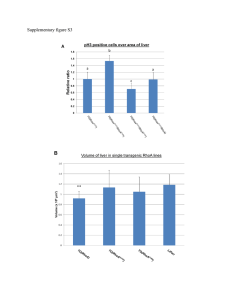
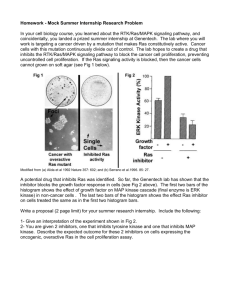
L SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM: PHYSIOLOGICAL AND PATHOLOGICAL ASPECTS
advertisement

Physiol Rev 93: 1659 –1720, 2013 doi:10.1152/physrev.00021.2012 SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM: PHYSIOLOGICAL AND PATHOLOGICAL ASPECTS Gervaise Loirand, Vincent Sauzeau, and Pierre Pacaud INSERM, UMR S1087; University of Nantes; and CHU Nantes, l’Institut du Thorax, Nantes, France Loirand G, Sauzeau V, Pacaud P. Small G Proteins in the Cardiovascular System: Physiological and Pathological Aspects. Physiol Rev 93: 1659 –1720, 2013; doi:10.1152/physrev.00021.2012.—Small G proteins exist in eukaryotes from yeast to human and constitute the Ras superfamily comprising more than 100 members. This superfamily is structurally classified into five families: the Ras, Rho, Rab, Arf, and Ran families that control a wide variety of cell and biological functions through highly coordinated regulation processes. Increasing evidence has accumulated to identify small G proteins and their regulators as key players of the cardiovascular physiology that control a large panel of cardiac (heart rhythm, contraction, hypertrophy) and vascular functions (angiogenesis, vascular permeability, vasoconstriction). Indeed, basal Ras protein activity is required for homeostatic functions in physiological conditions, but sustained overactivation of Ras proteins or spatiotemporal dysregulation of Ras signaling pathways has pathological consequences in the cardiovascular system. The primary object of this review is to provide a comprehensive overview of the current progress in our understanding of the role of small G proteins and their regulators in cardiovascular physiology and pathologies. L I. II. III. IV. V. VI. VII. INTRODUCTION SMALL G PROTEINS OF THE RAS... RAS PROTEINS IN HEART FUNCTION... RAS PROTEINS IN VASCULAR... “RASOPATHIES”-ASSOCIATED... THE RAS SUPERFAMILY PROTEINS... CONCLUDING REMARKS 1659 1659 1665 1682 1699 1703 1704 I. INTRODUCTION The cardiovascular system is a complicated multiorgan and multicellular system that constantly and instantaneously integrates, with an extreme accuracy, a considerable amount of input information to adjust blood circulation and therefore nutrient and oxygen delivery to the whole organism according to its demand. This adjustment involves coordinated control of a great number of different functions of cardiac and vascular cells and requires an extremely high level of spatial and temporal regulation of signaling pathways driving these cellular processes. Over the past two decades, biochemical, functional, and genetic analyses as well as observations from model organisms and human pathology support the notion that monomeric G proteins of the Ras superfamily are essential elements of the spatiotemporal fine tuning of numerous cardiovascular functions and also of cardiovascular development. Ras proteins act as binary molecular switches able to regulate a wide range of cellular processes including pro- liferation, differentiation, metabolism, contraction, motility, survival, and apoptosis. They form complex signaling networks that can be activated by a large array of external upstream stimuli and can achieve highly specific cellular effects thanks to a myriad of regulators and effectors. Accordingly, basal Ras protein activity is required for homeostatic functions in physiological conditions, but spatiotemporal dysregulation or sustained overactivation of Ras protein signaling has pathological consequences in the cardiovascular system. In this review, we focus on our current understanding of the roles the different subfamilies of Ras proteins in cardiovascular development, function, and diseases. The studies presented here will help reveal the major challenges in the field and the duality of Ras proteins, displaying both beneficial and deleterious effects in the cardiovascular system. II. SMALL G PROTEINS OF THE RAS SUPERFAMILY Ras proteins (H-, N-, and K-Ras) are the founding members of a large superfamily of monomeric small GTPases (20 –25 kDa). These proteins are best known for their ability to serve as molecular switches regulating diverse cellular processes that include cell cycle progression, cell survival, actin cytoskeletal organization, cell polarity and movement, and vesicular and nuclear transport (62, 161, 266). Both unicellular and multicellular organisms express Ras proteins. The human Ras superfamily is composed of 166 members, which is divided into five major 0031-9333/13 Copyright © 2013 the American Physiological Society 1659 LOIRAND, SAUZEAU, AND PACAUD branches: 36 Ras proteins, 20 Rho, 27 Arf/Sar, 1 Ran, 61 Rab, and 11 “unclassified” sequences (TABLE 1). Although the separation of these five protein families was an evolutionary event that predated the expansion of eukaryotes, all these proteins present sequence and functional similarities (201, 263, 397). A. Structures and Domains (Figure 1) The basic structure of Ras proteins consists of a central six-stranded mixed -sheet surrounded by five ␣-helices. The ⬃20 kDa core G domain (corresponding to Ras residues 4 –166) is conserved among all Ras superfamily proteins and is involved in GTP binding and hydrolysis (355). This domain is comprised of five polypeptides loops designated G1 through G5. The diphosphate-binding loop G1 (also known as P-loop), with the consensus sequence GXXXXGKS/T, connects the 1 strand to the ␣1 helix and contacts the ␣- and -phosphates of the guanine nucleotide. The connection between the ␣1 helix and the 2 strand corresponds to G2 and contains a conserved threonine residue (Thr35) involved in Mg2⫹ coordination. The G3 domain, at the NH2 terminus of the ␣2 helix, links the sites for binding Mg2⫹ and the ␥-phosphate of GTP. The G4 domain that links the 5 strand and the ␣4 helix recognizes the guanine ring. The G5 loop, located between 6 and helix ␣5, reinforces the guanine base recognition site (195, 266, 397). Structurally, the Rho family proteins are distinguished from the Ras family by the presence of an extra 10 –15 amino acids between helix ␣3 and strand 5 (47). The function of this insert region remains to be elucidated. The transition between the GTP- and the GDP-bound form of Ras proteins is accompanied by conformational changes that dramatically affect its affinity for downstream signaling molecules. The crystal and NMR structures, in the active and inactive state, revealed that structural differences are primarily confined to two highly mobile regions, designated as switch I (corresponding to Ras residues 30 –38) and switch II (corresponding to Ras residues 59 –76) (195, 266, 397, 523). Most proteins of the Ras superfamily are localized to membranes by virtue of posttranslational modification with lipid moieties and posttranslational lipid processing (8). The only significant differences in the sequence of Ras proteins resides in COOH-terminal 25 amino acids of the hypervariable domain (HVR) and probably account for diversity of the biological functions of Ras proteins The HVR contains the protein sequences that are involved in the association of Ras proteins with membranes. This domain commonly terminates with a CAAX motif that signals for farnesyl or geranylgeranyl isoprenoid addition to the cysteine residue. This process is catalyzed in the cytoplasm by either type I geranylgeranyltransferase (GGT) or farnesyltransferase (FT). Farnesyltransferase inhibitors (FTIs), by preventing isopre- 1660 nylation of normally farnesylated Ras proteins inhibit their activity. Rab proteins contain a COOH-terminal HVR that terminates with cysteine-containing motifs that are modified by addition of geranylgeranyl lipids, with some undergoing carboxymethylation (8, 62, 195, 266, 397). Unlike other members of Ras superfamily, Ran protein is not lipid modified but contains a COOH-terminal extension that is essential for its functions (500). B. The GDP-GTP Cycle and Regulatory Proteins Ras proteins are transducers that couple cell surface receptors to intracellular effector pathways. Ras-superfamily small GTPases act by a conserved mechanism (FIGURE 2) (47, 266, 283, 284). Ras proteins cycle between “on” and “off” conformations that are conferred by the binding of GTP and GDP, respectively. Ras functions require the participation of distinct regulatory proteins to control the GDP/GTP cycling rate. Indeed, the extent and duration of Ras activation in cells depends on the interplay between a variety of negative and positive regulators of the Ras cycle. Guanine dissociation inhibitors (GDIs) maintain small GTPases in the inactive state. GTP exchange factors (GEFs) promote the active GTP-bound state by facilitating the exchange of GDP for GTP. Ras proteins have only a low intrinsic GTPase activity that is increased by interaction with GTPase activating proteins (GAPs). 1. GAPs The basic function of GTPase-activating proteins is to promote GTP hydrolysis leading to inhibition of the active state of small G proteins. Based on the consensus sequence of the typical G domains, the human genome codes for ⬃150 potential GAPs. The sizes of these proteins range from 50 to 250 kDa. GAPs acting on Rho/Rac family of small G proteins are especially abundant. In humans, over 70 potential GAPs for Rho/Rac proteins have been identified, 38 GAPs for the Rab family, 15 GAPs for Ras proteins, 31 GAPs for Arf family, and 1 GAP for the Ran protein. It is interesting to note that in the Rho family, the number of GAPs is in almost threefold excess over Rho proteins, while in contrast, in the Ras and Rab families there are approximately two more G proteins than GAPs (266). In the native state, several GAPs are in a folded, autoinhibited conformation that limits the accessibility of the catalytic site. GAPs are regulated by several different mechanisms including phosphorylation/dephosphorylation, lipid-protein or proteinprotein interactions, and degradation/resynthesis (262, 265, 266). 2. GEFs GEFs provide a direct link between Ras protein activation and membrane receptors for soluble factors such as Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM Table 1. Ras protein superfamily Ras (36) Rho (20) Family members H-Ras N-Ras K-Ras E-Ras M-Ras/R-Ras3 R-Ras TC21/R-Ras2 Rit1 Rit2/Rin RalA RalB Rap1A Rap1B Rap2A Rap2B Rap2C Noey2 Di-Ras1 Di-Ras2 RasD1 RasD2 RRP17 RPR22 Rem1 Rem2 Gem/Kir Rad Rerg Ris RERGL RasL11A RasL11B Rheb Rheb2 NKIRas1 NKIRas2 RhoA RhoB RhoC Rnd1 Rnd2 Rnd3 RhoD Rif RhoG RhoH Rac1 Rac2 Rac3 Cdc42 TC10 TCL Wrch-1 Wrch-2 RhoBTB1 RhoBTB2 Posttranslational modification Number of GEFs Number of GAPs Number of GDIs Main downstream effectors Farnesyl 29 15 1 (GDI-like) Raf/MAPK PI3-kinase Ral-GDS Farnesyl or geranylgeranyl ⬎80 ⬎70 3 Rho kinases Paks Rab (64) Ran (1) Arf (28) Other (11) Rab1A, B Rab2A, B Rab3A-D Rab4A, B Rab5A-C Rab6A-C Rab7A, B Rab7L1 Rab8A, B Rab9A, B Rab10 Rab11 A, B Rab12 Rab13 Rab14 Rab15 Rab18 Rab17 Rab19 Rab21 Rab22A, B Rab23 Rab24 Rab25 Rab26 Rab28 Rab27A, B Rab30 Rab31 Rab32 Rab33A, B Rab34 Rab35 Rab36 Rab37 Rab38 Rab39A, B Rab40A-C Rab41 Rab42 Rab43 RabL4 RasEF Geranylgeranyl 53 38 3 Ran/TC4 Arf1 Arf3 Arf4 Arf4L Arf5 Arf6 ArfRP1 ArfRP2 FLJ22595 Arfd1 Arl1 Arl2 Arl2L1 Arl3 Arl4 Arl5 Arl6 Arl7 Arl8 Arl9 Arl10A Arl10B Arl10C Arl11 Sar1b Sar1a Arl16 Arl14 RabL2A RabL2B RabL5 SRPRB LOC401884 Arl10C Arl10B RabL3 Rab20 Miro1 Miro2 1 1 Myristoyl 18 31 Number of proteins is given in parentheses. See text for definitions. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1661 LOIRAND, SAUZEAU, AND PACAUD A B α4 β 6 β6 α4 β5 G4 Rho Rab Arf Ran β4 α5 G1 β1 G5 α2 β3 Mg2+ α1 G3 (Switch II) G2 (Switch I) FIGURE 1. A: typical structure of Ras protein illustrated by the structure of H-Ras in complex with GTP. The Ras 3-dimensional structure is composed by 6 -sheets and 5 ␣-helices. Conserved sequence motifs (G1–G5) involved in nucleotide binding are represented. B: similar representation of Rho, Rab, Arf, and Ran protein structure. neurotransmitters, cytokines, or growth factors and adhesion molecules (62, 284). The first mammalian Rho GEF, Dbl, was identified as a transforming gene in an NIH3T3 focus formation assay using DNA from a cell line of human diffuse B-cell lymphoma. Since then, over 70 distinct members of this Dbl family have been identified in humans (115). GEFs for the large number of different Ras superfamily proteins vary drastically in structure, but there are significant similarities. GEFs constitute subfamilies specific for the different subclasses of Ras proteins, characterized by a 20- to 30-kDa catalytic domain responsible for the GEF activity. Examples of such GEF domains are CDC25 for the Ras family, Dblhomology (DH) and Pleckstrin-homology (PH) for Rho family, and Sec7 for the Arf family GEFs. GEFs are multidomain proteins, and the regulation of their activity occurs via a variety of strategies. They include protein-protein interactions, lipid binding, interaction with second messenger molecules [cAMP, diacylglycerol (DAG), Ca2⫹], and posttranslational modification such as phosphorylation. Basically, when GEF proteins are activated, they interact with the switch I and switch II domains of Ras-related proteins, causing a series of side-chain rearrangements. These structural changes induce a 10,000-fold increase in the rate of GDP release and its replacement by GTP, the concentration of which in the cytosol is 10 times higher than that of GDP (400). 3. GDIs Ras-related proteins are regulated by a third class of proteins, the guanine nucleotide dissociation inhibitors 1662 (GDIs) thus named as they were first identified as inhibitors of GDP dissociation (126). GDI proteins have been identified for geranylgeranylated Rab and Rho GTPases, but PDE␦ that preferentially solubilizes farnesylated Ras subfamily proteins, has been qualified as a GDI-like factor (71, 297, 330) (TABLE 1). The most important effects of GDI are their capacity to solubilize membrane-associated small G proteins in the cytosol (62, 132, 161, 283). By binding to their prenylated COOH terminus, GDIs are able to extract GDP-bound small G proteins from the membrane and to keep them inactive in the cytosol by protecting the hydrophobic lipid tail from aqueous solvent. Binding of GDIs, GEFs, and GAPs is mutually exclusive, suggesting that cytosolic GDI-bound G proteins are protected from nucleotide alternation regulated by GEFs and GAPs (78). The dissociation of GDI from the small G protein is therefore a prerequisite for GEF- and GAP-mediated regulation of the activity of small G proteins. Nevetheless, the mechanisms by which GDIs deliver their target G proteins to membranes remain largely unknown. Current knowledge suggests that a variety of different mechanisms may be involved, including regulation by candidate GDI-displacement factors (GDFs) for RabGDI and PDE␦ and phosphorylations for RhoGDIs (78). GDFs, through allosteric regulation, may squeeze the lipid out by shrinking the lipid-binding pocket of GDI, thereby facilitating the release of prenylated G proteins and its translocation to membranes. Contrary to RabGDI and PDE␦, there is no GDF candidates identified for RhoGDIs, and accumulating evidence suggests that the affinity between RhoGDIs and Rho proteins, and thus their release, is regulated by phosphorylations (132). Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM A B Ras Ras GTP GDP Rho GTP GAP GDP Effector Pi GTP Rho Ras Rho GDP GDP Pi GDI GTP GDP GDI C D Cargo GTP GAP Effector GEF GEF Rho GTP GDI Importin Rab GAP Ran GDP GDF Nucleus REP Importin Cargo REP Rab Rab GDP Ran GDP Cytoplasm Rab GDP Rab GDP GDP Ran GTP GDP RCC1 Rab Rab GDP GTP GAP GEF REP GDP GTP Effector FIGURE 2. Activation cycle of small G proteins and their regulatory proteins. A: Ras GTPase cycle. Inactive Ras is GDP-bound and localized in the cytosol. Activation of Ras is mediated by a guanine nucleotide exchange factor (GEF). GTP loading induces the translocation of Ras from the cytosol to the plasmatic membrane and permits the interaction with effectors. To “turn off” the cycle, a GTPase-activating protein (GAP) accelerates the intrinsic GTPase activity of Ras, allowing Ras to return to its inactive state in the cytosol. Arf proteins have the same cycle of activation. B: the GDP-GTP cycle of Rho GTPases is similar to Ras proteins but a new regulator appears: the guanine nucleotide-dissociation inhibitor (GDI). This protein binds specifically to GDP-bound Rho, prolonging the inactive state and sequestering the GTPase in the cytosol. C: Ran protein is a regulator of nuclear import and export. Activation cycle of Ran occurs in the cytoplasm and in the nucleus. Nuclear Ran is maintained in the GTP-bound state through localization of GAP in the cytosol and the GEF RCC1 in the nucleus. Ran is the only small GTPase without posttranslational modification. D: Rab proteins include COOH-terminal prenylation signals. The Rab escort protein (REP) interacts with newly synthesized Rab and allows the Rab geranylgeranyl transferase (RabGGT) to add two geranylgeranyl lipid groups to the COOH terminus of Rab. Rab activation follows the prototypical GDP/GTP cycle of Rho proteins. Nevertheless, an additional step is needed to the localization of Rab. The insertion of Rab in the target membrane is mediated by a GDI dissociation factor (GDF) that releases the Rab from GDI. C. Subgroups of Ras Superfamily 1. Ras family The history of Ras proteins started in the early 1980s by the discovery that a common set of genes, termed ras (for rat sarcoma virus), are responsible for the oncogenic properties of the Harvey and Kirsten sarcoma viruses (97). The Harvey and Kirsten sarcoma virus-associated oncogenes were thus named H-ras and K-ras, respectively. Together with the subsequently discovered neuroblastoma ras oncogene (Nras), products of these genes constitute the founding members of the canonical Ras proteins (149, 478). Hormones, cytokines, and growth factors through the stimulation of plasma membrane receptors activate Ras proteins. This activation involves receptors tyrosine kinase and specific Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1663 LOIRAND, SAUZEAU, AND PACAUD GEFs (Sos, RasGRP, or RasGRF) (213). The effectors with which they interact determine the biological effects of Ras proteins, including gene expression, proliferation, survival, differentiation, cell cycle entry and cytoskeletal dynamics. The most important signaling pathways initiated by active Ras proteins are summarized in other reviews (116, 213, 374, 571). Dysregulation of these cellular functions is a hallmark of cancer. In humans, dominant gain-of-function mutations of these Ras sequences were discovered in many cancers of the bladder, colon, and lungs (374). The resulting amino acid replacements were usually found to affect the hot spot residue 12, and also less frequently residues 13 and 61. Oncogenic substitutions in residues G12 and G13 prevent the formation of van der Waals bonds between Ras and the GAP proteins, which results in the pronounced attenuation of GTP hydrolysis. Similarly, oncogenic mutations of Q61 reduce the rate of GTP hydrolysis by affecting the coordination of a water molecule thereby protecting from the nucleophilic attack on the ␥-phosphate (59, 424). 2. Rho family The Ras homologous (Rho) family of small GTPases contains 20 members in humans. The most studied members are RhoA, Rac1, and Cdc42. Rho subfamily proteins act as molecular switches within extracellular factor-activated intracellular signaling pathways that regulate multiple cellular functions. RhoA, Rac1, and Cdc42 are ubiquitously expressed, and the major function of these proteins is the regulation of the cytoskeleton. However, the impact of each member of Rho family on the cytoskeleton could be different. RhoA activation leads to the formation of actin stress fibers and focal adhesion, as well as membrane protusion by activating mDia and driving actin polymerization (162, 391). Stimulation of Rac1 induces actin polymerization at the cell periphery to generate protrusive actin-rich lamellipodia and membrane ruffling. Cdc42 initiates long parallel bundles of actin filaments to induce the formation of filopodia. These reorganizations of the actin cytoskeleton induced by Rho proteins have consequences on cell morphology, polarity, motility, and adhesion (47, 62, 161, 283). In addition to their regulation of the cytoskeleton, Rho proteins could act as regulators of enzymatic activity. For example, Rac1 controls the enzyme complex responsible for superoxide formation (NADPH oxidase) (421), whereas RhoA regulates the Rho-kinases (Rock1 and Rock2) to modulate cell contraction (283, 284). Last but not least, Rho proteins play a major role in additional process such as gene transcription and cell cycle progression (306, 353, 524). 3. Ran family The small GTPase Ran has functions in centrosome duplication, microtubule dynamics, chromosome alignment, ki- 1664 netochore attachment of microtubules, and nuclear-envelope dynamics, in addition to its characterized roles in nucleocytoplasmic transport. Ran was first identified as an essential cofactor for nuclear import of nuclear localization signal (NLS)-containing proteins that interacts with transport factors from the importin- superfamily, also known as the karyopherins (240). The NLS-containing protein-associated importin- binds directly GTP-bound Ran protein in the nucleus, and this interaction triggers the release of the NLS-containing protein from the imported complex. The Ran-GTP-importin- complex returns to the cytoplasm, and importin- disassembles from Ran-GTP upon Ran-GAP1-mediated hydrolysis of GTP to GDP. Free Ran-GDP is then imported into the nucleus by a mechanism that involves the nuclear transport factor-2 (390, 453), where it is loaded with GTP by the Ran guanine nucleotide-exchange factor RCC1 (46). RCC1 is restricted to the nucleus, while the Ran-GAP1 is concentrated in the cytoplasm. Due to this compartmentalization of Ran regulators, the concentration of GTP-bound form of Ran is predominantly localized in the nucleus while the GDP-bound form is prevalently cytoplasmic and the energy supplied by GTP hydrolysis in the cytosol drives nuclear transport (147, 572). 4. Rab and Arf families Originally described as Ras-like proteins in brain, Rab proteins constitute the largest family of small Ras-like GTPases with more than 60 members in humans. They are localized in the membrane of various subcellular organelles (463). Most intracellular vesicles contain more than one type of Rab preotein. Analysis of Rab-GTP structures shows the greatest structural heterogeneity in their switch domains. These structural differences could explain how different Rab proteins recruit specific sets of effectors to regulate precisely their respective pathways. Rab proteins are associated with vesicular trafficking functions, including ensuring transport specificity and demarcating organelle identity (156, 185, 463). Rab proteins use the classical GDP-GTP cycle, but use distinct additional regulatory protein. De novo synthesized Rab is GDP bound and inactive, with a very low affinity for RabGGT. Newly synthesized Rab protein associates with Rab escort protein (REP) to interact with RabGGT and to be prenylated. The insertion of the Rab-GDP into the target membrane is mediated by a GDF that releases the Rab from GDI (185). A large body of evidence indicates that the Arf family is also implicated in membrane trafficking. The Arf family includes the Arf proteins (ADP-ribosylation factor), the Sar proteins (secretion and Ras-related protein), and the Arl (Arf-like) proteins. Arfs are ubiquitously expressed, but they are mainly concentrated in the Golgi apparatus. In this Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM organelle, Arfs regulate the assembly of coat complexes around budding vesicles (195, 267). pressed in the heart cannot compensate for the loss of K-Ras function in K-Ras-deficient mice. Recent results suggest a cross talk between Rab and Arf proteins. For example, it is reported that the Arf6 GTPase negatively regulates Rab35 activation and hence the fast endocytic recycling pathway (79). Nevertheless, the embryonic heart development is normal in mice in which K-Ras has been replaced by H-Ras (371) and the introduction of H-Ras transgene in K-Ras-deficient mice rescued mice from embryonic lethality in association with correction of thin ventricular walls of the heart of K-Ras-deficient mice (327). These observations thus suggest that intrinsic activities of H-Ras and K-Ras are similar and that H-Ras can functionally replace K-Ras in embryonic heart development and cardiomyocyte proliferation (327). However, during the adult life, K-Ras function is not entirely rescued by H-Ras as adult mice in which K-Ras has been replaced by H-Ras develop dilated cardiomyopathy associated with arterial hypertension, suggesting that KRas has a unique role in cardiovascular homeostasis (371). III. RAS PROTEINS IN HEART FUNCTION AND DISEASES A. Embryonic Heart Development The heart is the first organ to be formed during embryogenesis in vertebrates. The primordium of this organ is derived from mesodermal cells that are involved in numerous interactions with cells from several embryonic origins (352). The constitution of the multichambered heart is a multistep process, starting by linear heart tube formation from paired cardiac fields, then followed by the formation of the cardiac chambers, heart looping, and septation (352). The epithelialmesenchymal transformation of the endocardial cells and invasion of the endocardial cells into the cardiac jelly are the major events involved in the formation of endocardial cushions that play a critical role in valve formation and cardiac septation, which separates the primitive heart tube into four chambers (352, 459). After cardiac looping, cardiac myocytes express muscle genes, the specific site of which is characteristic for each chamber, thereby leading to chamberspecific properties. These processes require precise temporal and region-specific regulation of cell proliferation, migration, death, and differentiation. Among the intracellular signaling molecules involved in this regulation, small G proteins and their effectors have been recognized as key actors of cardiac embryogenesis. 1. Ras proteins A) RAS. In situ hybridization analysis showed that all three ras genes (H-ras, K-ras, N-ras) are uniformly expressed in the embryonic heart (embryonic day E13.5–E18.5) (329). While H-Ras-deficient mice and N-Ras-deficient mice are born and grow normally, the K-Ras-deficient embryos die progressively between E12.5 and term (230). K-Ras-deficient embryos display heart abnormality on E15.5, with thin ventricular walls that can be reduced to a one-cellcompact layer (230). However, the hearts of the K-Rasdeficient embryos were without septal and trabecular defects, and cardiomyocyte-specific makers were expressed in ventricular myocytes. It is likely that the thin ventricular walls in the K-Ras-deficient embryos are not related to abnormal differentiation or cell death but are due to a deficiency in the proliferation of the cardiomyocytes (230). This indicates that K-Ras is essential for normal heart development and that other Ras members H-Ras and N-Ras ex- Activation of the Raf/MEK/ERK1/2 signaling pathway in endothelial cells is a major effector of Ras during cardiac development. The hearts of endothelial specific ERK1/2 knockout mice are much smaller than that of control mice, with highly reduced cellularity, and the endocardial layer was detached from myocardial trabeculae indicating defective heart development and function (458). This phenotype is associated with a reduced migration and proliferation of endothelial cells. The Ras exchange factor of K-Ras and H-Ras Sos1 has been identified as one potential regulator of Ras activity during embryonic heart development. Sos1-deficient embryos have an enlarged heart and distended pericardium (508). The myocardium of mutant embryos frequently showed reduced trabeculation. The orientation of the heart during development may also be abnormal in Sos1-deficient mice. Other aspects of cardiac development in Sos1-deficient mice appeared relatively normal, including the partitioning of the heart and the development of endocardial cuffs (508). This specific phenotype of Sos1-deficient mice has been related to the role of Sos1 downstream of the EGF receptor (508). The role of Ras in cardiac development has been further documented through indirect evidence arising from studies on neurofibrin encoded by the Nf1 gene. Neurofibrin is a Ras GAP and thus downregulates Ras signaling (294). Loss of function mutation of Nf1 in human leads to neurofibromatosis type1 (NF1) characterized by the appearance of benign and malignant tumors and cardiovascular defects such as pulmonary valvular stenosis (574). Mice in which the Nf1 gene has been inactivated have been useful to decipher the role of Ras signaling in the embryonic development of the heart, in particular valve formation. Homozygous deficient embryos die in mid gestation (E14) and exhibit signs of cardiac dysfunction with histological abnormalities such as an overabundance of tissue in endocardial cush- Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1665 LOIRAND, SAUZEAU, AND PACAUD ions of the outflow tract due to hyperproliferation and lack of normal apoptosis, and abnormal cardiac morphogenesis including a failure of proper conotruncal rotation resulting in double outlet right ventricle (247). Endothelial specific inactivation of Nf1 recapitulates the cardiovascular abnormalities involving endothelial cushions and myocardium (138). In contrast, endothelial specific expression of the Ras GAP-domain of neurofibromin rescued cardiac development in Nf1⫺/⫺ embryos (190). With the use of explants of endocardial cushion tissue, it has been also demonstrated that Nf1⫺/⫺ explants displayed enhanced epithelial-mesenchymal transformation that was corrected by the dominant negative Ras (Ras-N17) and mimicked by constitutively activated Ras (Ras-L61) in wild-type endocardial cushion explants (247). This indicates that Ras signaling is necessary and sufficient for endocardial-mesenchymal transformation and proliferation. B) RALA AND RALB. RalA and RalB are both expressed in the myocardium and endocardial cushions of developing heart in mice from E9.5 to E16.5. Higher levels of expression were seen in the apical edge of endocardial cushions in the fusion seam of the septa at E12.5 and E13.5, a critical period for the formation of septa that involved mechanism of apoptosis in atrioventricular cushions (584). C) RHEB. Rheb (Ras homolog enriched in brain)-deficient embryos die around midgestation because an impaired development of the cardiovascular system with thinning of the ventricular walls (141). 