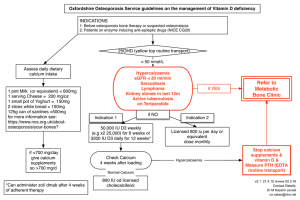
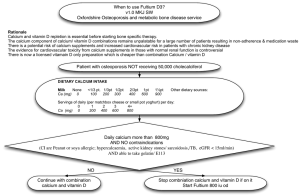
BONE DEVELOPMENT AND MINERAL HOMEOSTASIS IN THE FETUS AND NEONATE: PHOSPHOTROPIC HORMONES
advertisement

Physiol Rev 94: 1143–1218, 2014 doi:10.1152/physrev.00014.2014 BONE DEVELOPMENT AND MINERAL HOMEOSTASIS IN THE FETUS AND NEONATE: ROLES OF THE CALCIOTROPIC AND PHOSPHOTROPIC HORMONES Christopher S. Kovacs Faculty of Medicine-Endocrinology, Memorial University of Newfoundland, St. John’s, Newfoundland, Canada Kovacs CS. Bone Development and Mineral Homeostasis in the Fetus and Neonate: Roles of the Calciotropic and Phosphotropic Hormones. Physiol Rev 94: 1143–1218, 2014; doi:10.1152/physrev.00014.2014.—Mineral and bone metabolism are regulated differently in utero compared with the adult. The fetal kidneys, intestines, and skeleton are not dominant sources of mineral supply for the fetus. Instead, the placenta meets the fetal need for mineral by actively transporting calcium, phosphorus, and magnesium from the maternal circulation. These minerals are maintained in the fetal circulation at higher concentrations than in the mother and normal adult, and such high levels appear necessary for the developing skeleton to accrete a normal amount of mineral by term. Parathyroid hormone (PTH) and calcitriol circulate at low concentrations in the fetal circulation. Fetal bone development and the regulation of serum minerals are critically dependent on PTH and PTH-related protein, but not vitamin D/calcitriol, fibroblast growth factor-23, calcitonin, or the sex steroids. After birth, the serum calcium falls and phosphorus rises before gradually reaching adult values over the subsequent 24 – 48 h. The intestines are the main source of mineral for the neonate, while the kidneys reabsorb mineral, and bone turnover contributes mineral to the circulation. This switch in the regulation of mineral homeostasis is triggered by loss of the placenta and a postnatal fall in serum calcium, and is followed in sequence by a rise in PTH and then an increase in calcitriol. Intestinal calcium absorption is initially a passive process facilitated by lactose, but later becomes active and calcitriol-dependent. However, calcitriol’s role can be bypassed by increasing the calcium content of the diet, or by parenteral administration of calcium. L I. II. III. IV. V. VI. VII. VIII. IX. X. XI. XII. XIII. INTRODUCTION OVERVIEW OF FETAL MINERAL... OVERVIEW OF NEONATAL... PARATHYROID HORMONE-RELATED... PARATHYROID HORMONE ROLE OF PTHrP AND PTH... CALCIUM-SENSING RECEPTOR CALCITRIOL AND VITAMIN D CALCITONIN FIBROBLAST GROWTH FACTOR-23 SEX STEROIDS FETAL AND NEONATAL RESPONSES... CONCLUSIONS 1143 1145 1164 1168 1174 1178 1180 1182 1193 1194 1196 1196 1200 I. INTRODUCTION Normal calcium and bone homeostasis in the adult can be almost fully explained by the interactions of several regulatory hormones, including parathyroid hormone (PTH), calcitriol, fibroblast growth factor-23 (FGF23), calcitonin, and the sex steroids (estradiol and testosterone). Loss of any one of these hormones can have significant consequences for the adult. Hypoparathyroidism can cause fatalities due to hypocalcemia; it also causes hyperphosphatemia (leading to ectopic calcifications), hypercalciuria (leading to nephrolithiasis and nephrocalcinosis), low bone turnover, and possible neurological sequelae from basal ganglia calcifications. With vitamin D deficiency, the serum calcium may be normal or reduced (but not as low as in hypoparathyroidism), serum phosphorus is low, and undermineralization of the skeleton leads to rickets or osteomalacia. Loss of FGF23 causes hyperphosphatemia, extraskeletal calcifications, and early mortality. Loss of calcitonin increases bone resorption, an effect that may become most apparent during lactation. Deficiency of either of the sex steroids increases bone resorption and will lead to osteoporosis. The roles that these hormones play in regulating osteoblasts or osteoclasts have led to some of them (or their synthetic analogs) being used to treat osteoporosis and other skeletal disorders. This review will make clear that mineral and bone metabolism are regulated differently in utero such that many of these hormones do not appear to have the crucial roles that they have in the adult. 0031-9333/14 Copyright © 2014 the American Physiological Society 1143 CHRISTOPHER S. KOVACS The fetus has several developmental goals, which include the need to actively pump minerals across the placenta against concentration and electrochemical gradients, to maintain higher extracellular concentrations of minerals compared with the maternal circulation, and to adequately mineralize the skeleton before birth. During embryonic development the early pattern for the endochondral skeleton is laid down, but it is during the later stages of fetal development that rapid bone formation and mineralization create a significant demand for mineral. The adequacy of the mineral supply is a limiting factor for how much mineral can be accreted by the skeleton before birth, and the availability of mineral also influences the function and activity of osteoblasts and osteoclasts. A human fetus typically accumulates ⬃30 g of calcium by term, with 80% of that mineral content obtained in the third trimester (121, 224, 683, 733, 734). The kidneys, intestines, and skeleton (through bone turnover) are not dominant sources of mineral supply for the normal fetus as they are for the adult (FIGURE 1). Instead, the placenta meets the fetal need for mineral by actively transporting calcium, phosphorus, and magnesium from the maternal circulation. To obtain 30 g of calcium by term, the placenta must deliver 120 –150 mg·kg⫺1·day⫺1 of calcium during the third trimester, or more than 300 mg/day between weeks 35 and 38 (635, 734, 735, 761). The placenta appears capable of providing mineral even when faced with reduced concentrations of those same minerals in the maternal circulation. The fetus maintains higher mineral concentrations than in the mother or normal adult, and such high levels appear necessary for the skeleton to form and mineralize normally. As this review will demonstrate, fetal bone and mineral metabolism are critically dependent on PTH and PTH-related protein (PTHrP), whereas vitamin D/calcitriol, FGF23, calcitonin, and the sex steroids may not be required. The intestines become the main source of mineral after birth, while the kidneys begin to reabsorb mineral, and normal bone turnover contributes additional mineral to the circulation. This switch in the regulation of mineral homeostasis is triggered by a postnatal fall in serum calcium, and followed in sequence by a rise in PTH and then an increase in calcitriol. FGF23 then begins to play an important role in regulating serum phosphorus, renal phosphorus excretion, and calcitriol synthesis and catabolism. Intestinal calcium absorption is initially a passive process facilitated by lactose, but later becomes active and calcitriol-dependent. However, calcitriol’s role can be bypassed by increasing the calcium content of the diet, or by administering parenteral calcium, thereby preventing or correcting the skeletal changes of rickets or osteomalacia, independent of calcitriol or vitamin D sufficiency. There are only limited human data that directly examine regulation of mineral and bone homeostasis during fetal development. Much of those data consist of cord blood 1144 Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ Ca2+ FIGURE 1. Circulation of mineral within the fetal-placental unit. Calcium is represented here, but these statements apply to phosphorus (phosphate) and magnesium as well. At the top right, the main flux of mineral is across the placenta and through the fetal circulation into bone; however, some mineral returns to the maternal circulation (backflux). On the bottom right, the fetal kidneys filter the blood and excrete mineral into urine, which in turn makes up much of the volume of amniotic fluid. On the bottom left, amniotic fluid is swallowed, and its mineral content can be absorbed by the fetal intestines, thereby restoring it to the circulation. The renalamniotic-intestinal loop is likely a minor component for fetal mineral homeostasis. On the top left, although the net flux of mineral is into bone, some mineral is resorbed from the developing skeleton to reenter the fetal circulation. If placental delivery of mineral is deficient, fetal secondary hyperparathyroidism ensues, which causes more substantial resorption of mineral from the fetal skeleton, reduced skeletal mineral content, and possible fractures occurring in utero or during the birthing process. samples taken from normal and abnormal preterm and term fetuses and pathological examination of embryos and fetuses that died due to congenital abnormalities or obstetrical accidents. The main exception is with vitamin D and calcitriol, for which there have been randomized interventional trials, cohort studies, and associational studies, each examining possible effects of vitamin D deficiency on fetal and neonatal bone and mineral metabolism. In contrast to the limited human data, mineral and bone homeostasis have been extensively examined in several animal species using surgical, genetic, and pharmacological approaches. Regulation of fetal mineral homeostasis was first studied by administering pharmacological doses of hormones or antibodies to the mother or fetus, by making the mother severely vitamin D deficient before pregnancy, or by removing the thyroid or parathyroids from the mother, fetus, or both. But most calciotropic and phospho- Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM tropic hormones cannot be adequately studied by these approaches because they are produced by more than one tissue. Global and conditional gene deletion models have enabled us to study fetuses that lack PTHrP, PTH, calcitriol, the vitamin D receptor, calcitonin, or FGF23 (see TABLE 1 for a glossary of mouse models to be discussed). It is through consideration of these animal data that it becomes possible to theorize how fetal mineral homeostasis is regulated in the human, and how this regulation changes after birth. In this review, the term phosphorus is used for consistency and simplicity when referring to its presence in blood or bone. However, it is recognized that serum contains mainly inorganic phosphates (dihydrogen and monohydrogen phosphate), bone contains phosphorus largely in the form of hydroxyapatite, while the soft tissues and extracellular fluids contain organic phosphates in complex with carbohydrates, lipids, and proteins (37). To avoid any confusion between animal and human data, within each subsection this review will sequentially discuss animal data followed by the extant human data. The animal data will be used to infer how mineral and bone homeostasis are regulated during fetal and neonatal development, and the sometimes limited human data will reveal whether there is agreement or disagreement with what the animal models suggest. II. OVERVIEW OF FETAL MINERAL METABOLISM AND SKELETAL FORMATION This section describes the normal circulating concentrations (FIGURE 2) and sources (FIGURE 1) of minerals and hormones during fetal development, and the participation Table 1. Glossary of mouse models Name Protein(s) Encoded by the Gene Casr null CasrChon, Pre, Ob, Oc, Para null Calcium sensing receptor (CaSR) (257, 338) Calcium sensing receptor, conditionally deleted from chondrocytes, preosteoblasts, osteoblasts, osteocytes, and parathyroids, respectively (109, 169, 549) Calcitonin and calcitonin gene-related peptide-␣ (119, 260, 430, 744) Calcitonin receptor (134, 361) 1␣-Hydroxylase/Cyp27b1 (cytochrome P-450, family 27, subfamily B, polypeptide 1), which converts 25-hydroxyvitamin D into calcitriol (138–140) Fibroblast growth factor-23 (FGF23) (395, 626) Transcription factor glial cells missing (Gcm2), absence of which leads to loss of parathyroids (238, 384, 385, 623, 691) Glycoprotein 130 is a transmembrane protein and a receptor subunit common to many cytokines; it interacts with PTH (616, 753) Homeobox a3 ablation causes abnormalities in tissues derived from the third and fourth pharyngeal arches: loss of parathyroids and thymus, hypoplasia of the thyroid, loss of hyoid, and structural weakness in major arteries that lead hemorrhage at birth (114, 336, 341, 411) Phex (phosphate regulating endopeptidase homolog, X-linked) encodes an endopeptidase; an inactivating mutation in the gene leads to elevated FGF23 and models X-linked hypophosphatemic rickets (395, 484, 627). These are also known as Hyp mice due to the hypophosphatemia Parathyroid hormone (PTH) (310, 439, 440, 623) Double-mutant lacking PTH and calcitriol (747, 749) Double-mutant lacking both PTH and Hoxa3 (parathyroids) PTH1 receptor (PTH1R) also known as PTH/PTHrP receptor (336, 340, 341, 360, 714) Double mutant lacking PTH1 receptor and CaSR (341) Parathyroid hormone-related protein (PTHrP) (297, 336, 340, 570) Double mutant lacking PTHrP and CaSR (338) Double mutant lacking PTHrP and Hoxa3 (parathyroids) (336) Double mutant lacking PTHrP and PTH (439) TRPV6 (transient receptor potential cation channel, subfamily V, member 6) (659) Ctcgrp null Ctr null Cyp27b1 null Fgf23 null Gcm2 null Gp130 null Hoxa3 null Phex null Pth null Pth/Cyp27b1 Pth/Hoxa3 Pth1r null Pth1r/Casr Pthrp null Pthrp/Casr Pthrp/Hoxa3 Pthrp/Pth Trpv6 null VdrBos null VdrLeu, Tok, Mun null Vitamin D receptor (VDR); developed in Boston, the second zinc finger of the receptor DNAbinding domain has been deleted; there is no transcribed or translated receptor (24, 192, 342, 371–373) Vitamin D receptor (VDR); developed by three separate groups in Leuven, Tokyo, and Munich; each deleted the first zinc finger of the receptor DNA-binding domain, but a surrogate ATG downstream from the second zinc finger causes an aberrant VDR to be expressed which has normal ligand binding affinity but no DNA binding ability (175, 376, 574, 704, 755) Reference numbers are given in parentheses. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1145 CHRISTOPHER S. KOVACS of parathyroids, kidneys, intestines, placenta, and bone in regulating fetal mineral metabolism. The roles of the individual calciotropic and phosphotropic hormones are discussed in sections IV–XI. TOTAL Ca (mmol/l) 2.7 A. Serum Mineral Concentrations 1.6 1. Animal data IONOZED Ca (mmol/l) 2.1 A consistent finding among mammalian fetuses is that the serum calcium concentration is significantly higher than the simultaneous maternal calcium. Typically the fetal serum calcium is 0.30 – 0.50 mM or higher than the maternal level in rhesus monkeys (189, 524), lambs (148, 201, 435, 505), calves (201, 715), rodents (201, 338, 340, 342, 346, 347, 623), pigs (98, 101, 353), and foals (201). The ionized calcium is also increased 0.25– 0.50 mM above the maternal value in fetal rodents (148, 340, 422), confirming that true hypercalcemia is present relative to maternal and normal adult values. Although only ⬃50% of serum calcium is ionized in the adult rodent, within fetal rodents ⬃80% of calcium is free or ionized (148, 340). It is unknown how early in gestation the serum calcium becomes increased compared with the maternal value, but it has been demonstrated at all time points for which it has been technically possible to measure it. This includes the 35th day of gestation in lambs (93, 397), day 14 in rats (346, 347, 675), and day 15 in mice (330). Within rat fetuses, a small but progressive increase in total and ionized calcium occurs over the final 7 days, which may indicate that at an earlier time point there is no difference between embryonic/fetal and maternal calcium concentrations (346, 675). 0.9 PHOSPHATE (mmol/l) 1.7 0.9 PTH (pmol/l) 5.0 0.0 1, 25-D (pmol/l) 300 0.0 CALCITONIN (ng/l) 100 0.0 PTHrP (pmol/l) 5.0 0.0 0 1 2 3 4 DAYS POSTNATAL FIGURE 2. Schematic illustration of the longitudinal changes in calcium, phosphate, and calciotropic hormone levels that occur during the fetal and neonatal period in humans. Normal adult ranges are indicated by the shaded areas. The progression in PTHrP levels has been depicted by a dashed line to reflect that it is speculative. [From Kovacs and Kronenberg (339). Copyright 1997 The Endocrine Society; permission conveyed through the Copyright Clearance Center, Inc.] 1146 This level of blood calcium, both total and ionized, exceeds the value that the calcium sensing receptor (CaSR) sets in the adult through its regulation of PTH synthesis and secretion, and which is represented by the simultaneous maternal serum calcium concentration (338). CaSR likely acts in response to the high serum calcium to suppress release of PTH (FIGURE 3). When one or both alleles of Casr bear an inactivating mutation, the fetal serum calcium and PTH increase above normal fetal values, confirming that CaSR can raise the fetal serum calcium by increasing PTH (338). That CaSR requires PTH to raise the fetal serum calcium was confirmed by the finding that if the PTH/PTHrP receptor (PTH1R) is also ablated in Pth1r/Casr double mutants, thereby blocking the actions of PTH, then inactivating mutations of Casr fail to increase the fetal serum calcium (338). What sets the fetal blood calcium at its high target level in normal fetuses? One possibility is that novel calcium-sensing receptors are expressed during fetal development, which set the calcium concentration at specific values within the placental or fetal circulations. Alternatively, the active, forward flow of calcium across the placenta may be responsible for maintaining the high fetal blood calcium. If active Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM iCa2+ 1.75 mmol/l Fetal hypercalcemia due to CASR+/– • PTH with PTHrP iCa2+ 1.50 mmol/l Normal fetal ionized calcium • Combined effects of PTHrP and PTH iCa2+ 1.25 mmol/l Fetal hypocalcemia but normal adult level • Loss of PTHrP with compensatory PTH • Loss of PTH PTHrP iCa2+ 1.00 mmol/l Extreme fetal hypocalcemia • Loss of parathyroids • Loss of PTH and PTHrP • Loss of PTH1R FIGURE 3. Regulation of fetal ionized calcium. This schematic illustrates changes in ionized calcium as observed in murine fetuses and extrapolated to humans; the levels shown are meant as a guide for relative changes and are not meant to indicate precise values found in human cord blood. The normal ionized calcium level is higher in fetuses than in the normal adult, and is maintained by the combined effects of high concentrations of PTHrP and low levels of PTH in the circulation. CaSR likely suppresses PTH in response to the normal high level of calcium in fetal blood. The high fetal blood calcium may be set by a novel calcium-sensing receptor (other than the known CaSR), which regulates PTHrP release from the placenta and possibly other fetal tissues. In the absence of PTHrP, serum calcium falls but is now maintained at the normal adult level through the normal actions of CaSR to regulate PTH. Loss of PTH leads to a similar blood calcium value, approximately equal to the maternal calcium concentration, without a compensatory increase in circulating PTHrP. Combined loss of PTH and PTHrP, loss of parathyroids, or loss of PTH1R, each results in the lowest fetal ionized calcium values. Conversely, inactivating mutations of CASR cause PTH and the blood calcium to increase above normal fetal levels, in turn causing PTHrP to become decreased. placental calcium transfer is the explanation, then fetuses should maintain a relative gradient of serum calcium about the maternal level, and acute or chronic changes in maternal serum calcium should alter the fetal blood calcium. However, the fetus does not maintain a set gradient of serum calcium relative to the maternal serum calcium; instead, it sets a fixed level of calcium independent of the maternal calcium value. This value can be defended and maintained despite significant challenges. The evidence includes that fetal rats were normocalcemic despite maternal hypocalcemia induced by a calcium-restricted diet (377), vitamin D deficiency (74, 241, 443), or thyroparathyroidectomy (67, 276). Fetal lambs also maintained normal serum calcium levels despite maternal hypocalcemia induced by parathyroidectomy (564). In fetal mice both Casr (257) and Vdr (372) deletion models have revealed that fetuses will maintain the ionized calcium level determined by their genotype, regardless of whether the mother is normocalcemic, hypercalcemic, or hypocalcemic (FIGURE 4) (338, 342). In these models, a lower maternal serum calcium means that the calcium gradient from mother to fetus is increased, whereas a higher maternal serum calcium means that the gradient is decreased. But in all of these models the fetal ionized calcium was unchanged, confirming that it is the blood level of calcium that is set and not a relative gradient compared with the mother. These findings are consistent with calcium-sensing receptors that determine the fetal blood calcium level, as opposed to the fetal blood calcium being determined by active placental calcium transport. Moreover, acute alterations in the maternal blood calcium of rodents and primates (such as by infusions of calcium, calcitriol, calcitonin, PTH, or EDTA) had minimal or no effect on the fetal blood calcium level (87, 214, 345, 478, 547). However, although a high fetal blood calcium level was maintained in fetal rats between the 12th and 17th days of gestation after maternal parathyroidectomy, it declined during the last several days of gestation, the interval when the skeleton is most rapidly accreting mineral (106, 208, 220). What is the purpose of the fetus maintaining a higher blood concentration of calcium? The robust maintenance of this “fetal hypercalcemia” across numerous mammalian species suggests that it is physiologically important. But fetal hypocalcemia does not impair survival to the end of gestation, as shown by study of Pthrp null, Pth1r null, Hoxa3 null, Trpv6 null, Gp130 null, and Pthrp/Hoxa3 double mutant fetuses (336, 339, 341, 616, 659). Another possibility is that a high blood calcium level in utero may improve survival after birth. As discussed in section III, the postnatal onset of breathing, obligatory rise in pH, and loss of the placental calcium infusion contribute to a rapid fall in calcium within 12 h after birth. During the next 24 h, the serum calcium increases to the adult value. In rodents, the serum calcium drops more markedly by 40% after birth (201, 347). If the fetal blood calcium is lower than normal, conceivably the neonatal blood calcium will fall to an even lower value, thereby conferring an increased risk of hypocalcemic tetany and death. The early postnatal mortality of Pthrp null, Pth1r null, Pth null, Gp130 null, and Hoxa3 null fetuses is compatible with this possibility (297, 340, 341, 360, 616, 623). The high fetal calcium concentration also appears to be important for normal skeletal mineralization to be achieved, since hypocalcemia is associated with reduced ash weight and skeletal mineral content in Pth null, Hoxa3 null, Pth1r null, and Gcm2 null fetuses (see sects. VC and VIC) (336, 341, 623). The serum ionized calcium concentration differs significantly among fetuses obtained from different commercially available strains of mice; for example, fetuses from the commonly used inbred C57BL/6 strain maintain a significantly lower ionized calcium compared with fetuses from outbred Black Swiss (340). Therefore, unknown genes also contribute to the regulation of fetal mineral homeostasis, or specifically the set calcium level maintained by normal fetuses. Phosphorus plays key roles during endochondral bone development: it induces apoptosis of hypertrophic chondrocytes (158, 579) and is incorporated into osteoid before Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1147 CHRISTOPHER S. KOVACS A B Ionized calcium (mmol/L) 2.0 1.5 Casr +/- mothers Vdr +/- mothers WT mothers 1.0 0.5 Vdr null mothers 0.0 WT Vdr+/- Vdr null WT Casr+/- Casr null FIGURE 4. Fetal ionized calcium level is set independently of the maternal calcium value. Data have been merged for simplicity and schematically represented in this illustration. In A, the ionized calcium of Vdr⫹/⫺ and Vdr null mothers is represented by their respective horizontal lines ⫾ SE. Despite normocalcemia in Vdr⫹/⫺ mothers (who bore WT, Vdr⫹/⫺, and Vdr null fetuses) and hypocalcemia in Vdr null mothers (who bore Vdr⫹/⫺ and Vdr null fetuses), the maternal ionized calcium did not affect the ionized calcium level of their respective fetuses. The fetuses maintained a set level independent of the maternal ionized calcium, and that value was no different in Vdr null fetuses compared with WT and Vdr⫹/⫺ littermates. In B, the ionized calcium of WT and Casr⫹/⫺ mothers is represented by the respective horizontal lines (⫾ SE). Despite normocalcemia in WT mothers (who bore WT and Casr⫹/⫺ fetuses) and hypercalcemia in Casr⫹/⫺ mothers (who bore WT, Casr⫹/⫺, and Casr null fetuses), the maternal ionized calcium did not affect the ionized calcium level of their respective fetuses. The fetuses maintained a set level independent of the maternal ionized calcium, and that value was increased in Casr⫹/⫺ and Casr null fetuses compared with their WT littermates. The background strain was Black Swiss for all mice represented in this figure. [Original data in A were published as two separate panels in Kovacs et al. (342). Original data in B were also published as two separate panels in Kovacs et al. (338) and adapted with permission of the American Society for Clinical Investigation, copyright 1998.] calcium binds to it (487, 579, 759). Consequently, serum phosphorus is an important determinant of the formation and mineralization of bone during fetal development. It is typically 0.5–1.0 mM higher in the fetus than in the mother, as observed in rats (211, 213, 315, 346), mice (623), lambs (377, 403, 435), pigs (98, 353), calves (201), and foals (201). Fetuses maintained normal phosphorus levels despite hypophosphatemia in pregnant rats and mice (207, 395). Conversely, hyperphosphatemia in pregnant rats and sheep caused fetal hyperphosphatemia (41, 199, 206, 208). Serum magnesium also appears to be maintained independent of the maternal value, but variable results have been seen in animal models, including slightly increased (42, 100, 330, 435) and decreased (201, 377) concentrations compared with the maternal value in fetal lambs, modest increases (202) or no change (201) in fetal rats, a modestly increased level in foals (201), and small but statistically significant increases in fetal mice (430, 623). Maternal hypermagnesemia did not alter the fetal magnesium level in fetal lambs or rats (42, 200), and maternal hypomagnesemia did not alter the fetal magnesium concentration in fetal lambs (42). 2. Human data Human fetuses share the finding of an elevated serum calcium and ionized calcium, typically 0.30 – 0.50 mM above the maternal level (143, 148, 463, 523, 594, 595). This has 1148 been demonstrated at 15–20 wk of gestation by fetoscopy (453), and at delivery of preterm singleton and twin pregnancies (mean gestational age 33 wk) (152). Serum phosphorus is typically ⬃0.5 mM higher in fetal than maternal blood (143, 523, 544, 594). Since calcium and phosphorus are each raised above adult normal values, fetal blood has a high calcium x phosphorus product (Ca x P) that may facilitate spontaneous calcium-phosphate crystals to form and cause soft tissue calcifications. In adults, a Ca x P value ⬎5.6 mmol2/l2 or 70 mg2/dl2 is associated with calciphylaxis, coronary artery calcifications, and increased mortality (58, 115, 126, 724). However, this level may be needed to mineralize the fetal skeleton and may be nonhazardous in utero due to its relatively short duration. Serum magnesium is also set independently of the mother’s concentration but is typically raised only 0.05 mM higher than the maternal value (60, 143, 523, 544, 594). 3. Summary of key points Serum calcium, ionized calcium, and phosphorus concentrations are set at significantly higher concentration in fetuses than in the mother and can generally be maintained despite abnormal concentrations in the maternal circulation. Putative calcium-sensing receptors (other than the known CaSR) may set the fetal or placental calcium concentration at its high target value. This “fetal hypercalce- Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM mia” is not necessary for fetal viability, but may reduce the occurrence of neonatal hypocalcemia, and is required to mineralize the fetal skeleton. B. Calciotropic and Phosphotropic Hormone Concentrations 1. PTH A) ANIMAL DATA. PTH circulates at lower levels in the fetal circulation compared with the maternal value in rodents, lambs, and calves (118, 623, 675, 685, 715). These low values likely derive from the fetal sources because intact PTH does not cross the placenta of non-human primates, sheep, and rodents (196, 205, 341, 345, 478). The fetal parathyroids begin to express PTH by mid-gestation in rats and lambs (390, 602), and must be its dominant source because genetic ablation of the parathyroids (in Hoxa3 null fetal mice) results in undetectable serum PTH (336, 341). An additional source of PTH is the placenta, which in the mouse has been shown to express the Pth gene and may contribute a small amount of PTH to the circulation (623). The normally low concentration of PTH in fetal blood remains important for maintaining the blood calcium concentration because fetal mice lacking PTH or the PTH/PTHrP receptor are each hypocalcemic compared with their WT littermates (340, 341, 623). Why is PTH suppressed in fetal blood? As noted above, the high concentrations of total and ionized calcium in fetal blood likely activate CaSR on fetal parathyroids to suppress PTH synthesis and release (FIGURE 3). Several lines of evidence from fetal mice support this conclusion. Ablation of one or both alleles of Casr results in a progressive increase in PTH in fetal mice and an increase in the fetal blood calcium compared with WT fetal littermates (338). Conversely, in the absence of PTHrP (Pthrp null fetuses), the circulating PTH level rises but the fetal blood calcium is maintained at a level equal to the maternal value, rather A Casr+/- mothers B 100 80 80 PTH (pg/ml) PTH (pg/ml) 100 40 20 60 p<0.02 40 20 0 0 WT (4) Casr+/- Casr-/(10) (4) The low circulating level of PTH does not mean that the fetal parathyroids are unable to produce greater amounts of PTH. PTH increases not only with ablation of one or both alleles of Casr (336, 340), but in response to EDTA-induced hypocalcemia in fetal lambs (631), calves (715), and rhesus monkeys (524). However, a second study in rhesus monkeys found no increase in fetal PTH in response to EDTAinduced hypocalcemia (189). Active placental transport of calcium may contribute to the suppression of PTH by bringing calcium into the circulation through a route that does not require PTH. Furthermore, maternal hypercalcemia seems to enhance the forward flow of calcium to the fetus and further suppress the fetal PTH level, even if the fetal serum calcium does not change. For example, when wild-type (WT) and Casr⫹/⫺ sister females are mated to the same Casr⫹/⫺ males, the fetal PTH concentration is suppressed in offspring of hypercalcemic Casr⫹/⫺ females compared with their genetic counterparts from normocalcemic WT females, even though the fetal blood calcium remains unaffected by the mother’s genotype or serum calcium (FIGURE 5) (338). Conversely, maternal hypocalcemia may impair the forward flow of calcium from mother to fetus, and force the fetal-placental unit to upregulate the mechanisms that maintain the fetal blood calcium and placental calcium transport. Consistent with this, maternal parathyroidectomy or calcitonin infusions in pregnant rats have been shown to cause a marked increase in fetal PTH, parathyroid gland hyperplasia, and bone resorption; the skeleton is undermineralized (104, 206, 541, 542, 625). WT mothers p<0.001 60 than above it (336, 340). These data suggest that the fetal parathyroids are constrained by CaSR to maintain the blood calcium no higher than the adult level. Furthermore, when Casr is also ablated in Pthrp null fetuses to create Pthrp/Casr double-mutants, the fetal blood calcium rises well above the maternal level despite the absence of PTHrP (338). WT (6) FIGURE 5. Maternal hypercalcemia suppresses fetal PTH while fetal expression of CaSR determines the relative fetal PTH level. Ablation of one or both copies of Casr results in a stepwise increase in PTH concentrations in the fetal circulation (A and B), confirming the ability of CaSR to regulate PTH release from the fetal parathyroids. However, fetuses obtained from hypercalcemic Casr⫹/⫺ mothers (A) have lower PTH levels than in fetuses of the corresponding genotype obtained from normocalcemic WT mothers (B). Since maternal PTH cannot cross the placenta, the suppression of fetal PTH must be mediated by maternal serum calcium. The number of observations for each genotype is indicated in parentheses. [From Kovacs et al. (338). Copyright 1998 American Society for Clinical Investigation.] Casr+/(6) Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1149 CHRISTOPHER S. KOVACS B) HUMAN DATA. In human babies, PTH also circulates at low levels compared with the maternal circulation and normal adult values; very low (⬍0.5 pM) concentrations may be observed (20, 143, 168, 177, 255, 306, 501, 544, 571, 580, 598 – 600, 671, 736). This suppression of PTH has been observed as early as 19 wk of gestation in preterm babies (501, 571, 580, 599), although one study found unsuppressed PTH values in cord blood of infants aged 31–36 wk (152). The suppression of fetal PTH is considerable when it is realized that the maternal PTH value is also usually suppressed during pregnancy compared with normal adult values, at least in women from North America and Europe (328, 339). The parathyroids produce immunoreactive PTH beginning at 10 wk of gestation (367); whether the human placenta produces PTH has not been determined. Maternal hypercalcemia can result in a normal or slightly increased cord blood calcium (131, 278, 610, 700) and suppression of the fetal parathyroids, which will later be realized as neonatal hypoparathyroidism, hypocalcemia, and tetany (see sect. XII, A, B, and G). These findings have been observed in babies born of mothers whose hypercalcemia during pregnancy was caused by primary hyperparathyroidism (79, 149, 304, 393, 609, 718), inactivating mutations of Casr (529), or hypercalcemia of malignancy (9, 131, 278, 454, 529, 673, 674, 700). Conversely, maternal hypocalcemia caused by hypoparathyroidism (11, 76, 389, 586, 652, 710) or pseudohypoparathyroidism (226, 710) has been associated with fetal parathyroid gland hyperplasia, normal cord blood calcium, increased PTH, and effects on the fetal skeleton that include increased resorption, demineralization, and fractures occurring in utero or during delivery (see sect. XII, C and D). 2. Calcitriol A) ANIMAL DATA. Rodents and lambs have been most frequently used to study fetal mineral homeostasis, and in the data discussed thus far, they have yielded similar results. However, with respect to vitamin D physiology, there are some differences between fetal rodents and lambs. This may be in part due to the differences in placental structure and function that are discussed in section IIF2; in brief, rodents have hemochorial placentas (with similar structure and diffusional characteristics as human placentas), while lambs have epitheliochorial placentas (536). Calcitriol typically circulates at ⬍50% of the maternal value in fetal rodents (309, 342, 369, 709) and pigs (352, 353); in contrast, calcitriol significantly exceeds the maternal value in fetal lambs (4, 101, 155, 377, 505, 565). Radiolabeled calcitriol does not cross the rat placenta (475), which means that the low concentrations of calcitriol must be synthesized within the fetus or placenta. Placentas of lambs differ in that radiolabeled calcitriol crosses in both directions (155), and a high metabolic clearance rate of calcitriol within fetal lambs indicates that calcitriol produc- 1150 tion is markedly upregulated compared with their ewes (566). Maternal nephrectomy in rats did not alter fetal calcitriol levels, confirming that these metabolites are independently synthesized in the fetal-placental unit (369). Fetal kidneys and placenta each express the 1␣-hydroxylase (Cyp27b1), which converts 25-hydroxyvitamin D to calcitriol (234, 663, 729). Placental trophoblasts, yolk sac, and decidua also express 24-hydroxylase (Cyp24a1), which catabolizes 25-hydroxyvitamin D and calcitriol into inactive forms (30, 135, 728). The fetal kidneys contribute a significant amount of calcitriol to the fetal circulation because fetal nephrectomy reduced the fetal calcitriol level by 60% in fetal lambs (564). This may mean that ⬃40% of calcitriol in the fetal circulation is produced by the placenta, at least in lambs. Although the data from lambs indicate that calcitriol can cross the placenta from the feta to maternal circulations (155), comparatively little calcitriol produced by the placenta may reach the maternal circulation in rodents. This conclusion is supported by the finding that maternal kidneys in pregnant mice have 35-fold higher expression of Cyp27b1 than in the placentas (310). 25-Hydroxyvitamin D readily crosses the rat placenta; consequently, the low level of calcitriol cannot be due to inadequate supply of its precursor (240). It is likely that renal 1␣-hydroxylase expressed by fetal kidneys is suppressed by the high ionized calcium, high phosphorus, and low PTH concentrations typically observed in fetal blood. The 1␣hydroxylase is capable of responding to PTH in utero because Casr⫹/⫺ and Casr null fetuses demonstrated a stepwise increase in both PTH and calcitriol levels (338), but the calcitriol levels of Casr null fetuses remained lower than in adult WT females (338, 342). Calcitriol levels did reach the adult normal range in Vdr null fetuses which lacked the vitamin D receptor (VDR) (192). As noted below, PTHrP circulates at high levels in fetal blood and through its mimicry of PTH action on the PTH/PTHrP receptor it might stimulate high fetal calcitriol concentrations. However, systematic studies of PTH1R binding and signaling induced by NH2-terminal PTH and PTHrP, comparison of their crystal structures, and clinical studies of the effect of NH2-terminal PTH and PTHrP infusions, suggest that PTHrP is much less potent than PTH at stimulating the 1␣-hydroxylase (195, 269, 270, 740; reviewed in detail in Ref. 333). FGF23 conceivably contributes to low calcitriol levels because it inhibits 1␣-hydroxylase and stimulates the catabolic enzyme 24-hydroxylase. However, its effect during fetal development appears relatively insignificant because Fgf23 null fetuses had no change in serum calcitriol or the renal and placental expression of Cyp27b1 (395); however, Fgf23 null fetuses did have modest but significantly reduced renal expression of Cyp24a1 (395). Conversely, 11-fold increased FGF23 in Phex null male fetuses did cause a small Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM but statistically significant decrease in serum calcitriol, and this was accompanied by significantly increased expression of Cyp24a1 in placenta and fetal kidneys but no change in the expression of Cyp27b1 (395). As has been demonstrated in the fetal rat, low serum calcitriol in WT fetuses may also be due to preferential catabolism of 25-hydroxyvitamin D to 24,25-dihydroxyvitamin D, instead of synthesis of calcitriol from 25-hydroxyvitamin D (369, 475). This results in fetal levels of 24-hydroxylated forms that are up to 40-fold higher than calcitriol in fetal rats and sheep (369, 475, 506). The high expression of Cyp24a1 in murine placentas is approximately equal to that of the maternal kidneys (310), confirming that the placenta has significant capacity to catabolize 25-hydroxyvitamin D and calcitriol into 24-hydroxylated forms. B) HUMAN DATA. The human data are largely consistent with the rodent data mentioned above, and differ from data obtained from fetal lambs. Calcitriol circulates at low levels in fetal blood, typically ⬍50% of the maternal value (190, 264, 599, 642, 736). Umbilical artery values are higher than those obtained from the umbilical vein (736), which suggests that it is the fetal kidneys and not the placenta that contribute much calcitriol to the fetal circulation. As in the animal models, 25-hydroxyvitamin D readily crosses the placenta (254) and achieves cord blood levels that are typically 75–100% of the maternal value at term with a fetalmaternal correlation coefficient of 0.8 – 0.9 (190, 264, 599, 713, 736). Calcitriol synthesis in the fetus is likely suppressed by the high serum calcium, high phosphorus, and low PTH concentrations typically observed in cord blood. Human trophoblasts and maternal decidua express 1␣-hydroxylase and convert 25-hydroxyvitamin D to calcitriol (156, 728, 758). Trophoblasts, yolk sac, and decidua also express 24-hydroxylase (30, 728). In one study of human placentas, CYP27B1 mRNA expression was higher than in adjacent decidua while CYP24A1 mRNA was lower (176), and there is evidence that CYP24A1 is methylated in human placenta (479). These findings have prompted other researchers to conclude that trophoblast synthesis of calcitriol is unopposed or “unfettered” by catabolism into 24-hydroxylated forms (383, 479). However, the available functional data suggest the opposite conclusion, that human placentas preferentially metabolize 25-hydroxyvitamin D to 24,25-dihydroxyvitamin D instead of calcitriol (572), resulting in up to 40-fold higher concentrations of 24-hydroxylated forms compared with calcitriol in cord blood (152, 253). These findings are the same as in fetal rodents, as noted above. Maternal calcitriol increases two- to threefold during pregnancy, and the placenta has often been assumed to be the source of this increase. However, the placenta contributes little or no calcitriol to the maternal circulation, as revealed by an anephric woman on dialysis whose serum calcitriol remained low before, during, and after a pregnancy (698). Instead, it is the maternal kidneys that upregulate the synthesis of calcitriol during pregnancy. This is also similar to the rodent data mentioned earlier. 3. PTHrP A) ANIMAL DATA. Cytochemical assays have revealed that fetal blood contains high PTH-like bioactivity, which cannot be accounted for by the low immunoreactive levels of PTH (3, 20, 67). Following the discovery of PTHrP and the development of specific radioimmunoassays for it, studies in fetal pigs (7) and sheep (94, 396) determined that high immunoreactive levels of PTHrP correlated closely with, and likely accounted for, the high PTH-like bioactivity in fetal blood. PTHrP has a full length of PTHrP1–139 in rodents compared with splice variants PTHrP1–139, PTHrP1–141, or PTHrP1–173 in humans. But PTHrP is a prohormone that is processed into several circulating peptides. In the human these include PTHrP1–36, PTHrP38 –94, PTHrP38 –101, PTHrP107–139, and possibly PTHrP141–173, each of which may have its own receptor and distinct role (333, 490, 517, 745). The structures of these peptides were originally determined through study of tumor cell lines transfected with the human PTHrP gene. Which of these PTHrP peptides circulate in fetal life of animals or humans has not been determined. Most available assays have been designed to detect fragments that contain PTHrP1–34 or PTHrP1– 86. Notably, a PTHrP1–34 assay used in rodents or lambs will detect NH2-terminal PTHrP1–36 and full-length PTHrP1–139, whereas the PTHrP1– 86 assay will only detect full-length PTHrP1–139, a form that may not be abundant due to rapid cleavage into PTHrP1–36. Immunoreactive forms of PTHrP detected by a PTHrP1– 86 assay did not cross the placentas of sheep and goats (537), but whether smaller fragments can cross the placenta has not been determined. Full-length PTHrP1–139 has about twice the molecular weight of PTH, and since PTH does not cross the placenta (196, 205, 341, 345, 478), full-length PTHrP is also unlikely to cross. Numerous tissue sources may contribute to the circulating concentration of PTHrP, because in the embryo and fetus the gene is expressed by skeletal growth plates (299, 365), cardiac and vascular smooth muscle (89), trophoblasts (5, 336, 602), intraplacental yolk sac (336), amnion (68, 184), chorion (68), umbilical cord (185), and numerous other tissues and cell types. Venous umbilical PTHrP levels are higher than umbilical arterial levels in pigs, which suggests that the placenta may be an important source of circulating PTHrP in the fetus (7). As discussed in section IIC, the fetal parathyroids were originally proposed to be the source of PTHrP in fetal sheep, but the effect of fetal parathyroidectomy on the plasma PTHrP concentration was not reported. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1151 CHRISTOPHER S. KOVACS Loss of fetal parathyroids did not alter the plasma PTHrP1–34 concentration in Hoxa3 null and Gcm2 null fetuses (623). Pth1r null fetuses, which lack the PTH1R, have an 11-fold increase in PTHrP1–34 and increased placental expression of PTHrP mRNA and protein (341). These findings are compatible with the placenta being the dominant source of PTHrP in the circulation of fetal rodents. Whether fetal parathyroid glands in rodents produce PTHrP remains unresolved and is discussed in section IIC. B) HUMAN DATA. As with animal fetuses, prior to the discovery of PTHrP it was recognized that human cord blood contained low immunoreactive intact PTH concentrations but high levels of PTH-like bioactivity as measured in cytochemical bioassays (20, 21, 571). The discrepancy is now explained by high circulating levels of PTHrP (600, 671), which mimics many of the actions of PTH on the PTH1R. The concentrations of PTH and PTHrP are often reported in unrelated units, but when expressed in equivalent units (pM), human cord blood PTHrP levels are 2– 4 pM and up to 15-fold higher than the simultaneous levels of PTH (0.2– 0.5 pM) (168, 306, 671). A few studies have provided conflicting data by finding undetectable levels of PTHrP1– 86 in cord blood (592) or very low levels of PTHrP1– 86 that were no different among pregnant women, fetal samples obtained by cordocentesis, and cord blood samples (501). These results likely reflect several problems that are commonly found among clinical studies that included PTHrP measurements. First, PTHrP is subject to rapid cleavage and degradation in serum; consequently, blood samples are optimally collected as EDTA plasma with aprotinin (a protease inhibitor), kept chilled on ice, and promptly (within 15 min) centrifuged, separated, and frozen. Even when optimally collected as EDTA-aprotinin plasma and kept chilled on ice, PTHrP begins to degrade within 15 min of sample collection (275). Many studies did not approach this rigor of sample collection and preparation, and it is common to see use of stored sera that had been allowed to clot at room temperature for up to 60 min (678). All of these factors will result in failure to detect the full concentration of PTHrP that was present in the original blood sample, and may even result in undetectable PTHrP when it should be present. Second, the most commonly used assays in clinical studies have been those that purportedly measure PTHrP1– 86. This form is not predicted to exist in humans (333), but the assay should detect fulllength forms PTHrP1–139, PTHrP1–141, or PTHrP1–173 if they are present in the circulation. However, it will not detect PTHrP1–36, which is conceivably the more abundant NH2-terminal form of PTHrP because it derives from all full-length forms. Conversely, a PTHrP1–34 assay should detect each of PTHrP1–36, PTHrP1–139, PTHrP1–141, and PTHrP1–173. Standardization of sample requirements and PTHrP assays are sorely needed; in the meantime, it is preferable to use a PTHrP1–34 assay and to use only plasma that 1152 has been rigorously collected and processed under the optimal conditions described here. 4. FGF23 A) ANIMAL DATA. FGF23, produced largely by osteocytes and osteoblasts, is a phosphorus-regulating hormone that controls the supply of phosphorus at the mineralizing surface of bone through actions on distal tissues (622). It downregulates the expression of sodium-phosphate cotransporters 2a and 2c (NaPi2a and NaPi2c) in proximal renal tubules (614). It also inhibits 1␣-hydroxylase (Cyp27b1), increases expression of 24-hydroyxlase (Cyp24a1), and inhibits intestinal expression of NaPi2b (614). In adults, increased effective circulating levels of FGF23 lead to hypophosphatemia and renal phosphate wasting, while loss of FGF23 causes hyperphosphatemia and impaired renal phosphate excretion (52). FGF23 is predominantly expressed in rat fetal osteoblasts, in addition to thymus, liver, and kidney (754). Murine fetuses express FGF23 as early as embryonic day 12.5 in heart, liver, and somites; it later appears in bone (627). Using the sensitive method of real-time quantitative RTPCR, comparative study of WT, Fgf23 null, and Phex null placentas revealed that the murine placenta expresses a low level of Fgf23 mRNA, which is absent in Fgf23 null placentas but increased in Phex null placentas (395). An independent study used the less sensitive method of conventional RT-PCR and did not find visible Fgf23 amplification on an agarose gel (484). The concentration of intact FGF23 in fetal blood from WT fetuses was equal to the simultaneous maternal level in mice from the outbred Black Swiss strain for several mouse models (395), whereas the level was about half the maternal value in WT fetuses from the inbred C57BL/6 strain (194, 395). The maternal value in turn was approximately double the nonpregnant level as revealed through longitudinal studies in both strains of mice (194). The discrepancy in the fetal FGF23 blood levels between the Black Swiss and C57BL/6 mice, within the same laboratory and using the same intact FGF23 assay, indicates that the genetic background of the mice influences fetal FGF23 physiology. Phex null male fetuses and Phex⫹/⫺ female fetuses had markedly increased intact FGF23 concentrations that equaled the simultaneous maternal concentration from their Phex⫹/⫺ mothers (395), whereas in another study the FGF23 level in Phex null male fetuses far exceeded the maternal concentration (484). Although no formal studies have been done with radiolabeled FGF23, it evidently does not cross the murine placenta because Fgf23 null fetuses had undetectable FGF23 despite high levels present in the maternal circulation (395). Furthermore, the high FGF23 concentrations observed in Phex⫹/⫺ mothers did not alter the circulating FGF23 level Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM in their WT fetuses compared with WT fetuses born of Fgf23⫹/⫺ mothers (395). 6. Sex steroids A) ANIMAL DATA. The dominant sex steroids, testosterone and B) HUMAN DATA. Scant data are available pertaining to FGF23 in normal human fetuses. In two studies, intact FGF23 levels in cord blood were 0.15- to 0.30-fold the values obtained from unrelated healthy adults or their mothers (483, 661). But COOH-terminal FGF23 was approximately twofold the values of unrelated healthy adults (661). These divergent results may indicate that FGF23 is abundantly produced but rapidly cleaved in utero, thereby resulting in elevated levels of the nonfunctional COOH-terminal fragment. Cord blood levels of the FGF23 coreceptor Klotho were sixfold adult and neonatal values and may contribute to a reduction in the circulating intact FGF23 level (483). Human trophoblasts also express Klotho (483). Use of conventional RT-PCR did not reveal visible evidence of FGF23 expression in human trophoblasts (484). 5. Calcitonin A) ANIMAL DATA. Thyroid C-cells and calcitonin are detectable as early as the 30th day of an approximate 145-day gestation in fetal lambs (292), whereas in fetal mice and rats C-cells and calcitonin do not appear until day 15.5 or later out of a gestational period that lasts 19 –22 days, respectively (22, 203, 284, 474, 645, 679). Near term, fetal calcitonin concentrations are maintained at higher concentrations than in the maternal circulation in rodents (201, 430) and lambs (201, 204). Calcitonin within the fetal circulation must derive from fetal sources because maternal calcitonin does not cross the rat or mouse placenta (212, 430). Rat and mouse placentas also express calcitonin mRNA and protein (284, 293, 337). The high circulating concentration of calcitonin may occur in response to the increased serum and ionized calcium that is normally present in the fetal circulation. Consistent with this, experimentally induced hypercalcemia in fetal pigs, lambs, calves, and nonhuman primates caused a further, marked increase in calcitonin, the magnitude of which correlated with the change in serum calcium (101, 380, 548). The relative contribution of thyroid and placenta to circulating calcitonin has not been determined. The fetal thyroid is a significant source because infusion of calcium chloride into the left thyroid artery of fetal lambs caused a 2- to 10-fold increase in calcitonin secretion (215). B) HUMAN DATA. The thyroid C-cells differentiate around the 12th week of gestation (107), and calcitonin is detectable in those cells from as early as the 15th week of gestation (368). Serum calcitonin is increased to as much as twice the maternal level in both term and preterm babies (255, 343, 523, 583, 599, 736, 742). Trophoblasts of the placenta also express calcitonin and may, therefore, contribute to the amount in the fetal circulation (35). estradiol, are considered as calciotropic hormones within this review because of their importance in regulating postnatal skeletal development and bone turnover. Testosterone in male fetal rats surges over the last several days of gestation before declining on the last day (720, 730). It exceeds the concentration in female fetuses (239, 272, 720, 730) and was higher than the maternal value in three studies (239, 720, 730), and equal to maternal in another (272). Testosterone was also higher in male compared with female fetal lambs (18). Conversely estradiol concentrations are similar between male and female rat fetuses (239), and exceed the maternal concentration (146, 239). In fetal lambs, testosterone and estradiol exceeded the maternal concentration at all time points (117, 650, 757). B) HUMAN DATA. Human pituitary cells produce luteinizing hormone and follicle stimulating hormone from as early as 5 wk of gestation (619, 620), and the levels of both gonadotropins are higher in female fetuses throughout gestation (676). The gonadotropins and placenta-derived human chorionic gonadotropin drive the Leydig cells within the developing male testis to produce testosterone (29), whereas only negligible amounts of testosterone are produced in the ovaries or in the adrenals of both sexes (546). The ovaries, testes, and adrenals produce little or no estradiol until late in fetal life (91, 546). In male fetuses, testosterone exhibits higher concentrations between 11–17 wk of fetal age before subsequently declining to trough values (545, 739); at term, the concentration is modestly higher or no different than in female fetuses (301, 458, 509, 545, 682, 739). Estradiol levels are also similar between male and female fetuses (250, 509, 545, 682, 739). It is generally considered that in fetal humans estradiol and testosterone are present at low levels in cord blood near term compared with normal adult values (133, 676). Indeed, in most studies the fetal levels of estradiol and testosterone were much lower than simultaneous maternal concentrations at term (15, 179, 216, 458, 465, 509, 681, 682, 696). However, in a few studies either or both of testosterone and estradiol have been approximately equal to (250, 545) or significantly greater than (14, 357, 739) the maternal concentrations. These discrepancies in relative fetal-to-maternal sex steroid levels are likely the result of whether the umbilical vein, artery, or mixed cord blood was sampled. By the third trimester, the placenta accounts for nearly all of the estrogens (estradiol, estrone, and estriol) in the maternal circulation (437), and it synthesizes them through aromatization of androgens that arise about equally from the fetal and maternal adrenals (382, 618). The concentrations of these estrogens progressively increase during pregnancy and reach peak values in the maternal circulation during the third trimester (382, 480, 697). If the umbilical vein or mixed Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1153 CHRISTOPHER S. KOVACS cord blood are sampled, the levels will indicate high concentrations due to placental synthesis of sex steroids, whereas umbilical artery values reflect systemic levels within the fetus. Several studies have found that the umbilical artery estradiol or conjugated estrogen levels are significantly lower than the umbilical vein values (589, 617, 696). Such findings confirm that placental production of sex steroids confounds the cord blood measurements and that the umbilical artery values alone should be used to indicate systemic fetal concentrations. In the previously cited studies that reported elevated sex steroid levels in cord blood, it was mixed cord blood that had been sampled in two of them (14, 739), whereas the source was not specified in the third study (357). 