein fragment, AICD, remains embedded in the membrane (Figure 1). The alternate route for proteolytic cleavage of APP begins with -secretase cleavage after Met671. This cleavage results in the secretion of a large soluble ectodomain fragment (-APPs) and the retention of a 99-residue COOH-terminal fragment (APP-CTFor C99). APP-CTF is then cleaved by -secretase at either Val711 or Ala713. This cleavage is important because if C99 is cut at position 711 it will result in A40 while if APP is cut position 713 it will result in A424 (Figure 1). Cleavage also results in AICD. Figure 1: The cleavage of amyloid precursor protien by secretases.4 The amyloidogenic nature of A fragments is dependent upon the -secretase cleavage site. In studies of the brains of Down’s syndrome patients, who often develop the trademark plaques young in age, it was found that A42 aggregates much more 5 rapidly than A40. In patients fifty years or younger it was found that only 6.3% of senile plaques were A40, while in older brains, fifty or older, the amount of A40 increased to 42%5. A42 has two additional hydrophobic amino acids than A40; the presence of isoleucine and alanine increase the amyloidogenic property of the fragments leading to increased aggregation4. Tau forms Neurofibrillary Tangles Following Hyperphosphorylation: Tau proteins are a normal cellular product existing in six different isoforms. In healthy neurons, tau binds to and stabilizes microtubules (MTs) as well as promotes tubulin assembly and polymerization. Tau is most prominent in neurons because MTs form the axons that allow for intracellular transportation of neurotransmitters and nutrients. The disruption of microtubules leads to the inability to transport nutrients and other vital materials in the cell, and through the accumulation of neurofibrillary tau the ultimate result will be neuronal death. Tau is phosphorylated in areas that surround MT binding repeats; thus increased tau phosphorylation negatively affects tau’s ability to bind and stabilize MTs7. Hyperphosphorylation leads to both gain and loss of tau activity that results in neurotoxicity. Hyperphosphorylated tau destabilizes MT binding and increases tau-tau interactions. Destabilized MT does not transport the necessary nutrients to other parts of the neuron. An example of this is that destabilized MTs cannot transport neurotransmitters from the cell body to the synapse where they can be released. Cutting off a neuron’s ability to communicate with other cells effectively isolates it, causing it to die. The increased tau-tau interactions of hyperphosphorylated tau causes the formation of paired helical filaments that constitute neurofibrillary tangles (NFT’s). Paired helical 6 filaments are composed of two strands of filament twisted around one another with a periodicity of 80 nm and a width varying from 8 to 20 nm7. Cyclin dependent kinase 5 (Cdk5) is one of many kinases active in the hyperphosphorylation of Tau. A cyclin-like membrane protein, p35, regulates Cdk5’s phosphorylation activity by anchoring it to the plasma membrane. Calpain, a protease, cleaves p35 into two fragments, p10 and p25; P10 anchors the active p25-Cdk5 complex to the plasma membrane. Upon cleavage, the active p25-Cdk5 complex is unregulated and free to phosphorylate throughout the cytoplasm (Figure 2). The p25-Cdk5 complex increases phosphorylation and decreases the binding ability of tau on microtubules8,9. Tau that is phosphorylated by P25-Cdk5 has reduced binding to microtubules and causes the collapse of microtubule and formation of NFTs10. Figure 2: The Cdk5, a kinase responsible for phosphorylation of Tau, is regulated by cofactor p35.8 Risks Factors The largest risk factor for the development of AD is increasing age. After 65 years of age, the incidence and prevalence of Alzheimer’s disease doubles every 5 years. 7 Genetic studies have been able to isolate inheritable factors contributing to AD, as well as genetic mutations, which increase the risk for developing AD. Other than the fact that autosomally dominant variety of AD is early in onset, the phenotypic expression of familial versus sporadic AD is difficult to distinguish. There are four known genetic factors which are related to the amyloid- pathway, the inheritance of the E4 allele of apolipoprotein, or mutations in either Presenilin-1 (PS1), Presenilin-2(PS2), or APP11,12,13. Mutations in certain genes influence the development of A plaques Missense mutations in the APP gene, in the PS1gene, or in the PS2 gene increase the likelihood for developing Alzheimer’s disease. The gene encoding the amyloid precursor protein is located on chromosome 21; missense mutations arising in this gene that have been shown to confer an increased risk to AD. Missense mutations in the APP gene increase the overall amount of Athrough the alteration of cleavage sites for the processing of proteases (Figure 3). Missense mutations in APP, however, are considered very rare and are found in only about two dozen families. It is believed that these mutations effect proteolytic activity by the secretases because the mutations are all located either directly before or directly after the secretase cleavage sites4. 