2. Rho proteins Rho proteins have been found necessary for the development of normal heart. An original experimental approach consisting of cardiac overexpression of Rho GDI␣ using the ␣-myosin heavy chain promoter, active at embryonic day E8.0 during morphogenesis on the linear heart tube, has been used to analyze the role of Rho proteins in cardiac morphogeneis (520). Cardiac overexpression of RhoGDI␣ led to embryonic lethality. In these embryos, cardiac morphogenesis was disrupted, with incomplete looping, lack of chamber demarcation, hypocellularity with inhibition of cell proliferation, and lack of trabeculation. As the activities of RhoA, Rac1, and Cdc42 were all decreased in this model, the respective role of these three proteins cannot be determined. A) RHOA. RhoA transcripts and proteins have been shown to be markedly upregulated in the heart forming region in chick embryo (204). SiRNA-mediated RhoA silencing inhibited the fusion of the bilateral heart primordia, suggesting that RhoA is involved in the migration of cardiac precursors to the ventral midline and is necessary for the proper looping of the heart and the development of normal heart (204). During development, the initial myocardiumwide expression of RhoA becomes restricted to the right 1666 side of the sinus venosus myocardium including the sinoatrial node, suggesting a role of RhoA in the conduction system development (501). Rho-kinases appear as key effectors of RhoA signaling during cardiogenenesis. Rhokinase transcripts are enriched in cardiac mesoderm and Rho-kinase inhibition induced cardia bifida and precocious expression of early marker genes of cardiomyocytes differentiation (521). Rock1 and Rock2 genes are expressed in the atrioventricular endocardial cushions at all phases of cardiac development, and pharmacological blockade of Rho-kinases inhibits epithelial-mesenchymal transformation and cell migration, suggesting that Rhokinases play an essential role in endocardial cell differentiation and migration during endocardial cushion development (521, 583). Rho-kinases are also involved in the formation of initial heart myofibrillogenesis by means of actin-myosin assembly, and focal adhesion/costamere and cell-cell adhesion (410). Upstream to RhoA, the Rho GEF AKAP13 could account, at least in part, for the role of RhoA activity in the development of the heart (302). Developing hearts of AKAP13-null mice display thin cardiac walls, with cardiomyocytes showing deficient sarcomere formation, and mice die at embryonic day 10.5–11.0. This phenotype is likely to be due to a reduced RhoA-dependent activation of the transcription factors SRF and MEF2c and defect in actin assembly (302). B) RHOB. RhoB is mainly localized on endosomes and regulates cytokine trafficking and cell survival. RhoB levels vary through the cell cycle, and the rhoB transcript has a half-life of 30 min, which is substantially shorter than rhoA or rhoC and indicates that RhoB function requires its expression to be highly regulated (526). During cardiac embryogenesis, RhoB is expressed in the developing atrioventricular and outflow tract endocardial cushions throughout their development (163). Transcripts can first be detected in the endocardial cells overlying the cushions, prior to the delamination of cushion mesenchyme. RhoB increases in expression as epithelium to mesenchyme transformation begins, and reaches a peak of expression as the mesenchymal cells migrate into the cardiac jelly. RhoB expression is downregulated at E11.5 as the endocardial cushions begin their differentiation to form the valves and septa of the heart (163). Expression of RhoB in migrating cells seems likely to be associated with its role in organization of the actin cytoskeleton. C) RAC1. Rac1 is the only Rac gene expressed early in development. In mouse embryo, although the cardiac mesoderm was specified, the lack of Rac1 is associated with the absence of fusion of the heart at the midline, leading to cardia bifida (308). This defect has been ascribed to a defect in the actin remodeling required for the migration of the nascent mesoderm (308). Targeted deletion of Rac1 in the endothelial cells is sufficient to produce defects in cardiac development. Endothelial Rac1-deficient embryos lack a common Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM ventricular chamber of the heart but seemed to develop the atrial chamber and the atrioventricular canal (477). The atypical Rac exchange factor Dock180 was found to have a major role during cardiovascular development. Deletion of Dock180 in mouse induces major cardiac abnomalities such as submembranous ventricular septal defect and double outlet right ventricle (413). Mitral valve leaflets were also thickened and sometimes fused, leading to blood retention in left atrium. A similar phenotype was obtained by specific Dock180 deletion in endothelial cells, suggesting the endothelial origin of the cardiac defect in Dock180deficient mice. This effect has been ascribed to a major role of Dock180 in the regulation of Rac activation to control endothelial cell migration during cardiac development, but not in the epithelial-mesenchymal transformation of cardiac endothelial cells (413). D) CDC42. Indirect evidence also suggested that Cdc42 is required for normal heart formation as the absence of the Cdc42 effector p21-activated kinase (Pak) 4 led to lethality by embryonic day 11.5, due to a defect in the fetal heart associated with a thinning of the myocardium and a dilation of the atrium and sinus venosus, probably related to the role of Cdc42/Pak4 signaling in the regulation of cytoskeleton organization (377). B. Cardiac Excitation/Contraction Coupling: Contractility The transient change in tension and length in a working cardiac myocyte during the heart beat reflects the intregrated effects of signaling cascades regulating mechanisms controlling the dynamics and intensity of rise in cytoplasmic Ca2⫹ as well as the responsiveness of the sarcomeric proteins to Ca2⫹. The opening of cardiac voltage-dependent Na⫹ channel causes initial plasma membrane and t-tubule depolarization that induces activation of L-type voltage-dependent Ca2⫹ channel (CaV1.2) and Ca2⫹ entry into the cell. The resulting fast and localized Ca2⫹ rise in the subsarcolemmal space elicits the opening of sarcoplasmic reticulum Ca2⫹ release channels (ryanodine receptors, RyRs) leading to an amplified release of Ca2⫹ constituting the Ca2⫹ transient. The sarcoplasmic Ca2⫹-ATPase SERCA2 is responsible for sarcoplasmic reticulum Ca2⫹ reuptake after systole. The Ca2⫹-dependent contractile response is regulated by the binding of cytosolic Ca2⫹ to a single site on cardiac troponin C (cTnC), which strengthens the affinity of cTnC for cardiac troponin I (cTnI). Through change in cardiac troponin T (cTnT), these Ca2⫹-dependent conformational changes in cTnC-cTnI are transmsitted to tropomyosin, favoring the displacement of the troponin complex/tropomyosin on the thin filament away from the actin-bindin sites for myosin. Myosin cross-bridges then bind actin, and crossbridge cycling proceeds (229). Phosphorylation of myosin binding protein C (MyBP-C) and myosin regulatory light chain (RCL, MLC2) also control the Ca2⫹ sensitivity of the contraction (455). Cardiac excitation-contraction coupling is regulated by a variety of signaling molecules and receptors, one of the most significant being the -adrenergic stimulation coupled to cAMP pathway. Emerging evidence indicates that small G proteins, particularly by controlling intracellular Ca2⫹ signaling and phosphorylation/dephosphorylation processes, contribute to the regulation of cardiac excitation-contraction coupling and contraction. 1. Regulation of Ca2⫹ handling mechanisms A) RAS PROTEINS. I) Ras. The hypertrophic effect of Ras is associated with changes in the contractile properties of the myocardium. The cellular mechanism involved in Ras-mediated cardiac contractile defects has thus been the subject of a number of studies. Expression of constitutively active Ras (HRas-V12) in cultured neonatal cardiac myocytes reduced the amplitude and prolonged the decay phase of the contractile Ca2⫹ transients (177). It also decreased the expression of SERCA2 and striated myofibrils. This Ras-mediated regulation of intracellular Ca2⫹ in cardiac myocytes was ascribed to the activation of the Raf-MEK-ERK cascade (177). However, it was unexpectedly observed that Ca2⫹ loading of the reticulum was increased in H-Ras-V12 cardiomyocytes, suggesting that H-Ras-V12 affects the excitation-Ca2⫹ release coupling. In support of this hypothesis, H-Ras-V12 was shown to reduce the amplitude of the L-type calcium channel current in cardiac myocytes through a Raf-MEK-ERK-dependent decrease in the expression of the pore-forming ␣1C subunit (also called Cav1.2) of the Ca2⫹ channel (176). In vivo, direct hemodynamic measurements in mice demonstrated that selective expression of H-Ras-V12 in cardiac myocytes significantly decreases cardiac contractility as well as causes diastolic dysfunction associated with interstitial fibrosis and a blunted response to a -adrenergic agonist (585). At the ventricular myocyte level, these changes in contractility were associated with a reduction and a slowing of the decay of intracellular Ca2⫹ transient and a decrease of the sarcoplasmic reticulum Ca2⫹ content, confirming results obtained in cultured myocytes (402, 585). Only a modest reduction of the expression of SERCA2 was observed, and the expression and activity of the Na⫹/Ca2⫹ exchanger were found to be increased (402, 585). However, a strong decrease in Ser-16 phosphorylation of the SERCA2 regulator phospholamban (PLB) associated with an increase in the expression of protein phosphatase PP1␣, the major PLB phosphatase in the heart, without change in PP2A level was observed in H-Ras-V12 ventricular myocytes (402, 585). These results provide evidence to implicate Ras signaling in the regulation of cardiac contractility Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1667 LOIRAND, SAUZEAU, AND PACAUD through modulation of Ca2⫹ handling proteins expression and activity, including the L-type Ca2⫹ channel and SERCA2. B) RAP PROTEINS. A role of Rap1 protein in cardiac myocyte excitation-contraction coupling became evident from recent studies. Besides the activation of the canonical PKA signaling, cAMP activates other signaling molecules, including Rap1 and Rap2 via the recruitment of cAMP-activated exchange factors Epac (52). In cardiac myocytes, activation of Epac increases electrically evoked Ca2⫹ transient via PLC⑀, the catalytic activity of which is directly regulated by Rap (350). Furthermore, inhibition of Rap activity by expressing RapGAP significantly inhibited -adrenoceptordependent stimulation of Ca2⫹ release. Epac-mediated regulation of -adrenergic-dependent stimulation of CICR appears to be mediated by PLC⑀-dependent stimulation of CAM kinase II and subsequent increase in the phosphorylation of the type 2 RyR (RyR2) and phospholamban (349, 362). Further evidence also suggested that Rap is involved in the electrical coupling of cardiomyocytes through the regulation of gap junctions which are essential to coordinate cardiac contractions; Epac-Rap1 signaling redistributes connexin 43, increasing the accumulation at cell-cell contacts, and leads to the formation of new gap junctions. This redistribution of connexin 43 is related to Rap-mediated N-cadherin accumulation at the cell-cell contacts, the major component of adherens junctions that are responsible for mechanical coupling between cardiomyocytes (456). C) THE RGK SUBFAMILY OF RAS PROTEINS. A common feature of the RGK subfamily members (Rad, Rem1, Rem2, Gem/Kir) is their ability to potently inhibit L-type Ca2⫹ channels in the heart and to reduce left ventricular systolic function (324, 509, 552). In addition, it has been recently shown that Rem prevents the stimulatory action of cAMP/cAMP-activated kinase (PKA) on L-type Ca2⫹ channel, suggesting a physiological and/or pathophysiological role of RGK proteins in the modulation of -adrenergic regulation of cardiac function (551). This hypothesis is further supported by the observation that Rad overexpression negated -adrenergic effects on L-type Ca2⫹ current and Ca2⫹ transients (509). One prevailing explanation of this effect is that RGK proteins interfere with the trafficking of pore forming subunit ␣1C to the sarcolemma, possibly by buffering endogenous  subunits (1, 2, and 3), thereby reducing the surface density of the cardiac L-type Ca2⫹ channels (31, 367). However, recent data showing that Rem predominantly inhibits L-type Ca2⫹ channel current by arresting surface CaV1.2 channels in a low open probability gating mode emphasize the idea that RGK proteins use diverse mechanisms and determinants to inhibit L-type Ca2⫹ channel current (551). Similarly, whether or not the RGK proteins should be in the active GTP-bound state and targeted to the cell membrane to exert its effect on Cav1.2 remains controversial (89, 551, 561, 562). 1668 D) RHO PROTEINS. I) RhoA. Ventricular myocytes isolated from transgenic mice carrying a sixfold overexpression of Rho GDI␣ in the myocardium exhibited a 40% decrease in the density of the L-type Ca2⫹ channel current (565). As there was no change in the levels of mRNA and protein of the L-type Ca2⫹ channel, this inhibition is due to an effect on the activity of the channel. Expression of a dominant negative form of RhoA but not a dominant negative Rac1 or Cdc42 mimicked the effect of GDI␣ overexpression, indicating that the L-type Ca2⫹ channel is a downstream target of RhoA in cardiac myocytes (565). The exact molecular mechanism responsible for the positive action of RhoA on the L-type Ca2⫹ channel has not been precisely defined; however, as the density of channels in the plasma membrane was not modified, it seems to affect the activity of the channel through an actin-independent signal pathway (565). The progressive atrioventricular conduction defects displayed by GDI␣ overexpressing mice are in agreement with a loss of the positive regulatory role of RhoA on L-type Ca2⫹ channel activity (522). However, surprisingly, the ventricular contractile function was largely preserved in Rho GDI␣ transgenic hearts (522). In ventricular myocytes, L-type Ca2⫹ current constitute the major pathway for Ca2⫹ entry that initiates excitation-contraction coupling, and pharmacological inhibition of L-type Ca2⫹ channel expression or activity affects myocyte contractility. This thus suggests that functional compensation exists in GDI␣ overexpressing mice, allowing a normal Ca2⫹ transient and contractility of ventricular myocytes despite a reduced L-type Ca2⫹ current (522). RhoA/Rho-kinase signaling also affects intracellular Ca2⫹ store as in contracting neonatal ventricular myocytes, and pharmacological inhibition of Rho-kinase by Y27632 induces a 150% increase in SERCA2 mRNA expression (502). This effect has been shown to be due to a repressing effect of RhoA/Rho-kinase signaling on the SERCA2a promoter activity by transcription factor other than the wellknown RhoA/Rho-kinase-dependent transcription factor SRF, myocardin, or GATA4 (502). II) Rac and Cdc42. Although it has been suggested in other cell types that Rac and CdC42 inhibit L-type calcium current, direct evidence for such an effect in the heart does not exist (529). Nevertheless, the identification of the Rac/ Cdc42 Pak1 as a major regulator of the catalytic activity of the protein phosphatase 2A (PP2A) strongly argues for a role of Rac/Cdc42-Pak1 signaling in the regulation of cardiac contractility. It has been suggested that Pak1 forms a signaling module with PP2A in cardiomyocytes. Pak1 is highly expressed in the heart, and, as PP2A, localized to Z-disc, cell and nuclear membrane and intercalated disc of ventricular myocytes (220). In the resting state of the Pak1/ PP2A complex, PP2A is inactive and phosphorylated on Tyr-307 (221, 525). Activation of Rac1/Cdc42 and the subsequent association of the small G protein with the Rho binding domain of Pak1 promotes its activation and its Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM auto-phosphorylation at Thr-423 inducing a conformational change in PP2 that allows autodephosphorylation of Tyr-307 and stimulation of its catalytic activity towards its substrates such as L-type Ca2⫹ channel and Ryr2 (220, 221). Expression of constitutively active Pak1 in single, isolated adult rat ventricular myocytes significantly changes the decay and peak characteristics of spontaneous [Ca2⫹]i transients both under resting conditions and after -adrenergic stimulation (441). This occurs without changes in the Ca2⫹ content of the sarcoplasmic reticulum and in the phosphorylation of phospholomban. Although not demonstrated, it has been suggested that Pak1 activity reduced Ca2⫹ transient by altering L-type Ca2⫹ channels and/or RyR2 gating via dephosphorylation by PP2A (220, 441). In addition, Rac1 controls the electrical coupling of cardiomyocytes. In cardiac myocytes in vitro, activation of Rac1, downstream of N-cadherin, determined the localization of connexin 43 at the intercalated disks (300). E) RAB PROTEINS. Endosomal trafficking represents an important mechanism for maintaining proper plamalemmal expression of ion channels, including L-type Ca2⫹ channel. Endosomal transport is largely regulated by the Rab family of small G proteins (463). Among Rab proteins, Rab11b has been recently identified as a regulator of CaV1.2 L-type Ca2⫹ channel surface membrane density (37). Rab11b is expressed in ventricular myocardium and in cardiac myocytes; expression of a dominant-negative form of Rab11b increases the peak of L-type Ca2⫹ channel current by 98%, indicating that contrary to that expected, Rab11b limited rather than promoted the plasma membrane expression of CaV1.2. It has thus been proposed that, in cardiac myocytes, Rab11b regulates endocytic recycling of CaV1.2 by targeting the internalized channels toward a degradation pathway (37). 2. Regulation of contractile proteins Ras signaling through ERK1/2 to p90RSK (ribosomal S6 kinase) induces phosphorylation of cTnI on S23/S24 (194). In protein lysates from cardiomocytes, phosphorylation of cTnT is inhibited by the Raf inhibitor GW5074, and in vitro kinase assays indicated that Raf-1 phosphorylated cTnT on T206 that has been linked to myofilament function (363). A) RAS PROTEINS. B) RHO PROTEINS. I) RhoA. RhoA through Rho-kinase increases the phosphorylation of the 20-kDa myosin regulatory light chain (RCL), via direct phosphorylation of myosin phosphatase targeting peptide (MYPT2) and consequently, inhibition of the myosin light chain phosphatase (MLCP) (428). This Rho-kinase-mediated increase in RLC phosphorylation could participate in the positive inotropic effects of RhoA activating agent such as ␣-adrenoceptor or PGE2 receptor agonists (381). More recently, it has been demonstrated that Rho-kinase also directly phsophorylates cTnI (on S23, S24, and T144) and cTnT (on S278 and T287) (493). This effect is associated with a depression of the maximal tension and a decrease in the Ca2⫹ sensitivity (493). In addition, RhoA/Rho-kinase signaling has been proposed to participate in the maturation of myocardial contractile system through phosphorylation of its molecular targets in Z-discs and intercalated discs (464). II) Rac1/Cdc42. More recent evidence also suggests that activation of the Rac1 and Cdc42 effector Pak1 leads to decreased phosphorylation of TnI and MyBP-C through interaction with the phosphatase PP2A and stimulation of PP2A activity (221, 440). In single cardiac myocytes, Pak1 increased the Ca sensitivity of tension, consistent with PP2mediated dephosphorylation Ser-23/24 of cTnI (441). Although through a distinct mechanism, Pak3 has been shown to also increase the Ca sensitivity of cardiac myofilaments (61). This effect could be related to direct phosphorylation of cTnI on a new phosphorylation site identified as Ser-149 in the inhibitory domain, and of cTnC. Taken together, these studies suggest that Paks form signaling complexes in the heart that are active in cTnI regulation. C. Heart Rhythm and Arrhythmia (Table 2) The normal mechanical functioning of the heart depends on proper rhythmic electrical activity, originating from specialized “pacemaker” cells in the sinoatrial node (SAN) in the right atrium. Propagation of this initial excitation wave through intercellular gap junctions leads to the depolarization of adjacent atrial myocytes, ultimately resulting in excitation of the atria. Next, the excitation wave propagates via the atrioventricular node (AVN) and the Purkinje fibers to the ventricles, where ventricular myocytes are depolarized, leading to excitation of the ventricles. Rhythmic cardiac electrical activity is attributed to the generation of action potentials in individual cardiac cells. In atrial and ventricular myocytes, the resting potential between each action potential is stable and negative (around ⫺85 mV) due to the high conductance of the IK1 channel (15). Electrical impulses received from adjacent cells induces opening of Na⫹ channels and the generation of Na⫹ current responsible for the rapid depolarization phase of the action potential. Transient outward K⫹ current (ITO) is then responsible for the early repolarization phase. The plateau of the action potential results from a balance between depolarizing L-type Ca2⫹ channel and outward delayed rectifying K⫹ currents (IKur, IKr, and IKs). The repolarization phase is due to the closing of L-type Ca2⫹ channels and the maintenance of outward rectifying K⫹ current and then to K⫹ efflux through IK1 channels. The P wave and the QRS complex of the surface electrocardiogram represent atrial and ventricular excitation, respectively, and the T wave represents ventricular repolarization. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1669 LOIRAND, SAUZEAU, AND PACAUD Table 2. Effect of Ras proteins on cardiac ion channels and heart rhythm Ras Protein Ras Target Currents/Channels IK,Ach Effect Inhibition of activation by muscarinic receptor stimulation Effector/Signaling Involved Ras-GAP Mice Models H-Ras-V12 Increased Gi␣ signaling Kir2.1 L-type Ca2⫹ channel Gem L-type Ca2⫹ channels Inhibition by decreasing membrane density of the channel Inhibition by decreasing expression of the ␣1c subunit of the channel Inhibition by decreasing membrane density of the channel Rad L-type Ca2⫹ channels Rem L-type Ca2⫹ channels Rap1a IKAch RhoA IKAch Inhibition by decreasing expression of the ␣1c subunit of the channel Inhibition by maintaining the channel in a lox open probability gating mode Antagonize the effect of Ras on atrial IK,Ach Constitutive activation Kv1.2 Inhibition IKr IK1 L-type Ca2⫹ channel Inhibition of hERG Inhibition of Kir2.1 Stimulation of channel activity Rac1 Cdc42 Rab11b 1670 IKr Stimulation of hERG Kir2.1 Increased membrane density of the channel Alteration of gating properties Alteration of gating properties Target the channels toward degradation T-type Ca2⫹ channel T-type Ca2⫹ channel T-type Ca2⫹ channel In Vivo Phenotype Arrhythmia (sinus arrest, idioventricular rhythm, ventricular tachycardia, conduction block, and AF) Reference Nos. 402, 566 MAPK 137 Raf-MEK-ERK 176 Adenoviral delivery of Gem: - in the left ventricle - in the AV node Cardiac specific Rad-DN overexpression 324 - QT interval shortening - PR interval prolongation Arrhytmia (sinus node dysfunction, AV block) 509, 552 551 Cardiac specific RhoA overexpression Bradychardia with prolonged PR intervals, seconddegree AV block and AF Rho-kinase Alteration of electrical propagation (decreased expression of Cx40) NADPH oxidase (CTGF, N-cadherin, Cx43) Cardiac specific overexpression of GDI␣ Bradychardia, AV block Cardiac specific Rac-V12 overexpression AF Increased activity of PP5 65, 407 467 203 522, 565 2, 3 134, 467 54 Pak1/PP2A 220, 441 Pak1/PP2A 223, 441 Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 37 SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM In contrast to atrial and ventricular myocytes, the absence of IK1 in SAN and AVN myocytes allows a slow depolarization of the resting potential to occur between each action potential, via, though not exclusively, the activation of the inward pacemaker currents (If), the T-type Ca2⫹ current, and Cav1.3-dependent L-type Ca2⫹ current. As a consequence, most Na⫹ channels are inactivated and, in SAN and AVN myocytes, the depolarization phase of the action potential is mainly achieved by the L-type Ca2⫹ current (290). Heart rate is physiologically regulated by the autonomic nervous system that modulates pacemaker currents in SAN cells. Release of norepinephrine and -adrenergic stimulation following sympathetic stimulation increases the heart rate, primarily by modulating Ca2⫹ and Na⫹ permeability. In contrast, parasympathetic fiber stimulation decreases heart rate through the activation of IK,Ach (generated by Kir3.1 and Kir3.4) and the decrease of If pacemaker current and Cav1.3-dependent L-type Ca2⫹ current by acetylcholine-mediated M2 muscarinic receptor activation. Modifications of the properties, regulation, and/or expression of cardiac ion channels can modify action potential waveforms, synchronization, and/or propagation, thereby increasing susceptibility to arrhythmias. Atrial fibrillation (AF) is the most common cardiac arrhythmia, often associated with many forms of cardiac pathology. However, ventricular arrhythmias are the most common cause of sudden cardiac death in hypertrophic cardiomyopathy and other cardiac diseases, including heart failure. Numerous studies have shown that small G protein regulation of electrical coupling, ion channel function, location, or expression participates in the generation and maintenance of normal and/or abnormal heart rhythms (2, 203, 324, 402, 407, 522, 552). 1. Ras proteins A) RAS. Ras and Ras-GAP have been found to block IK,Ach activation by muscarinic receptor (566). This effect occurs by uncoupling of the receptor from the trimeric G protein rather than uncoupling of the G protein from the channel. It has been proposed that binding of GTP-bound Ras to RasGAP causes a conformational change that exposes the SH2SH3 domain of the NH2 terminus of Ras-GAP, allowing interaction with membrane phosphoproteins and finally uncoupling of muscarinic receptor from the trimeric G protein (295). Ras is also a negative regulator of the inwardly rectifying K⫹ channel Kir2.1 via a mechanism involving the MAPK cascade (137). Active Ras reduces IK1 current by reducing the amount of available Kir2.1 channel at the cell surface membrane. This effect is not due to a transcriptional regulation but to an action of the Ras-MAPK pathway on the trafficking of Kir2.1 molecules. In 1991, a tight linkage between H-ras gene on chromosome 11p and long QT syndrome in a large Utah family was reported (222). In agreement with its effect on IK,Ach, H-Ras is considered as a candidate gene for this autosomal dominant cardiac arrhythmia. However, both sequencing and more precise definition of the chromosomic region containing the LQT gene has excluded H-ras from the candidate genes for LQT syndromes (401). Nevertheless, some studies in transgenic mice model described a role of Ras in heart rhythm regulation and arrhythmia. In addition to cardiac hypertrophy, transgenic mice with a tamoxifen inducible cardiac specific expression of H-Ras-V12 also gradually developed severe arrhythymia (402). Cardiac arrhythmia in H-Ras-V12 transgenic mice started at around day 7 after tamoxifen induction of the trangene expression and was more pronounced at day 14. Electrocardiogram recordings indicated sinus arrest, idioventricular rhythm, ventricular tachycardia, conduction block, and AF. Severity of arrhythmia in H-Ras-V12 mice was influenced by gender-specific factors as females demonstrated a more consistent arrhythmia phenotype. The onset of arrhythmia in H-Ras-V12 mice was temporally correlated with the increase of action potential duration, an altered sarcoplasmic reticulum Ca2⫹ cycling leading to an increase in Na⫹-Ca2⫹ exchanger current, and a decrease in outward K⫹ current (402). In addition to these changes in electrophysiological properties of cardiac cells, H-Ras-V12 expression also led to induction of Gi␣1 expression and pertussis toxin (PTX) treatment, that blocks Gi␣ activity, normalized electrophysiological parameters, and reduced arrhythmias (402). This report thus suggests that contrary to hypertrophy, the electrophysiological remodeling induced by H-Ras-V12 expression in mouse heart does not seem to depend on a MAPK-dependent pathway, but strongly involves upregulation of Gi␣ signaling. B) RGK FAMILY. The role of RGK family in the regulation of heart rhythm is directly related to their actions on the L-type Ca2⫹ channel. In vivo adenovirus-mediated delivery of Gem, injected in the left ventricular cavity of guinea pig hearts, produces a significant shortening of the electrographic QT interval, due to decreased expression of L-type Ca2⫹ channels and the resulting abbreviation of action potential duration (324). Focal delivery of Gem adenovirus to AV node in pig prolongs the PR interval of the surface electrocardiogram, indicating slowed conduction in the AV node. Moreover, overexpression of Gem in the AV node caused a 20% decrease in the ventricular rate during AF (324). In contrast, cardiac ␣-myosin heavy chain promoter-driven overexpression of a dominant negative Rad mutant in mouse produced a prolongation of the QT interval on the surface electrocardiogram, without changes of others parameters. This was associated with an increased expression of the ␣1c subunit of the L-type Ca2⫹ channel and a pro- Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1671 LOIRAND, SAUZEAU, AND PACAUD longed duration of action potentials recorded from papillary muscle (552). Diverse arrhythmias such as sinus node dysfunction, atrioventricular block, and ventricular extrasystoles were observed in Rad transgenic mice (552). C) RAP1A. A microarray screening performed to search differentially expressed genes in samples of right appendages from patients with AF or sinusal rhythm has identified Rap1a as a gene potentially involved in the pathological process. Rap1a transcript expression is negatively correlated with AF (223). Further supporting the potential effect of Rap1a in the regulation of heart rhythm is its role as an antagonist of the inhibitory effect of Ras on atrial muscarinic K⫹ channels (567). 2. Rho proteins A) RHOA. Development of transgenic mouse models has allowed direct in vivo assessment of the role of RhoA in heart rhythm. Cardiac specific overexpression of RhoA induces a marked sinus and AV node dysfunction characterized by depression of heart rate, wandering pacemaker, prolonged PR intervals, second-degree AV block, and AF (407). While a marked positive chronotropic effect was induced by muscarinic receptor blockade by atropine in control mice, this effect was not observed in RhoA transgenic mice so that the bradycardia was not reversed by atropine (407). Since muscarinic receptors activate IK,ACh and slow heart rate via hyperpolarization of pacemaker cells in SA and AV nodes, an hypothesis to explain this phenotype and the bradycardia observed in RhoA transgenic mice would be that RhoA overexpression led to constitutive activation of IKACh, which thus became independent of muscarinic regulation. Another potential mechanism could involve delayed rectifier potassium channels that participate in the repolarization of the action potential. Supporting this hypothesis, it has been shown that RhoA physically associates with and inhibits the delayed rectifier K⫹ channel Kv1.2, originally cloned from rat heart atrium and found to physically interact with RhoA in mouse atria and ventricles (65, 407). Overexpression of RhoA would therefore be predicted to suppress Kv1.2 current leading to prolongation of action potential thereby decreasing SA nodal rate. AV conduction abnormalities at a young age, bradycardia, and severe atrial arrhythmia at old age were also observed in mice overexpressing the endogenous Rho protein inhibitor GDI␣ specifically in the heart (522). As the overexpression of GDI␣ resulted in the simultaneous reduction of the activity of RhoA, Rac1, and Cdc42, it is not possible to link the phenotype observed to a single Rho proteins. Electrocardiography and intracardiac electrophysiological recordings revealed first-degree AV block in GDI␣ mice at 1 wk of age, which progressed into second-degree AV block at 4 wk of age (522). These conduction defects were associated with a dramatic decrease in the expression of connexin 40 (Cx40), a connexin specifically expressed in the atrium and 1672 the conduction system, the ablation of which resulted in AV and ventricular conduction abnormalities (225). GDI␣ mice displayed normal positive chronotropic response to -adrenergic receptor stimulation, and the AV conduction defects could be partially rescued by -adrenoceptor stimulation (522). This effect of RhoGDI␣/Rho protein signaling on AV conduction was thus probably due to a primary effect on the expression and/or activity of cardiac proteins, including Cx40, involved in the propagation of the electrical activity. In addition to in vivo studies in transgenic mouse models, several studies directly analyzed the effects of Rho proteins on ion channels involved in electrical cardiac activity. An example of these channels is the human ether-a-go-go related gene K⫹ channels (h-ERG encoded by KCNH2 gene), responsible for IKr and the reduction of which leads to prolonged cardiac repolarization resulting in the development of long QT syndrome. The constitutively active RhoA-L63 inhibits h-ERG channels, and it has been suggested that this effect was due to Rho-kinase either by directly affecting the channel or indirectly by modulation of protein phosphatase activity (467). RhoA is also defined as a negative regulator of the IK1 current. Constitutively active RhoA inhibited Kir2.1, and in contrast, dominant negative form of RhoA prevented muscarinic receptor-induced inhibition of Kir2.1 (203). This suggests that RhoA is involved in the physiological regulation of Kir2.1 by activation of muscarinic receptor and that abnormal overactivation could potentially result in ventricular arrhythmia. This inhibitory effect of RhoA on Kir2.1 could also participate in the loss of the positive chonotopic effect of muscarinic receptor blockade observed in mice overexpressing RhoA in the heart (407). B) RAC. Evidence for a role of Rac1 in the regulation of heart rhythm comes from both in vivo and in vitro studies. One of the strategies used in mice was to overexpress a constitutively active Rac1 (Rac1-V12) under the control of the ␣-MHC promoter (2). Cardiac Rac1-V12 mice, showing a 30-fold increase in cardiac Rac1 activity, spontaneously developed AF with age. Electrocardiogram indicated that at the age of 10 and 16 mo, 44 and 75% of Rac1-V12 mice displayed AF, respectively. Aging of Rac1-V12 mice was associated with increased size of the atrium, but not the ventricles. A fourfold increase in atrial nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity and a significant enhancement of atrial collagen content was observed in Rac1-V12 mice. Treatment with statin strongly reduced the cardiac Rac1 and NADPH activities of Rac1V12 mice, without affecting the atrial dilation and interstitial fibrosis. Interestingly, a fourfold upregulation of Rac1 and NADPH oxidase activities was observed in samples of the left atrial appendage of patients with AF (2). A more recent study has suggested that the role of Rac1/NADPH Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM signaling in AF involves upregulation of connective tissue growth factor (CTGF) and the resulting modulation of Ncadherin and Cx43 that were all upregulated in Rac1-V12 mice and in left atrial myocardium from patients with AF (3). This study thus suggests that Rac1 and NADPH oxidase contributes to signal transduction during AF. It can be speculated that statin-induced inhibition of Rac1 could contribute to the prevention of postoperative AF obtained by statin treatment (515). Results obtained in guinea pig SAN pacemaker cells expressing constitutively active Pak1 indirectly suggest a role of Rac1 and/or Cdc42 in Ca2⫹ and K⫹ channels. Endogenous Pak1 protein is abundant in SAN, atrial, and ventricular tissue (219). Expression of active Pak1 does not modify the amplitude of the L-type Ca2⫹ current in SAN cells but strongly reduces the stimulatory effect of -adrenergic stimulation (219). Similarly, in the resting condition, the outward K⫹ current was not changed in SAN cells expressing active Pak1, but the -adrenergic stimulation-induced increase in K⫹ current amplitude and acceleration of the K⫹ current decay was almost abolished. In whole heart preparations, overexpression of active Pak1 attenuated the positive chronotropic action of -adrenergic stimulation (219). As the PP2A inhibitor okadaic acid partially reversed the suppressing effect of active Pak1 on the response to -adrenergic stimulation on Ca2⫹ and K⫹ currents, the effect of Pak1 is likely to be due to PP2A (219). These observations thus identify Pak1 as a potential target for therapeutic strategies against tachycardia triggered by cathecholamines in different pathological conditions. Several studies performed in non-cardiac cells can help to define the molecular identity of channels that underlie the effect of Rac1 on heart rhythm. For example, active Rac1 mutant stimulates ERG channel activity, via a stimulatory action on the serine-threonine phosphatase PP5 (134, 467). Dominant negative Rac1 has been shown to strongly increase Kir2.1 current, while the constitutively active Rac1 had no significant effect (54). This effect resulted from Rac1-mediated reduction of basal internalization of the channels, leading to an increase in the number of Kir2.1 channels at the plasma membrane, without change of its global expression level (54). These data thus suggest that Rac1-mediated regulation of ERG or Kir2.1 channels can participate in the arrhythmogenic effect of Rac1 activation. D. Cardiac Hypertrophy/Remodeling and Heart Failure (Table 3) The primary response of the heart to increased work load is cardiomyocyte growth in an attempt to enhance contractility and preserve cardiac function (170). Cardiac hypertrophy is a physiological adaptative response to normal postnatal maturation or physical exercice that is characterized by the maintenance of a normal myocardial structure and function. However, cardiac hypertrophy is also a common response to pathological stressors such as hypertension, myocardial infarction-induced defects in the efficiency of the contraction, neurohormonal stimulation, or oxidative stress. Although it is an initial compensatory response to maintain cardiac output, it evolves into a maladaptive process and a decompensated state with profound changes in cardiac structure and function, predisposing to arrythmia and sudden death, and leading to the development of heart failure (121, 175). Pathological hypertrophy corresponds to an abnormal enlargement of the heart muscle originating from an increase in cell size of myocytes, interstitial fibrosis, and proliferation of nonmuscle cells. Cardiomyocyte hypertrophy is associated with the enhancement of myofibril organization and change in gene expression including increased expression of immediate early genes (c-jun, c-fos, egrI) and reexpression of fetal genes such as atrial natriuretic factor (ANF) and B-type natriuretic peptide (BNP) as well as genes involved in cardiac contractility [-myosin heavy chain (-MHC), ␣-skeletal actin]. Since heart failure is the leading cause of hospitalization and mortality, the signaling mechanisms underlying cardiac hypertrophy and, even more importantly, the processes responsible for the transition between compensated hypertrophy to decompensated heart failure remain the subject of extensive research. The following section focuses on the role of small G protein in cardiac hypertrophy and remodeling, in particular Ras and Rho subfamilies, shown to play a key role in the hypertrophic signaling pathways initiated by the activation of G protein-coupled receptor. 