7. Summary of key points The human fetal circulation is characterized by low concentrations of PTH, calcitriol, and the sex steroids as well as high levels of PTHrP and calcitonin (FIGURE 2). Parathyroid CaSR likely suppresses PTH in response to the high fetal blood calcium. 25-Hydroxyvitamin D readily crosses the placenta whereas calcitriol does not. The low fetal calcitriol levels are likely due to suppression of the fetal renal 1␣hydroxylase (CYP27b1) by low PTH and high serum calcium and phosphorus, and increased activity of the catabolic enzyme 24-hydroxylase. C. Fetal Parathyroids 1. Animal data In rodents, the thymus and a single pair of parathyroids derive from the third pharyngeal pouch (56, 384). Parathyroids are friable such that fragments detach during embryonic migration, which likely accounts for parathyroid tissue within the thymus and adjacent tissues (232, 384, 385, 657, 705). Gcm2 is intensely expressed by the primordium derived from the third pharyngeal pouch in mice; therefore, deletion of Gcm2 should ablate the parathyroids and result in undetectable PTH, as occurs when Hoxa3 is deleted (232, 336, 341, 411). Consistent with this, the normal expression of PTH mRNA and protein in the common parathyroid/thymus primordium on embryonic day 11.5–12.5 is absent in Gcm2 null fetuses (385). However, a low level of PTH has been measured in the circulation of Gcm2 null fetuses even though the parathyroids (and parathyroid remnants in the thymus) have been eliminated (623). Pth is also expressed in normal placenta, and its expression there is upregulated in Gcm2 null fetuses compared with placentas from WT littermates (623), and so placental PTH may explain the low but detectable PTH blood levels in Gcm2 null fetuses. Adult Gcm2 null mice were originally reported to have normal circulating levels of PTH, a finding that is not easily explainable unless Gcm2-independent sources of PTH activate in the absence of parathyroids (238). 1154 PTH mRNA and protein can also be detected by mid-gestation in the parathyroids of fetal rats and lambs (390, 602). It has been suggested that fetal parathyroids secrete both PTH and PTHrP. However, the evidence that fetal parathyroids express PTHrP mRNA and protein is inconsistent and inconclusive. PTHrP was first detected in parathyroids of fetal lambs by immunocytochemical methods with older polyclonal antibodies that did not cross react with PTH (5, 390, 396). These studies have not been repeated with newer and more specific monoclonal antibodies, nor has PTHrP mRNA expression been examined in parathyroids from fetal lambs. RT-PCR and in situ hybridization failed to detect PTHrP mRNA in fetal or adult rat parathyroids (601, 695), whereas PTHrP mRNA was detected by RT-PCR in a third study of adult rat parathyroids (274). The more sensitive technique of real time quantitative RT-PCR has not been used on fetal parathyroids from rats or lambs, while murine fetal parathyroids have not been directly examined at all for their ability to express PTHrP. If the fetal parathyroids are a significant source of PTHrP in the fetal circulation, then fetal parathyroidectomy or genetic ablation of the parathyroids should reduce the circulating PTHrP level. However, the plasma PTHrP level was not measured in any of the studies in which fetal lambs and rats were parathyroidectomized. Instead, the parathyroid secretion of PTHrP was inferred from the finding that parathyroidectomy in fetal lambs caused a reduction in placental calcium transport that could be rescued by infusion of autologous blood or PTHrP, but not PTH (discussed in more detail in sects. IVB and VB). In contrast, genetic ablation of parathyroids in Hoxa3 null and Gcm2 null fetuses did not alter the plasma PTHrP level (341, 623), while an 11-fold upregulation of plasma PTHrP in Pth1r null mice was associated with upregulation of PTHrP mRNA and protein expression in the placenta but not in the region of the neck that contains the parathyroids (341). These findings in rats and mice suggest that the parathyroids are unlikely to be a significant source of PTHrP in the fetal circulation, whereas the data from fetal lambs remain consistent with parathyroid-localized expression of PTHrP. Despite a low level of PTH in the fetal circulation, the evidence is consistent across all animal models that fetal parathyroids are required to maintain the normally high level of blood calcium. Loss of parathyroids from any cause results in fetal hypocalcemia, as demonstrated in fetal lambs (thyroparathyroidectomy followed by replacement of thyroxine) (1, 2), rats (thyroparathyroidectomy or decapitation) (520, 521, 554), and mice (loss of parathyroids through genetic ablation of Hoxa3 or Gcm2, or loss of PTH alone in Pth null fetuses) (336, 341, 623). PTH and PTHrP both participate in the normal regulation of the fetal blood calcium, such that loss of either hormone results in fetal hypocalcemia, and loss of both leads to the Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM greatest decrease in blood calcium (336, 340, 341, 623). This is discussed in more detail in section VIA. Fetal parathyroids are also required to regulate magnesium and phosphorus, since thyroparathyroidectomized fetal lambs, and Hoxa3 null, Gcm2 null, and Pth null fetal mice have hypomagnesemia and hyperphosphatemia (47, 341, 397, 398, 623). Loss of PTHrP itself causes hyperphosphatemia with a normal serum magnesium concentration (341, and unpublished data). In sections IVB and VB, the evidence that the parathyroids regulate placental calcium transport is discussed. 2. Human data In humans, parathyroids and thymus share a common developmental origin within the third pharyngeal pouch, while a second pair of parathyroids originate from the fourth pouch (56, 222, 518, 669). As in the animal models, the friable nature of developing parathyroids leads to thymic and other ectopic locations for fragments that detach during embryonic migration. PTH is detectable by immunochemical methods within parathyroids beginning at 10 wk of gestation (367). Expression of PTHrP has not been sought in human fetal parathyroids, but PTHrP mRNA and protein have been detected in the oxyphil cells of adult normal, hyperplastic, and adenomatous parathyroids by a variety of methods (28, 137, 157, 277, 313, 314, 424, 552). Since adult parathyroids express PTHrP, it is conceivable that the fetal parathyroids do as well. 3. Summary of key points Fetal parathyroids produce low amounts of PTH, but whether or not they produce PTHrP is unclear. The evidence is stronger for PTHrP production by parathyroids of fetal lambs than it is for fetal rodents or humans; species differences may explain these discrepancies. D. Renal Mineral Reabsorption, Excretion, and the Amniotic Fluid 1. Animal data In the adult, the kidneys play key roles in the regulation of mineral homeostasis: 1) by adjusting the relative reabsorption and excretion of minerals in response to PTH, calcitriol, calcitonin, FGF23, and CaSR; and 2) as the dominant site of expression of 1␣-hydroxylase and synthesis of calcitriol. The fetal kidneys may be of lesser importance because the placenta assumes control over mineral handling and synthe- sizes calcitriol. Another reason is that excreted mineral is not lost to the organism, because urine forms a large component of the amniotic fluid which is swallowed (autourophagia), absorbed, and thereby recycled (563). Renal blood flow and glomerular filtration rate are also much lower during fetal development compared with after birth (237, 378, 605). Nevertheless, alterations in serum mineral concentrations and renal excretion of minerals have been observed in response to alterations in kidney function, or from manipulations in the factors that normally control renal tubular function. In fetal lambs, bilateral nephrectomy led to hypocalcemia, hyperphosphatemia, increased PTH, and a 60% fall in the circulating level of calcitriol (455, 564). Additional fetal lambs underwent a control procedure that consisted of bilateral ureteral sectioning to permit fetal urine to drain into the fetal peritoneal cavity while retaining functional kidneys in situ. Ureteral sectioning had no effect on fetal calcium or calcitropic hormone levels (455). The investigators concluded that loss of renal 1␣-hydroxylase and production of calcitriol resulted in altered fetal mineral homeostasis, rather than the loss of renal function to reabsorb and excrete minerals. However, other investigators performed bilateral nephrectomy in fetal lambs and observed an increase in serum phosphorus, no change in serum calcium, and a nonsignificant reduction in the excretion of calcium into the amniotic fluid and allantois (459). Bilateral nephrectomy in fetal rats did not alter the serum calcium or phosphorus either; calcitriol and PTH were not measured in those experiments (105). With respect to hormonal control of renal mineral handling, thyroparathyroidectomy in fetal lambs caused hypocalcemia, hyperphosphatemia, increased renal fractional excretion of calcium, and reduced renal phosphorus excretion (398). Pharmacological administration of NH2-terminal fragments of PTH or PTHrP increased serum calcium and reduced renal calcium excretion, but did not alter serum phosphorus or urine phosphorus excretion (142, 398). These results may indicate that thyroparathyroidectomy causes hypocalcemia in part through loss of the effects of parathyroid-derived PTH and PTHrP on the renal tubules. However, in contrast to fetal lambs, absence of parathyroids in Hoxa3 null and Gcm2 null fetal mice, loss of PTH1R in Pth1r null fetuses, and absence of PTH in Pth null fetuses, each caused hypocalcemia, hyperphosphatemia, and reduced amniotic fluid calcium, which is a measure of renal calcium excretion (341, 623; and unpublished data). The low blood calcium in these murine fetuses must cause a reduced renal filtered load of calcium, which in turn may explain reduced excretion of calcium into the amniotic fluid. Conversely, fetal mice with high blood calcium have increased excretion of calcium into the amniotic fluid, which is likely the result of an increased renal filtered load of calcium (338). Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1155 CHRISTOPHER S. KOVACS Whether calcitriol itself plays any important role in fetal kidney function is uncertain. Data have already been cited about how circulating calcitriol levels are low in fetal rats but increased in fetal sheep. As already noted, fetal nephrectomy in lambs reduced the serum calcitriol level by 60% and was accompanied by a fall in serum calcium and an increase in serum phosphorus in one study (455), but serum calcium remained normal in another study of nephrectomized fetal lambs (459), and serum calcium and phosphorus were normal in nephrectomized fetal rats (105). The investigators in the first study assumed that loss of renal production of calcitriol caused the hypocalcemia and hyperphosphatemia and were able to show that pharmacological treatment with calcitriol reversed these abnormalities (455). However, in Vdr null fetal mice, ionized calcium and phosphorus, and amniotic fluid calcium and phosphorus, were not altered compared with WT littermates (342). It appears likely that renal production of calcitriol is relatively unimportant for fetal calcium homeostasis in most mammals, although it is possible that the situation is different for fetal sheep because of high circulating levels of calcitriol. FGF23 increases renal phosphate excretion in the adult by downregulating expression of NaPi2a and NaPi2c in the renal tubules. Fetal kidneys show abundant expression of the FGF23 targets Klotho, NaPi2a and NaPi2c, Cyp27b1, Cyp24a1, and Fgfr1-Fgfr4 (395). But in Fgf23 null fetal mice, loss of FGF23 did not alter serum phosphorus or amniotic fluid phosphorus excretion, and there was no effect on expression of NaPi2 and NaPi2c in fetal kidneys (395). Conversely, in Phex null male fetuses, which have markedly increased circulating concentrations of FGF23, there was also no effect on serum phosphorus, amniotic fluid phosphorus excretion, and NaPi2 and NaPi2c expression in fetal kidneys (395). These results suggest that fetal kidneys do not share the key role in renal phosphate handling or responsiveness to FGF23 that the adult kidneys have. 2. Human data There are no available mineral or calciotropic hormone measurements or skeletal data from anephric human fetuses, apart from noting that affected babies can have scoliosis, rib anomalies, and absent toes, radii, or thumbs (528). Those skeletal abnormalities may reflect the underlying genetic anomalies rather than an effect of absent kidney function. Bilateral renal agenesis presents with very low amniotic fluid volumes (oligohydramnios or anhydramnios) due to the lack of fetal urine production (147, 528), confirming that urine comprises much of the normal volume of amniotic fluid. In turn, the lack of amniotic fluid causes pulmonary hypoplasia, which has been confirmed by noting that a monoamniotic twin with bilateral renal agenesis was born with normal pulmonary function, presumably because the twin’s kidneys provided the normal volume of amniotic fluid (511). The absent kidney function and pul- 1156 monary hypoplasia contribute to the uniform fatality of this condition in the newborn period. 3. Summary of key points The fetal kidneys are a likely source of the low level of calcitriol in the fetal circulation and are capable of excreting and reabsorbing minerals. Whether fetal kidneys contribute importantly to fetal mineral homeostasis is uncertain, especially since urine enters the amniotic fluid and can then be recycled through the process of autourophagia. E. Intestinal Mineral Absorption 1. Animal data Adult intestines are the main route of mineral entry into the organism, with calcium being absorbed in part through passive processes, and actively through calcitriol-dependent mechanisms. Fetal intestines must play a minor role in mineral homeostasis because the only material available to be swallowed and absorbed is the amniotic fluid. No studies have examined the relative contribution of the fetal intestines to mineral homeostasis. However, data from newborns likely reflect the function of the intestines in utero. Intestinal calcium absorption in newborn rats is largely passive, nonsaturable, and not dependent on calcitriol (218, 219, 243). The vitamin D receptor is undetectable within enterocytes at days 7 and 14 after birth (242), which explains its lack of responsiveness to calcitriol at earlier stages (243). 2. Human data Although the intestines are considered a minor player in the regulation of fetal mineral homeostasis, a significant volume of amniotic fluid (fetal urine) is normally swallowed and absorbed. When swallowing or absorption are not possible, such as in gastrointestinal atresias, an increased volume of amniotic fluid (polyhydramnios) is present at term (8, 141). Intestinal calcium absorption has not been studied in human fetuses. However, a preterm baby at birth is the equivalent of a fetus of the same gestational age, and the functional capacity of a preterm neonate’s intestines may inform about the functional capacity of the fetal intestines. Studies in preterm neonates have shown that calcium absorption is proportional to intake and does not reach a maximum (i.e., is nonsaturable); these findings suggest that passive absorption of calcium is the dominant mechanism by which calcium is absorbed in preterm fetuses (44, 221, 581). Calcium absorption did not vary by vitamin D intake in preterm infants, consistent with passive absorption being the dominant mechanism of absorption rather than active, calcitrioldependent mechanisms as seen in older infants and children Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM (75). The calcium requirement of the preterm neonate is very high at birth because the fetal skeleton is typically accreting ⬃300 mg or 120 –150 mg/kg of calcium daily at that age (761). Current guidelines recommend a daily calcium intake of 120 –200 mg/kg for preterm babies, which exceeds what the intestines should be capable of absorbing, so parenteral administration of calcium is needed (445). The postnatal changes in intestinal calcium absorption are discussed in more detail in section IIIE. 3. Summary of key points The fetal intestines are likely a minor player in fetal mineral homeostasis because only amniotic fluid is available to be ingested, but their precise contribution has not been determined. Study of preterm babies have suggested that calcium absorption in the fetal intestines is likely passive and not regulated by calcitriol. F. Placental Mineral Transport This section reviews the mechanisms through which minerals are thought to be transported across the placenta, the methods used to study placental mineral transport, and the evidence that maternal factors influence the rate of transport. The effect of fetal sources of the calciotropic and phosphotropic hormones on placental calcium transport is discussed later within the relevant portions of sections IV–X. 1. Overview of mechanisms A) ANIMAL DATA. Most of the calcium content of the newborn skeleton is obtained from the maternal circulation during the last third of gestation. In calves, 95% of the 673 g of calcium in the fetus is accreted during the last 3 mo of a 9-mo gestation, while in fetal rats 95% of the required 12.3 mg of calcium is accreted during the last 5 days of a 22-day gestation (121). Active placental calcium transport is necessary because diffusional flow is insufficient and the calcium exchange occurs against a substantial electrochemical gradient (95, 198). Additional studies have confirmed that it is an active process, dependent on Mg2⫹ and ATP (188). It has been estimated from study of perfused sheep placentas that active transport of calcium comprises two-thirds of the forward flow from mother to fetus (331). Calcium transport is thought to occur via mechanisms and pathways that are similar to calcium transport across intestinal cells. Gated calcium channels [such as transient receptor potential cation channel, subfamily V, member 6 (TRPV6)] open within maternal-facing basement membranes to allow calcium entry into the relevant cells, calcium then shuttles to the opposite basement membrane bound to binding proteins, and then Ca2⫹-ATPase at the fetal-facing basement membranes actively pumps calcium into the fetal circulation (95, 659). The relevant calcium transporting cells are the trophoblasts and probably the intraplacental yolk sac cells within rodent placentas (63, 188, 337). The flow of calcium is not unidirectional, but the magnitude of backflux is difficult to measure; it may be ⬍1% of the forward flow in sheep, rats, and mice (340, 507, 535, 660, 721), but 80% of the forward flow in primates (535). The calcium binding protein calbindin-D9k transports calcium within renal and intestinal cells, and likely plays a similar role in trophoblasts and yolk sac cells (288). It is expressed as early as day 10 of gestation in rodent placentas (607), and its expression increases markedly over the last week of gestation in rats (81, 151, 227) and mice (150, 659). Other calcium binding proteins are also expressed within trophoblasts and intraplacental yolk sac cells and show increased expression over the same interval (251, 252, 692). Ca2⫹-ATPase expression doubles during the last 7 days of gestation (227, 693). TRPV6 expression increases 14-fold during the last 4 days of gestation in the mouse (659). All three of these calciotropic genes are expressed by trophoblasts, but within rodent placentas, they are most intensely expressed within the intraplacental yolk sac, a unique structure that also expresses other calciotropic genes at much higher intensity than in the surrounding trophoblasts (FIGURE 6) (659). Varying levels of evidence support the role of calbindinD9k, Ca2⫹-ATPase, and TRPV6 in placental calcium transport. Pthrp null and Trpv6 null fetuses exhibit reduced placental calcium transport, and this is accompanied by reduced expression of calbindin-D9k within the intraplacental yolk sac of each model (337, 659), but whether the reduced expression caused or resulted from a lower rate of placental calcium transfer is unknown. Deletion of calbindin-D9k does not alter serum calcium or intestinal calcium absorption in adult mice, but fetal minerals and placental calcium transport have not been measured (51, 351, 364). Ca2⫹-ATPase has been shown to be key because inhibition of its activity by dinitrophenol, ouabain, quercetin, or a neutralizing antibody leads to a reduction in the rate of calcium transport in perfused placentas from rats (63, 95). Pmca1 encodes the dominant placental Ca2⫹-ATPase, but ablation of it results in embryonic lethality (486, 530); consequently, placental calcium transport cannot be measured. Pmca2 or Pmca4 are isoforms that are also expressed within trophoblasts (456, 649), and the knockout mice each have mild phenotypes, but the fetal skeleton and placenta have not been examined (486, 530). The lack of an adult phenotype does not preclude a significant fetal phenotype, as seen by Trpv6 null adult mice that have no impairment of intestinal calcium absorption (51), whereas Trpv6 null fetuses have severe hypocalcemia, a 40% reduction in placental calcium transfer, and a 50% reduction in skeletal ash weight (659). Overall, the available data are consistent with important roles for Ca2⫹-ATPase and TRPV6 in regulating Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1157 CHRISTOPHER S. KOVACS A Giant trophoblasts Amnion Labyrinthine trophoblasts mated to be ⬃7% of the forward (maternal to fetal) flow in lambs (660). The placenta expresses numerous phosphateregulating genes that are present in the intestines and kidneys, including the sodium-phosphate channels NaPi2a, NaPi2b, NaPi2c; the FGF23 coreceptor Klotho, and all four FGF receptors (395, 483, 484). A low level of FGF23 expression has also been demonstrated (395). 28 Visceral Yolk Sac Intraplacental Yolk Sac Parietal Yolk Sac Endoderm B Trophoblasts Maternal Vessels Reichert’s Membrane Parietal Yolk Sac Mg has been used to study active placental magnesium transport in lambs (47, 96), wherein the back-flow is ⬃30% of the forward flow (100). The factors that control magnesium transport are unknown. B) HUMAN DATA. The human skeleton accretes more than 80% of its 30 g of calcium during the third trimester (121, 224, 683, 733, 734). Human placentas have been perfused in vitro, and this has led to estimates that active transport of calcium comprises about one-third of the forward flow from mother to fetus (655). Human trophoblasts express the same genes (TRPV6, calbindin-D9k, and Ca2⫹-ATPase) and are expected to transport calcium in a manner similar to rodent placentas. C) SUMMARY OF KEY POINTS. Sinus of Duval Visceral Yolk Sac Fetal Vessels FIGURE 6. Anatomy of the intraplacental yolk sac. By the time the mature placenta has formed (A), compressed portions of the yolk sac line the uterine cavity (decidua) that is not in contact with the placenta, and it overlays the dome of the placenta. The parietal layer and Reichert’s membrane of the yolk sac are in contact with the decidua and the dome of the placenta, while the columnar or visceral yolk sac layer is in apposition to this layer, separated by the yolk sac cavity. The yolk sac bilayer that overlays the dome of the placenta forms fingerlike projections into the placenta near the insertion of the fetal vessels (boxed area in B, the intraplacental yolk sac). The intraplacental yolk sac is positioned between maternal and fetal blood spaces, as visualized in detail in B. Within this structure, the parietal layer and Reichert’s membrane overlay maternal blood spaces and vessels, while the columnar layer overlays fetal blood vessels. Between these layers is the sinus of Duval, which also communicates with the yolk sac cavity and the uterine lumen. The two layers of the intraplacental yolk sac have the most intense expression of calciotropic genes compared with the rest of the murine placenta and are thought to be a site of maternal-fetal calcium exchange. [From Kovacs et al. (337).] placental calcium transfer, while it remains unknown whether calbindin-D9k is essential. Fewer studies have examined placental transport of other minerals. 32P transport across the rat placenta increases until term, paralleling the increase in placental calcium transport (315, 701, 702). Back-flux of 32P has been esti- 1158 Active transport of calcium is required to meet the fetal requirement for mineral, and it is transported through mechanisms that are similar to those active within intestinal cells. The mechanisms governing phosphorus and magnesium absorption are not established. 2. Placental structure and expression of calciotropic and phosphotropic genes A) ANIMAL DATA. The placentas of monkeys, lambs, pigs, goats, rats, and mice have each been studied as models of human placental structure and function, but there are significant differences among their macro- and microstructures that must be considered (174, 311, 536, 553, 575, 644). Lambs, goats, and pigs have cotyledonary placentas, in which the structure is organized into 60 –70 cotyledons that spread out over the entire uterine wall. Microscopically the placenta is epitheliochorial, in which the maternal and fetal circulations remain separated by full thicknesses of maternal and fetal tissues (536, 575, 644). Calbindin-D9K and Ca2⫹-ATPase expression are concentrated in the interplacentomal region (457, 472, 743), which is thought to be an important site of calcium exchange between mother and fetus (291). Monkeys, rats, and mice have discoid placentas in which the tissue is confined to a single plate (575, 644). Microscopically the structure is hemochorial, in which maternal blood directly bathes the fetal chorion because the fetal trophoblasts have fully invaded the maternal (uterine) tissue. Consequently, structural barriers between the maternal and fetal circulations are significantly reduced in hemo- Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM chorial compared with epitheliochorial placentas (575, 644). Rodent placentas are hemotrichorial with three layers of trophoblasts interposed between the maternal and fetal circulations (404, 536, 644). Maternal blood circulates in a labyrinthine series of small channels created by the trophoblasts (129). Rodent placentas also contain the intraplacental yolk sac, a bilayered membrane formed when part of the yolk sac becomes enfolded within the placenta; it is positioned directly between fetal and maternal blood vessels (FIGURE 6) (166, 167, 289). This structure shows more intense expression of many calciotropic genes compared with the surrounding trophoblasts, including calbindin-D9k, Ca2⫹-ATPase, PTHrP, PTH1R, TRPV6, vitamin D receptor, calcitonin, calcitonin receptor, and CaSR (35, 63, 81, 83, 84, 95, 150, 151, 293, 337, 338, 356, 423, 471, 608, 656, 659). Its location between the maternal and fetal vessels is optimal for it to be a site of maternal-fetal calcium and other nutrient transport, independent of the trophoblast labyrinth (330, 337, 659). This route may enable rodents to meet proportionately larger calcium demands created by their large litters and short gestational interval. It has not been possible to determine the extent to which the intraplacental yolk sac may contribute to placental calcium transport. Platelet-derived growth factor-␣ encodes the intraplacental yolk sac, but deletion of this gene leads to embryonic lethality (482). However, Pthrp null and Trpv6 null fetuses have reduced placental calcium transfer (340, 659), and this is accompanied by reduced expression of calbindinD9k and Ca2⫹-ATPase within the intraplacental yolk sac of the Pthrp null, reduced expression of calbindin-D9k within the Trpv6 null, but normal expression of calbindin-D9k within the trophoblasts of the Pthrp null (337, 340, 659). The mouse placenta expresses numerous FGF23 target genes including Klotho, NaPi2a, NaPi2b, NaPi2c, Cyp27b1, Cyp24a1, Fgfr1, Fgfr2, Fgfr3 and Fgfr4; there was also low-level expression of Fgf23 (395). Cellular localization of these genes has not been determined. B) HUMAN DATA. Human placentas are discoid and hemochorial and are considered to have similar structure, function, and diffusional characteristics to placentas of mice and rats (174, 311, 536, 553). They are hemomonochorial, meaning that a single trophoblast cell layer separates maternal and fetal blood. Fingerlike projections (villi) protrude into a large intervillous maternal blood-filled space. There is no labyrinth as in the rodent placenta, nor is the intraplacental yolk sac present. The trophoblasts express numerous calciotropic and phosphotropic genes including calbindin-D9k, Ca2⫹-ATPase (isoforms 1 and 4), and TRPV6 (63, 95, 273), PTHrP (82, 162, 184), PTH1R (82, 183), CaSR (69), VDR (664), 1␣hydroxylase (156, 728, 758), calcitonin (35), calcitonin receptor (356, 471), and Klotho (483). C) SUMMARY OF KEY POINTS. The epitheliochorial placentas of sheep are structurally and functionally different from the hemochorial placentas of humans and rodents, but there are also differences between human and rodent placentas. The rodent placenta is considered a close functional match to the human placenta. All in vivo data about placental function come from study of non-human placentas, whereas there are limited in vitro human data of placental gene expression and mineral transport. 3. Methods for studying placental mineral transport Several methods have been used to assess placental mineral transport. There are significant differences among them that need to be reviewed to understand the inherent limitations in the data obtained from them. A) IN SITU PLACENTAL PERFUSION IN RATS AND LAMBS. The most rigorous technique is in situ placental perfusion, which has been used in lambs, goats, guinea pigs, and rats (97, 433, 554, 654, 723). The procedure begins by removing the fetus and connecting the placenta to a semi-closed circuit via the umbilical vessels (FIGURE 7). Autologous fetal blood or a blood substitute is used to perfuse the placenta in situ (i.e., within the mother), but the flow rate and perfusion pressure are maintained at arbitrary set levels. 45Ca and 51Cr-EDTA are infused together into the maternal circulation (32P or 28 Mg are substituted for 45Ca to measure placental phosphorus or magnesium transport, respectively). Repeated blood samples are taken from the maternal circulation and umbilical vein to calculate the radioisotope clearance rates. 51 Cr-EDTA is not actively transported but diffuses across these placentas, so its appearance within the umbilical vein is considered to reflect diffusion of isotope. The rate of active calcium transfer is calculated from formulas that incorporate the speed at which the 45Ca concentration declines in the maternal circulation, and the rate at which the concentration of 45Ca increases relative to 51Cr-EDTA in the umbilical vein (554). Hormones, drugs, or antibodies can be infused into the umbilical artery (placental in-flow) to test their effects on fetal regulation of placental calcium transport, or into the maternal circulation to determine if there are maternal effects on the regulation of placental calcium transport (723). Significant limitations of this technique include that it is done in the absence of fetal control, the mother is deeply anesthetized with potential effects on uterine and placental blood flow, a single placenta is measured with no simultaneous matched control, and the perfusion is done at a set pressure and rate that may not reflect physiological values. Indeed, when 32P accumulation in the placenta has been assessed before and up to 10 days after removal of the fetus and compared with an intact placenta and fetus within the same uterus, significant differences in phosphate concentration are presented at 1 h after the fetus is removed, are four to five times normal at 6 h, and become profound as the days pass (703). Significant advantages of this technique include that frequent sampling from the pla- Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1159 CHRISTOPHER S. KOVACS Venous sampling Pressure gauge Reservoir from umbilical veins to umbilical arteries Perfusion (in-flow) Pump FIGURE 7. Placental perfusion studies in fetal lambs and rats. The fetus is removed from the uterus sometime before the experiment begins, and the umbilical vessels are connected to a semi-closed circuit to enable perfusion of the placenta in situ. 45Ca (alternatively, 32P or 28Mg) and 51Cr-EDTA are infused in the anesthetized mother, and continuous measurements are made of the declining concentration of radioactivity in the maternal circulation, and the increasing concentration of radioactivity in the umbilical vein portion of the circuit. 51Cr-EDTA controls for diffusion of the isotopes and for blood flow differences from one placenta to the next. Test peptides or hormones are infused via the umbilical artery portion of the circuit, and changes in subsequent maternal-fetal transfer of 45Ca, 32P, or 28Mg are detected as an increase in the rate at which the isotope concentration increases within the umbilical vein portion of the circuit. The reservoir contains autologous fetal blood or a blood substitute. The same procedure has been miniaturized for use in murine placentas, but due to their small size it is possible to take only a single blood sample from the mother and the fetal circuit, it is necessary to dilate the placental and umbilical vasculature first, and the experiment is done without 51 Cr-EDTA. [From Kovacs (330), with permission from Elsevier.] cental circuit and the mother enable similar experimental conditions to be maintained from one pregnant dam to the next. The same method has been used to study placental phosphorus and magnesium transport. Studies with magnesium have been very limited due to the short half-life of the isotope, hence the need to have it custom-synthesized locally and at high cost. B) PLACENTAL TRANSPORT WITHIN INTACT MICE. The second method was developed for use in mice (FIGURE 8). An intracardiac injection of 45Ca and 51Cr-EDTA is administered to a pregnant mouse that remains anesthetized from isoflurane for ⬍30 s. Between 5 and 30 min after the injection, a C-section is done to remove the fetuses, and the radioactivity within each fetus is quantified. 51Cr-EDTA controls for differences in diffusion, blood flow, or size of the individual placentas because it crosses the placenta by passive diffusion only. The relative rate of placental calcium transfer is calculated by normalizing the 45Ca activity to 51Cr-EDTA within each fetus; the 45Ca activity alone can also be compared (340). Additional adjustments can be made for placental weight, but these do not add significantly to the adjustment for 51Cr-EDTA. The utility of 51Cr-EDTA has been shown by a two- to threefold variation in its accumulation that can be present among WT fetuses of the same mother, while the 45Ca/51Cr activity of the WT fetuses remains unchanged (unpublished data). This likely indicates that blood flow to any one placenta within the same murine uterus is constantly changing and will not be simultaneously equal among all placentas in a litter. Significant limitations of this procedure include that a relative and not an 1160 absolute rate of calcium transfer is obtained among the fetuses within each litter, and that after a blind intracardiac injection the amount of isotope reaching the maternal circulation may differ somewhat between experiments. However, each fetus within a litter is exposed to the same ratio of 45Ca to 51Cr-EDTA in the maternal blood. Significant strengths include that the dam is only briefly anesthetized, the fetuses remain intact, placental blood flow or perfusion pressure has not been changed, all placentas within a litter received the same concentrations of isotopes, and the 8 –12 normal, heterozygous, and null fetuses within each litter serve as controls for each other, as well as enabling comparisons from one litter to the next. The fetuses can also be treated with hormones prior to the intracardiac injection of isotopes (FIGURE 8). The procedure has also been adapted to study placental phosphorus transport (395). C) IN SITU PLACENTAL PERFUSION ADAPTED TO MICE. The third method has adapted the in situ placental perfusion technique for the small scale of murine placentas. Mouse placentas are ⬍1 cm in diameter with tiny umbilical vessels, which represents substantial technical challenges for a perfusion experiment. One placenta is selected, and its fetus is removed. The placental and umbilical vasculature are first maximally vasodilated with nitroglycerin in order that the umbilical vessels can be successfully catheterized and hooked up to a semi-closed circuit (61, 62). 45Ca is infused alone, without 51Cr-EDTA or another isotope to control for diffusional differences or placental size. Numerous maternal or umbilical blood samples cannot be collected because Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM A B FIGURE 8. Placental mineral transfer studies in fetal mice. The pregnant mother receives an intracardiac injection of isotopes [45Ca or 32P combined with 51Cr-EDTA] (A). After 5, 15, or 30 min, the fetuses are removed from the uterus, and the activity of each isotope is measured within each fetus. Net transfer of 45Ca or 32P is expressed relative to the accumulation of 51Cr-EDTA for each pup. To test the effect of hormones and drugs to regulate the rate of placental mineral transfer, a laparotomy is performed and selected fetuses are given an intra-abdominal injection of the test substance or diluent (B). After a time course of 60 min or longer (to allow the hormone or drug to have its effects), the placental calcium transfer experiment proceeds with the intracardiac injection of isotopes administered to the mother as in A. [From Kovacs (330), with permission from Elsevier.] the recoverable blood volumes of adult and fetal mice are too small, so maternal-fetal clearance of 45Ca cannot be directly calculated. Instead, the rate of disappearance of 45 Ca from the maternal circulation was determined by sampling a single time point from different mothers, and creating a disappearance curve from the aggregate results (61, 62). This curve allows the maternal-fetal 45Ca clearance to be estimated under the assumption that experimental conditions remain identical from mouse to mouse. Significant strengths of this procedure are that it provides a quantitative estimate of the rate of placental calcium transfer in mice, and that it may be possible to study the effects of different hormones or drugs. This technique shares the significant weaknesses mentioned earlier for the in situ perfusion technique in fetal lambs and rats. Additional significant concerns include that an artifactual result may occur from dilating the placental vessels with nitroglycerin, especially if placental vasculature, blood pressure, or responsiveness to vasodilators differ between WT and mutant placentas. Furthermore, the lack of repeated blood sampling and the use of a derived clearance curve mean that the amount of isotope in the maternal and placental circulations are assumed rather than directly measured. As noted above, blood flow can vary two- to threefold from one murine placenta to the next within the same uterus based on the 51Cr activity in each fetus after its administration to the mother, but this technique requires a single placenta and an arbitrary perfusion pressure and rate. D) INDIRECT MEASURES. A fourth technique has involved measuring the calcium or phosphorus content of the fetal skeleton and inferring the rate of placental mineral transport from that. In one approach, the pregnant ewe receives a single infusion of 45Ca or 32P 7 days before slaughter followed by daily treatment with a calciotropic hormone, and at the end of the experiment, the 45Ca or 32P content of the fetal skeleton is measured (38, 165). In a second approach, the pregnant ewe is immobilized and catheterized, and calciotropic hormones or diluent are injected three times daily into fetal lambs for 12 days. At the end of the experiment, the ash weight and mineral content of the fetal skeleton are determined (40). At best, these two approaches are measures of net accretion of mineral by the fetal skeleton, and only very indirect measures of what the rate of placental calcium or phosphorus transport might have been during the experiment. The amount of mineral in the skeleton is a composite result from cycling within several compartments of the fetus (blood, amniotic fluid, soft tissues, and bone), the effect of the hormones to directly stimulate or inhibit bone formation and mineralization independent of placental mineral transport, and back-flux of mineral across the placenta into the maternal circulation (330). Some investigators have interpreted a fall in fetal blood calcium to the maternal level or below (so-called “reversal of the maternal-fetal gradient”) to indicate that placental calcium transport has decreased. This is not a valid approach; as results will show in sections IV–X, the blood calcium level is regulated independently of the rate of placental calcium transport. E) ISOLATED PLACENTAL MEMBRANES AND VESICLES. The fetalfacing basement membranes of human syncytiotrophoblasts have been isolated and induced to form vesicles in vitro, with the fetal side facing inward. These can then be used to study the effects of calciotropic hormones and inhibitors to induce signal transduction or stimulate 45Ca accumulation within the vesicle (649). In such studies, accumulation of 45Ca within the cultured vesicles reflects the transport of 45Ca across the basement membrane, so it is a surrogate for measurement of placental calcium transport in the intact animal. Other investigators have created vesicles from either maternal-facing or fetal-facing basement membranes, but with each membrane oriented so that calcium will be transported outward into the media. The vesicle is loaded with 45Ca and then efflux is measured under the influence of PTHrP, PTH, competitive antagonists of PTH or PTHrP, and other hormones (178). Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1161 CHRISTOPHER S. KOVACS Alternatively, other investigators have used isolated maternalfacing brush-border membranes, and fetal-facing basement plasma membranes of syncitiotrophoblasts, to study the expression of calciotropic hormone and their receptors, and the effects of calciotropic hormones and inhibitors on signal transduction within these cells (354, 355). No estimate of placental calcium transport is provided with these approaches. F) SUMMARY OF KEY POINTS. Several methods exist for estimating the rate of placental mineral transport, and each has advantages and disadvantages that must be considered in interpreting the data. The placental perfusion technique in rats and lambs provides the most quantitative data but occurs in the absence of fetal control, and placental function begins to deteriorate within an hour after removal of the fetus. The placental transport technique within intact mice enables relative transport among fetal genotypes within a litter to be accurately determined while each fetus remains undisturbed and in control of placental function. 4. Maternal regulation of placental mineral transport A) ANIMAL DATA. The fetal-placental unit regulates placental mineral transport, but it remains unclear to what extent (if any) maternal hormones or other factors provide any direct regulation of placental mineral transport. Indirect maternal regulation may occur because the concentration of minerals within the maternal circulation represents the supply from which the placenta must extract the necessary minerals. For example, if the maternal blood calcium is reduced by diet or altered maternal hormone levels, it might be anticipated to reduce the rate of placental calcium transport. However, several studies suggest that placental calcium transport is normal even in the presence of maternal hypocalcemia or specific hormone deficiencies. For example, calcium transport across perfused placentas of sheep remained normal despite significant maternal hypocalcemia induced by parathyroidectomy or a severely calcium-deficient diet (99, 723). Similarly, hypocalcemia in Vdr null and Pth null mothers did not impair placental calcium transfer or reduce the skeletal mineral content in their WT or mutant fetuses (342, 623). Loss of calcitonin in pregnant Ctcgrp null mice did not alter placental calcium transfer or the ash weight and calcium content of the fetal skeleton (430). Excess FGF23 and hypophosphatemia in Phex⫹/⫺ females did not alter placental phosphorus transport to any of the fetuses nor the phosphorus content of the skeleton (395). On the other hand, when pregnant ewes were made hypocalcemic by a very low calcium diet, their fetal lambs had slightly delayed ossification and ash weight, despite no change in fetal levels of calcium, phosphorus, PTH, calcitriol, and a bone resorption marker (377). Similarly, when pregnant rats were hypocalcemic due to thyroparathyroidectomy or calcitonin infusions, the fetal bones showed increased resorption when subsequently cultured in vitro 1162 (541, 542), the fetal parathyroids enlarged (206, 625), and fetal femur length and mineral ash content were reduced (104). These findings in lambs and rats are compatible with a reduction in placental calcium transport that is sufficient to delay primary mineralization of bone or induce secondary hyperparathyroidism in the fetuses, while enabling the fetuses to maintain normal concentrations of minerals within the circulation. Conversely, acute (295) and chronic (338) maternal hypercalcemia in rodents have been shown to suppress the fetal PTH level (FIGURE 5). This suggests that maternal hypercalcemia increases the flow of calcium across the placenta, thereby suppressing the fetal parathyroids. Furthermore, when the perfusate calcium concentration is increased on the fetal side of a perfused placenta, the rate of active transport is not decreased in the perfused guinea pig placenta (433). This means that hypercalcemia in the fetus does not suppress placental calcium transport, which implies that placental calcium transport is regulated independently of the fetal blood calcium. With placental calcium transport being nonsuppressible by fetal hypercalcemia, this may explain why maternal hypercalcemia can have adverse consequences on fetal parathyroid function (see sect. XII, A, B, and G). The impact that maternal hypercalcemia and hypocalcemia have on fetal bone metabolism in intact fetuses contradicts the normal placental calcium transport results obtained in the study of isolated, perfused placentas of sheep and rats. The problem may be that the placental perfusion model is not sensitive enough to detect a small reduction in placental calcium transport, one that may be sufficient over the course of gestation to cause fetal secondary hyperparathyroidism and skeletal resorption. Alternatively, the absence of the fetus may lead to an artifactually normal result that does not reflect what occurs when the fetus remains in control of placental function. When the mother is hypocalcemic, placental mechanisms may upregulate to extract the needed mineral from a lower maternal concentration. Consistent with this, when a maternal calcium infusion was administered to pregnant rats, it caused an acute and marked rise in the serum calcium of fetuses whose mothers were hypocalcemic from thyroparathyroidectomy, but caused no increase in serum calcium of fetuses from normal pregnant rats (106). The effect of maternal hormone deficiencies on placental mineral transport has also been inferred by determining the net skeletal calcium content of vitamin D-deficient rats (228), thyroidectomized, thyroxine-supplemented (“calcitonin-deficient”) sheep (38, 163), and sheep that received daily administration of prolactin or bromocriptine (39). In each study, the fetal skeleton had normal mineral content, implying that placental mineral transport had been normal in utero. The lack of an effect of maternal vitamin D or calcitonin deficiency has been confirmed by direct measure- Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM ment of placental calcium transport in Vdr null and Ctcgrp null mothers compared with their WT sisters, as discussed below in sections VIIIB and IXB (342, 430). supply context for the subsequent sections that describe specific skeletal actions of the calciotropic and phosphotropic hormones. A competitive but weak PTHrP antagonist, PTHrP7–34, was infused via osmotic mini-pumps into the circulation of pregnant rats between day 8 and 15 of gestation and was found to decrease fetal weight, inconsistently reduce placental weight, and to increase the number of apoptotic cells within the placenta (677). The investigators inferred that administration of PTHrP7–34 causes fetal-placental growth restriction, but these findings have not been independently replicated. Since the Pthrp null fetal mouse does not show evidence of growth restriction or an alteration in placental size or weight (62, 340), and PTHrP7–34 is unlikely to cross the placenta, these purported actions of PTHrP7–34 must be exerted on the maternal side of the placenta. Patterning of the early embryonic skeleton has been studied in most detail within the mouse. Numerous genes and signaling pathways are required, include Hox genes, Wnts, Hedgehogs, bone morphogenetic proteins, fibroblast growth factors, Notch/Delta, and others (751). Mesenchymal cells are first laid down where bones of the axial and appendicular skeletons will form. In a few places, such as the flat bones of the skull, these mesenchymal cells differentiate directly into osteoblasts that then form intramembranous bones. But most of the skeleton forms through the multistep process of endochondral bone formation, in which mesenchymal progenitors differentiate first into chondroblasts and chondrocytes. A relatively avascular cartilaginous template of each bone is formed, with proliferating chondrocytes at each end that lengthen the template away from the center. The proliferating and early differentiating cells respond to growth factors and hormonal signals such as PTHrP, which prevent chondrocytes from terminally differentiating. As the distance increases from the zone of proliferating chondrocytes, older chondrocytes no longer receive the signals that prevent them from undergoing terminal differentiation, so they become hypertrophic and then undergo apoptosis. Dead chondrocytes and matrix are removed by chondroclasts, new blood vessels form, and osteoblasts begin to lay down the primary spongiosa of bone. Primary ossification centers are present in most endochondral bones of the mouse at term (an exception being small bones within the digits), while most secondary ossification centers develop after birth. B) HUMAN DATA. There are, of course, no direct measurements in human babies of placental mineral transport. However, there are indirect indications that placental calcium transport may be altered when maternal serum calcium is significantly increased or decreased. Maternal hypercalcemia due to primary hyperparathyroidism or familial hypocalciuric hyercalcemia causes suppression of the fetal parathyroid glands that can last for months after birth or be permanent (54, 79, 393, 417, 493, 529, 609, 674). Maternal hypercalcemia likely increases the flow of calcium across the placenta, thereby suppressing the fetal parathyroids. Conversely, maternal hypocalcemia caused by hypoparathyroidism or pseudohypoparathyroidism has been associated with intrauterine fetal hyperparathyroidism, skeletal demineralization, intrauterine fractures, and bowing of the long bones (226, 389, 652, 710). This is compatible with reduced placental calcium transfer that prompts secondary hyperparathyroidism to develop in the fetus. C) SUMMARY OF KEY POINTS. There is no direct evidence that maternal hormones regulate placental mineral transport. Study of isolated perfused placentas of sheep suggest that maternal hypocalcemia does not reduce placenta calcium transport. However, the functional outcomes in animals and humans (hypoparathyroidism in fetuses from hypercalcemic mothers, and secondary hyperparathyroidism in fetuses from hypocalcemic mothers) indicate that significant disturbances in maternal serum mineral levels must alter placental mineral transport despite failure of the perfused placentas to demonstrate it. G. Endochondral Bone Formation, Mineralization, and Remodeling 1. Animal data The details of fetal skeletal formation are well beyond the scope of this review; instead, a brief overview is provided to It is not until primary ossification centers have formed that the skeleton begins to accrete substantial mineral content; simultaneous with this, the placenta ramps up the expression of calciotropic genes and the rate of mineral delivery. As previously mentioned, the fetal rat skeleton accretes 95% of its mineral content during the last 5 days of a 22-day gestation (121). While the net flow of mineral is into bone to support bone modeling, some turnover or remodeling of the newly formed bone occurs in utero, which allows calcium to reenter the circulation and maintain the serum calcium level. This concept is supported by the presence of osteoclasts and expression of osteoclast-specific genes within fetal bones (336, 359) and the presence of bone resorption markers in serum and amniotic fluid (338). As described in detail in sections IVA and VIA, Pthrp null and Pth1r null fetuses are hypocalcemic, which may be due in part to loss of normal skeletal responsiveness or bone turnover. In support of this, expression of a constitutively active PTH1R (Jansen receptor) within the endochondral skeleton increased the fetal serum calcium, including in double-mutants that also Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1163 CHRISTOPHER S. KOVACS lacked Pthrp (330, 596). Furthermore, the fetal skeleton can undergo increased osteoclast-mediated resorption in response to such factors as maternal hypocalcemia (104, 377) or an inactivating mutation of CaSR (338), and present with an undermineralized skeleton or fractures at birth. 