8 Figure 3: Missense mutations occurring in APP which lead to the production of A4 Double Mutations occurring before the -secretase site (K670N and L671M) induce -secretase cleavage, and allows for increased amounts of the A precursor. Five mutations occur on the COOH-terminal side of the -secretase cleavage site(I714T, V715M, I716V, and I,G,F,L717V); these mutations increase the production of A, which is the A form that most readily forms into plaques. Three APP mutations occur inside the and cleavage sites (A692G, Q693E, and N,K,G694D); these mutations increase the aggregation properties of all Aresidues Missense mutations in the presenilin genes also increase the risk for developing AD. So far, there are 75 known missense mutations in the PS-1, which is located on chromosome 14, and only three to PS-2, located on chromosome 1, which are known to cause AD. Mutations in the PS-1 gene cause the most common form of dominant early 9 onset familial AD. PS-1 mutations selectively increase precursor C99 cleavage by secretase to produce more A42.; A42 is a more amyloidogenic peptide compared with A4014. In its complete and functional form, -secretase consists of PS-1 or PS-2, nicastrin (nct), Aph-1 and PEN-2. PS-1 is found to play a vital role in the enzymatic cleavage of APP by -secretase. PS-1 is made up of 9 transmembrane domains, and is activated as the result of endoproteolysis. Endoproteolysis occurs in domain 7 of presenilin, and creates the N-terminal fragment (NTF) and the C-terminal fragment (CTF). The full length PS-1 protein is short lived, wheras the processed protein has a much longer half life, suggesting that cleavage is necessary for function15. Determining the structure of -secretase The necessity of all four of these proteins for -secretase activity was proven following the expression of the genes encoding for these proteins in Saccharomyces cerevisiae by Steiner et al16. S. cerevisiae does not contain -secretase, so it is possible to address the question of which components of the complex are necessary for activity by selectively introducing the four components. An APP based protein that contains a sequence analogous to the -secretase cleavage site was synthesized in order to report secretase activity. This protein is bound to GAL4, a transcription factor for βgalactosidase (β-gal). Cleavage of C1–55–GAL4 after co-expression of functional γsecretase is expected to liberate GAL4 from the cytosolic side of the membrane. GAL4 then translocates to the nucleus where it activates transcription of the Escherichia coli LacZ gene, which encodes β-gal14. β-gal breaks down the sugar lactose, producing a blue 10 color that can be measured and quantified. In this experiment, the presence and intensity of the blue coloring is a test for -secretase cleavage of C1–55–GAL4 (Figure: 4). Figure 4: B-Gal activity to determine components of functional -secretase.14 GAL4: Expression of GAL4 protein as a control shows high B-Gal activity PS1wt: Expression of the PS-1 without Nct, APH-1, or Pen 2 showed no b-gal activity PS1wt: Expression of the PS-1 with Nct, APH-1 and Pen 2 showed high b-gal activity PS1d3854A: Expression of a functionally inactive PS-1 with Nct, APH-1, and Pen 2 showed almost no activity No NCT: Expression of PS-1, APH-1, and Pen 2 without NCT showed almost no activity No APH-1: Expression of PS-1, Nct, and Pen 2 without APH-1 showed almost no activity No PEN-2: Expression of PS-1, Nct, and APH-1 without PEN-2 showed almost no activity No PS1: Expression of Nct, APH-1, and PEN2 without PS-1 showed no activity. The only complex that showed significant cleavage of the APP-like substrate leading to -galactoside activity was the complete complex containing PS-1, nicastrin (nct), Aph-1 and PEN-2. When the complex lacked even one of these components, 11 activity was significantly decreased16. In further experiments with mice, the results showed that deleting PS-1 leads to