1. Ras proteins A) RAS. Transient upregulation of H-Ras has been observed in association with reexpression of c-myc in a variety of experimental models of cardiac hypertrophy in adult rats, suggesting that it may participate in hypertrophy of heart (198, 232). In agreement with this observation in animal models, H-Ras is also upregulated in patients with hypertrophic cardiomyopathy and cardiomyocyte size correlated with the expression level of H-Ras (205). Moreover, patients suffering from “the RAS/MAPK syndromes” caused by mutations of molecules in the RAS/MAPK cascade (Noonan, LEOPARD, Costello, and CFC syndromes and neurofibromatosis type I) exhibit hypertrophic cardiomyopathy (18). Both in vitro and animal studies support this role of H-Ras in cardiac hypertrophy (TABLE 3). In primary neonatal rat ventricular myocytes, microinjection of active H-Ras-V12 stimulates hypertrophic response, attested by an increase in cell surface and the induction of the expression of c-fos and ANF (483). Expression of a H-Ras-V12 mutant that selectively activates Raf or a constitutively active Raf mutant induces the same phenotype as H-Ras-V12, suggesting that Raf is the downstream effector involved in the hypertrophic Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1673 LOIRAND, SAUZEAU, AND PACAUD Table 3. Genetically modified mice and cardiac hypertrophy Target Type of Modification Ras Cardiac specific overexpression (␣-MHC promoter) Nf1 Myocardial specific deletion RASSF1A Deficiency (KO) Raf-1 Cardiac specific overexpression (␣-MHC promoter) Transgene/Mouse Active H-Ras mutant (H-Ras-V12) Dominant-negative Raf1 mutant (DN-Raf1) Cardiac specific deletion Rad Deficiency (KO) Rab1 Cardiac specific overexpression (␣-MHC promoter) Cardiac specific overexpression (␣-MHC promoter) RhoA Rock1 Haploinsuficiency (Rock1⫹/⫺) Deficiency (Rock1⫺/⫺) RhoA Active RhoA mutant (RhoA-L63) Phenotype Reference Nos. Cardiac muscle hypertrophy, with increased ventricular mass and myocardial cell size Myofibrillar disarray Fetal cardiac gene expression Abnormal left ventricular diastolic function Normal systolic functions remained normal Progressive cardiac hypertrophy, fibrosis, and cardiac myocytes enlargement Hyperactivation of Ras/Erk signaling in the heart Massive cardiac hypertrophy after transverse aortic constriction Cardiac fibrosis Left ventricular dilatation Fetal cardiac gene expression 2 of pressure overlaod-induced cardiac hypertrophy 2 of pressure overlaod-induced fetal cardiac gene expression Increased cardiomyocyte apoptosis Decreased ERK1/2 activity and cell growth Left ventricular dysfunction and dilatation Increased number of apoptotic cardiac cells Increased susceptibility to cardiac hypertrophy Increased CaM-kinase II activity Enhanced cardiac fibrosis and increased CTGF expression 142, 184, 402 Cardiac hypertrophy 542 Premature death No increase in ventricular mass 407 No increase in cardiomyocyte size Increased expression of ANF and -MHC Cardiac conduction defects No change in heart structure and function No change in ANG II- or L-NAMEinduced cardiac hypertrophy 2of ANG II-, L-NAME- or pressure overload-induced cardiac fibrosis 2of pressure-overload-induced cardiac fibrosis 2of pressure-overload-induced cardiomyocyte apoptosis 548 346 186 554 73 393 563, 582 Continued 1674 Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM Table 3.—Continued Target Type of Modification Deficiency (Rock1⫺/⫺) Cardiac specific overexpression (␣-MHC promoter) Rac1 Cardiac-specific overexpression (␣-MHC promoter) Cardiac-specific overexpression (␣-MHC promoter) Transgene/Mouse In mice overexpressing G␣q in cardiomyocytes Rock1: in mice overexpressing G␣q in cardiomyocytes Active Cter-truncated Rock1 Active Rac1 mutant (Rac1-V12) Cardiomyocyte-specific Rac1 deficiency (c-Rac1⫺/⫺) Tamoxifen-inducible Cremediated deletion of floxed Rac1 gene in cardiomyocytes Pak1 Cardiomyocyte-specific Pak1 deficiency (c-Pak1⫺/⫺) Cre-mediated deletion of floxed Pak1 gene in cardiomyocytes Cdc42 Cardiomyocyte-specific Cdc42 deficiency (c-Cdc42⫺/⫺) Cre-mediated deletion of floxed Cdc42 gene in cardiomyocytes effect of Ras (177). However, other evidence indicates that activation of Raf/MEK1/2/ERK1/2 alone is insufficient to initiate a complete hypertrophic response and that other Ras effectors are required. Indeed, it has been shown that in addition to Raf, the Ras effectors RalGDS, an exchange factor for RalA and RalB, and phosphatidylinositol (PI) 3-kinase participate in Ras-mediated hypertrophic response (128). RalGDS and Raf are mediators of specific hypertrophic transcriptional responses, whereas PI 3-kinase elicits a global effect on gene expression in cardiac myocytes (128). Other signaling molecules have been proposed to participate in Ras-mediated hypertrophy including MEKK/JNK pathway (383) and Ras-GAP (1). Ras has emerged as a central signaling protein required for cardiac hypertrophy induced by different external stimuli such as mechanical overload or neurohumoral factors in- Phenotype No effect on cardiac hypertrophy Improved survival Preserved left ventricular structure and contractile function Decreased cardiomyocyte apoptosis Accelerate hypertrophic decompensation Reference Nos. 443 442 Extensive cardiac fibrosis 563 Neonatal lethal dilated cardiomyopathy Transient hypertrophy in young mice (hypercontractile heart) 2of ANG II-induced cardiac hypertrophy 473 of pressure-overload- or ANG IIinduced cardiac hypertrophy of pressure-overload- or ANG IIinduced cardiac fibrosis of pressure-overload-induced cardiomyocyte apoptosis of pressure-overload-induced cardiac fetal gene expression vulnerability to transition to heart failure of pressure-overload- or ANG II, PhE-induced cardiac hypertrophy of exercice-induced cardiac hypertrophy vulnerability to transition to heart failure 279 415 282 cluding phenylephrine (PhE) or angiotensin II (ANG II) (86, 404, 483, 558). G␣q downstream of ␣1-adrenergic or type 1 ANG II (AT1) receptors is required for Ras activation associated with cardiomyocyte hypertrophic response to PhE and ANG II, in a protein kinase C-dependent or -independent manner (86, 171, 404). Furthermore, ␥-subunits of Gi protein-dependent activation of Ras has also been shown to be critical for ANG II-dependent cardiac fibroblast proliferation that also participates in cardiac hypertrophy in vivo (557). While Ras GEFs linking trimeric G protein to Ras activation following hypertrophic agonist stimulation of cardiomyocytes are unknown, ␣1-adrenergic receptormediated cardiac hypertrophy has been ascribed to reactive oxygen species-dependent Ras activation (512, 545). Stimulation of ␣1-adrenergic receptor causes the oxidative posttranslational modification of Ras leading to the decrease in free Ras thiols and, thereby, Ras activation (243). Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1675 LOIRAND, SAUZEAU, AND PACAUD In mice, cardiac specific expression of H-Ras-V12 leads to cardiac muscle hypertrophy, with increased ventricular mass and myocardial cell size, myofibrillar disarray, and reactivation of ANF gene expression (142, 184) (TABLE 3). Functionally, mice expressing cardiac H-Ras-V12 display abnormal left ventricular diastolic function, but basal and -adrenergic stimulated systolic functions remained normal (184). This hypertrophic action of H-Ras-V12 does not seem to secondarily result from the overactivation of Ras signaling during embryonic development because induction of H-Ras-V12 transgene expression in adult mice heart also leads to significant myocyte hypertrophy already observed 1 wk after induction as posterior wall thickness of the left ventricle, increased heart weight and single-cell capacitance, and fetal cardiac gene expression (402). Cardiac hypertrophy thus appears as an immediate downstream effect of Ras activation in adult hearts. This was confirmed by a recent study showing the reversibility of Ras-induced cardiac hypertrophy in a mouse model of temporary expression of H-Ras-V12 in myocardium (517). Myocardial specific deletion of the Ras GAP Nf1 in mice promotes progressive cardiac hypertrophy, fibrosis, and cardiac myocytes enlargement associated with an hyperactivation of Ras/Erk signaling in the heart, suggesting a role of Nf1 to limit Ras activity in cardiomyocytes of the adult heart (548) (TABLE 3). These findings are in agreement with observations made in Ras-association domain family protein isoform 1A (RASSF1A) knockout mice, another mice model with defective Ras signaling (346). RASSF1A is a potent tumor suppressor ubiquitously expressed that inhibits the Ras-Raf1-ERK1/2 pathway in cardiomyocytes and prevents PhE-induced hypertrophy (346). RASSF1A knockout mice, after transverse aortic constriction, exhibited massive cardiac hypertrophy that was almost double that observed in control mice, associated with cardiac fibrosis and increased expression of fetal genes and left ventricular dilatation (346). A role of Ras signaling in cardiac hypertrophy is further supported by the discovery that Ras GAP and the Ras protein Rheb would be potential targets of miR1, a miRNA involved in cardiac hypertrophy (423). Taken together, these data underlie the importance of endogenous molecular systems that maintain a low level of Ras activity under physiological conditions. Transgenic mice with cardiac specific expression of a dominant negative form of Raf (DN-Raf-1) has been generated to define the requirement of Raf-1, downstream to Ras in cardiac hypertrophy in vivo (TABLE 3). Under basal conditions, DN-Raf-1 mice have normal cardiac structure and function (155). However, cardiac hypertrophy and hypertrophic gene induction in response to transverse aortic constriction-induced pressure overload are reduced in DN-Raf mice, associated with an increased cardiomyocyte apoptosis and a decreased ERK1/2 activity and cell growth (155). A more pronounced phenotype was obtained by cardiac 1676 specific deletion of Raf-1 in mice that, in absence of external stress, leads to left ventricular dysfunction and dilatation, with an increased number of apoptotic cells (554). These data are thus consistent with an antiapoptotic effect of Ras/ Raf signaling, independent of ERK1/2 but related to the antagonistic action of Raf against the propapoptotic kinase ASK1 (554). These findings also indicate that the role of Ras/Raf signaling in cardiac hypertrophy is mainly due to its effect on cardiomyocyte hypertrophy and apoptosis resistance. Very recently, a proteomic analysis of heart samples from the transgenic mouse model of cardiac specific expression of H-Ras-V12 has suggested that activation of the Wnt/catenin canonical pathway occurs secondary to Ras activation in the course of pathogenic myocardial hypertrophic transformation and may also participate in the hypertrophic effect of Ras activation (518). addition to its inhibitory role on Ca2⫹ channels, Rad has been shown to play a protecting role in cardiac hypertrophy. Rad is abundantly expressed in human heart, and its expression is strongly decreased in left ventricular myocardium samples from patients with heart failure that displayed severe hypertrophic morphology (73). Similarly, Rad has been shown to be decreased in the model of PhE-induced cardiomyocyte hypertrophy (73). Silencing of Rad expression by siRNA in rat neonatal cardiomyocytes strongly increased protein synthesis and ANF expression, whereas overexpression of Rad significantly reduced PhE-induced protein synthesis and ANF expression, and decreased CaM kinase II activity (73). In vivo, Rad-deficient mice were more susceptible than normal mice to cardiac hypertrophy, with increased CaM kinase II activity (73) and more severe cardiac fibrosis related to an increase in CTGF expression (579). Rad is thus identified as an endogenous cardioprotective mediator that prevents cardiac hypertrophy through the inhibition of CaM kinase II activity and cardiac fibrosis through the inhibition of CTGF. B) RAD. In C) RAP. In addition to their role in the regulation of excitation-contraction coupling, Rap proteins could also participate in signaling cascades involved in cardiac hypertrophy. Rap1 has been shown to mediate the activation of B-Raf/ ERK1/2 induced by M1 muscarinic receptor activation, via the activation of the Rap1 GEF Ca2⫹ and diacylglycerolregulated GEF I (CalDAG-GEFI) (146). In neonatal ventricular myocytes, cAMP/PKA-dependent Rap1 activation is responsible for ERK1/2 activation and cell growth downstream of the PGE2 receptor EP4 (160). In the same cellular model, the role of Rap1 in hypertrophy has been ascribed to cAMP-mediated activation of the Rap1 GEF Epac1, leading to Rap1-dependent inhibition of ERK5 (106). Nevertheless, surprisingly, more recent in vivo and in vitro studies suggested that Epac1 hypertrophic effects, including those in- Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM duced by -adrenergic receptor stimulation, are independent of Rap1 but rather involve Ras and Rac, calcineurin, and CaM kinase II (305, 319). Both Rap1 and Epac1 knockout mice will be useful to decipher the role of Rap1 signaling in cardiac hypertrophy. D) RAB. Rab1 regulates protein transport, specifically from the endoplasmic reticulum to the Golgi apparatus. Overexpression of Rab1 induced hypertrophy of neonatal cardiomyocytes in culture (117). Transgenic expression of Rab1 in the myocardium promotes cardiac hypertrophy in mouse that has been ascribed to an abnormal expression and subcellular localization of proteins, such as PKC␣ (542). E) RAL. RalA and RalB are among the closest relatives of Ras, and Ral proteins have been shown to be recruited by Ras. Activated Ras binds to the Ras-binding domain of Ral GEFs such as Ral-GDS, Ral guanine nucleotide exchange factor-like factor (Rlf), RGL1, and RGL2, leading to its translocation to the membrane where it produces GDP/ GTP exchange on Ral (299). Since the identification of RalGDS as an effector of Ras involved in Ras-mediated ventricular myocytes hypertrophy (128), further studies supported the role of Ral signaling in cardiac hypertrophy. Overexpression of Ral-GDS, constitutively active Ral mutant, or Rlf stimulates promoter activity of early and embryonic genes in rat neonatal myocytes, synergically with activated Ras (218, 370). Dominant-negative Ral partially inhibited active Ras-induced promoter activation (218). Stimulation of ␣1-adrenergic receptor induced activation of Ral in neonatal ventricular myocyte (370), and Ral activity is increased in hypertrophied heart (218). 2. Rho proteins A) RHOA. RhoA has been implicated in the morphological changes and induction of gene expression associated with hypertrophic response in cardiac myocytes. In vitro, stimulation of cultured neonatal ventricular myocytes with hypertrophic factors such as PhE, endothelin-1, or ANG II induced activation of RhoA (16, 84). Transfection or infection of neonatal ventricular myocytes with active RhoA mutant (RhoA-V14) stimulates ANF expression and myofibrillogenesis (16, 181, 406, 484). Conversely, expression of the dominant negative RhoA-N19, Rho-GDI, or the selective RhoA inhibitor Clostridium botulitum C3 exoenzyme inhibits hypertrophic responses induced by soluble stimuli such as PhE, ANG II, or lysophosphatidic acid (10, 16, 168, 406) or by mechanical stress (12). Mechanisms linking extracellular stimuli to RhoA activation in neonatal myocytes are still not firmly established. Trimeric G proteins play a key role in the activation of RhoA by agonists of G protein-coupled receptor, probably through activation of Rho GEFs that remain to be identified. G␣q was initially found to mediate RhoA activation following PhE-induced ␣1-adrenergic receptor stimulation (171, 406), but G␣12/13 was also shown to be responsible for this activation (296). ANG II-induced RhoA activation depends on G␣12/13 signaling (337), whereas activation of RhoA by lysophosphatidic acid involves G␣i (168). In response to mechanical stretch, activation of RhoA depends on PKC␣ (356). Rho-kinases have been identified as the RhoA effector mediating RhoA-dependent hypertrophic responses both in vitro and in vivo. In neonatal ventricular myocytes, pharmacological inhibition of Rho-kinase (fasudil or Y27632) or transfection of dominant negative forms of Rho-kinase prevents morphological and cytoskeleton changes, protein synthesis, and ANF expression associated with response induced by hypertrophic factors such as PhE, endothelin-1, or leptin (181, 245, 576). In neonatal ventricular myocytes, Ras activation by hypertrophic factors is also essential to their hypertrophic effects (see above), suggesting that Ras/ MAPK and RhoA induce a hypertrophic response by separate, complementary, and synergistic pathways (58, 292). In fact, RhoA/Rho-kinase-mediated actin cytoskeleton remodeling has been shown to be necessary for the translocation of ERK to the nucleus, providing a mechanism for the synergy between RhoA and Ras signaling pathways regulating gene transcription and inducing hypertrophy. The precise downstream mechanisms by which RhoA/Rho-kinase activate the hypertrophic gene program remain obscure, however. Activation of the cardiac transcription factor GATA-4 is identified as downstream nuclear mediators of Rho-kinase during myocardial cell hypertrophy (560). Recently, myocardin-related transcription factor A (MRTF-A), and the subsequent transcriptional activation of serum response factor (SRF)-dependent fetal cardiac genes, have been defined as critical downstream mediators of RhoA/Rho-kinase-dependent hypertrophic response in cardiac myocytes (244). Another RhoA effector, protein kinase N (PKN), has also been involved in the SRF-dependent transcriptional responses associated with cardiomyocytes hypertrophy (321). In vivo, pressure overload induces a rapid activation of Rho-kinases in the adult rat myocardium, suggesting that it could participate in mechanisms of initial as well as chronic adaptative responses of cardiac myocytes to mechanical stress (488). While both Rock1 and Rock2 are expressed in the myocardium, the increased Rho-kinase activity induced by pressure overload has been ascribed to Rock1 (582). In Dahl salt-sensitive hypertensive rats, the ventricular hypertrophy and function are significantly ameliorated by Rhokinase inhibition possibly through an upregulation of the downregulated endothelial nitric oxide synthase (eNOS) and the reduction of oxidative stress through the inhibition of NAD(P)H oxidase and LOX-1 expression (228, 314). Chronic Rho-kinase inhibition also markedly limits pathological cardiac hypertrophy induced by pressure overload in rats and improves diastolic function in rats (23, 364), but has no effect on chronic swimming-induced physiological Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1677 LOIRAND, SAUZEAU, AND PACAUD cardiac hypertrophy (23). Several neurohormonal factors, such as ANG II, are suspected to play a role in ventricular hypertrophy and the transition to heart failure. Long-term treatment with the Rho-kinase inhibitor fasudil reduces the ANG II-induced cardiomyocyte hypertrophy in mice (166, 514). Rho-kinase activity is also involved in the pathogenesis of left ventricular remodeling after myocardial infarction (158). All these findings support the involvement of RhoA/Rho-kinase signaling in the development of cardiac hypertrophy in vivo. Genetically modified mouse models have then been useful to further refine the role of RhoA and Rho-kinases in cardiac remodeling and hypertrophy (TABLE 3). Surprisingly, although ventricular expression of some hypertrophic marker genes such as ANF and -MHC are increased in mice that overexpressed RhoA in a cardiac specific manner, they did not develop an obvious cardiac hypertrophy (407). In contrast, they display progressive left ventricular chamber dilation and decreased contractility. The role of RhoA during cardiac development and the potential of compensatory mechanisms could explain why in vivo overexpression of RhoA did not mimic its in vitro hypertrophic effect. Haploinsufficiency of Rock1 in mice does not alter cardiac structure and function under basal conditions (393). All indexes of cardiac hypertrophy, i.e., wall thickness, left ventricular mass, cardiomyocyte size, or expression of ANF, induced by ANG II or L-NAME were similar in control and Rock1⫹/⫺ mice (393). However, a marked reduction of cardiac fibrosis is observed in Rock1⫹/⫺ mice chronically treated with ANG II or L-NAME, or after transaortic constriction, compared with control mice (393). This decreased fibrosis is associated with a reduced expression of markers of fibrosis including transforming growth factor- (TGF), CTGF, and type III collagen. Similar reduction of fibrosis has been observed in Rock1 knockout mice (582), together with a decreased cardiomyocyte apoptosis following pressure overload by aortic banding (563). These results thus indicate that in vivo, Rock1 contributes to the development of cardiac fibrosis but not hypertrophy in response to various pathological conditions. This was recently confirmed in a transgenic mice expressing a truncated activate form of Rock1 in cardiomyocytes (563) These mice displayed extensive cardiac fibrosis due to upregulation of TGF- and cytokine release in cardiomyocytes that promoted myofibroblast differentiation (563). In a mouse model with cardiac specific overexpression of G␣q, that develops compensated cardiac hypertrophy at young ages and progressing to heart failure, transgenic overexpression of Rock1 increases cardiomyocyte apopotosis and accelerates hypertrophic decompensation (443). In contrast, although it does not prevent the development of cardiac hypertrophy, deletion of Rock1 improves survival, inhibits cardiomyocytes apoptosis, and preserves left ventricular structure and function in old G␣q mice (442, 443). 1678 These data thus support the absence of a role of Rho-kinase in pathological hypertrophy but provide evidence for a crucial role of Rock1 in the transition from cardiac hypertrophy to heart failure. The difference in the cardiac phenotype resulting from genetic modifications of RhoA or Rock1 in mice could be related to the relative independence of Rock1 activation from RhoA activity. Rock1 can be constitutively activated by caspase-3-mediated proteolytic cleavage of its inhibitory COOH terminus (432). Accumulation of this COOH terminus-truncated form of Rock1 has been described in failing human heart and genetic murine models of heart failure (72). This active truncated Rock1 stimulates caspase-3 and phosphatase and tensin homolog deleted on chromosome ten (PTEN) activities. The resulting repression of the PI 3-kinase/Akt cell survival pathway thus sets on a positive feed-forward regulatory loop that favors cardiomyocyte apoptosis (72). Because cardiomyocyte apoptosis is a major cellular event involved in the transition from compensated hypertrophy to dilated cardiomyopathy and failure, this mechanism can support the observed important role of Rock1 in hypertrophic decompensation (443). Finally, the inhibitory effect of pharmacological inhibition of Rho-kinases on cardiac hypertrophy is not mimicked by Rock1 deletion. That might suggest that Rock2 could be involved in cardiac hypertrophy or the antihypertrophic effect of Rho-kinase inhibitors did not result from Rhokinase inhibition. Cardiac specific deletion of Rock2 in mice would be useful to address this question. B) RAC1. As for other small G proteins, first evidence for a role of Rac1 in cardiac hypertrophy was provided by in vitro analysis in neonatal ventricle myocytes. Expression of active V12-Rac1 results in sarcomeric reorganization, increase in cell size, and atrial natriuretic peptide (ANP) secretion (372). In contrast, the dominant negative N17-Rac1 expression inhibits the morphological changes and the increased protein synthesis induced by PhE (372). In agreement with this observation, hypertrophic factors including PhE, ANG II, and endothelin-1 have been shown to induce rapid Rac1 activation in cardiac myocytes, and N17-Rac1 inhibits the generation of reactive oxygen species (ROS) and the stimulation of ERK cascade, but not the activation of p38MAPK induced by these stimuli (84, 93, 172). Mechanical stretch also induces Rac1 activation in cardiomyocytes via a signaling involving PKC␦ (84, 356). In contrast to that observed after stimulation with soluble hypertrophic factors, expression of dominant negative form of Rac1 prevents stretch-induced p38MAPK activation, increased protein synthesis, and intracellular ROS generation (13). Stretch-induced p38MAPK kinase activation is also inhibited by treatment with the antioxidant N-acetyl-L-cysteine (NAC), indicating that stretch-induced Rac1-dependent activation of p38MAPK was mediated by ROS production Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM (13). Indeed, the stimulation of the superoxide generating NADPH oxidase by Rac1 and the subsequent generation ROS generation are important mediators of the cellular responses downstream of Rac1. The use of Rac1-V12 and Rac1-N17 mutant expression in cardiomyocytes has demonstrated that, in addition to p38MAPK, Rac1-mediated ROS is responsible for the activation of the transcription factor NFB. It was further shown that this activation involved ROS-mediated activation of apoptosis signal-regulating kinase 1 (ASK1) and that this pathway is essential for Rac1-induced hypertrophic effects (167). This Rac1-ROS signaling pathway has been also shown to be upstream to JNK activation and cardiomyocyte death, in particular in the context of -adrenoceptor-stimulated apoptosis (192). In vivo, transaortic constriction-induced cardiac hypertrophy in mice is associated with activation of Rac1 NADPH oxidase in left ventricular myocardium (93). To further address the role of Rac1 activation in the heart, transgenic mice that express constitutively activated Rac1 specifically in the myocardium have been developed (473). These mice display two different cardiac phenotypes: a neonatal lethal dilated cardiomyopathy and a transient hypertrophy in young mice that resolved with age. Hypertrophic hearts do not show signs of myofibril disarray and are hypercontractile in working heart analyses (473). These results thus unexpectedly suggest that both phenotypes result from deregulation of signaling by Rac1 and Pak, followed by alteration of cellular adhesion. The use of mice harboring cardiomyocyte-specific, tamoxifen-inducible Cre recombinase circumvented the problem of the embryonic lethality of Rac1 deletion to generate cardiomyocyte-specific Rac1 knockout (c-Rac1⫺/⫺) mice (415). Cardiac specific deletion of Rac1 prevents ANG II infusion-induced myocardial oxidative stress, NADPH activation, and cardiac hypertrophy. In agreement with in vitro studies, this mouse model supports the notion that Rac1-mediated NADPH oxidase activity and ROS production, but not some other effects of Rac1, are the primary regulators of cardiac hypertrophy in response to ANG II (415). This idea has been recently supported by data obtained in cardiomyocyte-specific Pak1 knockout mice (c-Pak1⫺/⫺) (279). These mice develop enhanced cardiac hypertrophy and fibrosis in response to pressure overload or ANG II infusion, compared with control, and they are more vulnerable to the transition to heart failure. This phenotype is associated with an increase in cardiomyocyte apoptosis and fetal gene expression. In c-Pak1⫺/⫺ mice myocardium, pressure overload does not induce the activation of MKK4/MKK7-JNK pathway, whereas activation of p38MAPK and ERK1/2 are normal. In ANG II-treated mice, ROS production is comparable in control and c-Pak1⫺/⫺ mice (279). These mice thus allow to uncover the cadioprotective role of Pak1/JNK pathway in attenuating cardiac hypertrophy and halting the transition to heart failure. In addition, the opposed phenotype of c-Rac1⫺/⫺ and c-Pak1⫺/⫺ mice provides further evidence that Pak1 is not a major effector of Rac1 in cardiomyocytes and supports an important role of ROS in the hypertrophic effect of Rac1. This also suggests that upstream activators of Pak1 other that Rac1 regulate Pak1 activity in cardiomyocytes, with Cdc42 as a good candidate. All these in vivo data, together with the threefold increase of Rac1 activity and the upregulation of NADPH oxidasemediated ROS release observed in myocardium of patients with ischemic or dilated cardiomyopathy, suggest that Rac1/ROS signaling contributes to the pathophysiology (286). C) CDC42. Very little is known about the function of Cdc42 in cardiomyocytes. In fact, in vitro, Cdc42 was found to have hypertrophic effects in neonatal cardiomyocytes that appear morphologically distinct from those exerted by Rac1 or RhoA (328). Expression of dominant active mutant of Cdc42 promoted myofibril assembly in series and increased the length/width ratio of cardiomyocytes while RhoA and Rac1 induced parallel assembly of sarcomeric units and did not affect the length/width ratio (328). Dominant active mutant of Cdc42 expression also increased p38MAPK activity in neonatal cardiomyocytes (13). An increase in Cdc42 activity is rapidly observed (10 min) in cardiomyocytes stimulated by hypertrophic stimuli such as leukocyte inhibitory factor (LIF), ANG II, PhE, or isoproterenol (288), and expression of a dominant negative form of Cdc42 inhibits LIF-induced sarcomere organization in series and length-to-width ratio increase (328). Furthermore, silencing of Cdc42 by shRNA also reduces MEKK1/ MKK4/7 and JNK activation induced by LIF or isoproterenol in neonatal cardiomyocytes (288). The observation that dominant negative Cdc42 suppressed stretch-induced p38MAPK activation indirectly suggests that mechanical stretch also activates Cdc42 (13). These data thus support pro-hypertrophic effect of Cdc42 that may involve MEKK1/MKK4/7/JNK and p38MAPK pathways. However, this pro-hypertrophic effect of Cdc42 observed in vitro has not been confirmed by in vivo studies. In fact, mice with cardiac-specific deletion of Cdc42 developed exacerbated pressure overload hypertrophy, greater cardiac hypertrophy in response to neuroendocrine agonist infusion (ANG II and PhE), and enhanced exercise-induced hypertrophy and sudden death (288). They transitioned more quickly from hypertrophy to heart failure than control mice. This phenotype is associated with the suppression of MEKK1/MKK4/7/JNK pathway activity, resulting in the upregulation of calcineurin-nuclear factor of activated T cells (NFAT) signaling (288). The causal role of the suppression of the JNK pathway in the cardiac phenotype of c-Cdc42⫺/⫺ mice has been demonstrated by the reversion of the enhanced cardiac growth when they were crossed with MKK7 transgenic mice (288). These results thus identify Cdc42 as a antihypertrophic and protective mediator during stress stimulation. Interestingly, the phenotype of Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1679 LOIRAND, SAUZEAU, AND PACAUD c-Cdc42⫺/⫺ mice is very similar to that of c-Pak1⫺/⫺ mice, thereby supporting the idea that Cdc42 is the endogenous activator of Pak1 and that the Cdc42/Pak1/JNK signaling pathway is cardioprotective, in limiting hypertrophy and transition to heart failure. Other clues speaking in favor of a role of Cdc42 in cardiac hypertrophy were recently supplied by studies of microRNAs. Cdc42 was identified as a target of, and negatively regulated by miR-1 and miR-133 (that also targets RhoA), and a decreased expression of both miR-133 and miR-1 was observed in mouse and human models of cardiac hypertrophy (66, 376). E. Ischemia/Reperfusion Damage, Preconditioning Myocardial ischemia, due to a reduced blood supply, induces severe tissue injury in the heart, and large effort has been made to understand the mechanisms causing cellular damage during ischemia. It is now recognized that upon reperfusion, exposure of ischemic myocardium to molecular oxygen initiates a cascade of events that paradoxically augments myocardial cell dysfunction and death, with ROS as essential mediators of ischemia/reperfusion injury. Nevertheless, the heart and cardiac myocytes have developed powerful endogenous mechanisms to protect themselves against ischemia/reperfusion injury which can be activated by brief period of ischemia and reperfusion preceding a more severe one through a phenomenon known as ischemic preconditioning. Signaling mechanisms mediating both ischemia/reperfusion injury and cardioprotection are the subject of intense investigation as they may represent molecular targets that could be used to mimic ischemic preconditionning by pharmacological interventions. Members of the Ras superfamilly of small G proteins represent potential good candidates for such an objective. However, despite intensive research in this field, data are still inconsistent. Furthermore, studies are essentially performed in rat or mice models of cardiac ischemia/reperfusion while the temporal and spatial development of myocardial ischemia/reperfusion injury in these animal species is different from that seen in humans (165). Therefore, although small rodent heart models are useful to decipher molecular mechanisms and identify potentially interesting targets, the confirmation of such novel mechanisms in larger animal (such as pig or dog) models of myocardial ischemia/reperfusion that more closely resembles the human situation (124) appears mandatory before translation to the clinic. 