2. Human data In human fetuses, a complete cartilaginous scaffold of the endochondral skeleton forms by the 8th week of gestation. Primary ossification centers form in vertebrae and long bones between the 8th to 12th wk, but most mineralization does not occur until the third trimester when 80% of the ash weight and mineral content is accreted (121, 224, 683, 733, 734). Secondary ossification centers form in the femurs by the 34th week. The rate of mineral accretion accelerates from ⬃60 mg/day at week 25 to ⬎300 mg/day from the 35th through 38th weeks, before tapering off slightly in the last 2 wk (635, 734, 735, 761). Normal skeletal turnover likely contributes to maintaining the high level of calcium within the fetal circulation, and skeletal resorption will increase in response to severe maternal hypocalcemia caused by hypoparathyroidism. calcium then rises steadily to reach adult levels by 7–14 days after birth (201, 422). Ionized calcium displays a bimodal pattern, increasing to ⬃80% of the high fetal value during the subsequent 12–36 h, and then declining to normal adult levels over the subsequent 7 days (422). Albumin increases steadily such that the albumin-bound fraction will account for ⬃50% of the serum calcium, in contrast to only 20% of the total during fetal life (422). Neonatal lambs differ significantly by maintaining a high serum calcium, similar to the fetal value, until 3– 4 mo of age (94, 201). Phosphorus follows a similar time course in rodents, dropping significantly in the first 2– 4 h after birth, increasing significantly by 6 h after birth, and declining again over the subsequent day (201). The early decline is consistent with the loss of the placental phosphorus infusion, while the subsequent rise corresponds to a transient hypoparathyroid state of the newborn. Conversely in lambs, and similar to what occurs with calcium, serum phosphorus increases over the first 24 h and stays elevated for at least a week (403). 3. Summary of key points 2. Human data The skeleton is developing and mineralizing during fetal development, but still participates in the normal regulation of serum mineral levels, and will be adversely resorbed if the mineral supply from the placenta is insufficient. After birth, the decline in serum and ionized calcium are typically 20 –30% over the first 12–24 h (32, 143, 387, 594). In one study, the ionized calcium fell from the mean umbilical cord level of 1.45 mM to a mean of 1.20 mM by 24 h after birth (387). After that, the serum and ionized calcium follow a pattern similar to the rodent data, achieving normal adult values after several days. A single study reported that delivery by elective C-section resulted in a lower blood calcium and higher PTH levels at birth compared with babies delivered vaginally (31). Most studies have not distinguished mode of delivery among the neonatal blood values. The use of formula or breast milk influences the postnatal time course of calcium as well. The fall is greater in infants fed formula (32, 686) because the high phosphorus content of formula causes hyperphosphatemia and complexes calcium within the circulation. III. OVERVIEW OF NEONATAL MINERAL HOMEOSTASIS The alterations in circulating concentrations of minerals and hormones that occur during the neonatal period are depicted in FIGURE 2. This section describes the changes in function and roles that are invoked at birth in parathyroids, kidneys, intestines, and endochondral skeleton. A. Serum Mineral Concentrations 1. Animal data The regulation of bone and mineral metabolism changes significantly after birth. Severing the umbilical cord means that the placental mineral pump is lost, and the neonate must initiate mechanisms to absorb mineral from the intestines and reabsorb mineral in the kidney tubules. The onset of breathing induces a rise in pH from relatively acidotic levels in utero, and this will contribute to a fall in the ionized calcium. The serum calcium and ionized calcium fall 40% during the first 6 –12 h after birth in rodents (201, 347). Total 1164 Phosphorus rises over the first 24 – 48 h after delivery (32, 582, 594); after that, it declines toward adult values, consistent with resolution of transient hypoparathyroidism in the newborn. 3. Summary of key points The early neonatal period in humans and rodents is characterized by a significant fall in ionized and total calcium and a rise in phosphorus over the first 12 h, which largely corrects over the succeeding 24 h. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM B. Calciotropic and Phosphotropic Hormone Concentrations 1. Animal data In newborn rats, PTH increases markedly by 6 –12 h after birth (199, 675), with the peak level corresponding to the trough level in serum calcium (675). After that, PTH declines toward adult values over the subsequent 2 wk (199, 675). Calcitriol does not increase until during the third week after birth (near weaning); during that time, 24-hydroxylase actively produces the inactive metabolite 24,25-dihydroxyvitamin D while little or no calcitriol is produced (201, 729). Similar findings were found in newborn lambs up to 11 days after birth with preferential catabolism of 25-hydroxyvitamin D and no production of calcitriol, not even in response to PTH administration (201, 326). These studies suggest that the neonatal kidney is initially resistant to PTH and sluggish in its ability to produce calcitriol. PTHrP becomes undetectable in the adult circulation, but exactly when this occurs after birth has not been rigorously determined. The placenta and extraembryonic membranes are sources of PTHrP secretion that are lost at birth; if these are the main sources, then a prompt fall in PTHrP may be another trigger that causes the parathyroids to upregulate PTH synthesis and secretion. However, neonatal rat pups were found to have persistently high PTHrP1–34 concentrations, in the range of 20 – 40 pM (401). Neonatal lambs also have persistent PTHrP immunoreactivity in the parathyroid glands, and serum bioactivity attributable to PTHrP, over the first several months after birth (94, 396); however, circulating PTHrP measurements were not done to confirm this. Data cited earlier from fetal lambs indicated that the parathyroid gland may be a key source of PTHrP; the persistently elevated serum calcium and bioactivity attributable to PTHrP in neonates may be an indication of persistent secretion of PTHrP by the parathyroids. Calcitonin increases during the newborn period in rats (209), calves (201), and foals (201, 210). In the neonatal lamb it increases and persists for at least 6 wk after birth before falling to adult levels (204, 215). There is a surge in testosterone level of neonatal rats and mice within the first 2–3 h after birth, which rapidly declines to baseline by 6 h (124, 461). After this, testosterone is equivalent between male and female mice for the rest of the newborn period (498, 499, 730). Estradiol is also low in both sexes (498). 2. Human data The postnatal time course of PTH was originally delineated using older COOH-terminal assays, and it was found that PTH remained low over the first 12–24 h, and did not reach peak levels until 48 h or later (32, 143, 177, 595, 598, 685). Subsequent studies used intact PTH assays but obtained measurements solely at birth and 24 h later, and demonstrated that PTH rose significantly (31, 386, 388, 416, 444, 473, 580). Those studies did not determine when the PTH value increased or whether the peak level was achieved at 24 h or later. Calcitriol is low in cord blood and increases to adult values during the first 2 days after birth, likely in response to the rise in PTH (144, 642). A similar increase to peak levels by the fourth day after birth occurs in preterm infants supplemented with vitamin D (231, 582). Milk contains very high concentrations of PTHrP which, if absorbed into the neonatal circulation, could also explain persistently high circulating levels of PTHrP. Neonatal goats that consumed mother’s milk had significantly higher PTHrP compared with kids that received only formula, which may imply that PTHrP had been absorbed into the circulation (559). No study has examined a time course of PTHrP levels in neonatal blood after suckling, or used radiolabeled PTHrP in milk to determine if it reaches the neonatal bloodstream. It is not known whether the neonatal parathyroids synthesize PTHrP, or how soon after birth the circulating PTHrP level becomes undetectable. A single study reported that the plasma PTHrP1–34 level was reduced 50% between birth and day 1 in healthy newborns, but it then increased back to the cord blood level by day 2 (72). As in the animal models, human milk is a very potent source of PTHrP that could support the circulating level if absorbed; by comparison, PTHrP concentrations are low to absent in infant formulas (306, 392, 488). The breast milk content of PTHrP doubled between 2 and 10 days after delivery, but serum PTHrP was very low in the neonates and did not change between those time points (678). The investigators concluded that PTHrP was not absorbed from milk; however, the blood collections were not timed to breastfeeding, and the authors did not consider that PTHrP has a short half-life in blood. Moreover, the assay requires plasma, but the authors measured PTHrP1– 86 in stored sera, so the low PTHrP values may reflect the inevitable degradation of PTHrP in sera, and use of an assay that does not detect PTHrP1–34. Scant data are available pertaining to serum FGF23 levels soon after birth. In WT mice, FGF23 was 10-fold higher than adult values at 12 h after birth (34). Intact FGF23 increased fourfold between birth and 4 –5 days of age (483, 661), whereas COOH-terminal FGF23 maintained the high values of fetal life (661). Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1165 CHRISTOPHER S. KOVACS Calcitonin may increase up to 10-fold over cord blood levels during the first 48 h after birth (32, 53, 444, 595), before gradually declining over the subsequent days or weeks (32, 583). Estradiol remains very low in male and female newborns and stays that way until puberty (91, 739). Testosterone remains very low in females, but in males it surges within the first 24 h (124), and again in the second week after birth until about 4 mo of age, after which it declines to very low levels and remains that way until puberty (133, 739). first 24 h after birth and contributes to the development of postnatal hypocalcemia and hyperphosphatemia. The postnatal awakening of parathyroid function explains the subsequent increase in calcium and calcitriol, and decline in serum phosphorus. D. Renal Mineral Reabsorption and Excretion 1. Animal data 3. Summary of key points Calciotropic hormones levels rapidly progress from fetal values to levels that are similar to the adult (FIGURE 2). PTH, calcitriol, and intact FGF23 increase; PTHrP disappears from the circulation after an uncertain time course; and calcitonin may stay elevated for several weeks. C. Neonatal Parathyroid Function 1. Animal data The parathyroids are important for control of serum mineral concentrations in the neonate. Parathyroidectomy in the newborn rat pup caused a fall in serum calcium and an increase in serum phosphorus (199, 344). Notably the length, morphology, trabecular content, and calcium and phosphorus content of the neonatal skeleton were unaffected by parathyroidectomy in the neonate compared with sham controls, indicating that the development of the skeleton and accretion of mineral is not dependent on PTH during this time period (344). Whether the neonatal parathyroids produce PTHrP is uncertain, but suggestive evidence for it comes from neonatal lambs which maintain increased serum calcium and high PTH-like bioactivity, similar to what is present during fetal development. 2. Human data The increase in PTH follows the early postnatal drop in the ionized calcium, and precedes (and is likely responsible for) the subsequent rise in ionized calcium and calcitriol, and decline in phosphorus. Consistent with the concept that the neonatal parathyroids recover from suppression experienced during fetal development, in the 48 h after birth the parathyroids are sluggish in their response to acute hypocalcemia, such as due to exchange transfusion with citrated blood (143, 595, 685, 686). This sluggishness resolves with increasing postnatal age (685, 686) but becomes prolonged after maternal hypercalcemia during pregnancy (see sect. XII, A and B). 3. Summary of key points A transient phase of relative hypoparathyroidism or sluggish responsiveness by the parathyroids occurs during the 1166 The neonatal kidneys begin to actively reabsorb and excrete minerals. Over the first 24 h there is reduced cAMP responsiveness to PTH in vitro compared with the response obtained during fetal life (675). The rat kidney does not show expression of VDR or responsiveness to calcitriol until the third week after birth (633). TRPV6 and calbindin-D9k show a progressive increase in expression, reaching a maximum at 3 wk after birth (634). There is low level of expression of CaSR at birth that increases after the first postnatal day in mice (112). 2. Human data Urine calcium excretion is low at birth despite the high level of serum calcium in cord blood and the suppressed PTH level. After the first 5–7 days, urine calcium excretion increases over the subsequent weeks despite a lower serum calcium and higher PTH concentration (298, 473). The change in renal calcium excretion is likely due to the developmental increase in renal blood flow and glomerular filtration rate that occurs over the same interval (171, 237, 298). Additional factors include loss of the effects of PTHrP to reabsorb calcium, increased expression of CaSR, and increased responsiveness to calcitriol. Phosphorus excretion is also low at birth and increases with postnatal age (378, 605). This likely reflects not only the increasing level of PTH in the circulation, but (as in the rodent) a developmental increase in renal responsiveness to PTH that has been demonstrated in term and preterm infants (378, 406). The rapid rise in calcitriol after birth likely arises from synthesis in the kidneys, which are stimulated by PTH. 3. Summary of key points The neonatal kidneys undergo developmental changes that progressively increase their responsiveness to PTH, calcitriol, and CaSR. Sluggish kidney function over the first several days after birth likely contributes to the development of neonatal hypocalcemia. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM E. Intestinal Mineral Absorption 1. Animal data In newborn rodent pups, calcium absorption occurs mainly through passive, nonsaturable processes that are not dependent on calcitriol (218, 219, 243, 510 –512). Milk contains a high content of lactose, which directly increases paracellular calcium absorption and net absorption of dietary calcium (86, 366, 500). As the days pass, intestinal cells begin to express VDR (242), increase their expression of calbindin-D9k (80, 634) and TRPV6 (634), and display evidence of calcitriol-dependent active absorption of calcium (243). Passive transport of calcium progressively declines, such that by the time of weaning (3 wk of age) calcitriol-dependent active transport of calcium is the dominant means of intestinal calcium absorption (218, 219, 243), while both calbindin-D9k and TRPV6 have reached maximally upregulated expression compared with the day of birth (634). 2. Human data As noted earlier, preterm human infants demonstrate calcium absorption that is proportional to intake, with a linear relationship between intake and absorption; this is consistent with passive absorption of calcium (44, 46, 221, 581). Absorption did not vary by vitamin D intake in preterm infants, which is also consistent with passive, non-vitamin D-dependent absorption (75). An interventional study found that preterm infants showed no calcemic response to short courses of supraphysiological doses of calcitriol (0.1– 3.0 g/kg), which suggests that the preterm intestines are refractory to calcitriol (708). For perspective, the adult requirement in hypoparathyroidism is typically a total daily dose of 0.25–1.0 g. A second interventional study used 4.0 g/kg of calcitriol administered intravenously for three consecutive days and found a significant increase in serum calcium (325). However, that effect was not the result of an alteration in intestinal calcium absorption but likely reflected an increase in bone resorption: oral intake was minimal, osteocalcin increased significantly, and urine calcium/ creatinine ratio did not change (325). A small study of preterm neonates compared human milk, human milk plus 1,200 IU vitamin D, and human milk plus 1,200 IU vitamin D and phosphorus, and found that only the group receiving phosphorus supplementation had a significant increase in calcium retention (603). This is also consistent with a lack of effect of vitamin D/calcitriol to increase calcium absorption and retention in the preterm intestines. The preterm infant is also limited by reduced intestinal absorption of vitamin D that increases with postnatal age (263). Another study supplemented the preterm neonate with 1,200 IU vitamin D or 0.5 g calcitriol, both of which increased intestinal absorption of calcium but required up to 4 wk to achieve the full effect, with use of calcitriol being more effective than vitamin D (604). Similar to the data from rodents, calcium absorption in neonates is enhanced by the lactose content of milk (317, 318). Intestinal absorption and retention of calcium increase with both postnatal age and gestational age, with additional variations superimposed by the composition of the milk (45, 221, 256, 485, 604, 613). These findings suggest a maturational effect toward an active, calcitriol-dependent, saturable mechanism of intestinal calcium absorption as seen in older children and adults. It may be inferred from these data that, similar to the rodent intestine, the human neonate’s intestines undergo progressive upregulation in expression of the vitamin D receptor. However, longitudinal changes in expression of VDR have not been studied in human neonatal intestines. Similarly, it is unclear when the neonatal intestines change from passive to active absorption, although the responsiveness to calcitriol at 4 wk of age in one clinical study may be an indication that active absorption has become more prominent by that age (604). The preterm infant must supply a high rate of calcium to the skeleton as if it were in utero, but this exceeds what can be achieved with breast milk or standard infant formulas. In the first week after birth, most nutrition is provided parenterally because the intestines are not able to absorb a sufficient amount (445). As oral feeds are implemented, special formulas high in calcium are needed to meet the preterm neonate’s need for mineral. In the very low birth weight preterm neonate, a daily calcium intake of 120 –200 mg/kg is usually recommended with much of that being given parenterally (445). As the postnatal days pass for the preterm baby, the skeletal demand for mineral declines from the high fetal rate and can eventually be met with breast milk or formula. The efficiency of intestinal calcium absorption is influenced greatly by the nutrition that the neonate receives. Absorption of calcium is more efficient from breast milk than from infant formula regardless of vitamin D supplementation (223, 604, 732). More than 90% of the phosphate content of breast milk and formula is absorbed (604), but the higher phosphate content of formula can lead to double the serum phosphorus level compared with neonates that consume breast milk (32, 223, 604). High serum phosphate lowers serum calcium and is a cause of neonatal hypocalcemia (686). The differences between breast milk and formula have had clinical effects, with breast-fed infants having higher serum calcium levels and a lower risk of hypocalcemia compared with formula-fed infants (32, 223). 3. Summary of key points A developmentally programmed maturation alters intestine calcium absorption from passive, nonsaturable mechanisms to an active, calcitriol-dependent process. This has been well established in the neonatal rodent, but there is sufficient evidence to accept that it probably occurs in human Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1167 CHRISTOPHER S. KOVACS neonates as well. This explains why preterm babies are dependent on passive absorption of mineral until the intestines are capable of responding to calcitriol. F. Skeletal Metabolism 1. Animal data The efficiency of intestinal mineral absorption has significant effects on the neonatal skeleton. Neonatal rodents are largely protected from abnormalities due to the effects of lactose to enhance absorption of calcium. Consequently, despite significant abnormalities such as hypoparathyroidism, vitamin D deficiency, or loss of VDR, the neonatal rodent maintains normal bone mineralization until the time of weaning. It is at that point in time, when the intestines are dependent on calcitriol-regulated active intestinal calcium absorption, that the skeleton begins to develop deficits in mineral content and altered development (see sects. VD and VIIID). Normal peak bone density and skeletal calcium content differ markedly among mouse strains (48), with the commonly used inbred C57BL/6 strain having a significantly lower bone mass than the outbred Black Swiss strains (192, 309, 310, 744). Serum and ionized calcium are also significantly lower in C57BL/6 compared with Black Swiss mice (340), but there are no obvious differences in calciotropic and phosphotropic hormone levels between the two strains. Unidentified genes and proteins that affect bone metabolism and development likely explain this interstrain variability. 2. Human data The late term fetus accumulates skeletal calcium at a rate of over 300 mg/day or ⬃150 mg·kg⫺1·day⫺1 (635, 734, 735, 761), whereas this drops to ⬃30 – 40 mg·kg⫺1·day⫺1 in the normal neonate (683). The skeleton is highly dependent on the intestines to supply needed mineral; any deficiency in the diet or in the ability to absorb mineral will cause impaired bone mineralization and increased resorption of the skeleton. This is especially true for the preterm neonate for whom breast milk or standard formula would provide insufficient amount of calcium. However, the ability of the intestines to passively absorb mineral in proportion to intake enables special formulas that are high in calcium to provide the needed mineral, after an initial phase in which most of the nutrition is administered parenterally. A consequence of inadequate intestinal delivery of calcium and phosphorus is rickets of prematurity. This is not due to vitamin D deficiency because it is not prevented by supplementation of the mother during pregnancy, nor is it responsive to treatment of the neonate with vitamin D or calcitriol (10, 92, 323, 432, 510). It is precipitated by loss of the 1168 placental calcium pump at a time when skeletal demand for calcium is very high, normal oral calcium intake is insufficient to meet the demand, and the intestines are developmentally immature, and likely lack expression of VDRs, to deliver calcium through active absorption (92, 510). Bone mineral content is initially appropriate for gestational age at birth, but it fails to increase appropriately if left untreated (235, 236, 324, 446, 640, 641, 690). Physical and radiological signs of rickets of prematurity may develop, including rachitic rosary, chest deformity, osteopenia, pathological (especially rib) fractures, metaphyseal stippling, and flaring and widening of the epiphyses of long bones (92, 513, 514). Special oral formulas or parenteral preparations containing high calcium and phosphorus content will correct the bone demineralization and allow the skeleton to develop normally (10, 92, 323, 432, 641, 643). Follow-up at 3– 4 yr of age has shown normal bone mineral content in children who earlier had rickets or low bone mineral content of prematurity (266). 3. Summary of key points The neonatal skeleton accrues mineral at a rapid rate and but will quickly become demineralized if intestinal mineral delivery is inadequate. Infants that are preterm, severely vitamin D deficient, lack calcitriol or the VDR, or are hypoparathyroid/aparathyroid, are especially prone to this complication. IV. PARATHYROID HORMONE-RELATED PROTEIN In the decades that followed the development of specific PTH assays, it was recognized that fetal blood contains high PTH-like bioactivity but low immunoreactive levels of PTH (3, 20, 21, 67, 571). PTHrP was cloned in 1987 (410, 646, 658), and subsequent work confirmed that it mimics many of the biological actions of PTH (333), circulates at high levels in fetal blood compared with PTH (168, 306, 600, 671), and accounts for the high PTH-like bioactivity in fetal blood (7, 94, 396). The relative difference in circulating levels of PTHrP and PTH may even be underestimated because the commonly used PTHrP1– 86 assay does not detect biologically active PTHrP1–36. These data led many to hypothesize that PTHrP is the main calcium-and-bone-regulating hormone of fetal life, and that the suppressed levels of PTH indicate that it is relatively unimportant until after birth. Given this context, PTHrP’s role in fetal and neonatal mineral metabolism will be discussed first, followed by PTH in section V. A. Regulation of Serum Minerals Comparison of Pthrp null fetuses to their WT and Pthrp⫹/⫺ littermates, which share the same intrauterine environment, Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM has confirmed the importance of PTHrP in regulating aspects of fetal mineral and bone metabolism. Pthrp null fetuses have a hypoparathyroid-like phenotype characterized by hypocalcemia (both total and ionized levels), and a serum phosphorus that is raised even further above the normally high fetal phosphorus level (62, 340, 341, 694). Serum magnesium remains normal (330). Notably the fetal ionized calcium falls to a level equal to the maternal ionized calcium (340, 694). Serum PTH increases threefold in Pthrp null fetuses (336), likely through CaSR-mediated effects to increase serum calcium to the normal adult level but no higher. This conclusion is supported by additional observations in double-mutant fetuses. When one or both alleles of CaSR are inactivated in Pthrp null fetuses (i.e., Pthrp null/ Casr⫹/⫺ fetuses and Pthrp/Casr double mutants), serum PTH and ionized calcium levels increase significantly (338). However, when the NH2-terminal actions of PTH and PTHrP are blocked by deletion of PTH1R (Pth1r null fetuses), simultaneous ablation of one or both alleles of CaSR does not alter the ionized calcium (i.e., Pth1r null/Casr⫹/⫺ fetuses and Pth1r/Casr double mutants) (338). Section V that follows notes that hypocalcemia occurs in several models lacking PTH or parathyroids, but within each there is no compensatory increase in plasma PTHrP (341, 623). Therefore, PTHrP is unresponsive to falls in the ambient fetal blood calcium, which may imply that it is autonomously produced and regulated by, and in response to, local factors within the placenta such as the flow of calcium. The source of PTHrP in the fetal circulation cannot be determined from the Pthrp null model because PTHrP is absent in all fetal tissues. B. Regulation of Placental Mineral Transport Several lines of evidence indicate that PTHrP is an important regulator of the active transport of calcium and magnesium across the placenta from mother to fetus. Fetal lambs were thyroparathyroidectomized and treated with thyroid hormone; several days later, the fetus was removed from the uterus and its placenta was studied using the in situ placental perfusion model (see details in section IIF3). The rate of increase in the concentration of 45Ca/ 51 Cr-EDTA in the umbilical vein portion of the circuit (placental outflow) was significantly reduced after prior fetal thyroparathyroidectomy, compared with the rate observed in placentas from separate ewes whose fetuses had been intact (97, 723). Furthermore, infusion of autologous blood from intact fetal lambs (sometimes the twin from the same ewe) restored the rate of increase in 45Ca/51Cr-EDTA to normal values (97). These results showed that prior fetal thyroparathyroidectomy reduced the basal rate of placental calcium transport when measured several days later but, importantly, in the absence of the fetus which had been removed hours before. The investigators concluded that fetal parathyroids produce a factor that stimulates placental calcium transport, such that its absence causes the rate of transport to fall. The same investigators went on to infuse specific peptide sequences of PTHrP or PTH into the umbilical artery, to determine their effects on placental calcium transport. Peptides that contained a mid-region portion of PTHrP (PTHrP1–141, PTHrP1– 86, and PTHrP67– 86) increased the rate of change in the concentration of 45Ca/51Cr-EDTA in the umbilical vein, whereas PTHrP1–34 and PTH1–34 had no effect (6, 96, 556). When PTHrP38 –94 was later determined to be the structure of mid-regional PTHrP (490, 517), these investigators repeated the experiments and confirmed that PTHrP38 –94 also significantly increased the rate of placental calcium transport in perfused placentas from previously thyroparathyroidectomized fetal lambs (745). This approach has also been used to show that mid-molecular PTHrP stimulates placental magnesium transport, but not phosphate transport (43, 47, 96, 653). The conclusion of these experiments was twofold: 1) that PTHrP stimulates placental calcium and magnesium transport and 2) that the relevant source of PTHrP is the fetal parathyroids. However, no measurements of plasma PTHrP were done in thyroparathyroidectomized lambs, so it remains unknown whether fetal parathyroidectomy reduced the circulating level of PTHrP. The in situ placental perfusion model has also been used in rats, and at first glance the evidence obtained seems inconsistent with the data obtained from perfused placentas of lambs. Decapitation was done to crudely mimic parathyroidectomy, and (similar to the effects of thyroparathyroidectomy in fetal lambs) this resulted in a fall in placenta calcium transport (554). That deficit in placental calcium transport was increased slightly (but remained well below the normal value) after infusion of PTH1–34 (554); no PTHrP peptides were tested in the decapitation model. However, in perfused placentas from intact fetuses, PTH1– 1–34 , and PTHrP67– 86 each had no significant ef34, PTHrP fect on placental calcium transport (554, 611). These data do not contradict the findings in ovine placentas because mid-molecular PTHrP was not tested in the decapitated fetal rat model, and the efficacy of PTH1–34 in the decapitation model was very modest compared with that of midmolecular PTHrP in placentas from thyroparathyroidectomized fetal sheep. Furthermore, the failure of mid-molecular PTHrP to stimulate placental calcium transport in placentas from intact rats may be the result of PTHrP-mediated effects already being maximally exerted. Data described in the next paragraph corroborate this conclusion. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1169 CHRISTOPHER S. KOVACS The creation of Pthrp null fetuses (297) enabled a different approach to measure placental calcium transport within intact fetuses using the methodology described in section IIF3. 45Ca and 51Cr-EDTA were administered by intracardiac injection to pregnant mice, and the intact fetuses were removed 5, 15, or 30 min later. Compared with their WT and Pthrp⫹/⫺ littermates, 45Ca/51Cr-EDTA accumulation in Pthrp null fetuses was 25% lower after 5 min and 40% lower after 30 min (340). To confirm that this reduction was specific to loss of PTHrP, a laparotomy was done to administer an intra-abominal injection of a specific peptide form of PTHrP, PTH, or diluent to several fetuses. Sixty minutes later, 45Ca and 51Cr-EDTA were administered to the mother by intracardiac injection, and the fetuses were removed 30 min after that. PTHrP1– 86 and PTHrP67– 86 increased the accumulation of 45Ca/51Cr-EDTA to a value not different from WT littermates, whereas PTH1– 84 and PTHrP1–34 had no significant effect (340). Importantly, injections of mid-molecular PTHrP had no effect in WT fetuses, which suggests that the WT fetus may already have maximally stimulated PTHrP responses on the placenta, such that pharmacological treatment had no additional effect. This is consistent with the data cited in the preceding paragraph, in which mid-molecular PTHrP failed to stimulate placental calcium transport in placentas from intact rats. These results from intact Pthrp null fetuses, and the aforementioned data from in situ perfused placentas from thyroparathyroidectomized fetal lambs, both showed that midmolecular forms of PTHrP stimulate placental calcium transport, whereas peptides of PTHrP that contain only the NH2-terminal form, and PTH itself, have no significant effect. Analysis of Pthrp null placentas revealed reduced mRNA and protein levels of Ca2⫹-ATPase and calbindinD9k within the intraplacental yolk sac compared with placentas from WT littermates (337), which may indicate that PTHrP acts in part through the stimulation of Ca2⫹-ATPase and calbindin-D9k expression. Alternatively, the reductions in Ca2⫹-ATPase and calbindin-D9k expression could be a consequence of reduced placental calcium transport rather than its cause. That these reductions in Ca2⫹-ATPase and calbindin-D9k expression may be physiologically significant is supported by Trpv6 null fetuses, which have markedly reduced placental calcium transport, and loss of Trpv6, Ca2⫹-ATPase, and calbindin-D9k expression within the intraplacental yolk sac (659). Notably, the reduction of Ca2⫹ATPase and calbindin-D9k expression within the Pthrp null placenta was limited to the intraplacental yolk sac which normally expresses PTHrP (337). No difference in Ca2⫹ATPase and calbindin-D9k mRNA or protein expression was found within the adjacent trophoblasts or when the entire placenta was analyzed by Western blot (62, 337). The results described so far are consistent in revealing a role for PTHrP to stimulate placental calcium transport in fetal 1170 lambs and mice. Contrary evidence comes from the in situ placental perfusion model, miniaturized for use in fetal mice (see details in sect. IIF3) (61, 62). A fetal mouse was removed, and nitroglycerin was used to dilate the placental vasculature before cannulating the umbilical vessels and performing the placental perfusion technique. 45Ca was administered alone, and a single sample was later obtained from the maternal circulation and the placental circuit. With the use of this technique, Pthrp null placentas had an increased rate of placental 45Ca transport compared with perfused WT placentas from separate mothers. The investigators concluded that PTHrP inhibits rather than stimulates placental calcium transfer. There are several possible reasons why the results of these experiments were the opposite of what the prior studies found in lambs and mice had revealed. The likeliest is that the use of nitroglycerin to dilate the placental vasculature may have created an artifactual result when the vascular expression and actions of PTHrP are considered. PTHrP is expressed within vascular smooth muscle wherein it has actions to relax smooth muscle and thereby cause vasodilation and increased blood flow (517, 551). PTHrP and PTH share vasodilatory effects when injected into living animals (120, 127, 247, 464, 497), and overexpression of Pthrp or Pth1r within vascular smooth muscle cells of mice leads to lower blood pressure, increased perfusion pressure, and reduced vascoconstriction in response to challenge with angiotensin II (476). When PTHrP was deleted from vascular smooth muscle cells by using a vascular smooth muscledriven promoter for Cre in Pthrp floxed mice, the mice showed reduced renal perfusion and glomerular filtration, consistent with renovascular constriction (533). Therefore, a similar increase in vascular constriction and reduced blood flow may be present in the Pthrp null placenta compared with the WT and Pthrp⫹/⫺ placentas. Consequently, nitroglycerin may have differential vasodilatory effects in Pthrp null placentas which, combined with placental function being studied well after the fetus has been removed, may have artifactually increased the apparent transport of calcium across the perfused placenta. The expression of PTHrP within vascular smooth muscles raises the possibility that one of the mechanisms through which PTHrP controls placental calcium transport may be through regulating blood flow to and within the placenta. In the absence of PTHrP, intraplacental blood flow may be reduced due to increased vasoconstriction, thereby reducing the rate at which maternal and fetal blood can exchange minerals and nutrients. However, altering blood flow dynamics is at best a partial explanation because PTHrP and PTH share these vasodilatory effects through their actions on PTH1R, whereas it is mid-molecular PTHrP, which does not act on PTH1R, that has been clearly shown to stimulate placental calcium transport. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM That PTHrP stimulates rather than inhibits placental calcium transport is supported by studies in other mouse models wherein the placental calcium transport technique for intact fetal mice was used. Pth1r null fetuses (360) have greater than 10-fold increases in circulating levels of both PTHrP and PTH (336, 341). They lack PTH1R and so cannot respond to NH2-terminal actions of PTH or PTHrP, but should be responsive to actions of mid-molecular PTHrP on its (as yet uncloned) receptor(s). The placental calcium transport rate was increased in Pth1r null fetuses to 150% of the value of their WT fetal littermates (340), in keeping with anticipated effects of high plasma PTHrP concentrations to increase the rate of placental calcium transport. Aparathyroid Hoxa3 null fetuses and Pth null fetuses each have normal plasma PTHrP concentrations and normal rates of placental calcium transport (336, 341, 623). Casr null mice have significantly reduced plasma PTHrP levels and reduced placental calcium transport (330, 338). The concordance between plasma PTHrP concentrations and placental calcium transport among these different mouse models (Pthrp null, Pth1r null, Hoxa3 null, Pth null, and Casr null fetuses) are consistent with the conclusion that PTHrP’s role is to stimulate placental calcium transport. Previously the increased fetal blood mineral concentrations were considered to be proof of active transport of calcium across the placenta, and elimination of the maternal-fetal gradients in the relative concentrations of calcium or magnesium (such as by fetal thyroparathyroidectomy) were considered to be evidence that active transport had been eliminated (97, 100, 198). However, study of these murine gene deletion models have revealed that placental calcium transport is not a direct determinant of the fetal blood calcium level or skeletal mineral content. The placenta supplies the mineral which may then be incorporated into bone or soft tissues, remain in the circulation or extracellular fluids, be excreted into the urine and amniotic fluid, or be transported back across the placenta and into the maternal circulation. The blood levels of calcium and incorporation of calcium into bone are regulated separately from the placental delivery of calcium. Consequently, despite hypocalcemia and reduced placental calcium transport, Pthrp null fetuses have a normal or even increased ash weight and mineral content (the reason for this is addressed in the next section) (62, 336, 694). In contrast, Pth null and aparathyroid Hoxa3 null fetuses have a normal rate of placental calcium transport, but are hypocalcemic with significantly undermineralized skeletons. These findings point to the role of PTH in regulating blood calcium and stimulating mineralization of the fetal skeleton independent of PTHrP and the rate of placental calcium transport. Casr null fetuses have a decreased rate of placental calcium transport and a reduced mineral content of the skeleton, despite a higher than normal serum calcium that should otherwise facilitate mineralization of the developing skeleton (338). Although the Pthrp null model does not address whether fetal parathyroids produce PTHrP or even if parathyroidderived PTHrP regulates placental calcium transfer, when considered together with results from Hox3 null and Pth1r null fetuses (sects. V and VI), the studies in fetal mice are more consistent with a placental source of PTHrP. The studies in fetal sheep and mice both found that a mid-molecular form of PTHrP stimulates placental calcium transfer, but differ on what its source might be. These seemingly discrepant results between fetal lambs and mice might be due to species difference; there are no comparable human data. An indirect approach to estimating placental calcium transport was carried out in fetal lambs that received thrice-daily injections of PTHrP1–34 or PTHrP1– 86, with or without calcitonin, for a total of 12 days. Each form of PTHrP significantly increased the mineral content of the fetal skeleton compared with control (diluent), and the simultaneous use of calcitonin blocked the effect of PTHrP (40). The authors inferred that both forms of PTHrP stimulated placental calcium transport and that calcitonin countered PTHrP’s actions on the placenta. However, these results can be explained by the effects of both NH2-terminal forms of PTHrP to stimulate bone formation and mineralization, and for calcitonin to inhibit bone resorption (and, secondarily, bone formation), without implicating any direct effect of calcitonin on placental calcium transport. Magnesium and phosphorus are each actively transported across the in situ perfused placentas of rats (612, 653) and lambs (47, 96, 397). Mid-molecular PTHrP stimulated magnesium transport across in situ perfused ovine placentas (47, 96, 397) but not rodent placentas (611), and it did not affect placental phosphorus transfer (43, 397, 653). Placental transport of these minerals has not been examined in Pthrp null fetuses. C. Regulation of Endochondral Bone Formation PTHrP is produced by perichondrial cells and proliferating chondrocytes, and its receptor (PTH1R) is expressed further down the growth plate, within prehypertrophic chondrocytes, preosteoblasts, and osteoblasts. PTHrP acts in concert with Indian hedge hog and Wnt signaling to delay terminal differentiation and hypertrophy of chondrocytes (300, 714). PTHrP’s role is clearly illustrated by the Pthrp null skeleton, which has premature chondrocyte hypertrophy, apoptosis, and initiation of primary bone formation; the result is a chondrodysplasia with shortened limbs (FIGURE 9) (297, 300, 360). The normally cartilaginous portions of the ribs turn into bone, and bones that normally mineralize after birth become mineralized in utero (297). In contrast to the effects of loss of PTHrP, overexpression of PTHrP or PTH1R Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1171 CHRISTOPHER S. KOVACS A B C D E F WT Pthrp null H p<0.02 120 Ash Calcium (Normailzed to Pthrp+/-) Ash Calcium (Normailzed to Hoxa3+/-) G Hoxa3 null 100 80 60 40 20 0 WT Hoxa3+/- Hoxa3-/- has the opposite effect of delaying chondrocyte hypertrophy, resulting in a cartilaginous skeleton at birth that slowly undergoes bone formation and mineralization over the succeeding weeks (596, 726). Although PTHrP’s anabolic actions on preosteoblasts and osteoblasts have been lost, Pthrp null fetuses show normal to increased expression of osteoblast-specific genes and proteins, including collagen ␣1(I), collagenase-3, osteocalcin, and osteopontin (297, 359). This is likely because the three- 1172 WT Pthrp+/- Pthrp-/- FIGURE 9. Comparison of the effects of loss of PTH vs. loss of PTHrP on the fetal skeleton. Hoxa3 null fetuses lack parathyroids and PTH but have normal circulating PTHrP, while Pthrp null fetuses lack PTHrP and have upregulated PTH in the circulation. A–C: whole mounts of fetal skeletons (ED 18.5) stained with alizarin red S (mineral) and alcian blue (cartilage). Crownrump length, lengths of long bones, and mineralization pattern appear unaltered in the Hoxa3 null fetus (A) compared with its WT sibling (B). Note the normal (blue) cartilaginous portions of the ribs and sternum in the WT and Hoxa3 null. Contrast the Pthrp null from the same genetic background (C), which is shorter, has a rounded vault of the skull, shortened mandible (hence protruding tongue), shortened and thickened long bones, advanced mineralization, and abnormal mineralization of the cartilaginous portion of the ribs and sternum. D–F: von Kossa-stained sections of the fetal tibias counterstained with methyl green. The overall morphology and lengths of the upper portions of the tibia (both chondrocytic and osseous zones) are normal in the Hoxa3 null (E), compared with the progressively shortened and disorganized growth plates of the Pthrp null (F). The double-headed arrows compare the lengths of the chondrocytic growth plate from the proliferating through hypertrophic zones. The amount of mineral (black from von Kossa stain) appears to be markedly reduced in the Hoxa3 null but normal in the Pthrp null. G shows the calcium content (by atomic absorption spectroscopy) of Hoxa3 null fetuses compared with WT and Hoxa⫹/⫺ littermates, while H compares the mineral content of Pthrp null fetuses compared with WT and Pthrp⫹/⫺ littermates. The calcium content is significantly reduced in Hoxa3 nulls, but normal in Pthrp nulls due to the accelerated mineralization of bone and abnormal mineralization of cartilaginous structures. Ash weight and magnesium content were also reduced in Hoxa3 nulls but normal in Pthrp nulls (not shown). [Adapted from Kovacs et al. (336). Copyright 2001 The Endocrine Society; permission conveyed through the Copyright Clearance Center, Inc.] fold compensatory increase in circulating PTH (336) stimulates osteoblasts and prevents the deficits in osteoblast function that loss of PTHrP should otherwise have caused. This conclusion is borne out by careful comparison of Pthrp null to Pth1r null fetuses, as described in section VIC. Even though increased PTH seemingly ameliorates the skeletal effects of PTHrP deletion prior to birth, mice that lack PTHrP expression only within preosteoblasts and osteoblasts have reduced bone formation and bone mass by 6 wk of age (438). Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM In striking contrast to the physiological findings of reduced placental calcium transport and hypocalcemia, the ash weight and mineral content of the Pthrp null skeleton has been variably reported to be normal or modestly increased (FIGURE 9) (62, 336, 340, 694). This seemingly contradictory finding is likely explained by the early, accelerated, and abnormal calcification that occurs within the Pthrp null skeleton. As described above, bones that normally do not mineralize until after birth become prematurely mineralized in utero; also, normally cartilaginous structures (such as the medial aspects of the ribs and the sternum) are transformed into bone (297). Consequently, the Pthrp null skeleton matures faster and accretes more mineral sooner, and in normal and abnormal locations, thereby leading to a seemingly normal or increased mineral content despite its slightly smaller size and reduced rate of placental calcium transport. purpose? Passive immunization of day-old neonatal mice with antibodies to NH2-terminal PTHrP did not affect serum calcium or the calcium content of their ashed skeletons at 48 h (348), but a longer duration may have been needed to definitively determine if the skeleton would become abnormal. Oral administration of PTHrP1–34 to neonatal mice had no effect, whereas subcutaneously administered PTHrP1–34 increased serum calcium significantly (348); from these data, investigators concluded that PTHrP is not absorbed into the circulation of the neonatal mouse. Deletion of the Pthrp gene from mammary tissue at the onset of lactation resulted in milk that lacks detectable PTHrP, but the growth rate of the pups was qualitatively described as no different than expected (706). Collectively, these results suggest that eliminating PTHrP from milk is of no consequence to the neonate. Complete loss of PTHrP causes death often within minutes of birth, although a few pups have lived for 5 days. The cause of death may be multifactorial, including severe hypocalcemia with cardiac arrhythmias, a rigid rib cage that restricts ventilation, and stiff lungs due to abnormal development of alveolar type II cells and low surfactant (297, 340, 570). However, the same investigators recently examined pups that nursed from mothers in which none, one, or two of the Pthrp alleles had been deleted from mammary tissue at the onset of lactation (409). Deletion of one Pthrp allele reduced the milk PTHrP content, whereas loss of both alleles eliminated PTHrP, but the calcium content of milk was unaltered (409). At day 12, there was an inverse dose-response relationship between the PTHrP content of milk and the ash calcium content of the pups (409), with loss of milk PTHrP leading to increased skeletal calcium. Does PTHrP reduce net intestinal absorption of calcium? That seems unlikely given the previously cited evidence that the calcium content of milk is more readily bioavailable than from formula, and results in a higher serum calcium in neonates. Does some functional portion of PTHrP get absorbed into the neonatal circulation and act on osteoblasts to reduce mineral accretion? Such a pathway could explain how milk consumption might lead to a higher serum calcium but a lower mineral content of the skeleton. It would give PTHrP the evolutionary role of protecting against neonatal hypocalcemia by preventing the skeleton from accreting too much of the mineral in the circulation. D. Role in the Neonate The abrupt fall in serum calcium experienced by most mammals after birth is consistent with loss of placental PTHrP, but there are insufficient data to confirm that PTHrP has been lost from the neonatal circulation. Conversely neonatal lambs do not experience a fall in serum calcium after birth, but maintain the same high levels that were present during fetal life until 3– 4 mo of age. When the calcium level finally declines in lambs, it corresponds to the age when increased PTH-like bioactivity attributable to PTHrP finally subsides (94). This result seemingly indicates that parathyroids of neonatal lambs produce PTHrP for some months after birth. PTHrP continues to be produced in the growth plates of endochondral bone wherein it delays terminal differentiation of chondrocytes, and within osteoblasts wherein it directs bone formation and mineralization, and inhibits apoptosis. Its importance has been illustrated by several mouse models. Posnatally, Pthrp⫹/⫺ mice develop modestly accelerated endochondral ossification and deficits in bone microarchitecture and mass (23). When Pth null mice are made haploinsufficient for Pthrp, osteoprogenitor cell recruitment decreases, osteoblast apoptosis increases, bone formation diminishes, and trabecular bone mass is reduced (442). Conditional deletion of Pthrp from osteoblasts leads to an osteoporotic skeleton by the age of maturity (438, 441). The very high concentration of PTHrP in milk seemingly declares that it is important, but for what physiological E. Human Data Possible effects of PTHrP on human trophoblast function or placental calcium transport have been investigated in vitro. Vesicles prepared from fetal-facing basement membranes of syncytiotrophoblasts (32–37 wk of gestation) showed a linear increase in ATP-dependent accumulation of 45Ca within the vesicles (649), which is consistent with a progressively increasing rate of active placental calcium transport in late gestation. Mid-molecular PTHrP38 –94 stimulated ATP-dependent uptake of 45Ca calcium in a concentration-dependent fashion starting at the physiological dose of 5 pg/ml (648), while PTH, PTHrP1–34, PTHrP67– 86, and calcitonin had only modest effects at pharmacological doses (50 or 250 pg/ml) (648). 