the reduction of -secretase activity16. The vital discovery that PS-1 was required for producing A led researchers to question how PS-1 cleaves APP. Wolfe et al. determined that two aspartate residues on PS1 were required for the endoproteolytic and γ-secretase function. Introducing mutations to two intermembrane aspartates in PS-1, D275A and D385A or D275E, reduced the production of both A40 and A42 while increasing the amounts of A precursor substrates C83 and C99. The Asp to Ala double mutations led to substantially less total A (mean 57 +/- 3%) and A42(53 +/- 4%) than cell lines expressing wild-type PS-117. D275E mutations was done to see if removing the negative charge of the Asp had a detrimental effect on protein folding that could have resulted in decreased cleavage. However, Glutamate, with its negative charge still resulted in decreased A production 17. An increase in concentrations of both APP-CTFand -APPs shows that both the and secretases are still active, while the decreased concentrations of A means that -secretase is unable to process these fragments, contributing evidence that the aspartates are required for proteolytic activity. The double mutations of the aspartates also halted endoproteolytic activity of presenilin17. Levels of A produced from cells with aspartate double mutations were found to be similar to levels where PS1 was deleted17. Furthermore, the activity of the enzyme requires a slightly acidic pH, which contributes more evidence to this theory of PS-1 as an aspartyl protease18. 12 Current Treatment: Current therapeutics are insufficient for the treatment of AD. The most popular drugs used to treat AD are acetylcholinesterase (AChE) inhibitors. AChE inhibitors block the enzyme responsible for the degradation of the neurotransmitter acetycholine in the synapse. As a result of neuronal loss associated with AD, patients suffer from decreased levels of this neurotransmitter, thus inhibiting the activity of AChE, more acetylcholine will be present at the synapse. The second method used for treating AD is memantine, which is an uncompetitive NMDA receptor antagonist. Memantine regulates glutamate, a messenger chemical required for information processing, storage and retrieval. Glutamate binds to NMDA receptors, and allows calcium to enter nerve cells in controlled amounts. Calcium influx is required for information storage, however, too much glutamate overstimulates NMDA receptors and allows too much calcium into cells, disrupting cells and causing death. Memantine protects against excess glutamate activity by partially blocking NMDA receptors. The current treatments for AD only serve to ameliorate the side-effects without curing the disease, therefore researchers seek novel methods for curing AD1. -Secretase Inhibitors Research targeting the enzyme -secretase as a potential target for combating AD has shown some promise as well as some setbacks. Inhibiting -secretase cleavage of APP would prevent the second step in the sequential cleavage of the Aandthus decrease the amount of amyloid plaque developing in the brain. Research has focused on two approaches for altering -secretase activity. The first method is by inhibiting the creation of A of all lengths, while another is to increase the formation of shorter, less 13 pathogenic Afragments. Several inhibitors have been found to be successful in halting A production; developers, however, hit a roadblock when complications arose due to the fact that -secretase is also being responsible for cleaving other membrane proteins vital to cellular survival. LY-450139, the first -secretase inhibitor to go through clinical trials was found to decrease plasma levels of A however; it was unable to lower cerebrospinal fluid levels19. While this drug was found to be partially effective and well tolerated, LY-450139 lacked the desired specificity for the cleavage of APP and dosages had to be kept low because of the detrimental side effects that it would have have. secretase is active in the cleavage of several substrates other than APP, including Notch, a transmembrane cellular protein active in cell signaling. Notch first requires a signal to bind to its extracellular domain that will cause cleavage and the release of this domain. Notch then undergoes transmembrane cleavage by -secretase, which releases an intracellular fragment termed, Notch Intracellular Domain, NICD, which enters the nucleus and causes transcriptional changes in the cell20-21. In vivo studies of a -secretase inhibitor, LY-411,575, by Wong et al., showed decreased production