1. Ras proteins (Figure 3) A) RAS. Oxidative stress activates Ras in the cardiomyocytes and in the heart (85, 469). Numerous in vitro and in vivo studies clearly identified the Ras/Raf/MEK1/2/ERK1/2 pathway as a prosurvival pathway that protects cardiomyocytes from apoptotic death following oxidative stress, 1680 IR/ROS Ras ↑ Raf-1 Rad ↑ Rassf1A ↑ MEK ↓ ASK1, Mst2 ↑ Mst1 ↑ ERK1/2 ↓ JNK/p38 ↑ JNK/p38 ↓ apoptosis cardioprotection ↑apoptosis FIGURE 3. Involvement of Ras proteins in ischemia/reperfusioninduced myocardium injury. Ras and Rab signaling activated by ishemia/reperfusion (IR) or reactive oxygen species (ROS) leading to decrease myocardial cell survival and increase apoptosis pathways. thus identifying ERK1/2 as major components of the reperfusion injury salvage kinase (RISK) pathway (11, 325, 399, 573). Results from pharmacological studies confirmed these data by showing that activation of ERK1/2 signaling significantly contributes to the cardioprotective effects of -adrenergic receptor antagonists that are largely used in the clinical management of human ischemic heart diseases (237). Genetic manipulations of MEK1/2, ERK1/2, and Raf-1 in mice nicely demonstrated the anti-apoptotic action of the Raf/MEK1/2/ERK1/2 signaling leading to cardioprotection during reperfusion (155, 269). However, cardiac specific disruption of the c-raf-1 gene indicated that the beneficial effect of Raf-1 on cardiomyocytes survival is not mediated by the MEK1/2-ERK1/2 pathway but involves repression of ASK1, Mst2, and JNK-p38MAPK activity (76, 325, 554). More recent data also suggest that Ras, through the activation of its other downstream effector Rassf1A, has a proapoptotic effect through the kinase Mst1 already known to be activated by ischemia/reoxygenation in the heart (347, 556). Rassf1A has been identified as an endogenous activator of Mst1 in the heart, and the Rassf1A/Mst1 pathway promoted apoptosis in cardiomyocytes (98). It therefore appears that although Ras signaling exerts potent cardioprotective effects through Raf-dependent and -independent mechanisms, stimulation of Raf-independent apototic signaling also occurred downstream of Ras. Accordingly, the dynamic balance of this dual signaling would be critical in determining cardiomyocyte fate subsequent to reperfusion injury. The observation that inhibition of Ras proteins by FTIs limits ischemia/reperfusion-induced injury even suggests that the Raf-independent pro-apoptotic Ras effects, probably through Mst1 and JNK, predominates Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM over its Raf-ERK1/2-mediated cardioprotective action (35, 358). B) RAD. Although its physiological or pathophysiological role has not been yet assessed, it has been recently shown in vitro in neonatal rat ventricular myocytes that Rad induces cardiomyocyte apoptosis through p38MAPK activation (472). 2. Rho proteins (Figure 4) A) RHOA. RhoA has been initially described as both a mediator of survival and a trigger of cell death. High levels of expression of activated RhoA in cardiomyocytes initially elicit a hypertrophic response, but sustained expression leads to the development of apoptosis, via a mechanism involving Rho-kinase, p53-dependent upregulation of the pro-apoptotic protein Bax and the consequent induction of the mitochondrial death pathway (100). A special role has been ascribed to Rock1 as this isoform, cleaved by caspase 3, becomes irreversibly active and leads to a positive feedback on caspase 3 activity, activation of PTEN, and inhibition of Akt in cardiomyocytes (72). This truncated form is absent in normal heart but was found in human failing heart (72). In contrast to this pro-apoptotic effect of RhoA/ Rho-kinase signaling, moderate expression of activated RhoA, at levels close to physiological levels of RhoA activation, protects cardiomyocytes from ROS-induced apo- IR Rac-1 ROS RhoA ↑ Rock1 ↑ FAK ↑ PDK ↑ PKN ↑ Caspase 3 ↑ PTEN ↓ Akt ↑ apoptosis ↓ survival ↑ PI3-K ↑ Akt ↓ apoptosis ↑ survival FIGURE 4. Involvement of Rho proteins in ischemia/reperfusioninduced myocardium injury. RhoA and Rac signaling activated by ishemia/reperfusion (IR) or reactive oxygen species (ROS) leading to decrease myocardial cell survival and increase apoptosis pathways. ptosis through FAK phosphorylation, association of FAK with PI 3-kinase, and finally, Akt activation (99). Confirming these findings, inhibition of endogenous RhoA by C3 exoenzyme has been shown to decrease Akt activity and stimulate cardiomyocyte apoptosis (238). Thereafter, several studies have been designed to investigate in vivo whether RhoA signaling is beneficial or detrimental to cardiomyocyte survival during ischemia/reperfusion. The first in vivo data reported were based on the effects of the pharmacological inhibition of the RhoA effector Rhokinase on heart damages caused by ischemia/reperfusion in rats or mice. RhoA/Rho-kinase activity is not modified by ischemia, but an early and sustained activation is observed in the ischemic myocardium during reperfusion (25, 150). Rho-kinase inhibition with Y27632 or fasudil reduces ischemia/reperfusion-induced cardiomyocytes apoptosis and improves postischemic cardiac function through the activation of PI 3-kinase/Akt/NO signaling (25, 150, 538) and the inhibition of JNK (580). All these pharmacological data thus support a deleterious effect of Rho-kinase activity in myocardial ischemia/reperfusion injury. However, recent results obtained in mice with cardiomyocyte-specific conditional expression of low levels of activated RhoA (RhoAL63) unexpectedly reveal that RhoA protects the heart against ischemia/reperfusion injury (544). Both in perfused heart experiments and in vivo, expression of active RhoA strongly reduced ischemia/reperfusion-induced myocardial cell damage, evidenced by a reduction of the infarct size and lactate dehydrogenase and cytochrome c release, thus showing that active RhoA in cardiomyocytes is protective against ischemia/reperfusion injury (544). Conversely, in mice with cardiomyocyte specific deletion of RhoA, infarct size and lactate dehydrogenase release following ischemia/ reperfusion are significantly increased compared with control mice, thus confirming the beneficial effect of RhoA in cardiomyocytes (544). Downstream of RhoA, all the potential effectors involved such as Akt, FAK, ERK1/2, or PKN are not modified in hearts expressing active RhoA compared with control hearts, while PKD activity is markedly increased (544). A causal role of the PKD activation in the cardioprotective effect of RhoA is supported in vitro by the similar potentiation of H2O2-induced cardiomyocyte death by RhoA and PKD inhibition, and in vivo by the complete reversion of the cardioprotection conferred by active RhoA expression by PKD blockade (544). It is of interest to mention recent data regarding the other RhoA effector PKN. Hypotonic swelling of cardiomyocytes, a condition found in pathological contexts such as ischemia-reperfusion, leads to RhoA and PKN activation that mediates cardiac myocyte survival (206). In vivo, cardiomyocyte specific expression and deletion of the RhoA effector PKN in mice demonstrate that activation of PKN plays a cell-protective role in cardiac myocytes during ischemia/reperfusion (475). Although a direct link with RhoA was not made, this study is however Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1681 LOIRAND, SAUZEAU, AND PACAUD in agreement with a cardioprotective role of RhoA against ischemia/reperfusion injury in vivo. This discrepancy between pharmacological studies and genetic manipulation of RhoA could result from multiple reasons, including 1) the targeting of Rho-kinase in pharmacological studies and RhoA in mice models, 2) the specific manipulation of RhoA signaling in cardiomyocytes in genetically modified mice while pharmacological inhibition affects every cell types with potential effects on neurohumoral feedback loops, and 3) the possible lack of selectivity of the pharmacological inhibitors. Similarly, inconsistent results have been reported regarding the role of RhoA/Rho-kinase in preconditioning. Ischemic preconditioning reduces the activity of Rho-kinase in the myocardium, while, in contrast, the ERK1/2 signaling is increased (578). This downregulation of the RhoA/Rhokinase signaling as well as the decrease in cardiomyocyte apoptosis induced by ischemic preconditioning is lost after ERK1/2 inhibition (578). These data suggest a relationship between ERK1/2 and RhoA/Rho-kinase, such that ERK1/2 activity is required in ischemic preconditioning to oppose the effect of RhoA/Rho-kinase signaling on apoptosis. This is in agreement with the observation that inhibition of Rhokinase by Y27632 or high dose of fasudil mimicks the effect of ischemic preconditioning in rats (101, 102). However, in opposition to these results, it has also been suggested that RhoA/Rho-kinase activity participates in the cardioprotective effect of ischemic preconditioning since inhibition of Rho-kinase by Y27632 in perfused hearts or by low doses of fasudil in vivo in rats reduces the protective effects of ischemic preconditioning (101, 409). This cardioprotective role of RhoA is also supported by its involvement in the preconditioning effect of adenosine, coupling A3 adenosine receptor to phospholipase D activation (259, 322). In short, these data highlight the difficulties to definitively define whether RhoA signaling is deleterious or protective during ischemia/reperfusion and preconditioning, and much remains to be done before considering RhoA/Rhokinase inhibition as a potential therapy of ischemia/reperfusion injury. B) RAC. As a component of the NADPH complex, Rac1 has been suggested to play a dominant role in ROS generation after ischemia/reperfusion. Nevertheless, a limited number of studies have addressed the role of Rac in cardiac ischemia/reperfusion injury, and there are no data on cardiac preconditioning. Expression of the dominant negative Rac1-N17 mutant results in protection of cardiomyocytes from hypoxia/reoxygenation-induced cell death (224). In vivo, ischemia/reperfusion increases Rac1 in the heart and ROS production, and ischemia/reperfusion-induced ROS production and apoptosis are strongly decreased in hearts from mice with cardiac specific deletion of Rac1, compared 1682 with controls, resulting in a limitation of the infarct size and preservation of myocardial function (439). Pharmacological inhibition of Rac1 by NSC23766 gives the same results (439), thus providing evidence that Rac1 is involved in myocardial damage produced by ischemia/reperfusion and that strategy aiming at decreasing Rac1 activity in these circumstances could represent new potential therapy. IV. RAS PROTEINS IN VASCULAR FUNCTION AND DISEASES A. Embryonic Vascular Development (Vasculogenesis and Angiogenesis) Vascular development in vertebrates is a complex phenomenon, initiated in the embryo at different places and times, and underlaid by two processes, vasculogenesis and angiogenesis. The first blood vessels of the mouse begin to form in the yolk sac at E6 – 6.5. Later in development, vasculogenesis is initiated with the embryo proper, with blood vessels appearing in the following order: endocardium, primary vascular network lateral to the midline, paired dorsal aortae, head, and cardinal vessels. Vasculogenesis corresponds to de novo formation of vessels from endothelial cell precursors or angioblasts that differentiate into endothelial cells and form lumenized tubes organized in primitive continuous vascular network (after E8.5 in mouse embryo). Angiogenesis is then responsible for the formation of secondary vessels by endothelial cells sprouting from the primary vessels. Endothelial cell proliferation, migration, and differentiation, essential to form mature vasculature are guided by extracellular cues that include soluble growth factors, extracellular matrix, and cell-cell interactions (249, 425). When the endothelial cells are assembled into vascular tubes, they become surrounded by mural cells of the smooth muscle cell lineage, referred to as pericytes (in capillaries, small venules, and immature blood vessels) and vascular smooth muscle cells (in mature and large blood vessels). Pericytes and smooth muscle cells differentiate from the mesenchyme and migrate around the growing blood vessels. Signaling molecules such as small G proteins that regulate differentiation, proliferation, and migration have been shown to play important role in vasculogenesis and angiogenesis. 1. Ras proteins A) RAS. Mouse embryos lacking p120-Ras GAP exhibited major defects in their circulatory system organization. The yolk sac of p120-RasGAP deficient embryos was abnormal, with delayed organization of endothelial cells that aggregated into a honeycombed pattern but subsequently failed to organize into a vascular network (164). Within p120RasGAP deficient embryos, the dorsal aorta was thinned with aberrant ventral branches that resemble intersegmen- Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM tal arteries. In late stage embryo (E9.5), the embryonic vascular exhibited local ruptures causing leakage of blood into the body cavity. This phenotype is likely to be due to abnormal endothelial cell migration and organization, related to inappropriate regulation of Ras activity by upstream signaling that controlled Ras activity through p120-RasGAP. The important role of p120-RasGAP in the regulation of Ras activity during vasculogenesis/angiogenesis has been further supported by the discovery of mutations in RASA1, the gene encoding p120-RasGAP in human (108). These mutations, leading to the loss of the GAP activity towards Ras proteins, cause capillary and arteriovenous malformations (CVM-AVM). This indicates that p120-RasGAP plays specific roles in the regulation of Ras signaling during vessel development that cannot be compensated for by other RasGAPs such as RAS2, RASAL, and NF1. R-Ras GAP (also called GAP1IP4BP), which is membrane associated and displays a stronger GAP activity against RRas than H-Ras, has also been described as an important regulator of embryonic vascular development, in particular in blood barrier establishment (197). Homozygous mice expressing a R-Ras GAP mutant lacking its catalytic activity die at E12.5–13.5 of massive subcutaneous and intraparencymal bleeding, probably due to underdeveloped adherens junctions between capillary endothelial cells, suggesting that Ras proteins are involved in the implementation of cell-cell junctions during vascular development. Mechanisms and/or exchange factors upstream to Ras that ensure the control of Ras activity during the vascular development have been little addressed. Nevertheless, deletion of the H-Ras and K-Ras exchange factor Sos1 induces embryo death at mid-gestation with cardiovascular defects (508). In addition to an enlarged heart, the major vessels of Sos1deficient embryos are distended, with areas of extensive hemorrhage. However, whether these changes are a secondary consequence of an earlier developmental failure or a direct result of loss of Sos1 protein has not been determined. Effect of Ras during vascular development may involve at least in part its effector Braf identified as a critical signaling factor in the formation of the vascular system (537). Brafdeficient embryos die of vascular defects during mid-gestation. They had enlarged and irregularly shaped large blood vessels that were incompletely lined with endothelial cells. Apoptotic cells were particularly abundant in the endothelium of Braf-deficient embryos. Braf thus plays a critical and indispensable role downstream of Ras in the integration of mechanisms that regulate the differentiation of angioblasts, development and integrity of large vessels, and the survival of cells of the endothelial lineage (537). In fact, inactivation of Erk1 and Erk2 in endothelial cells during embryonic development results in embryonic lethality due to strongly altered angiogenesis with a reduced vas- cular complexity of the vascular network, with only large unbranched vessels (458). This phenotype has been ascribed to a redundant role of ERK1 and ERK2 in the regulation of endothelial cell proliferation and migration, two processes that are critical for angiogenesis. The transcription factors Ets1 and Ets2, defined as targets of ERK signaling could mediate, at least in part, the effect of Ras/Raf/ ERK1/2 signaling during angiogenesis (519). However, the rescue of the abnormal blood vessel organization in K-ras knock-down by Akt provided evidence that PI 3-kinase/Akt also plays an important role in mediating Ras signaling during embryonic vascular development (275). Downstream to Ras, the Ras interacting protein 1 (Rasip1/ Rain), identified as an endomembrane Ras effector, has been shown to be exclusively expressed in the endothelium of the developing vasculature in both frog and mice and involved in vasculogenesis (550). Although angioblasts emerged relatively normally, blood vessel development was severely inhibited in the Rasip1 knock-down frog embryo, due to a defect in branching. This suggests a role of Ras/ Rasip1 signaling in angioblasts and endothelial cells after their initial angioblast specification, possibly during their migration, cord formation, or tubulogenesis. However, as rescue experiments demonstrate that failure of tubulogenesis in the absence Rasip1, can be fully restored by dominant-negative RhoA or siRhoA treatment, it appears that Rasip1 suppresses RhoA (known to inhibit lumen formation, see below) and promotes activation of Cdc42 and Rac1 (both known to positively mediate lumen formation, see below) probably secondary to the downregulation of RhoA activity (550). B) RAP1. Total deletion of either Rap1a or Rap1b in mice leads to an inhibition of angiogenesis in vivo that has been ascribed to an endothelial cell defect (80, 81, 559). This defective angiogenesis in Rap1b-knockout mice resulted from an altered VEGF response as Rap1 has been shown to be required for VEGF-mediated VEGFR2 activation, in part via an integrin ␣v3-dependent mechanism (248). A role in the stabilization of endothelial cell-cell junctions is also ascribed to Rap1 and its specific effector KRIT1 (274). Mouse homozygous null mutations of the Krit1 gene are embryonic lethal due to vascular defects including dilation of precursor vessels in the brain, enlargement and increased endothelial proliferation of the caudal dorsal aorta, as well as variable narrowing of the branchial arch arteries and proximal dorsal aorta (528). In humans, loss-of-function mutations of this gene causes autosomal dominant familial cerebral cavernous malformations (246). Mechanistically, it has been demonstrated that Rap1/KRIT1 interaction leads to the translocation of microtubule-sequestrated KRIT1 to cell-cell junctions, thereby supporting junctional integrity of endothelial cells and vascular development (139, 274). Interestingly, a recent study points to a role of Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1683 LOIRAND, SAUZEAU, AND PACAUD the Rap-activated GAP ARAP3 in developmental angiogenesis (131). ARAP3 is a GAP for both Arf6 and RhoA, and its deletion in mice resulted in a major defect in developmental sprouting angiogenesis. ARAP3 could thus represent the molecular link between Rap1/KRIT1 and RhoA/Rho-kinase signaling, involving the repression of RhoA/Rho-kinase activity normally exerted by KRIT1 (see below). Upstream to Rap1 protein, the guanine exchange factor C3G has been identified as a key regulator of vascular development. C3G stimulates guanine nucleotide exchange predominantly on Rap1 and, to a lesser extent, on R-Ras (187). C3G is a molecular link between Rap1 and cell-cell junction molecules such as cadherins, and induces activation of Rap1 upon dissociation of junctions (24). C3G mutant embryos died of vascular defect around E11.5 due to hemorrhage and loss of vascular integrity (503). Vascular endothelial cells appeared to be functioning normally, and the defects were related to abnormal blood vessel maturation with aberrant recruitment/migration, adhesion, and differentiation of mural cells. These data thus suggest that the role of Ras protein during vascular development is not restricted to the regulation of endothelial cell functions. 2. Rho proteins A) RHOA. Part of the evidence for a role of RhoA in murine embryonic vascular development indirectly arises from the analysis of experimental models of cerebral cavernous malformation (CCM). CCM disease results from autosomal dominant loss of function mutations in the genes KRIT1 (CCM1), CCM2 (malcavernin, MGC4607), or PDCD10 (CCM3). Homozygous null mutations in KRIT1 or CCM2 results in lethal vascular and cardiac phenotype in mice and zebrafish, ascribed to essential dysfunction of endothelium causing embryonic lethality at midgestation cells (270, 408, 527). While initial vascular patterning is normal, targeted deletion of CCM2 in endothelial cells severely affects angiogenesis in mice, leading to morphogenic defects in the major arterial and venous blood vessels (53). KRIT1 and CCM2 (and also CCM3) physically interact, suggesting that they converge on the same downstream signaling/effector and that the mechanisms altered by the mutations in these three genes causing the pathological phenotype are the same (504, 575). Indeed, KRIT1, CCM2, or CCM3 mutations induce activation of RhoA/Rho-kinase signaling, responsible for reorganization of endothelial cell actin cytoskeleton, loss of endothelial cell junctions, and increased vascular permeability (51, 466). The phenotype of endothelial cells from CCM2 mutant mice was rescued by pharmacological inhibition of Rho protein by simvastatin (466, 527). RhoA and Rho-kinase activity in endothelial cells thus appears to have a leading role during vascular morphogenesis and that a tight spatial and temporal inhibition of this pathway in endothelial cells is essential for normal cell migration, lumen formation, and normal angiogenesis. This 1684 concept is in agreement with in vitro studies that provide evidence that RhoA and Rho-kinase need to be actively inhibited during endothelial tube lumen formation and tube maintenance events (96). The role of RhoA/Rho-kinase axis in the developmental angiogenesis was further supported recently by the observation that homozygous mice deficient in both isoforms of Rho-kinase (Rock1⫺/⫺;Rock2⫺/⫺) display defective vascular remodeling in the yolk sac and die in utero before E9.5 (208). These results also suggest a redundant role of Rho-kinase isoform functions in vascular development during embryogenesis. Loss-of-function studies in zebrafish and mouse point to a specific role of the RhoA GEF Syx (also called Tech, GEF720, PLEKHG5) in angiogenic sprouting in the developing vascular bed. Syx is not involved in vasculogenesis and angioblast differentiation, but in vascular sprouting. In zebrafish, Syx depletion induced the truncation and even the absence of intersomitic vessels (133). In syx knockout mice, arterial networks are sparse, with deficiency in small-diameter vessels and capillaries, whereas major arteries are intact (133). These phenotypes suggest evolutionay conservation of Syx function. At the molecular level, Syx has been identified as a regulator of the directional endothelial cell migration required for angiogenic sprouting through its interaction with angiomotin (Amot) to precisely localize RhoA activity to the leading front of migrating cells (110, 133). Among RhoA activity regulators, a recent study pointed out a role of the RhoA GAP ARAP3, activated by PI 3-kinase and Rap. Deleting Arap3 in the mouse causes embryonic death in mid-gestation due to an endothelial defect in sprouting angiogenesis (131). However, as ARAP3 is also a GAP for Arf6, in addition to altered RhoA activity, change in the activity of Arf6 can also participate in the effect of Arap3 deletion on angiogenesis. In addition to its role in endothelial cells, RhoA/Rho-kinase signaling in vascular smooth muscle cells is important for their recruitment to nascent vessels and vessel stabilization. Adapted regulation of RhoA and Rho-kinase activity is required for normal coverage of endothelial tubes with vascular smooth muscle, through the control of vascular smooth muscle cell contraction, spreading, polarity, and direct migration (317). B) RHOB. RhoB plays a critical role in vascular development. Retinas of newborn RhoB-deficient mice display signs of retardation in the outgrowth of the primary plexus (6). The mechanism responsible for this effect is related to RhoBmediated regulation of Akt stability and trafficking into the nucleus of endothelial cells. This regulation of Akt confers on RhoB a stage-specific role in the survival of sprouting endothelial cells that contributes to blood vessel assembly during development (6). Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM C) RAC1. The role of Rac1 in vascular development has been addressed thanks to the generation of mice with endothelial-specific deletion of Rac1. Endothelial-specific Rac1-deficient mouse embryos die at midgestation (around E9.5). They show an alteration of the development of major vessels, and small branched vessels were completely absent both in the embryos and their yolk sacs (477). In vitro functional studies of Rac1-deficient endothelial cells functions demonstrated the essential role of Rac1 in the transduction of the effects of vascular endothelial growth factor (VEGF) such as migration, tube formation, adhesion, and permeability. Rac1-deficient endothelial cells are not able to form lamellipodia structures and focal adhesions, or to reorganize their cell-cell contacts (477). Rac1 is not strictly required for the initial differentiation or recruitment of angioblasts from the mesoderm and blood islands but essential for subsequent migration of angioblasts to promote the establishment of capillary-like networks (477). These data support an important role of Rac1 in developmental angiogenesis rather than in vasculogenesis. Furthermore, Rac1 is necessary for the transition from blood endothelial cells to lymphatic endothelial cells required for endothelial cells to separate and form lymphatic vasculature and is therefore an essential part of lymphangiogenesis (94). D) CDC42. Ablation of Cdc42 in mouse embryonic stem cells does not affect endothelial lineage differentiation but completely blocks vascular network assembly because of an impaired directional migration (375). PKC and GSK-3 have been identified as downstream effectors of Cdc42 during vascular morphogenesis (375). In zebrafish Cdc42 is involved in intracellular vacuole formation, the fusion of which plays a fundamental role in endothelial cell lumen formation of intersegmental vessels (207). These data are in agreement with results obtained in mice lacking the specific Cdc42 effector Pak4 that showed abnormalities in vasculature throughout the extraembryonic tissue and the epiblast (486). In the knockout embryos, large vessels could still be detected, but smaller branching vessels were completely absent. The Pak4 knockout yolk sac did stain positively for endothelial cells, showing the presence of the vascular plexus, but did not have organized vessels. These data suggest that Cdc42/Pak4 signaling affects angiogenesis and branching, rather than the initial formation of vessels (486). 3. Other Rho signaling molecules Pak2a, another member of the Pak family that can be activated by Cdc42 or Rac1, also participates in embryonic vessel formation. In zebrafish, Pak2a was identified as a downstream effector of the Rac1 and Cdc42 exchange factor Pix (273). Loss of Pix or Pak2 leads to loss of vascular integrity and hemorrhage in the head but not in other vascular bed. The Pix/Pak2a axis is thus involved in cerebro- vascular stabilization, an effect that is likely to be due to a role in support cells (vascular smooth muscle cells and pericytes) surrounding the cranial vasculature (273). B. Blood Pressure Regulation and Hypertension In general, three major mechanisms contribute to long-term blood pressure control: the contraction of small arteries, the control of blood volume in which the kidney is a key player, and the regulation of the cardiac output. The first part of this section focuses on the involvement of Ras proteins and their regulators in the control of vascular tone, with a focus on their role in arterial smooth muscle cell contractility, either directly or indirectly through modulation of endothelial cell functions. The following sections will address the role of Ras proteins in the central control of blood pressure, and in the kidney-mediated regulation of water-electrolytes balance. 1. Role of Ras protein superfamily members in arterial smooth muscle cell contractility and vascular tone The regulation of arterial contractility is essential for blood pressure control and represents a typical multicellular function regulated by Rho signaling pathways. Phosphorylation of 20-kDa myosin light chain (MLC) is the key event of the regulation of vascular smooth muscle contraction (373). MLC is phosphorylated by the Ca2⫹/calmodulin-activated MLC kinase (MLCK) and dephosphorylated by the Ca2⫹independent MLC phosphatase (MLCP). Agonists (ANG II, norepinephrine, endothelin-1, thromboxane A2 . . .) that bind to G protein-coupled receptors produce contraction by increasing both 1) the cytosolic Ca2⫹ concentration and 2) the Ca2⫹ sensitivity of the contractile apparatus. The rise in cytosolic Ca2⫹ concentration results from the stimulation of Ca2⫹ entry through L-type voltage-dependent Ca2⫹ channels and Ca2⫹ release from the sarcoplasmic reticulum through IP3-gated Ca2⫹ channels activated by PLC-generated inositol 1,4,5-trisphosphate (IP3). The increased sensitivity of vascular smooth muscle toward Ca2⫹ results from inhibition of MLCP activity leading to increased MLC phosphorylation and tension at a constant Ca2⫹ concentration. In contrast, vasorelaxing agents decrease both the cytosolic Ca2⫹ concentration and the Ca2⫹ sensitivity of the contractile apparatus. Surprisingly, while Rho family proteins emerged as key regulators of smooth muscle cell contraction, the role of the other subfamilies of Ras proteins in the regulation of vascular smooth muscle contraction has not been extensively addressed. 