45Ca uptake was inhibited by a protein kinase C inhibitor, while PTHrP38 –94 increased inositol tri- Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1173 CHRISTOPHER S. KOVACS sphosphate production and protein kinase C phosphorylation. PTHrP may also promote trophoblast survival since PTHrP1–34 prevented cytotrophoblast apoptosis while PTHrP67– 86 had no effect (128). In contrast to those findings, other investigators created similar vesicles from maternal-facing and fetal-facing basement membranes of human syncytiotrophoblasts, but with the direction of calcium transport oriented outwards into the media. The vesicles were loaded with 45Ca and then efflux of the isotope was measured. PTHrP1–34 was most potent to increase 45Ca efflux; PTH1–34 required a fourfold higher dose; PTHrP67–94 was without effect (178). The NH2-terminal antagonist PTHrP7–34 significantly reduced the effect of both peptides. These results suggest that NH2terminal PTHrP can stimulate transmembrane calcium transport while the mid-molecular form cannot. As noted previously, cord blood NH2-terminal PTHrP concentrations are 15-fold higher than PTH when expressed in equivalent pM units (330), although this may underestimate the true concentration of PTHrP depending on the assay and how the blood sample was collected. The plasma concentration of mid-molecular PTHrP38 –94 was increased almost twofold in cord blood of intrauterine growth restricted babies compared with healthy neonates at term (647). When vesicles of fetal-facing basement membranes of synciotrophoblasts were made from the placentas of these same growth-restricted babies, 45Ca uptake increased 50% into the vesicles from IUGR babies over those from healthy babies (647). These data positively correlate an increase in the concentration of mid-molecular PTHrP with an in vitro measure of placental calcium transport. The effects of NH2-terminal PTHrP fragments have also been studied on isolated maternal-facing and fetal-facing basement membranes from term human placentas. PTHrP1–34 stimulated phosphoinositide turnover, phospholipase C, and protein kinase C in both membrane fractions (355, 362). It was equipotent to PTH1–34 on the phospholipase C pathway but more potent than PTH at stimulating protein kinase C activity (362). An equivalent to the murine Pthrp null state has not yet been identified in human fetuses, but is expected to cause intrauterine death. An autosomal dominant microdeletion in the human PTHrP gene (PTHLH) has been linked to brachydactyly type E, which is characterized by short stature and shortened metacarpals and metatarsals (316). This finding is consistent with a role for PTHrP in regulating the formation of the endochondral skeleton in humans. Whether the very high concentrations of PTHrP in human milk have effects in the neonate to alter intestinal calcium absorption or bone metabolism has not been directly investigated. Studies during the first 6 to 12 mo after birth have 1174 shown variable results of higher, lower, or no difference in bone mineral content gains achieved by human milk versus formula-fed babies (90, 145, 636, 722). However, several studies have found consistent and significant positive correlations between the duration that an infant received human milk versus formula and the bone mass measured by DXA as a child, adolescent, or young adult (186, 290, 450). These correlations do not prove causation nor do they directly implicate the PTHrP content of human milk. However, the results are intriguing, especially given that the PTHrP content is one of the most striking differences in the composition of human milk versus formula. F. Summary of Key Points The bulk of the available data are consistent with PTHrP playing several key roles in fetal bone and mineral metabolism, including the regulation of serum calcium and phosphorus, stimulation of placental calcium and magnesium transport, control of endochondral bone development by delaying terminal differentiation of chondrocytes, and several effects that stimulate formation, recruitment, activity, and survival of osteoblasts. Although PTHrP is needed to maintain a normal serum calcium in the fetus, PTHrP does not increase in response to systemic hypocalcemia. Animal and human data support the conclusion that it is a midmolecular form of PTHrP, likely PTHrP38 –94, which stimulates placental calcium transfer through actions on an as yet unidentified receptor. It remains unclear whether placenta, parathyroids, or both are the key source of PTHrP in the fetal circulation, nor is it certain when PTHrP disappears from the postnatal circulation. Through its local production in chondrocytes and osteoblasts, PTHrP continues to play key roles in regulating skeletal development in the neonate, infant, and child. Exactly what role the high content of PTHrP in milk has is unknown, but there are intriguing clues that it may modulate intestinal calcium absorption or mineral accretion by the skeleton. V. PARATHYROID HORMONE The low levels of PTH in late term fetuses, which rise significantly after birth, suggested that PTH may be unimportant for fetal bone and mineral homeostasis. However, this is now known to be incorrect. A. Regulation of Serum Minerals The most specific evidence that PTH regulates fetal blood calcium is that hypocalcemia occurs when Pth is ablated in fetal mice (623), and when PTH antiserum is infused into fetal rats (197). Hypocalcemia also occurs after parathyroidectomy in fetal lambs and rats (1, 2, 97, 105, 520), and genetic ablation of parathyroids in mice (341); however, hypocalcemia due to loss of parathyroids may be caused in Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM part by loss of other parathyroid-derived factors such as PTHrP. Consistent with this possibility, in fetal mice loss of parathyroids due to Hoxa3 ablation causes the ionized calcium level to fall well below the maternal calcium level (336), whereas loss of PTH alone in Pth null fetuses leads to a blood calcium equal to the maternal level (623). In thyroparathyroidectomized fetal lambs, the blood calcium also falls below the maternal level (2), but in thyroparathyroidectomized or decapitated fetal rats the serum calcium remains equal to or above the maternal level (105, 520). There is no “PTH-null” equivalent in lambs and rats. In section IVA, it was noted that in Pthrp null fetuses, serum PTH increases and likely maintains the blood calcium at the normal adult level. But in several mouse models that are hypocalcemic due to PTH deficiency (Pth null, Gcm2 null, and aparathyroid Hoxa3 null fetuses), there is no compensatory increase in plasma PTHrP (341, 623). These findings confirm that PTH is responsive to falls in the ambient fetal blood calcium whereas PTHrP is not. Although thyroparathyroidectomy or decapitation in fetal rats causes the blood calcium to fall to a level equal to or above the maternal level, when the mother was also thyroparathyroidectomized, the fetal serum calcium remained significantly higher than the maternal serum calcium (105, 519). This was interpreted by the investigators to imply persistence of active transport of calcium in the absence of maternal and fetal parathyroids. The experiments were done prior to the discovery of PTHrP and may indicate persistent actions of placental PTHrP on serum calcium and placental calcium transport. The low level of PTH in the fetal circulation also regulates serum phosphorus and magnesium because hyperphosphatemia and hypomagnesemia occur in Pth null fetuses (623). Hyperphosphatemia and a modest reduction in serum magnesium also occur in thyroparathyroidectomized fetal lambs and rats, and aparathyroid fetal mice (105, 341, 397, 398, 520, 623). Notably calcitriol levels do not change in thyroparathyroidectomized fetal lambs (1, 97), indicating that fetal production of calcitriol is not dependent on PTH or parathyroids. Therefore, PTH is a key regulator of fetal blood calcium, phosphorus, and magnesium, despite the low circulating levels that originally suggested otherwise. In fact, PTH may have a greater role in determining the serum calcium level than PTHrP, as revealed through study of double-mutants (see section VI). B. Regulation of Placental Mineral Transport The PTH1R is expressed by murine trophoblasts but is most intensely expressed within the intraplacental yolk sac (337). That intense expression implies that NH2-terminal PTH (⫾N-terminal PTHrP) has some function within the placenta, but most in vivo studies have failed to find an effect of PTH on placental mineral transport. These included studies of in situ perfused placentas from thyroparathyroidectomized fetal sheep (6, 96, 556), and placental calcium transfer in intact Pthrp null and WT fetuses (340). However, Pthrp null fetuses have threefold higher PTH which may have maximized any effect that exogenously administered PTH could have had. Exogenous PTH1–34 did modestly stimulate calcium transport in perfused placentas from rat fetuses that had been decapitated to crudely mimic parathyroidectomy (554), but it had no effect in perfused placentas from intact fetuses (554, 611). Placentas from thyroparathyroidectomized rat fetuses were not studied. Comparative study of Pth null and Gcm2 null fetuses within a similar genetic background has provided evidence of a possible role for PTH in stimulating placental mineral transport (623). Placental 45Ca transfer appeared normal at baseline in Pth null fetuses, but microarray and real-time quantitative RT-PCR analysis of Pth null placentas revealed significantly reduced expression in Trpv6, Sg100 (Calbindin-D9k), Vdr and other cation transporters (623). Pth null fetuses treated in utero with PTH1– 84 responded with a 30% increased rate of 45Ca accumulation, confirming that exogenous PTH can stimulate placental calcium transfer in vivo when endogenous PTH is absent (623). Gcm2 null fetuses have a similar phenotype to Pth nulls with hypocalcemia and hyperphosphatemia, but differ by having a low but detectable concentration of circulating PTH compared with their WT littermates. They also differ from Pth nulls by having a significantly increased rate of placental calcium transfer, and increased placental expression of Trpv6, Sg100, and Vdr relative to WT littermates (623). These observations led to the discovery that there is a low level of expression of Pth mRNA within WT placentas, which is absent in Pth null placentas, and significantly increased in Gcm2 null placentas (623). Therefore, PTH may regulate placental calcium transfer through local expression of PTH and PTH1R, in addition to PTH having possible effects from within the systemic circulation. PTH does not regulate phosphorus transfer in perfused placentas from fetal lambs (330), and it probably does not regulate magnesium transport either because parathyroid extracts and PTHrP1–34 failed to have effects (47, 96). Placental phosphorus and magnesium transfer have not been studied in aparathyroid or Pth null fetal mice. Overall, PTH may act systemically and locally within placenta to regulate the transfer of calcium and other solutes. Its effects may be weaker or secondary to that of PTHrP, since Pthrp null fetuses have reduced placental calcium Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1175 CHRISTOPHER S. KOVACS transfer despite a threefold increase in endogenous PTH. If so, then loss of both PTH and PTHrP should cause a greater decline in placental calcium transport than loss of PTHrP alone, but this has not been investigated. 0.25 mM higher serum calcium, and adults have higher bone mass, compared with C57BL/6 mice (192, 309, 310, 340, 744). These and other differences between the background strains may explain why absence of PTH causes variable effects on bone. C. Regulation of Endochondral Bone Formation Overall, PTH is a key regulator of skeletal mineralization in the fetus, as observed in lambs and mice. This may occur indirectly through PTH’s actions to support the fetal blood calcium; when serum calcium falls, substrate availability is reduced and mineral accretion is impaired. PTH may also directly regulate osteoblast development and function during fetal development, with its effects varying by genetic background. Thyroparathyroidectomized fetal lambs displayed reduced mineralization of the fetal skeleton at term (2), but whether this is due to loss of PTH, PTHrP, or both could not be determined. Specifically, examination of the fourth lumbar vertebrae revealed reduced ash weight, eroded surface, and mineralizing surface, but increased bone volume, trabecular thickness, osteoid thickness, and osteoid surface. This was described as a rachitic phenotype by the investigators, but shows the effect of loss of parathyroids and PTH to reduce skeletal mineralization, thereby resulting in the accumulation of unmineralized osteoid. Study of the more recently developed murine models has shown that PTH is an important determinant of skeletal mineralization during fetal development, but its effects on endochondral bone development appear weaker and somewhat variable compared with loss of PTHrP (624). Hoxa3 null fetuses lack parathyroid glands and PTH and (within the Black Swiss outbred background) have very low skeletal mineral content, but normal cartilaginous and osseous components of the endochondral skeleton (FIGURE 9) (336, 341). These include normal expression of chondrocyte and osteoblast-specific genes, lengths of the long bones, and cellular morphology and lengths of the tibial growth plate and trabecular compartment (336, 341). The skeleton is essentially normal except for a significant deficit in mineral content (FIGURE 9). In contrast, Pth null fetuses (within the inbred C57BL/6 background) were initially reported to have slightly shorter tibial metaphyses, shortened metacarpals and metatarsals, smaller vertebrae, reduced trabecular bone volumes, and fewer osteoclasts and osteoblasts (439). These structural changes largely affected osseous and not chondrocytic parameters, including reduced expression of osteoblast-specific genes. Why should loss of PTH lead to a bone phenotype that loss of parathyroids does not share? To resolve this, Pth nulls and PTH-deficient Gcm2 nulls were back-crossed into the outbred Black Swiss strain to match that of Hoxa3 nulls (623). In this strain Pth nulls and Gcm2 nulls each had normal skeletal lengths and morphology, and a modestly reduced mineral content that was not as severe as in aparathyroid Hoxa3 nulls (336, 623). Therefore, loss of PTH caused an osteoblast-deficiency phenotype only in the C57BL/6 strain. Black Swiss fetuses and their mothers have 1176 D. Role in the Neonate PTH becomes a major controller of mineral and bone homeostasis in the hours after birth. The parathyroids increase synthesis and secretion of PTH, which acts to raise the blood calcium, lower phosphorus, stimulate calcitriol synthesis, inhibit calcitriol catabolism, reabsorb calcium in the kidney tubules, and upregulate bone formation. The importance of PTH is most clearly evident through the study of Pth null mice, which are born in the expected Mendelian ratios at birth. They are hypocalcemic and hyperphosphatemic at birth, but it is around the time of weaning at 3 wk of age that sporadic sudden deaths begin to occur from presumed hypocalcemia-induced tetany and arrhythmias (310). The timing is consistent with the maturation of intestinal calcium absorption from a passive to an active, calcitriol-dependent process, and with Pth nulls being unable to normally stimulate calcitriol synthesis. These deaths are prevented by using a calcium-phosphate-lactoseenriched “rescue diet” that increases passive absorption of mineral (310). A similar sporadic mortality occurs in Gcm2 null mice when maintained in outbred background strains, but mortality is 70 –100% in the inbred C57BL/6 strain for reasons that appear unrelated to loss of PTH (384). By 2 wk after birth, Pth null mice in the C57BL/6 background are slightly smaller than their WT siblings due to shortening of the long bones. Histomorphometric assessment has shown that chondrocytic parameters are still largely normal, but abnormalities within the osseous component include a significant reduction in mineral apposition rate, trabecular bone volume, and mineralization (as assessed by von Kossa) (748). The hypocalcemia in Pth nulls contributes to their increased mortality and reduced skeletal mineralization independent of any effect of PTH to stimulate the formation of calcitriol. This has been tested in Pth/Cyp27b1 double mutants that lack PTH and calcitriol, are hypocalcemic, hyperphosphatemic, and hypercalciuric, have shortened skeletal lengths Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM and reduced trabecular bone volumes, and die before weaning at 3 wk of age (748). When PTH or PTHrP injections were administered daily starting at day 4 after birth to increase the serum calcium, survival improved such that the pups could be analyzed at 1 mo of age (749). The serum calcium increased significantly, hyperphosphatemia persisted, urine calcium excretion decreased, and there were significantly improved skeletal lengths, trabecular bone volumes, and mineral content (749). These results could not be mediated by calcitriol because the double-mutants lacked the ability to stimulate calcitriol synthesis in response to PTH or PTHrP treatment (749). In a separate report, Pth/ Cyp27b1 double mutants were treated with exogenous calcitriol, and the results were quite similar with improved survival, increased serum calcium, lowered serum phosphorus, reduced urine calcium excretion, and improved skeletal lengths, trabecular bone volumes, and mineral content (747). These effects were not mediated by PTH since the double-mutants lacked it. Since similar outcomes occurred when the serum calcium of Pth/Cyp27b1 double mutants was raised by treatment with PTH, PTHrP, or calcitriol, these results may indicate the importance of serum calcium to support osteoblast function and skeletal mineralization, as well as to prevent deaths due to hypocalcemia. Calcium should exert its effects through the CaSR expressed by chondrocytes, osteoblasts, and osteocytes (see sect. VIIC). E. Human Data Human trophoblasts express PTH1R (82, 183), which makes it likely that PTH exerts biological effects within the placenta. PTH1–34 stimulated 45Ca accumulation into vesicles prepared from fetal-facing basement membranes of human syncytiotrophoblasts, but the minimum dose needed was 10-fold or more than that needed for mid-molecular PTHrP38 –94 (648). In vesicles oriented to transport calcium outward, PTH1–34 stimulated 45Ca efflux but required a fourfold greater dose than PTHrP1–34 (178). These findings indicate that PTH has the ability to stimulate calcium transport in human trophoblasts when studied in vitro, but that it is less potent than PTHrP. NH2-terminal PTHrP may be the endogenous ligand for the PTH1R expressed within human trophoblasts, unless PTH is produced locally within the human placenta. Congenital hypoparathyroidism can occur from genetic deletion of the parathyroids (DiGeorge and other 22q11.2 deletion syndromes, ablation of Gcm2, etc.) and from activating mutations of the calcium sensing receptor (70). The animal data predict that hypoparathyroid babies should have hypocalcemia, hyperphosphatemia, and impaired skeletal mineralization at birth. However, due to the sporadic nature of these conditions and lack of symptoms at birth, no cord blood or skeletal radiographs are available. Symptomatic hypocalcemia is not inevitable after genetic loss of parathyroids: in a large series of 22q11.2 deletions, only 60% of affected individuals developed hypocalcemia at some point in their lives (578). Many presented as neonates, but in others, the presentation was in childhood or as late as 18 years of age (578). In another series of 12 cases with confirmed hypocalcemia, only 4 were symptomatic; 10 were diagnosed before 1 mo of age, and the remaining 2 presented at 3 mo and 12 yr of age (71). Serum phosphorus was inconsistently measured and not always elevated. In another series of 10 cases with confirmed hypoparathyroidism, the age at diagnosis ranged from 9 days to 13 yr (12). It seems likely that when hypocalcemia begins in utero as a result of failure of the parathyroids to form, the individual adapts to the low calcium and is less likely to become symptomatic after birth. This contrasts to the marked symptoms that occur after surgically induced hypoparathyroidism in children or adults. Pseudohypoparathyroidism or PTH resistance also causes hypocalcemia, but it develops later in childhood and is usually preceded by hyperphosphatemia and elevated PTH (412, 470, 687). Consequently, pseudohypoparathyroid newborns should have normal cord blood calcium and are unlikely to become hypocalcemic as neonates. Neonatal or infantile primary hyperparathyroidism has been described in older literature (57, 405, 562), but these cases likely represented neonatal severe primary hyperparathyroidism mediated by homozygous or compound heterozygous inactivating mutations of CaSR (see sect. VIID). The earliest that conventional primary hyperparathyroidism caused by adenomas or 4-gland hyperplasia has been reported is 4 years of age, and these are usually in the setting of multiple endocrine neoplasia types 1 or 2, or familial primary hyperparathyroidism (321). F. Summary of Key Points Despite its low levels in the fetal circulation, PTH is an important determinant of serum calcium and phosphorus. It may play a modest role in stimulating placental calcium transport by reaching trophoblasts from the systemic circulation or through localized expression within the placenta. Alternatively, expression of PTH1R within trophoblasts may indicate a role for NH2-terminal PTHrP that is only fulfilled by PTH when secondary hyperparathyroidism occurs in utero. PTH contributes to endochondral bone development in the fetus mainly by supporting the mineralization of bone. This occurs by PTH maintaining the normal concentration or supply of minerals in the circulation, as well as through possible direct effects of PTH on osteoblasts that may be independent of the higher circulating concentrations of PTHrP. Postnatally, PTH increases after the first 12 h and assumes its well-known roles as a dominant regulator of bone and mineral status, through actions in osteoblasts, kidneys, and (indirectly by stimulating formation of calcitriol) the intestines. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1177 CHRISTOPHER S. KOVACS VI. ROLE OF PTHrP AND PTH CONSIDERED IN COMBINATION PTHrP and PTH overlap in their contribution to the regulation of fetal bone and mineral metabolism. This has been made most clear by comparative study of double mutants that lack either PTHrP and PTH, or PTHrP and the parathyroids, and by ablation of their common NH2-terminal receptor, PTH1R. A. Regulation of Serum Minerals It is clear that PTH and PTHrP each participates in the regulation of the fetal blood calcium, such that loss of either hormone causes fetal hypocalcemia (336, 340, 341, 623). Their individual effects appear additive as revealed by studies of a double-mutant colony in which PTHrP, parathyroids, or both were deleted, and compared with the findings in Pth1r null fetuses that lack the PTH1R (FIGURE 10). Compared with WT littermates, Pthrp null fetuses had a modestly reduced blood calcium (equal to the maternal level), aparathyroid Hoxa3 null fetuses had a markedly reduced blood calcium (well below maternal values), and Pthrp/Hoxa3 double mutants had the lowest blood calcium (336). The ionized calcium level caused by simultaneous loss of parathyroids and PTHrP was equivalent to the ionized calcium level of Pth1r null fetuses (336, 340). Taken together, these data suggest that the ionized calcium level is largely determined by the combined NH2-terminal effects p < 0.001 p < 0.01 2.00 Ionized Calcium (mmol/l) p < 0.05 1.50 1.00 0.50 0.00 WT Pthrp null Hoxa3 null Double Mutant FIGURE 10. PTH and PTHrP additively regulate the fetal blood calcium. Mean serum ionized calcium is shown for WT, Hoxa3 null, Pthrp null, and Pthrp/Hoxa3 double mutant fetuses. For clarity, the other five possible genotypes have been omitted from the figure. The upper dashed line indicates the mean maternal ionized calcium level in this genetic background (equal to Pthrp null), while the lower dashed line indicates the mean ionized calcium level in Pth1r null fetuses in a similar genetic background. [From Kovacs et al. (336). Copyright 2001 The Endocrine Society; permission conveyed through the Copyright Clearance Center, Inc.] 1178 of both PTHrP and PTH, since loss of the PTH1R led to the same reduction in blood calcium as loss of PTHrP and parathyroids combined (336). The greater decline in ionized calcium within aparathyroid Hoxa3 null fetuses (336, 623) might imply that parathyroid-produced PTHrP has been lost; however, plasma PTHrP is unchanged among WT, Hoxa3⫹/⫺, and Hoxa3 null fetal littermates (336, 341). It is possible that the parathyroids contribute to fetal mineral homeostasis directly or indirectly through the release of other factors. For example, the chromogranins have been estimated to represent 50% of the protein output of the parathyroids (305, 606). Chromogranin expression increases with hypocalcemia and decreases with hypercalcemia (160, 467), and chromogranins have been shown to inhibit PTH synthesis and release (26, 576); therefore, chromogranins could conceivably have distant effects to regulate the production of PTHrP by the placenta (248, 319). However, the possible role that nonPTH secretory products of the parathyroids may have on mineral homeostasis has not been studied in the fetus or adult. Pth/Hoxa3 double mutants should reveal whether loss of parathyroids adds to the phenotype caused by loss of PTH, but that model has not been studied. Similarly, although Pthrp/Pth double mutants have been examined for their effect on skeletal development (439), no measurements of serum calcium or phosphorus have been reported. As noted earlier in section VA, Pth null and aparathyroid Gcm2 null fetuses in the same Black Swiss strain each had an ionized calcium that was equal to the maternal level, and not reduced well below it as in aparathyroid Hoxa3 null fetuses (623). Pth nulls have hyperplastic parathyroids that conceivably could produce PTHrP and compensate for the loss of PTH (439); however, no increase in expression of PTHrP was observed in the Pth null fetuses (623). Upregulation of placental PTH expression was found in aparathyroid Gcm2 null fetuses, and that may have blunted the fall in blood calcium caused by loss of parathyroids (623). The fact that the ionized calcium of Pthrp null fetuses in the same background is equal to the maternal level should not be taken as evidence that the magnitude of PTHrP’s effect is to raise the blood calcium above the maternal level. Pthrp null fetuses have a significant compensatory increase in PTH; without it, the serum calcium of Pthrp null fetuses would likely have been much lower and approaching the level observed in Pthrp/Hoxa3 null double mutants (336). Pth1r null fetuses have the lowest ionized calcium level, and the absence of PTH1R is accompanied by markedly increased circulating concentrations of PTH and PTHrP (336, 340, 341). Although the parathyroids of Pth1r null fetuses were not directly examined, there was no increase in PTHrP mRNA expression in the neck region that contains the parathyroids (341). However, there was increased expression of PTHrP mRNA (by Northern blot and in situ hybridization) Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM and protein (by immunohistochemistry) within placenta and liver of Pth1r null fetuses (340, 341). These findings provide additional evidence that the fetal parathyroids are unlikely to be a dominant source of PTHrP in the fetus, and that placenta and possibly liver may provide much of the PTHrP that is normally present in the fetal circulation. B. Regulation of Placental Mineral Transport Pth1r null fetuses exhibit a significantly increased relative rate of placental calcium transport compared with their WT littermates (340). Since PTHrP circulates at 11-fold higher levels in these fetuses compared with WT (341), and loss of the PTH1R blocks NH2-terminal actions of PTHrP, it seems likely that high circulating levels of mid-molecular PTHrP are acting on a mid-molecular receptor to stimulate placental calcium transport (336, 340). These results support earlier findings that mid-molecular PTHrP stimulated placental calcium transport in fetal lambs and mice (sect. IVB). The mid-molecular receptor for PTHrP has yet to be cloned. C. Regulation of Endochondral Bone Formation Pthrp/Hoxa3 double mutants have an overall appearance and skeletal phenotype that combines the Pthrp null chondrodysplasia with the undermineralized skeleton of the Hoxa3 null; they are also globally smaller in size (336). Combining these two genetic deletions confirms that loss of fetal parathyroids leads to undermineralization of the fetal skeleton without affecting the underlying cartilaginous and osseous structure. In contrast, loss of PTHrP alone causes marked acceleration in chondrocyte differentiation and initiation of primary ossification centres, and both accelerated and abnormal mineralization of bone (336). Pthrp/Pth double mutants also show the Pthrp null chondrodysplasia combined with undermineralized skeleton of the Pth null; they too are globally smaller than WT siblings (439). However, the Pth null has mild defects in osteoblast parameters that appear only within the C57BL/6 genetic background and not the Black Swiss background (see sect. VC). Pth1r nulls have a similar global phenotype as the Pthrp nulls, including accelerated endochondral ossification, dysplasia, and ossification of the cartilaginous portions of ribs; however, they are globally much smaller than their WT siblings (360). They also have a greater deficit in skeletal mineral content than that caused by loss of either ligand alone (336, 340). This may result from their much lower ionized calcium level, combined with loss of the effects of PTHrP or PTH to stimulate osteoblasts. There are other differences in the skeletal phenotypes of Pth1r null and Pthrp null fetuses. Whereas Pthrp null fetuses have normal expression of osteoblast genes, and normal or increased mineralization (359), Pth1r null fetuses show reduced expression of collagenase-3, osteocalcin, and osteopontin and have significantly reduced mineralization (359). The explanation may be the increased circulating levels of PTH in Pthrp null fetuses. The chondrocytic portion of the growth plate is avascular, which will prevent systemic PTH from compensating for loss of PTHrP production and its local actions within chondrocytes. But systemically high PTH levels should easily reach the highly vascular bone compartment to maintain apparently normal expression of osteoblast genes. Such actions of PTH are lost in Pth1r nulls, resulting in downregulation of these osteoblast-specific genes. Furthermore, the Pth1r null skeleton is undermineralized despite a relative upregulation in placental calcium transfer (as described in sect. VIB). This likely indicates that, notwithstanding relatively increased delivery of calcium from the maternal circulation in Pth1r nulls compared with their WT littermates, calcium cannot be incorporated into bone without NH2-terminal actions of PTH or PTHrP, so it returns to the maternal circulation. An activating mutation of the PTH1R creates the opposite phenotype of the Pth1r null fetuses, exaggerating what are likely the normal NH2-terminal actions of PTHrP to delay chondrocyte hypertrophy. The result is a largely cartilaginous at birth (596). Its appearance is similar to the effect of PTHrP overexpression within the developing skeleton (726). D. Role in the Neonate Pthrp/Pth double mutants and Pthrp/Hoxa3 double mutants die soon after birth (336, 341, 439). This is likely a consequence of the lower blood calcium that these double mutants must have, in addition to the early mortality conferred by ablation of Pthrp or Hoxa3 alone. In the original C57BL/6 background, Pth1r null fetal mice die at E11.5– 12.5 due to widespread cardiomyocyte apoptosis, which implies important roles for PTH1R in early embryogenesis (532). When crossed into the outbred Black Swiss strain, they survive to the end of gestation before dying promptly in a manner similar to Pthrp nulls (340, 360). Consequently, there are no neonatal data on the effects of combined loss of PTH and PTHrP. E. Human Data As noted earlier, human trophoblasts express PTH1R (82, 183), which implies that PTH or PTHrP exert NH2-terminal biological effects within the placenta. Loss of PTH1R causes Blomstrand chondrodysplasia, which is characterized by a chondrodysplasia quite similar to what has been Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1179 CHRISTOPHER S. KOVACS found in Pth1r null fetal mice (296, 489). It is embryonically lethal; therefore, there are no cord blood measurements of calcium, phosphorus, etc. F. Summary of Key Points PTH and PTHrP have additive effects on the serum calcium level, and in turn affect mineralization of the fetal skeleton. Loss of PTH or PTHrP have equivalent effects on the serum calcium, whereas loss of parathyroids in Hoxa3 null fetuses or loss of PTH1R causes more significant hypocalcemia. PTHrP has a more profound effect on regulating endochondral bone development, primarily by regulating the fate of chondrocytes and the initiation of osteoblast activity. PTH has more modest effects on osseous portions of the skeleton, but these become much more apparent after birth. Combining absence of PTHrP with deletion of PTH results in a skeleton that bears the Pthrp-null chondrodysplasia but is also undermineralized as in the Pth null or Hoxa3 null skeleton. Upregulation of PTH likely compensates for loss of NH2-terminal actions of PTHrP on bone within Pthrp null fetuses. VII. CALCIUM-SENSING RECEPTOR CaSR is considered here because it plays a pivotal role in regulating mineral and bone homeostasis in the adult, through its regulation of PTH synthesis and release, and renal tubular handling of calcium. Furthermore, calcium functions like a calciotropic hormone by binding to its receptor in different tissues, including parathyroids, bone, kidneys, and placenta. Calcium appears to have direct actions that influence the function of osteoblasts and chondrocytes. A. Regulation of Serum Minerals As discussed in section II, A and B, CaSR does not set the high level of calcium found in normal fetuses, but it suppresses PTH in response to it. Ablation of one (Casr⫹/⫺) or both (Casr null) alleles of CaSR results in an equal increase in the fetal blood calcium above the WT value (FIGURE 3), and a dose-dependent increase in circulating PTH (FIGURE 4) and calcitriol (338). The increase in calcium depends on PTH because simultaneous ablation of the PTH1R in Pth1r/ Casr double mutants prevents Casr ablation from increasing the blood calcium above the low level present in Pth1r null fetuses (338). It is puzzling that ionized calcium of Casr null fetuses is not higher than in Casr⫹/⫺ fetuses; the intrauterine environment may constrain Casr null fetuses from achieving the higher blood calcium level that they reach after birth. CaSR is likely responsible for the ionized calcium level of Pthrp null fetuses being maintained at the maternal level rather than falling below it, through its ac- 1180 tions to stimulate PTH to achieve the adult set point of calcium (see also sects. IVA and VA) (338, 340). It is evident from data discussed earlier that CaSR does not stimulate PTHrP to maintain the fetal blood calcium, because in Hoxa null fetuses, the ionized calcium is reduced well below the maternal concentration, PTH is undetectable, and the plasma PTHrP level does not increase in compensation (336, 341). However, CaSR may have effects to downregulate the expression of PTHrP, because circulating plasma PTHrP is significantly lower in Casr null fetal mice compared with their siblings (334). The increased ionized calcium in Casr nulls may be a factor, or CaSR may directly regulate PTHrP through their colocalized expression within trophoblasts and intraplacental yolk sac. In the adult, ablation of CaSR decreases renal calcium clearance in a dose-dependent fashion (257), leading to hypercalcemia combined with hypocalciuria. However, Casr⫹/⫺ and Casr null fetal mice each have increased amniotic fluid calcium with no difference between them (338). This striking difference from the adult phenotype is likely the result of two factors. First, fetal kidneys express very low levels of Casr mRNA until the first postnatal day (112); consequently, the renal phenotype potentially caused by Casr ablation is absent. Second, the renal filtered load of calcium should be increased equally in both genotypes due to their similar increments in ionized calcium. Therefore, in the absence of modulation by CaSR, the increased filtered load of calcium simply leads to increased renal excretion of calcium into the amniotic fluid. CaSR has been selectively ablated from parathyroid glands (CasrPara null), but the effect that this may have on fetal mineral homeostasis has not been reported (109). An activating mutation of Casr has been described in Nuf mice (271). No fetal data have been published, but it is anticipated that they should have a similar phenotype as Pth null fetuses, with reduced skeletal mineralization and low PTH. B. Regulation of Placental Mineral Transport As noted earlier, CaSR is expressed in murine trophoblasts and intraplacental yolk sac cells (337). Placental transport of 45Ca is significantly and dose-dependently reduced in Casr⫹/⫺ and Casr null fetuses compared with their WT siblings (338). It is unknown why placental calcium transfer is reduced. If the CaSR senses calcium flow within the placenta, or regulates placental PTHrP synthesis, CaSR ablation could explain a decrease in placental calcium transport. The lower plasma PTHrP levels in Casr null fetuses could result from either postulated mechanism. Alternatively, the elevated serum calcium or secondary hyperparathyroidism Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM that occur in Casr null fetuses might also lead to downregulation of placental calcium transfer. Additional studies of Pthrp/Casr double mutant fetuses revealed that although ablation of Casr reduced placental calcium transport in fetuses that had two normal Pthrp alleles, ablation of one or both alleles of Casr had no effect on placental calcium transport when both Pthrp alleles were ablated (338). This result is compatible with downregulation of Pthrp explaining reduced placental calcium transport in both Pthrp null and Casr null fetuses. There have been no studies of the Casr activating mutation from Nuf mice of placental calcium transport. C. Regulation of Endochondral Bone Formation CaSR is expressed by chondrocytes and osteoblasts, with its highest expression seen within the hypertrophic chondrocytes (109, 110). Chondrocyte-specific ablation of CaSR (CasrChon null) caused embryonic lethality with severely delayed cartilage and bone development (109), but when the chondrocyte-specific deletion was induced between embryonic day 16 and 18 by use of a tamoxifen-driven promoter, the fetal skeleton was shortened and undermineralized due to delayed terminal differentiation of chondrocytes (109). These results indicate that CaSR promotes chondrocyte differentiation and that its absence delays differentiation, actions that oppose those of PTHrP within the growth plate. This is consistent with the known effect of high extracellular calcium concentrations to promote chondrocyte differentiation (108, 111, 257); ablating CaSR must block this effect. Ablation of CaSR has also been carried out in preosteoblasts, immature osteoblasts, mature osteoblasts, and osteocytes (CasrPre, CasrOb and CasrOc null), and in each the fetal skeleton is unremarkable, although skeletal abnormalities develop later (see below) (109, 169, 549). Bone resorption is increased in global Casr-null fetuses, as suggested by the increased circulating PTH levels, increased excretion of the bone resorption marker deoxypyridinoline into amniotic fluid, and a significant reduction in the ash weight and mineral content of the fetal skeleton (331, 338). Since Casr ablation reduces placental calcium transport and increases excretion of calcium into the amniotic fluid, the increased circulating calcium concentration must be maintained through resorption of mineral from the fetal skeleton, thus compromising bone strength and mineral content before birth. The endochondral skeleton has normal lengths and microscopic morphology at birth in Casr null mice apart from the reduced mineral content, and this contrasts with the significant abnormalities found in the chondrocyte-specific knockout of Casr. The explanation for this discrepancy is thought to be that, although exon 5 is deleted in the global Casr null, a splice variant lacking exon 5 is produced which is able to form heterodimers and provide calcium sensing in certain tissues of the global Casr null (187). The chondrocyte-specific deletion of Casr does not produce that splice variant. Casr has also been deleted selectively from parathyroid cells (CasrPara null), but the fetal phenotype of this model has not been reported (109). It is unremarkable in size at birth but will likely be found to have hypercalcemia and a demineralized skeleton in utero. D. Role in the Neonate Postnatally, global Casr nulls develop severe hyperparathyroidism with grossly enlarged parathyroids, hypercalcemia, fluid depletion (calcium diuresis), and progressive growth failure (257). Most die before 3 wk of age, but surgical or genetic deletion of the parathyroids will enable them to survive (327). Casr nulls are the murine equivalent of neonatal severe primary hyperparathyroidism in humans. Casr⫹/⫺ neonates are hypercalcemic but have normal life spans and fertility, analogous to familial benign hypercalcemia (familial hypercalciuric hypercalcemia, FHH). The skeleton is normal at birth when Casr is ablated from early osteoblasts (CasrPre null), but the mice fail to thrive, develop multiple fractures, and die at 1 mo (109, 169). There are severe abnormalities including decreased osteoblasts, osteoblast apoptosis, osteoclasts, and increased osteoid, all of which contribute to a soft and disorganized bone structure (109, 169). In contrast, deletion of Casr from mature osteoblasts and osteocytes (CasrOb null and CasrOc null) lead to viable mice that later develop osteoporosis with increased numbers of osteoclasts (109, 549). Postnatally the parathyroid-specific knockout of CaSR (CasrPara null) develops severe hypercalcemia, markedly elevated PTH, growth retardation, multiple fractures, a severely demineralized skeleton, and marked delay in osteoblast differentiation (109). These findings suggest that much of the phenotype of the global Casr null is mediated by hyperparathyroidism, with little or no effect from Casr ablation in chondrocytes, osteoblasts, and kidney tubules. The neonatal phenotype of Nuf mice has not been reported. At 4 wk of age (the earliest time point described) they have hypocalcemia, hyperphosphatemia, low PTH, and ectopic calcifications in multiple tissues; they are also prone to sudden death (271). E. Human Data No case reports have described blood calcium or PTH values in fetuses or newborns with inactivating mutations of Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1181 CHRISTOPHER S. KOVACS CASR. These births are sporadic, and serum calcium is not routinely measured in healthy newborns. The findings from Casr⫹/⫺ fetal mice predict that human babies with FHH will be hypercalcemic in utero and that the cord blood calcium will be increased. Since hypercalcemia begins during fetal development, this likely explains why individuals with FHH are adapted to a high ionized calcium, and feel hypocalcemic if their blood calcium is reduced to “normal” values. This parallels the findings in disorders causing hypocalcemia during fetal development, in which affected neonates, children, and adults may remain asymptomatic for hypocalcemia (see sect. VE). Neonatal severe primary hyperparathyroidism is usually caused by homozygous or compound heterozygous inactivating mutations of CASR (193, 394), and occasionally by heterozygous mutations through a presumed dominantnegative effect (33, 481). It is usually not identified at birth, but the findings from Casr null fetal mice predict that hypercalcemia will be present in utero and in cord blood. The neonate is at very high risk to develop severe and life-threatening hypercalcemia, dehydration, failure to thrive, neurological disturbances, nephrocalcinosis, and skeletal demineralization with fractures. The presentation can be delayed as much as a month after birth (557), in part because maternal hypercalcemia has a suppressive or constraining effect on parathyroid function in utero (see sect. XIIB). Parathyroids are grossly enlarged and serum PTH is quite high. This is an almost invariably fatal condition unless parathyroidectomy is urgently performed (57), although some very rare cases have been reported to spontaneously resolve into milder hypercalcemia that did not require surgery (57, 405, 562, 719). Bisphosphonates and the calcimimetic drug Cinacalcet have been used to lower the serum calcium temporarily to enable the newborn to have surgery at an older age (719, 737). Activating Casr mutations cause autosomal dominant hypocalcemia with hyperphosphatemia, and normal (inappropriately low) PTH; hypercalciuria is variably present (508, 526). The presentation is expected to be similar to disorders causing hypoparathyroidism, as described in section VE. Cord blood calcium was in the adult-normal range in one newborn, but below the expected fetal value, and recurrent apneic seizures began shortly after birth (670). In another affected neonate, the cord blood calcium and ionized calcium were both low, but the baby showed no hypocalcemic symptoms (217). The variable postnatal presentation is exemplified by the development of hypocalcemic seizures at day 7 of age in a baby carrying an activating mutation of CaSR (113). Subsequent investigations confirmed that his sister (age 3 yr), mother (age 31), and grandfather (age 63) all had the same activating mutation causing hypocalcemia, low PTH, hypercalciuria, nephrolithiasis, and nephrocalcinosis (113). The sister subsequently developed tetany but only during episodes of gastroenteritis. Fur- 1182 thermore, a survey of 25 affected patients found that only 2 had developed hypocalcemia within the first 30 days after birth (540). The overall mean age that hypocalcemia was diagnosed was 28 yr and only 50% had ever been symptomatic (540). F. Summary of Key Points Global inactivating mutations of the CaSR cause hypercalcemia in utero. If homozygous or compound heterozygous mutations are present, then skeletal demineralization may develop prior to birth. Postnatally, single inactivating mutations of CaSR cause benign life-long hypercalcemia (FHH), whereas two inactivating mutations (and occasionally a single mutated allele) cause lifethreatening severe hypercalcemia and hyperparathyroidism. The mutations may occur de novo or be inherited, in which case the mother may also have FHH; the effect of maternal hypercalcemia on the fetus and neonate is discussed in section XIIB. Activating mutations resemble hypoparathyroidism with hypocalcemia, hyperphosphatemia, and inappropriately normal or low PTH; these individuals are indistinguishable from those with genetic causes of hypoparathyroidism, unless they are tested to demonstrate their abundant PTH secretory reserve, or genetic testing reveals CASR mutations. VIII. CALCITRIOL AND VITAMIN D A. Regulation of Serum Minerals Consistent evidence from multiple species and models confirms that vitamin D, calcitriol, and the vitamin D receptor are not required for the fetus to be able to maintain normal serum mineral concentrations. This includes severely vitamin D deficient rats (74, 228, 241, 443), 1␣-hydroxylase-null pigs (352, 353), and VdrBos and VdrLeu null fetuses (342, 376), in which serum calcium, ionized calcium, and phosphorus were all normal in utero (FIGURE 4). PTH was also normal in VdrBos and VdrLeu null fetuses, indicating that absence of VDR did not induce compensatory secondary hyperparathyroidism, as occurs after birth (342, 376). Four groups independently made Vdr null mice, of which only the VdrBos null truly lacks VDRs. The three other models (VdrLeu, VdrMun, and VdrTok) each deleted the exon that encodes the first zinc finger of VDR, but a splice variant is secreted which has normal ligand binding ability but no DNA binding ability (175, 376, 574, 704, 755). Of these four models, studies have been performed in VdrBos null and VdrLeu fetuses. Inconsistent data have been obtained from the VdrLeu model that expresses an aberrant but functional Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM VDR (376, 574), and these data contrast somewhat with data obtained from VdrBos null fetuses that lack VDRs (342, 372). In the first fetal study that used VdrLeu mice, the investigators obtained VdrLeu null mothers by mating VdrLeu⫹/⫺ females to VdrLeu null males, and then mated those VdrLeu null mothers to WT males so that all fetuses were VdrLeu⫹/⫺. The progeny of those studies were compared with unrelated WT females mated to WT males. The authors observed the fetal plasma calcium to be paradoxically increased in VdrLeu⫹/⫺ fetuses compared with WT fetuses from unrelated mothers, but the serum calcium was normalized (reduced) in VdrLeu⫹/⫺ fetuses whose mothers had been placed on the calcium-phosphate-lactose-enriched “rescue diet” (574). The ionized calcium (the physiologically relevant fraction) was not measured, nor were the unrelated WT control mothers and their fetuses studied on the enriched diet. It is not possible to determine from that study whether the maternal genotype or the fetal genotype led to the apparent changes in serum calcium on the normal diet and a reduction in serum calcium on the rescue diet, since WT and VdrLeu⫹/⫺ fetuses each came from different mothers. However, the VdrLeu⫹/⫺ fetuses may also have aberrant signaling since the alternatively spliced VDR has normal ligand binding ability and may be able to form functional heterodimers with RXR. Consequently, the unexpected phenotype of hypercalcemia in the VdrLeu⫹/⫺ fetuses may be due in part to the addition of an aberrant VDR as opposed to resulting from loss of one normal VDR allele. Moreover, the fact that the VdrLeu⫹/⫺ and WT mothers were unrelated raises the concern that the apparent differences in fetal serum calcium were artifacts resulting from the comparison of unrelated mice. Such artifactual results have been seen before in other models when unrelated controls have been used. These concerns appear to have been reaffirmed by a second study in which VdrLeu null and WT mothers were properly generated as first degree relatives of each other (376). VdrLeu null mothers were mated to VdrLeu⫹/⫺ males while the WT mothers were mated to WT males. In this new study, with proper related controls, there was no difference in serum calcium, phosphorus, or PTH of VdrLeu⫹/⫺ or VdrLeu null fetuses from VdrLeu⫹/⫺ mothers compared with WT fetuses from WT mothers (376). The results contradict the earlier study from the same group because VdrLeu⫹/⫺ fetuses were normocalcemic in the new study (376) but hypercalcemic in the earlier study (574). These recent results are identical to those obtained with the VdrBos model as noted above, in which WT, VdrLeu⫹/⫺, and VdrLeu null fetuses each have normal serum calcium, phosphorus, and PTH. However, in this recent study the investigators reported that while VdrLeu null pups survived, their VdrLeu⫹/⫺ siblings died soon after birth (376). An increased postnatal mortality of Vdr⫹/⫺ pups has not been seen in the VdrBos model (192, 342), and in fact VdrBos null mothers nurse the same number of pups as their WT sisters (192). Increased mortality of VdrLeu⫹/⫺ mice was also not reported in the first study from the same group (574). If VdrLeu⫹/⫺ neonates have a real increase in mortality, it is conceivable that aberrant signaling and physiology created by the mutant VdrLeu is responsible for it. However, the investigators speculated that high maternal levels of calcitriol cross the placenta and lead to increased calcitriol levels in the VdrLeu⫹/⫺ and VdrLeu null fetuses, which might be toxic to VdrLeu⫹/⫺ fetuses but not to VdrLeu null fetuses that cannot respond to calcitriol. Such speculation is not consistent with the stepwise increase in fetal calcitriol levels that were seen when WT, VdrBos⫹/⫺, and VdrBos null fetuses were compared with each other within the same litters; notably, calcitriol levels remained below the maternal value in all except VdrBos null fetuses, and the WT calcitriol level was no different from the value seen in other WT fetuses from other knockout models in the same genetic background (342). In short, there was no evidence of transplacental passage of calcitriol in the VdrBos model, and earlier cited studies determined that calcitriol does not cross the rodent placenta (see sect. IIB) (475). Therefore, it seems unlikely that maternal calcitriol caused increased mortality in VdrLeu⫹/⫺ mice. These data from VdrLeu fetal mice need to be interpreted cautiously because of possible aberrant signaling in VdrLeu⫹/⫺ fetuses, and because unrelated controls were used in the first study. The studies in VdrBos fetuses are not affected by aberrant VDRs and have compared WT to VdrBos null fetuses, and VdrBos⫹/⫺ to VdrBos null fetuses, within their respective litters. Other investigators have found possible calcitriol-mediated effects on the fetal serum calcium level. Pharmacological doses of calcitriol given to pregnant guinea pigs increased the fetal serum calcium and phosphorus (164). However, the observed effect was likely indirect through a documented increase in maternal serum calcium and phosphorus; a direct effect on the fetus is unlikely because calcitriol does not cross the rodent placenta (475). Surprisingly, vitamin D deficiency in fetal guinea pigs lowered calcitriol but caused a paradoxical increased serum calcium (573). Their pregnant mothers had 25-hydroxyvitamin D levels of ⬃ 400 nM (573), which is in a toxic range for humans or rodents and, therefore, is not relevant for vitamin D physiology in those species. Furthermore, with a maternal 25-hydroxyvitamin D level of 400 nM compared with a level of ⬃20 nM on a vitamin D-deficient diet, the guinea pig studies should be considered as contrasting the effects of maternal hypervitaminosis D to vitamin D deficiency, as opposed demonstrating effects of vitamin D sufficiency vs. deficiency. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1183 CHRISTOPHER S. KOVACS Infusion of an antiserum to calcitriol into fetal lambs for 72 h reportedly lowered the blood calcium level; however, no data, controls, or details about the antiserum were shown to confirm that brief statement (564). Direct injection of calcitriol into decapitated rats from thyroparathyroidectomized mothers caused an increase in serum calcium, a decrease in serum phosphorus, and an increase in ash weight; there was no effect in intact or decapitated fetuses of normal mothers (103). What those pharmacological effects of calcitriol in decapitated rat fetuses might mean for normal fetal physiology is not clear. Bilateral nephrectomy in fetal sheep reduced serum calcitriol by 60% and also lowered the ionized and total calcium, and increased serum phosphorus and PTH (455). The abnormal serum calcium and phosphorus could be corrected by direct administration of calcitriol to the fetus (455). The investigators inferred that loss of renal production of calcitriol explained the results rather than uremia from loss of kidney function. However, the calcitriol-induced changes in serum calcium and phosphorus are pharmacological effects that were not tested in intact lambs. Furthermore, the control procedure involved leaving the fetal kidneys in situ while diverting the ureters so that the urine drained into the fetal peritoneal cavity instead of the amniotic fluid. Consequently, it is difficult to interpret these results and the suitability of the renal diversion procedure as a control for bilateral nephrectomy. In the control procedure, the pooling of urine in the peritoneum means that fluid, solute, and waste products are retained in a compartment that may not lead to the equivalent effects that uremia or absent renal function may have on the fetus. The results in nephrectomized fetal lambs contrast with those from nephrectomized fetal rats, in which the serum calcitriol level fell, but there was no effect on the serum calcium (105). Species differences may explain why nephrectomy causes hypocalcemia in lambs but not rats. In summary, vitamin D deficiency in fetal rats, loss of 1␣hydroxylase in fetal pigs, loss of VDR in VdrBos null fetal mice, and loss of VDR in a recent study from VdrLeu null fetal mice, have shown that the serum calcium, phosphorus, PTH, and renal excretion of calcium and phosphorus are unchanged despite the absence of vitamin D, calcitriol, or VDR. Data from VdrLeu⫹/⫺ fetal mice are inconsistent between the two studies reported by the same group (376, 574), but it is only their most recent study that provided data from VdrLeu null fetuses (376). Some of the problems with the VdrLeu⫹/⫺ fetal data include the presence of a mutant VDR that may cause aberrant signaling, and use of unrelated control mice in the first study. The remaining animal data provide evidence that pharmacological doses of calcitriol can have effects in fetuses that may not be relevant to normal physiology, while the suitability of a control pro- 1184 cedure for bilateral nephrectomy in fetal lambs clouds the interpretation of the data from that study. B. Regulation of Placental Mineral Transport Where placental calcium transport has been directly measured, it is clear that calcitriol is not required because the calcium transport rate was normal in severely vitamin D deficient rats (228), and increased in both VdrBos and VdrLeu null fetal mice (342, 376). The placentas of severely vitamin D deficient rats and both VdrBos and VdrLeu null fetuses displayed normal expression of calbindinD-9k and Ca2⫹-ATPase (228, 342, 413, 574, 709), while PTHrP and TRPV6 were upregulated in both VdrBos and VdrLeu null placentas (342, 376). These findings indicate that the mechanisms that regulate transplacental movement of calcium do not require calcitriol. Furthermore, the findings in VdrBos and VdrLeu null fetuses may indicate that the normal role of calcitriol is to inhibit placental calcium transport, such that the rate increases when VDR has been ablated. In contrast, Care (95) nephrectomized five fetal lambs and judged that the rate of placental calcium transport fell across their in situ perfused placentas compared with the placentas from three intact lambs. A pharmacological dose of calcitriol was reported to increase the rate of calcium transfer in three placentas from the nephrectomized lambs (95). Surprisingly, the data were published only in a review article and without a statistical analysis, but this author’s assessment of the raw data has revealed that there was no statistically significant difference among the three groups (P ⫽ 0.28), nor in the comparison of the placental calcium transfer rates before and after treatment (P ⫽ 0.14). The effect of calcitriol was not assessed in the placentas of intact lambs. Therefore, the conclusion that nephrectomy reduced placental calcium transport and that it was rescued by calcitriol is not justified by the available data. An indirect approach to measuring placental mineral transport was carried out in pregnant ewes. A single infusion of 45 Ca or 32P was followed by daily treatment with pharmacological doses of 1␣-hydroxycholecalciferol or diluent. At the end of a week-long treatment period, the amount of 45 Ca and 32P was higher in fetal tissues from ewes that had received the calcitriol treatments (165). The same investigators tested pharmacological doses of calcitriol or diluent administered to pregnant guinea pigs for 10 days, after which it was found that the mineral content of the fetuses was higher in the offspring of calcitriol-treated animals than in those from diluent-treated animals (164). A dose of 45Ca was also given to the pregnant guinea pigs, and the amount of radioactivity in fetal blood samples was increased 30 –90 min later in the fetuses whose mothers had previously received treatment with calcitriol. In both reports, the investigators inferred that placental calcium and phosphorus Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM transport had been increased, but the approaches were too indirect to support that conclusion. Both treatments caused a significant increase in maternal serum calcium and phosphorus, and in turn that would have increased the availability of mineral to be transferred to the fetuses without invoking any direct actions of 1␣-hydroxycholecalciferol or calcitriol on trophoblasts. A simple calcium infusion has been shown to acutely increase the flow of calcium from mother to fetus (106), so a role for calcitriol in stimulating placental calcium transport cannot be inferred when use of calcitriol raises the maternal blood calcium. C. Regulation of Endochondral Bone Formation Calcitriol is not required for the formation and mineralization of the endochondral skeleton during fetal development. Severely vitamin D-deficient rats (74, 241, 443) and both VdrBos and VdrLeu null fetuses (342, 376) have normal skeletal lengths, morphology, ash weight, and mineral content at term (FIGURE 11). In the two remaining models, VdrMun and VdrTok null, the fetuses were also described as normal at birth and until the end of weaning, but no fetal data have been published (175, 755). Chondrocyte and osteoblast gene expression pertinent to endochondral bone development were also normal within the growth plates and trabecular compartment of VdrBos null fetal tibias (342). Additional studies mated VdrBos⫹/⫺ and VdrBos null sisters to the same VdrBos⫹/⫺ males to determine whether the maternal Vdr null status influenced the fetal phenotype. WT, VdrBos⫹/⫺, and VdrBos null fetuses born of VdrBos⫹/⫺ mothers were indistinguishable from each other, but had a slightly larger size, weight, and skeletal mineral content than their VdrBos⫹/⫺ and VdrBos null counterparts born of VdrBos null mothers (342). When adjusted for their smaller size or weight, the ash weight and mineral content of the VdrBos null mothers’ offspring were no different from that of VdrBos⫹/⫺ mothers (342). VdrBos null mothers were also placed on the calcium-phosphate-lactose-enriched “rescue diet” and compared with VdrBos null mothers that consumed the regular diet, and this had no effect on the size, weight, or mineral content of the offspring. Thus fetal loss of VDR in the VdrBos model had no effect on skeletal development or mineral content compared with respective littermates, maternal loss of VDR resulted in a slightly smaller fetal size (but proportionately normal mineral content) that was independent of the fetal genotype, and the maternal diet had no influence on fetal size or skeletal ash weight and mineral content (342). These results contrast with those obtained from the VdrLeu null model (574), which, as described in section VIIIA, in the first study mated VdrLeu null mothers to WT males so that all fetuses were VdrLeu⫹/⫺, and compared them with fetuses from unrelated WT females mated to WT males. Fetal skeletal mineralization was reduced in VdrLeu⫹/⫺ fetuses compared with unrelated WT fetuses, but improved when their mothers were treated during pregnancy with the “rescue diet” (574). This change was in the opposite direction of the fetal serum calcium, which was reduced when the VdrLeu null mother was placed on the enriched diet. But in the second study, the authors mated VdrLeu null mothers to VdrLeu⫹/⫺males so that the offspring were VdrLeu null and VdrLeu⫹/⫺, and compared them to WT fetuses from related WT females mated to WT males. Fetal skeletal mineralization was normal in VdrLeu null fetuses but reduced in VdrLeu⫹/⫺ fetuses from VdrLeu null mothers, while the previous finding of reduced serum calcium was not seen. When the mothers were placed on the “rescue diet” during pregnancy, the skeletal mineralization was normal in VdrLeu⫹/⫺ fetuses. As previously postulated in section VIIIA, it is possible that the reduced mineralization of VdrLeu⫹/⫺ compared with VdrLeu null and WT fetuses, which was not seen in the VdrBos null model, is due to aberrant VDR signaling introduced by the alternately spliced receptor that is expressed by VdrLeu⫹/⫺ mice. It seems a likely explanation since the Vdr null skeleton was normal in both the VdrBos and VdrLeu null null fetuses; it was only the VdrLeu⫹/⫺ skeleton that differed between the two models (342, 376). This would be made clearer if VdrLeu⫹/⫺ ⫻ VdrLeu⫹/⫺ matings were done so that WT, VdrLeu⫹/⫺, and VdrLeu null fetuses can be compared within the same litters. If abnormal VDR signaling explains the phenotype of VdrLeu⫹/⫺ fetuses, then WT and VdrLeu null fetuses will have normal skeletons and mineral content while VdrLeu⫹/⫺ fetuses will continue to be abnormal. Furthermore, to test the investigators’ hypothesis that the VdrLeu⫹/⫺ skeletal phenotype is caused by increased calcitriol in the circulation of VdrLeu null mothers, VdrLeu⫹/⫺ fetuses obtained from VdrLeu null mothers should be compared with VdrLeu⫹/⫺ fetuses obtained from WT mothers. Fetal guinea pigs differ from the results described for fetal rats and mice, in that 8 wk of a vitamin D-deficient diet caused reduced fetal body weight and total body bone mineral content, and increased osteoid with expansion of the hypertrophic zone of chondrocytes (573). However, as noted earlier, the comparison may not be physiologically relevant because the maternal 25-hydroxyvitamin D was 400 nM on the vitamin D-replete diet and ⬃20 nM on the D-deficient diet, with fetuses having a 25-hydroxyvitamin D level of ⬃200 nM when their mothers were on the vitamin D-replete diet. It is a comparison of the skeletal effects of hypervitaminosis D to vitamin D deficiency, rather than vitamin D sufficiency to vitamin D deficiency. Furthermore, the vitamin D-replete and vitamin D-deficient diets were made by different manufacturers and differed in more than just vitamin D content, including higher vitamin C and lower phosphorus content in the vitamin D-deficient diet. Since the standard and control diet differed in more than Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1185 CHRISTOPHER S. KOVACS just the nutrient being studied, it is possible that dietary differences influenced the phenotypes of vitamin D “replete” and deficient guinea pigs. A Overall, while there are some differences among the models, vitamin D deficient rats and VdrBos and VdrLeu null null fetal mice show normal endochondral bone development and mineralization by term. This is very convincing evidence that calcitriol is not required during fetal development to regulate skeletal development and mineralization. B D. Role in the Neonate C Multiple models of disrupted vitamin D physiology have been studied during the neonatal period, including severely vitamin D-deficient rats (74, 241, 443), four different Vdr null mice (175, 372, 704, 755), and two different models of 1␣-hydroxylase null (Cyp27b1 null) mice (138, 496). All of these appear normal at birth, although chemistries and ash weights have yet to be reported in any of the Cyp27b1 null models at birth. All models remain phenotypically normal through most of the succeeding 3 wk that the mother is lactating. As described earlier, near the time of weaning the intestines become less permeable to passive absorption of calcium, and more dependent on active, calcitriol-dependent intestinal calcium absorption. It is at this point in time that all of these models begin to develop hypocalcemia, hypophosphatemia, secondary hyperparathyroidism, and skeletal evidence of rickets (138, 241, 371, 372, 443, 496). However, a calcium-phosphate-lactose-enriched “rescue diet” bypasses the need for vitamin D/calcitriol or its receptor and allows each of these models to maintain normal serum chemistries, skeletal morphology, and skeletal mineral content (24, 64, 65, 140, 154, 175, 371, 372, 496, 638, 639, 704, 755). D E These animal data clearly demonstrate that vitamin D, calcitriol, and its receptor are not required for normal fetal calcium homeostasis, skeletal development, and mineralization. It is at the time of weaning, when the intestines become the route of mineral delivery, that calcitriol becomes required. Furthermore, since arguably all of the skeletal man- 100 Calcium (mg/g ash) 90 80 70 60 50 40 30 20 10 0 F WT (14) Vdr+/(23) Vdr null (17) WT (14) Vdr+/(23) Vdr null (17) Magnesium (mg/g ash) 20 18 16 14 12 10 8 6 4 2 0 1186 FIGURE 11. Skeletal morphology and mineral content in Vdr null fetuses. A and B show images of fetal skeletons (ED 18.5) stained with alizarin red (for mineral) and alcian blue (for cartilage). Skeletal morphology of the Vdr null was consistently normal, as shown by the normal crown-rump length, lengths of long bones, and mineralization pattern of the Vdr null (B) and its WT sibling (A). C and D are von Kossa preparations (counterstained with methyl green) of the upper halves of fetal tibias (ED 18.5), which include the growth plates and part of the tibial shafts. The overall morphology of the tibias, the lengths of the growth plates, and periosteal thickness were normal in Vdr null (D) compared with WT (C), and a normal amount of mineral (black) was present in the shafts of the tibias. E shows the calcium content (by atomic absorption spectroscopy) of Vdr null fetuses compared with WT and Vdr⫹/⫺ littermates, while F compares the magnesium content among the littermates. The calcium and magnesium content were normal in Vdr nulls. Ash weight was also normal (not shown). Scale bar indicates 100 m for C and D. [Adapted from Kovacs et al. (342).] Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM ifestations of vitamin D-related rickets can be prevented by use of a high-calcium diet, these findings suggest that the main role of calcitriol with respect to the skeleton is not through direct actions on chondrocytes or osteoblasts, but indirect through upregulation of intestinal mineral delivery to provide mineral for chondrocytes and osteoblasts to use. This point has been confirmed by the observation that selective expression of Vdr in intestinal cells rescued the skeletal phenotype of Vdr null mice (375, 746), whereas after selective ablation of Vdr from intestinal cells a rachitic phenotype develops (375), and that phenotype can also be produced by dietary calcium restriction (375). Moreover, ablating Vdr or Cyp27b1 from cells within the skeleton does not cause rickets either. The skeleton is normal if Vdr is selectively ablated from chondrocytes, mature osteoblasts, or osteocytes (375, 421), and a high bone mass phenotype with reduced resorptive surfaces occurs when Vdr is ablated from osteoblasts (750). Ablating Cyp27b1 from chondrocytes does not reproduce the rachitic growth plate, whereas low phosphorus will cause the widened, irregular growth plate (466). The only evidence from mice supporting a possible direct role for calcitriol or VDR in bone cells is that Cyp27b1/Vdr null double mutants have a skeletal phenotype that is not completely prevented by the calcium-phosphate-lactose-enriched “rescue diet” (495). E. Human Data 1. Observational studies and case reports The most rigorous assessment of the fetal skeleton at birth involved babies that had died of obstetrical accidents: seven from mothers with clinically diagnosed osteomalacia and vitamin D deficiency and eight from otherwise healthy mothers (427). This 1925 report has been cited as confirming the existence of prenatal rickets, despite the fact that the paper concluded otherwise. The investigators obtained plain radiographs of the fetal long bones, and reduced the femurs to ash to measure the mineral content by atomic absorption spectroscopy. One “osteomalacic parent” baby was born prematurely at 7.5 mo and had an ash weight that was less than half of the other 14 near-term or term fetuses, which is a normal finding given that more than 80% of the mineral content of the human skeleton is deposited during the third trimester (224, 683, 734). But that single fetus’s low ash weight led to the investigators’ impression that “osteomalacic parent” babies had softer bone and lower ash weight compared with normal babies. No statistical analysis was done, but the raw data were presented in a table that permits such an analysis by modern methods. The calcium content (mean ⫾ SE) was 374.2 ⫾ 3.0 per thousand in “osteomalacic parent” babies versus 371.8 ⫾ 13.9 per thousand in normal babies. Similarly, the phosphorus content was 189.0 ⫾ 1.1 per thousand in “osteomalacic parent” babies versus 189.6 ⫾ 1.6 per thousand in normal babies. The radiographs revealed no signs of rickets, and centers of ossification were normal (427). The authors speculated about “a curious cupping” at the ulnar ends in two babies born to osteomalacic mothers, but that is now known to be a normal variant (225). Maxwell et al. (427) concluded “there is no evidence of prenatal rickets” despite his initial enthusiasm that he would find such evidence. Rickets could not be present with normal skeletal mineral content because unmineralized osteoid is pathognomonic for the condition. Epidemiological data show that hypocalcemia and rickets due to vitamin D deficiency are not usually diagnosed until weeks to months after birth; the peak incidence is between 6 and 18 mo, even in regions where vitamin D deficiency is endemic (17, 49, 332, 510). Clinically recognized cases within the first few weeks after birth are rare and the subject of individual case reports. Vitamin D deficiency is endemic in India, so clinical suspicion for its presence is high. Over 16 years Teotia et al. (666) examined 165 babies born of women with severe osteomalacia. Of these, only six babies showed abnormalities: three had postnatal hypocalcemia and three had postnatal rickets without hypocalcemia (666). In another paper summarizing their experience, Teotia et al. (667) wrote that congenital rickets is “uncommon and develops only when maternal mineral and vitamin D stores have been completely exhausted.” This clinical experience of the natural history of rickets concurs with the results from the animal models in that the fetal skeleton should be normal at birth with rickets developing later. As explained earlier, intestinal calcium absorption is largely passive at birth but becomes calcitriol dependent as the baby matures (330, 332, 335); this is why vitamin D-dependent rickets usually develops after calcitriol becomes required to maintain the supply of calcium for the developing skeleton. Vitamin D-deficiency rickets also needs to be distinguished from rickets of prematurity, which is the result of the preterm skeleton having a much higher demand for calcium than what the preterm intestines can absorb from a normal intake of milk or formula (see sect. IIIE). A few rare, isolated case reports from the past 100 years have demonstrated that physical, radiographic, or histological changes consistent with rickets may be detected at birth (50, 279, 400, 426, 429, 448, 577, 741), although only two of these reports have biochemical confirmation of low 25hydroxyvitamin D levels (279, 448), while one measured serum vitamin D which is normally low in cord blood (400). This is an important distinction because skeletal evidence of rickets is not synonymous with vitamin D deficiency. Instead, rickets means an undermineralized skeleton that is primarily caused by deficiency of calcium or phosphorus, of which vitamin D deficiency or genetic disturbances of vitamin D physiology are but a few of the causes (539). The differential diagnosis of the rachitic-appearing fetal and Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1187 CHRISTOPHER S. KOVACS neonatal skeleton encompasses at least 50 different conditions (525), including fetal hypoparathyroidism, skeletal dysplasias (249), metaphyseal chondrodysplasias (525), hypophosphatasias (555, 584), type V osteogenesis imperfecta (27), and scurvy (73). Consequently, it is possible that some of these cases of rickets were not due to vitamin D deficiency. For example, in a 1932 case report, the cord blood calcium was normal (2.65 mM) in the presence of an apparently undermineralized skeleton (426). In other cases, the babies hemorrhaged from multiple orifices before dying, which suggests other severe disturbances such as scurvy or vitamin K deficiency (73, 426, 429, 543). In other reports that described rickets as being present “at birth,” the diagnosis was actually made within the first or second week after birth, so the skeletal status at birth remains unknown (50, 59, 191, 451, 502, 585, 586, 666). “Neonatal rickets” is a more appropriate term for such cases, as opposed to “congenital rickets,” because the latter implies that the condition was present at birth. Since the neonate normally adds ⬃30 – 40 mg/kg of calcium per day to the skeleton while maintaining appropriate blood calcium and phosphorus levels, any impairment in intestinal absorption of mineral can quickly lead to progressive demineralization of the skeleton, and hypocalcemia. This is what happens in rickets of prematurity, for example, which is not caused by vitamin D deficiency but by the skeletal demand for calcium being far higher in the preterm baby than in the normal neonate (see sect. IIIE). In one illustrative case of possible neonatal vitamin D-deficiency rickets, the clinicians anticipated from the maternal history that rickets might be present in the infant (585). However, convincing radiographic findings were not present at day 2 but had developed by day 16, confirming that radiographic evidence can develop rapidly after birth (585). In many cases of neonatal rickets attributed by the authors to vitamin D deficiency, the cause was clearly not isolated vitamin D deficiency. Instead, the mothers had such problems as severe anorexia, malnutrition, malabsorption from celiac disease or pancreatic insufficiency, lactose intolerance, extreme avoidance of calcium and dairy food sources, use of drugs that interfere with vitamin D metabolism, or very low intakes of both calcium and vitamin D (50, 279, 426, 428, 448, 543, 632, 666). This fits with Teotia’s impression that rickets will be present at birth only in extreme cases of complete nutritional exhaustion (667), or as Maxwell described in 1935, “women who have, in the matter of diet, been living near the starvation-line.” Vitamin D deficiency itself is normally not sufficient to adversely affect the development or mineralization of the fetal skeleton. But combined with other significant maternal nutritional deficiencies, an abnormal skeleton may be present at birth. Additional case reports and case series have used questionable criteria to make a diagnosis of rickets, whether or not 1188 it is due to vitamin D deficiency. Craniotabes (the subjective determination that the skull is softer than normal) is a common criterion used alone to make the diagnosis of rickets in many of these reports, but public health data from several countries have shown craniotabes to be present in 30 –50% of healthy infants, and to have no correlation with maternal or neonatal 25-hydroxyvitamin D levels or skeletal evidence of rickets (55, 122, 136, 320, 515, 516). Consequently, craniotabes is not considered a reliable indicator of rickets or of vitamin D deficiency; its presentation may be predicted more by ethnicity and gestational age than vitamin D sufficiency. As mentioned earlier, distal ulnar cupping is a normal variant that should not be used to diagnose rickets (225). Although a large or slowly closing anterior fontanelle can be seen with rickets (512, 689), these are nonspecific changes shared among more than 25 metabolic, genetic, and skeletal disorders (308, 569). Use of widened fontanelle alone to indicate vitamin D deficiency led to misdiagnosis and maltreatment of a newborn with hypophosphatasia (449). Among the questionable reports that claimed vitamin D-deficient rickets to be present at birth are those that relied on the normal variant of ulnar cupping (491, 534, 577, 752) or craniotabes (191, 752). One report noted craniotabes at birth but the skeletal radiographs were normal, but by 2 wk of age the baby had developed hypocalcemia, hyperphosphatemia, elevated alkaline phosphatase; the hyperphosphatemia indicates hypoparathyroidism and not vitamin D deficiency (191). The biochemical abnormalities normalized in the months following initiation of vitamin D treatment, but this likely reflected improvement in the hypoparathyroidism rather than being proof of vitamin D deficiency. A single-authored report from Japan (491) is particularly interesting for the claim that 60 of 255 healthy newborn infants had radiographic evidence of rickets at the wrist. The serum calcium, ionized calcium, phosphorus, and alkaline phosphatase were normal in cord blood and in the newborns over the first 8 days after birth, with no differences between the babies with “rickets” versus those without. The normal chemistries (especially calcium and alkaline phosphatase) are an indication that if the radiographic findings (ulnar cupping, etc.) represented true abnormalities, they were unlikely to be the result of vitamin D deficiency. Similarly, a single observer reported that rachitic rosaries and ulnar cupping were present in 75 of 858 consecutive newborn infants in Kuwait, who also had normal serum calcium, phosphorus, and alkaline phosphatase (534). Would other observers have agreed with that single observer’s subjective impression that rachitic rosary was present? 25-Hydroxyvitamin D was measured only in the infants with the suspected rib cage abnormality, so it is unknown whether the postulated rib cage abnormality correlated with objective evidence of vitamin D deficiency. The normal calcium, phosphorus, and alkaline phosphatase is evidence that vitamin D deficiency was not the explanation Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM for the apparent findings. Another report from Japan suggested that the presence of craniotabes in 22% of consecutive newborns meant rickets, but since larger surveys have already shown that craniotabes is present in up to 30 –50% of healthy newborns and to have no association with vitamin D sufficiency, that conclusion is not supportable (46). A curious report from China examined 124 babies that had died either before or sometime after birth, with no data provided about the cause of death, the mothers’ health or obstetrical history, or the percentage that these babies represented of the catchment population (381). Sixteen fetuses and 28 neonates were said to have histological evidence of rickets (381). However, the two main criteria used by the investigator included an island (apophysis) of chondrocytes in the diaphysis, and an irregular growth plate (381). Islands or protuberances of cartilage into bone and an irregular growth plate are not standard or defining criteria for rickets and can be seen in a variety of other congenital bone disorders, including achondroplasias, osteopetrosis, collagen disorders, and other skeletal dysplasias (16, 88, 268, 503, 550, 680). The defining histological criterion for rickets is increased osteoid (unmineralized bone), but the report did not describe this. Nor were any data provided to support the hypothesis that the abnormalities were due to vitamin D deficiency and not any of the other conditions on the differential. Consequently, vitamin D-deficiency rickets cannot be confirmed to be present with any confidence from those data. Overall, out of 100 years of published literature, these reports represent a very uncommon occurrence. For those cases that may have been truly due to vitamin D deficiency, it should be clear that these are exceptional cases and not reflective of vitamin D deficiency during pregnancy that is commonly seen today. Severe hypocalcemia does not usually occur in vitamin D deficiency because secondary hyperparathyroidism minimizes the fall in serum calcium. But the descriptions of the mothers in the case reports above underscore how extreme their conditions were, with serum calcium levels often half normal, signs of globally poor nutrition, carpopedal spasms, positive Chvostek’s signs, hypotonia, severe osteomalacia, severe back pain, waddling gait, etc. The confounding presence of other nutritional deficiencies makes it difficult to be certain the degree to which vitamin D deficiency contributed to the fetal or neonatal phenotype. But it seems likely that vitamin D deficiency alone did not explain these rare presentations. Additional cohort studies have reported that bone mineral content in newborns did not differ by measures of vitamin D sufficiency in the mother or baby (122, 725). Congdon et al. (122) studied infants born to 45 untreated Asian women, 19 Asian women who had received 1,000 IU vitamin D during the third trimester, and 12 unsupplemented Caucasian women. Mean cord blood concentrations of 25-hy- droxyvitamin D were 5.9, 15.2, and 33.4 nM, respectively (122). There were no significant differences in serum calcium, phosphorus, and alkaline phosphatase, while bone mineral content of the forearm, measured within 5 days of birth, did not differ among groups (122). Weiler et al. (725) measured bone mineral content of the lumbar spine, femur, and whole body by DXA within 15 days after birth in 50 healthy term infants. Mean cord blood 25-hydroxyvitamin D was 73 and 36 nM in the sufficient and insufficient Caucasians, 44 nM in Asians, and 27 nM in First Nations infants. Differences in bone mineral content were seen that were attributable to ethnicity but not vitamin D sufficiency (725). Viljakainen et al. (712) divided a cohort of 87 children into two groups above and below the median cord blood 25hydroxyvitamin D, and measured DXA soon after birth and at 14 mo. There were no differences in anthropometric parameters or tibial bone mineral density at either time point. Tibial bone mineral content and cross-sectional area of the tibia were slightly lower at baseline in the cohort with lower 25-hydroxyvitamin D; the difference in bone mineral content was gone at 14 mo while a difference in crosssectional area persisted. Notably bone mineral content is affected by bone size or area, whereas bone mineral density corrects for this. The investigators noted disparities in sex and size of the newborns above and below the median 25hydroxyvitamin D level, and those disparities may account for the transient difference in bone mineral content noted at birth. If vitamin D deficiency alone could readily cause prenatal rickets, then even more severe abnormalities would be expected to occur in fetuses and neonates bearing inherited mutations of vitamin D physiology, including the inability to synthesize calcitriol (1␣-hydroxylase deficiency; pseudovitamin D deficiency rickets; vitamin D dependent rickets type I; VDDR-I) or the expression of non-functional vitamin D receptors (hereditary vitamin D-resistant rickets; vitamin D dependent rickets type II; VDDR-II). But these babies appear normal at birth (apart from alopecia in VDDR-II) and have normal calcium levels (66, 180, 312, 408, 538, 621, 662, 665). In both conditions the child is fated to develop hypocalcemia, hypophosphatemia, and rickets. VDDR-I presents within the first 3–12 mo after birth; VDDR-II can also present in infancy but has often been diagnosed during the second year or even later (36, 66, 230, 258, 312, 538, 621, 662, 665, 727). Mortality can also occur in infancy or childhood due to the severity of the hypocalcemia (36, 374). Pseudofractures and fractures can occur in both conditions, but bowing of the long bones and metaphyseal widening are much more common (36, 66, 180, 230, 258, 312, 408, 538, 621, 662, 665, 727). Available clinical data from pediatric and older age VDDR type II children are also consistent with the animal data in Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1189 CHRISTOPHER S. KOVACS that calcitriol’s role to regulate intestinal calcium absorption can be bypassed. Repeated intravenous or intracaval calcium infusions, or high oral dose calcium, have been shown to normalize calcium, phosphorus, PTH, alkaline phosphatase; rickets was prevented or healed in these children (36, 259, 312, 727). In one case series, the use of parenteral calcium caused growth velocity to increase, bone pain to decrease, and biochemical abnormalities to normalize within a year; it also enabled some affected children to walk for the first time ever (259). Rickets healed more rapidly in younger patients and when parenteral calcium was used instead of high dose oral calcium. For VDDR type I, the high-calcium approach is usually not needed because physiological doses of calcitriol or 1␣-hydroxyvitamin D should normalize intestinal calcium absorption (662). Once weight bearing begins, the natural history of untreated vitamin D-deficiency rickets includes progressive deformities of the long bones (enlargement of the wrist and bowing of the distal radius and ulna, and lateral bowing of the femur and tibia), distorted growth plates, and short stature (513, 514). These changes are the result of the softer, malleable structure of rachitic bone which bends in response to weight-bearing activity, and the resulting varus and valgus deformities can be very dramatic. Notably, fractures of the long bones are not a dominant feature of rickets, but when they do occur they are single and not multiple. A study of 736 Nigerian children aged 18 mo or older found that previous fracture was present in 9% with active rickets, 7% with early or nearly healed rickets, and 9% of normal children (668). The other clinical features that predicted rickets were age ⬍5, height-for-age Z-score ⬍2 standard deviations, leg pain during walking, wrist enlargement, and costochondral enlargement (668). Oddly, despite the evidence that overt rickets is not associated with an increased risk of fracture, vitamin D insufficiency has been proposed to cause marked skeletal fragility in infants such that it would mimic the multiple fractures seen in physically abused infants and others with osteogenesis imperfecta (303). Moreover, it has been suggested that for a 6-mo window of time the infant is transiently susceptible to fragility from vitamin D insufficiency in what has been termed “temporary brittle bone disease” (504). Since severe vitamin D deficiency causes slowly developing deformities of the limbs without more fractures than in normal children (668), how could the more modest condition of vitamin D insufficiency be a cause of overt skeletal fragility with multiple atraumatic fractures? This seems a theory that lacks credible evidence to support it; consequently, it has been thoroughly discredited by others (181, 182, 286, 287, 414, 629, 637, 651). The Society for Pediatric Radiology and the European Society of Paediatric Radiology jointly wrote that the diagnosis fails to meet minimum standards of reliable scientific information that can be presented in the courts of law “because it lacks appropriate grounding 1190 in scientific methods and proceduresѧit is not generally accepted within the field of radiology, but is instead based on the unsupported speculation and subjective beliefs of a very small number of medical professionals” (436). Overall, the human genetic disorders, the population surveillance data on the incidence of rickets, 100 years of case reports of extreme cases, and much of the clinical observational studies pertaining to vitamin D deficiency, are consistent with the animal models in that hypocalcemia and rickets are not likely to be present at birth. Instead, hypocalcemia and rickets will develop postnatally in children with severe vitamin D deficiency or genetic disorders causing loss of calcitriol or VDR, with the peak age of presentation occurring not at birth but between 6 and 18 mo of age. Furthermore, calcitriol’s role in stimulating intestinal calcium absorption can be bypassed by enriching the diet or administering calcium parenterally. Left untreated, the long bones are malleable, resulting in limb deformities, short stature, and eventually osteoarthritic changes from the deformed joints. 2. Interventional studies One of the earliest randomized trials of vitamin D supplementation still stands as the most definitive because it involved women who were severely vitamin D deficient by today’s standards (77). Brooke et al. (77) studied 126 Asian women living in England, 59 who were treated with vitamin D and 67 controls. The mean 25-hydroxyvitamin D level was 20 nM at baseline and the treatment group achieved a level of 168 nM at term; this marked difference was ideal for determining if correction of vitamin D deficiency resulted in any maternal or fetal benefits. The treatment group nominally received 1,000 IU vitamin D per day but must have received 10,000 IU per day or more to have reached that increment in 25-hydroxyvitamin D. The mean cord blood 25-hydroxyvitamin D level was 10 nM in control babies and 138 nM in babies born of the treated mothers. Cord blood calcium and phosphorus showed no statistically significant differences between the control and treated babies. Treatment also had no effect on birth weight, crown-heel length, forearm length, triceps skinfold thickness, and head circumference, but a small decrease in fontanelle diameter was observed (77). There was also a higher alkaline phosphatase level in control babies, but fractionation confirmed that this was entirely accounted for by the placental fraction. The babies were examined radiologically, and there were no signs of rickets in either group (77). Interestingly, at days 3 and 6 after birth, the control group had a significantly lower blood calcium than in the vitamin D-replete babies (2.18 ⫾ 0.04 vs. 2.30 ⫾ 0.04 mM, P ⬍ 0.05) (77). Five infants in the control group had symptomatic hypocalcemia (irritability and serum calcium ⬍1.8 mM), whereas none of vitamin D-replete babies became hypocalcemic (77). These results mirror the findings in the Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM previously discussed animal studies, in which serum minerals will be normal at birth but fall postnatally when severe vitamin D deficiency is present. In one of two follow-up analyses of the same data (which have been mistaken as separate trials by other reviewers), a slightly greater increment in weight and length over the subsequent 12 mo was seen in the babies of vitamin D-supplemented mothers (78, 425). Cockburn et al. (116) randomized 164 women to receive either 400 IU vitamin D beginning at the 12th week of pregnancy or nothing. Cord blood 25-hydroxyvitamin D was 28 versus 20 nM in the treated versus controls, and there was no significant difference in cord blood calcium or phosphorus. However, by the sixth day, calcium was significantly higher while phosphorus was significant lower in the infants whose mothers had received vitamin D compared with controls. A lower phosphorus is not understandable in the context of a higher 25-hydroxyvitamin D level. In a smaller study of 77 women from France, Mallet et al. (407) administered 1,000 IU vitamin D2 daily during the third trimester, or a single dose of 200,000 IU vitamin D2 at the start of the third trimester, or no supplementation (407). The two treatment arms raised cord blood 25-hydroxyvitamin D to 15.7 and 18.2 nM compared with 5.3 nM in controls. There were no significant differences in maternal, cord blood, and day 2 or 6 neonatal calcium levels, hypocalcemic tetany, and skeletal or anthropometric parameters of the babies. A randomized study by Delvin of 40 women in France compared 1,000 IU vitamin D3 supplementation daily to placebo starting at 6 mo of pregnancy, and raised the cord blood 25-hydroxyvitamin D from 17.5 to 45 nM (153). There were no effects on skeletal parameters or on the cord blood calcium, phosphorus, or PTH. However, at 5 days of age, the treated group had a significantly higher serum and ionized calcium (153). Marya performed two studies in vitamin D-deficient Asian women in India (419, 420). In the first study there were three treatment arms: 800,000 IU vitamin D2 was given in the 7th and 8th months of pregnancy to 20 women, 1,200 IU vitamin D2 per day were given to 25 women during the third trimester, and 75 women received nothing. There was a small increase in uncorrected cord calcium in babies of high-dose mothers compared with babies of untreated mothers (2.68 vs. 2.52 mM in untreated), but no change in serum phosphorus or other benefit (420). The second study administered 600,000 IU vitamin D2 in the 7th and 8th months of pregnancy to 100 women and gave nothing to 100 others. A significant increase in uncorrected cord blood calcium was observed in the treatment group (2.77 vs. 2.57 mM) with no difference in cord blood phosphorus (419). A slightly greater birth weight, crown-heel length, and head circumference were also found in the babies from treated mothers (419). Cord blood 25-hydroxyvitamin D was not measured in either study. The lack of disclosure of methodological details in the two studies, especially whether and how the subjects were truly randomized, leaves the results somewhat questionable. Yu et al. (756) conducted a randomized unblinded trial in London of 180 women, with three arms beginning at week 27 of pregnancy: 800 IU vitamin D2 per day, a single dose of 200,000 IU of vitamin D2, or no treatment (756). Mean cord 25-hydroxyvitamin D was significantly higher in the two treatment groups compared with the controls (26, 25, and 17 nM, respectively). However, there were no significant differences in gestational age at delivery, birth weight, or incidence of low birth weight. Roth et al. (567) completed a trial in Bangladesh (NCT01126528) in which 160 women were randomized to 35,000 IU/wk vitamin D3 or placebo beginning at 26 –29 wk until delivery. Achieved cord blood levels of 25-hydroxyvitamin D were 103 versus 30 nM, respectively. There was no effect on length, weight, head circumference, and femur length at birth (568). Cord blood calcium levels were done but have not been reported. A similar study by Hashemipour et al. (246) in Iran randomized 160 women to receive 50,000 IU vitamin D per week or 400 IU daily beginning at 26 –28 wk for 8 wk (246). Cord blood 25-hydroxyvitamin D was 69 versus 27 nM, and cord blood calcium (not corrected for albumin) was also significantly higher in the vitamin D-treated babies at 2.48 versus 2.28 mM. There was no difference in the incidence of hypocalcemia between the groups. Small but statistically significant increases in newborn weight, length, and head circumference were seen in the vitamin D-treated babies. Grant et al. (233) completed a study in New Zealand (ACTRN12610000483055) that randomized 260 women to placebo, 1,000 IU vitamin D daily, or 2,000 IU vitamin D daily. Cord blood 25-hydroxyvitamin D levels were 33, 60, and 65 nM, respectively. There was no difference in serum calcium among the three groups at 2.58, 2.62, and 2.60 mM, respectively (233). Postnatally the infants continued in their treatment arms by receiving placebo, 400 IU, or 800 IU of vitamin D daily. 25-Hydroxyvitamin D rose faster in the high-dose group, but by 6 mo of age the values were no different among the three groups of infants (78, 85, and 84 nM, respectively), and there was no difference in serum calcium at any postnatal time point (233). Kalra et al. (294) completed a trial in India in which 97 women were randomized to one dose of 60,000 IU vitamin D in the second trimester or 120,000 IU vitamin D in each of the second and third trimesters; 43 other women who Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1191 CHRISTOPHER S. KOVACS received standard care served as controls. Achieved cord blood 25-hydroxyvitamin D median values were 28.2, 24.1, and 18.5 nM, respectively, and there was no difference in cord blood calcium across the groups (294). Small increments in length, head circumference, and weight, and a slight decrease in anterior fonatelle length, were seen in the both vitamin D groups compared with the nonrandomized controls (294). Hollis et al. (262) recently completed two larger trials. The first (NCT00292591) randomized women at 12–16 wk of gestation to receive 400, 2,000, or 4,000 IU vitamin D3 per day; 350 completed the study out of the planned 516 enrollment (262). The primary outcome of the first study was a combination of the 25-hydroxyvitamin D levels and bone mineral density of both mother and infant; however, the BMD values have not been published. Achieved cord blood 25-hydroxyvitamin was 46, 57, and 66 nM, respectively. The second study (NCT00412087) randomized women at 12–16 wk of gestation to receive 2,000 or 4,000 IU vitamin D3 per day; 160 completed this study out of the planned 559 (717). Achieved cord blood 25-hydroxyvitamin D was 55 and 68 nM, respectively. The cord blood calcium values for both studies, and the bone density results for the first study, have been described verbally by the authors in public presentations as showing no significant differences (716). In the first study, 4,000 IU vitamin D had no effect on gestational age at delivery, birth weight, mode of delivery, need for level II or III neonatal care, cord blood calcium, preterm birth, preterm labor, preeclampsia, infection, or BMD (261, 262, 716). Post-hoc analyses of both studies have suggested that some combinations of nonskeletal obstetric or neonatal outcomes may show a trend to improvement with higher achieved 25-hydroxyvitamin D level, but only after arbitrary exclusion of certain races from the analysis (261, 262, 265, 716). Whether vitamin D supplementation during pregnancy has any effect on neonatal bone mass may be determined by the Maternal Vitamin D Osteoporosis Study (MAVIDOS), in which more than 1,000 women have been randomized to receive 1,000 IU vitamin D or placebo daily from 14 wk of gestation until term (245). The primary outcome is the whole body bone mineral content of the neonates as assessed by DXA. Overall, these trials have not shown any compelling evidence that higher dose vitamin D supplementation achieves any benefit related to calcium and bone metabolism prior to birth, although there is likely a decreased risk of hypocalcemia after birth when the starting 25hydroxyvitamin D level is very low, as in the Brooke study. When the basal level of 25-hydroxyvitamin D is higher than that, there may be no additional benefit from supplementation. 1192 3. Associational studies A number of associational studies have recently drawn attention for their inferences that higher intakes of vitamin D may be needed during pregnancy to ensure optimal bone health in the fetus, infant, and child. These studies have examined associations between estimated maternal intake of vitamin D or single measurements of maternal 25-hydroxyvitamin D during pregnancy, and various skeletal outcomes in the offspring. In agreement with data from observational studies and randomized trials discussed above, there were no significant associations found with weight, skeletal lengths, or bone mineral density at birth (281, 285, 363, 399, 460, 713). A study by Morley et al. (460) concluded that maternal 25-hydroxyvitamin D below 28 nM predicted a slightly shorter knee-heel length in newborns as assessed by a handheld device. However, left unstated in the abstract was that the association was not statistically significant after correcting for gestational age of the babies. Seemingly opposing results were obtained in studies by Mahon et al. and Viljakainen et al. in which increased metaphyseal area of the femur or tibia was associated with lower (399) or higher (713) maternal 25-hydroxyvitamin D during pregnancy. Curiously, for each study the investigators concluded that an adverse effect of low 25-hydroxyvitamin D had been found. Mahon et al. (399) considered greater cross-sectional area of the femoral metaphysis to be evidence of prenatal rickets in 242 mother-baby pairs assessed by high-resolution ultrasound, while Viljakainen et al. (713) considered greater cross-sectional area of the tibial metaphysis to be an indication of stronger bone in 125 mother-baby pairs assessed by pQCT. More recently Ioannou, together with Mahon and colleagues, adapted their ultrasound technique to calculate femoral volumes in 357 mother-baby pairs (281). Maternal 25-hydroxyvitamin below 75 nM was associated with slightly smaller femur length, width, and volume, an association that is in the opposite direction from the original report by Mahon and co-workers (281). Although maternal 25-hydroxyvitamin D level predicted fetal femoral volume in a univariate analysis, there was no association after adjusting for maternal height and skinfold thickness (280). The seemingly contradictory results in the studies by Mahon, Viljakainen, and Ioannou could indicate that vitamin D has complex, sitespecific effects on skeletal development, or these could be chance findings with no true causal link between maternal 25-hydroxyvitamin D during pregnancy and the areas or volumes of fetal long bones. A study by Javaid et al. (285) has drawn much attention. In 198 mother-child pairs there were no significant associations between maternal serum 25-hydroxyvitamin D during pregnancy and birth weight, length, placental weight, abdominal circumference, head circumference, or cord blood Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM calcium (285). There were also no associations of maternal 25-hydroxyvitamin D with skeletal and anthropometric parameters in the infants at age 9 mo. But at 9 years of age, children whose mothers had had a 25(OH)D below 27.5 nM during pregnancy had a slightly but significantly lower bone mineral content by DXA than children of mothers whose 25-hydroxyvitamin D had been ⱖ50 nM. These data confirm the theory that vitamin D exposure during fetal development programs bone mass attained later in childhood (123). However, a recent study by Lawlor et al. (363) analyzed ⬃20 times the number of mother-child pairs than Javaid (3,960 vs. 198) and found no significant association between maternal 25-hydroxyvitamin D during pregnancy and offspring bone mass at age 9 years, as assessed by DXA. Lawlor et al. (363) had almost eight times the number of women with 25-hydroxyvitamin D below 27.5 nM (220 vs. 28 mothers) and should have had greater power to detect an association. But the contradictions continue, because Lawlor’s colleagues originally reported from the same database that estimated maternal UVB exposure in the third trimester of pregnancy in 6,995 mother-offspring pairs positively predicted offspring bone mass at age 9 years as assessed by DXA (593). The difference between the two studies by Lawlor and colleagues may be that estimated UVB exposure is a less accurate variable than the measurement of 25-hydroxyvitamin D. Overall, it is challenging to conclude anything at present from these contradictory studies, which could be reporting chance associations rather than indications of causal links. These associational studies are confounded by factors that contribute to low maternal 25-hydroxyvitamin D, including maternal obesity, lower socioeconomic status, poorer nutrition, and lack of exercise, prenatal care, vitamin supplementation, etc. Consequently, a lower value of 25-hydroxyvitamin D may simply predict a less healthy pregnant woman, rather than revealing cause-and-effect results of low maternal 25-hydroxyvitamin D on fetal bone health. Furthermore, the mother’s 25-hydroxyvitamin D value in pregnancy may predict factors that will remain unchanged and be shared or held in common with the child (lower socioeconomic status, poorer nutrition, obesity, etc.), in turn influencing childhood growth and bone mass at age 9 years without invoking a causative role for 25-hydroxyvitamin D exposure prior to birth. Some of the associational studies have much larger sample sizes than in the clinical trials of vitamin D supplementation, and therefore may be detecting real differences that the trials could not. Alternatively, the associational studies may simply be reporting “noise” caused by residual confounding, which in turn could explain the contradictory findings. A basic principle of associational studies is that causation cannot be proven from the results, but hypotheses may be raised that warrant further testing. F. Summary of Key Points Overall, the available animal and human data indicate that cord blood calcium, phosphorus, and skeletal morphology and mineral content should be normal at birth despite vitamin D deficiency, VDDR-I, or VDDR-II. In rare instances where the mother is more profoundly unwell due to vitamin D deficiency combined with global malnutrition or other factors, the baby may develop skeletal signs of rickets that are detectable at birth. Consistently, it is after birth that hypocalcemia and rickets will develop if severe vitamin D deficiency persists, or VDDR-I and VDDR-II are not recognized and treated. The peak incidence of vitamin D deficiency rickets is at 6 –18 mo after birth. Larger-scale clinical trials of vitamin D supplementation during pregnancy that systematically measure skeletal mineral content and morphology in newborns, and larger and carefully executed associational studies, are needed to be certain whether vitamin D deficiency in utero has any effects on skeletal mineralization at term or later in childhood. Several systematic reviews agree with the conclusion that current data are insufficient to know if vitamin D supplementation during pregnancy confers any skeletal or nonskeletal benefits on fetuses (244, 560, 561). IX. CALCITONIN Ctcgrp encodes both calcitonin and calcitonin gene-related peptide-␣, and ablation of it results in Ctcgrp null fetuses appearing in the expected Mendelian ratios. In contrast, ablation of the calcitonin receptor (Ctr) causes embryonic lethality at mid-gestation for unknown reasons (134, 361). Ctcgrp null mice live normal lifespans and are fertile, but show a small but significant reduction in the number of viable fetuses the day before expected birth compared with Ctcgrp⫹/⫺ mothers (430). Calcitonin is expressed in the endometrium during the time of implantation (349), and attenuation of its expression blocks implantation of the early embryo (760). Taken together, these findings may indicate that calcitonin plays critical roles during implantation and during development until mid-gestation. That Ctcgrp null fetuses do not experience embryonic lethality may indicate that maternal calcitonin compensates for its absence in the early embryo before the placental barrier is in place, whereas such rescue would not be possible in Ctr nulls due to lack of the receptor. Alternatively, Ctr may also mediate signaling by unknown ligands that are critical during embryonic development. A. Regulation of Serum Minerals There is some evidence for calcitonin having a role in regulating fetal mineral concentrations. Infusion of calcitonin antiserum into fetal rats at day 21.5 of gestation caused a 0.16 mM increase in blood calcium 1 h later (200), while Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1193 CHRISTOPHER S. KOVACS pharmacological doses of calcitonin injected into fetal rats or lambs caused hypocalcemia and hypophosphatemia (41, 211). However, when calcitonin deficiency was created by fetal thyroidectomy followed by thyroxine replacement, the blood calcium in fetal lambs was unaltered (97). The potential role of calcitonin has been investigated in Ctcgrp null fetal mice. Serum calcium and phosphorus were normal, but serum magnesium was significantly reduced compared with the littermates (430). This finding also occurred in Ctcgrp null fetuses from both Ctcgrp⫹/⫺ and Ctcgrp null mothers (430) and indicates that loss of either calcitonin or calcitonin gene-related peptide-␣ affects magnesium homeostasis. Calcium, phosphorus, and magnesium content of amniotic fluid were normal in Ctcgrp null fetuses. PTH showed a nonsignificant increase among Ctcgrp⫹/⫺ and Ctcgrp null fetuses compared with WT. B. Regulation of Placental Mineral Transport A reduction in placental mineral transport has been inferred from intact lambs in which pharmacological doses of calcitonin blunted the effect of PTHrP1–34 and PTHrP1–36 to increase the mineral content of the skeleton at term (40). However, that indirect measurement could have resulted from reduced incorporation of calcium into bone, as opposed to a specific reduction in placental calcium transport. In contrast, fetal thyroidectomy with subsequent thyroxine replacement did not alter placental calcium transport in sheep (298). Placental transport of calcium was unaltered in Ctcgrp null fetuses from both Ctcgrp⫹/⫺ and Ctcgrp null mothers (430). Magnesium transport has not been measured. C. Regulation of Endochondral Bone Formation Endochondral bone development appeared normal in Ctcgrp null fetuses as assessed by examination of whole mount fetuses, tibial histomorphometry, and gene expression within the developing endochondral skeleton (430). The ash weight and calcium content were also normal, but there was a modest but statistically significant reduction in skeletal magnesium content of Ctcgrp null fetuses which paralleled the reduction in serum magnesium (430). D. Role in the Neonate No disturbances in mineral metabolism have been reported in Ctcgrp null neonates. 