of A, but also showed deleterious effects in mice. Large impairments of the spleen, thymus, and intestine were observed. The most pronounced side effects of the -secretase inhibitor were caused by the inhibition of Notch cleavage, ultimately leading to gastrointestinal toxicity and interference with the maturation of B- and T-lymphocytes21-22. From these initial studies into the inhibition of secretase, a new direction must be taken for drugs aimed at this enzyme. The new direction must be one that can inhibit the production of amyloid plaques without affecting the cleavage of other -secretase targets. Two approaches will be discussed below, one will be the use of NSAID’s which aim to alter 14 the cleavage site of -secretase and thus produce shorter, less amyoidal plaques while the second will seek to inhibit -secretase activity on APP through allosteric inhibition of ATP binding. Nonsteroidal Anti-Inflammatory Drugs The formation of less amyloidogenic plaques is an approach taken by researchers. Instead of inhibiting the secretion of all amyloid plaques, certain drugs will aim at decreasing the percentage of the longer, and more amyloidogenic plaques. In order to circumvent the problems associated with the lack of inhibition specificity for APP by -secretase inhibitors, a new generation of more specific inhibitors are being investigated. These drugs exhibit greater selectivity towards the inhibition of APP cleavage and allow for normal enzymatic cleavage towards Notch. NSAIDs have been shown to decrease the amount of the more amyloidogenic A42 by altering the binding site of -secretase. In a study conducted by Beher et al., sulindac sulfide, an NSAID, and R-flurbiprofen, a NSAID-like compound were tested for their ability to specifically inhibit -secretase activity on APP. A B Figure 5: Structure of sulindac sulfide (A) and R-flurbiprofen (B).23 The two compounds decreased overall production of both A40 and A42. (Figure: 5, A: sulindac acid and B: R-flurbiprofen). For R-flurbiprofen, A42 production was reduced by 75% when compared to the control. The study also showed that 15 increasing the concentration of either R-flurbiprofen decreased the rate of production of both A40 and A42 (Figure 6:C and D). Kinetic assays were undertaken to determine the mechanism of R-flurbiprofen inhibition, which showed that secretase enzyme velocity decreased as the concentration of drug increased. A Lineweaver-Burke plot from the inhibition kinetic assays of A42 by R-flurbiprofen demonstrated a noncompetitive mode of inhibition. As the concentration of drug was increased, the plot showed a decrease in Vmax with an while the Km remained the same, data consistent with noncompetitive inhibition (Figure 6:E). These data suggest that the NSAIDs bind at a site independent of the substrate binding site23. Figure 6: Certain NSAIDs decrease the rate of A production via noncompetitive inhibition.23 A) Decrease in A levels from treatment with Sulindac Sulfide B) Decrease in A levels from treatment with R-flurbiprofen C) -secretase velocity (production of A40) decreases upon treatments with Rflurbiprofen D) -secretase velocity (production of A42) decreases upon treatments with Rflurbiprofen E) Lineweaver-Burke Plot showing decrease in Vmax while Km remains the same 16 Further evidence that the binding of NSAIDs occurs at a site other than the catalytic site was performed using a transition state radioligand, which bind to the catalytic site of the enzyme. Competition studies using tritiated Merck A, the transition state analog inhibitor, showed that both R-flurbiprofen and sulindac sulfide displace the radioligand. The noncompetitive nature of these two compounds suggests that they cause conformational changes to the enzyme complex, which lead to decreased binding of the transition state inhibitor (Figure 7). A B Figure 7. -secretase displacement assays.23 The displacement of a transition state analog, tritiated Merck A, by Sulindac Sulfide (A) and R-flurbiprofen (B). A proposed mechanism for this action was proposed by Hyman et al, who found that the NSAIDs affect the conformation of PS-1, which leads to altered APP-PS1 conformation. The distance between the N-terminus and C-terminus was found to be increased after the addition of s-ibuprofen, indomethacin, or flurbiprofen. (Figure 7) The addition of these drugs causes a decrease in A42 and an increase in the less amyloidogenic A3824. 