2. Ras protein-mediated regulation of smooth muscle cell contraction, vascular tone and blood pressure (Figure 4) A) RAS. The first demonstration of a role of Ras in the regulation of smooth muscle contraction was provided by the Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1685 LOIRAND, SAUZEAU, AND PACAUD observation that active H-Ras induces tension of permeabilized arteries by increasing the Ca2⫹ responsiveness in a concentration-dependent manner (416). Downstream of Ras, ERK1/2 activation triggers MLCK phosphorylation, leading to an increase in its sensitivity to calmodulin and, consequently, its ability to phosphorylate MLC (227). In addition to this stimulation of MLCK activity, the RasERK1/2 signaling was also shown to positively regulate the expression of MLCK in arterial smooth muscle by stimulating MLCK promoter (151, 152). This signaling pathway is activated by ANG II, both in vitro and in vivo in rats (152), and is also involved in the upregulation of MLCK in the arteries of spontaneously hypertensive rats (SHR) (151). The attenuation of high blood pressure produced by Ras FTIs in SHR or in ANG II-induced hypertension in rats suggested that activation of Ras signaling can contribute to the development of hypertension (34, 326). All these data, suggesting a vasoconstricting and hypertensive action of Ras-ERK1/2 pathway, have been confirmed in vivo in genetically modified mice. Knock-in mice in which K-Ras has been replaced by H-Ras develop arterial hypertension (371). More recently, high blood pressure was also observed in a strain of genetically modified mice carrying the activating G12V mutation within their endogenous HRas locus (H-Ras-V12) (427). In contrast, H-Ras⫺/⫺ mice show lower blood pressure than control animals, probably related to an upregulation of the NO-cGMP pathway (70). B) RAL. Although a direct role of Ral proteins has not been provided, recent evidence suggests that RalA and RalGDS Rap1 but not RalB is involved in the coupling of AT1 receptor to the activation of IP3 signal transduction (140). C) RAP. A recent study reported the existence of a cross-talk between Rap1 and RhoA signaling pathways in vascular smooth muscle. Activation of Rap1 causes relaxation by decreasing the Ca2⫹ sensitization of force in smooth muscle through downregulation of RhoA activity (588). Rap1 activation is triggered by the exchange factor Epac, activated by the rise in cAMP induced by the stimulation of vasorelaxing receptors such as -adrenergic or prostglandine I2 receptors. The mechanism by which Rap1 inactivates RhoA has not been identified but, by analogy to that found in endothelial cells and in neurons, it was suggested that a Rap1-activated RhoGAP such as RA-RhoGAP or ARAP3, shown to be directly stimulated by Rap1 (239, 553), may downregulate RhoA activity in smooth muscle cells (588). 3. Rho protein-mediated regulation of smooth muscle cell contraction, vascular tone and blood pressure (Figure 5) A) RHOA. The major role of RhoA in smooth muscle contrac- tion has been unequivocally demonstrated over the past 15 years. The Ca2⫹-sensitizing effect of vasoconstrictors is ascribed to RhoA activation and the subsequent stimulation of its target Rho-kinase that phosphorylates MYPT-1, the regulatory subunit of MLCP, and inhibits its activity (490). While Rock1 and Rock2 are both expressed in vascular smooth muscle cells, Rock2 plays a prominent role in the regulation of vascular smooth muscle contractility (513). This mechanism of Rho-kinase-mediated Ca2⫹ sensitiza- RhoA (Rock2) H-Ras (ERK1/2) RalA Vasodilation MLC MLCP MLCK ↑ Cai IP3 P-MLC Vasoconstriction Rac1 (Pak1) Caldesmon Rac1 (Pak3) FIGURE 5. Role of Ras proteins in the regulation of vascular smooth muscle cell contraction. Contraction is determined by the level of myosin light chain (MLC) phosphorylation (P-MLC), controlled by the activity of the MLC kinase (MLCK) and the MLC phosphatase (MLCP). 1686 Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM tion is thus responsible for the tonic component of vascular smooth muscle cell contraction. For in-depth coverage of RhoA/Rho-kinase signaling-mediated Ca2⫹ sensitization and regulation of smooth muscle contraction, the reader is referred to more comprehensive reviews (282, 373, 457). Following the discovery of the key role of RhoA/Rho-kinase signaling in vascular smooth muscle cell contraction, numerous studies have addressed the role of this pathway in vivo in the control of blood pressure and hypertension (283). In SHR or in rats and mice treated with ANG II, L-NAME, or DOCAsalt, the development of hypertension is associated with an increase in RhoA and Rho-kinase activity in arteries (145, 216, 431, 435), thus showing that RhoA/Rho-kinase signaling activation is a common component for the pathogenesis of hypertension (282, 587). The causal relationship between the activation of RhoA and Rho-kinase and the increase in blood pressure has been indirectly addressed by the use of pharmacological Rho-kinase inhibitors. Rho-kinase inhibitors such as Y27632, fasudil, or SAR407899 reduce blood pressure in animal models of hypertension [ANG II (281); L-NAME (216, 281, 435); DOCA-salt (281, 490); SHR (241, 323)]. In addition to their blood pressure-lowering effect, Rho-kinase inhibitors also prevent the hypertension-associated cardiovascular remodeling, fibrosis, and inflammation (166, 189, 210, 216, 323). All vasoconstrictors acting through G protein-coupled receptors induce RhoA activation, and different types of hypertension are all associated with an increased arterial RhoA activity. It is therefore likely that different Rho GEFs couple vasoconstrictor receptors to RhoA activation and are differently involved in the overactivation of Rho during hypertension. Nevertheless, only a few studies have addressed Rho GEFs expression, regulation, and activation in vascular smooth muscle cells. They mainly focused on the RGS-containing Rho GEF subfamily of Rho GEFs grouping Arhgef1 (p115-RhoGEF), Arhgef11 (PDZ-RhoGEF), and Arhgef12 (Larg) that are supposed to link G protein-coupled receptors to RhoA activation. All three RGS-containing Rho GEFs are expressed in arteries (169, 202, 569). Arhgef11 was described as the most expressed RGS-RhoGEF in rat aorta and mesenteric artery, while in mice aorta, the more abundant RGS-Rho GEF was Arhgef12 (531). Arhgef12 transcript and protein are expressed in vascular smooth muscle of murine embryos from E14 to later stages of development and in adulthood (30). The specific RhoA GEF p63Rho GEF is the most expressed Rho GEF in arterial smooth muscle cells (67, 316). It has been recently demonstrated both in vivo and in vitro that Arhgef1 is involved in ANG II-induced RhoA activation and contraction in vascular smooth muscle cells and is activated in arteries from ANG II-infused rats or mice (145). ANG II-induced Arhgef1 activation depends on AT1 receptor stimulation and subsequent JAK2-mediated Arhgef1 phosphorylation (145). Two other RhoGEFs, p63RhoGEF and Arhgef11, have also been shown to participate in the early RhoA activation induced by ANG II stimulation of vascular smooth muscle cells (543, 568). The RhoGEF p63RhoGEF is also involved in RhoA activation and contraction induced by PhE and endothelin-1, by selectively coupling G␣q/11 protein to RhoA activation (316). The RhoA exchange factors Lbc and Arhgef12 have been identified as mediator of RhoA activation in vascular smooth muscle cells stimulated by 5-HT and sphingosine 1-phosphate, respectively (28, 303) (FIGURE 6). Genetic targeting of Rho protein signaling in mice shed light on the role of Rho GEFs and RhoA/Rho-kinase signaling in the regulation of blood pressure and hypertension. Rock1 haploinsufficient and deficient mice have normal basal blood pressure, and the rise in blood pressure induced by ANG II or L-NAME treatment is similar to that observed in control mice but vascular fibrosis is reduced (393, 582). Given the effect of Rho-kinase inhibitors in ANG II- and L-NAME-treated animals, this observation might suggest that Rock2 is the main isoform of Rho-kinase mediating the vasoconstricting effect of RhoA signaling in vascular smooth muscle cells. Unfortunately, since Rock2 deficiency is embryonically lethal, the cardiovascular phenotype of Rock2-deficient mice has not been assessed. Upstream to RhoA and Rho-kinase, genetic ablation of Arhgef1 and Arhgef12 has confirmed in vivo the role of these Rho GEFs in the regulation of vascular tone and blood pressure and highlighted their specific involvement in different pathways. While specific Arhgef1 deficiency in smooth muscle renders the mice resistant to ANG II- and L-NAME-induced hypertension (145), deletion of Arhgef12 induces insensitivity to salt-dependent hypertension (531). B) RAC1. Rac1 is rapidly activated in vascular smooth smooth muscle cells in response to stimulation with various vasoconstrictors such as ANG II, endothelin-1, and thromboxane A2. Nevertheless, in contrast to RhoA, its role in the regulation of smooth muscle contraction and the control of vascular tone remains unclear (452). The Rac effector Pak1 has been shown to phosphorylate and inhibit MLCK, thereby decreasing MLC phosphorylation and smooth muscle cell contraction (412, 532). This suggests that Rac action antagonizes the effect of RhoA on MLC phsophorylation, and negatively regulates vascular smooth muscle tone. However, opposite effects have also been reported as the other Rac effector Pak3 has been described to enhance smooth muscle cell Ca2⫹ sensitivity and contraction through phosphorylation of the thin filament regulatory protein caldesmon (119). In vivo, chronic ANG II infusion induces Rac1 activation, essentially described in cardiac tissue (385, 415) but probably also in arteries (516). By triggering the activation of NADPH oxidase and redox signaling, Rac1 is a major mediator of the cardiac and vascular hypertrophic effect of Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1687 LOIRAND, SAUZEAU, AND PACAUD PhE Ang II α1R AT1R Arhgef1 Arhgef11 ET1 5HT S1P Lbc Arhgef12 ETAR p63RhoGEF RhoA Rock2 MLCP MLC P-MLC Vasoconstriction FIGURE 6. RhoA GEFs involved in the activation of RhoA and Rock2 by the stimulation of G protein-coupled receptor by vasoconstrictors: ANG II and type 1 ANG II receptor (AT1R), phenylephrine (PhE) and ␣1-adrenoceptor (␣1R), endothelin-1 (ET1) and endothelin receptor A (ETAR), serotonin (5HT), and sphingosine 1-phosphate. Contraction of vascular smooth muscle cells is determined by the level of myosin light chain (MLC) phosphorylation (P-MLC), controlled by the activity of the MLC kinase (MLCK) and the MLC phosphatase (MLCP). ANG II (385, 421). Furthermore, it has been recently demonstrated that Rac1 is responsible for the oxidative stress and vascular dysfunction induced by mechanical stress of the arterial wall caused by high intraluminal pressure levels (499). Mechanical stress activates via integrin-linked kinase 1 and the Rac1 GEF betaPIX. Adenoviral transfection of a dominant-negative Rac1 decreases high blood pressure-induced NADPH oxidase activity and oxidative stress in carotid arteries, and normalizes vascular function. These data thus support a role of Rac1 in oxidative stress and its consequent vascular damages associated with hypertension (499). This role of Rac1 has been confirmed in mice with specific overexpression of the constitutively active mutant of Rac1 (Rac1-V12) in smooth muscle cells. These mice develop moderate hypertension compared with wild-type littermates, caused by enhanced superoxide production (157). Conversely, mice with heterozygous inactivation of Rac1 in endothelial cells (EC-Rac1⫹/⫺) develop moderate hypertension and display decreased eNOS expression and 1688 activity as well as impaired endothelium-dependent vasorelaxation (422). These observations suggest that endothelial Rac1 indirectly participates in the regulation of vascular tone and blood pressure through its effects on eNOS. Pharmacological strategies aiming to enhance endothelial Rac1 function might be of therapeutic interest for preserving endothelial function and normal blood pressure. 4. Rho protein-dependent endothelial-smooth muscle cells crosstalk The maintenance of vascular homeostasis is largely dependent on the functional crosstalk existing between endothelial and smooth muscle cells within the arterial wall. The endothelium is a key determinant of vascular tone and resistance that acts by releasing vasoactive factors among which endothelium-derived nitric oxide (NO) is the most important. Both endothelial NO production and NO effects in vascular smooth muscle cells are regulated by RhoA and Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM Rac1 signaling (283). RhoA, through Rho-kinase activation, downregulates eNOS expression by destabilizing eNOS mRNA (256, 587) and decreases eNOS catalytic activity by inhibiting and stimulating PI 3-kinase/Akt pathway (538) and arginase activity (313), respectively. Rac1 antagonizes RhoA/Rho-kinase effects on eNOS. Rac1 upregulates eNOs expression by increasing eNOS mRNA stability and Pak1-mediated stimulation of transcription, and stimulates eNOS activity (422, 436). In vascular smooth muscle cells, it has been shown that cGMP-dependent protein kinase (PKG) inhibits RhoA/ Rho-kinase signaling by phosphorylating Ser188 of RhoA (417). This inhibitory phosphorylation, by decreasing the Ca2⫹ sensitivity of contractile proteins, plays a major role in the vasodilator effect of NO (417). However, recent findings indicate that Rac1 participates in the control of cGMP level in vascular smooth muscle cells. Activation of Rac1 increases intracellular cGMP concentration via Pak1-mediated inhibition of the activity of type 5 phosphodiesterase (PDE5) (418). The reduced cGMP and vasodilatory response to NO donors observed in arteries from Vav2-deficient mice supports a role of the Rho GEF Vav2 and the tyrosine kinase Src as the upstream regulators of Rac1 in vascular smooth muscle cells (418). 5. Role of Rho protein superfamily members in central blood pressure regulation The baroreceptor reflex is one of the main physiological mechanism that contributes to the stability of blood pressure (365). The specific areas involved in this nervous regulation of blood pressure are located in the brain stem (365). Afferent fibers from arterial baroreceptors project in the nucleus tractus solitarii (NTS). Signals are transmitted from the NTS to the rostral ventrolateral medulla (RVLM) from which sympathetic nerves conduct them to heart and vessels (365). adenovirus vector encoding a dominant-negative Rac1 reduces the increase in blood pressure induced by ANG II (589). Similarly, the intracisternal infusion of the Rhokinase inhibitor Y27632 reduces the central pressure action of ANG II (405). These observations thus provide evidence that ANG II-mediated Rac1 and RhoA/Rhokinase activation in the brain stem contributes to hypertension induced by ANG II infusion. 6. Role of Ras protein superfamily members on kidney-mediated blood pressure regulation The kidney has a central role in the control of blood pressure by regulating the maintenance of water-electrolyte balance, mainly through the reabsorption of filtered Na⫹. Na⫹ flux through epithelial Na⫹ channels (ENaCs) expressed in the apical plasma membrane of the renal collecting duct is the final renal adjustment step for Na⫹ balance. ENaC thus plays a critical role in modulating the homeostasis of Na⫹ and blood pressure. This channel is an end-effector in the renin-angiotensin-aldosterone system, and several regulatory pathways, some of them involving Ras proteins, modulate its activity and its expression (FIGURE 7). 7. Role of Ras proteins in the regulation of ENaC A) RAS. In renal epithelia, aldosterone induces expression and activation of K-Ras (465). Moreover, K-Ras is both necessary and sufficient for activation of ENaC (465). KRas increases the activity of channels already present in the High-salt Aldosterone ↑ MR ↑ SGK1 ↓ AS160 In vivo studies in the rat revealed that RhoA/Rho-kinase signaling in the NTS is involved in the sympathetic nervous system-dependent regulation of basal blood pressure. Inhibition of Rho-kinase in the NTS by adenoviral transfection of dominant-negative Rho-kinase or local infusion of the Rho-kinase inhibitor Y27632 reduces mean blood pressure and urinary excretion of norepinephrine (191). Blockade of Rac1 signaling in the NTS does not affect mean blood pressure or urinary norepinephrine excretion, although it decreases NADPH oxidase activity (344). These data suggest that RhoA- but not Rac1-dependent pathways in the NTS participate in the physiological regulation of blood pressure via the central nervous system. In contrast, both proteins play a role in ANG II-dependent hypertension. ANG II infusion increases NADPH oxidase activity and superoxide production and activates RhoA/Rho-kinase in the brain stem through an AT1 receptor-dependent mechanism (405, 590). Transfection into the brain of K-Ras Rac1 Rab RhoA ↑ Rock ↑ PI3-kinase ↑ WAVE1/2 ↑ PI(4)P5-kinase ↑ PI(4,5)P2 ↑ ENAc activity ↑ ENAc expresssion ↑ Na+ reabsorption ↑ Blood pressure FIGURE 7. Ras protein-mediated regulation of blood pressure through modification of activity and/or expression of the renal epithelial Na⫹ channels (ENaC). Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1689 LOIRAND, SAUZEAU, AND PACAUD membrane by increasing channel open probability (461, 462, 465). Inhibition of PI 3-kinase by wortmannin blocks, while in contrast overexpression of active PI 3-kinase mimics the stimulation of ENaC induced by K-Ras (461, 462). This wortmannin-sensitive stimulation of ENaC is also observed in cells expressing an effector-specific mutant of Ras capable of only activating PI 3-kinase (462). These data indicate ENaC is activated by K-Ras in a PI 3-kinase signaling-dependent manner and that this K-Ras-dependent pathway may participate in the control of systemic Na⫹ balance and blood pressure. B) RAB. Rab proteins have been recently identified as a component of aldosterone-dependent ENaC trafficking, thus regulating the ENaC density the apical membrane of epithelial cell of the collecting duct and Na⫹ absorption (63, 264). Under basal conditions, the nonphosphorylated RabGAP AS160, by maintaining Rap proteins in an inactive GDP-bound state, stabilizes ENaC within an intracellular compartment (264). Aldosterone stimulation of Na⫹ transport, associated with increased apical ENaC density, involves the transcriptional induction of serum- and glucocorticoid-induced kinase (SGK1), which phosphorylates AS160 and inhibits its GAP activity leading to Rab activation. This relieves the AS160-mediated intracellular retention of ENaC and permits forward trafficking of the channel to the apical surface to augment Na⫹ absorption (264). As ENaC recycles in polarized kidney epithelial cells, it is likely that the channel traverses a number of Rab-dependent compartments en route to the apical membrane, involving different Rab proteins. In fact, Rab 4, 11, and 27 have been shown to participate in ENaC regulation, and recent data indicate the absolute requirement for Rab11b to deliver ENaC to the apical membrane (63, 64). 8. Role of Rho proteins in the regulation of ENaC A) RHOA. As for Ras proteins, evidence for a role of RhoA in the regulation of ENaC has been obtained in cell sytems in vitro. Expression of wild-type RhoA and constitutively active V14-RhoA markedly increases ENaC activity, while in contrast the dominant negative RhoA-N19 decreases it (460). The effects of RhoA are primarily to increase the density of ENaC on the plasma membrane. Activation of ENaC in response to RhoA is sensitive to inhibition of Rhokinase and disruption of PI(4,5)P2 synthesis and is mimicked by overexpressing phosphatidylinositol-4-phosphate 5-kinase [PI(4)P5-kinase], indicating that activation of Rho-kinase and its known effector PI(4)P5-kinase, and the consequent increase in PI(4,5)P2 levels mediate the increase in membrane levels of ENaC (460). It was further demonstrated that activation of RhoA rapidly increases the membrane levels of ENac by promoting translocation and insertion of the channel. In addition to PI(4,5)P2, RhoA-induced targeting of vesicles containing ENaC to the plasma membrane also involves cytoskeletal actin and tubulin filaments (214, 366). 1690 B) RAC1. Expression of wild-type Rac1 or constitutively active Rac1 significantly increases ENaC activity, while CdC42 has no effect (360, 460). In contrast, the Rac inhibitor NSC23766 markedly decreases ENaC activity in cortical collecting duct (215). Knock-down of Rac1 also decreases ENaC-mediated Na⫹ reabsorption by decreasing the number of channels at the apical membrane (215). The effect of Rac1 on ENaC depends on WAVE1 and WAVE2, two members of the Wiskott-Aldrich syndrome proteins known to stimulate Arp2/3-mediated actin polymerization (215). The Rac1/WAVE signaling complex might thus play a central role in blood pressure control in the kidney, although the exact mechanisms involved are not defined yet. In support of such a role of Rac1, however, it has been recently suggested that Rac1 in kidneys is essential for saltsensitive hypertension, via a mineralocorticoid receptor-dependent pathway (123, 446). High-salt loading triggers Rac1 activation in the kidneys of the Dahl rat model of salt-sensitive hypertension, and NSC23766 treatment significantly reduces the blood pressure of the rats, indicating a causal role of Rac1 activity in the salt-dependent elevation of blood pressure (446). Along with the repression of Rac1 activity, the treatment of Dahl rats with NSC23766 also significantly reduces salt loading-induced increase in mineralocorticoid receptor expression as well as SGK1 upregulation, indicating that the effect of Rac1 activation on blood pressure involved the activation mineralocorticoid receptor signaling in the kidney (446, 447). 9. Role of Rho proteins on sodium-hydrogen exchanger In addition to the role of ENaC in the renal collecting duct, the sodium-hydrogen exchanger isoform 3 (NHE3) expressed in the apical membrane of the proximal convoluted tubules is one of the major transporters involved in the renal reabsorption of sodium under physiological conditions. Inhibition of Rho proteins by toxin B from Clostridium difficile or Rho-kinase by Y27632 results in reduced NHE3 activity, due to a marked decrease in the surface expression of NHE3 and relocation of the exchanger to an intracellular compartment (159). In fact, RhoA and Rho-kinase, by controlling the phosphorylation of MLC and, ultimately, the organization of the actin cytoskeleton, are important for NHE3 association with ezrin and actin, and this interaction is required for maintenance of the exchanger at the apical surface (159, 474). In SHR, hypertension is associated with a marked increase in NHE3 activity and a decreased Na⫹ excretion (338). Inhibition of Rho-kinase by Y27632 reduces apical NHE3 activity and enhances Na⫹ excretion and total urine volume (338). Because, in general, a natriuresis in patients with hypertension reduces blood pressure, it has been suggested that the natriuresis induced by Y27632 may contribute to its blood pressure lowering effect (474). Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 10. Evidence for a role of Rho protein signaling in high blood pressure in humans prevent or limit vascular remodeling associated with cardiovascular diseases. Both pharmacological data and genetic studies indirectly support a potential role of Rho proteins in the generation of high blood pressure in humans. The first data suggesting a role of RhoA/Rho-kinase in the pathogenesis of human hypertension were obtained by showing that the increase in forearm blood flow and the decrease in forearm vascular resistance produced by the Rho-kinase inhibitor fasudil are significantly stronger in patients with essential hypertension than in the normotensive group of patients, suggesting that hypertension-associated vascular dysfunction depended, at least in part, on Rho-kinase activation (298). The altered vasodilation and increased resistance of the forearm observed in patients with heart failure has been also ascribed to Rho-kinase activation (226). More recently, data obtained in healthy male subjects indicated that the Rho-kinase activity correlates with oxidative stress and aortic stiffness (340). 1. Ras proteins In addition to these functional evidences, genetic analyses have shown that Rho-kinase polymorphism influences blood pressure. This has been demonstrated for the nonsynonymous Thr431Asn Rock2 variant (430). A high basal blood pressure and increased systemic vascular resistance is associated with the Asn/Asn genotype (compared with Asn/ Thr or Thr/Thr). More recently, a lower risk of high blood pressure is associated with a major haplotype block at the Rock2 locus in a recessive manner (384). In addition, recent data from genome-wide association studies have identified the association between diastolic blood pressure and common variants in eight loci, with one of them containing two genes related to Rho protein signaling: 1) RTKN2 encoding the RhoA effector rhotekin-2 and 2) RHOBTB1 encoding RhoBTB (333). Nevertheless, the potential role of these two gene products in the regulation of blood pressure is completely unknown. C. Vascular Remodeling Vascular remodeling occurs during normal development and participates in various physiological processes. However, vascular smooth muscle cell hypertrophy, proliferation, and migration also occur in hypertension, atherosclerosis, and restenosis after angioplasty or stent implantation, leading to pathophysiological remodeling of the arterial wall. These processes are possible because, in contrast to the majority of differentiated cells, smooth muscle cells retain the capacity to modulate their phenotype, to lose their contractile properties, to proliferate, and to migrate in response to a variety of pathological stimuli. By making analogies between vascular proliferative disorders and cancer, studies of Ras proteins have led to the discovery of their roles in cell processes underlying pathological arterial wall remodeling, and suggested that targeting these small G proteins may A) RAS. A role for Ras protein in the regulation of vascular smooth muscle cell proliferation has been proposed more than 20 years ago. H-Ras is expressed in smooth muscle cells, with a peak expression in proliferating cells in mid to late G1 phase (403). Growth factors such as PDGF activate the Ras-MAPK pathway in vascular smooth muscle cells, and Ras inhibition by FTI or expression of dominant negative H-Ras mutant reduces proliferation (235, 491). In contrast, introduction of the dominant active H-Ras-V12 in vascular smooth muscle cells induces a growth arrest with phenotypic characteristics of cellular senescence, suggesting that H-Ras signaling contributes to age-related vascular disorders (312). The other member of the Ras family, KiRas2A, is identified as a mediator of ANG II-induced vascular smooth muscle cell proliferation and senescence (310, 311). Accordingly, it has been proposed that different levels of Ras expression and activity may be one of the mechanisms that regulate the switch between growth/proliferation and senescence signaling in vascular cells (309). Evidence for a role of Ras signaling in the regulation of vascular smooth muscle cell in vivo was then provided by studies showing that plasmid- or virus-mediated local expression of H-Ras dominant negative mutant significantly reduced neointimal formation in balloon-injured rat carotid arteries (188, 491) or pig coronary artery (541). In the same in vivo models, prevention of posttranslational modifications and thereby, inhibition of Ras activity by FTIs or S-adenosylhomocysteine hydrolase inhibitors (433, 540), or S-trans,trans-farnesylthiosalicylic acid (136) has the same inhibitory action on balloon injury-induced neointimal proliferation. Genetically modified mice models have further confirmed the in vivo role of Ras signaling in vascular remodeling and vascular cell proliferation, in particular models of haploinsufficiency or tissue specific deletion. Haploinsufficient nf1 mice (nf1⫹/⫺) have increased neointima formation, vascular inflammation, excessive vessel wall cell proliferation, and ERK activation after vascular injury (252, 253). A similar phenotype is observed in mice with smooth muscle cell specific deletion of nf1 but is rescued by smooth muscle cell expression of the GAP domain of NF1, indicating the causative role of excessive Ras activity in the increased vascular smooth muscle cell proliferation produced by nf1 deletion (549). Upstream to Ras, the Grb2 adaptator, linking growth factor receptors to the Ras exchange factor Sos, has been shown to be expressed in vascular smooth muscle cells and recruited to the membrane by stimulation with PDGF, ANG II, or mechanical stretch, suggesting that Grb2/Sos could be re- Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1691 LOIRAND, SAUZEAU, AND PACAUD sponsible for the activation of Ras (33, 182, 268). This hypothesis is supported in vivo by the resistance to the development of neointima thickening in injured carotid of heterozygous Grb2⫹/⫺ mice (581). The Ras/MAPK pathway is also the target of physiological processes that inhibit vascular smooth muscle cell proliferation and maintain vascular wall homeostasis. Vascular smooth muscle cells expressed endogenous negative regulator of Ras that, under normal conditions, maintain a low activity of the Ras-Raf-MAPK signaling, thus preserving differentiation and quiescence. Such a regulation is responsible for the antiproliferative effect of the contractile phenotype marker SM22␣. SM22␣ binds Ras, resulting in downregulation of Ras-Raf-MAPK signaling and blockade of G1-S phase cycle progression in vascular smooth muscle cells (107). In vivo, this mechanism is responsible for SM22␣ overexpression-induced inhibition of neointimal hyperplasia induced by balloon injury (107). In the same way, R-Ras is a member of Ras protein family that antagonizes H-Ras signaling (88, 438). R-Ras is abundantly expressed in normal mature blood vessels but significantly downregulated in hyperplastic neointimal smooth muscle cells (231). R-Ras knockout mice develop exaggerated neointimal thickening in injured femoral artery due to sustained smooth muscle cell proliferation (231). These results thus indicate the in vivo role of R-ras as a negative regulator of vascular smooth muscle cell hyperplasia, probably through inhibition of H-Ras signaling. A similar role has been ascribed to mitofusin-2, initially called hyperplasia suppressor gene (HSG). Mitofusin expression is decreased in hyperplastic smooth muscle cells from SHR arteries and from balloon-injured rat carotids compared with controls (77). It is also reduced in vitro upon stimulation of vascular smooth muscle cells by mitogenic factors such as PDGF, FGF, or ET-1 (77). Overexpression of mitofusin-2 by adenoviral infection almost totally prevents balloon injuryinduced neointima formation and restenosis in rat carotid arteries (77). Mechanistically, mitofusin-2 binds Ras leading to inhibition of Raf-MAPK signaling and G1/G0 cell cycle arrest (77). B) RAD. Rad expression has been shown to be increased during vascular lesion formation after balloon injury (122). Adenovirus-mediated in vivo Rad gene delivery in carotid artery induces a marked reduction in neointimal formation after balloon injury, while in contrast, expression of a dominant negative form of Rad increases neointimal thickening. Rad was thus identified as an endogenous inhibitor of vascular lesion formation, acting by reducing migration of vascular smooth muscle cells, through at least in part, inhibition of Rho-kinase signaling (122). C) RAP. Rap1a and Rap1b proteins have been shown to be upregulated by PDGF in vascular smooth muscle cells, and this upregulation during the smooth muscle cell cycle is 1692 directly linked to cell proliferation (378). PDGF and FGF-2 also induce an increase in Rap2 during late S phase and G2/M phase of the smooth muscle cell cycle (368). Silencing of Rap2 indicated that Rap2 is required for migration but not for vascular smooth muscle cell proliferation. Despite these data, the role of Rap proteins in vascular smooth muscle cells and vascular remodeling is not known, and in vivo studies are now necessary to address this question. 