1194 E. Human Data Genetic calcitonin deficiency has not been described in humans. A few early studies suggested that the normal high calcitonin levels in the neonate could contribute to hypocalcemia in preterm infants (558, 583, 595). However, subsequent studies found that gestational age at birth was a more significant determinant of neonatal calcitonin levels than the postnatal fall in serum calcium or the development of hypocalcemia (444, 558, 707). Therefore, the available human data suggest that calcitonin may have little role in mineral homeostasis of the neonate. F. Summary of Key Points Calcitonin may play critical roles during implantation and early embryonic development, which could explain the embryonic lethality caused by loss of CTR. Loss of calcitonin and calcitonin gene-related peptide-␣ caused a selective reduction in serum magnesium and skeletal magnesium content, but no other disturbance in bone and mineral metabolism. Whether loss of calcitonin or calcitonin gene-related peptide-␣ exerts this effect on magnesium is unknown. X. FIBROBLAST GROWTH FACTOR-23 Fibroblast growth factor-23 (FGF23) plays a key role in regulating phosphorus metabolism in postnatal mice and humans. FGF23 forms a complex with membrane-bound or soluble forms of its co-receptor Klotho and one of four FGF receptors (usually FGFR1c), and downregulates expression of sodium-phosphate cotransporters 2a and 2c (NaPi2a and NaPi2c) within the proximal renal tubules (350, 614, 699, 731). It also inhibits the renal 1␣-hydroxylase (Cyp27b1), increases expression of 24-hydroxylase (Cyp24a1), inhibits PTH, and inhibits intestinal expression of NaPi2b (161, 267, 415, 614). Loss of FGF23 leads to impaired renal phosphorus excretion, hyperphosphatemia, increased calcitriol, increased intestinal phosphorus absorption, skeletal abnormalities, extraskeletal calcifications, and early mortality (161, 267, 415). Conversely, excess FGF23 causes enhanced renal phosphorus excretion, hypophosphatemia, low calcitriol, reduced intestinal phosphorus absorption, and rickets or osteomalacia (19, 161, 267). A. Regulation of Serum Minerals Intact FGF23 is undetectable in Fgf23 null fetal sera, but serum phosphorus and calcium, amniotic fluid phosphorus and calcium, calcitriol, and PTH are all normal (395). Conversely, Phex null male fetuses (also known as Hyp fetuses) have 7.8-fold increased intact FGF23 in one study (395) and 1,000-fold increased in another (484). Despite elevated FGF23, Phex null males have normal serum phosphorus and calcium, amniotic fluid phosphorus and calcium, and Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM PTH (395, 484). Serum calcitriol was halved in Phex null males compared with WT fetuses (395, 484). Fetal kidneys showed abundant expression of Klotho, NaPi2a and NaPi2c, Cyp27b1, Cyp24a1, and Fgfr1-Fgfr4 (395). But in Fgf23 null fetal kidneys, the only change in expression was downregulation of Cyp24a1 (395). Conversely, in Phex null male fetal kidneys there was significantly increased expression of Cyp24a1 (395, 484) and decreased expression of Klotho and Fgfr2 (395), whereas all other FGF23 targets showed no changes (395, 484). These results show that neither loss nor excess of FGF23 disturbs serum phosphorus or renal excretion of phosphorus into amniotic fluid, but excess FGF23 did lower calcitriol, likely through increased Cyp24a1-mediated catabolism. The relative lack of responsiveness to deficiency or excess of FGF23 was not due to failure of expression of FGF23 target genes. B. Regulation of Placental Mineral Transport Murine placentas showed abundant expression of Klotho, NaPi2a, NaPi2b, NaPi2c (NaPi2b expression was greater than NaPi2a and NaPi2c), Cyp27b1, Cyp24a1, and Fgfr1Fgfr4; there was also low level expression of Fgf23 (395). But in Fgf23 null fetal placentas the only significant change was loss of Fgf23 (395). Conversely, in Phex null male placentas, there was significantly increased expression of Cyp24a1 (395, 484) and a threefold increase in Fgf23 (P ⫽ 0.054) (395), whereas all other FGF23 targets showed unchanged expression (395, 484). Placental transport of 32P was assessed using the procedure for intact fetuses. Fgf23 null and Phex null male fetuses showed a normal rate of placental 32P transport at 5 min, confirming that neither absence nor excess of FGF23 affected placental phosphorus transport (395). Therefore, FGF23 does not regulate placental phosphorus transport, despite expression of the relevant FGF23 target genes within the placenta. C. Regulation of Endochondral Bone Formation In Fgf23 null and Phex null male fetuses, endochondral development appears normal the day prior to birth. Tibias showed no alteration in the length or cellular morphology of the cartilaginous or osseous compartments, and a normal distribution of mineral (395). Skeletal ash weight and ash mineral content (calcium, magnesium, phosphorus) were also unchanged in Fgf23 null and Phex null male fetuses compared with their WT siblings (395). D. Role in the Neonate Scant data are available pertaining to the role of FGF23 in phosphorus metabolism of the neonate. In WT mice, FGF23 spikes to 10-fold higher than adult values within 12 h after birth, concurrent with a transient increase in serum phosphorus (34). But this increase appears nonessential because Fgf23 null mice have normal serum calcium and phosphorus at delivery (395) and 1 day after birth (615). Hyperphosphatemia and increased calcitriol are evident in Fgf23 nulls at 10 days after birth, and hypercalcemia by 2 wk (615). Reduced renal phosphorus excretion is present at 3 wk after birth in Fgf23 nulls (the earliest time point studied) (626). Fgf23 nulls are born with normal length and weight but are smaller by 2 wk of age (615). By 3 wk of age, the skeleton is undermineralized and progressive limb deformities develop, but whole body mineral content is increased due to excessive mineralization of soft tissues, including heart, lungs, and kidneys (468, 615, 626, 627). Mortality is 100% by 10 –12 wk (468, 615, 626, 627). Phex null mice are similarly indistinguishable from their WT siblings at birth, and as adults they are commonly identified by low serum phosphorus rather than genotyped. Although serum phosphorus is normal at birth (395), Phex null pups surprisingly develop modestly increased serum phosphorus compared with WT from 2 h until 10 h of age (34). Hypophosphatemia begins at 24 h and is accompanied by increased FGF23, low calcitriol, and high PTH (34). By 72 h, the tibias display slight shortening, normal bone volumes, but fewer osteoblasts and osteoclasts within the trabecular compartment (34). The neonatal findings reveal that absence or excess FGF23 exert powerful effects on mineral metabolism in the neonate, in striking contrast to a lack of effect in the fetus. E. Human Data Intact FGF23 rises from the low levels in cord blood to reach adult values by 5 days after birth (661). Genetic loss of FGF23 action causes familial hyperphosphatemic tumor calcinosis, which commonly presents during the first two decades of life with calcific tumor masses, hyperphosphatemia, elevated calcitriol, and normal or elevated serum calcium (19, 161, 267, 494, 531). It has presented as early as 18 days after birth with a calcific mass and hyperphosphatemia (527, 628) and as late as age 65 (494). Specific causes include missense mutations in FGF23, inactivating mutations in GALNT3 which normally O-glycosylates FGF23 to be prevent its premature cleavage and inactivation, and inactivating mutations in KLOTHO which make FGF23 unable to activate its receptor (19, 161, 267). Hereditary disorders of excessive FGF23 action include Xlinked hypophosphatemic rickets, autosomal dominant hy- Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1195 CHRISTOPHER S. KOVACS pophosphatemic rickets, autosomal recessive hypophosphatemic rickets, and McCune-Albright syndrome (267). All display hypophosphatemia, relatively high FGF23, inappropriately normal or low calcitriol, normal to low PTH, normal calcium, myopathy, and rickets or osteomalacia (19, 161, 267). X-linked hypophosphatemic rickets is the seminal example of excess FGF23 action. It can be diagnosed during the first year when suspected by family history and early onset of hypophosphatemia (452), but in the absence of family history it more often presents at 2 or 3 years of age with bowing of the long bones or growth failure, or at later ages with short stature (102, 172, 588). In four cases in which affected parents prompted an early diagnosis, serum phosphorus was low in all four between 2 and 6 wk of age, tubular reabsorption of phosphorus was low at 9 days of age in one but normal in the others until 6 mo, and radiographic findings of rickets did not develop until 3– 6 mo (452). explain why loss of the estrogen receptor has no obvious effect during fetal development. Deletion of RANKL results in osteopetrotic changes in mice by 2 days after birth, whereas growth and serum calcium, phosphorus, and alkaline phosphatase remain normal until weaning (322). Deletion of RANK similarly leads to a normal appearance at birth. However, the mice are stunted and osteopetrotic by 3 wk of age, tooth eruption fails to occur, and they also develop hypocalcemia, hypophosphatemia, and secondary hyperparathyroidism (370). An opposing phenotype of severe osteoporosis with arterial calcifications occurs in mice lacking OPG (85, 447). Although grossly normal until a month of age, slight trabecular and cortical thinning are evident in femurs and vertebral bodies within 7 days after birth, and fractures of spine and long bones can occur before weaning (85). No studies have been done in fetuses. F. Summary of Key Points The only human data come from case reports, such as a man with an inactivating mutation of the estrogen receptor who had normal birth weight, length, and early development (630). He later presented with a severe form of osteoporosis. FGF23 does not contribute importantly to fetal regulation of serum phosphorus, placental phosphorus transport, or skeletal development and mineralization. Its effects may be curtailed by the limited role that the fetal kidneys play in mineral homeostasis. FGF23 begins to have significant effects within days after birth such that deficiency or excess of FGF23 alter serum phosphorus and calcitriol, although clinical manifestations may not appear until years later. XI. SEX STEROIDS A. Animal Data Mice lacking estrogen receptor alpha or beta, or the aromatase, have normal skeletal lengths at birth and do not develop altered skeletal metabolism until several weeks after birth (125, 391, 462, 711, 738). At 3 wk of age (the first postnatal measurement) in male estrogen receptor knockout mice, there was no difference in total weight, crownrump length, femur length, and bone density at multiple skeletal sites (711). Therefore, it is likely that no difference is present at the end of fetal development and that mineral metabolism is undisturbed through at least 3 wk of age. However, no fetal studies have been done. Estradiol exerts its effects on osteoblasts in part by downregulating the expression of receptor activator of nuclear factor kappa-B ligand (RANKL) and upregulating the expression of its decoy receptor osteoprotegerin (OPG). The RANKL-OPG-RANK system plays a critical role in regulating the formation, recruitment, and function of osteoclasts within the adult. However, it may be relatively unimportant from the perspective of the fetal skeleton and regulation of mineral homeostasis, and in turn, this may 1196 B. Human Data C. Summary of Key Points Overall, although estradiol and testosterone play important roles in regulating calcium and bone metabolism in the adult, in part through regulating RANKL and OPG, the available evidence is compatible with these hormones (and the RANKL-OPG system) being unimportant for mineral and bone metabolism in the fetus and neonate. Detailed study of bone and mineral metabolism in these models during fetal development is needed. XII. FETAL AND NEONATAL RESPONSES TO MATERNAL MINERAL DISTURBANCES A. Primary Hyperparathyroidism in the Mother 1. Animal data Maternal hypercalcemia adversely affects fetal parathyroid function. An acute infusion of calcium raised the serum calcium of pregnant ewes from 2.25 to 3.75 mM, and this in turn caused an increase in the fetal serum calcium from 3.00 to 3.5 mM and a ⬎50% fall in the fetal PTH level (295). In contrast, exogenous PTH administration to the mother failed to raise the fetal serum calcium level (295), consistent with the failure of PTH to be transported across the placenta. These results suggest that maternal hypercalcemia Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM will increase the fetal blood calcium and, in turn, suppress the fetal parathyroids. Furthermore, hypercalcemia within the fetal circulation did not suppress active transport of calcium across the perfused guinea pig placenta (433). The lack of a compensatory decrease in placental calcium transport means that the effect of maternal hypercalcemia can be magnified to cause fetal hypercalcemia and suppression of fetal parathyroid function. 2. Human data Maternal hypercalcemia likely increases the flow of calcium across the placenta, and in turn it may lead to a higher than normal cord blood calcium (610). Importantly, maternal hypercalcemia suppresses the fetal parathyroids and can cause significant fetal morbidity and mortality in up to 80% of cases (597). These outcomes include stillbirth, miscarriage, and neonatal tetany. Comparison of older to more recent case series suggest that these outcomes have improved (609); however, a recent sobering report found that fetal death occurred during the second or third trimester in 30 of 62 medically managed cases, representing a 3.5-fold increased risk of fetal mortality compared with surgically treated cases (477). After experiencing higher than normal calcium levels in utero, the neonatal calcium level may decline more slowly after birth (610). Failure of the neonate’s parathyroids to upregulate promptly after birth may precipitate hypocalcemia and tetany. In several case series 50% of neonates had tetany or other complications of maternal hypercalcemia, while 25–30% of them died (149, 304, 393, 718). Hypoparathyroidism normally resolves by 3–5 mo after birth (609), but permanent hypoparathyroidism has also occurred (79, 393, 609). Some cases have had delayed onset or recognition until several weeks or months after birth (282, 469, 688). A common clinical impression is that mild maternal hypercalcemia during pregnancy may be unlikely to cause fetal or neonatal problems. However, neonatal hypocalcemia and tetany have occurred after seemingly mild cases of primary hyperparathyroidism (522). A twin pregnancy complicated by maternal hypercalcemia provides a seminal example of the variability: one neonate remained normocalcemic while the other had hypocalcemic seizures (431). B. Familial Hypocalciuric Hypercalcemia in the Mother 1. Animal data Study of Casr⫹/⫺ mice, the murine equivalent of FHH, has shown that fetal PTH is suppressed compared with the PTH values obtained in fetuses from WT mothers (338). 2. Human data Despite the seemingly benign nature of hypercalcemia in adults with FHH, maternal hypercalcemia during pregnancy has caused suppression of the fetal parathyroids, and subsequent neonatal hypocalcemia and tetany (529, 673, 674). This includes the heterozygous neonate who may still develop symptomatic hypocalcemia and tetany, before eventually becoming hypercalcemic when the parathyroids gain full function and achieve the higher calcium level set by the mutated CaSR (672). C. Hypoparathyroidism and Activating Mutations of the Calcium-Sensing Receptor in the Mother 1. Animal data Maternal hypocalcemia compromises the supply of mineral available to the fetus. Thyroparathyroidectomy has been used in rats and ewes to mimic the effects of maternal hypocalcemia due to hypoparathyroidism during pregnancy. These fetal rats (67, 276) and lambs (564) had normal serum calcium levels despite severe maternal hypocalcemia, although the serum calcium declined during the last several days of gestation in other studies of parathyroidectomized rats (106, 208, 220). A normal serum calcium likely requires upregulation of placental calcium transfer to maintain it, and this is supported by the observation that an acute calcium infusion caused a marked rise in serum calcium of fetuses from thyroparathyroidectomized rats, but not in fetuses from thyroidectomized rats (106). The fetal skeleton may also be compromised by resorbing itself to maintain the normal high level of calcium in fetal blood, as seen by enlargement of the fetal parathyroids (206, 625), increased resorption in fetal rat bones that requires intact fetal parathyroids for it to occur (542), and reduced femur length and mineral ash content (104). But even with overt hypocalcemia throughout pregnancy, the neonate can have a perfectly normal skeleton (217). 2. Human data When maternal hypocalcemia during pregnancy has been prolonged and severe, fetal hyperparathyroidism has developed, in turn causing a severely demineralized skeleton, and fractures in utero or during birth (11, 76, 389, 586, 652). Spontaneous abortion, stillbirth, and neonatal death have also occurred (25, 170, 302). The cord blood calcium is variably normal, low, or even increased, whereas the fetal parathyroids may be enlarged and hyperplastic. Although this is secondary hyperparathyroidism in the fetus, the function remains autonomous for a time, such that even if the fetus is normal at birth, hypercalcemia and skeletal demineralization can develop in the neonate before the parathyroid function subsides to nor- Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1197 CHRISTOPHER S. KOVACS mal (672). Avoiding the magnitude of hypocalcemia that can occur with hypoparathyroidism should prevent these outcomes during pregnancy. The same issues apply to activating mutations of the CaSR in the mother. In two pregnancies of an affected woman whose serum calcium was below normal despite supplemental calcium and calcitriol, her babies (who did not inherit the mutation) had low-normal serum calcium and elevated PTH at birth (492). This suggests that some secondary hyperparathyroidism had developed in utero. But in another report of three pregnancies complicated by maternal hypocalcemia, the babies were described as normal at birth, although no cord blood calcium or PTH values were provided (13). Maintaining the maternal calcium concentration in or near the normal range prevents the development of fetal hyperparathyroidism (217). D. Pseudohypoparathyroidism in the Mother 1. Animal data No animal models have been studied during pregnancy. 2. Human data In women, hypocalcemia due to maternal pseudohypoparathyroidism during pregnancy can lead to the same fetal outcomes that have been associated with maternal hypoparathyroidism, including parathyroid hyperplasia, skeletal demineralization, and fractures (226, 710). Postnatally, the relatively autonomous parathyroid function can cause neonatal hypercalcemia before the parathyroids involute. Maintaining normocalcemia in the mother during pregnancy should prevent these complications. E. Vitamin D-Related Disorders in the Mother 1. Animal data Vitamin D deficiency in the mother will cause low levels of 25-hydroxvitamin D in the fetus, and the consequences of this have already been addressed in section VIII. The fetuses show normal serum minerals, PTH, and skeletal lengths and mineral content. Despite marked hypocalcemia in VdrBos null mice during pregnancy, the only abnormality that their fetuses have shown is a somewhat smaller size compared with their counterparts from VdrBos⫹/⫺ mothers, with a normal ash weight and mineral content after adjustment for their smaller size (342). Serum calcium, phosphorus, and PTH are all normal (342). Similarly hypocalcemic 1␣-hydroxy- 1198 lase null pigs bore fetuses that had normal serum mineral concentrations (352, 353). These results indicate that the fetus will be able to maintain normal serum mineral concentrations and skeletal development despite maternal hypocalcemia. The benign outcome differs from the fetal hyperparathyroidism that can develop in response to maternal hypoparathyroidism, and this is likely because maternal hypocalcemia is more modest with disorders of vitamin D physiology because secondary hyperparathyroidism offsets the fall in serum calcium. With a more modest decline in maternal serum calcium, the fetus is less likely to be adversely affected. 2. Human data The potential effect of maternal vitamin D deficiency during pregnancy has been addressed in detail in section VIII. Pregnancies have been uneventful in women who lack 1␣hydroxylase (VDDR-I) (229). Treatment with calcium and physiological doses of calcitriol or 1␣-cholecalciferol have been adjusted as needed to maintain a near-normal ionized or albumin-corrected serum calcium. Pregnancy has also been uneventful in women who lack functional vitamin D receptors (VDDR-II) (408). Depending on the nature of the mutation, there may be a partial response to calcitriol or cholecalciferol or complete loss of calcitriol-induced effects. The calcium content of the diet may need to be increased to maintain a near-normal ionized or albumin-corrected serum calcium. If maternal hypercalcemia occurs from excess consumption of calcitriol, 1␣-cholecalciferol, or vitamin D (hypervitaminosis D), then fetal parathyroid suppression and neonatal hypocalcemia and hypoparathyroidism can result. This effect is mediated by the high maternal calcium concentration, and not directly by the vitamin D metabolites. If women consume modestly high doses of vitamin D during pregnancy but remain normocalcemic, the neonate is unlikely to be adversely affected based on available clinical data. This was shown by a randomized clinical trial that administered 400, 2,000 or 4,000 IU of vitamin D daily to women during the second half of pregnancy (262). No maternal or fetal hypercalcemia was reported during the study. Maternal 25-hydroxyvitamin D at the end of pregnancy ranged from 79 nM in the low-dose group to 111 nM in the highest dose group, while fetal 25-hydroxyvitamin D values in the corresponding offspring were 46 and 66 nM (262). Intense placental activity of 24-hydroxylase causes preferential catabolism of 25-hydroxyvitamin D to 24,25-dihydroxyvitamin D (as described earlier in sect. IIB2), and likely explains why the fetal rise in 25-hydroxyvitamin D was more modest than in the mothers. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM F. Maternal Magnesium Infusions (Tocolytic Therapy) Intravenous magnesium is used in pregnant women to inhibit uterine contractions of premature labor and to prevent seizures from eclampsia. The infusion causes fetal hypermagnesemia, suppressed PTH and parathyroid responsiveness, and variable effects on the total and ionized calcium of neonates (130, 159). Prolonged exposure has also caused defective ossification of bone and enamel in the teeth (358), and abnormal mineralization within the metaphyses of long bones (132, 402, 587). Severe hypermagnesemia (⬎7 mg/ dl) is more likely to cause hypotonia, respiratory depression, and bone abnormalities (159, 379, 402, 591). G. Hypercalcemia of Malignancy in the Mother Maternal hypercalcemia in these cases is more extreme and likely to suppress fetal parathyroid function, thereby provoking neonatal hypocalcemia, tetany, and possibly permanent hypoparathyroidism. Hypercalcemia may be present in cord blood and the first few postnatal samples (131, 278, 700), but the calcium will fall and may reach severely hypocalcemic values with attendant risk of respiratory distress, tetany, and seizures (9, 454). One of four babies died where the outcome was reported (454). H. Pseudohyperparathyroidism in the Mother Excess maternal production of PTHrP causes pseudohyperparathyroidism, a less common cause of maternal hypercalcemia during pregnancy. It can occur as a result of excess mammary production of PTHrP and present with hypercalcemia during late pregnancy or in the puerperium (283, 307, 590); it has also occurred in nonpregnant women with large breasts (418). Placental PTHrP can also cause pseudohyperparathyroidism, as shown by a pregnant woman who presented with severe hypercalcemia (5.25 mM), undetectable PTH, and a serum PTHrP of 21 pM (173). Six hours after an urgent C-section, she was profoundly hypocalcemic with undetectable PTHrP and elevated PTH. Her breasts were of unremarkable size and she did not breast feed. The rapid change in her status after delivery is most consistent with the placenta being the source of PTHrP (173). In all cases of pseudohyperparathyroidism, hypercalcemia may be present in cord blood (590), and in turn maternal hypercalcemia increases the risk of neonatal hypocalcemia and hypoparathyroidism. I. Maternal Diabetes Causing Fetal and Neonatal Hypoparathyroidism Poorly controlled maternal diabetes during pregnancy can cause hypocalcemia, seizures, and tetany within the first FIGURE 12. Relative roles of PTH, PTHrP, and calcitriol during fetal and neonatal life. The placenta is the main source of mineral during fetal life. PTH and PTHrP are expressed within the placenta but may also act on it from systemic sources to stimulate calcium transfer. The intestines are a trivial source of mineral in the fetus but are the main source for the neonate. Intestinal calcium absorption is initially passive but later becomes active, saturable, and calcitriol-dependent in the infant. Within the endochondral skeleton, PTHrP is produced by proliferating chondrocytes and perichondrial cells (arrowheads) and delays terminal differentiation of prehypertrophic chondrocytes. PTHrP is also produced within preosteoblasts and osteoblasts and stimulates bone formation (semicircular arrows). During fetal life PTHrP and PTH act together to maintain high blood calcium and phosphorus levels to facilitate mineralization; loss of either PTHrP or PTH causes hypocalcemia and hyperphosphatemia. Calcitriol is not required to regulate blood calcium, endochondral bone formation, or skeletal mineralization in the fetus. [From Kovacs (329), with permission from Elsevier.] Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1199 CHRISTOPHER S. KOVACS 24 –72 h after birth. Hyperphosphatemia is commonly present, indicating that transient neonatal hypoparathyroidism is contributing to the hypocalcemia. The mechanism for this is unknown. One case series found high ionized and total serum calcium levels in cord blood of infants of diabetic mothers (684), which could account for suppression of the fetal parathyroids. But why maternal diabetes should cause fetal hypercalcemia is unclear, unless some aspect of the maternal diabetic state leads to increased maternal-fetal flow of calcium. Another possibility is that glucosuria caused by uncontrolled diabetes during pregnancy may cause renal wasting of magnesium and maternal hypomagnesemia, which in turn could cause fetal hypomagnesemia and parathyroid suppression. However, a small clinical trial found no effect of postnatal magnesium supplementation on the incidence of neonatal hypocalcemia in infants of diabetic mothers (434). Other factors that contribute to hypocalcemia in babies born of diabetic mothers are associated increased risks of preterm birth, lung immaturity, and asphyxia. XIII. CONCLUSIONS The fetal and neonatal skeletons require adequate mineral delivery to develop and mineralize normally (FIGURE 12). Placental mineral transfer requires PTHrP and possibly PTH, but not calcitriol, calcitonin, or FGF23. PTH and PTHrP work together to maintain serum calcium and phosphorus at high levels in utero for optimal bone mineralization, while calcitriol, calcitonin, and FGF23 are not required. PTH and PTHrP are required to regulate endochondral bone development during gestation, whereas calcitriol, calcitonin, FGF23, and the sex steroids are not. During the neonatal period, intestinal calcium absorption becomes dependent on vitamin D/calcitriol. This means that skeletal development and mineralization in turn become dependent on vitamin D/calcitriol as well. However, this is not an absolute requirement because the role of calcitriol can be bypassed by increasing the calcium content of the diet or by administering parenteral calcium. ACKNOWLEDGMENTS Address for reprint requests and other correspondence: C. Kovacs, Health Sciences Centre, 300 Prince Philip Dr., St. John’s, NL A1B 3V6, Canada (e-mail: ckovacs@mun.ca). GRANTS This work was supported by Canadian Institutes of Health Research Grants 133413, 126469, and 84253 as well as by Research & Development Corporation of Newfoundland and Labrador Grant 5404.1145.102. 1200 DISCLOSURES No conflicts of interest, financial or otherwise, are declared by the author. REFERENCES 1. Aaron JE, Abbas SK, Colwell A, Eastell R, Oakley BA, Russell RG, Care AD. Parathyroid gland hormones in the skeletal development of the ovine foetus: the effect of parathyroidectomy with calcium and phosphate infusion. Bone Miner 16: 121–129, 1992. 2. Aaron JE, Makins NB, Caple IW, Abbas SK, Pickard DW, Care AD. The parathyroid glands in the skeletal development of the ovine foetus. Bone Miner 7: 13–22, 1989. 3. Abbas SK, Caple IW, Care AD, Loveridge N, Martin TJ, Pickard DW, Rodd C. The role of the parathyroid glands in fetal calcium homeostasis in the sheep (Abstract). J Physiol 386: 27P, 1987. 4. Abbas SK, Care AD, Van Baelen H, Bouillon R. Plasma vitamin D-binding protein and free 1,25-dihydroxyvitamin D3 index in pregnant ewes and their fetuses in the last month of gestation. J Endocrinol 115: 7–12, 1987. 5. Abbas SK, Pickard DW, Illingworth D, Storer J, Purdie DW, Moniz C, Dixit M, Caple IW, Ebeling PR, Rodda CP. Measurement of parathyroid hormone-related protein in extracts of fetal parathyroid glands and placental membranes. J Endocrinol 124: 319 – 325, 1990. 6. Abbas SK, Pickard DW, Rodda CP, Heath JA, Hammonds RG, Wood WI, Caple IW, Martin TJ, Care AD. Stimulation of ovine placental calcium transport by purified natural and recombinant parathyroid hormone-related protein (PTHrP) preparations. Q J Exp Physiol 74: 549 –552, 1989. 7. Abbas SK, Ratcliffe WA, Moniz C, Dixit M, Caple IW, Silver M, Fowden A, Care AD. The role of parathyroid hormone-related protein in calcium homeostasis in the fetal pig. Exp Physiol 79: 527–536, 1994. 8. Abele H, Starz S, Hoopmann M, Yazdi B, Rall K, Kagan KO. Idiopathic polyhydramnios and postnatal abnormalities. Fetal Diagnosis Therapy 32: 251–255, 2012. 9. Abraham P, Ralston SH, Hewison M, Fraser WD, Bevan JS. Presentation of a PTHrPsecreting pancreatic neuroendocrine tumour, with hypercalcaemic crisis, pre-eclampsia, and renal failure. Postgrad Med J 78: 752–753, 2002. 10. Abrams SA. Calcium and vitamin D requirements of enterally fed preterm infants. Pediatrics 131: e1676 –1683, 2013. 11. Aceto T Jr, Batt RE, Bruck E, Schultz RB, Perz YR. Intrauterine hyperparathyroidism: a complication of untreated maternal hypoparathyroidism. J Clin Endocrinol Metab 26: 487– 492, 1966. 12. Adachi M, Tachibana K, Masuno M, Makita Y, Maesaka H, Okada T, Hizukuri K, Imaizumi K, Kuroki Y, Kurahashi H, Suwa S. Clinical characteristics of children with hypoparathyroidism due to 22q11.2 microdeletion. Eur J Pediatr 157: 34 –38, 1998. 13. Adeniyi JO, Esen U, Parr J. Pregnancy outcome in women with autosomal dominant hypocalcaemic hypercalciuric nephrocalcinosis. J Matern Fetal Neonatal Med. In press. 14. Adeyemo O, Jeyakumar H. Plasma progesterone, estradiol-17 beta and testosterone in maternal and cord blood, and maternal human chorionic gonadotropin at parturition. African J Med Medical Sci 22: 55– 60, 1993. 15. Adlercreutz H, Luukkainen T. Identification and determination of oestrogens in various biological materials in pregnancy. Ann Clin Res 2: 365–380, 1970. 16. Akawi NA, Ali BR, Al-Gazali L. Stuve-Wiedemann syndrome and related bent bone dysplasias. Clin Genet 82: 12–21, 2012. 17. Al-Mustafa ZH, Al-Madan M, Al-Majid HJ, Al-Muslem S, Al-Ateeq S, Al-Ali AK. Vitamin D deficiency and rickets in the Eastern Province of Saudi Arabia. Ann Trop Paediatr 27: 63– 67, 2007. 18. Alexander BM, Singh P, Austin KJ, Cockrum RR, Cammack KM, Hess BW, Moss GE, Nathanielsz PW, Ford SP. Effect of maternal fatness on fetal steroids and semiquantitative real-time PCR expression of receptor genes in sheep. Anim Reprod Sci 116: 58 – 64, 2009. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM 19. Alizadeh Naderi AS, Reilly RF. Hereditary disorders of renal phosphate wasting. Nat Rev Nephrol 6: 657– 665, 2010. 20. Allgrove J, Adami S, Manning RM, O’Riordan JL. Cytochemical bioassay of parathyroid hormone in maternal and cord blood. Arch Dis Child 60: 110 –115, 1985. 21. Allgrove J, Manning RM, Adami S, Chayen J, O’Riordan JL. Biologically active parathyroid hormone in foetal and maternal plasma (Abstract). Clin Sci 60: 11P, 1981. 22. Alumets J, Hakanson R, Lundqvist G, Sundler F, Thorell J. Ontogeny and ultrastructure of somatostatin and calcitonin cells in the thyroid gland of the rat. Cell Tissue Res 206: 193–201, 1980. 23. Amizuka N, Karaplis AC, Henderson JE, Warshawsky H, Lipman ML, Matsuki Y, Ejiri S, Tanaka M, Izumi N, Ozawa H, Goltzman D. Haploinsufficiency of parathyroid hormone-related peptide (PTHrP) results in abnormal postnatal bone development. Dev Biol 175: 166 –176, 1996. 24. Amling M, Priemel M, Holzmann T, Chapin K, Rueger JM, Baron R, Demay MB. Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology 140: 4982– 4987, 1999. 25. Anderson GW, Musselman L. The treatment of tetany in pregnancy. Am J Obstet Gynecol 43: 547–567, 1942. 26. Angeletti RH, Mints L, Aber C, Russell J. Determination of residues in chromogranin A-(16 – 40) required for inhibition of parathyroid hormone secretion. Endocrinology 137: 2918 –2922, 1996. 27. Arundel P, Offiah A, Bishop NJ. Evolution of the radiographic appearance of the metaphyses over the first year of life in type V osteogenesis imperfecta: clues to pathogenesis. J Bone Miner Res 26: 894 – 898, 2011. 28. Asa SL, Henderson J, Goltzman D, Drucker DJ. Parathyroid hormone-like peptide in normal and neoplastic human endocrine tissues. J Clin Endocrinol Metab 71: 1112– 1118, 1990. 40. Barlet JP, Davicco MJ, Coxam V. Calcitonin modulates parathyroid hormone related peptide-stimulated calcium placental transfer. In: Calcium Regulating Hormones and Bone Metabolism: Basic and Clinical Aspects, edited by Cohn DV, Gennari C, and Tashjian AHJ. Amsterdam: Elsevier, 1992, p. 124 –128. 41. Barlet JP, Davicco MJ, Lefaivre J, Garel JM. Endocrine regulation of plasma phosphate in sheep fetuses with catheters implanted in utero. In: Homeostasis of Phosphate and Other Minerals, edited by Massry SG, Ritz E, and Rapado A. New York: Plenum, 1978, p. 243–256. 42. Barlet JP, Davicco MJ, Moncaup M, Lefaivre J. Foetal plasma magnesium levels during maternal hypo- or hypermagnesaemia in ewes. Br J Nutr 42: 559 –566, 1979. 43. Barlet JP, Davicco MJ, Rouffet J, Coxam V, Lefaivre J. Short communication: parathyroid hormone-related peptide does not stimulate phosphate placental transport. Placenta 15: 441– 444, 1994. 44. Barltrop D, Mole RH, Sutton A. Absorption and endogenous faecal excretion of calcium by low birthweight infants on feeds with varying contents of calcium and phosphate. Arch Dis Child 52: 41– 49, 1977. 45. Barltrop D, Oppe TE. Absorption of fat and calcium by low birthweight infants from milks containing butterfat and olive oil. Arch Dis Child 48: 496 –501, 1973. 46. Barltrop D, Oppe TE. Calcium and fat absorption by low birthweight infants from a calcium-supplemented milk formula. Arch Dis Child 48: 580 –582, 1973. 47. Barri M, Abbas SK, Pickard DW, Hammonds RG, Wood WI, Caple IW, Martin TJ, Care AD. Fetal magnesium homeostasis in the sheep. Exp Physiol 75: 681– 688, 1990. 48. Beamer WG, Donahue LR, Rosen CJ, Baylink DJ. Genetic variability in adult bone density among inbred strains of mice. Bone 18: 397– 403, 1996. 49. Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, Jensen TK. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol 160: 491– 497, 2009. 29. Aslan AR, Kogan BA, Gondos B. Testicular development. In: Fetal and Neonatal Physiology (3rd ed.), edited by Polin RA, Fox WW, and Abman SH. Philadelphia, PA: Saunders, 2004, p. 1950 –1955. 50. Begum R, Coutinho ML, Dormandy TL, Yudkin S. Maternal malabsorption presenting as congenital rickets. Lancet 1: 1048 –1052, 1968. 30. Avila E, Diaz L, Halhali A, Larrea F. Regulation of 25-hydroxyvitamin D3 1alphahydroxylase, 1,25-dihydroxyvitamin D3 24-hydroxylase and vitamin D receptor gene expression by 8-bromo cyclic AMP in cultured human syncytiotrophoblast cells. J Steroid Biochem Mol Biol 89 –90: 115–119, 2004. 51. Benn BS, Ajibade D, Porta A, Dhawan P, Hediger M, Peng JB, Jiang Y, Oh GT, Jeung EB, Lieben L, Bouillon R, Carmeliet G, Christakos S. Active intestinal calcium transport in the absence of transient receptor potential vanilloid type 6 and calbindin-D9k. Endocrinology 149: 3196 –3205, 2008. 31. Bagnoli F, Bruchi S, Garosi G, Pecciarini L, Bracci R. Relationship between mode of delivery and neonatal calcium homeostasis. Eur J Pediatr 149: 800 – 803, 1990. 52. Bergwitz C, Juppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med 61: 91–104, 2010. 32. Bagnoli F, Bruchi S, Sardelli S, Buonocore G, Vispi L, Franchi F, Bracci R. Calcium homeostasis in the first days of life in relation to feeding. Eur J Pediatr 144: 41– 44, 1985. 53. Bertelloni S, Baroncelli GI, Pelletti A, Battini R, Saggese G. Parathyroid hormonerelated protein in healthy pregnant women. Calcif Tissue Int 54: 195–197, 1994. 33. Bai M, Pearce SH, Kifor O, Trivedi S, Stauffer UG, Thakker RV, Brown EM, Steinmann B. In vivo and in vitro characterization of neonatal hyperparathyroidism resulting from a de novo, heterozygous mutation in the Ca2⫹-sensing receptor gene: normal maternal calcium homeostasis as a cause of secondary hyperparathyroidism in familial benign hypocalciuric hypercalcemia. J Clin Invest 99: 88 –96, 1997. 34. Bai X, Miao D, Goltzman D, Karaplis AC. Early lethality in Hyp mice with targeted deletion of Pth gene. Endocrinology 148: 4974 – 4983, 2007. 35. Balabanova S, Kruse B, Wolf AS. Calcitonin secretion by human placental tissue. Acta Obstet Gynecol Scand 66: 323–326, 1987. 36. Balsan S, Garabedian M, Larchet M, Gorski AM, Cournot G, Tau C, Bourdeau A, Silve C, Ricour C. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest 77: 1661–1667, 1986. 37. Bansal VK. Serum inorganic phosphorus. In: Clinical Methods: The History, Physical, and Laboratory Examinations, edited by Walker HK, Hall WD, and Hurst JW. Boston: Butterworths, 1990. 38. Barlet JP. Calcitonin may modulate placental transfer of calcium in ewes. J Endocrinol 104: 17–21, 1985. 39. Barlet JP. Prolactin and calcium metabolism in pregnant ewes. J Endocrinol 107: 171– 175, 1985. 54. Better OS, Levi J, Grief E, Tuma S, Gellei B, Erlik D. Prolonged neonatal parathyroid suppression. A sequel to asymptomatic maternal hyperparathyroidism. Arch Surg 106: 722–724, 1973. 55. Bille BS. Non-rachitic craniotabes. Acta Paediatr 44: 185–202, 1955. 56. Blackburn CC, Manley NR. Developing a new paradigm for thymus organogenesis. Nat Rev Immunol 4: 278 –289, 2004. 57. Blair JW, Carachi R. Neonatal primary hyperparathyroidism: a case report and review of the literature. Eur J Pediatr Surg 1: 110 –114, 1991. 58. Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium ⫻ phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 31: 607– 617, 1998. 59. Blond MH, Gold F, Pierre F, Berger C, Guerois M, Queru MS, Ramponi N. Nutritional fetal rickets. A case report. J Gynecol Obstet Biol Reprod 26: 834 – 836, 1997. 60. Bogden JD, Thind IS, Kemp FW, Caterini H. Plasma concentrations of calcium, chromium, copper, iron, magnesium, and zinc in maternal and cord blood and their relationship to low birth weight. J Lab Clin Med 92: 455– 462, 1978. 61. Bond H, Baker B, Boyd RD, Cowley E, Glazier JD, Jones CJ, Sibley CP, Ward BS, Husain SM. Artificial perfusion of the fetal circulation of the in situ mouse placenta: methodology and validation. Placenta 27 Suppl A: S69 –75, 2006. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1201 CHRISTOPHER S. KOVACS 62. Bond H, Dilworth MR, Baker B, Cowley E, Requena Jimenez A, Boyd RD, Husain SM, Ward BS, Sibley CP, Glazier JD. Increased maternofetal calcium flux in parathyroid hormone-related protein-null mice. J Physiol 586: 2015–2025, 2008. 83. Bruns ME, Kleeman E, Mills SE, Bruns DE, Herr JC. Immunochemical localization of vitamin D-dependent calcium-binding protein in mouse placenta and yolk sac. Anat Rec 213: 514 –517, 1985. 63. Borke JL, Caride A, Verma AK, Kelley LK, Smith CH, Penniston JT, Kumar R. Calcium pump epitopes in placental trophoblast basal plasma membranes. Am J Physiol Cell Physiol 257: C341–C346, 1989. 84. Bruns ME, Wallshein V, Bruns DE. Regulation of calcium-binding protein in mouse placenta and intestine. Am J Physiol Endocrinol Metab 242: E47–E52, 1982. 64. Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C, Demay M. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev 29: 726 –776, 2008. 65. Bouillon R, Lieben L, Mathieu C, Verstuyf A, Carmeliet G. Vitamin D action: lessons from VDR and Cyp27b1 null mice. Pediatr Endocrinol Rev 10 Suppl 2: 354 –366, 2013. 66. Bouillon R, Verstuyf A, Mathieu C, Van Cromphaut S, Masuyama R, Dehaes P, Carmeliet G. Vitamin D resistance. Best Pract Res Clin Endocrinol Metab 20: 627– 645, 2006. 67. Bourdeau A, Manganella G, Thil-Trubert CL, Sachs C, Cournot G. Bioactive parathyroid hormone in pregnant rats and fetuses. Am J Physiol Endocrinol Metab 258: E549 – E554, 1990. 68. Bowden SJ, Emly JF, Hughes SV, Powell G, Ahmed A, Whittle MJ, Ratcliffe JG, Ratcliffe WA. Parathyroid hormone-related protein in human term placenta and membranes. J Endocrinol 142: 217–224, 1994. 69. Bradbury RA, Sunn KL, Crossley M, Bai M, Brown EM, Delbridge L, Conigrave AD. Expression of the parathyroid Ca2⫹-sensing receptor in cytotrophoblasts from human term placenta. J Endocrinol 156: 425– 430, 1998. 70. Brandi ML. Genetics of hypoparathyroidism and pseudohypoparathyroidism. J Endocrinol Invest 34: 27–34, 2011. 71. Brauner R, Le Harivel de Gonneville A, Kindermans C, Le Bidois J, Prieur M, Lyonnet S, Souberbielle JC. Parathyroid function and growth in 22q11.2 deletion syndrome. J Pediatr 142: 504 –508, 2003. 72. Briana DD, Boutsikou M, Baka S, Hassiakos D, Gourgiotis D, Marmarinos A, Iacovidou N, Malamitsi-Puchner A. N-terminal parathyroid hormone-related protein levels in human intrauterine growth restricted pregnancies. Acta Obstet Gynecol Scand 86: 945–949, 2007. 73. Brickley M, Ives R. Skeletal manifestations of infantile scurvy. Am J Physical Anthropol 129: 163–172, 2006. 74. Brommage R, DeLuca HF. Placental transport of calcium and phosphorus is not regulated by vitamin D. Am J Physiol Renal Fluid Electrolyte Physiol 246: F526 –F529, 1984. 75. Bronner F, Salle BL, Putet G, Rigo J, Senterre J. Net calcium absorption in premature infants: results of 103 metabolic balance studies. Am J Clin Nutr 56: 1037–1044, 1992. 76. Bronsky D, Kiamko RT, Moncada R, Rosenthal IM. Intra-uterine hyperparathyroidism secondary to maternal hypoparathyroidism. Pediatrics 42: 606 – 613, 1968. 77. Brooke OG, Brown IR, Bone CD, Carter ND, Cleeve HJ, Maxwell JD, Robinson VP, Winder SM. Vitamin D supplements in pregnant Asian women: effects on calcium status and fetal growth. Br Med J 280: 751–754, 1980. 85. Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12: 1260 –1268, 1998. 86. Buchowski MS, Miller DD. Lactose, calcium source and age affect calcium bioavailability in rats. J Nutr 121: 1746 –1754, 1991. 87. Burnette JC, Simpson DM, Chandler DC Jr, Bawden JW. Fetal blood calcium response to maternal parathyroid and vitamin D administration in guinea pigs. J Dent Res 47: 444 – 446, 1968. 88. Burns C, Powell BR, Hsia YE, Reinker K. Dyggve-Melchior-Clausen syndrome: report of seven patients with the Smith-McCort variant and review of the literature. J Pediatr Orthop 23: 88 –93, 2003. 89. Burton DW, Brandt DW, Deftos LJ. Parathyroid hormone-related protein in the cardiovascular system. Endocrinology 135: 253–261, 1994. 90. Butte NF, Wong WW, Hopkinson JM, Smith EO, Ellis KJ. Infant feeding mode affects early growth and body composition. Pediatrics 106: 1355–1366, 2000. 91. Byskov AG, Westergaard LG. Differentiation of the ovary. In: Fetal and Neonatal Physiology (3rd ed.), edited by Polin RA, Fox WW, and Abman SH. Philadelphia, PA: Saunders, 2004, p. 1941–1949. 92. Campbell DE, Fleischman AR. Rickets of prematurity: controversies in causation and prevention. Clin Perinatol 15: 879 – 890, 1988. 93. Caple IW, Heath JA, Care AD, Heaton C, Farrugia W, Wark JD. The regulation of osteoblast activity in fetal lambs by placental calcium transport and the fetal parathyroid glands. In: Fetal and Neonatal Development, edited by Jones CT. Ithaca, NY: Perinatal Press, 1988, p. 90 –93. 94. Care AD. Development of endocrine pathways in the regulation of calcium homeostasis. Baillieres Clin Endocrinol Metab 3: 671– 688, 1989. 95. Care AD. The placental transfer of calcium. J Dev Physiol 15: 253–257, 1991. 96. Care AD, Abbas SK, Pickard DW, Barri M, Drinkhill M, Findlay JB, White IR, Caple IW. Stimulation of ovine placental transport of calcium and magnesium by mid-molecule fragments of human parathyroid hormone-related protein. Exp Physiol 75: 605– 608, 1990. 97. Care AD, Caple IW, Abbas SK, Pickard DW. The effect of fetal thyroparathyroidectomy on the transport of calcium across the ovine placenta to the fetus. Placenta 7: 417– 424, 1986. 98. Care AD, Caple IW, Singh R, Peddie M. Studies on calcium homeostasis in the fetal Yucatan miniature pig. Lab Anim Sci 36: 389 –392, 1986. 99. Care AD, Dutton A, Mott JC, Robinson JS, Ross R. Studies of the transplacental calcium gradient in sheep (Abstract). J Physiol 290: 19P–20P, 1979. 78. Brooke OG, Butters F, Wood C. Intrauterine vitamin D nutrition and postnatal growth in Asian infants. Br Med J 283: 1024, 1981. 100. Care AD, Pickard DW, Weatherley A, Appleby D. The measurement of transplacental magnesium fluxes in the sheep. Res Vet Sci 27: 121–122, 1979. 79. Bruce J, Strong JA. Maternal hyperparathyroidism and parathyroid deficiency in the child, with account of effect of parathyroidectomy on renal function, and of attempt to transplant part of tumor. Q J Med 24: 307–319, 1955. 101. Care AD, Ross R, Pickard DW, Weatherley AJ, Garel JM, Manning RM, Allgrove J, Papapoulos S, O’Riordan JL. Calcium homeostasis in the fetal pig. J Dev Physiol 4: 85–106, 1982. 80. Bruns ME, Bruns DE, Avioli L. Vitamin D-dependent calcium-binding protein of rat intestine: changes during postnatal development and sensitivity to 1,25-dihydroxycholecalciferol. Endocrinology 105: 934 –938, 1979. 102. Carpenter TO. The expanding family of hypophosphatemic syndromes. J Bone Miner Metab 30: 1–9, 2012. 81. Bruns ME, Fausto A, Avioli LV. Placental calcium binding protein in rats. Apparent identity with vitamin D-dependent calcium binding protein from rat intestine. J Biol Chem 253: 3186 –3190, 1978. 82. Bruns ME, Ferguson IIJE, Bruns DE, Burton DW, Brandt DW, Juppner H, Segre GV, Deftos LJ. Expression of parathyroid hormone-related peptide and its receptor messenger ribonucleic acid in human amnion and chorion-decidua: implications for secretion and function. Am J Obstet Gynecol 173: 739 –746, 1995. 1202 103. Chalon S, Garel JM. 1,25-Dihydroxyvitamin D3 injections into rat fetuses: effects on fetal plasma calcium, plasma phosphate and mineral content. Reprod Nutr Dev 23: 567–573, 1983. 104. Chalon S, Garel JM. Plasma calcium control in the rat fetus. I. Influence of maternal hormones. Biol Neonate 48: 313–322, 1985. 105. Chalon S, Garel JM. Plasma calcium control in the rat fetus. II. Influence of fetal hormones. Biol Neonate 48: 323–328, 1985. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM 106. Chalon S, Garel JM. Plasma calcium control in the rat fetus. III. Influence of alterations in maternal plasma calcium on fetal plasma calcium level. Biol Neonate 48: 329 –335, 1985. 127. Crass MF 3rd, Jayaseelan CL, Darter TC. Effects of parathyroid hormone on blood flow in different regional circulations. Am J Physiol Regul Integr Comp Physiol 253: R634 –R639, 1987. 107. Chan AS, Conen PE. Ultrastructural observations on cytodifferentiation of parafollicular cells in the human fetal thyroid. Lab Invest 25: 249 –259, 1971. 128. Crocker I, Kaur M, Hosking DJ, Baker PN. Rescue of trophoblast apoptosis by parathyroid hormone-related protein. BJOG 109: 218 –220, 2002. 108. Chang W, Tu C, Bajra R, Komuves L, Miller S, Strewler G, Shoback D. Calcium sensing in cultured chondrogenic RCJ3.1C518 cells. Endocrinology 140: 1911–1919, 1999. 129. Cross JC, Rossant J. Development of the embryo. In: Fetal Growth and Development, edited by Harding R, Bocking AD. Cambridge, UK: Cambridge Univ. Press, 2001, p. 1–16. 109. Chang W, Tu C, Chen TH, Bikle D, Shoback D. The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Science Signaling 1: ra1, 2008. 110. Chang W, Tu C, Chen TH, Komuves L, Oda Y, Pratt SA, Miller S, Shoback D. Expression and signal transduction of calcium-sensing receptors in cartilage and bone. Endocrinology 140: 5883–5893, 1999. 111. Chang W, Tu C, Pratt S, Chen TH, Shoback D. Extracellular Ca2⫹-sensing receptors modulate matrix production and mineralization in chondrogenic RCJ3.1C518 cells. Endocrinology 143: 1467–1474, 2002. 112. Chattopadhyay N, Baum N, Bai M, Riccardi D, Hebert SC, Harris EW, Brown EM. Ontogeny of the extracellular calcium-sensing receptor in rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol 271: F736 –F743, 1996. 113. Chikatsu N, Watanabe S, Takeuchi Y, Muraosa Y, Sasaki S, Oka Y, Fukumoto S, Fujita T. A family of autosomal dominant hypocalcemia with an activating mutation of calcium-sensing receptor gene. Endocr J 50: 91–96, 2003. 114. Chisaka O, Capecchi MR. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature 350: 473– 479, 1991. 115. Cianciolo G, La Manna G, Donati G, Persici E, Dormi A, Cappuccilli ML, Corsini S, Fattori R, Russo V, Nastasi V, Coli L, Wratten M, Stefoni S. Coronary calcifications in end-stage renal disease patients: a new link between osteoprotegerin, diabetes and body mass index? Blood Purification 29: 13–22, 2010. 116. Cockburn F, Belton NR, Purvis RJ, Giles MM, Brown JK, Turner TL, Wilkinson EM, Forfar JO, Barrie WJ, McKay GS, Pocock SJ. Maternal vitamin D intake and mineral metabolism in mothers and their newborn infants. Br Med J 281: 11–14, 1980. 117. Colas AE, Curet LB. Testosterone, dehydroepiandrosterone and 4-androstene-3, 17-dione concentrations in maternal and fetal plasma in the last days of ovine gestation. Biol Reprod 18: 434 – 440, 1978. 118. Collignon H, Davicco MJ, Barlet JP. Calcitonin mRNA expression and plasma calciotropic hormones in fetal lambs. Domest Anim Endocrinol 13: 269 –276, 1996. 119. Collins JN, Kirby BJ, Woodrow JP, Gagel RF, Rosen CJ, Sims NA, Kovacs CS. Lactating Ctcgrp nulls lose twice the normal bone mineral content due to fewer osteoblasts and more osteoclasts, whereas bone mass is fully restored after weaning in association with up-regulation of Wnt signaling and other novel genes. Endocrinology 154: 1400 – 1413, 2013. 130. Cruikshank DP, Pitkin RM, Reynolds WA, Williams GA, Hargis GK. Effects of magnesium sulfate treatment on perinatal calcium metabolism. I. Maternal and fetal responses. Am J Obstet Gynecol 134: 243–249, 1979. 131. Culbert EC, Schfirin BS. Malignant hypercalcemia in pregnancy: effect of pamidronate on uterine contractions. Obstet Gynecol 108: 789 –791, 2006. 132. Cumming WA, Thomas VJ. Hypermagnesemia: a cause of abnormal metaphyses in the neonate. Am J Roentgenol 152: 1071–1072, 1989. 133. Cuttler L, Palmert MR. Luteinizing hormone and follicle-stimulating hormone secretion in the fetus and newborn infant. In: Fetal and Neonatal Physiology (3rd ed.), edited by Polin RA, Fox WW, Abman SH. Philadelphia, PA: Saunders, 2004, p. 1896 –1906. 134. Dacquin R, Davey RA, Laplace C, Levasseur R, Morris HA, Goldring SR, GebreMedhin S, Galson DL, Zajac JD, Karsenty G. Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. J Cell Biol 164: 509 –514, 2004. 135. Danan JL, Delorme AC, Mathieu H. Presence of 25-hydroxyvitamin D3 and 1,25dihydroxyvitamin D3 24-hydroxylase in vitamin D target cells of rat yolk sac. J Biol Chem 257: 10715–10721, 1982. 136. Dancaster CP, Jackson WP. Studies in rickets in the Cape Peninsula. I. Cranial softening in a coloured population and its relationship to the radiological and biochemical changes of rickets. S African Medical J 34: 776 –780, 1960. 137. Danks JA, Ebeling PR, Hayman JA, Diefenbach-Jagger H, Collier FM, Grill V, Southby J, Moseley JM, Chou ST, Martin TJ. Immunohistochemical localization of parathyroid hormone-related protein in parathyroid adenoma and hyperplasia. J Pathol 161: 27– 33, 1990. 138. Dardenne O, Prud’homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D3-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology 142: 3135–3141, 2001. 139. Dardenne O, Prud’homme J, Hacking SA, Glorieux FH, St-Arnaud R. Correction of the abnormal mineral ion homeostasis with a high-calcium, high-phosphorus, highlactose diet rescues the PDDR phenotype of mice deficient for the 25-hydroxyvitamin D-1alpha-hydroxylase (CYP27B1). Bone 32: 332–340, 2003. 140. Dardenne O, Prudhomme J, Hacking SA, Glorieux FH, St-Arnaud R. Rescue of the pseudo-vitamin D deficiency rickets phenotype of CYP27B1-deficient mice by treatment with 1,25-dihydroxyvitamin D3: biochemical, histomorphometric, and biomechanical analyses. J Bone Miner Res 18: 637– 643, 2003. 120. Collip JB, Clark EP. Further studies on the physiological action of a parathyroid hormone. J Biol Chem 64: 485–507, 1925. 141. Dashe JS, McIntire DD, Ramus RM, Santos-Ramos R, Twickler DM. Hydramnios: anomaly prevalence and sonographic detection. Obstet Gynecol 100: 134 –139, 2002. 121. Comar CL. Radiocalcium studies in pregnancy. Ann NY Acad Sci 64: 281–298, 1956. 142. Davicco MJ, Coxam V, Lefaivre J, Barlet JP. Parathyroid hormone-related peptide increases urinary phosphate excretion in fetal lambs. Exp Physiol 77: 377–383, 1992. 122. Congdon P, Horsman A, Kirby PA, Dibble J, Bashir T. Mineral content of the forearms of babies born to Asian and white mothers. Br Med J 286: 1233–1235, 1983. 123. Cooper C, Westlake S, Harvey N, Javaid K, Dennison E, Hanson M. Review: developmental origins of osteoporotic fracture. Osteoporos Int 17: 337–347, 2006. 143. David L, Anast CS. Calcium metabolism in newborn infants. The interrelationship of parathyroid function and calcium, magnesium, and phosphorus metabolism in normal, sick, and hypocalcemic newborns. J Clin Invest 54: 287–296, 1974. 124. Corbier P, Edwards DA, Roffi J. The neonatal testosterone surge: a comparative study. Arch Int Physiol Biochim Biophys 100: 127–131, 1992. 144. Davis OK, Hawkins DS, Rubin LP, Posillico JT, Brown EM, Schiff I. Serum parathyroid hormone (PTH) in pregnant women determined by an immunoradiometric assay for intact PTH. J Clin Endocrinol Metab 67: 850 – 852, 1988. 125. Couse JF, Curtis SW, Washburn TF, Lindzey J, Golding TS, Lubahn DB, Smithies O, Korach KS. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol 9: 1441–1454, 1995. 145. De Curtis M, Pieltain C, Studzinski F, Moureau V, Gerard P, Rigo J. Evaluation of weight gain composition during the first 2 months of life in breast- and formula-fed term infants using dual energy X-ray absorptiometry. Eur J Pediatr 160: 319 –320, 2001. 126. Cozzolino M, Dusso AS, Slatopolsky E. Role of calcium-phosphate product and boneassociated proteins on vascular calcification in renal failure. J Am Soc Nephrol 12: 2511–2516, 2001. 146. De Lauzon S, Uhrich F, Vandel S, Cittanova N, Jayle MF. Determination of progesterone and of free and conjugated estrogens in pregnant and peudo-pregnant rats. Steroids 24: 31– 40, 1974. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1203 CHRISTOPHER S. KOVACS 147. De NC, Harper JR. Renal agenesis and pulmonary hypoplasia. Br Med J 3: 696 – 697, 1972. 169. Dvorak-Ewell MM, Chen TH, Liang N, Garvey C, Liu B, Tu C, Chang W, Bikle DD, Shoback DM. Osteoblast extracellular Ca2⫹-sensing receptor regulates bone development, mineralization, and turnover. J Bone Miner Res 26: 2935–2947, 2011. 148. Delivoria-Papadopoulos M, Battaglia FC, Bruns PD, Meschia G. Total, protein-bound, and ultrafilterable calcium in maternal and fetal plasmas. Am J Physiol 213: 363–366, 1967. 170. Eastell R, Edmonds CJ, de Chayal RC, McFadyen IR. Prolonged hypoparathyroidism presenting eventually as second trimester abortion. Br Med J 291: 955–956, 1985. 149. Delmonico FL, Neer RM, Cosimi AB, Barnes AB, Russell PS. Hyperparathyroidism during pregnancy. Am J Surg 131: 328 –337, 1976. 171. Edelmann CM Jr, Spitzer A. The maturing kidney. A modern view of well-balanced infants with imbalanced nephrons. J Pediatr 75: 509 –519, 1969. 150. Delorme AC, Danan JL, Ripoche MA, Mathieu H. Biochemical characterization of mouse vitamin D-dependent calcium-binding protein. Evidence for its presence in embryonic life. Biochem J 205: 49 –57, 1982. 172. Ekpebegh CO, Blanco-Blanco E. Familial hypophosphataemic rickets affecting a father and his two daughters: a case report. West African J Med 29: 271–274, 2010. 151. Delorme AC, Marche P, Garel JM. Vitamin D-dependent calcium-binding protein. Changes during gestation, prenatal and postnatal development in rats. J Dev Physiol 1: 181–194, 1979. 152. Delvin EE, Glorieux FH, Salle BL, David L, Varenne JP. Control of vitamin D metabolism in preterm infants: feto-maternal relationships. Arch Dis Child 57: 754 –757, 1982. 153. Delvin EE, Salle BL, Glorieux FH, Adeleine P, David LS. Vitamin D supplementation during pregnancy: effect on neonatal calcium homeostasis. J Pediatr 109: 328 –334, 1986. 154. Demay MB. Physiological insights from the vitamin D receptor knockout mouse. Calcif Tissue Int 92: 99 –105, 2013. 155. Devaskar UP, Ho M, Devaskar SU, Tsang RC. 25-Hydroxy-1alpha, 25-dihydroxyvitamin D. Maternal-fetal relationship and the transfer of 1,25-dihydroxyvitamin D3 across the placenta in an ovine model. Dev Pharmacol Ther 7: 213–220, 1984. 156. Diaz L, Sanchez I, Avila E, Halhali A, Vilchis F, Larrea F. Identification of a 25-hydroxyvitamin D3 1alpha-hydroxylase gene transcription product in cultures of human syncytiotrophoblast cells. J Clin Endocrinol Metab 85: 2543–2549, 2000. 173. Eller-Vainicher C, Ossola MW, Beck-Peccoz P, Chiodini I. PTHrP-associated hypercalcemia of pregnancy resolved after delivery: a case report. Eur J Endocrinol 166: 753–756, 2012. 