17 A) B) C) Figure 7: Structures of s-ibuprofen (A), indomethacin (B) flurbiprofen (C).24 Inhibition of secretase via the ATP Binding Site Another approach to selectively inhibit -secretase enzymatic cleavage of APP is by blocking the ATP binding domain. Wolfe et al. determined that production of Aand AICD by -secretase cleavage of APP was dose-dependent on ATP concentrations. The incubation of 1uM C100Flag (an APP based substrate), purified -secretase, and different concentrations of ATP produced AICD-Flag, a fragment analogous to AICD, which is produced in the normal pathway. As the concentration of ATP increases, the amount AICD was found to increases (Figure 8). Using the ELISA technique with anti-A antibodies, it was determined that as the ATP concentration increased from 1mM to 5mM, a 1.75 fold increase in A occurred. 18 Figure 8: ATP increases the hydrolysis of APP-based substrate.25 The group next sought to determine whether ATP concentration had an effect on the production of a Notch substrate. Wolfe et al. found that increasing the concentration of ATP did not increase the production of Notch fragments. This critical discovery led the researchers to believe -secretase cleavage of APP could be selectively inhibited without affecting the cleavage of other membrane targets, which was a problematic with active site inhibition of -secretase. The next step was to determine how ATP was responsible for -secretase activity. The high energy released upon the cleavage of ATP’s terminal phosphate group is the source of energy for many enzyme reactions. To see if -secretase requires cleaving ATP into ADP or AMP, the enzyme was incubated with ATP in the presence of and in the absence of an APP-based substrate. Following incubation, the amounts of ADP and AMP were measured and it was found that increased hydrolysis did not occur in the sample incubated with the substrate (Figure 9). This suggests that ATP hydrolysis is not required for the activity of -secretase25. 19 Figure 9: -secretase does not require ATP hydrolysis.25 Lanes 1-5 show fragments of ATP that have been hydrolyzed by phosphotase to form ADP and AMP. Samples 6-10 were incubated with ATP, and -secretase. Upon electrophoresis, it was shown that no hydrolysis occurred. Samples 21-25 were incubated with ATP, -secretase, and an APP-like substrate. Upon electrophoresis, it was shown that no hydrolysis of ATP occurred. Contributing further evidence for this hypothesis was a study that found a 1mM concentration of ATPS, a nonhydrolyzable ATP analog, increased production of A25. This meant that merely the binding of ATP is necessary for -secretase cleavage. The researchers were unable to find the exact effect that ATP has on the protein, however they suggest that nucleotide binding causes a conformational change that allows the protein to recognize APP. Wolfe’s research group also investigated potential ligands for the inhibition of APP cleavage. They found that tyrosine kinase inhibitors are potentially potent therapeutics because they bind to ATP-binding sites and block the cleavage of an APPbased substrate. Compound 1367, one of the tyrosine kinase inhibitors, was also found to stimulate the cleavage of a Notch-like substrate25. 20 Figure 10: Compound 1367 inhibits APP cleavage and induces Notch cleavage.25 At concentrations higher than 30m, Compound 1367 was found to be a potent inhibitor of -secretase cleavage of an APP-like substrate, AICD-FLAG. Further studies showed that 1367 actually increased Notch cleavage (NICD-Flag) This approach shows much promise because of its ability to limit the production of A peptides without inhibiting the cleavage of Notch, and in fact stimulating its production. CONCLUSION The wealth of knowledge concerning Alzheimer’s disease is rapidly increasing through the use of many biochemical techniques that aim at determining the root causes of this debilitative neurological disorder. From Alois Alzheimer’s detection of abnormal protein deposits in brain slides, both the -amyloid plaques and neurofibrillary tangles have been researched extensively in the hopes of finding cures to halt their progressive debilitating effects. Aprotein fragments accumulate in intercellular spaces and cause neuronal death. These fragments are created through the action of a family of proteases termed secretases. While their activity produces a natural product, p3, the pathological causes of AD are traced to the incorrect cleavage of the amyloid precursor protein by -secretase. Through 21 many well designed and well thought out experiments, it was found that -secretase is a mulitpass transmembrane aspartyl protease, which gave further insight into the mechanism for A production. Understanding the biochemical mechanisms allows researchers to create therapeutic agents that could halt -secretase activity, and thus the progression of AD. Currently, two major approaches are in clinical trials to test their efficacy towards halting A production. The first method is through the use of Nonsteroidal anti inflammatory drugs, which induce cleavage of the amyloid precursor protein at a locus that will produce a smaller fragment that are less prone to accumulation. The second approach came from the knowledge that -secretase contains an ATP binding domain. By blocking this site, it was found that the production of Aproteins can be halted without stopping other important biological processes of secretase, including Notch cleavage. The secondary cause of AD, one that has also been found in several other neurological diseases, is the accumulation of neurofibrillary tangles within the neuron. NFTs results from the hyperphosphorylation of the tau protein. Tau normally binds to, and stabilizes microtubules in neurons, but when they are phosphorylated in the segments that usually bind the tubulin filaments their binding ability is decreased. Destabilized tau proteins begin accumulating in the cell and disrupting cellular transportation and communication. Continuing knowledge is being amassed concerning the kinases responsible for the hyperphosphorylation so that appropriate drugs can be designed to halt their phosphorylation activity. As the wealth of knowledge concerning AD continues to be amassed the possibility for creating newer and more effective drugs is increased. Significant findings 22 concerning the inheritability factors are also being researched. While the largest contributing factor to developing AD is increasing age, there are several inheritable risk factors that increase the risk for developing AD. These too are being researched in order to tailor specific drugs and treatments for these problems. References: 1 Alzheimer’s Association. www.alz.org. December 27, 2006. 2 Goedert M.; Grazia Spillantini M. Science. 2006, 314, 777-781. (3) Argellati, F.; Massone, S.; d'Abramo, C.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C.; Ricciarelli, R. IUBMB Life. 2006, 58, 103-106. (4) Selkoe, D.J; Physiol. Rev. 2001, 81, 741-766. (5) Iwatsubo, T.; Mann, D.M.; Odaka, A.; Suzuki, N.; Ihara. Y. Ann Neurol. 1995, 37, 287-8. (6) Haass C; Hung AY; Schlossmacher MG; Teplow DB; Selkoe DJ. J Biol Chem, 1993, 268, 3021-3024. (7) Crowther, R.A.; Wischik, C.M. EMBO J. 1985, 4, 3661–3665. (8) Mandelkow, E.; Nature. 1999, 402, 588-589. (9) Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte S.; Dikkesk, P.; Tsai, L.H. Nature. 402, 1999, 615-22. (10) Kusakawa, G.; Saito, T.; Onuki R.; Ishiguro, K.; Kishimoto, T.; Hisanaga, S; J. Biol Chem. 275. 2000, 17166–17172. (11) McCullagh, C.D.; Craig D.; McIlroy, S.P.; Passmore, A.P. Adv. In Psych. Treatment. 2001, 7, 24-31. 23 (12) Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Proc. Natl. Acad. Sci. USA. 2006, 103, 5644-5651. (13) Stittmatter, W.J.; Roses, A.D. Proc. Natl. Acad. Sci. USA. 1995, 92, 4725-4727. (14) Lemere, C.A.; Lopera, F.; Kosik, K.S.; Lendon, C.L.; Ossa, J.; Saido, T.C.; Yamaguchi, H.; Ruiz, A.; Martinez, A.; Madrigal, L.; Hincapie, L.; Arango, J.C.; Anthony, D.C.; Koo, E.H.; Goate, A.M.; Selkoe, D.J.; Arango, J.C. Nat Med. 1996, 2,1146-50. (15) Ratovitski, T.; Slunt, H.H.; Thinakaran, G.; Price, D.L.; Sisodia, S.S.; Borchelt, D.R. J. Biol. Chem. 1997, 272, 24536–24541. (16) Edbauer, D.; Winkler,E.; Regula, JT.; Pesold, B.; Steiner H.; Haass, C. Nature Cell Biology. 2003, 5, 486-488. (17)Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Nature. 1999, 398, 513-7. (18) De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Gundula, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Nature. 1998, 391, 387-390. (20) Roberson, E.D.; Mucke, L. Science. 2006, 314, 781-784. (21) Struhl G.; Adachi, A. Molecular Cell. 2000, 6, 625-636. (22) Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, G.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.J.J.; Zhang, L.; Higgins, G.A.; Parker, E.M. J. Biol. Chem, 2004 279, 12876-12882. (23) Beher, D.; Clarke, E.E.; Wrigley, J.D.J.; Martin, A.C.L.; Nadin, A.; Churcher, I.; Shearman, M.S.; J. Biol. Chem. 2004, 279, 43419-43426. (24) Lleo, A., Berezovska, O., Herl, L., Raju, S., Deng A., Bacskai BJ., Frosch, MP., Irizary M., Hyman BT. Nature Medicine. 2004, 10, 1065-1066. 24 (25) Wolfe, M.S.; Slekoe, D.J.; Fraering P.C.; Ye, W.; LaVoie, M.J.; Ostaszewski, B.L.; J. Biol. Chem. 2005, 280, 41987-41996. 25