174. Enders AC. A comparative study of the fine structures of the trophoblast in several hemochorial placentas. Am J Anat 116: 29 – 68, 1965. 175. Erben RG, Soegiarto DW, Weber K, Zeitz U, Lieberherr M, Gniadecki R, Moller G, Adamski J, Balling R. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol Endocrinol 16: 1524 –1537, 2002. 176. Evans KN, Bulmer JN, Kilby MD, Hewison M. Vitamin D and placental-decidual function. J Soc Gynecol Invest 11: 263–271, 2004. 177. Fairney A, Jackson D, Clayton BE. Measurement of serum parathyroid hormone, with particular reference to some infants with hypocalcaemia. Arch Dis Child 48: 419 – 424, 1973. 178. Farrugia W, de Gooyer T, Rice GE, Moseley JM, Wlodek ME. Parathyroid hormone(1–34) and parathyroid hormone-related protein(1–34) stimulate calcium release from human syncytiotrophoblast basal membranes via a common receptor. J Endocrinol 166: 689 – 695, 2000. 157. Docherty HM, Ratcliffe WA, Heath DA, Docherty K. Expression of parathyroid hormone-related protein in abnormal human parathyroids. J Endocrinol 129: 431– 438, 1991. 179. Faupel-Badger JM, Wang Y, Karumanchi SA, Stanczyk F, Pollak M, McElrath T, Hoover RN, Troisi R. Associations of pregnancy characteristics with maternal and cord steroid hormones, angiogenic factors, and insulin-like growth factor axis. Cancer Causes Control 22: 1587–1595, 2011. 158. Donohue MM, Demay MB. Rickets in VDR null mice is secondary to decreased apoptosis of hypertrophic chondrocytes. Endocrinology 143: 3691–3694, 2002. 180. Feldman D, Malloy PJ. Mutations in the vitamin D receptor and hereditary vitamin D-resistant rickets. Bone Key Rep 3: 2014. 159. Donovan EF, Tsang RC, Steichen JJ, Strub RJ, Chen IW, Chen M. Neonatal hypermagnesemia: effect on parathyroid hormone and calcium homeostasis. J Pediatr 96: 305–310, 1980. 181. Feldman K. Commentary on “congenital rickets” article. Pediatr Radiol 39: 1127– 1130, 2009. 160. Drees BM, Hamilton JW. Processing of chromogranin A by bovine parathyroid secretory granules: production and secretion of N-terminal fragments. Endocrinology 134: 2057–2063, 1994. 161. Drezner MK. Phosphorus homeostasis and related disorders. In: Principles of Bone Biology (3rd ed.), edited by Bilezikian JP, Raisz LG, and Martin TJ. San Diego, CA: Academic, 2008, p. 465– 486. 162. Dunne FP, Ratcliffe WA, Mansour P, Heath DA. Parathyroid hormone related protein (PTHrP) gene expression in fetal and extra-embryonic tissues of early pregnancy. Hum Reprod 9: 149 –156, 1994. 163. Durand D, Barlet JP. Influence de la calcitonine maternelle sur la calcémie foetale chez la brebis. Ann Endocrinol 42: 8C, 1981. 164. Durand D, Barlet JP, Braithwaite GD. The influence of 1,25-dihydroxycholecalciferol on the mineral content of foetal guinea pigs. Reprod Nutr Dev 23: 235–244, 1983. 165. Durand D, Braithwaite GD, Barlet JP. The effect of 1␣-hydroxycholecalciferol on the placental transfer of calcium and phosphate in sheep. Br J Nutr 49: 475– 480, 1983. 166. Duval M. Le placenta des rongeurs. Trosième partie Le placenta de la souris et du rat. J Anat Physiol Norm Pathol Homme Animaux 27: 24 –73, 344 –395, 515– 612, 1891. 167. Duval M. Le placenta des ronguers. Quatrième partie Le placenta du cochon d’Inde. J Anat Physiol Norm Pathol Homme Animaux 28: 58 –98, 333– 453, 1892. 168. Dvir R, Golander A, Jaccard N, Yedwab G, Otremski I, Spirer Z, Weisman Y. Amniotic fluid and plasma levels of parathyroid hormone-related protein and hormonal modulation of its secretion by amniotic fluid cells. Eur J Endocrinol 133: 277–282, 1995. 1204 182. Feldman KW, Done S. Vitamin D deficiency rickets and allegations of non-accidental injury. Acta Paediatr 99: 486 – 487, 2010. 183. Ferguson IIJE, Seaner RM, Bruns DE, Iezzoni JC, Bruns ME. Expression and specific immunolocalization of the human parathyroid hormone/parathyroid hormone-related protein receptor in the uteroplacental unit. Am J Obstet Gynecol 179: 321–329, 1998. 184. Ferguson JE, Gorman JV, Bruns DE, Weir EC, Burtis WJ, Martin TJ, Bruns ME. Abundant expression of parathyroid hormone-related protein in human amnion and its association with labor. Proc Natl Acad Sci USA 89: 8384 – 8388, 1992. 185. Ferguson JE, Seaner R, Bruns DE, Redick JA, Mills SE, Jüppner H, Segre GV, Bruns ME. Expression of parathyroid hormone-related protein and its receptor in human umbilical cord: evidence for a paracrine system involving umbilical vessels. Am J Obstet Gynecol 170: 1018 –1024, 1994. 186. Fewtrell MS, Williams JE, Singhal A, Murgatroyd PR, Fuller N, Lucas A. Early diet and peak bone mass: 20 year follow-up of a randomized trial of early diet in infants born preterm. Bone 45: 142–149, 2009. 187. Finney B, Wilkinson WJ, Searchfield L, Cole M, Bailey S, Kemp PJ, Riccardi D. An exon 5-less splice variant of the extracellular calcium-sensing receptor rescues absence of the full-length receptor in the developing mouse lung. Exp Lung Res 37: 269 –278, 2011. 188. Fisher GJ, Kelley LK, Smith CH. ATP-dependent calcium transport across basal plasma membranes of human placental trophoblast. Am J Physiol Cell Physiol 252: C38 –C46, 1987. 189. Fleischman AR, Lerman S, Oakes GK, Epstein MF, Chez RA, Mintz DH. Perinatal primate parathyroid hormone metabolism. Biol Neonate 27: 40 – 49, 1975. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM 190. Fleischman AR, Rosen JF, Cole J, Smith CM, DeLuca HF. Maternal and fetal serum 1,25-dihydroxyvitamin D levels at term. J Pediatr 97: 640 – 642, 1980. 191. Ford JA, Davidson DC, McIntosh WB, Fyfe WM, Dunnigan MG. Neonatal rickets in Asian immigrant population. Br Med J 3: 211–212, 1973. 192. Fudge NJ, Kovacs CS. Pregnancy up-regulates intestinal calcium absorption and skeletal mineralization independently of the vitamin D receptor. Endocrinology 151: 886 – 895, 2010. 193. Fujimoto Y, Hazama H, Oku K. Severe primary hyperparathyroidism in a neonate having a parent with hypercalcemia: treatment by total parathyroidectomy and simultaneous heterotopic autotransplantation. Surgery 108: 933–938, 1990. 194. Fukumoto S. Phosphate metabolism and vitamin D. Bone Key Rep 3: 2014. 195. Gardella T, Jüppner H, Bringhurst FR, Potts JT Jr. Receptors for parathyroid hormone (PTH) and PTH-related protein. In: Principles of Bone Biology (3rd ed.), edited by Bilezikian JP, Raisz LG, Martin TJ. New York: Academic, 2008, p. 555–576. 196. Garel JM. Distribution of labeled parathyroid hormone in rat fetus. Horm Metab Res 4: 131–132, 1972. 215. Garel JM, Savajol H, Barlet JP, Care AD. Dosage radio-immunologique de la calcitonine chez le foetus de mouton. C R Acad Sci Hebd Seances Acad Sci D 217: 217–220, 1973. 216. Gemer O, Sevillia J, Zalis J, Segal S. Umbilical cord androgens in infants of diabetic mothers. Arch Gynecol Obstet 259: 139 –141, 1997. 217. Gherman RB, Bowen E, Eggleston MK, Teague KE, Sayles T, Brown EM, Pollak MR. Successful pregnancy outcome in a woman with a gain-of-function mutation of the calcium-sensing receptor. A case report. J Reprod Med 44: 745–747, 1999. 218. Ghishan FK, Jenkins JT, Younoszai MK. Maturation of calcium transport in the rat small and large intestine. J Nutr 110: 1622–1628, 1980. 219. Ghishan FK, Parker P, Nichols S, Hoyumpa A. Kinetics of intestinal calcium transport during maturation in rats. Pediatr Res 18: 235–239, 1984. 220. Gilbert M, Besnard P, Garel JM. Effects of maternal parathyroid glands and vitamin D3 metabolites on fetal growth and fetal liver glycogen stores in thyro-parathyroidectomized pregnant rats. Biomedicine 32: 93–99, 1980. 197. Garel JM. Effect of the injection of an anti-parathyroid hormone serum in the rat fetus. C R Acad Sci Hebd Seances Acad Sci D 271: 2364 –2366, 1970. 221. Giles MM, Fenton MH, Shaw B, Elton RA, Clarke M, Lang M, Hume R. Sequential calcium and phosphorus balance studies in preterm infants. J Pediatr 110: 591–598, 1987. 198. Garel JM. Hormonal control of calcium metabolism during the reproductive cycle in mammals. Physiol Rev 67: 1– 66, 1987. 222. Gilmour JR. The embryology of the parathyroid glands, the thymus and certain associated remnants. J Pathol Bacteriol 45: 507–522, 1937. 199. Garel JM. Parathyroid hormone, calcitonin and mineral metabolism in the mammalian fetus and neonate. In: Perinatal Calcium and Phosphorus Metabolism, edited by Hollick MF, Anast CS, and Gray TK. Amsterdam: Excerpta Medica, 1983, p. 71–104. 223. Gittleman IF, Pincus JB. Influence of diet on the occurrence of hyperphosphatemia and hypocalcemia in the newborn infant. Pediatrics 8: 778 –787, 1951. 200. Garel JM, Barlet JP. Calcitonin in the mother, fetus and newborn. Ann Biol Anim Biochim Biophys 18: 53– 68, 1978. 201. Garel JM, Barlet JP. Calcium metabolism in newborn animals: the interrelationship of calcium, magnesium, and inorganic phosphorus in newborn rats, foals, lambs, and calves. Pediatr Res 10: 749 –754, 1976. 202. Garel JM, Barlet JP. The effects of calcitonin and parathormone on plasma magnesium levels before and after birth in the rat. J Endocrinol 61: 1–13, 1974. 203. Garel JM, Besnard P, Rebut-Bonneton C. C cell activity during the prenatal and postnatal periods in the rat. Endocrinology 109: 1573–1577, 1981. 224. Givens MH, Macy IC. The chemical composition of the human fetus. J Biol Chem 102: 7–17, 1933. 225. Glaser K. Double contour, cupping and spurring in roentgenograms of long bones in infants. Am J Roentgenol Radium Ther 61: 482– 492, 1949. 226. Glass EJ, Barr DG. Transient neonatal hyperparathyroidism secondary to maternal pseudohypoparathyroidism. Arch Dis Child 56: 565–568, 1981. 227. Glazier JD, Atkinson DE, Thornburg KL, Sharpe PT, Edwards D, Boyd RD, Sibley CP. Gestational changes in Ca2⫹ transport across rat placenta and mRNA for calbindin9K and Ca2⫹-ATPase. Am J Physiol Regul Integr Comp Physiol 263: R930 – R935, 1992. 204. Garel JM, Care AD, Barlet JP. A radioimmunoassay for ovine calcitonin: an evaluation of calcitonin secretion during gestation, lactation and foetal life. J Endocrinol 62: 497– 509, 1974. 228. Glazier JD, Mawer EB, Sibley CP. Calbindin-D9K gene expression in rat chorioallantoic placenta is not regulated by 1,25-dihydroxyvitamin D3. Pediatr Res 37: 720 –725, 1995. 205. Garel JM, Dumont C. Distribution and inactivation of labeled parathyroid hormone in rat fetus. Horm Metab Res 4: 217–221, 1972. 229. Glorieux FH, Edouard T, St-Arnaud R. Pseudo-vitamin D deficiency. In: Vitamin D (3rd ed.), edited by Feldman D, Pike JW, Adams JS. San Diego, CA: Academic, 2011, p. 1187–1195. 206. Garel JM, Geloso-Meyer A. Fetal hyperparathyroidism in rats following maternal hypoparathyroidism. Rev Eur Etud Clin Biol 16: 174 –178, 1971. 230. Glorieux FH, Pettifor JM. Vitamin D/dietary calcium deficiency rickets and pseudovitamin D deficiency rickets. Bone Key Rep 3: 2014. 207. Garel JM, Gilbert M. Dietary calcium and phosphorus manipulations in thyroparathyroidectomized pregnant rats and fetal liver glycogen stores. Reprod Nutr Dev 21: 969 –977, 1981. 231. Glorieux FH, Salle BL, Delvin EE, David L. Vitamin D metabolism in preterm infants: serum calcitriol values during the first five days of life. J Pediatr 99: 640 – 643, 1981. 208. Garel JM, Gilbert M, Besnard P. Fetal growth and 1,25-dihydroxyvitamin D3 injections into thyroparathyroidectomized pregnant rats. Reprod Nutr Dev 21: 961–968, 1981. 232. Gordon J, Bennett AR, Blackburn CC, Manley NR. Gcm2 and Foxn1 mark early parathyroid- and thymus-specific domains in the developing third pharyngeal pouch. Mech Dev 103: 141–143, 2001. 209. Garel JM, Jullienne A. Plasma calcitonin levels in pregnant and newborn rats. J Endocrinol 75: 373–382, 1977. 210. Garel JM, Martin-Rosset W, Barlet JP. Plasma immunoreactive calcitonin levels in pregnant mares and newborn foals. Horm Metab Res 7: 429 – 432, 1975. 233. Grant CC, Stewart AW, Scragg R, Milne T, Rowden J, Ekeroma A, Wall C, Mitchell EA, Crengle S, Trenholme A, Crane J, Camargo CA Jr. Vitamin D during pregnancy and infancy and infant serum 25-hydroxyvitamin D concentration. Pediatrics 133: e143– 153, 2014. 211. Garel JM, Milhaud G, Jost A. Hypocalcemic and hypophosphatemic action of thyrocalcitonin in fetal rats. C R Acad Sci Hebd Seances Acad Sci D 267: 344 –347, 1968. 234. Gray TK, Lester GE, Lorenc RS. Evidence for extra-renal 1␣-hydroxylation of 25hydroxyvitamin D3 in pregnancy. Science 204: 1311–1313, 1979. 212. Garel JM, Milhaud G, Sizonenko P. Thyrocalcitonin and the placental barrier in rats. C R Acad Sci Hebd Seances Acad Sci D 269: 1785–1787, 1969. 235. Greer FR. Determination of radial bone mineral content in low birth weight infants by photon absorptiometry. J Pediatr 113: 213–219, 1988. 213. Garel JM, Pic P. Evolution of phosphatemia in the rat fetus during the late stages of gestation. Biol Neonate 21: 369 –374, 1972. 236. Greer FR, McCormick A. Bone growth with low bone mineral content in very low birth weight premature infants. Pediatr Res 20: 925–928, 1986. 214. Garel JM, Pic P, Jost A. Action de la parathormone chez de foetus de rat. Ann Endocrinol 32: 253–262, 1971. 237. Guignard JP, Torrado A, Da Cunha O, Gautier E. Glomerular filtration rate in the first three weeks of life. J Pediatr 87: 268 –272, 1975. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1205 CHRISTOPHER S. KOVACS 238. Günther T, Chen ZF, Kim J, Priemel M, Rueger JM, Amling M, Moseley JM, Martin TJ, Anderson DJ, Karsenty G. Genetic ablation of parathyroid glands reveals another source of parathyroid hormone. Nature 406: 199 –203, 2000. 239. Habert R, Picon R. Testosterone, dihydrotestosterone and estradiol-17 beta levels in maternal and fetal plasma and in fetal testes in the rat. J Steroid Biochem 21: 193–198, 1984. 240. Haddad JG Jr, Boisseau V, Avioli LV. Placental transfer of vitamin D3 and 25-hydroxycholecalciferol in the rat. J Lab Clin Med 77: 908 –915, 1971. 241. Halloran BP, De Luca HF. Effect of vitamin D deficiency on skeletal development during early growth in the rat. Arch Biochem Biophys 209: 7–14, 1981. 242. Halloran BP, DeLuca HF. Appearance of the intestinal cytosolic receptor for 1,25dihydroxyvitamin D3 during neonatal development in the rat. J Biol Chem 256: 7338 – 7342, 1981. 243. Halloran BP, DeLuca HF. Calcium transport in small intestine during early development: role of vitamin D. Am J Physiol Gastrointest Liver Physiol 239: G473–G479, 1980. 244. Harvey NC, Holroyd C, Ntani G, Javaid K, Cooper P, Moon R, Cole Z, Tinati T, Godfrey K, Dennison E, Bishop NJ, Baird J, Cooper C. Vitamin D supplementation in pregnancy: A systematic review. Health Technol Assess. In press. 245. Harvey NC, Javaid K, Bishop N, Kennedy S, Papageorghiou AT, Fraser R, Gandhi SV, Schoenmakers I, Prentice A, Cooper C. MAVIDOS Maternal Vitamin D Osteoporosis Study: study protocol for a randomized controlled trial. The MAVIDOS Study Group Trials 13: 13, 2012. 246. Hashemipour S, Lalooha F, Zahir Mirdamadi S, Ziaee A, Dabaghi Ghaleh T. Effect of vitamin D administration in vitamin D-deficient pregnant women on maternal and neonatal serum calcium and vitamin D concentrations: a randomised clinical trial. Br J Nutr 110: 1611–1616, 2013. 258. Hochberg Z, Benderli A, Levy J, Vardi P, Weisman Y, Chen T, Feldman D. 1,25Dihydroxyvitamin D resistance, rickets, and alopecia. Am J Med 77: 805– 811, 1984. 259. Hochberg Z, Tiosano D, Even L. Calcium therapy for calcitriol-resistant rickets. J Pediatr 121: 803– 808, 1992. 260. Hoff AO, Catala-Lehnen P, Thomas PM, Priemel M, Rueger JM, Nasonkin I, Bradley A, Hughes MR, Ordonez N, Cote GJ, Amling M, Gagel RF. Increased bone mass is an unexpected phenotype associated with deletion of the calcitonin gene. J Clin Invest 110: 1849 –1857, 2002. 261. Hollis BW. Vitamin D supplementation in pregnancy & breastfeeding– effectiveness and safety/The vitamin D requirement during pregnancy and lactation http://www.youtube.com/watch?v⫽O0elnh4D08g [October 12, 2011]. 262. Hollis BW, Johnson D, Hulsey TC, Ebeling M, Wagner CL. Vitamin D supplementation during pregnancy: double-blind, randomized clinical trial of safety and effectiveness. J Bone Miner Res 26: 2341–2357, 2011. 263. Hollis BW, Lowery JW, Pittard WB, Guy DG, Hansen JW. Effect of age on the intestinal absorption of vitamin D3-palmitate and nonesterified vitamin D2 in the term human infant. J Clin Endocrinol Metab 81: 1385–1388, 1996. 264. Hollis BW, Pittard WB. Evaluation of the total fetomaternal vitamin D relationships at term: evidence for racial differences. J Clin Endocrinol Metab 59: 652– 657, 1984. 265. Hollis BW, Wagner CL. Vitamin D and pregnancy: skeletal effects, nonskeletal effects, and birth outcomes. Calcif Tissue Int 92: 128 –139, 2013. 266. Hori C, Tsukahara H, Fujii Y, Kawamitsu T, Konishi Y, Yamamoto K, Ishii Y, Sudo M. Bone mineral status in preterm-born children: assessment by dual-energy X-ray absorptiometry. Biol Neonate 68: 254 –258, 1995. 267. Hori M, Shimizu Y, Fukumoto S. Minireview: fibroblast growth factor 23 in phosphate homeostasis and bone metabolism. Endocrinology 152: 4 –10, 2011. 247. Hashimoto K, Nakagawa Y, Shibuya T, Satoh H, Ushijima T, Imai S. Effects of parathyroid hormone and related polypeptides on the heart and coronary circulation of dogs. J Cardiovasc Pharmacol 3: 668 – 676, 1981. 268. Horton WA, Scott CI. Dyggve-Melchior-Clausen syndrome. A histochemical study of the growth plate. J Bone Joint Surg 64: 408 – 415, 1982. 248. Helle KB, Aunis D. A physiological role for the granins as prohormones for homeostatically important regulatory peptides? A working hypothesis for future research. Adv Exp Med Biol 482: 389 –397, 2000. 269. Horwitz MJ, Tedesco MB, Sereika SM, Hollis BW, Garcia-Ocana A, Stewart AF. Direct comparison of sustained infusion of human parathyroid hormone-related protein-(1–36). J Clin Endocrinol Metab 88: 1603–1609, 2003. 249. Heo JS, Choi KY, Sohn SH, Kim C, Kim YJ, Shin SH, Lee JM, Lee J, Sohn JA, Lim BC, Lee JA, Choi CW, Kim EK, Kim HS, Kim BI, Choi JH. A case of mucolipidosis II presenting with prenatal skeletal dysplasia and severe secondary hyperparathyroidism at birth. Korean J Pediatr 55: 438 – 444, 2012. 270. Horwitz MJ, Tedesco MB, Sereika SM, Syed MA, Garcia-Ocana A, Bisello A, Hollis BW, Rosen CJ, Wysolmerski JJ, Dann P, Gundberg C, Stewart AF. Continuous PTH and PTHrP infusion causes suppression of bone formation and discordant effects on 1,25(OH)2 vitamin D. J Bone Miner Res 20: 1792–1803, 2005. 250. Hercz P, Kazy Z, Siklos P, Ungar L. Quantitative comparison of serum steroid and peptide hormone concentrations in male and female fetuses in the maternal-fetoplacental system during the 28th-40th weeks of pregnancy. Eur J Obstet Gynecol Reprod Biol 30: 201–204, 1989. 271. Hough TA, Bogani D, Cheeseman MT, Favor J, Nesbit MA, Thakker RV, Lyon MF. Activating calcium-sensing receptor mutation in the mouse is associated with cataracts and ectopic calcification. Proc Natl Acad Sci USA 101: 13566 –13571, 2004. 251. Hershberger ME, Tuan RS. Functional analysis of placental 57-kDa Ca2⫹-binding protein: overexpression and downregulation in a trophoblastic cell line. Dev Biol 215: 107–117, 1999. 252. Hershberger ME, Tuan RS. Placental 57-kDa Ca2⫹-binding protein: regulation of expression and function in trophoblast calcium transport. Dev Biol 199: 80 –92, 1998. 253. Higashi T, Mitamura K, Ohmi H, Yamada N, Shimada K, Tanaka K, Honjo H. Levels of 24,25-dihydroxyvitamin D3, 25-hydroxyvitamin D3 and 25-hydroxyvitamin D3 3-sulphate in human plasma. Ann Clin Biochem 36: 43– 47, 1999. 272. Houtsmuller EJ, de Jong FH, Rowland DL, Slob AK. Plasma testosterone in fetal rats and their mothers on day 19 of gestation. Physiol Behav 57: 495– 499, 1995. 273. Howard A, Legon S, Walters JR. Plasma membrane calcium pump expression in human placenta and small intestine. Biochem Biophys Res Commun 183: 499 –505, 1992. 274. Huan J, Olgaard K, Nielsen LB, Lewin E. Parathyroid hormone 7– 84 induces hypocalcemia and inhibits the parathyroid hormone 1– 84 secretory response to hypocalcemia in rats with intact parathyroid glands. J Am Soc Nephrol 17: 1923–1930, 2006. 254. Hillman LS, Haddad JG. Human perinatal vitamin D metabolism. I. 25-Hydroxyvitamin D in maternal and cord blood. J Pediatr 84: 742–749, 1974. 275. Hutchesson AC, Hughes SV, Bowden SJ, Ratcliffe WA. In vitro stability of endogenous parathyroid hormone-related protein in blood and plasma. Ann Clin Biochem 31: 35–39, 1994. 255. Hillman LS, Slatopolsky E, Haddad JG. Perinatal vitamin D metabolism. IV. Maternal and cord serum 24,25-dihydroxyvitamin D concentrations. J Clin Endocrinol Metab 47: 1073–1077, 1978. 276. Ibrahim MM, Thomas ML, Forte LR. Maternal-fetal relationships in the parathyroidectomized rat. Intestinal calcium transport, serum calcium, immunoreactive parathyroid hormone and calcitonin. Biol Neonate 46: 89 –97, 1984. 256. Hillman LS, Tack E, Covell DG, Vieira NE, Yergey AL. Measurement of true calcium absorption in premature infants using intravenous 46Ca and oral 44Ca. Pediatr Res 23: 589 –594, 1988. 277. Ikeda K, Weir EC, Mangin M, Dannies PS, Kinder B, Deftos LJ, Brown EM, Broadus AE. Expression of messenger ribonucleic acids encoding a parathyroid hormone-like peptide in normal human and animal tissues with abnormal expression in human parathyroid adenomas. Mol Endocrinol 2: 1230 –1236, 1988. 257. Ho C, Conner DA, Pollak MR, Ladd DJ, Kifor O, Warren HB, Brown EM, Seidman JG, Seidman CE. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat Genet 11: 389 –394, 1995. 1206 278. Illidge TM, Hussey M, Godden CW. Malignant hypercalcaemia in pregnancy and antenatal administration of intravenous pamidronate. Clin Oncol 8: 257–258, 1996. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM 279. Innes AM, Seshia MM, Prasad C, Al Saif S, Friesen FR, Chudley AE, Reed M, Dilling LA, Haworth JC, Greenberg CR. Congenital rickets caused by maternal vitamin D deficiency. Paediatr Child Health 7: 455– 458, 2002. 301. Keelan JA, Mattes E, Tan H, Dinan A, Newnham JP, Whitehouse AJ, Jacoby P, Hickey M. Androgen concentrations in umbilical cord blood and their association with maternal, fetal and obstetric factors. PLoS One 7: e42827, 2012. 280. Ioannidis G, Papaioannou A, Hopman WM, Akhtar-Danesh N, Anastassiades T, Pickard L, Kennedy CC, Prior JC, Olszynski WP, Davison KS, Goltzman D, Thabane L, Gafni A, Papadimitropoulos EA, Brown JP, Josse RG, Hanley DA, Adachi JD. Relation between fractures and mortality: results from the Canadian Multicentre Osteoporosis Study. CMAJ 181: 265–271, 2009. 302. Kehrer E. Die geburtschilflich-gynäkologische bedeutung der tetanie. Arch Gynaek 99: 372– 447, 1913. 281. Ioannou C, Javaid MK, Mahon P, Yaqub MK, Harvey NC, Godfrey KM, Noble JA, Cooper C, Papageorghiou AT. The effect of maternal vitamin D concentration on fetal bone. J Clin Endocrinol Metab 97: E2070 –2077, 2012. 282. Ip P. Neonatal convulsion revealing maternal hyperparathyroidism: an unusual case of late neonatal hypoparathyroidism. Arch Gynecol Obstet 268: 227–229, 2003. 283. Jackson IT, Saleh J, van Heerden JA. Gigantic mammary hyperplasia in pregnancy associated with pseudohyperparathyroidism. Plastic Reconstructive Surg 84: 806 – 810, 1989. 284. Jarzab B, Kokot F, Baldys A. Immunoreactive calcitonin content in foetal thyroid glands and in placentae of rats. Acta Endocrinol 105: 567–570, 1984. 303. Keller KA, Barnes PD. Rickets vs abuse: a national and international epidemic. Pediatr Radiol 38: 1210 –1216, 2008. 304. Kelly TR. Primary hyperparathyroidism during pregnancy. Surgery 110: 1028 –1034, 1991. 305. Kemper B, Habener JF, Rich A, Potts JT Jr. Parathyroid secretion: discovery of a major calcium-dependent protein. Science 184: 167–169, 1974. 306. Khosla S, Johansen KL, Ory SJ, O’Brien PC, Kao PC. Parathyroid hormone-related peptide in lactation and in umbilical cord blood. Mayo Clin Proc 65: 1408 –1414, 1990. 307. Khosla S, van Heerden JA, Gharib H, Jackson IT, Danks J, Hayman JA, Martin TJ. Parathyroid hormone-related protein and hypercalcemia secondary to massive mammary hyperplasia (Letter). N Engl J Med 322: 1157, 1990. 308. Kiesler J, Ricer R. The abnormal fontanel. Am Family Physician 67: 2547–2552, 2003. 285. Javaid MK, Crozier SR, Harvey NC, Gale CR, Dennison EM, Boucher BJ, Arden NK, Godfrey KM, Cooper C. Maternal vitamin D status during pregnancy and childhood bone mass at age 9 years: a longitudinal study. Lancet 367: 36 – 43, 2006. 286. Jenny C. Multiple unexplained fractures in infants–the need for clear thinking. Acta Paediatr 99: 491– 493, 2010. 309. Kirby BJ, Ardeshirpour L, Woodrow JP, Wysolmerski JJ, Sims NA, Karaplis AC, Kovacs CS. Skeletal recovery after weaning does not require PTHrP. J Bone Miner Res 26: 1242–1251, 2011. 287. Jenny C. Rickets or abuse? Pediatr Radiol 38: 1219 –1220, 2008. 310. Kirby BJ, Ma Y, Martin HM, Buckle Favaro KL, Karaplis AC, Kovacs CS. Upregulation of calcitriol during pregnancy and skeletal recovery after lactation do not require parathyroid hormone. J Bone Miner Res 28: 1987–2000, 2013. 288. Johnson JA, Kumar R. Renal and intestinal calcium transport: roles of vitamin D and vitamin D-dependent calcium binding proteins. Semin Nephrol 14: 119 –128, 1994. 311. Kirby DR, Bardbury S. The hemo-chorial mouse placenta. Anat Rec 152: 279 –282, 1965. 289. Jollie WP. Development, morphology, and function of the yolk-sac placenta of laboratory rodents. Teratology 41: 361–381, 1990. 312. Kitanaka S, Takeyama K, Murayama A, Sato T, Okumura K, Nogami M, Hasegawa Y, Niimi H, Yanagisawa J, Tanaka T, Kato S. Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med 338: 653– 661, 1998. 290. Jones G, Riley M, Dwyer T. Breastfeeding in early life and bone mass in prepubertal children: a longitudinal study. Osteoporos Int 11: 146 –152, 2000. 291. Jones GV, Taylor KR, Morgan G, Wooding FB, Care AD. Aspects of calcium transport by the ovine placenta: studies based on the interplacentomal region of the chorion. Placenta 18: 357–364, 1997. 292. Jordan RK, McFarlane B, Scothorne RJ. An electron microscopic study of the histogenesis of the ultimobranchial body and of the C-cell system in the sheep. J Anat 114: 115–136, 1973. 293. Jousset V, Legendre B, Besnard P, Segond N, Jullienne A, Garel JM. Calcitonin-like immunoreactivity and calcitonin gene expression in the placenta and in the mammary gland of the rat. Acta Endocrinol 119: 443– 451, 1988. 294. Kalra P, Das V, Agarwal A, Kumar M, Ramesh V, Bhatia E, Gupta S, Singh S, Saxena P, Bhatia V. Effect of vitamin D supplementation during pregnancy on neonatal mineral homeostasis and anthropometry of the newborn and infant. Br J Nutr 108: 1052–1058, 2012. 295. Kaplan EL, Burrington JD, Klementschitsch P, Taylor J, Deftos L. Primary hyperparathyroidism, pregnancy, and neonatal hypocalcemia. Surgery 96: 717–722, 1984. 296. Karaplis AC, He B, Nguyen MT, Young ID, Semeraro D, Ozawa H, Amizuka N. Inactivating mutation in the human parathyroid hormone receptor type 1 gene in Blomstrand chondrodysplasia. Endocrinology 139: 5255–5258, 1998. 313. Kitazawa R, Kitazawa S, Fukase M, Fujita T, Kobayashi A, Chihara K, Maeda S. The expression of parathyroid hormone-related protein (PTHrP) in normal parathyroid: histochemistry and in situ hybridization. Histochemistry 98: 211–215, 1992. 314. Kitazawa R, Kitazawa S, Maeda S, Kobayashi A. Expression of parathyroid hormonerelated protein (PTHrP) in parathyroid tissue under normal and pathological conditions. Histol Histopathol 17: 179 –184, 2002. 315. Klem KK. Placental transmission of 32P in late pregnancy and in experimental prolongation of pregnancy in rats. Acta Obstet Gynecol Scand 35: 445– 454, 1956. 316. Klopocki E, Hennig BP, Dathe K, Koll R, de Ravel T, Baten E, Blom E, Gillerot Y, Weigel JF, Kruger G, Hiort O, Seemann P, Mundlos S. Deletion and point mutations of PTHLH cause brachydactyly type E. Am J Hum Genet 86: 434 – 439, 2010. 317. Kobayashi A, Kawai S, Obe Y, Nagashima Y. Effects of dietary lactose and lactase preparation on the intestinal absorption of calcium and magnesium in normal infants. Am J Clin Nutr 28: 681– 683, 1975. 318. Kocian J, Skala I, Bakos K. Calcium absorption from milk and lactose-free milk in healthy subjects and patients with lactose intolerance. Digestion 9: 317–324, 1973. 319. Koeslag JH, Saunders PT, Wessels JA. The chromogranins and the counter-regulatory hormones: do they make homeostatic sense? J Physiol 517: 643– 649, 1999. 297. Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, Kronenberg HM, Mulligan RC. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev 8: 277–289, 1994. 320. Kokkonen J, Koivisto M, Lautala P, Kirkinen P. Serum calcium and 25-OH-D3 in mothers of newborns with craniotabes. J Perinat Med 11: 127–131, 1983. 298. Karlén J, Aperia A, Zetterstrüm R. Renal excretion of calcium and phosphate in preterm and term infants. J Pediatr 106: 814 – 819, 1985. 321. Kollars J, Zarroug AE, van Heerden J, Lteif A, Stavlo P, Suarez L, Moir C, Ishitani M, Rodeberg D. Primary hyperparathyroidism in pediatric patients. Pediatrics 115: 974 – 980, 2005. 299. Karmali R, Schiffmann SN, Vanderwinden JM, Hendy GN, Nys-DeWolf N, Corvilain J, Bergmann P, Vanderhaeghen JJ. Expression of mRNA of parathyroid hormone-related peptide in fetal bones of the rat. Cell Tissue Res 270: 597– 600, 1992. 300. Karsenty G, Kronenberg HM, Settembre C. Genetic control of bone formation. Annu Rev Cell Dev Biol 25: 629 – 648, 2009. 322. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveirados-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397: 315–323, 1999. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1207 CHRISTOPHER S. KOVACS 323. Koo WW, Sherman R, Succop P, Ho M, Buckley D, Tsang RC. Serum vitamin D metabolites in very low birth weight infants with and without rickets and fractures. J Pediatr 114: 1017–1022, 1989. 324. Koo WW, Sherman R, Succop P, Oestreich AE, Tsang RC, Krug-Wispe SK, Steichen JJ. Sequential bone mineral content in small preterm infants with and without fractures and rickets. J Bone Miner Res 3: 193–197, 1988. 325. Koo WW, Tsang RC, Poser JW, Laskarzewski P, Buckley D, Johnson R, Steichen JJ. Elevated serum calcium and osteocalcin levels from calcitriol in preterm infants. A prospective randomized study. Am J Dis Child 140: 1152–1158, 1986. 326. Kooh SW, Vieth R. 25-Hydroxyvitamin D metabolism in the sheep fetus and lamb. Pediatr Res 14: 360 –363, 1980. 327. Kos CH, Karaplis AC, Peng JB, Hediger MA, Goltzman D, Mohammad KS, Guise TA, Pollak MR. The calcium-sensing receptor is required for normal calcium homeostasis independent of parathyroid hormone. J Clin Invest 111: 1021–1028, 2003. 328. Kovacs CS. Calcium and bone metabolism disorders during pregnancy and lactation. Endocrinol Metab Clin N Am 40: 795– 826, 2011. 329. Kovacs CS. Fetal control of calcium and phosphate homeostasis: lessons from mouse models. In: Genetics of Bone Biology and Skeletal Disease, edited by Thakker RV, Whyte MP, Eisman JA, and Igarashi T. San Diego: Academic Press/Elsevier, 2012, p. 205–220. 330. Kovacs CS. Fetal Mineral Homeostasis. In: Pediatric Bone: Biology and Diseases (2nd ed.), edited by Glorieux FH, Pettifor JM, and J̈uppner H. San Diego: Elsevier/Academic, 2011, p. 247–275. 331. Kovacs CS. Fetal mineral homeostasis. In: Pediatric Bone: Biology and Diseases, edited by Glorieux FH, Pettifor JM, J̈uppner H. San Diego: Academic, 2003, p. 271–302. 332. Kovacs CS. Fetus, neonate and infant. In: Vitamin D (3rd ed.), edited by Feldman D, Pike WJ, and Adams JS. New York: Academic, 2011, p. 625– 646. 333. Kovacs CS. Interactions of PTHrP with receptors and signaling. In: The Parathyroids (3rd ed.), edited by Bilezikian JP, Marcus R, Levine MA, Marcocci C, Potts JT Jr, Silverberg SJ. San Diego, CA: Elsevier, 2014. 334. Kovacs CS. The role of PTHrP in regulating mineral metabolism during pregnancy, lactation, and fetal/neonatal development. Clin Rev Bone Miner Metab 12: 142–164, 2014. 335. Kovacs CS. The role of vitamin D in pregnancy and lactation: insights from animal models and clinical studies. Annu Rev Nutr 32: 9.1.27142–9, 2012. 336. Kovacs CS, Chafe LL, Fudge NJ, Friel JK, Manley NR. PTH regulates fetal blood calcium and skeletal mineralization independently of PTHrP. Endocrinology 142: 4983– 4993, 2001. 337. Kovacs CS, Chafe LL, Woodland ML, McDonald KR, Fudge NJ, Wookey PJ. Calcitropic gene expression suggests a role for intraplacental yolk sac in maternal-fetal calcium exchange. Am J Physiol Endocrinol Metab 282: E721–E732, 2002. 338. Kovacs CS, Ho-Pao CL, Hunzelman JL, Lanske B, Fox J, Seidman JG, Seidman CE, Kronenberg HM. Regulation of murine fetal-placental calcium metabolism by the calcium-sensing receptor. J Clin Invest 101: 2812–2820, 1998. 339. Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium and lactation. Endocr Rev 18: 832– 872, 1997. 340. Kovacs CS, Lanske B, Hunzelman JL, Guo J, Karaplis AC, Kronenberg HM. Parathyroid hormone-related peptide (PTHrP) regulates fetal-placental calcium transport through a receptor distinct from the PTH/PTHrP receptor. Proc Natl Acad Sci USA 93: 15233–15238, 1996. 341. Kovacs CS, Manley NR, Moseley JM, Martin TJ, Kronenberg HM. Fetal parathyroids are not required to maintain placental calcium transport. J Clin Invest 107: 1007–1015, 2001. 342. Kovacs CS, Woodland ML, Fudge NJ, Friel JK. The vitamin D receptor is not required for fetal mineral homeostasis or for the regulation of placental calcium transfer. Am J Physiol Endocrinol Metab 289: E133–E144, 2005. 343. Kovarik J, Woloszczuk W, Linkesch W, Pavelka R. Calcitonin in pregnancy (Letter). Lancet 1: 199 –200, 1980. 344. Krukowski M, Kahn AJ. The role of parathyroid hormone in mineral homeostasis and bone modeling in suckling rat pups. Metab Bone Dis Relat Res 2: 257–260, 1980. 1208 345. Krukowski M, Lehr D. Parathyroid hormone and the placental barrier. Arch Int Pharmacodyn 146: 245–265, 1963. 346. Krukowski M, Smith JJ. Acidosis, hypercalcemia, and hyperphosphatemia in rat fetuses near term and effects of maternal acid/base loading. Proc Soc Exp Biol Med 162: 359 –364, 1979. 347. Krukowski M, Smith JJ. pH and the level of calcium in the blood of fetal and neonatal albino rats. Biol Neonate 29: 148 –161, 1976. 348. Kukreja SC, D’Anza JJ, Melton ME, Wimbiscus SA, Grill V, Martin TJ. Lack of effects of neutralization of parathyroid hormone-related protein on calcium homeostasis in neonatal mice. J Bone Miner Res 6: 1197–1201, 1991. 349. Kumar S, Zhu LJ, Polihronis M, Cameron ST, Baird DT, Schatz F, Dua A, Ying YK, Bagchi MK, Bagchi IC. Progesterone induces calcitonin gene expression in human endometrium within the putative window of implantation. J Clin Endocrinol Metab 83: 4443– 4450, 1998. 350. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281: 6120 – 6123, 2006. 351. Kutuzova GD, Akhter S, Christakos S, Vanhooke J, Kimmel-Jehan C, Deluca HF. Calbindin D(9k) knockout mice are indistinguishable from wild-type mice in phenotype and serum calcium level. Proc Natl Acad Sci USA 103: 12377–12381, 2006. 352. Lachenmaier-Currle U, Breves G, Harmeyer J. Role of 1,25-(OH)2D3 during pregnancy; studies with pigs suffering from pseudo-vitamin D-deficiency rickets, type I. Q J Exp Physiol 74: 875– 881, 1989. 353. Lachenmaier-Currle U, Harmeyer J. Placental transport of calcium and phosphorus in pigs. J Perinat Med 17: 127–136, 1989. 354. Lafond J, Auger D, Fortier J, Brunette MG. Parathyroid hormone receptor in human placental syncytiotrophoblast brush border and basal plasma membranes. Endocrinology 123: 2834 –2840, 1988. 355. Lafond J, Ayotte N, Brunette MG. Effect of (1–34) parathyroid hormone-related peptide on the composition and turnover of phospholipids in syncytiotrophoblast brush border and basal plasma membranes of human placenta. Mol Cell Endocrinol 92: 207–214, 1993. 356. Lafond J, Simoneau L, Savard R, Lajeunesse D. Calcitonin receptor in human placental syncytiotrophoblast brush border and basal plasma membranes. Mol Cell Endocrinol 99: 285–292, 1994. 357. Lagiou P, Samoli E, Okulicz W, Xu B, Lagiou A, Lipworth L, Georgila C, Vatten L, Adami HO, Trichopoulos D, Hsieh CC. Maternal and cord blood hormone levels in the United States and China and the intrauterine origin of breast cancer. Ann Oncol 22: 1102–1108, 2011. 358. Lamm CI, Norton KI, Murphy RJ, Wilkins IA, Rabinowitz JG. Congenital rickets associated with magnesium sulfate infusion for tocolysis. J Pediatr 113: 1078 –1082, 1988. 359. Lanske B, Divieti P, Kovacs CS, Pirro A, Landis WJ, Krane SM, Bringhurst FR, Kronenberg HM. The parathyroid hormone/parathyroid hormone-related peptide receptor mediates actions of both ligands in murine bone. Endocrinology 139: 5192–5204, 1998. 360. Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize L, Ho C, Abou-Samra AB, Jüppner H, Segre GV, Kronenberg HM. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 273: 663– 666, 1996. 361. Laplace C, Li X, Goldring SR, Galson DL. Homozygous deletion of the murine calcitonin receptor gene is an embryonic lethal (Abstract). J Bone Miner Res 17 Suppl: S166, 2002. 362. Laramee M, Simoneau L, Lafond J. Phospholipase C axis is the preferential pathway leading to PKC activation following PTH or PTHrP stimulation in human term placenta. Life Sci 72: 215–225, 2002. 363. Lawlor DA, Wills AK, Fraser A, Sayers A, Fraser WD, Tobias JH. Association of maternal vitamin D status during pregnancy with bone-mineral content in offspring: a prospective cohort study. Lancet 381: 2176 –2183, 2013. 364. Lee GS, Lee KY, Choi KC, Ryu YH, Paik SG, Oh GT, Jeung EB. Phenotype of a calbindin-D9k gene knockout is compensated for by the induction of other calcium transporter genes in a mouse model. J Bone Miner Res 22: 1968 –1978, 2007. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM 365. Lee K, Deeds JD, Segre GV. Expression of parathyroid hormone-related peptide and its receptor messenger ribonucleic acids during fetal development of rats. Endocrinology 136: 453– 463, 1995. 385. Liu Z, Yu S, Manley NR. Gcm2 is required for the differentiation and survival of parathyroid precursor cells in the parathyroid/thymus primordia. Dev Biol 305: 333– 346, 2007. 366. Leichter J, Tolensky AF. Effect of dietary lactose on the absorption of protein, fat and calcium in the postweaning rat. Am J Clin Nutr 28: 238 –241, 1975. 386. Loughead JL, Mimouni F, Ross R, Tsang RC. Postnatal changes in serum osteocalcin and parathyroid hormone concentrations. J Am Coll Nutr 9: 358 –362, 1990. 367. Leroyer-Alizon E, David L, Anast CS, Dubois PM. Immunocytological evidence for parathyroid hormone in human fetal parathyroid glands. J Clin Endocrinol Metab 52: 513–516, 1981. 387. Loughead JL, Mimouni F, Tsang RC. Serum ionized calcium concentrations in normal neonates. Am J Dis Child 142: 516 –518, 1988. 368. Leroyer-Alizon E, David L, Dubois PM. Evidence for calcitonin in the thyroid gland of normal and anencephalic human fetuses: immunocytological localization, radioimmunoassay, and gel filtration of thyroid extracts. J Clin Endocrinol Metab 50: 316 –321, 1980. 369. Lester GE, Gray TK, Lorenc RS. Evidence for maternal and fetal differences in vitamin D metabolism. Proc Soc Exp Biol Med 159: 303–307, 1978. 370. Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, McCabe S, Elliott R, Scully S, Van G, Kaufman S, Juan SC, Sun Y, Tarpley J, Martin L, Christensen K, McCabe J, Kostenuik P, Hsu H, Fletcher F, Dunstan CR, Lacey DL, Boyle WJ. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA 97: 1566 – 1571, 2000. 371. Li YC, Amling M, Pirro AE, Priemel M, Meuse J, Baron R, Delling G, Demay MB. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology 139: 4391– 4396, 1998. 372. Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. Targeted ablation of the vitamin D receptor: an animal model of vitamin D dependent rickets type II with alopecia. Proc Natl Acad Sci USA 94: 9831–9835, 1997. 373. Li YC, Pirro AE, Demay MB. Analysis of vitamin D-dependent calcium-binding protein messenger ribonucleic acid expression in mice lacking the vitamin D receptor. Endocrinology 139: 847– 851, 1998. 374. Liberman UA, Samuel R, Halabe A, Kauli R, Edelstein S, Weisman Y, Papapoulos SE, Clemens TL, Fraher LJ, O’Riordan JL. End-organ resistance to 1,25-dihydroxycholecalciferol. Lancet 1: 504 –506, 1980. 375. Lieben L, Masuyama R, Torrekens S, Van Looveren R, Schrooten J, Baatsen P, LafageProust MH, Dresselaers T, Feng JQ, Bonewald LF, Meyer MB, Pike JW, Bouillon R, Carmeliet G. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J Clin Invest 122: 1803–1815, 2012. 376. Lieben L, Stockmans I, Moermans K, Carmeliet G. Maternal hypervitaminosis D reduces fetal bone mass and mineral acquisition and leads to neonatal lethality. Bone 57: 123–131, 2013. 377. Lima MS, Kallfelz F, Krook L, Nathanielsz PW. Humeral skeletal development and plasma constituent changes in fetuses of ewes maintained on a low calcium diet from 60 days of gestation. Calcif Tissue Int 52: 283–290, 1993. 378. Linarelli LG. Newborn urinary cyclic AMP and developmental renal responsiveness to parathyroid hormone. Pediatrics 50: 14 –23, 1972. 379. Lipsitz PJ. The clinical and biochemical effects of excess magnesium in the newborn. Pediatrics 47: 501–509, 1971. 380. Littledike ET, Arnaud CD, Whipp SC. Calcitonin secretion in ovine, porcine, and bovine fetuses. Proc Soc Exp Biol Med 139: 428 – 433, 1972. 388. Loughead JL, Mimouni F, Tsang RC, Khoury JC. A role for magnesium in neonatal parathyroid gland function? J Am Coll Nutr 10: 123–126, 1991. 389. Loughead JL, Mughal Z, Mimouni F, Tsang RC, Oestreich AE. Spectrum and natural history of congenital hyperparathyroidism secondary to maternal hypocalcemia. Am J Perinatol 7: 350 –355, 1990. 390. Loveridge N, Caple IW, Rodda C, Martin TJ, Care AD. Further evidence for a parathyroid hormone-related protein in fetal parathyroid glands of sheep. Q J Exp Physiol 73: 781–784, 1988. 391. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA 90: 11162–11166, 1993. 392. Lubetzky R, Weisman Y, Dollberg S, Herman L, Mandel D. Parathyroid hormonerelated protein in preterm human milk. Breastfeed Med 5: 67– 69, 2010. 393. Ludwig GD. Hyperparathyroidism in relation to pregnancy. N Engl J Med 267: 637– 642, 1962. 394. Lutz P, Kane O, Pfersdorff A, Seiller F, Sauvage P, Levy JM. Neonatal primary hyperparathyroidism: total parathyroidectomy with autotransplantation of cryopreserved parathyroid tissue. Acta Paediatr Scand 75: 179 –182, 1986. 395. Ma Y, Samaraweera M, Cooke-Hubley S, Kirby BJ, Karaplis AC, Lanske B, Kovacs CS. Neither absence nor excess of FGF23 disturbs murine fetal-placental phosphorus homeostasis or prenatal skeletal development and mineralization. Endocrinology 155: 1596 –1605, 2014. 396. MacIsaac RJ, Caple IW, Danks JA, Diefenbach-Jagger H, Grill V, Moseley JM, Southby J, Martin TJ. Ontogeny of parathyroid hormone-related protein in the ovine parathyroid gland. Endocrinology 129: 757–764, 1991. 397. MacIsaac RJ, Heath JA, Rodda CP, Moseley JM, Care AD, Martin TJ, Caple IW. Role of the fetal parathyroid glands and parathyroid hormone-related protein in the regulation of placental transport of calcium, magnesium and inorganic phosphate. Reprod Fertil Dev 3: 447– 457, 1991. 398. MacIsaac RJ, Horne RS, Caple IW, Martin TJ, Wintour EM. Effects of thyroparathyroidectomy, parathyroid hormone, and PTHrP on kidneys of ovine fetuses. Am J Physiol Endocrinol Metab 264: E37–E44, 1993. 399. Mahon P, Harvey N, Crozier S, Inskip H, Robinson S, Arden N, Swaminathan R, Cooper C, Godfrey K. Low maternal vitamin D status and fetal bone development: cohort study. J Bone Miner Res 25: 14 –19, 2010. 400. Maiyegun SO, Malek AH, Devarajan LV, Dahniya MH. Severe congenital rickets secondary to maternal hypovitaminosis D: a case report. Ann Trop Paediatr 22: 191–195, 2002. 401. Major BJ, Ho PM, Moseley JM, Martin TJ, Leaver DD. Circulating levels of parathyroid hormone related protein in neonates (Abstract). Bone 16: 204S, 1995. 381. Liu D. Pathological and X-ray study on bony specimens of rickets from 124 fetal and infantile autopsies. Zhonghua yi xue za zhi 71: 385–387, 328, 1991. 402. Malaeb SN, Rassi AI, Haddad MC, Seoud MA, Yunis KA. Bone mineralization in newborns whose mothers received magnesium sulphate for tocolysis of premature labour. Pediatr Radiol 34: 384 –386, 2004. 382. Liu JH. Endocrinology of pregnancy. In: Creasy and Resnik’s Maternal-Fetal Medicine: Principles and Practice (7th ed.), edited by Creasy RK, Resnik R, Iams JD, Lockwood CJ, Moore TR, Greene MF. Philadelphia, PA: Elsevier, 2014, p. 100 –111. 403. Malan AJ. Studies in mineral metabolism. VIII. Comparison of phosphorus partition in the blood of calf foetus, sheep foetus, and lambs, with corresponding maternal blood. J Agric Sci 18: 397– 400, 1928. 383. Liu NQ, Hewison M. Vitamin D, the placenta and pregnancy. Arch Biochem Biophys 253: 37– 47, 2012. 404. Malassine A, Frendo JL, Evain-Brion D. A comparison of placental development and endocrine functions between the human and mouse model. Hum Reprod Update 9: 531–539, 2003. 384. Liu Z, Farley A, Chen L, Kirby BJ, Blackburn CC, Kovacs CS, Manley NR. Thymusassociated parathyroid hormone has two cellular origins with distinct endocrine and immunological functions. PLoS Genet 6: e1001251, 2010. 405. Mallet E. Primary hyperparathyroidism in neonates and childhood. The French experience (1984 –2004). Horm Res 69: 180 –188, 2008. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1209 CHRISTOPHER S. KOVACS 406. Mallet E, Basuyau JP, Brunelle P, Devaux AM, Fessard C. Neonatal parathyroid secretion and renal receptor maturation in premature infants. Biol Neonate 33: 304 – 308, 1978. 428. Maxwell JP, Pi HT, Lin HA, Kuo CC. Further studies in adult rickets (osteomalacia) and foetal rickets: section of obstetrics and gynaecology. Proc R Soc Med 32: 287–297, 1939. 407. Mallet E, Gugi B, Brunelle P, Henocq A, Basuyau JP, Lemeur H. Vitamin D supplementation in pregnancy: a controlled trial of two methods. Obstet Gynecol 68: 300 – 304, 1986. 429. Maxwell JP, Turnbull HM. Two cases of foetal rickets. J Pathol Bacteriol 33: 327–332, 1930. 408. Malloy PJ, Tiosano D, Feldman D. Hereditary 1,25-dihydroxyvitamin-D-resistant rickets. In: Vitamin D (3rd ed.), edited by Feldman D, Pike JW, Adams JS. San Diego, CA: Academic, 2011, p. 1197–1232. 409. Mamillapalli R, VanHouten J, Dann P, Bikle D, Chang W, Brown E, Wysolmerski J. Mammary-specific ablation of the calcium-sensing receptor during lactation alters maternal calcium metabolism, milk calcium transport, and neonatal calcium accrual. Endocrinology 154: 3031–3042, 2013. 410. Mangin M, Webb AC, Dreyer BE, Posillico JT, Ikeda K, Weir EC, Stewart AF, Bander NH, Milstone L, Barton DE, Francke U, Broadus AE. Identification of a cDNA encoding a parathyroid hormone-like peptide from a human tumor associated with humoral hypercalcemia of malignancy. Proc Natl Acad Sci USA 85: 597– 601, 1988. 411. Manley NR, Capecchi MR. The role of Hoxa-3 in mouse thymus and thyroid development. Development 121: 1989 –2003, 1995. 412. Mantovani G. Clinical review: pseudohypoparathyroidism: diagnosis and treatment. J Clin Endocrinol Metab 96: 3020 –3030, 2011. 413. Marche P, Delorme A, Cuisinier-Gleizes P. Intestinal and placental calcium-binding proteins in vitamin D-deprived or -supplemented rats. Life Sci 23: 2555–2561, 1978. 414. Marcovitch H, Mughal MZ. Cases do not support temporary brittle bone disease. Acta Paediatr 99: 485– 486, 2010. 415. Marsell R, Jonsson KB. The phosphate regulating hormone fibroblast growth factor23. Acta Physiol 200: 97–106, 2010. 430. McDonald KR, Fudge NJ, Woodrow JP, Friel JK, Hoff AO, Gagel RF, Kovacs CS. Ablation of calcitonin/calcitonin gene related peptide-␣ impairs fetal magnesium but not calcium homeostasis. Am J Physiol Endocrinol Metab 287: E218 –E226, 2004. 431. McDonnell CM, Zacharin MR. Maternal primary hyperparathyroidism: discordant outcomes in a twin pregnancy. J Paediatr Child Health 42: 70 –71, 2006. 432. McIntosh N, De Curtis M, Williams J. Failure of mineral supplementation to reduce incidence of rickets in very-low-birthweight infants (Letter). Lancet 2: 981–982, 1986. 433. McKercher HG, Derewlany LO, Radde IC. Free calcium concentrations in KrebsRinger bicarbonate buffer: effects on 45Ca- and 32P-transport across the perfused guinea pig placenta. Biochem Biophys Res Commun 105: 841– 846, 1982. 434. Mehta KC, Kalkwarf HJ, Mimouni F, Khoury J, Tsang RC. Randomized trial of magnesium administration to prevent hypocalcemia in infants of diabetic mothers. J Perinatol 18: 352–356, 1998. 435. Mellor DJ, Matheson IC. Variations in the distribution of calcium, magnesium and inorganic phosphorus within chronically catheterized sheep conceptuses during the last eight weeks of pregnancy. Q J Exp Physiol Cogn Med Sci 62: 55– 63, 1977. 436. Mendelson KL. Critical review of “temporary brittle bone disease.” Pediatr Radiol 35: 1036 –1040, 2005. 437. Menon RK, Sperling MA. Developmental endocrinology in the fetal-placental unit. In: Principles of Perinatal-Neonatal Metabolism (2nd ed.), edited by Cowett RM. New York: Springer-Verlag, 1998, p. 425– 436. 416. Martinez ME, Catalan P, Lisbona A, Sanchez-Cabezudo MJ, Pallardo F, Jans I, Bouillon R. Serum osteocalcin concentrations in diabetic pregnant women and their newborns. Horm Metab Res 26: 338 –342, 1994. 438. Miao D, He B, Jiang Y, Kobayashi T, Soroceanu MA, Zhao J, Su H, Tong X, Amizuka N, Gupta A, Genant HK, Kronenberg HM, Goltzman D, Karaplis AC. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1–34. J Clin Invest 115: 2402–2411, 2005. 417. Marx SJ, Attie MF, Levine MA, Spiegel AM, Downs RW Jr, Lasker RD. The hypocalciuric or benign variant of familial hypercalcemia: clinical and biochemical features in fifteen kindreds. Medicine 60: 397– 412, 1981. 439. Miao D, He B, Karaplis AC, Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J Clin Invest 109: 1173–1182, 2002. 418. Marx SJ, Zusman RM, Umiker WO. Benign breast dysplasia causing hypercalcemia. J Clin Endocrinol Metab 45: 1049 –1052, 1977. 440. Miao D, He B, Lanske B, Bai XY, Tong XK, Hendy GN, Goltzman D, Karaplis AC. Skeletal abnormalities in Pth-null mice are influenced by dietary calcium. Endocrinology 145: 2046 –2053, 2004. 419. Marya RK, Rathee S, Dua V, Sangwan K. Effect of vitamin D supplementation during pregnancy on foetal growth. Indian J Med Res 88: 488 – 492, 1988. 420. Marya RK, Rathee S, Lata V, Mudgil S. Effects of vitamin D supplementation in pregnancy. Gynecol Obstet Invest 12: 155–161, 1981. 421. Masuyama R, Stockmans I, Torrekens S, Van Looveren R, Maes C, Carmeliet P, Bouillon R, Carmeliet G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest 116: 3150 –3159, 2006. 422. Math F, Davrainville JL. Postnatal variations of extracellular free calcium levels in the rat. Influence of undernutrition. Experientia 35: 1355–1356, 1979. 423. Mathieu CL, Burnett SH, Mills SE, Overpeck JG, Bruns DE, Bruns ME. Gestational changes in calbindin-D9k in rat uterus, yolk sac, and placenta: implications for maternal-fetal calcium transport and uterine muscle function. Proc Natl Acad Sci USA 86: 3433–3437, 1989. 424. Matsushita H, Hara M, Honda K, Kuroda M, Usui M, Nakazawa H, Hara S, Shishiba Y. Inhibition of parathyroid hormone-related protein release by extracellular calcium in dispersed cells from human parathyroid hyperplasia secondary to chronic renal failure and adenoma. Am J Pathol 146: 1521–1528, 1995. 441. Miao D, He B, Tong XK, Goltzman D, Karaplis AC. Conditional knockout of PTHrP in osteoblasts leads to premature osteoporosis (Abstract). J Bone Miner Res 17, Suppl 1: S138, 2003. 442. Miao D, Li J, Xue Y, Su H, Karaplis AC, Goltzman D. Parathyroid hormone-related peptide is required for increased trabecular bone volume in parathyroid hormone-null mice. Endocrinology 145: 3554 –3562, 2004. 443. Miller SC, Halloran BP, DeLuca HF, Jee WS. Studies on the role of vitamin D in early skeletal development, mineralization, and growth in rats. Calcif Tissue Int 35: 455– 460, 1983. 444. Mimouni F, Loughead JL, Tsang RC, Khoury J. Postnatal surge in serum calcitonin concentrations: no contribution to neonatal hypocalcemia in infants of diabetic mothers. Pediatr Res 28: 493– 495, 1990. 445. Mimouni FB, Mandel D, Lubetzky R, Senterre T. Calcium, phosphorus, magnesium and vitamin D requirements of the preterm infant. World Rev Nutr Dietetics 110: 140 –151, 2014. 446. Minton SD, Steichen JJ, Tsang RC. Bone mineral content in term and preterm appropriate-for-gestational-age infants. J Pediatr 95: 1037–1042, 1979. 426. Maxwell JP, Hu CH, Turnbull HM. Foetal rickets. J Pathol Bacteriol 35: 419 – 440, 1932. 447. Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, Gomibuchi T, Yano K, Shima N, Washida N, Tsuda E, Morinaga T, Higashio K, Ozawa H. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun 247: 610 – 615, 1998. 427. Maxwell JP, Miles LM. Osteomalacia in China. J Obstet Gynaecol Br Empire 32: 433– 473, 1925. 448. Mohapatra A, Sankaranarayanan K, Kadam SS, Binoy S, Kanbur WA, Mondkar JA. Congenital rickets. J Trop Pediatr 49: 126 –127, 2003. 425. Maxwell JD, Ang L, Brooke OG, Brown IR. Vitamin D supplements enhance weight gain and nutritional status in pregnant Asians. Br J Obstet Gynaecol 88: 987–991, 1981. 1210 Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM 449. Mohn A, De Leonibus C, de Giorgis T, Mornet E, Chiarelli F. Hypophosphatasia in a child with widened anterior fontanelle: lessons learned from late diagnosis and incorrect treatment. Acta Paediatr 100: e43– 46, 2011. 450. Molgaard C, Larnkjaer A, Mark AB, Michaelsen KF. Are early growth and nutrition related to bone health in adolescence? The Copenhagen Cohort Study of infant nutrition and growth. Am J Clin Nutr 94: 1865s–1869s, 2011. 451. Moncrieff M, Fadahunsi TO. Congenital rickets due to maternal vitamin D deficiency. Arch Dis Child 49: 810 – 811, 1974. 452. Moncrieff MW. Early biochemical findings in familial hypophosphataemic, hyperphosphaturic rickets and response to treatment. Arch Dis Child 57: 70 –72, 1982. 453. Moniz CF, Nicolaides KH, Tzannatos C, Rodeck CH. Calcium homeostasis in second trimester fetuses. J Clin Pathol 39: 838 – 841, 1986. 454. Montoro MN, Paler RJ, Goodwin TM, Mestman JH. Parathyroid carcinoma during pregnancy. Obstet Gynecol 96: 841, 2000. 455. Moore ES, Langman CB, Favus MJ, Coe FL. Role of fetal 1,25-dihydroxyvitamin D production in intrauterine phosphorus and calcium homeostasis. Pediatr Res 19: 566 – 569, 1985. 456. Moreau R, Daoud G, Masse A, Simoneau L, Lafond J. Expression and role of calciumATPase pump and sodium-calcium exchanger in differentiated trophoblasts from human term placenta. Mol Reprod Dev 65: 283–288, 2003. 457. Morgan G, Wooding FB, Care AD, Jones GV. Genetic regulation of placental function: a quantitative in situ hybridization study of calcium binding protein (calbindin-D9k) and calcium ATPase mRNAs in sheep placenta. Placenta 18: 211–218, 1997. 458. Morisset AS, Dube MC, Drolet R, Pelletier M, Labrie F, Luu-The V, Tremblay Y, Robitaille J, John Weisnagel S, Tchernof A. Androgens in the maternal and fetal circulation: association with insulin resistance. J Matern Fetal Neonatal Med 26: 513–519, 2013. 459. Moritz KM, Macris M, Talbo G, Wintour EM. Foetal fluid balance and hormone status following nephrectomy in the foetal sheep. Clin Exp Pharmacol Physiol 26: 857– 864, 1999. 460. Morley R, Carlin JB, Pasco JA, Wark JD. Maternal 25-hydroxyvitamin D and parathyroid hormone concentrations and offspring birth size. J Clin Endocrinol Metab 91: 906 –912, 2006. 461. Motelica-Heino I, Castanier M, Corbier P, Edwards DA, Roffi J. Testosterone levels in plasma and testes of neonatal mice. J Steroid Biochem 31: 283–286, 1988. 462. Mueller SO, Korach KS. Estrogen receptors and endocrine diseases: lessons from estrogen receptor knockout mice. Curr Opin Pharmacol 1: 613– 619, 2001. 463. Mull JW, Bill AH. Inorganic phosphorus content of prenatal and post-partum serum. Am J Obstet Gynecol 23: 807– 814, 1932. 464. Musso MJ, Plante M, Judes C, Barthelmebs M, Helwig JJ. Renal vasodilatation and microvessel adenylate cyclase stimulation by synthetic parathyroid hormone-like protein fragments. Eur J Pharmacol 174: 139 –151, 1989. somatic mosaicism in a mother with two affected sons. Am J Med Genet Part A 152: 2784 –2790, 2010. 471. Nicholson GC, D’Santos CS, Evans T, Moseley JM, Kemp BE, Michelangeli VP, Martin TJ. Human placental calcitonin receptors. Biochem J 250: 877– 882, 1988. 472. Nikitenko L, Morgan G, Kolesnikov SI, Wooding FB. Immunocytochemical and In situ hybridization studies of the distribution of calbindin D9k in the bovine placenta throughout pregnancy. J Histochem Cytochem 46: 679 – 688, 1998. 473. Nishioka T, Yasuda T, Niimi H. A discordant movement in urine calcium excretion in relation to serum calcium and parathyroid function occurring immediately after birth. Acta Paediatr Scand 80: 590 –595, 1991. 474. Nishiyama I, Fujii T. Developmental change in calcitonin secretory capacity of fetal rat thyroid C-cells. Jpn J Pharmacol 52: 149 –153, 1990. 475. Noff D, Edelstein S. Vitamin D and its hydroxylated metabolites in the rat. Placental and lacteal transport, subsequent metabolic pathways and tissue distribution. Horm Res 9: 292–300, 1978. 476. Noonan WT, Qian J, Stuart WD, Clemens TL, Lorenz JN. Altered renal hemodynamics in mice overexpressing the parathyroid hormone (PTH)/PTH-related peptide type 1 receptor in smooth muscle. Endocrinology 144: 4931– 4938, 2003. 477. Norman J, Politz D, Politz L. Hyperparathyroidism during pregnancy and the effect of rising calcium on pregnancy loss: a call for earlier intervention. Clin Endocrinol 71: 104 –109, 2009. 478. Northrop G, Misenhimer HR, Becker FO. Failure of parathyroid hormone to cross the nonhuman primate placenta. Am J Obstet Gynecol 129: 449 – 453, 1977. 479. Novakovic B, Sibson M, Ng HK, Manuelpillai U, Rakyan V, Down T, Beck S, Fournier T, Evain-Brion D, Dimitriadis E, Craig JM, Morley R, Saffery R. Placenta-specific methylation of the vitamin D 24-hydroxylase gene: implications for feedback autoregulation of active vitamin D levels at the fetomaternal interface. J Biol Chem 284: 14838 –14848, 2009. 480. O’Leary P, Boyne P, Flett P, Beilby J, James I. Longitudinal assessment of changes in reproductive hormones during normal pregnancy. Clin Chem 37: 667– 672, 1991. 481. Obermannova B, Banghova K, Sumnik Z, Dvorakova HM, Betka J, Fencl F, Kolouskova S, Cinek O, Lebl J. Unusually severe phenotype of neonatal primary hyperparathyroidism due to a heterozygous inactivating mutation in the CASR gene. Eur J Pediatr 168: 569 –573, 2009. 482. Ogura Y, Takakura N, Yoshida H, Nishikawa SI. Essential role of platelet-derived growth factor receptor alpha in the development of the intraplacental yolk sac/sinus of Duval in mouse placenta. Biol Reprod 58: 65–72, 1998. 483. Ohata Y, Arahori H, Namba N, Kitaoka T, Hirai H, Wada K, Nakayama M, Michigami T, Imura A, Nabeshima Y, Yamazaki Y, Ozono K. Circulating levels of soluble alphaKlotho are markedly elevated in human umbilical cord blood. J Clin Endocrinol Metab 96: 943–947, 2011. 465. Nagata C, Iwasa S, Shiraki M, Shimizu H. Estrogen and alpha-fetoprotein levels in maternal and umbilical cord blood samples in relation to birth weight. Cancer Epidemiol Biomarkers Prevention 15: 1469 –1472, 2006. 484. Ohata Y, Yamazaki M, Kawai M, Tsugawa N, Tachikawa K, Koinuma T, Miyagawa K, Kimoto A, Nakayama M, Namba N, Yamamoto H, Okano T, Ozono K, Michigami T. Elevated fibroblast growth factor 23 exerts its effects on placenta and regulates vitamin D metabolism in pregnancy of Hyp mice. J Bone Miner Res. In press. 466. Naja RP, Dardenne O, Arabian A, St Arnaud R. Chondrocyte-specific modulation of Cyp27b1 expression supports a role for local synthesis of 1,25-dihydroxyvitamin D3 in growth plate development. Endocrinology 150: 4024 – 4032, 2009. 485. Okamoto E, Muttart CR, Zucker CL, Heird WC. Use of medium-chain triglycerides in feeding the low-birth-weight infant. Am J Dis Child 136: 428 – 431, 1982. 467. Nakajima K, Okazaki T, Okamoto T, Kimura H, Takano K, Sato K. Genes up- or down-regulated by high calcium medium in parathyroid tissue explants from patients with primary hyperparathyroidism. Endocr J 57: 153–159, 2010. 468. Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. FASEB J 23: 433– 441, 2009. 469. Naru T, Khan RS, Khan MA. Primary hyperparathyrodism in pregnancy and review of literature. J Pakistan Med Assoc 61: 401– 403, 2011. 470. Ngai YF, Chijiwa C, Mercimek-Mahmutoglu S, Stewart L, Yong SL, Robinson WP, Gibson WT. Pseudohypoparathyroidism type 1a and the GNAS p. R231H mutation: 486. Okunade GW, Miller ML, Pyne GJ, Sutliff RL, O’Connor KT, Neumann JC, Andringa A, Miller DA, Prasad V, Doetschman T, Paul RJ, Shull GE. Targeted ablation of plasma membrane Ca2⫹-ATPase (PMCA) 1 and 4 indicates a major housekeeping function for PMCA1 and a critical role in hyperactivated sperm motility and male fertility for PMCA4. J Biol Chem 279: 33742–33750, 2004. 487. Omelon S, Georgiou J, Henneman ZJ, Wise LM, Sukhu B, Hunt T, Wynnyckyj C, Holmyard D, Bielecki R, Grynpas MD. Control of vertebrate skeletal mineralization by polyphosphates. PLoS One 4: e5634, 2009. 488. Onda K, Yamaguchi M, Ohashi M, Sato R, Ochiai H, Iriki T, Wada Y. Modification of the analysis of parathyroid hormone-related protein in milk and concentrations of this protein in commercial milk and milk products in Japan. J Dairy Sci 93: 1861–1867, 2010. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1211 CHRISTOPHER S. KOVACS 489. Oostra RJ, van der Harten JJ, Rijnders WP, Scott RJ, Young MP, Trump D. Blomstrand osteochondrodysplasia: three novel cases and histological evidence for heterogeneity. Virchows Arch 436: 28 –35, 2000. 490. Orloff JJ, Reddy D, de Papp AE, Yang KH, Soifer NE, Stewart AF. Parathyroid hormone-related protein as a prohormone: posttranslational processing and receptor interactions. Endocr Rev 15: 40 – 60, 1994. 511. Perez-Brayfield MR, Kirsch AJ, Smith EA. Monoamniotic twin discordant for bilateral renal agenesis with normal pulmonary function. Urology 64: 589, 2004. 512. Pettifor JM. Nutritional rickets. In: Pediatric Bone: Biology and Diseases (2nd ed.), edited by Glorieux FH, Pettifor JM, and J̈uppner H. San Diego: Elsevier/Academic, 2011, p. 625– 654. 491. Owada C. Are there New-born Rickets? Tohoku J Exp Med 64: 37– 43, 1956. 513. Pettifor JM. Vitamin D and/or calcium deficiency rickets in infants and children: a global perspective. Indian J Med Res 127: 245–249, 2008. 492. Pagan YL, Hirschhorn J, Yang B, D’Souza-Li L, Majzoub JA, Hendy GN. Maternal activating mutation of the calcium-sensing receptor: implications for calcium metabolism in the neonate. J Pediatr Endocrinol Metab 17: 673– 677, 2004. 514. Pettifor JM. Vitamin D deficiency and nutritional rickets in children. In: Vitamin D (3rd ed.), edited by Feldman D, Pike WJ, and Adams JS. New York: Academic, 2011, p. 1107–1128. 493. Page LA, Haddow JE. Self-limited neonatal hyperparathyroidism in familial hypocalciuric hypercalcemia. J Pediatr 111: 261–264, 1987. 515. Pettifor JM, Isdale JM, Sahakian J, Hansen JD. Diagnosis of subclinical rickets. Arch Dis Child 55: 155–157, 1980. 494. Palmer PE. Tumoural calcinosis. Br J Radiol 39: 518 –525, 1966. 516. Pettifor JM, Pentopoulos M, Moodley GP, Isdale JM, Ross FP. Is craniotabes a pathognomonic sign of rickets in 3-month-old infants? S Afr Med J 65: 549 –551, 1984. 495. Panda DK, Miao D, Bolivar I, Li J, Huo R, Hendy GN, Goltzman D. Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem 279: 16754 –16766, 2004. 517. Philbrick WM, Wysolmerski JJ, Galbraith S, Holt E, Orloff JJ, Yang KH, Vasavada RC, Weir EC, Broadus AE, Stewart AF. Defining the roles of parathyroid hormone-related protein in normal physiology. Physiol Rev 76: 127–173, 1996. 496. Panda DK, Miao D, Tremblay ML, Sirois J, Farookhi R, Hendy GN, Goltzman D. Targeted ablation of the 25-hydroxyvitamin D 1alpha -hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci USA 98: 7498 – 7503, 2001. 497. Pang PK, Tenner TE Jr, Yee JA, Yang M, Janssen HF. Hypotensive action of parathyroid hormone preparations on rats and dogs. Proc Natl Acad Sci USA 77: 675– 678, 1980. 498. Pang SF, Caggiula AR, Gay VL, Goodman RL, Pang CS. Serum concentrations of testosterone, oestrogens, luteinizing hormone and follicle-stimulating hormone in male and female rats during the critical period of neural sexual differentiation. J Endocrinol 80: 103–110, 1979. 499. Pang SF, Tang F. Sex differences in the serum concentrations of testosterone in mice and hamsters during their critical periods of neural sexual differentiation. J Endocrinol 100: 7–11, 1984. 500. Pansu D, Chapuy MC, Milani M, Bellaton C. Transepithelial calcium transport enhanced by xylose and glucose in the rat jejunal ligated loop. Calcif Tissue Res 21 Suppl: 45–52, 1976. 501. Papantoniou NE, Papapetrou PD, Antsaklis AJ, Kontoleon PE, Mesogitis SA, Aravantinos D. Circulating levels of immunoreactive parathyroid hormone-related protein and intact parathyroid hormone in human fetuses and newborns. Eur J Endocrinol 134: 437– 442, 1996. 502. Park W, Paust H, Kaufmann HJ, Offermann G. Osteomalacia of the mother: rickets of the newborn. Eur J Pediatr 146: 292–293, 1987. 503. Parnell SE, Phillips GS. Neonatal skeletal dysplasias. Pediatr Radiol 42 Suppl 1: S150 – 157, 2012. 504. Paterson CR. Temporary brittle bone disease: fractures in medical care. Acta Paediatr 98: 1935–1938, 2009. 518. Phitayakorn R, McHenry CR. Incidence and location of ectopic abnormal parathyroid glands. Am J Surg 191: 418 – 423, 2006. 519. Pic P. Maintenance of an elevated fetal blood calcium in the absence of maternal and fetal parathyroid glands in rats. C R Seances Soc Biol Fil 162: 1043–1047, 1968. 520. Pic P. Role of the foetal parathyroids in the regulation of calcemia and phosphatemia of the rat foetus (author’s transl). Ann d’Endocrinol 34: 621– 645, 1973. 521. Pic P, Maniey J, Jost A. Facteurs endocriniens reglant de la calcemie foetale. Indications sur le role des parathyroides. C R Seances Soc Biol Fil 159: 1274 –1277, 1965. 522. Pieringer H, Hatzl-Griesenhofer M, Shebl O, Wiesinger-Eidenberger G, Maschek W, Biesenbach G. Hypocalcemic tetany in the newborn as a manifestation of unrecognized maternal primary hyperparathyroidism. Wien Klin Wochenschr 119: 129 –131, 2007. 523. Pitkin RM, Cruikshank DP, Schauberger CW, Reynolds WA, Williams GA, Hargis GK. Fetal calcitropic hormones and neonatal calcium homeostasis. Pediatrics 66: 77– 82, 1980. 524. Pitkin RM, Reynolds WA, Williams GA, Kawahara W, Bauman AF, Hargis GK. Maternal and fetal parathyroid hormone responsiveness in pregnant primates. J Clin Endocrinol Metab 51: 1044 –1047, 1980. 525. Pitt MJ. Rickets and osteomalacia are still around. Radiol Clin N Am 29: 97–118, 1991. 526. Pollak MR, Brown EM, Estep HL, McLaine PN, Kifor O, Park J, Hebert SC, Seidman CE, Seidman JG. Autosomal dominant hypocalcaemia caused by a Ca2⫹-sensing receptor gene mutation. Nat Genet 8: 303–307, 1994. 527. Polykandriotis EP, Beutel FK, Horch RE, Grunert J. A case of familial tumoral calcinosis in a neonate and review of the literature. Arch Orthop Trauma Surg 124: 563–567, 2004. 528. Potter EL. Bilateral renal agenesis. J Pediatr 29: 68 –76, 1946. 505. Paulson SK, DeLuca HF, Battaglia F. Plasma levels of vitamin D metabolites in fetal and pregnant ewes. Proc Soc Exp Biol Med 185: 267–271, 1987. 506. Paulson SK, Langman CB. Plasma vitamin D metabolite levels in pregnant and nonpregnant ewes. Comp Biochem Physiol A 96: 347–349, 1990. 507. Payne JM, Sansom BF. The susceptibility of various groups of rats to experimental hypocalcemia. J Physiol 168: 554 –563, 1963. 508. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, Lewis-Barned N, McCredie D, Powell H, Kendall-Taylor P, Brown EM, Thakker RV. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med 335: 1115–1122, 1996. 509. Penny R, Parlow AF, Frasier SD. Testosterone and estradiol concentrations in paired maternal and cord sera and their correlation with the concentration of chorionic gonadotropin. Pediatrics 64: 604 – 608, 1979. 510. Pereira GR, Zucker AH. Nutritional deficiencies in the neonate. Clin Perinatol 13: 175–189, 1986. 1212 529. Powell BR, Buist NR. Late presenting, prolonged hypocalcemia in an infant of a woman with hypocalciuric hypercalcemia. Clin Pediatr 29: 241–243, 1990. 530. Prasad V, Okunade G, Liu L, Paul RJ, Shull GE. Distinct phenotypes among plasma membrane Ca2⫹-ATPase knockout mice. Ann NY Acad Sci 1099: 276 –286, 2007. 531. Prince MJ, Schaeffer PC, Goldsmith RS, Chausmer AB. Hyperphosphatemic tumoral calcinosis: association with elevation of serum 1,25-dihydroxycholecalciferol concentrations. Ann Intern Med 96: 586 –591, 1982. 532. Qian J, Colbert MC, Witte D, Kuan CY, Gruenstein E, Osinska H, Lanske B, Kronenberg HM, Clemens TL. Midgestational lethality in mice lacking the parathyroid hormone (PTH)/PTH-related peptide receptor is associated with abrupt cardiomyocyte death. Endocrinology 144: 1053–1061, 2003. 533. Raison D, Coquard C, Hochane M, Steger J, Massfelder T, Moulin B, Karaplis AC, Metzger D, Chambon P, Helwig JJ, Barthelmebs M. Knockdown of parathyroid hormone related protein in smooth muscle cells alters renal hemodynamics but not blood pressure. Am J Physiol Renal Physiol 305: F333–F342, 2013. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM 534. Ramavat LG. Vitamin D deficiency rickets at birth in Kuwait. Indian J Pediatr 66: 37– 43, 1999. roid glands and sheep placenta: comparisons with a similar protein implicated in humoral hypercalcaemia of malignancy. J Endocrinol 117: 261–271, 1988. 535. Ramberg CF Jr, Delivoria-Papadopoulos M, Crandall ED, Kronfeld DS. Kinetic analysis of calcium transport across the placenta. J Appl Physiol 35: 682– 688, 1973. 557. Rodrigues LS, Cau AC, Bussmann LZ, Bastida G, Brunetto OH, Correa PH, Martin RM. New mutation in the CASR gene in a family with familial hypocalciuric hypercalcemia (FHH) and neonatal severe hyperparathyroidism (NSHPT). Arquivos brasileiros de Endocrinologia e metabologia 55: 67–71, 2011. 536. Ramsey EM. The Placenta: Human and Animal. New York: Praeger, 1982. 537. Ratcliffe WA, Thompson GE, Abbas SK, Care AD. Studies on the metabolic clearance of parathyroid hormone-related protein in pregnant, nonpregnant and fetal animals. In: Calcium Regulating Hormones and Bone Metabolism: Basic and Clinical Aspects, edited by Cohn DV, Gennari C, and Tashjian AH, Jr. Amsterdam: Excerpta Medica, 1992, p. 69 –75. 538. Rauch F. Etiology and treatment of hypocalcemic rickets in children. In: UpToDate 173, edited by Rose BD. Welesley, MA: UpToDate, 2009. 539. Rauch F, Schoenau E. Skeletal development in premature infants: a review of bone physiology beyond nutritional aspects. Arch Dis Child Fetal Neonatal Ed 86: F82– 85, 2002. 540. Raue F, Pichl J, Dorr HG, Schnabel D, Heidemann P, Hammersen G, Jaursch-Hancke C, Santen R, Schofl C, Wabitsch M, Haag C, Schulze E, Frank-Raue K. Activating mutations in the calcium-sensing receptor: genetic and clinical spectrum in 25 patients with autosomal dominant hypocalcaemia: a German survey. Clin Endocrinol 75: 760 – 765, 2011. 541. Rebut-Bonneton C, Demignon J, Amor B, Miravet L. Effect of calcitonin in pregnant rats on bone resorption in fetuses. J Endocrinol 99: 347–353, 1983. 542. Rebut-Bonneton C, Garel JM, Delbarre F. Parathyroid hormone, calcitonin, 1,25dihydroxycholecalciferol, and basal bone resorption in the rat fetus. Calcif Tissue Int 35: 183–189, 1983. 543. Rector JM. Prenatal influence in rickets. J Pediatr 6: 161–166, 1935. 544. Reitz RE, Daane TA, Woods JR, Weinstein RL. Calcium, magnesium, phosphorus, and parathyroid hormone interrelationships in pregnancy and newborn infants. Obstet Gynecol 50: 701–705, 1977. 545. Reyes FI, Boroditsky RS, Winter JS, Faiman C. Studies on human sexual development. II. Fetal and maternal serum gonadotropin and sex steroid concentrations. J Clin Endocrinol Metab 38: 612– 617, 1974. 546. Reyes FI, Winter JS, Faiman C. Studies on human sexual development. I. Fetal gonadal and adrenal sex steroids. J Clin Endocrinol Metab 37: 74 –78, 1973. 547. Reynolds WA, Pitkin RM, Wezeman FH. Calcitonin effects in primate pregnancy. Am J Obstet Gynecol 122: 212–218, 1975. 548. Reynolds WA, Pitkin RM, Williams GA, Bauman AF, Hargis GK, Beluhan FZ, Kawahara W. Calcitonin responsiveness in the primate fetus. J Clin Endocrinol Metab 51: 595– 598, 1980. 549. Riccardi D, Brennan SC, Chang W. The extracellular calcium-sensing receptor, CaSR, in fetal development. Best Pract Res Clin Endocrinol Metab 27: 443– 453, 2013. 550. Richette P, Bardin T, Stheneur C. Achondroplasia: from genotype to phenotype. Joint Bone Spine 75: 125–130, 2008. 551. Riddle RC, Macica CM, Clemens TL. Vascular, cardiovascular, and neurologic actions of parathyroid-related protein. In: Principles of Bone Biology (3rd ed.), edited by Bilezikian JP, Raisz LG, and Martin TJ. New York: Academic, 2008, p. 733–748. 552. Ritter CS, Haughey BH, Miller B, Brown AJ. Differential gene expression by oxyphil and chief cells of human parathyroid glands. J Clin Endocrinol Metab 97: E1499 –1505, 2012. 553. Robinson NR, Atkinson DE, Jones CJ, Sibley CP. Permeability of the near-term rat placenta to hydrophilic solutes. Placenta 9: 361–372, 1988. 554. Robinson NR, Sibley CP, Mughal MZ, Boyd RD. Fetal control of calcium transport across the rat placenta. Pediatr Res 26: 109 –115, 1989. 555. Rockman-Greenberg C. Hypophosphatasia. Pediatr Endocrinol Rev 10 Suppl 2: 380 – 388, 2013. 556. Rodda CP, Kubota M, Heath JA, Ebeling PR, Moseley JM, Care AD, Caple IW, Martin TJ. Evidence for a novel parathyroid hormone-related protein in fetal lamb parathy- 558. Romagnoli C, Zecca E, Tortorolo G, Diodato A, Fazzini G, Sorcini-Carta M. Plasma thyrocalcitonin and parathyroid hormone concentrations in early neonatal hypocalcaemia. Arch Dis Child 62: 580 –584, 1987. 559. Rong H, Hydbring E, Olsson K, Burtis WJ, Rankin W, Grill V, Bucht E. Parathyroid hormone-related protein in neonatal and reproductive goats determined by a sensitive time-resolved immunofluorometric assay. Eur J Endocrinol 136: 546 –551, 1997. 560. Rosen CJ, Adams JS, Bikle D, Black DM, Demay MB, Manson JE, Murad MH, Kovacs CS. The nonskeletal effects of vitamin D: an Endocrine Society Scientific Statement. Endocr Rev 33: 456 – 492, 2012. 561. Ross AC, Abrams SA, Aloia JF, Brannon PM, Clinton SK, Durazo-Arvizu RA, Gallagher JC, Gallo RL, Jones G, Kovacs CS, Manson JE, Mayne ST, Rosen CJ, Shapses SA. Dietary Reference Intakes for Calcium and Vitamin D. Washington, DC: Institute of Medicine, 2011. 562. Ross AJ, Cooper A, Attie MF, Bishop HC. Primary hyperparathyroidism in infancy. J Pediatr Surg 21: 493– 499, 1986. 563. Ross MG, Beall MH. Amniotic fluid dynamics. In: Creasy and Resnik’s Maternal-Fetal Medicine: Principles and Practice (7th ed.), edited by Creasy RK, Resnik R, Iams JD, Lockwood CJ, Moore TR, and Greene MF. Philadelphia, PA: Elsevier, 2014, p. 47–52e43. 564. Ross R, Care AD, Robinson JS, Pickard DW, Weatherley AJ. Perinatal 1,25-dihydroxycholecalciferol in the sheep and its role in the maintenance of the transplacental calcium gradient. J Endocrinol 87: 17P–18P, 1980. 565. Ross R, Dorsey J. Postnatal changes in plasma 1,25-dihydroxyvitamin D3 in sheep: role of altered clearance. Am J Physiol Endocrinol Metab 261: E635–E641, 1991. 566. Ross R, Halbert K, Tsang RC. Determination of the production and metabolic clearance rates of 1,25-dihydroxyvitamin D3 in the pregnant sheep and its chronically catheterized fetus by primed infusion technique. Pediatr Res 26: 633– 638, 1989. 567. Roth DE, Al Mahmud A, Raqib R, Akhtar E, Perumal N, Pezzack B, Baqui AH. Randomized placebo-controlled trial of high-dose prenatal third-trimester vitamin D3 supplementation in Bangladesh: the AViDD trial. Nutr J 12: 47, 2013. 568. Roth DE, Perumal N, Al Mahmud A, Baqui AH. Maternal vitamin D3 supplementation during the third trimester of pregnancy: effects on infant growth in a longitudinal follow-up study in Bangladesh. J Pediatr 163: 1605–1611, 2013. 569. Rothman SM, Lee BC. What bulges under a bulging fontanel? Arch Pediatr Adolesc Med 152: 100 –101, 1998. 570. Rubin LP, Kovacs CS, De Paepe ME, Tsai SW, Torday JS, Kronenberg HM. Arrested pulmonary alveolar cytodifferentiation and defective surfactant synthesis in mice missing the gene for parathyroid hormone-related protein. Dev Dyn 230: 278 –289, 2004. 571. Rubin LP, Posillico JT, Anast CS, Brown EM. Circulating levels of biologically active and immunoreactive intact parathyroid hormone in human newborns. Pediatr Res 29: 201–207, 1991. 572. Rubin LP, Yeung B, Vouros P, Vilner LM, Reddy GS. Evidence for human placental synthesis of 24,25-dihydroxyvitamin D3 and 23,25-dihydroxyvitamin D3. Pediatr Res 34: 98 –104, 1993. 573. Rummens K, van Bree R, Van Herck E, Zaman Z, Bouillon R, Van Assche FA, Verhaeghe J. Vitamin D deficiency in guinea pigs: exacerbation of bone phenotype during pregnancy and disturbed fetal mineralization, with recovery by 1,25(OH)2D3 infusion or dietary calcium-phosphate supplementation. Calcif Tissue Int 71: 364 –375, 2002. 574. Rummens K, van Cromphaut SJ, Carmeliet G, van Herck E, van Bree R, Stockmans I, Bouillon R, Verhaeghe J. Pregnancy in mice lacking the vitamin D receptor: normal maternal skeletal response, but fetal hypomineralization rescued by maternal calcium supplementation. Pediatr Res 54: 466 – 473, 2003. 575. Rurak DW. Development and function of the placenta. In: Fetal Growth and Development, edited by Harding R, Bocking AD. Cambridge, UK: Cambridge Univ. Press, 2001, p. 17– 43. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1213 CHRISTOPHER S. KOVACS 576. Russell J, Gee P, Liu SM, Angeletti RH. Inhibition of parathyroid hormone secretion by amino-terminal chromogranin peptides. Endocrinology 135: 337–342, 1994. 597. Schnatz PF, Curry SL. Primary hyperparathyroidism in pregnancy: evidence-based management. Obstet Gynecol Surv 57: 365–376, 2002. 577. Russell JG, Hill LF. True fetal rickets. Br J Radiol 47: 732–734, 1974. 598. Seino Y, Ishida M, Yamaoka K, Ishii T, Hiejima T, Ikehara C, Tanaka Y, Matsuda S, Shimotsuji T, Yabuuchi H, Morimoto S, Onishi T. Serum calcium regulating hormones in the perinatal period. Calcif Tissue Int 34: 131–135, 1982. 578. Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brondum-Nielsen K, Scambler PJ. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet 34: 798 – 804, 1997. 579. Sabbagh Y, Carpenter TO, Demay MB. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci USA 102: 9637–9642, 2005. 599. Seki K, Furuya K, Makimura N, Mitsui C, Hirata J, Nagata I. Cord blood levels of calcium-regulating hormones and osteocalcin in premature infants. J Perinat Med 22: 189 –194, 1994. 600. Seki K, Wada S, Nagata N, Nagata I. Parathyroid hormone-related protein during pregnancy and the perinatal period. Gynecol Obstet Invest 37: 83– 86, 1994. 601. Selvanayagam P, Graves K, Cooper C, Rajaraman S. Expression of the parathyroid hormone-related peptide gene in rat tissues. Lab Invest 64: 713–717, 1991. 580. Saggese G, Baroncelli GI, Bertelloni S, Cipolloni C. Intact parathyroid hormone levels during pregnancy, in healthy term neonates and in hypocalcemic preterm infants. Acta Paediatr Scand 80: 36 – 41, 1991. 602. Senior PV, Heath DA, Beck F. Expression of parathyroid hormone-related protein mRNA in the rat before birth: demonstration by hybridization histochemistry. J Mol Endocrinol 6: 281–290, 1991. 581. Salle B, Senterre J, Putet G, Rigo J. Effects of calcium and phosphorus supplementation on calcium retention and fat absorption in preterm infants fed pooled human milk. J Pediatr Gastroenterol Nutr 5: 638 – 642, 1986. 603. Senterre J, Putet G, Salle B, Rigo J. Effects of vitamin D and phosphorus supplementation on calcium retention in preterm infants fed banked human milk. J Pediatr 103: 305–307, 1983. 582. Salle BL, Glorieux FH, Delvin EE, David LS, Meunier G. Vitamin D metabolism in preterm infants. Serial serum calcitriol values during the first four days of life. Acta Paediatr Scand 72: 203–206, 1983. 604. Senterre J, Salle B. Calcium and phosphorus economy of the preterm infant and its interaction with vitamin D and its metabolites. Acta Paediatr Scand Suppl 296: 85–92, 1982. 583. Samaan NA, Anderson GD, Adam-Mayne ME. Immunoreactive calcitonin in the mother, neonate, child and adult. Am J Obstet Gynecol 121: 622– 625, 1975. 605. Senterre J, Salle B. Renal aspects of calcium and phosphorus metabolism in preterm infants. Biol Neonate 53: 220 –229, 1988. 584. Samson GR. Skeletal dysplasias with osteopenia in the newborn: the value of alkaline phosphatase. J Matern Fetal Neonatal Med 17: 229 –231, 2005. 606. Setoguti T, Inoue Y, Wild P. The biological significance of storage granules in rat parathyroid cells. Microscopy Res Technique 32: 148 –163, 1995. 585. Sann L, David L, Frederich A, Bovier-Lapierre M, Bourgeois J, Romand-Monier M, Bethenod M. Congenital rickets. Study of the evolution of secondary hyperparathyroidism. Acta Paediatr Scand 66: 323–327, 1977. 607. Shamley DR, Opperman LA, Buffenstein R, Ross FP. Ontogeny of calbindin-D28K and calbindin-D9K in the mouse kidney, duodenum, cerebellum and placenta. Development 116: 491– 496, 1992. 586. Sann L, David L, Thomas A, Frederich A, Chapuy MC, Francois R. Congenital hyperparathyroidism and vitamin D deficiency secondary to maternal hypoparathyroidism. Acta Paediatr Scand 65: 381–385, 1976. 608. Shamley DR, Veale G, Pettifor JM, Buffenstein R. Trophoblastic giant cells of the mouse placenta contain calbindin-D9K but not the vitamin D receptor. J Endocrinol 150: 25–32, 1996. 587. Santi MD, Henry GW, Douglas GL. Magnesium sulfate treatment of preterm labor as a cause of abnormal neonatal bone mineralization. J Pediatr Orthop 14: 249 –253, 1994. 609. Shangold MM, Dor N, Welt SI, Fleischman AR, Crenshaw MC Jr. Hyperparathyroidism and pregnancy: a review. Obstet Gynecol Surv 37: 217–228, 1982. 588. Santos F, Fuente R, Mejia N, Mantecon L, Gil-Pena H, Ordonez FA. Hypophosphatemia and growth. Pediatr Nephrol 28: 595– 603, 2013. 610. Shani H, Sivan E, Cassif E, Simchen MJ. Maternal hypercalcemia as a possible cause of unexplained fetal polyhydramnion: a case series. Am J Obstet Gynecol 199: 410 e411– 415, 2008. 589. Sarda IR, Gorwill RH. Hormonal studies in pregnancy. I. Total unconjugated estrogens in maternal peripheral vein, cord vein, and cord artery serum at delivery. Am J Obstet Gynecol 124: 234 –238, 1976. 590. Sato K. Hypercalcemia during pregnancy, puerperium, and lactation: review and a case report of hypercalcemic crisis after delivery due to excessive production of PTH-related protein (PTHrP) without malignancy (humoral hypercalcemia of pregnancy). Endocr J 55: 959 –966, 2008. 591. Savory J, Monif GR. Serum calcium levels in cord sera of the progeny of mothers treated with magnesium sulfate for toxemia of pregnancy. Am J Obstet Gynecol 110: 556 –559, 1971. 592. Saxe A, Dean S, Gibson G, Pandian MR, Levy J. Parathyroid hormone and parathyroid hormone-related peptide in venous umbilical cord blood of healthy neonates. J Perinat Med 25: 288 –291, 1997. 593. Sayers A, Tobias JH. Estimated maternal ultraviolet B exposure levels in pregnancy influence skeletal development of the child. J Clin Endocrinol Metab 94: 765–771, 2009. 594. Schauberger CW, Pitkin RM. Maternal-perinatal calcium relationships. Obstet Gynecol 53: 74 –76, 1979. 595. Schedewie HK, Odell WD, Fisher DA, Krutzik SR, Dodge M, Cousins L, Fiser WP. Parathormone and perinatal calcium homeostasis. Pediatr Res 13: 1– 6, 1979. 596. Schipani E, Lanske B, Hunzelman J, Luz A, Kovacs CS, Lee K, Pirro A, Kronenberg HM, Jüppner H. Targeted expression of constitutively active PTH/PTHrP receptors delays endochondral bone formation and rescues PTHrP-less mice. Proc Natl Acad Sci USA 94: 13689 –13694, 1997. 1214 611. Shaw AJ, Mughal MZ, Maresh MJ, Sibley CP. Effects of two synthetic parathyroid hormone-related protein fragments on maternofetal transfer of calcium and magnesium and release of cyclic AMP by the in-situ perfused rat placenta. J Endocrinol 129: 399 – 404, 1991. 612. Shaw AJ, Mughal MZ, Mohammed T, Maresh MJ, Sibley CP. Evidence for active maternofetal transfer of magnesium across the in situ perfused rat placenta. Pediatr Res 27: 622– 625, 1990. 613. Shaw JC. Evidence for defective skeletal mineralization in low-birthweight infants: the absorption of calcium and fat. Pediatrics 57: 16 –25, 1976. 614. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19: 429 – 435, 2004. 615. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113: 561–568, 2004. 616. Shin HI, Divieti P, Sims NA, Kobayashi T, Miao D, Karaplis AC, Baron R, Bringhurst R, Kronenberg HM. Gp130-mediated signaling is necessary for normal osteoblastic function in vivo and in vitro. Endocrinology 145: 1376 –1385, 2004. 617. Shutt DA, Smith ID, Shearman RP. Oestrone, oestradiol-17beta and oestriol levels in human foetal plasma during gestation and at term. J Endocrinol 60: 333–341, 1974. 618. Siiteri PK, MacDonald PC. The utilization of circulating dehydroisoandrosterone sulfate for estrogen synthesis during human pregnancy. Steroids 2: 713–730, 1963. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM 619. Siler-Khodr TM, Khodr GS. Studies in human fetal endocrinology. II. LH and FSH content and concentration in the pituitary. Obstet Gynecol 56: 176 –181, 1980. 640. Steichen JJ, Asch PA, Tsang RC. Bone mineral content measurement in small infants by single-photon absorptiometry: current methodologic issues. J Pediatr 113: 181–187, 1988. 620. Siler-Khodr TM, Morgenstern LL, Greenwood FC. Hormone synthesis and release from human fetal adenohypophyses in vitro. J Clin Endocrinol Metab 39: 891–905, 1974. 641. Steichen JJ, Gratton TL, Tsang RC. Osteopenia of prematurity: the cause and possible treatment. J Pediatr 96: 528 –534, 1980. 621. Silver J, Landau H, Bab I, Shvil Y, Friedlaender MM, Rubinger D, Popovtzer MM. Vitamin D-dependent rickets types I and II. Diagnosis and response to therapy. Isr J Med Sci 21: 53–56, 1985. 642. Steichen JJ, Tsang RC, Gratton TL, Hamstra A, DeLuca HF. Vitamin D homeostasis in the perinatal period: 1,25-dihydroxyvitamin D in maternal, cord, and neonatal blood. N Engl J Med 302: 315–319, 1980. 622. Silver J, Naveh-Many T. FGF23 and the parathyroid glands. Pediatr Nephrol 25: 2241– 2245, 2010. 643. Steichen JJ, Tsang RC, Greer FR, Ho M, Hug G. Elevated serum 1,25 dihydroxyvitamin D concentrations in rickets of very low-birth-weight infants. J Pediatr 99: 293–298, 1981. 623. Simmonds CS, Karsenty G, Karaplis AC, Kovacs CS. Parathyroid hormone regulates fetal-placental mineral homeostasis. J Bone Miner Res 25: 594 – 605, 2010. 624. Simmonds CS, Kovacs CS. Role of parathyroid hormone (PTH) and PTH-related protein (PTHrP) in regulating mineral homeostasis during fetal development. Crit Rev Eukaryot Gene Expr 20: 235–273, 2010. 625. Sinclair JG. Fetal rat parathyroids as affected by changes in maternal serum calcium and phosphorus through parathyroidectomy and dietary control. J Nutr 23: 141–152, 1942. 626. Sitara D, Kim S, Razzaque MS, Bergwitz C, Taguchi T, Schuler C, Erben RG, Lanske B. Genetic evidence of serum phosphate-independent functions of FGF-23 on bone. PLoS Genet 4: e1000154, 2008. 627. Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Jüppner H, Lanske B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol 23: 421– 432, 2004. 628. Slavin RE, Wen J, Barmada A. Tumoral calcinosis–a pathogenetic overview: a histological and ultrastructural study with a report of two new cases, one in infancy. Int J Surg Pathol 20: 462– 473, 2012. 629. Slovis TL, Chapman S. Evaluating the data concerning vitamin D insufficiency/deficiency and child abuse. Pediatr Radiol 38: 1221–1224, 2008. 630. Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 331: 1056 –1061, 1994. 631. Smith FG Jr, Alexander DP, Buckle RM, Britton HG, Nixon DA. Parathyroid hormone in foetal and adult sheep: the effect of hypocalcaemia. J Endocrinol 53: 339 –348, 1972. 632. Soler-Bel J, Veganzones I, Navarro A, Ramos F, Serra-Buxeda E, Ferreres JC. Fatal rickets in the fetus and undiagnosed maternal celiac disease. Gastroenterol Hepatol 34: 678 – 682, 2011. 633. Somjen D, Weisman Y, Berger E, Earon Y, Kaye AM, Binderman I. Developmental changes in the responsiveness of rat kidney to vitamin D metabolites. Endocrinology 118: 354 –359, 1986. 634. Song Y, Peng X, Porta A, Takanaga H, Peng JB, Hediger MA, Fleet JC, Christakos S. Calcium transporter 1 and epithelial calcium channel messenger ribonucleic acid are differentially regulated by 1,25 dihydroxyvitamin D3 in the intestine and kidney of mice. Endocrinology 144: 3885–3894, 2003. 635. Sparks JW. Human intrauterine growth and nutrient accretion. Semin Perinatol 8: 74 –93, 1984. 636. Specker BL, Beck A, Kalkwarf H, Ho M. Randomized trial of varying mineral intake on total body bone mineral accretion during the first year of life. Pediatrics 99: E12, 1997. 644. Steven DH. Anatomy of the placental barrier. In: Comparative Placentation: Essays in Structure and Function, edited by Steven DH. London: Academic, 1975, p. 25–57. 645. Stoeckel ME, Porte A. Embryonic origin and secretory differentiation of calcitonin cells (C cells) in the fetal rat thyroid. Electron microscopic study. Zeitschrift fur Zellforschung und mikroskopische Anatomie 106: 251–268, 1970. 646. Strewler GJ, Stern PH, Jacobs JW, Eveloff J, Klein RF, Leung SC, Rosenblatt M, Nissenson RA. Parathyroid hormonelike protein from human renal carcinoma cells. Structural and functional homology with parathyroid hormone. J Clin Invest 80: 1803– 1807, 1987. 647. Strid H, Bucht E, Jansson T, Wennergren M, Powell TL. ATP dependent Ca2⫹ transport across basal membrane of human syncytiotrophoblast in pregnancies complicated by intrauterine growth restriction or diabetes. Placenta 24: 445– 452, 2003. 648. Strid H, Care A, Jansson T, Powell T. Parathyroid hormone-related peptide (38 –94) amide stimulates ATP-dependent calcium transport in the basal plasma membrane of the human syncytiotrophoblast. J Endocrinol 175: 517–524, 2002. 649. Strid H, Powell TL. ATP-dependent Ca2⫹ transport is up-regulated during third trimester in human syncytiotrophoblast basal membranes. Pediatr Res 48: 58 – 63, 2000. 650. Strott CA, Sundel H, Stahlman MT. Maternal and fetal plasma progesterone, cortisol, testosterone and 17beta-estradiol in preparturient sheep: response to fetal ACTH infusion. Endocrinology 95: 1327–1339, 1974. 651. Strouse PJ. “Keller & Barnes” after 5 years: still inadmissible as evidence. Pediatr Radiol 43: 1424, 2013. 652. Stuart C, Aceto T Jr, Kuhn JP, Terplan K. Intrauterine hyperparathyroidism. Postmortem findings in two cases. Am J Dis Child 133: 67–70, 1979. 653. Stulc J, Stulcova B. Placental transfer of phosphate in anaesthetized rats. Placenta 17: 487– 493, 1996. 654. Stulc J, Stulcova B. Transport of calcium by the placenta of the rat. J Physiol 371: 1–16, 1986. 655. Stulc J, Stulcova B, Smid M, Sach I. Parallel mechanisms of Ca2⫹ transfer across the perfused human placental cotyledon. Am J Obstet Gynecol 170: 162–167, 1994. 656. Stumpf WE, Sar M, Narbaitz R, Huang S, DeLuca HF. Autoradiographic localization of 1,25-dihydroxyvitamin D3 in rat placenta and yolk sac. Horm Res 18: 215–220, 1983. 657. Su D, Ellis S, Napier A, Lee K, Manley NR. Hoxa3 and Pax1 regulate epithelial cell death and proliferation during thymus and parathyroid organogenesis. Dev Biol 236: 316 – 329, 2001. 637. Spivack BS, Otterman GJ. Does temporary brittle bone disease exist? Not by the evidence offered. Acta Paediatr 99: 486, 2010. 658. Suva LJ, Winslow GA, Wettenhall RE, Hammonds RG, Moseley JM, Diefenbach-Jagger H, Rodda CP, Kemp BE, Rodriguez H, Chen EY, Hudson PJ, Martin TJ, Wood WI. A parathyroid hormone-related protein implicated in malignant hypercalcemia: cloning and expression. Science 237: 893– 896, 1987. 638. St-Arnaud R, Arabian A, Travers R, Barletta F, Raval-Pandya M, Chapin K, Depovere J, Mathieu C, Christakos S, Demay MB, Glorieux FH. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology 141: 2658 –2666, 2000. 659. Suzuki Y, Kovacs CS, Takanaga H, Peng JB, Landowski CP, Hediger MA. Calcium TRPV6 is involved in murine maternal-fetal calcium transport. J Bone Miner Res 23: 1249 –1256, 2008. 639. St-Arnaud R, Naja RP. Vitamin D metabolism, cartilage and bone fracture repair. Mol Cell Endocrinol 347: 48 –54, 2011. 660. Symonds HW, Sansom BF, Twardock AR. The measurement of the transfer of calcium and phosphorus from foetus to dam in the sheep using a whole body counter. Res Vet Sci 13: 272–275, 1972. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1215 CHRISTOPHER S. KOVACS 661. Takaiwa M, Aya K, Miyai T, Hasegawa K, Yokoyama M, Kondo Y, Kodani N, Seino Y, Tanaka H, Morishima T. Fibroblast growth factor 23 concentrations in healthy term infants during the early postpartum period. Bone 47: 256 –262, 2010. 683. Trotter M, Hixon BB. Sequential changes in weight, density, and percentage ash weight of human skeletons from an early fetal period through old age. Anat Rec 179: 1–18, 1974. 662. Takeda E, Yamamoto H, Taketani Y, Miyamoto K. Vitamin D-dependent rickets type I and type II. Acta Paediatr Jpn 39: 508 –513, 1997. 684. Tsang RC, Chen I, Friedman MA, Gigger M, Steichen J, Koffler H, Fenton L, Brown D, Pramanik A, Keenan W, Strub R, Joyce T. Parathyroid function in infants of diabetic mothers. J Pediatr 86: 399 – 404, 1975. 663. Tanaka Y, Halloran B, Schnoes HK, DeLuca HF. In vitro production of 1,25-dihydroxyvitamin D3 by rat placental tissue. Proc Natl Acad Sci USA 76: 5033–5035, 1979. 664. Tanamura A, Nomura S, Kurauchi O, Furui T, Mizutani S, Tomoda Y. Purification and characterization of 1,25(OH)2D3 receptor from human placenta. J Obstet Gynaecol 21: 631– 639, 1995. 665. Teotia M, Teotia SP. Nutritional and metabolic rickets. Indian J Pediatr 64: 153–157, 1997. 666. Teotia M, Teotia SP, Nath M. Metabolic studies in congenital vitamin D deficiency rickets. Indian J Pediatr 62: 55– 61, 1995. 667. Teotia M, Teotia SPS, Singh RK. Metabolism of fluoride in pregnant women residing in endemic fluorosis areas. Fluoride 12: 58 – 64, 1979. 668. Thacher TD, Fischer PR, Pettifor JM. The usefulness of clinical features to identify active rickets. Ann Trop Paediatr 22: 229 –237, 2002. 669. Thakker RV. Genetic regulation of parathyroid gland development. In: Principles of Bone Biology (3rd ed.), edited by Bilezikian JP, Raisz LG, and Martin TJ. New York: Academic, 2008, p. 1415–1429. 670. Theman TA, Collins MT, Dempster DW, Zhou H, Reynolds JC, Brahim JS, Roschger P, Klaushofer K, Winer KK. PTH(1–34) replacement therapy in a child with hypoparathyroidism caused by a sporadic calcium receptor mutation. J Bone Miner Res 24: 964 –973, 2009. 671. Thiébaud D, Janisch S, Koelbl H, Hanzal E, Jacquet AF, Leodolter S, Burckhardt P, Pecherstorfer M. Direct evidence of a parathyroid related protein gradient between the mother and the newborn in humans. Bone Miner 23: 213–221, 1993. 672. Thomas AK, McVie R, Levine SN. Disorders of maternal calcium metabolism implicated by abnormal calcium metabolism in the neonate. Am J Perinatol 16: 515–520, 1999. 673. Thomas BR, Bennett JD. Late-onset neonatal hypocalcemia as an unusual presentation in an offspring of a mother with familial hypocalciuric hypercalcemia. Clin Pediatr 36: 547–550, 1997. 674. Thomas BR, Bennett JD. Symptomatic hypocalcemia and hypoparathyroidism in two infants of mothers with hyperparathyroidism and familial benign hypercalcemia. J Perinatol 15: 23–26, 1995. 675. Thomas ML, Anast CS, Forte LR. Regulation of calcium homeostasis in the fetal and neonatal rat. Am J Physiol Endocrinol Metab 240: E367–E372, 1981. 676. Thorpe-Beeston JG. Fetal endocrinology. In: Fetal Medicine: Basic Science and Clincial Practice, edited by Rodeck CH, Whittle MJ. London: Churchill Livingstone, 1999, p. 197–206. 677. Thota CS, Reed LC, Yallampalli C. Effects of parathyroid hormone like hormone (PTHLH) antagonist, PTHLH(7–34), on fetoplacental development and growth during midgestation in rats. Biol Reprod 73: 1191–1198, 2005. 678. Tov AB, Mandel D, Weissman Y, Dollberg S, Taxir T, Lubetzky R. Changes in serum parathyroid hormone-related protein in breastfed preterm infants. Breastfeed Med 7: 50 –53, 2012. 679. Treilhou-Lahille F, Cressent M, Taboulet J, Moukhtar MS. Immunohistochemical demonstration of calcitonin cells in the fetal thyroid of mice. Comptes Rendus des seances de l’Academie des sciences Serie D, Sciences Naturelles 289: 421– 423, 1979. 685. Tsang RC, Chen IW, Friedman MA, Chen I. Neonatal parathyroid function: role of gestational age and postnatal age. J Pediatr 83: 728 –738, 1973. 686. Tsang RC, Light IJ, Sutherland JM, Kleinman LI. Possible pathogenetic factors in neonatal hypocalcemia of prematurity. The role of gestation, hyperphosphatemia, hypomagnesemia, urinary calcium loss, and parathormone responsiveness. J Pediatr 82: 423– 429, 1973. 687. Tsang RC, Venkataraman P, Ho M, Steichen JJ, Whitsett J, Greer F. The development of pseudohypoparathyroidism. Involvement of progressively increasing serum parathyroid hormone concentrations, increased 1,25-dihydroxyvitamin D concentrations, and “migratory” subcutaneous calcifications. Am J Dis Child 138: 654 – 658, 1984. 688. Tseng UF, Shu SG, Chen CH, Chi CS. Transient neonatal hypoparathyroidism: report of four cases. Acta Paediatr Taiwan 42: 359 –362, 2001. 689. Tserendolgor U, Mawson JT, Macdonald AC, Oyunbileg M. Prevalence of rickets in Mongolia. Asia Pacific J Clin Nutr 7: 325–328, 1998. 690. Tsukahara H, Sudo M, Umezaki M, Fujii Y, Kuriyama M, Yamamoto K, Ishii Y. Measurement of lumbar spinal bone mineral density in preterm infants by dual-energy X-ray absorptiometry. Biol Neonate 64: 96 –103, 1993. 691. Tu Q, Pi M, Karsenty G, Simpson L, Liu S, Quarles LD. Rescue of the skeletal phenotype in CasR-deficient mice by transfer onto the Gcm2 null background. J Clin Invest 111: 1029 –1037, 2003. 692. Tuan RS. Ca2⫹-binding protein of the human placenta. Characterization, immunohistochemical localization and functional involvement in Ca2⫹ transport. Biochem J 227: 317–326, 1985. 693. Tuan RS, Bigioni N. Ca2⫹-activated ATPase of the mouse chorioallantoic placenta: developmental expression, characterization and cytohistochemical localization. Development 110: 505–513, 1990. 694. Tucci J, Hammond V, Senior PV, Gibson A, Beck F. The role of fetal parathyroid hormone-related protein in transplacental calcium transport. J Mol Endocrinol 17: 159 –164, 1996. 695. Tucci J, Russell A, Senior PV, Fernley R, Ferraro T, Beck F. The expression of parathyroid hormone and parathyroid hormone-related protein in developing rat parathyroid glands. J Mol Endocrinol 17: 149 –157, 1996. 696. Tulchinsky D. Placental secretion of unconjugated estrone, estradiol and estriol into the maternal and the fetal circulation. J Clin Endocrinol Metab 36: 1079 –1087, 1973. 697. Tulchinsky D, Hobel CJ. Plasma human chorionic gonadotropin, estrone, estradiol, estriol, progesterone, and 17 alpha-hydroxyprogesterone in human pregnancy. 3. Early normal pregnancy. Am J Obstet Gynecol 117: 884 – 893, 1973. 698. Turner M, Barre PE, Benjamin A, Goltzman D, Gascon-Barre M. Does the maternal kidney contribute to the increased circulating 1,25-dihydroxyvitamin D concentrations during pregnancy? Miner Electrolyte Metab 14: 246 –252, 1988. 699. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444: 770 –774, 2006. 680. Trivedi A. Low calcium. J Paediatr Child Health 48: 70 – 81, 2012. 700. Usta IM, Chammas M, Khalil AM. Renal cell carcinoma with hypercalcemia complicating a pregnancy: case report and review of the literature. Eur J Gynaecol Oncol 19: 584 –587, 1998. 681. Troisi R, Potischman N, Roberts J, Siiteri P, Daftary A, Sims C, Hoover RN. Associations of maternal and umbilical cord hormone concentrations with maternal, gestational and neonatal factors (United States). Cancer Causes Control 14: 347–355, 2003. 701. Van Bogaert EC, Gueuning CO, Graff GL. Phosphate metabolism and foetal growth in the rat. I. Net transfer of 31P and 32P phosphate from the maternal plasma to the normal placenta. Arch Int Physiol Biochim 89: 303–312, 1981. 682. Troisi R, Potischman N, Roberts JM, Harger G, Markovic N, Cole B, Lykins D, Siiteri P, Hoover RN. Correlation of serum hormone concentrations in maternal and umbilical cord samples. Cancer Epidemiol Biomarkers Prevention 12: 452– 456, 2003. 702. Van Bogaert EC, Gueuning CO, Graff GL. Phosphate metabolism and foetal growth in the rat. II. Net transfer of 31P and 32P inorganic phosphate from the maternal plasma to the normal foetus. Arch Int Physiol Biochim 90: 337–346, 1982. 1216 Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org FETAL AND NEONATAL MINERAL METABOLISM 703. Van Bogaert EC, Gueuning CO, Graff GL. Phosphate metabolism and foetal growth in the rat. III. Net transfer of 31P and 32P inorganic phosphate from the maternal plasma to the placenta after foetectomy. Arch Int Physiol Biochim 94: 135–149, 1986. 723. Weatherley AJ, Ross R, Pickard DW, Care AD. The transfer of calcium during perfusion of the placenta and intact and thyroparathyroidectomized sheep. Placenta 4: 271–277, 1983. 704. Van Cromphaut SJ, Dewerchin M, Hoenderop JG, Stockmans I, Van Herck E, Kato S, Bindels RJ, Collen D, Carmeliet P, Bouillon R, Carmeliet G. Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci USA 98: 13324 –13329, 2001. 724. Weenig RH, Sewell LD, Davis MD, McCarthy JT, Pittelkow MR. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol 56: 569 –579, 2007. 705. Van Dyke JH. Aberrant parathyroid tissue and the thymus: postnatal development of accessory parathyroid glands in the rat. Anat Rec 134: 185–203, 1959. 706. VanHouten JN, Dann P, Stewart AF, Watson CJ, Pollak M, Karaplis AC, Wysolmerski JJ. Mammary-specific deletion of parathyroid hormone-related protein preserves bone mass during lactation. J Clin Invest 112: 1429 –1436, 2003. 707. Venkataraman PS, Blick KE, Fry HD, Rao RK. Postnatal changes in calcium-regulating hormones in very-low-birth-weight infants. Effect of early neonatal hypocalcemia and intravenous calcium infusion on serum parathyroid hormone and calcitonin homeostasis. Am J Dis Child 139: 913–916, 1985. 708. Venkataraman PS, Tsang RC, Steichen JJ, Grey I, Neylan M, Fleischman AR. Early neonatal hypocalcemia in extremely preterm infants. High incidence, early onset, and refractoriness to supraphysiologic doses of calcitriol. Am J Dis Child 140: 1004 –1008, 1986. 725. Weiler HA, Fitzpatrick-Wong SC, Schellenberg JM. Bone mass in First Nations, Asian and white newborn infants. Growth Dev Aging 71: 35– 43, 2008. 726. Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci USA 93: 10240 –10245, 1996. 727. Weisman Y, Bab I, Gazit D, Spirer Z, Jaffe M, Hochberg Z. Long-term intracaval calcium infusion therapy in end-organ resistance to 1,25-dihydroxyvitamin D. Am J Med 83: 984 –990, 1987. 728. Weisman Y, Harell A, Edelstein S, David M, Spirer Z, Golander A. 1alpha,25-Dihydroxyvitamin D3 and 24,25-dihydroxyvitamin D3 in vitro synthesis by human decidua and placenta. Nature 281: 317–319, 1979. 729. Weisman Y, Sapir R, Harell A, Edelstein S. Maternal-perinatal interrelationships of vitamin D metabolism in rats. Biochim Biophys Acta 428: 388 –395, 1976. 709. Verhaeghe J, Thomasset M, Brehier A, Van Assche FA, Bouillon R. 1,25(OH)2D3 and Ca-binding protein in fetal rats: relationship to the maternal vitamin D status. Am J Physiol Endocrinol Metab 254: E505–E512, 1988. 730. Weisz J, Ward IL. Plasma testosterone and progesterone titers of pregnant rats, their male and female fetuses, and neonatal offspring. Endocrinology 106: 306 –316, 1980. 710. Vidailhet M, Monin P, Andre M, Suty Y, Marchal C, Vert P. Neonatal hyperparathyroidism secondary to maternal hypoparathyroidism. Arch Fr Pediatr 37: 305–312, 1980. 731. White KE, Econs MJ. Fibroblast growth factor-23. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism (7th ed.), edited by Rosen CJ. Washington, DC: ASBMR, 2008, p. 112–116. 711. Vidal O, Lindberg MK, Hollberg K, Baylink DJ, Andersson G, Lubahn DB, Mohan S, Gustafsson JA, Ohlsson C. Estrogen receptor specificity in the regulation of skeletal growth and maturation in male mice. Proc Natl Acad Sci USA 97: 5474 –5479, 2000. 732. Widdowson EM. Absorption and excretion of fat, nitrogen, and minerals from “filled” milks by babies one week old. Lancet 2: 1099 –1105, 1965. 712. Viljakainen HT, Korhonen T, Hytinantti T, Laitinen EK, Andersson S, Makitie O, Lamberg-Allardt C. Maternal vitamin D status affects bone growth in early childhood–a prospective cohort study. Osteoporos Int 22: 883– 891, 2011. 713. Viljakainen HT, Saarnio E, Hytinantti T, Miettinen M, Surcel H, Makitie O, Andersson S, Laitinen K, Lamberg-Allardt C. Maternal vitamin D status determines bone variables in the newborn. J Clin Endocrinol Metab 95: 1749 –1757, 2010. 714. Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Indian hedgehog and parathyroid hormone-related protein regulate the rate of cartilage differentiation. Science 273: 613– 622, 1996. 715. Wadsworth JC, Kronfeld DS, Ramberg CF Jr. Parathyrin and calcium homeostasis in the fetus. Biol Neonate 41: 101–109, 1982. 716. Wagner CL. Vitamin D supplementation during pregnancy: impact on maternal outcomes. In: Centers for Disease Control and Prevention Conference on Vitamin D Physiology in Pregnancy: Implications for Preterm Birth and Preeclampsia. Atlanta, GA: Centers for Disease Control and Prevention, 2011. 717. Wagner CL, McNeil R, Hamilton SA, Winkler J, Rodriguez Cook C, Warner G, Bivens B, Davis DJ, Smith PG, Murphy M, Shary JR, Hollis BW. A randomized trial of vitamin D supplementation in 2 community health center networks in South Carolina. Am J Obstet Gynecol 208: 137.e131101–113, 2013. 718. Wagner G, Transhol L, Melchior JC. Hyperparathyroidism and pregnancy. Acta Endocrinol 47: 549 –564, 1964. 719. Waller S, Kurzawinski T, Spitz L, Thakker R, Cranston T, Pearce S, Cheetham T, van’t Hoff WG. Neonatal severe hyperparathyroidism: genotype/phenotype correlation and the use of pamidronate as rescue therapy. Eur J Pediatr 163: 589 –594, 2004. 720. Ward IL, Weisz J. Differential effects of maternal stress on circulating levels of corticosterone, progesterone, and testosterone in male and female rat fetuses and their mothers. Endocrinology 114: 1635–1644, 1984. 733. Widdowson EM. Changes in body composition during growth. In: Scientific Foundations of Paediatrics, edited by Davis JA, Dobbing J. London: William Heinemann, 1981, p. 330 –342. 734. Widdowson EM, Dickerson JW. Chemical composition of the body. In: Mineral Metabolism: An Advanced Treatise. The Elements, edited by Comar CL, and Bronner F. New York: Academic Press, 1964, vol. II, pt. A, p. 1330 –247. 735. Widdowson EM, McCance RA. The metabolism of phosphorus, calcium, magnesium and strontium. Pediatr Clin North Am 12: 595– 614, 1965. 736. Wieland P, Fischer JA, Trechsel U, Roth HR, Vetter K, Schneider H, Huch A. Perinatal parathyroid hormone, vitamin D metabolites, and calcitonin in man. Am J Physiol Endocrinol Metab 239: E385–E390, 1980. 737. Wilhelm-Bals A, Parvex P, Magdelaine C, Girardin E. Successful use of bisphosphonate and calcimimetic in neonatal severe primary hyperparathyroidism. Pediatrics 129: e812– 816, 2012. 738. Windahl SH, Andersson G, Gustafsson JA. Elucidation of estrogen receptor function in bone with the use of mouse models. Trends Endocrinol Metab 13: 195–200, 2002. 739. Winter JS, Hughes IA, Reyes FI, Faiman C. Pituitary-gonadal relations in infancy. 2. Patterns of serum gonadal steroid concentrations in man from birth to two years of age. J Clin Endocrinol Metab 42: 679 – 686, 1976. 740. Wittelsberger A, Rosenblatt M. Parathyroid hormone-receptor interactions. In: Principles of Bone Biology (3rd ed.), edited by Bilezikian JP, Raisz LG, and Martin TJ. New York: Academic, 2008, p. 595– 637. 741. Wolfe JJ. Teeth in fetal rickets. Am J Dis Children 49: 905–911, 1935. 742. Woloszczuk W, Kovarik J, Pavelka P. Calcitonin in pregnant women and in cord blood. Gynecol Obstet Invest 12: 272–276, 1981. 721. Wasserman RH, Comar CL, Nold MM, Lengemann FW. Placental transfer of calcium and strontium in the rat and rabbit. Am J Physiol 189: 91–97, 1957. 743. Wooding FBP, Morgan G, Jones GV, Care AD. Calcium transport and the localisation of calbindin-D9k in the ruminant placenta during the second half of pregnancy. Cell Tissue Res 285: 477– 489, 1996. 722. Wauben IP, Atkinson SA, Shah JK, Paes B. Growth and body composition of preterm infants: influence of nutrient fortification of mother’s milk in hospital and breastfeeding post-hospital discharge. Acta Paediatr 87: 780 –785, 1998. 744. Woodrow JP, Sharpe CJ, Fudge NJ, Hoff AO, Gagel RF, Kovacs CS. Calcitonin plays a critical role in regulating skeletal mineral metabolism during lactation. Endocrinology 147: 4010 – 4021, 2006. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org 1217 CHRISTOPHER S. KOVACS 745. Wu TL, Vasavada RC, Yang K, Massfelder T, Ganz M, Abbas SK, Care AD, Stewart AF. Structural and physiologic characterization of the mid-region secretory species of parathyroid hormone-related protein. J Biol Chem 271: 24371–24381, 1996. 746. Xue Y, Fleet JC. Intestinal vitamin D receptor is required for normal calcium and bone metabolism in mice. Gastroenterology 136: 1317–1327, 2009. 747. Xue Y, Karaplis AC, Hendy GN, Goltzman D, Miao D. Exogenous 1,25-dihydroxyvitamin D3 exerts a skeletal anabolic effect and improves mineral ion homeostasis in mice that are homozygous for both the 1alpha-hydroxylase and parathyroid hormone null alleles. Endocrinology 147: 4801– 4810, 2006. 748. Xue Y, Karaplis AC, Hendy GN, Goltzman D, Miao D. Genetic models show that parathyroid hormone and 1,25-dihydroxyvitamin D3 play distinct and synergistic roles in postnatal mineral ion homeostasis and skeletal development. Hum Mol Genet 14: 1515–1528, 2005. 749. Xue Y, Zhang Z, Karaplis AC, Hendy GN, Goltzman D, Miao D. Exogenous PTHrelated protein and PTH improve mineral and skeletal status in 25-hydroxyvitamin D-1alpha-hydroxylase and PTH double knockout mice. J Bone Miner Res 20: 1766 – 1777, 2005. 750. Yamamoto Y, Yoshizawa T, Fukuda T, Shirode-Fukuda Y, Yu T, Sekine K, Sato T, Kawano H, Aihara K, Nakamichi Y, Watanabe T, Shindo M, Inoue K, Inoue E, Tsuji N, Hoshino M, Karsenty G, Metzger D, Chambon P, Kato S, Imai Y. Vitamin D receptor in osteoblasts is a negative regulator of bone mass control. Endocrinology 154: 1008 – 1020, 2013. 751. Yang Y. Skeletal morphogenesis during embryonic development. Crit Rev Eukaryot Gene Expr 19: 197–218, 2009. 752. Yorifuji J, Yorifuji T, Tachibana K, Nagai S, Kawai M, Momoi T, Nagasaka H, Hatayama H, Nakahata T. Craniotabes in normal newborns: the earliest sign of subclinical vitamin D deficiency. J Clin Endocrinol Metab 93: 1784 –1788, 2008. 1218 753. Yoshida K, Taga T, Saito M, Suematsu S, Kumanogoh A, Tanaka T, Fujiwara H, Hirata M, Yamagami T, Nakahata T, Hirabayashi T, Yoneda Y, Tanaka K, Wang WZ, Mori C, Shiota K, Yoshida N, Kishimoto T. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc Natl Acad Sci USA 93: 407– 411, 1996. 754. Yoshiko Y, Wang H, Minamizaki T, Ijuin C, Yamamoto R, Suemune S, Kozai K, Tanne K, Aubin JE, Maeda N. Mineralized tissue cells are a principal source of FGF23. Bone 40: 1565–1573, 2007. 755. Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, Kawakami T, Arioka K, Sato H, Uchiyama Y, Masushige S, Fukamizu A, Matsumoto T, Kato S. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet 16: 391–396, 1997. 756. Yu CK, Sykes L, Sethi M, Teoh TG, Robinson S. Vitamin D deficiency and supplementation during pregnancy. Clin Endocrinol 70: 685– 690, 2009. 757. Yu HK, Cabalum T, Jansen CA, Buster JE, Nathanielsz PW. Androstenedione, testosterone, and estradiol concentrations in fetal and maternal plasma in late pregnancy in the sheep. Endocrinology 113: 2216 –2220, 1983. 758. Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, Hewison M. Extrarenal expression of 25-hydroxyvitamin D3-1 alpha-hydroxylase. J Clin Endocrinol Metab 86: 888 – 894, 2001. 759. Zhang R, Lu Y, Ye L, Yuan B, Yu S, Qin C, Xie Y, Gao T, Drezner MK, Bonewald LF, Feng JQ. Unique roles of phosphorus in endochondral bone formation and osteocyte maturation. J Bone Miner Res 26: 1047–1056, 2011. 760. Zhu LJ, Bagchi MK, Bagchi IC. Attenuation of calcitonin gene expression in pregnant rat uterus leads to a block in embryonic implantation. Endocrinology 139: 330 –339, 1998. 761. Ziegler EE, O’Donnell AM, Nelson SE, Fomon SJ. Body composition of the reference fetus. Growth 40: 329 –341, 1976. Physiol Rev • VOL 94 • OCTOBER 2014 • www.prv.org