2. Rho proteins A) RHOA. Numerous studies provide evidence for a direct link between RhoA/Rho-kinase signaling and pathological vascular remodeling, by interfering with several processes such as vascular smooth muscle cell differentiation, migration, and proliferation. RhoA/Rho-kinase signaling has been demonstrated to be a critical mechanism for controlling smooth muscle differentiation through the regulation of the actin cytoskeleton that selectively controls differentiation marker gene expression by modulating SRF-dependent transcription (272, 287). In vitro, most mitogen factors for vascular smooth muscle cells activate RhoA and Rho-kinase, and inhibition of Rhokinase reduces vascular smooth muscle cell proliferation induced by mechanical stretch or by several growth factors and vasocontrictors including PDGF, thrombin, ANG II, and 5-HT (209, 210, 280, 345, 420, 429, 555, 577). RhoA/ Rho-kinase activation accelerates cell cycle progression through downregulation of p27Kip1 expression (257, 420). Conversely, the upregulation of p27Kip1 expression induced by Rho-kinase inhibitors accounts for their antiproliferative effects (210, 420). In addition to this effect on p27Kip1, the necessary role of Rho-kinase for the nuclear translocation of ERK1/2 may also participate in Rho-kinase-mediated regulation of smooth muscle cell proliferation (280, 345, 577). As RhoA and Rho-kinase positively regulate two incompatible processes, differentiation and proliferation, it is possible that functions of RhoA/Rho-kinase signaling depend on the differentiation status of smooth muscle cells. This hypothesis is supported by the differential effect of Rho-kinase inhibition on the migration of proliferative and differentiated arterial smooth muscle cells. Inhibition of the RhoA/Rho-kinase signaling by pharmacological agents or expression of dominant negative limits vascular smooth muscle cell migration induced by several factors such as PDGF, thrombin, or lysophosphatidic acid (9, 420, 431), while in contrast, it increases cell motility of differentiated aortic smooth muscle cells (74). In vivo studies have confirmed the role of RhoA/Rho-kinase in vascular remodeling. Rho-kinase inhibitors reduce neointimal formation induced by balloon injury in rat (420, 444) and pig (114, 332) or by stent implantation in pig (301). In these models, both an antiproliferative action through downregulation of the cyclin-dependent kinase inhibitor p27Kip1 (420) and stimulation of apoptosis (444, Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 445) have been involved in the beneficial effect of Rhokinase inhibitors. Hypertension-associated medial hypertrophy and perivascular fibrosis of small coronary artery of SHRs are also suppressed by long-term treatment with Rhokinase inhibitors (323). Heterozygous deletion of Rock1 and Rock2 alleles further demonstrated the major role of Rock2 in arterial remodeling. Neointima formation induced by carotid ligation was markedly reduced in Rock1⫹/⫺ mice compared with wild-type or Rock2⫹/⫺ mice (341). This correlates with decreased vascular smooth muscle cell migration, proliferation, and survival and reduced leukocyte infiltration (341). The RhoA/Rho-kinase signaling also participates in ageinduced arterial wall remodeling and stiffening developing with ageing. A threefold increase in RhoA expression and activity has been observed in artery of old (19 mo) compared with young (2 mo) rats (307). Indirect evidence suggested that this upregulation also exists in human artery as it has been observed that RhoA/Rho-kinase-mediated coldinduced cutaneous vascoconstriction is augmented with age (482). Increased RhoA and Rho-kinase activity could thus be part of the mechanisms underlying the hypersensitive response of the vessel wall to injury and its increased susceptibility to develop hypertension and advanced atherosclerotic lesions with age. Indeed, it has been recently demonstrated that age-dependent increase in extracellular matrix stiffness enhances vascular cell contractility through RhoA/Rho-kinase activation (186). In endothelial cells, this process leads to mechanical destabilization of cell-cell junctions, increasing endothelial permeability and thus susceptibility to atherosclerosis. B) RHOB. While RhoB is very similar to RhoA and shares its downstream effectors, in particular Rho-kinase, the role of RhoB in the vascular wall is, so far, almost not investigated. However, recently a role of RhoB in vascular smooth muscle cell migration induced by PDGF has been reported. Stimulation of the PDGF receptor- on primary vascular smooth muscle cells results in RhoB-dependent trafficking of endosome-bound Cdc42 from the perinuclear region to the cell periphery, where the Rho GEF Vav2 and Rac are also recruited to drive formation of circular dorsal and peripheral ruffles necessary for cell migration (183). Analysis of vascular remodeling in RhoB-deficent mice would be certainly of great interest. C) RAC1. Compared with RhoA, the function of Rac1 in vascular remodeling is less well understood. However, through its role in NADPH oxidase-dependent ROS generation, Rac1 plays an important role in the development of vascular smooth muscle hypertrophy and migration (180, 285). This role of Rac1 is involved, in particular, in vascular remodeling associated with increased oxidative stress as observed in high blood pressure (499). Moreover, Pak1 also appears as a downstream effector of Rac1 in vascular smooth muscle as expression of a dominant negative Rac1 suppresses ANG II-induced Pak1 activation, hypertrophy (539), and migration (173). Activation of the Rac1 and the subsequent engagement of Pak1 and JNK also participate in the EGF-induced vascular smooth muscle cell proliferation (32). Only a few studies have addressed the role of Rac1 in vascular remodeling in vivo. Transgenic overexpression of dominant active Rac1-V12 confirmed the role of Rac1 as a regulator of the redox state of blood vessel (157). In the balloon injury model of vascular remodeling, Pak1 is found to be activated in neointima of injured rat carotid artery, and a marked reduction of the neointimal thickening is observed in dominant-negative Pak1 or Rac1 adenovirustreated artery (49, 173). In diabetes-induced vascular injury linked to an increased oxidative stress and Rac1/Pak1 activity, dominant-negative Rac1 gene transfer has also been shown to be protective (498). In short, all these data suggest that Rac1/Pak1 activation participates in pathophysiological vascular remodeling. While Rac2 was initially found to have a restricted expression in hematopoietic cells, Rac2 expression and activity are induced upon stimulation of vascular smooth muscle cell with inflammatory cytokines (485). Retroviral overexpression of Rac2 in vascular smooth muscle cells leads to increased migration, activation of the NADPH oxidation cascade, and increased activation of Pak1 and its proximal effectors, ERK1/2, and p38MAPK. Rac2 was thus identified as a potential mediator of inflammation-driven vascular smooth muscle response to injury (485). Further studies, in particular in vivo analyses, are now required to clarify the role of Rac isoforms in arterial remodeling. D) CDC42. Almost nothing is known regarding the role of Cdc42 in the arterial wall. A very recent work reports that the regulation of collagen I secretion by PKC␦ in arterial smooth muscle cells is dependent on Cdc42 (261). Collagen type I is the most abundant component of extracellular matrix in the arterial wall, and a tight regulation of collagen is critical to homeostasis of the arterial wall. These data thus suggest that Cdc42, in addition to RhoA and Rac proteins, could also participate in the control of arterial wall structure. D. Vascular Permeability In addition to its role in the regulation of vascular tone and blood flow, a major function of the vascular endothelium is to constitute a semi-permable barrier that controls passage of macromolecules and fluids between circulating blood and the body tissues. Endothelial barrier integrity is governed by the junctions between adjacent endothelial cells that line blood vessels. Both tight junctions that are particularly developed in the blood-brain barrier and adherens junctions are responsible for endothelial cell-cell adhesion. Most of the factors affecting vascular permeability modify Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1693 LOIRAND, SAUZEAU, AND PACAUD the endothelial barrier function through the regulation of endothelial cell junctions. This regulation is particularly important during physiological processes that require transient breakdown of the endothelial barrier followed by resealing such as acute inflammation or angiogenesis (see sect. IVA). In contrast, dysregulated endothelial barrier function characterizes a pathological situation such as chronic inflammatory diseases and atherosclerosis (see sect. IVE). Vascular endothelial (VE)-cadherin plays a key role in the regulation of adherens junctions. VE-cadherin is a transmembrane protein that forms homotypic interactions between neighboring endothelial cells. Its intracellular part interacts with the actin cytoskeleton through both ␣- and -catenin and p120catenin (29). Because Rho proteins are potent regulators of the actin cytoskeleton organization, numerous studies have addressed the role of RhoA, Rac1, and Cdc42 in the control of endothelial permeability (369, 536). Rap1 has also been recognized as a regulator of the endothelial barrier, and more recently, a role of other small G proteins such as Ras has also been described (359, 369). 1. Ras proteins A) RAS. In endothelial cells, endogenous R-Ras physically interacts with filamin A, a member of the nonmuscle actin binding protein family (143). Knockdown of either R-Ras or filamin A promotes increased vascular permeability and disorganization of VE-cadherin at adherens junctions, through an increase in Src-dependent phosphorylation of VE-cadherin at Tyr731 (143). R-Ras, through its interaction with filamin A, is therefore required for maintaining endothelial barrier function. An analysis of tumor blood vessel formation in R-Ras knockout mice has confirmed its critical role in vessel integrity and function and particularly in the regulation of vascular permeability (419). Plasma leakage is increased, endothelial cell-cell junctions are disrupted, and VE-cadherin immunostaining is decreased in tumor vessels in R-Ras knockout mice. It was further demonstrated that expression of the constitutively active R-RasV38 in confluent endothelial cell cultures induced accumulation of VE-cadherin and -catenin at the cell-to-cell interface, thus improving endothelial barrier function (419). This effect has been ascribed to the suppression of VEcadherin internalization through inhibition of its phosphorylation on Ser665. Expression of the dominant negative R-Ras-N43 or knockdown of endogenous R-Ras has opposite effects (419). In contrast, in vitro in human umbilical endothelial cells, H-Ras signaling has been shown to disrupt endothelial barrier integrity (148, 437). H-Ras/Raf1-dependent activation of MEK/ERK signaling results in increased phosphorylation of MLC, which leads to the recruitment of Src to the VE-caherin, Src-mediated VE-cadherin phosphorylation, and dissociation of adherens junctions (148). Nevertheless, activation of PI 3-kinase/Akt path- 1694 way downstream of H-Ras is also described to be sufficient for H-Ras-induced vascular permeability (437). B) RAP1. In vitro activation of Epac1/Rap1 in endothelial monolayer decreases basal and thrombin-induced permeability by potentiating VE-cadherin-mediated endothelial cell-cell contacts (92, 125, 234). This Epac1/Rap1 signaling pathway participates in the cAMP-dependent increase in endothelial cell-cell contacts induced by physiological factors such as prostaglandins or ANP (44, 45). In vivo, activation of the Epac/Rap1 signaling strongly reduces the baseline and platelet-activating factor-induced acute increase in endothelial permeability in intact microvessels and stabilizes the endothelial-endothelial junction complex (5). Through this regulation of the endothelial barrier function, the Epac1/Rap1 signaling pathway was thus identified as an essential mediator of the anti-inflammatory effects of cAMP and cAMP-elevating agents. In addition to the AMPc/Epac1 activating system, Rap1 is also activated by homophilic VE-cadherin interaction upon cell-cell contact formation by the Rap1 GEF PDZ-GEF1 via the protein adaptator MAGI (411). This result thus reveals a reciprocal interaction between Rap1 and VE-cadherin, with Rap1 stabilizating VE-cadherin-dependent adhesion and cell-cell contact inducing Rap1 activation. Recently, it has been described that Rap1 isoforms differentially regulated endothelial cell junctions. Although Rap1B accounts for more than 90% of the total Rap1 expressed in endothelial cells, Rap1A is the predominant isoform involved in the formation and maintenance of endothelial cell barrier properties, and Rap1B is not able to substitute to Rap1A for these functions (534). Downstream of Rap1, several different effectors are involved in the effects on endothelial barrier function. Among them is Rac1 as activation of Epac1/Rap1 in endothelial cells results in the activation of the Rac1 GEFs Tiam1 and Vav2 (44, 45). The resulting activation of Rac1 leads to enhancement of peripheral actin cytoskeleton and adherens junctions. CCM1 was also identified as a Rap1 effector for endothelial cell-cell junction regulation (139). Activated Rap1 interacts with CCM1, which targets CCM1 to cellcell junctions through physical interaction with junction proteins (-catenin and the junctional scaffold protein AF-6) and finally leads to stabilization of endothelial junctions (139). Interestingly, AF6 is also considered as a Rap1 effector as it specifically binds Rap1-GTP (468) and preferentially the Rap1A isoforms (534). Recently, Raf1 has been identified as an essential component of Epac1/Rap1-mediated endothelial cell cohesion. Upon Rap1 activation, Raf1 is recruited to VE-cadherin complexes, which is required to bring Rock2 to VE-cadherin containing junctions to provide a relevant control of actomyosin activity in these structures (530). Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 2. Rho proteins A) RHOA. RhoA was first described for its role in endothelial cell contraction and hyperpermeability. Both in vitro and in vivo data demonstrated that RhoA activation plays an essential role in the barrier-disrupting effect of various agents such as thrombin, tumor necrosis factor (TNF)-␣, lipopolysaccharirde (LPS), and lysophosphatidic acid (LPA) (83, 111, 112, 495, 496, 535). In addition, RhoA and its target Rho-kinase also have a negative role in the regulation of basal endothelial barrier function. Inhibition of Rho-kinase reduced baseline vascular permeability in vivo and in different cultured endothelial cells in vitro (4). Generation of actomyosin contractility and the resulting isometric tension in endothelial cells are the main mechanisms through which RhoA/Rho-kinase signaling induces junctional breakdown and induction of permeability. RhoA-stimulated Rho-kinase activity phosphorylates and inhibits MLCP, leading to an increased phosphorylation of MLC and acto-myosin interactions (111). In contrast, it was also reported in confluent endothelial cell cultures that intrinsic basal activity of Rho-kinase is required for barrier maintenance through an action on VEcadherin endosomal recycling (494). Nevertheless, the relevance of these data has not been demonstrated in vivo. Several mechanisms have been proposed to trigger the activation of RhoA by agents that increase endothelial permeability. It has been first described that thrombin/PAR-1induced RhoA activation resulted from PKC␣-mediated Rho-GDI phosphorylation and inhibition of its activity, leading to increased RhoA-GDP/GTP turnover and RhoA activation (304). Arhgef1 has then been identified as the RhoA GEF mediating activation of RhoA in response to thrombin/PAR-1 receptor stimulation in a G␣12/13- and PKC␣-dependent fashion (40, 179). This first phase of Arhgef1-mediated RhoA activation is followed by a second phase, dependent on microtubule disassembly. It has been proposed that, in addition to its role on actomyosin contractility, Rho-kinase phosphorylates the microtubule regulatory protein tau, causing peripheral microtubule disassembly and the subequent release of microtubule-associated GEFs such as p190-RhoGEF and Arghef2 (GEF-H1) (39, 40). These events induce a more sustained RhoA activation, cytoskeletal changes, and endothelial barrier dysfunction in response to thrombin (39, 40). More recently, Arghef1 was also identified as the RhoA GEF mediated-RhoA activation and permeability induced by TNF-␣ (361) and LPS (546). Activation of Arhgef1 by TNF-␣ is mediated by PKC␣ phosphorylation of Arhgef1 (361). On the other hand, concomitantly to Rac1/Cdc42 activation, downregulation of RhoA activity is essential for both the maintenance of basal endothelial barrier function and recovery after disruption. The GAP p190-RhoGAP that inactivates RhoA has been recognized as a key signaling mol- ecule for this process. The activity of p190-RhoGAP is regulated by Src-mediated tyrosine phosphorylation, and in the basal condition, the tyrosine phosphatase SHP2 participates in the maintenance of a low RhoA activity by regulating p190-RhoGAP phosphorylation (144). Phosphorylation of p190-RhoGAP on serine/threonine residues also participates in the regulation of its activity (318), and kinases such as PKC␦ and FAK also positively control p190RhoGAP activity to limit basal RhoA activity and to stop RhoA activation during thrombin stimulation, respectively (154, 178). Regulation of p190-RhoGAP expression provides an additional level of regulation of RhoA activity as the enhancement of endothelial barrier function induced by cAMP elevating agents involves downregulation of RhoA activity through stimulation of p190RhoGAP-A expression at a transcriptional level (75). B) RAC1 AND CDC42. Contrary to RhoA, Rac and Cdc42 reinforce endothelial barrier function. Rac1 is required for the formation and the maturation of endothelial junctions. Rac1 activity is increased during junction assembly, and Rac1-deficient endothelial cells are unable to form cell-cell contacts (250, 343, 477). Consistent with this role of Rac1 for maintaining cell-cell junctions, expression of dominant negative Rac1 increases endothelial permeability and disturbs tight and adherens junctions (27, 535). Inhibition of Rac1, in association with RhoA activation, is involved in the increase in endothelial permeability and loss of cell-cell junctions induced by thrombin (505, 535). Thrombin-induced Rac1 inhibition results from proteinase-activated receptor 1/G␣i-mediated inactivation of adenylate cyclase, decrease of cAMP level, and subsequent inhibition of the Epac1/Rap1/Vav2, Tiam/Rac1 cascade (41, 507). On the other hand, factors that increase endothelial barrier function such as mechanical forces, sphingosine 1-phosphate, ANP, prostacyclin, oxidized phospholipids, or hepatocyte growth factor are thought to act via activation of Rac1 downstream of either Epac1/Rap1 or PI3-kinase signaling pathways (42, 45, 271, 449, 506). In addition to these mechanical and soluble factors, “ouside-in” signaling from VE-cadherin also regulates Rac1 activity by a positive feedback mechanism that is essential for maximal endothelial barrier function and recovery of endothelial cell monolayer integrity (43). Cdc42 does not participate in the effect of thrombin on endothelial permeability, but it contributes to the increased actomyosin contractility subsequent to thrombin stimulation as dominant negative Cdc42 prevents thrombin-induced stress fiber formation (535). Moreover, Cdc42 is activated at sites of junctional complex formation, suggesting its role in the regulation of cadherin junctions (343). Cdc42 has then been shown to promote association of ␣-catenin with E-cadherin/-catenin complex and its consequent interaction with the actin cytoskeleton (56). In vivo expression of a dominant active Cdc42 prevents RhoA-dependent increase in endothelial permeability, thus protecting barrier Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1695 LOIRAND, SAUZEAU, AND PACAUD function (382). Adherens junctions themselves control Cdc42 activity (369). Following loss of homotypic interaction, the cytoplasmic domain of VE-cadherin triggers the activation of Cdc42 (369). The mechanisms responsible for this VE-cadherin effect are not identified but might involve either a direct activation via a specific Cdc42 GEF or an indirect mechanism involving the recruitement of p190RhoGAP by p120-catenin in disassembled junctions, the inactivation of RhoA followed by the activation of Cdc42 (57, 236). This activation of Cdc42 by junction disassembly plays an essential role in the restoration of endothelial barrier integrity. inactivates cofilin, which finally leads to accumulation of cortical actin (506). Focal adhesion proteins have also been proposed to participate in Rac1/Cdc42-mediated regulation of endothelial permeability. Rac1/Cdc42 activation allows redistribution of focal adhesion proteins and association with adherens junction protein complexes through paxillin/-catenin interactions (42). Although the molecular mechanism involved is not completely understood, it might involve Pak-induced phosphorylation of paxillin on Ser273 (331). E. Atherosclerosis, Atherogenesis (Figure 8) The mechanisms by which Rac1 and Cdc42 contribute to junction assembly are still speculative but probably involve IQGAP1. Active Rac and Cdc42 bind IQGAP1, thus releasing -catenin from IQGAP and allowing -catenin/VE-cadherin association and stabilization of the junctional protein complex (242, 369). Another possibility is that Rac1 and Cdc42 indirectly control adherens junction through local actin cytoskeleton regulation (271, 369). Active Rac1 recruits Pak and its target Lim kinase that phosphorylates and Monocyte LDL In recent years, great efforts have been devoted to unraveling the basic mechanisms of atherosclerosis, the underlying pathology of cardiovascular diseases. An imbalanced lipid metabolism with increased circulating levels of low-density lipoprotein (LDL) and a maladaptive immune response entailing a chronic inflammation of the arterial wall are clearly identified as major triggering factors. Atherogenesis typically starts with dysfunction and activation of endothelial T-cell Adhesion receptor expression Permeability increase Ras RhoA Rac1 RhoA Rac1 Cdc42 ROS formation Rac1 LDL oxidation Cytokine/ chemokine release Endothelium Chemotaxis, migration T cell proliferation RhoA Rac1 Cdc42 RhoA Cholesterol efflux RhoA Cdc42 oxLDL uptake RhoA Contraction RhoA Foam cell formation Macrophage retention RhoA Rac1 Smooth muscle cells FIGURE 8. Involvement of Ras/Rho proteins in atherosclerotic lesion formation. Protein names are indicated under a brief description of the process in which they are involved in a green background for a positive effect and a red background for a negative action. 1696 Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM cells that triggers recruitment of proinflammatory circulating cells. The resulting local inflammation induces leukocyte chemotaxis and adhesion to the damaged endothelium, leading in turn to an increased vascular permeability for plasma lipids. Monocytes located in the arterial intima accumulate these lipids and transform into macrophage-derived foam cells, thus constituting early plaques. By secreting cytokines and growth factors that stimulate vascular cells, these early plaques generate their own growth and progress into mature plaques. Therefore, in addition to vascular wall cells, circulating immune cells including monocytes, lymphocytes, and platelets contribute to the disease initiation and progression. By regulating numerous functions in these different cell types, members of Ras protein superfamily, in particular Rho proteins, are involved in this complex pathological process. 1. Ras proteins A) RAS. Cellular senescence and inflammation are classical features of atherosclerosis. The enhancement of vascular inflammation and senescence induced in vivo by adenovirus-mediated expression of dominant active H-Ras in rat arteries has led to the suggestion of a potential role of Ras in atherogenesis (312). This is in agreement with the attenuation of atherosclerotic plaques in ApoE⫺/⫺ mice treated by FTI, in association with a reduced Ras activity in arteries (135, 471). FTI has no effect on plaque T lymphocyte and macrophage content, but vascular cell adhesion protein-1 (VCAM-1) and NFB expression, and oxidative stress are markedly reduced in treated ApoE⫺/⫺ mice compared with controls (135, 471). This suggests that Ras activity has a pro-atherogenic action, via mechanisms that are not elucidated. It has been proposed that the downstream Ras effector early growth response-1 (Egr-1) that regulates transcription of proinflammatory and procoagulant genes could be involved. Oxidized LDL induces expression of Egr-1 in endothelial and smooth muscle cells and in macrophages at least in part via the Ras-ERK1/2 pathway (153). Recently, pieces of evidence supporting a role of Ras proteins in atherosclerosis in human have been provided by genome-wide association studies (109). They identified a coronary artery disease locus in M-Ras. The gene encodes for the M-Ras protein that is widely expressed in all tissues, with a very high expression in the cardiovascular system. Although these data do not allow anticipating the role of M-Ras in atherogenesis, it could be related to its involvement in adhesion signaling (570). 2. Rho proteins Given the key role of Rho proteins in the regulation of vascular cell migration and proliferation, the recognition of their involvement, in particular RhoA and its effector Rhokinase, in atherosclerosis is not surprising. However, recent data also revealed that Rho proteins contribute to vascular inflammation, leukocytes infiltration, and lipid uptake/efflux in macrophages. A) RHOA. RhoA expression and activity increase with cellular infiltration during the progression of atherosclerotic lesions (351). Downstream RhoA, Rho-kinases have been also shown to be upregulated at inflammatory arteriosclerotic lesions in both a porcine model of coronary artery spasm (211) and arteriosclerotic human arteries (212). In agreement with this observation, in vitro studies have shown that oxidized LDL and its compound LPA activate RhoA in vascular smooth muscle and endothelial cells, suggesting that hyperlipidemia could contribute to the increased RhoA activity in the wall of atherosclerotic arteries (130, 470). A causal role of RhoA/Rho-kinase signaling in the progression of plaques was first provided by pharmacological analyses that showed that in vivo chronic treatment with Rhokinase inhibitors strongly reduced atherosclerotic lesions and spasms in pig (320) and in ldlr⫺/⫺ mice (289). NF-B activation downstream to RhoA/Rho-kinase and modulation of T-lymphocyte proliferation have been suggested to contribute to the pro-atherosclerotic effect of RhoA/Rhokinase signaling (289). Together with the indirect evidence that inhibition of Rho protein isoprenylation mediates, at least in part, the anti-inflammatory and anti-arteriosclerotic properties of statins, these observations thus confirm the pro-inflammatory and pro-arteriosclerotic effects of RhoA/ Rho-kinase signaling (334). Upregulation of RhoA in atherosclerotic vascular wall could also account for endothelial cell activation and dysfunction associated with atheroma plaque formation. Activation of RhoA/Rho-kinase signaling decreases expression and activity of eNOS and, by increasing endothelial cell contractile forces, enhances permeability, thus allowing easier passage of molecules, such as LDL, and leukocytes from the bloodstream into the vessel wall intimal layer (186, 254, 533). Many different mechanisms are likely to be involved in the overactivation of RhoA, including the increased oxidative stress and ROS production in atherosclerotic arteries. Effects of ROS on Rho proteins may be cell specific and depend on the localization and the activity of a particular Rho protein. Indeed, both ROS-mediated activation and inhibition of RhoA have been described (7, 336). Under conditions where ROS are generated at increased pathological levels, ROS can directly activate RhoA by oxidation of cysteine 16 and 20 (7). Leukocyte adhesion is further favored by RhoA-mediated direct positive regulation of leukocyte adhesion to the endothelium. RhoA controls both the expression of leukocyte adhesion receptors such as E-selectin, VCAM-1, and intercellular adhesion molecule-1 (ICAM-1) on endothelial cell membrane at least in part through NF-B signaling, and downstream signals from these receptors (69). In fact, en- Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1697 LOIRAND, SAUZEAU, AND PACAUD gagement of ICAM-1 following leukocyte adhesion induces RhoA activation and recruitment of ERM (ezrin/radixin/ moesin) to sites of ICAM-1 clustering. ERM proteins thus participate to further increase RhoA activity in endothelial cells by binding to and recruiting the Rho GEF Dbl, and by binding to RhoGDI which releases its inhibition of RhoA (533). Furthermore, RhoA/Rho-kinase activation also favors recruitment of circulating cells into atherosclerotic lesions by inducing release of cytokines and chemokines such as monocyte chemotactic protein-1 (MCP-1, also known as chemokine ligand 2, CCL2) and interleukin-6 by vascular cells (129, 193). On the other hand, adhesion to the endothelium also induces upregulation of Rock1 in leukocytes (120), and accumulating evidence also suggests that RhoA signaling in infiltrating leukocytes, in particular in monocyte/macrophages, plays a key role in the progression of atheroma plaques. Indeed, atherosclerosis is decreased in mutant mice with specific Rock1 deficiency in bone marrow-derived cells on a LDL receptor deficiency background (510). Lack of Rock1 in bone marrow-derived cell decreases macrophages, T cells, and lipid accumulation in the atherosclerotic lesions. In vitro, Rock1-deficient macrophages exhibit impaired chemotaxis and reduced oxidized LDL uptake and foam cell formation. By promoting macrophages chemotaxis and lipid accumulation, Rock1 in macrophages is therefore critical to the development of atherosclerosis. Whether or not Rock2 plays a similar role remains to be determined. Recently, although the involvement of RhoA activation has not been addressed, it has been shown that the Vav family of Rho GEFs regulate CD36-dependent uptake of oxidized LDL in macrophages and foam cell formation (380). The pro-atherosclerotic effect of RhoA/Rho-kinase signaling and its positive action of foam cell formation could also be related to its inhibitory effect on macrophage cholesterol efflux. RhoA, by inhibiting PPAR␥ activity, negatively controls ABCA1 expression and ApoA1-mediated cholesterol efflux, thus favoring foam cell formation (21, 293, 564). B) RAC1. As RhoA, Rac1 expression and activity is increased in arteries from atherogenic animal models (196, 255, 351). Interestingly, Rac1 expression and activity are reduced by estrogen in vascular smooth muscle and monocytes, suggesting that this effect could participate in the atheroprotective action of estrogen (255). The involvement of Rac1 activation in atherogenesis has not been directly assessed, although a number of results suggest it could play a role. Rac1 is part of the NADPH oxidases NOX1 and NOX2, which play a major role in ROS formation and subsequent processes involved in atheroma plaque formation such as vascular smooth muscle hypertrophy and migration (180, 285) and VEcadherin-dependent endothelial cell-cell adhesion (29, 1698 497). Rac1 also regulates ICAM-1 and VCAM-1 expression in endothelial cells and could thus participate in the recruitment of inflammatory cells (69). Moreover, VCAM-1 activation induces Rac1 signaling in endothelial cells leading to NADPH-derived ROS-dependent loss of cell-cell junction that facilitates paracellular leukocyte transendothelial migration (497, 533). A tight control of temporal and spatial activation of RhoA and Rac proteins is essential to coordinate leukocyte and macrophage migration. Active Rac at the leading edge of lamellipodia mediates local actin reorganization, producing lamellar extension and forward cell movement, while active RhoA at the rear produces contraction and detachement of the tail. The balance between the opposing activities of these proteins is therefore a key determinant of cell morphology and movement that is also regulated by Rac1-mediated ROS generation (336). Rac1 induces downregulation of RhoA activity through production of ROS that inhibits the low-molecular-weight protein phosphatase and then increases the tyrosine phosphorylation and activation of p190RhoGAP which in turn inactivates RhoA (336). This mechanism is essential for the induction and maintenance of lamellipodia in migrating cells (336). Macrophage chemotaxis is induced by chemokines and cytokines produced during local inflammation at sites of plaque formation. Macrophage migration triggered by such chemotactic factors, including MCP-1 and macrophage colony-stimulating factor (M-CSF), is dependent on Rac1 activation with Pak1 and ERK1/2 as downstream Rac1 effectors (392, 454). Although local activation of Rac1 in the lamellipodia is required for cell movement, strong enhancement of Rac activity and imbalanced Rac1/RhoA activities disrupt cell migration (14). Such a mechanism has been recently identified as responsible for the inability of macrophage foam cells to leave the artery, thus causing progression of atheroslerosic lesions. Cholesterol loading of macrophages leads to increased plama membrane localization and activation of Rac1, and abrogation of migration toward various chemotactic stimuli (354). Conversely, it has been suggested that the ability of high-density lipoproteins (HDL) to promote regression of atherosclerosis resulted from the inhibition of this mechanism and consequent emigration of macrophages from lesions. HDL, in conjunction with the ATP-binding cassette transporters ABCA1 and ABCG1, serve to prevent formation of cholesterol-rich domains on the inner side of the plasma membrane of cholesterolloaded macrophages and thus limit Rac1 activity (354). This antiatherogenic effect is further reinforced by the inhibitory action of HDL lysosphingolipids and type 3 sphingosine 1-phosphate receptors on Rac1 activity, ROS generation, and MCP-1 production by vascular smooth muscle Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM cells, which limits macrophage entry into atherosclerotic lesions (487). A role for Rac signaling in human atherosclerosis has been recently provided by a genetic association mapping study showing associations of single nucleotides polymorphisms (SNPs) in the KALRN gene with early onset of coronary arterial disease (511). Kalirin, the product of the KALRN gene, is a GEF for Rac (and RhoG). The strongest association was found for the SNP rs9289231 that resides in the first intron of an alternative transcript of the KALRN gene. Furthermore, the risk allele of rs9289231 was significantly correlated with atherosclerosis burden in human aortas (511). Strinkingly, the same study also identified a SNP in the CDGAP gene encoding for the Rac/Cdc42 GAP CDGAP that displays evidence of association with coronary artery disease. These data thus strongly suggest that dysregulation of Rac signaling contributes to the coronary artery disease susceptibility linked to KALRN SNP. Functional analyses of the consequence of kalirin mutations are now required to understand its role in the development of atherothrombotic diseases. C) CDC42. Again, like for Rac, the role of Cdc42 in atherosclerosis has never been directly assessed, although several observations suggest that it could participate in various processes involved in atherogenesis. First, Cdc42 activity is essential for endothelial barrier repair, adherens junctions stability, and restoration of permeability (57). Therefore, dysregulated Cdc42 activity might contribute to the failure to restore endothelial barrier function associated with atheroslerosis. The mechanisms involved in this Cdc42 function are not fully understood, although it probably involves its effect on actin remodeling. Upstream receptors as well as Cdc42 regulators and effectors engaged during endothelial barrier repair remain to be identified. Second, although in contrast to Rac and Rho, Cdc42 is not required for cell locomotion, it is essential for polarizing in the direction of the chemotactic gradient. Macrophages expressing a dominant negative Cdc42 mutant are able to migrate but are unable to polarize in the direction of the gradient of MCSF and move randomly (14). Cdc42 activity is thus probably involved in chemottractant-induced monocyte recruitment to sites of vascular inflammation during the formation and the progression of atherosclerosis. Third, Cdc42 is involved in cholesterol movement in macrophages. As mentioned above, HDL exerts atheroprotection via multiple mechanisms, but one of the most important is cholesterol efflux. Cholesterol efflux from the cells is the rate-limiting step in regulating the intracellular cholesterol content as well as raft structure in the plasma membrane. By this mechanism, that involved ABCA1 and ABCG1, excess cellular cholesterol is released from cells and transferred to HDL particles. This is the first step of reverse cholesterol transport that carries cholesterol from peripheral tissues to the liver for eventual elimination. The role of Cdc42 in cholesterol efflux has been discovered thanks to studies on Tangier disease, a hereditary HDL deficiency caused by Abca1 mutations characterized by the presence of defective cellular cholesterol efflux and development of premature atherosclerosis (174). Expression of Cdc42 is markedly decreased in macrophages, and also in fibroblasts from patients with Tangier disease. Expression of a dominant negative Cdc42 mutant reduces while in contrast, expression of an active Cdc42 mutant increases lipid efflux, suggesting that defective Cdc42 signaling is enough to alter lipid efflux (174). It was further demonstrated that binding of apoA-I, the major HDL apolipoprotein, to the plasma membrane induced activation of Cdc42 and JNK has been identified as a downstream Cdc42 effector involved in apoA-I/HDL-induced cholesterol efflux (174, 339). By promoting rearrangement of actin cytoskeleton and activation of JNK, Cdc42 activation could allow trafficking of cholesterol vesicles from the intracellular site to the plasma membrane where ABCA1 assembles apoA-I and cholesterol for its efflux from cells. Cdc42 thus plays an important role in cellular cholesterol efflux, and its dysregulation might lead to the development of atherosclerotic cardiovascular disease All these data point out the need to combine in vitro and in vivo approaches using relevant animal models to advance our understanding of Cdc42 role in the initiation and the progression of atherosclerosis. V. “RASOPATHIES”-ASSOCIATED CARDIOVASCULAR DYSFUNCTIONS The Rasopathies form a group of nine developmental syndromes caused by germline mutations in genes encoding Ras proteins as well as Ras protein regulators or effectors, leading to Ras/MAPK pathway dysregulation. (Table 4 is available online as supplementary data due to its size.) Therefore, although each syndrome displays unique phenotypic features, they share a common pathophysiology and many clinical signs including characteristic facial features, heart defects, skin anomalies, mental retardation or learning difficulties, growth deficit, and an increased risk of developing tumors. This part thus deals with the cardiovascular features of the major rasopathies. A. Noonan Syndrome 1. Clinical features Noonan syndrome (NS; OMIM 163950) is a relatively common autosomal dominant inherited disorder with an estimated prevalence of 1 in 1,000 to 1 in 2,500 live births Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1699 LOIRAND, SAUZEAU, AND PACAUD (342). The NS phenotype, which becomes less pronounced with age, includes a typical dysmorphic appearance, developmental delay and learning problems, visual abnormalities, auditory problems, orthopedic defects, and short stature (103, 398, 481). Congenital heart defects are found in 50 – 80% of patients with NS (60, 291, 398). Newborns and children suspected of developing NS need a careful cardiac evaluation. Pulmonary valve stenosis and hypertrophic cardiomyopathy are the most common forms of cardiac disease. Atrial septals and partial atrioventricular canal defects are also associated with the NS. Fifty percent of patients with NS have an unusual electrocardiographic pattern characterized by left-axis deviation, an abnormal R/S ratio over the left precordial precordium, and an abdnormal Q wave (379). 2. Molecular pathogenesis Tartaglia et al. (479) identified missense mutations in the Ptpn11 gene in ⬃50% of NS patients (479). It is more prevalent among NS patients with pulmonary valve stenosis and short stature, and less common in subjects with hypertrophic cardiomyopathies and/or severe cognitive deficits. The Ptpn11 gene encodes Src homology 2 containing protein tyrosine phosphatase 2 (SHP2), a nonreceptor protein tyrosine phosphatase. This ubiquitous protein is involved in the effects of numerous physiological mediators such as growth factors, hormones, or cytokines. In particular, by modulating phosphorylation of proteins within multiple signaling pathways, including the Ras/MAPK cascade, SHP2 controls intracellular transduction mechanisms that regulate cell cycle progression, growth, and migration (38, 95). Most of the mutations of Ptpn11 in NS patients disrupt an autoinhibitory interaction between the NH2-terminal domain and the catalytic phosphatase domain. These amino acid substitutions promote SHP2 gain-of-function leading to increased binding affinity for signaling partners of Ras/MAPK pathway. Various strains of mice have been generated to understand the role of SHP2 in cardiac development and physiology. Mice expressing a constitutively active form of SHP2 (Ptpn11 D61G allele) exhibit defects similar to those seen in NS patients including short stature, facial dysmorphia, valvular hyperplasia, and septal defects (20). By studying an inducible Ptpn11 allele, they demonstrated that the expression of mutant SHP2 only in endocardium is sufficient to induce all cardiac defects in NS (19). The cardiac-specific deletion of Ptpn11 rapidly induces a compensated dilated cardiomyopathy in mice without an intervening hypertrophic phase (233). In this study, the authors demonstrated in vitro and in vivo an abnormal ERK/ MAPK and RhoA/Rho-kinase activation in SHP2-deleted cardiomyocytes. Ex vivo, inhibition of these signaling pathways rescues the dilated phenotype, suggesting a critical role of RhoA and ERK/MAPK in the dilated cardiomyopathy of NS patients. Recently, Langdon et al. (251) have confirmed this hypothesis by introduction of Noonan SHP-2 mutation (SHP-2N308D) into Xenopus embryos. Em- 1700 bryos expressing this mutation are characterized by an over activity of Rho-kinase leading to abnormal arrangement and polarity of cardiac actin fibers, and consequently cardiac development defects. The second most frequent gene mutated identified in NS patient (20%) is Sos1 (258, 395, 480). The Sos1 gene encodes the Ras and Rac GEF Son of Sevenless 1 protein. Mutations in Sos1 affect principally two domains, the Pleckstrin homology (PH) and the Ras exchanger motif (REM) domains, which are not implicated in the exchange activity but rather in the intrinsic autoinhibitory function of Sos1. These mutations lead to the maintenance of Sos1 in a noninhibited form, resulting in a permanent activation of the Rac and Ras/ERK pathways. Although some GEFs have been found to be mutated or aberrantly expressed in human cancer, NS-associated germline mutations of Sos1 were the first described example of activating GEF mutations linked with a human genetic disease (395, 480). The extent to which Sos1 gain-of-function mutations affect different Rasdependent or Rac-dependent signals remains to be elucidated. In 2006, a germline K-Ras mutation was found in a NS patient who presented a severe clinical phenotype. Subsequently, K-Ras mutations were identified in ⬍2% of NS patients but generally associated with more significant learning issues and developmental delays (68, 426). Cardiac involvement was present in up to 85% of affected individuals, including pulmonary stenosis, hypertrophic cardiomyopathy, atrial septal defect, patent ductus arteriosus, and ventricle septal defect. Most mutations of K-Ras cluster in the first and second coding exons, which are shared by the two K-Ras isoforms K-RasA and K-RasB. Mutations affecting exon 6 encoding residues at the COOH terminus of K-RasB but not K-RasA have also been reported. NS-associated K-Ras mutations affect highly conserved amino acid residues and are assumed to confer mild gain of function. In 2007, heterozygous missense mutations in Raf1 were reported in 20% of NS patients (357). Whereas hypertrophic cardiopathies are rare (18%) in NS caused by Ptpn11 or Sos1 mutations, they are frequently observed (76%) in association with Raf1 mutations. These observations highlight differences in genotype/phenotype correlations, in particular for hypertrophic cardiopathies, and suggest an important role of Raf1 in pathophysiological modulation of cardiac hypertrophy. However, the role of Raf1 in cardiac hypertrophy obtained with animal experimental models remains controversial. In mice, cardiac expression of dominant-negative Raf1 was shown to prevent ERK activation and to strongly reduce pressure overload-induced cardiac hypertrophy (155). In contrast, cardiac-specific deletion of Raf1 had no effect on ERK expression/activation or heart growth (554). Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM B. LEOPARD Syndrome 2. Molecular pathogenesis 1. Clinical features Aoki et al. (17) identified heterozygous de novo missense mutations of H-Ras gene in CS patients. The patient missense mutations result in amino acid substitutions of a glycine residue in position 12 or 13 of the protein product. These particular amino acids are located at the GTP binding site, and mutations at these sites have previously been shown to cause constitutive activation of H-Ras, in turn causing increased activation of downstream effectors in MAPK signaling pathways controlling cell proliferation and differentiation. Interestingly, these H-Ras germline mutations affect the same amino acid residues that are mutated in cancer. LEOPARD syndrome (LS; OMIM 151100) is an acronym for multiple lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, growth retardation, and sensorineural deafness. LS was first reported by Zeisler and Becker in 1936 (577). Diagnostic clues to LS include multiple lentigines, café-au-lait macules, deafness, and hypertrophic cardiomyopathy. LS patients show retardation of growth and a delayed puberty. Electrocardiographic anomalies and progressive conduction anomalies are the most common heart defects, including left or biventricular hypertrophy, often in association with Q waves, prolonged QTc, and repolarization abnormalities. Hypertrophic cardiomyopathy is the most frequent anomaly (80%), which in general is asymmetric. Less frequent heart defects are pulmonary stenosis 25% and aortic/mitral valve abnormalities in some LS patients (414). 2. Molecular pathogenesis It has recently been reported that LS is related to a missense mutation in the Ptpn11 gene (105). As described in the previous paragraph, germinal mutations in the Ptpn11 gene are also responsible for ⬃40 –50% of NS. But biochemical in vitro studies point to a decrease of SHP-2 catalytic activity for LEOPARD-associated mutations, in contrast to the enhanced SHP-2 activity found for NS. Curiously, LS and NS are associated with the same gene and lead to similar clinical phenotype. To date, no explanation is provided to understand how the gain of function or the loss of function of SHP-2 could induce the same phenotype. In addition, Raf1 gene mutations were identified in LS patients (357, 426), indicating that LEAOPARD syndrome cannot simply be understood by invoking reduced Ras/MAPK signaling. A strain of mice carrying a germline G12V mutation within their endogenous H-Ras locus has been generated to shed light on the role of this gain of function of H-Ras in the developmental and physiological defects associated with CS (427). H-RasG12V mice are viable and closely phenocopy some of the defects observed in CS patients, including a number of cardiovascular dysfunctions. Four-month-old mutant mice develop cardiomyopathy characterized by general enlargement of all heart chambers, concentric left ventricle hypertrophy, and fibrosis. The authors have observed consistent thickening of the aortic valve without any alterations in the pulmonary or atrioventricular valves. In addition, H-RasG12V mice exhibit systemic arterial hypertension resulting from activation of the ANG II pathway. High blood pressure is not common in CS patients, but it has been described in 10% of CS patients with the mutation 34G (113). H-RasG12V mice thus appear as a good animal model to understand the etiology of cardiovascular CS symptoms and to evaluate potential therapeutic strategies. D. Cardio-Facio-Cutaneous Syndrome 1. Clinical features C. Costello Syndrome 1. Clinical features Costello syndrome (CS; OMIM 218040) was first described in 1971 by Costello. The clinical manifestations included postnatal growth retardation, distinct facial appearance with large head, curly hair, nasal papillomata, hyperextensible fingers with loose integuments of the hands and feet, and hyperpigmented skin (22, 90, 91). This rare congenital disorder affects multiple organ systems encompassing severe failure to thrive, cardiac anomalies, tumor predisposition, and cognitive impairment. More than 60% of patients develop cardiac defects including hypertrophic cardiomyopathies, congenital malformations, usually pulmonary valve stenosis, and arrhythmia, especially atrial tachycardia (103). The cardio-facio-cutaneous syndrome (CFCS; OMIM 115150) is a rare sporadic occurring disorder (389). Short stature, mental retardation, global developmental defects, and delayed language acquisition are found in most patients (⬎75%) (103, 217, 394). Children with CFC have typical facial characteristics similar to those noted in NS: high forehead with bitemporal constriction, palpebral ptosis, and a short nose with relatively broad nasal base. Cutaneous and adnexal abnormalities detected in CFCS are similar to those reported in the NS and CS. Cardiac abnormalities are detected in 75% of patients. Affected individuals often present with pulmonary stenosis (45%), hypertrophic cardiomyopathy (40%), and atrial septal defect (22%). Life expectancy of CFC patients is probably shortened on average, due to the severe cardiovascular defects. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1701 LOIRAND, SAUZEAU, AND PACAUD 2. Molecular pathogenesis CFCS has been first considered as a severe form of NS and CS because these three syndromes share several manifestations, and “borderline” cases do exist, usually in infants. However, Ptpn11 and H-Ras mutations were not found in CFC patients, indicating a specific etiology and genetic entity of this syndrome. The final evidence came in 2006, with the discovery among 23 patients with the CFC, of 11 different mutations of the B-Raf gene in 18 of them, mutations of MEK1 in 2 and of MEK2 in 1 patient (396). These mutations have then been rapidly confirmed by other authors (335). Two clusters of mutations were identified in the B-Raf gene: in the cysteine-rich domain in the exon 6 and in the protein kinase domain. All of them are missense mutations, suggesting a gain-of-function effect and a consequent over-activation of the B-Raf/MEK1/2 signaling. A mouse strain that expresses a constitutive active form of B-Raf (B-RafV600E) has been recently generated (492). This animal model mimics some characteristic defects found in CFC patients, including reduced life span, small size, facial dysmorphism, and epileptic seizures. In addition, B-RafV600E mice develop cataracts and upregulation of some sympathetic functions. With regard to the cardiovascular system, no gross alterations in the histological cardiac structure (auricules, ventricles, aortic and pulmonary valves) and sign of tissue fibrosis are observed in mutant mice. However, a cardiomegaly is detected, due to an increase in the total number of cardiomyocytes. Heart function from B-RafV600E mice is altered, including lower end-systolic and end-diastolic volumes. Systolic arterial pressure is not modified in the mutant animals, despite a significant increase in the heart ejection fraction. The other cardiovascular alterations typical of CFC patients, such as pulmonary valve stenosis as well as septal and aortic defects are not observed in the mutant mice. B-RafV600E mice are a suitable animal model of the RASopathy CTC and may serve as a tool to evaluate the potential therapeutic efficacy of B-Raf inhibitor, such as PLX4032 described recently as a potent inhibitor of B-Raf kinase activity (48, 118). E. Neurofibromatosis 1 1. Clinical features Neurofibromin is encoded by a large gene spanning over 350 kb in the chromosomal region 17q11.2 (26, 434). The protein product of this gene functions as a Ras GTPaseactivating protein (GAP) to negatively regulate Ras activity (294, 547). More than 240 different mutations have been described within the Nf1 gene (386). The majority of mutations lead to a truncated or no protein product. Reduction or complete loss of neurofibromin induces activation of Ras, which in turn activates mitogenic signaling leading to increased cellular proliferation or differentiation. For this 1702 reason, neurofibromin is considered as a tumor suppressor (260). Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease (OMIM 611431), is the condition most commonly associated with mutations in the Nf1 gene. It is a dominant disorder affecting 1 in 3,000 individuals. Most adults with NF1 develop café-au-lait macules, neurofibromas, optic glioma, osseous lesions, Lisch nodules, and axillary or inguinal freckling. In addition, NF1 patients present prominent cognitive impairments (attention deficit, hyperactivity disorder, spatial deficit, and language delay) and musculoskeletal abnormalities (scoliosis, long bone dysplasia, and osteopenia). NF1 is also remarkable for its association with occlusive (stenoses) or aneurysmal arterial disease affecting predominantly the aorta and renal, mesenteric and carotid-vertebral arteries. One of the more common vascular lesions in NF1 patients is renal artery stenosis with consequent hypertension (200, 253, 348). The frequency of vasculopathy among NF1 patients in general has not been determined because most of the time NF1-associated vascular lesions are asymptomatic. 2. Molecular pathogenesis NF1 vasculopathy lesions are characterized by an accumulation of vascular smooth muscle cells within the intima layer of the blood vessel resulting in neointima formation. The pathogenesis of this vascular lesion formation remains to be undertsood, whereas they contribute to excess morbidity and mortality of young NF1 patients (387). To shed light on the role of neurofibromin in vascular lesions, murine models of NF1 vasculopathy have been generated. Homozygous deficient embyros die in mid gestation (approximately E13.5). Mutant embryos display hyperplasia of neural crest-derived sympathetic ganglia and exhibit signs of cardiac dysfunction including over abundant tissue that accumulates and obstructs blood flow in the hearts due to hyperproliferation and lack of normal apoptosis of endocardial cushions (55, 199, 247). Endothelial-specific inactivation of Nf1 recapitulates the complete null cardiovascular phenotype providing evidence that neurofibromin is essential for endocardial cushion formation by modulating Ras activity in endothelial cells (138). In contrast, no apparent defects were observed in mice with specific Nf1 deletion in smooth muscle, and no structural abnormalities were detected in the vascular system (549). Nevertheless, the loss of Nf1 in vascular smooth muscle leads to an exaggerated vascular remodeling in response to vascular injury (carotid artery ligation) ascribed to an over-activation of Ras and its downstream effectors, ERK, S6K, and mTOR (549). Surprisingly, the same results were obtained in Nf1⫹/⫺ mice that also display vascular inflammation (252) but not in heterozygous inactivation of Nf1 in vascular smooth muscle cells or endothelial cells alone (253). These results suggest the role of another cell lineage in NF1 vascular diseases. This hypothesis has been confirmed by showing that specific heterozygous inactivation of Nf1 in bone Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM marrow-derived cells was sufficient for increased neointima formation in response to vascular injury (253). In vitro studies demonstrate that direct contact of Nf1⫹/⫺ macrophages with vascular smooth muscle cells stimulates vascular smooth muscle cell proliferation through a Ras/Erkdependent pathway, and this effect is further increased with Nf1⫹/⫺ vascular smooth muscle cells (253). This observation underlies the potential role of inflammatory cells in the pathogenesis of NF1 vasculopathy. This hypothesis is reinforced by the fact that NF1 patients have increased levels of circulating monocytes and proinflammatory cytokines known as biomarkers linked to human vasoocclusive disease (IL-1 and IL-6) compared with the healthy population (253). In short, these recent data provide evidence for an important role of vascular inflammation in NF1 patients and provide a framework for understanding the pathogenesis of NF1 vasculopathy and developing potential therapeutic and/or diagnostic strategies. VI. THE RAS SUPERFAMILY PROTEINS AND EFFECTORS AS PHARMACOLOGICAL TARGETS As frequently mentioned throughout this review, pharmacological compounds that with a more or less good speci- ficity targeting Ras proteins or their regulators have been extensively used in vitro and in animal models and have proven efficient to demonstrate the involvement of Ras protein signaling in numerous studies. Among these compounds, some of them are already used against cardiovascular diseases in humans, and development of new inhibitors targeting these signaling pathways for a potential therapeutical application is the subject of intensive research. A. Targeting Ras Protein Lipidation Ras protein activity absolutely requires their anchoring to cellular membranes via a lipophyllic prenyl group covalently bound to the COOH terminus of these proteins (FIGURE 9). For instance, enzymatic attachment of farnesyl isoprenoid by the action of FT is required for Ras to bind membranes, transform cells, and induce tumorigenesis. Over the past decade, intensive efforts have been devoted to developing FTIs as anticancer agents (36, 489). To date, four types of FTIs have been developed: peptidomimetics, farnesylpyrophosphate analogs, bisubstrate analogs that combine the two former features, and small-molecule inhibitors. In cells, FTIs inhibit H-Ras farnesylation and membrane association, block the activation of mitogen-activated protein ki- Acetyl CoA HMG CoA HMG CoA reductase statins mevalonate Geranyl pyrophosphate Farnesyl pyrophosphate synthase FTI farnesyl pyrophosphate Farnesyl transferase Geranylgeranyl pyrophosphate synthase Ras proteins RhoB Rho kinase inhibitors GGTI geranylgeranyl pyrophosphate Rho proteins Rab proteins Effectors Geranylgeranyl transferase FIGURE 9. Biosynthesis pathway of isoprenoids leading to prenylation of Ras proteins. Statins are specific inhibitors of HMG-CoA reductase (the rate-limiting enzyme of cholesterol biosynthesis). After prenylation, the proteins can move to the cellular membranes where they exert their signaling function. Inhibition of their activity could thus be obtained by farnesyl transferase inhibitors (FTI) or geranylgeranyl transferase inhibors (GGTI). Alternatively, Ras protein signaling pathways can be interrupted by specific inhibition of their target proteins, as shown for Rho-kinase inhibitors. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1703 LOIRAND, SAUZEAU, AND PACAUD nase and PI 3-kinase/Akt pathways, and reduce the growth of a wide variety of human cancer cell lines (388). However, results obtained from nearly 75 clinical trials of 4 different FTIs (lonafarnib, tipifarnib, BMS-214662, and L-778123) reveal that these agents have no or minimal efficacy in the treatment of hematopoietic cancers or solid tumors (36). With regard to the cardiovascular system, unwanted cell growth is involved in the pathogenesis of cardiovascular diseases. Proliferation of vascular smooth muscle cells participates in the development of atherosclerotic lesions and is the main mechanism leading to restenosis of the coronary artery after balloon angioplasty. Thus inhibition of vascular smooth muscle cell proliferation would logically have beneficial effects in these pathological contexts. Although Ras proteins are important, geranylgeranylated proteins such as Rho proteins may also be required for the proliferation of these cells (188). Accordingly, GGT has also been defined as a relevant target for the development of inhibitors of smooth muscle cell proliferation (FIGURE 9). While a great number of preclinical studies showed that FTI and GGTI inhibit vascular smooth muscle growth, and despite the possibility of local delivery with drug releasing stents in the case of prevention of restenosis (540), they did not lead to clinical trials (87). These compounds thus appear to be far from reaching clinical application for cardiovascular diseases. Nevertheless, the therapeutic interest of inhibition of Ras protein lipidation for cardiovascular diseases is strongly supported by the effect of statins (FIGURE 9). Statins are considered as the gold standard therapy for lowering serum LDL-cholesterol and improving cardiovascular morbidity/ mortality. Interestingly, results from recent experimental and clinical studies suggest that their cholesterol-lowering effects could not account for all their beneficial effects on the cardiovascular system (50). Actually, inhibition of the isoprenoid synthesis and the resulting downregulation of Ras protein signaling pathways are responsible for most of the cholesterol-independent pleiotropic effects of statins (586). Although the involvement of inhibition of the Ras subfamily in the beneficial effect of statins is not documented, numerous studies point out the important role of Rho protein inhibition. Indeed, statins, at clinically relevant concentrations that are used to reduce LDL-cholesterol, have been shown to inhibit RhoA isoprenylation (82) and Rho-kinase activity in humans (476). Furthermore, a recent clinical study demonstrates that therapeutic doses of statins mainly exhibit a pleiotropic effect through the inhibition of Rac1 (385). The analogies between the effects of RhoA/Rhokinase and Rac1 inhibition observed in preclinical/basic studies and the effects of statins in humans, including increased expression of atheroprotective genes, inhibition of pro-inflammatory mediators, endothelial protective effects, enhanced stability of atherosclerotic plaques, and inhibition of vascular smooth muscle proliferation, strongly argue of an important role of statin-mediated inhibition of Rho proteins in the benefits of statin treatments. In fact, measure- 1704 ment of leukocyte Rho-kinase activity is currently being utilized to identify patients at cardiovascular risk and to gauge statin efficacy on vascular function that is independent of cholesterol lowering effects (276 –278). On the basis of these observations, Rho protein inhibitors are thus considered as promising treatment for cardiovascular diseases, and the current search for small-molecule inhibitors of RhoA could be highly valuable for developing clinically useful agents (104). B. Rho-Kinase Inhibitors As the major downstream RhoA effector and a key regulator of vascular smooth muscle cell contraction but also cell migration, proliferation, differentiation, apoptosis and survival, and gene transcription, Rho-kinase is identified as an interesting target for cardiovascular pharmacology, and strong efforts are made for the development of Rho-kinase inhibitors. Numerous studies have demonstrated the potential beneficial effects of Rho-kinase inhibitors in animals and humans (282, 451). They have been shown to be effective for the treatment of vascular smooth muscle hyperconstriction-induced disorders such as coronary and cerebral vasospasm as well as systemic and pulmonary hypertension. Some data suggested that in addition, Rho-kinase inhibitors might be useful for treating arteriosclerotic diseases, including arteriosclerosis, restenosis, cardiac allograft vasculopathy and vein graft disease ischemia-reperfusion injury, CCM, myocardial hypertrophy, and heart failure. The Rho-kinase inhibitor fasudil was initially developed as an intracellular calcium antagonist. Fasudil has been approved in Japan since 1995 for the treatment of cerebral vasospasm after aneurysmal subarachnoid hemorrhage and to improve blood flow after acute ischemic stroke (448). More recently, clinical trials have been performed to assess the effectiveness of fasudil in other cardiovascular diseases such as stable effort angina pectoris where it was found to reduce myocardial ischemia (315, 450). Intra-arterial administration of fasudil was also shown to decrease the forearm vascular resistance in patients with heart failure (226). Moreover, intravenous fasudil treatment significantly reduces pulmonary vascular resistance in patients with severe pulmonary hypertension (127). Clinical trials with Rho-kinase inhibitors are in progress in the United States, Europe, and Japan, in various indications, including angina, pulmonary hypertension, atherosclerosis, and reperfusion injury (451). VII. CONCLUDING REMARKS Recent findings have considerably increased our understanding of the role of Ras proteins in cardiovascular phys- Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM iology and physiopathology. These proteins are not only oncogenes but, in physiological situation, are major regulators of cardiovascular cell functions, so long as activation of Ras proteins occurs at the right time and at the right place. Sustained or dysregulated spatiotemporal activation of Ras proteins is most of the time deleterious, leading to pathological situations. Thus, while these proteins are identified as potential targets for the development of new therapeutic strategies in cardiovascular medicine, efforts should be made to understand the mechanisms and identify the molecular partners that regulate their activity. They are likely to be different according to the cell types, upstream activating factors, and even not the same in diverse subcellular compartments. Much remains to be done to understand the role of the numerous Ras proteins expressed in the cardiovascular system. However, thanks to the rapidly increasing availability of mouse models of deletion or expression of active mutants of Ras proteins and regulators with inducible and tissue-selective targeting, our knowledge on the role of Ras protein signaling in cardiovascular functions and diseases should continuously improve, with the potential discovery of targets for the development of new therapeutic strategies. ACKNOWLEDGMENTS We thank Dr. Flavien Charpentier (UMR_S1087, Nantes) for critical reading of the manuscript and Dr. Agnès Quemener (UMR_S892, Nantes) for the Ras superfamily proteins 3D structure. growth factor regulates connexin 43 and N-cadherin expression in atrial fibrillation. J Am Coll Cardiol 55: 469 – 480, 2010. 4. Adamson RH, Curry FE, Adamson G, Liu B, Jiang Y, Aktories K, Barth H, Daigeler A, Golenhofen N, Ness W, Drenckhahn D. Rho and rho kinase modulation of barrier properties: cultured endothelial cells and intact microvessels of rats and mice. J Physiol 539: 295–308, 2002. 5. Adamson RH, Ly JC, Sarai RK, Lenz JF, Altangerel A, Drenckhahn D, Curry FE. Epac/Rap1 pathway regulates microvascular hyperpermeability induced by PAF in rat mesentery. Am J Physiol Heart Circ Physiol 294: H1188 –H1196, 2008. 6. Adini I, Rabinovitz I, Sun JF, Prendergast GC, Benjamin LE. RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev 17: 2721–2732, 2003. 7. Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One 4: e8045, 2009. 8. Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol 13: 39 –51, 2012. 9. Ai S, Kuzuya M, Koike T, Asai T, Kanda S, Maeda K, Shibata T, Iguchi A. Rho-Rho kinase is involved in smooth muscle cell migration through myosin light chain phosphorylation-dependent and independent pathways. Atherosclerosis 155: 321–327, 2001. 10. Aikawa R, Komuro I, Nagai R, Yazaki Y. Rho plays an important role in angiotensin II-induced hypertrophic responses in cardiac myocytes. Mol Cell Biochem 212: 177– 182, 2000. 11. Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest 100: 1813–1821, 1997. 12. Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Zhu W, Kadowaki T, Yazaki Y. Rho family small G proteins play critical roles in mechanical stress-induced hypertrophic responses in cardiac myocytes. Circ Res 84: 458 – 466, 1999. 13. Aikawa R, Nagai T, Tanaka M, Zou Y, Ishihara T, Takano H, Hasegawa H, Akazawa H, Mizukami M, Nagai R, Komuro I. Reactive oxygen species in mechanical stress-induced cardiac hypertrophy. Biochem Biophys Res Commun 289: 901–907, 2001. Address for reprint requests and other correspondence: G. Loirand, Inserm UMR_S1087, IRS-UN, 8 quai Moncousu, 44007, Nantes, France (e-mail: gervaise.loirand@univ-nantes.fr). 14. Allen WE, Zicha D, Ridley AJ, Jones GE. A role for Cdc42 in macrophage chemotaxis. J Cell Biol 141: 1147–1157, 1998. GRANTS 16. Aoki H, Izumo S, Sadoshima J. Angiotensin II activates RhoA in cardiac myocytes: a critical role of RhoA in angiotensin II-induced premyofibril formation. Circ Res 82: 666 – 676, 1998. This work was supported by French Agence Nationale de la Recherche Grants ANR-08-GENO-040-01, ANR-09-JCJC0115-01, and ANR-11-BSV1-013-01 and by Fondation pour la Recherche Médicale Grant DEQ20090515416. DISCLOSURES No conflicts of interest, financial or otherwise, are declared by the authors. 15. Amin AS, Tan HL, Wilde AA. Cardiac ion channels in health and disease. Heart Rhythm 7: 117–126, 2010. 17. Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, Matsubara Y. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet 37: 1038 –1040, 2005. 18. Aoki Y, Niihori T, Narumi Y, Kure S, Matsubara Y. The RAS/MAPK syndromes: novel roles of the RAS pathway in human genetic disorders. Hum Mutat 29: 992–1006, 2008. 19. Araki T, Chan G, Newbigging S, Morikawa L, Bronson RT, Neel BG. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc Natl Acad Sci USA 106: 4736 – 4741, 2009. 20. Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, Neel BG. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med 10: 849 – 857, 2004. REFERENCES 1. Abdellatif M, MacLellan WR, Schneider MD. p21 Ras as a governor of global gene expression. J Biol Chem 269: 15423–15426, 1994. 2. Adam O, Frost G, Custodis F, Sussman MA, Schafers HJ, Bohm M, Laufs U. Role of Rac1 GTPase activation in atrial fibrillation. J Am Coll Cardiol 50: 359 –367, 2007. 3. Adam O, Lavall D, Theobald K, Hohl M, Grube M, Ameling S, Sussman MA, Rosenkranz S, Kroemer HK, Schafers HJ, Bohm M, Laufs U. Rac1-induced connective tissue 21. Argmann CA, Edwards JY, Sawyez CG, O’Neil CH, Hegele RA, Pickering JG, Huff MW. Regulation of macrophage cholesterol efflux through hydroxymethylglutarylCoA reductase inhibition: a role for RhoA in ABCA1-mediated cholesterol efflux. J Biol Chem 280: 22212–22221, 2005. 22. Axelrad ME, Schwartz DD, Katzenstein JM, Hopkins E, Gripp KW. Neurocognitive, adaptive, and behavioral functioning of individuals with Costello syndrome: a review. Am J Med Genet C Semin Med Genet 157: 115–122, 2011. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1705 LOIRAND, SAUZEAU, AND PACAUD 23. Balakumar P, Singh M. Differential role of rho-kinase in pathological and physiological cardiac hypertrophy in rats. Pharmacology 78: 91–97, 2006. 24. Balzac F, Avolio M, Degani S, Kaverina I, Torti M, Silengo L, Small JV, Retta SF. E-cadherin endocytosis regulates the activity of Rap1: a traffic light GTPase at the crossroads between cadherin and integrin function. J Cell Sci 118: 4765– 4783, 2005. 25. Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, Ma XL, Willette RN, Yue TL. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res 61: 548 –558, 2004. 26. Barker D, Wright E, Nguyen K, Cannon L, Fain P, Goldgar D, Bishop DT, Carey J, Baty B, Kivlin J. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science 236: 1100 –1102, 1987. 27. Baumer Y, Spindler V, Werthmann RC, Bunemann M, Waschke J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J Cell Physiol 220: 716 –726, 2009. 28. Bear MD, Li M, Liu Y, Giel-Moloney MA, Fanburg BL, Toksoz D. The Lbc Rho guanine nucleotide exchange factor alpha-catulin axis functions in serotonin-induced vascular smooth muscle cell mitogenesis and RhoA/ROCK activation. J Biol Chem 285: 32919 – 32926, 2010. 29. Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost 103: 40 –55, 2010. 30. Becknell B, Shen T, Maghraby E, Taya S, Kaibuchi K, Caligiuri MA, Marcucci G. Characterization of leukemia-associated Rho guanine nucleotide exchange factor (LARG) expression during murine development. Cell Tissue Res 314: 361–366, 2003. 31. Beguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, Ozaki N, Geering K, Iwanaga T, Seino S. Regulation of Ca2⫹ channel expression at the cell surface by the small G-protein kir/Gem. Nature 411: 701–706, 2001. 32. Beier I, Dusing R, Vetter H, Schmitz U. Epidermal growth factor stimulates Rac1 and p21-activated kinase in vascular smooth muscle cells. Atherosclerosis 196: 92–97, 2008. 33. Benjamin CW, Jones DA. Platelet-derived growth factor stimulates growth factor receptor binding protein-2 association with Shc in vascular smooth muscle cells. J Biol Chem 269: 30911–30916, 1994. 34. Benter IF, Francis I, Khan I, Cojocel C, Juggi JS, Yousif MH, Canatan H, Alshawaf EH, Akhtar S. Signal transduction involving Ras-GTPase contributes to development of hypertension and end-organ damage in spontaneously hypertensive rats-treated with L-NAME. Pharmacol Res 52: 401– 412, 2005. 35. Benter IF, Juggi JS, Khan I, Akhtar S. Inhibition of Ras-GTPase, but not tyrosine kinases or Ca2⫹/calmodulin-dependent protein kinase II, improves recovery of cardiac function in the globally ischemic heart. Mol Cell Biochem 259: 35– 42, 2004. 36. Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer 11: 775–791, 2011. 37. Best JM, Foell JD, Buss CR, Delisle BP, Balijepalli RC, January CT, Kamp TJ. Small GTPase Rab11b regulates degradation of surface membrane L-type Cav1.2 channels. Am J Physiol Cell Physiol 300: C1023–C1033, 2011. 38. Binder G. Noonan syndrome, the Ras-MAPK signalling pathway and short stature. Horm Res 71 Suppl 2: 64 –70, 2009. 39. Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AD. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 290: L540 –L548, 2006. 40. Birukova AA, Birukov KG, Smurova K, Adyshev D, Kaibuchi K, Alieva I, Garcia JGN, Verin AD. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J 18: 1879 –1890, 2004. 41. Birukova AA, Burdette D, Moldobaeva N, Xing J, Fu P, Birukov KG. Rac GTPase is a hub for protein kinase A and Epac signaling in endothelial barrier protection by cAMP. Microvasc Res 79: 128 –138, 2010. 42. Birukova AA, Malyukova I, Poroyko V, Birukov KG. Paxillin-beta-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol 293: L199 –L211, 2007. 1706 43. Birukova AA, Tian Y, Dubrovskyi O, Zebda N, Sarich N, Tian X, Wang Y, Birukov KG. VE-cadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. J Cell Physiol 227: 3405–3416, 2012. 44. Birukova AA, Zagranichnaya T, Alekseeva E, Bokoch GM, Birukov KG. Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J Cell Physiol 215: 715–724, 2008. 45. Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE2 and PGI2 promote endothelial barrier enhancement via PKAand Epac1/Rap1-dependent Rac activation. Exp Cell Res 313: 2504 –2520, 2007. 46. Bischoff FR, Ponstingl H. Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature 354: 80 – 82, 1991. 47. Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J 348: 241–255, 2000. 48. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KY, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D’Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu X, Nathanson KL, Nolop K. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467: 596 –599, 2010. 49. Bond M, Wu YJ, Sala-Newby GB, Newby AC. Rho GTPase, Rac1, regulates Skp2 levels, vascular smooth muscle cell proliferation, and intima formation in vitro and in vivo. Cardiovasc Res 80: 290 –298, 2008. 50. Bonetti PO, Lerman LO, Napoli C, Lerman A. Statin effects beyond lipid lowering: are they clinically relevant? Eur Heart J 24: 225–248, 2003. 51. Borikova AL, Dibble CF, Sciaky N, Welch CM, Abell AN, Bencharit S, Johnson GL. Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J Biol Chem 285: 11760 –11764, 2010. 52. Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci 31: 680 – 686, 2006. 53. Boulday G, Blecon A, Petit N, Chareyre F, Garcia LA, Niwa-Kawakita M, Giovannini M, Tournier-Lasserve E. Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations. Dis Model Mech 2: 168 –177, 2009. 54. Boyer SB, Slesinger PA, Jones SV. Regulation of Kir2.1 channels by the Rho-GTPase, Rac1. J Cell Physiol 218: 385–393, 2009. 55. Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crestderived tissues. Genes Dev 8: 1019 –1029, 1994. 56. Broman MT, Kouklis P, Gao X, Ramchandran R, Neamu RF, Minshall RD, Malik AB. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ Res 98: 73– 80, 2006. 57. Broman MT, Mehta D, Malik AB. Cdc42 regulates the restoration of endothelial adherens junctions and permeability. Trends Cardiovasc Med 17: 151–156, 2007. 58. Brown JH, Del Re DP, Sussman MA. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ Res 98: 730 –742, 2006. 59. Buhrman G, Holzapfel G, Fetics S, Mattos C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc Natl Acad Sci USA 107: 4931– 4936, 2010. 60. Burch M, Sharland M, Shinebourne E, Smith G, Patton M, McKenna W. Cardiologic abnormalities in Noonan syndrome: phenotypic diagnosis and echocardiographic assessment of 118 patients. J Am Coll Cardiol 22: 1189 –1192, 1993. 61. Buscemi N, Foster DB, Neverova I, Van, Eyk JE. p21-activated kinase increases the calcium sensitivity of rat triton-skinned cardiac muscle fiber bundles via a mechanism potentially involving novel phosphorylation of troponin I. Circ Res 91: 509 –516, 2002. 62. Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29: 356 –370, 2007. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 63. Butterworth MB, Edinger RS, Frizzell RA, Johnson JP. Regulation of the epithelial sodium channel by membrane trafficking. Am J Physiol Renal Physiol 296: F10 –F24, 2009. 82. Cicha I, Schneiderhan-Marra N, Yilmaz A, Garlichs CD, Goppelt-Struebe M. Monitoring the cellular effects of HMG-CoA reductase inhibitors in vitro and ex vivo. Arterioscler Thromb Vasc Biol 24: 2046 –2050, 2004. 64. Butterworth MB, Edinger RS, Silvis MR, Gallo LI, Liang X, Apodaca G, Fizzell RA, Johnson JP. Rab11b regulates the trafficking and recycling of the epithelial sodium channel (ENaC). Am J Physiol Renal Physiol 302: F581–F590, 2012. 83. Clements RT, Minnear FL, Singer HA, Keller RS, Vincent PA. RhoA and Rho-kinase dependent and independent signals mediate TGF--induced pulmonary endothelial cytoskeletal reorganization and permeability. Am J Physiol Lung Cell Mol Physiol 288: L294 –L306, 2005. 65. Cachero TG, Morielli AD, Peralta EG. The small GTP-binding protein RhoA regulates a delayed rectifier potassium channel. Cell 93: 1077–1085, 1998. 66. Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med 13: 613– 618, 2007. 67. Cario-Toumaniantz C, Ferland-McCollough D, Chadeuf G, Toumaniantz G, Rodriguez M, Galizzi J, Lockhart B, Bril A, Scalbert E, Loirand G, Pacaud P. RhoA guanine exchange factor expression profile in arteries: evidence for a Rho kinase-dependent negative feedback in angiotensin II-dependent hypertension. Am J Physiol Cell Physiol 302: C1394 –C1404, 2012. 68. Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, Digilio C, Palleschi A, Pizzuti A, Grammatico P, Zampino G, Dallapiccola B, Gelb BD, Tartaglia M. Germline missense mutations affecting KRAS Isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet 79: 129 –135, 2006. 69. Cernuda-Morollon E, Ridley AJ. Rho GTPases and leukocyte adhesion receptor expression and function in endothelial cells. Circ Res 98: 757–767, 2006. 70. Chamorro-Jorganes A, Grande MT, Herranz B, Jerkic M, Griera M, Gonzalez-Nunez M, Santos E, Rodriguez-Puyol D, Lopez-Novoa JM, Rodriguez-Puyol M. Targeted genomic disruption of h-ras induces hypotension through a NO-cGMP-PKG pathwaydependent mechanism. Hypertension 56: 484 – 489, 2010. 71. Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, Ismail SA, Hedberg C, Hanzal-Bayer M, Venkitaraman AR, Wittinghofer A, Bastiaens PI. The GDIlike solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol 14: 148 –158, 2011. 72. Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, Schwartz RJ. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci USA 103: 14495– 14500, 2006. 73. Chang L, Zhang J, Tseng YH, Xie CQ, Ilany J, Bruning JC, Sun Z, Zhu X, Cui T, Youker KA, Yang Q, Day SM, Kahn CR, Chen YE. Rad GTPase deficiency leads to cardiac hypertrophy. Circulation 116: 2976 –2983, 2007. 74. Chang Y, Ceacareanu B, Dixit M, Sreejayan N, Hassid A. Nitric oxide-induced motility in aortic smooth muscle cells: role of protein tyrosine phosphatase SHP-2 and GTPbinding protein Rho. Circ Res 91: 390 –397, 2002. 75. Chava KR, Tauseef M, Sharma T, Mehta D. Cyclic AMP response element-binding protein prevents endothelial permeability increase through transcriptional controlling p190RhoGAP expression. Blood 119: 308 –319, 2012. 76. Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc Natl Acad Sci USA 98: 7783–7788, 2001. 77. Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol 6: 872– 883, 2004. 78. Cherfils J, Zeghouf M. Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 93: 269 –309, 2013. 79. Chesneau L, Dambournet D, Machicoane M, Kouranti I, Fukuda M, Goud B, Echard A. An ARF6/Rab35 GTPase cascade for endocytic recycling and successful cytokinesis. Curr Biol 22: 147–153, 2012. 84. Clerk A, Pham FH, Fuller SJ, Sahai E, Aktories K, Marais R, Marshall C, Sugden PH. Regulation of mitogen-activated protein kinases in cardiac myocytes through the small G protein Rac1. Mol Cell Biol 21: 1173–1184, 2001. 85. Clerk A, Sugden PH. Ras: the stress and the strain. J Mol Cell Cardiol 41: 595– 600, 2006. 86. Clerk A, Sugden PH. Small guanine nucleotide-binding proteins and myocardial hypertrophy. Circ Res 86: 1019 –1023, 2000. 87. Cohen LH, Pieterman E, van Leeuwen RE, Overhand M, Burm BE, van der Marel GA, van Boom JH. Inhibitors of prenylation of Ras and other G-proteins and their application as therapeutics. Biochem Pharmacol 60: 1061–1068, 2000. 88. Cole AL, Subbanagounder G, Mukhopadhyay S, Berliner JA, Vora DK. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a cAMP-dependent R-Ras/PI3-kinase pathway. Arterioscler Thromb Vasc Biol 23: 1384 –1390, 2003. 89. Correll RN, Pang C, Finlin BS, Dailey AM, Satin J, Andres DA. Plasma membrane targeting is essential for Rem-mediated Ca2⫹ channel inhibition. J Biol Chem 282: 28431–28440, 2007. 90. Costello JM. A new syndrome. NZ Med J 74: 374, 1971. 91. Costello JM. A new syndrome: mental subnormality and nasal papillomata. Aust Paediatr J 13: 114 –118, 1977. 92. Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105: 1950 –1955, 2005. 93. Custodis F, Eberl M, Kilter H, Bohm M, Laufs U. Association of RhoGDIalpha with Rac1 GTPase mediates free radical production during myocardial hypertrophy. Cardiovasc Res 71: 342–351, 2006. 94. D’Amico G, Jones DT, Nye E, Sapienza K, Ramjuan AR, Reynolds LE, Robinson SD, Kostourou V, Martinez D, Aubyn D, Grose R, Thomas GJ, Spencer-Dene B, Zicha D, Davies D, Tybulewicz V, Hodivala-Dilke KM. Regulation of lymphatic-blood vessel separation by endothelial Rac1. Development 136: 4043– 4053, 2009. 95. Dance M, Montagner A, Salles JP, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal 20: 453– 459, 2008. 96. Davis GE, Stratman AN, Sacharidou A, Koh W. Molecular basis for endothelial lumen formation and tubulogenesis during vasculogenesis and angiogenic sprouting. Int Rev Cell Mol Biol 288: 101–165, 2011. 97. DeFeo D, Gonda MA, Young HA, Chang EH, Lowy DR, Scolnick EM, Ellis RW. Analysis of two divergent rat genomic clones homologous to the transforming gene of Harvey murine sarcoma virus. Proc Natl Acad Sci USA 78: 3328 –3332, 1981. 98. Del Re DP, Matsuda T, Zhai P, Gao S, Clark GJ, Van Der Weyden L, Sadoshima J. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J Clin Invest 120: 3555–3567, 2010. 99. Del Re DP, Miyamoto S, Brown JH. Focal adhesion kinase as a RhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J Biol Chem 283: 35622–35629, 2008. 80. Chrzanowska-Wodnicka M. Regulation of angiogenesis by a small GTPase Rap1. Vasc Pharmacol 53: 1–10, 2010. 100. Del Re DP, Miyamoto S, Brown JH. RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J Biol Chem 282: 8069 – 8078, 2007. 81. Chrzanowska-Wodnicka M, Kraus AE, Gale D, White GC 2nd, Vansluys J. Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice. Blood 111: 2647–2656, 2008. 101. Demiryurek S, Kara AF, Celik A, Babul A, Tarakcioglu M, Demiryurek AT. Effects of fasudil, a Rho-kinase inhibitor, on myocardial preconditioning in anesthetized rats. Eur J Pharmacol 527: 129 –140, 2005. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1707 LOIRAND, SAUZEAU, AND PACAUD 102. Demiryurek S, Kara AF, Celik A, Tarakcioglu M, Bagci C, Demiryurek AT. Effects of Y-27632, a selective Rho-kinase inhibitor, on myocardial preconditioning in anesthetized rats. Biochem Pharmacol 69: 49 –58, 2005. 120. Fox R, Nhan TQ, Law GL, Morris DR, Liles WC, Schwartz SM. PSGL-1 and mTOR regulate translation of ROCK-1 and physiological functions of macrophages. EMBO J 26: 505–515, 2007. 103. Denayer E, de Ravel T, Legius E. Clinical and molecular aspects of RAS related disorders. J Med Genet 45: 695–703, 2008. 121. Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation 109: 1580 –1589, 2004. 104. Deng J, Feng E, Ma S, Zhang Y, Liu X, Li H, Huang H, Zhu J, Zhu W, Shen X, Miao L, Liu H, Jiang H, Li J. Design and synthesis of small molecule RhoA inhibitors: a new promising therapy for cardiovascular diseases? J Med Chem 54: 4508 – 4522, 2011. 122. Fu M, Zhang J, Tseng YH, Cui T, Zhu X, Xiao Y, Mou Y, De Leon H, Chang MM, Hamamori Y, Kahn CR, Chen YE. Rad GTPase attenuates vascular lesion formation by inhibition of vascular smooth muscle cell migration. Circulation 111: 1071–1077, 2005. 105. Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, Dallapiccola B. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet 71: 389 –394, 2002. 123. Fujita T. Mineralocorticoid receptors, salt-sensitive hypertension, and metabolic syndrome. Hypertension 55: 813– 818, 2010. 106. Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437: 574 –578, 2005. 107. Dong LH, Wen JK, Liu G, McNutt MA, Miao SB, Gao R, Zheng B, Zhang H, Han M. Blockade of the Ras-extracellular signal-regulated kinase 1/2 pathway is involved in smooth muscle 22 alpha-mediated suppression of vascular smooth muscle cell proliferation and neointima hyperplasia. Arterioscler Thromb Vasc Biol 30: 683– 691, 2010. 108. Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, Vanwijck R, Vikkula M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet 73: 1240 –1249, 2003. 109. Erdmann J, Grosshennig A, Braund PS, Konig IR, Hengstenberg C, Hall AS, LinselNitschke P, Kathiresan S, Wright B, Tregouet DA, Cambien F, Bruse P, Aherrahrou Z, Wagner AK, Stark K, Schwartz SM, Salomaa V, Elosua R, Melander O, Voight BF, O’Donnell CJ, Peltonen L, Siscovick DS, Altshuler D, Merlini PA, Peyvandi F, Bernardinelli L, Ardissino D, Schillert A, Blankenberg S, Zeller T, Wild P, Schwarz DF, Tiret L, Perret C, Schreiber S, El Mokhtari NE, Schafer A, Marz W, Renner W, Bugert P, Kluter H, Schrezenmeir J, Rubin D, Ball SG, Balmforth AJ, Wichmann HE, Meitinger T, Fischer M, Meisinger C, Baumert J, Peters A, Ouwehand WH, Deloukas P, Thompson JR, Ziegler A, Samani NJ, Schunkert H. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet 41: 280 –282, 2009. 110. Ernkvist M, Luna Persson N, Audebert S, Lecine P, Sinha I, Liu M, Schlueter M, Horowitz A, Aase K, Weide T, Borg JP, Majumdar A, Holmgren L. The Amot/Patj/Syx signaling complex spatially controls RhoA GTPase activity in migrating endothelial cells. Blood 113: 244 –253, 2009. 111. Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem 273: 21867–21874, 1998. 112. Essler M, Staddon JM, Weber PC, Aepfelbacher M. Cyclic AMP blocks bacterial lipopolysaccharide-induced myosin light chain phosphorylation in endothelial cells through inhibition of Rho/Rho kinase signaling. J Immunol 164: 6543– 6549, 2000. 113. Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA. HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A 140: 8 –16, 2006. 114. Eto Y, Shimokawa H, Hiroki J, Morishige K, Kandabashi T, Matsumoto Y, Amano M, Hoshijima M, Kaibuchi K, Takeshita A. Gene transfer of dominant negative Rho kinase suppresses neointimal formation after balloon injury in pigs. Am J Physiol Heart Circ Physiol 278: H1744 –H1750, 2000. 124. Fujiwara H, Matsuda M, Fujiwara Y, Ishida M, Kawamura A, Takemura G, Kida M, Uegaito T, Tanaka M, Horike K. Infarct size and the protection of ischemic myocardium in pig, dog and human. Jpn Circ J 53: 1092–1097, 1989. 125. Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N. Cyclic AMP potentiates vascular endothelial cadherinmediated cell-cell contact to enhance endothelial barrier function through an EpacRap1 signaling pathway. Mol Cell Biol 25: 136 –146, 2005. 126. Fukumoto Y, Kaibuchi K, Hori Y, Fujioka H, Araki S, Ueda T, Kikuchi A, Takai Y. Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene 5: 1321–1328, 1990. 127. Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, Abe K, Takeshita A, Shimokawa H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 91: 391–392, 2005. 128. Fuller SJ, Finn SG, Downward J, Sugden PH. Stimulation of gene expression in neonatal rat ventricular myocytes by Ras is mediated by Ral guanine nucleotide dissociation stimulator (Ral.GDS) and phosphatidylinositol 3-kinase in addition to Raf. Biochem J 335: 241–246, 1998. 129. Funakoshi Y, Ichiki T, Shimokawa H, Egashira K, Takeda K, Kaibuchi K, Takeya M, Yoshimura T, Takeshita A. Rho-kinase mediates angiotensin II-induced monocyte chemoattractant protein-1 expression in rat vascular smooth muscle cells. Hypertension 38: 100 –104, 2001. 130. Galle J, Mameghani A, Bolz SS, Gambaryan S, Gorg M, Quaschning T, Raff U, Barth H, Seibold S, Wanner C, Pohl U. Oxidized LDL and its compound lysophosphatidylcholine potentiate AngII-induced vasoconstriction by stimulation of RhoA. J Am Soc Nephrol 14: 1471–1479, 2003. 131. Gambardella L, Hemberger M, Hughes B, Zudaire E, Andrews S, Vermeren S. PI3K signaling through the dual GTPase-activating protein ARAP3 is essential for developmental angiogenesis. Sci Signal 3: ra76, 2010. 132. Garcia-Mata R, Boulter E, Burridge K. The “invisible hand”: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol 12: 493–504, 2011. 133. Garnaas MK, Moodie KL, Liu ML, Samant GV, Li K, Marx R, Baraban JM, Horowitz A, Ramchandran R. Syx, a RhoA guanine exchange factor, is essential for angiogenesis in vivo. Circ Res 103: 710 –716, 2008. 134. Gentile S, Darden T, Erxleben C, Romeo C, Russo A, Martin N, Rossie S, Armstrong DL. Rac GTPase signaling through the PP5 protein phosphatase. Proc Natl Acad Sci USA 103: 5202–5206, 2006. 115. Eva A, Vecchio G, Rao CD, Tronick SR, Aaronson SA. The predicted DBL oncogene product defines a distinct class of transforming proteins. Proc Natl Acad Sci USA 85: 2061–2065, 1988. 135. George J, Afek A, Keren P, Herz I, Goldberg I, Haklai R, Kloog Y, Keren G. Functional inhibition of Ras by S-trans,trans-farnesyl thiosalicylic acid attenuates atherosclerosis in apolipoprotein E knockout mice. Circulation 105: 2416 –2422, 2002. 116. Fehrenbacher N, Bar-Sagi D, Philips M. Ras/MAPK signaling from endomembranes. Mol Oncol 3: 297–307, 2009. 136. George J, Sack J, Barshack I, Keren P, Goldberg I, Haklai R, Elad-Sfadia G, Kloog Y, Keren G. Inhibition of intimal thickening in the rat carotid artery injury model by a nontoxic Ras inhibitor. Arterioscler Thromb Vasc Biol 24: 363–368, 2004. 117. Filipeanu CM, Zhou F, Wu G. Analysis of Rab1 function in cardiomyocyte growth. Methods Enzymol 438: 217–226, 2008. 118. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363: 809 – 819, 2010. 119. Foster DB, Shen LH, Kelly J, Thibault P, Van Eyk JE, Mak AS. Phosphorylation of caldesmon by p21-activated kinase. Implications for the Ca2⫹ sensitivity of smooth muscle contraction. J Biol Chem 275: 1959 –1965, 2000. 1708 137. Giovannardi S, Forlani G, Balestrini M, Bossi E, Tonini R, Sturani E, Peres A, Zippel R. Modulation of the inward rectifier potassium channel IRK1 by the Ras signaling pathway. J Biol Chem 277: 12158 –12163, 2002. 138. Gitler AD, Zhu Y, Ismat FA, Lu MM, Yamauchi Y, Parada LF, Epstein JA. Nf1 has an essential role in endothelial cells. Nat Genet 33: 75–79, 2003. 139. Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol 179: 247–254, 2007. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 140. Godin CM, Ferreira LT, Dale LB, Gros R, Cregan SP, Ferguson SS. The small GTPase Ral couples the angiotensin II type 1 receptor to the activation of phospholipase C-delta 1. Mol Pharmacol 77: 388 –395, 2010. 160. He Q, Harding P, LaPointe MC. PKA, Rap1, ERK1/2, and p90RSK mediate PGE2 and EP4 signaling in neonatal ventricular myocytes. Am J Physiol Heart Circ Physiol 298: H136 –H143, 2010. 141. Goorden SM, Hoogeveen-Westerveld M, Cheng C, van Woerden GM, Mozaffari M, Post L, Duckers HJ, Nellist M, Elgersma Y. Rheb is essential for murine development. Mol Cell Biol 31: 1672–1678, 2011. 161. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690 –701, 2008. 142. Gottshall KR, Hunter JJ, Tanaka N, Dalton N, Becker KD, Ross J Jr, Chien KR. Rasdependent pathways induce obstructive hypertrophy in echo-selected transgenic mice. Proc Natl Acad Sci USA 94: 4710 – 4715, 1997. 143. Griffiths GS, Grundl M, Allen JS 3rd, Matter ML. R-Ras interacts with filamin a to maintain endothelial barrier function. J Cell Physiol 226: 2287–2296, 2011. 144. Grinnell KL, Casserly B, Harrington EO. Role of protein tyrosine phosphatase SHP2 in barrier function of pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol 298: L361–L370, 2010. 145. Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, Henrion D, Scalbert E, Bril A, Torres RM, Offermanns S, Pacaud P, Loirand G. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med 16: 183–190, 2010. 146. Guo FF, Kumahara E, Saffen D. A CalDAG-GEFI/Rap1/B-Raf cassette couples M(1) muscarinic acetylcholine receptors to the activation of ERK1/2. J Biol Chem 276: 25568 –25581, 2001. 147. Guttler T, Gorlich D. Ran-dependent nuclear export mediators: a structural perspective. EMBO J 30: 3457–3474, 2011. 148. Haidari M, Zhang W, Chen Z, Ganjehei L, Warier N, Vanderslice P, Dixon R. Myosin light chain phosphorylation facilitates monocyte transendothelial migration by dissociating endothelial adherens junctions. Cardiovasc Res 92: 456 – 465, 2011. 149. Hall A, Marshall CJ, Spurr NK, Weiss RA. Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nature 303: 396 – 400, 1983. 150. Hamid SA, Bower HS, Baxter GF. Rho kinase activation plays a major role as a mediator of irreversible injury in reperfused myocardium. Am J Physiol Heart Circ Physiol 292: H2598 –H2606, 2007. 151. Han YJ, Hu WY, Chernaya O, Antic N, Gu L, Gupta M, Piano M, de Lanerolle P. Increased myosin light chain kinase expression in hypertension: regulation by serum response factor via an insertion mutation in the promoter. Mol Biol Cell 17: 4039 – 4050, 2006. 152. Han YJ, Hu WY, Piano M, de Lanerolle P. Regulation of myosin light chain kinase expression by angiotensin II in hypertension. Am J Hypertens 21: 860 – 865, 2008. 153. Harja E, Bucciarelli LG, Lu Y, Stern DM, Zou YS, Schmidt AM, Yan SF. Early growth response-1 promotes atherogenesis: mice deficient in early growth response-1 and apolipoprotein E display decreased atherosclerosis and vascular inflammation. Circ Res 94: 333–339, 2004. 154. Harrington EO, Shannon CJ, Morin N, Rowlett H, Murphy C, Lu Q. PKCdelta regulates endothelial basal barrier function through modulation of RhoA GTPase activity. Exp Cell Res 308: 407– 421, 2005. 155. Harris IS, Zhang S, Treskov I, Kovacs A, Weinheimer C, Muslin AJ. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation 110: 718 –723, 2004. 162. Heasman SJ, Ridley AJ. Multiple roles for RhoA during T cell transendothelial migration. Small GTPases 1: 174 –179, 2010. 163. Henderson DJ, Ybot-Gonzalez P, Copp AJ. RhoB is expressed in migrating neural crest and endocardial cushions of the developing mouse embryo. Mech Dev 95: 211– 214, 2000. 164. Henkemeyer M, Rossi DJ, Holmyard DP, Puri MC, Mbamalu G, Harpal K, Shih TS, Jacks T, Pawson T. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 377: 695–701, 1995. 165. Heusch G, Skyschally A, Schulz R. The in-situ pig heart with regional ischemia/reperfusion: ready for translation. J Mol Cell Cardiol 50: 951–963, 2011. 166. Higashi M, Shimokawa H, Hattori T, Hiroki J, Mukai Y, Morikawa K, Ichiki T, Takahashi S, Takeshita A. Long-term inhibition of Rho-kinase suppresses angiotensin IIinduced cardiovascular hypertrophy in rats in vivo: effect on endothelial NAD(P)H oxidase system. Circ Res 93: 767–775, 2003. 167. Higuchi Y, Otsu K, Nishida K, Hirotani S, Nakayama H, Yamaguchi O, Hikoso S, Kashiwase K, Takeda T, Watanabe T, Mano T, Matsumura Y, Ueno H, Hori M. The small GTP-binding protein Rac1 induces cardiac myocyte hypertrophy through the activation of apoptosis signal-regulating kinase 1 and nuclear factor-kappa B. J Biol Chem 278: 20770 –20777, 2003. 168. Hilal-Dandan R, Means CK, Gustafsson AB, Morissette MR, Adams JW, Brunton LL, Heller Brown J. Lysophosphatidic acid induces hypertrophy of neonatal cardiac myocytes via activation of Gi and Rho. J Mol Cell Cardiol 36: 481– 493, 2004. 169. Hilgers RH, Todd J Jr, Webb RC. Increased PDZ-RhoGEF/RhoA/Rho kinase signaling in small mesenteric arteries of angiotensin II-induced hypertensive rats. J Hypertens 25: 1687–1697, 2007. 170. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med 358: 1370 –1380, 2008. 171. Hines WA, Thorburn A. Ras and rho are required for galphaq-induced hypertrophic gene expression in neonatal rat cardiac myocytes. J Mol Cell Cardiol 30: 485– 494, 1998. 172. Hingtgen SD, Tian X, Yang J, Dunlay SM, Peek AS, Wu Y, Sharma RV, Engelhardt JF, Davisson RL. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics 26: 180 –191, 2006. 173. Hinoki A, Kimura K, Higuchi S, Eguchi K, Takaguri A, Ishimaru K, Frank GD, Gerthoffer WT, Sommerville LJ, Autieri MV, Eguchi S. p21-activated kinase 1 participates in vascular remodeling in vitro and in vivo. Hypertension 55: 161–165, 2010. 174. Hirano K, Ikegami C, Zhang Z. Contribution of Cdc42 to cholesterol efflux in fibroblasts from Tangier disease and Werner syndrome. Methods Enzymol 439: 159 –169, 2008. 175. Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol 22: 6A-159 –13A, 1993. 156. Harris KP, Littleton JT. Vesicle trafficking: a Rab family profile. Curr Biol 21: R841– 843, 2011. 176. Ho PD, Fan JS, Hayes NL, Saada N, Palade PT, Glembotski CC, McDonough PM. Ras reduces L-type calcium channel current in cardiac myocytes. Corrective effects of L-channels and SERCA2 on [Ca2⫹]i regulation and cell morphology. Circ Res 88: 63– 69, 2001. 157. Hassanain HH, Gregg D, Marcelo ML, Zweier JL, Souza HP, Selvakumar B, Ma Q, Moustafa-Bayoumi M, Binkley PF, Flavahan NA, Morris M, Dong C, GoldschmidtClermont PJ. Hypertension caused by transgenic overexpression of Rac1. Antioxid Redox Signal 9: 91–100, 2007. 177. Ho PD, Zechner DK, He H, Dillmann WH, Glembotski CC, McDonough PM. The Raf-MEK-ERK cascade represents a common pathway for alteration of intracellular calcium by Ras and protein kinase C in cardiac myocytes. J Biol Chem 273: 21730 – 21735, 1998. 158. Hattori T, Shimokawa H, Higashi M, Hiroki J, Mukai Y, Tsutsui H, Kaibuchi K, Takeshita A. Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation 109: 2234 –2239, 2004. 178. Holinstat M, Knezevic N, Broman M, Samarel A, Malik A, Mehta D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoAGAP: role in regulation of endothelial permeability. J Biol Chem 281: 2296 –2305, 2006. 159. Hayashi H, Szaszi K, Coady-Osberg N, Furuya W, Bretscher AP, Orlowski J, Grinstein S. Inhibition and redistribution of NHE3, the apical Na⫹/H⫹ exchanger, by Clostridium difficile toxin B. J Gen Physiol 123: 491–504, 2004. 179. Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. Protein kinase Calphainduced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem 278: 28793–28798, 2003. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1709 LOIRAND, SAUZEAU, AND PACAUD 180. Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res 98: 453– 462, 2006. 200. Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med 12: 1–11, 2010. 181. Hoshijima M, Sah VP, Wang Y, Chien KR, Brown JH. The low molecular weight GTPase Rho regulates myofibril formation and organization in neonatal rat ventricular myocytes. Involvement of Rho kinase. J Biol Chem 273: 7725–7730, 1998. 201. Jiang SY, Ramachandran S. Comparative and evolutionary analysis of genes encoding small GTPases and their activating proteins in eukaryotic genomes. Physiol Genomics 24: 235–251, 2006. 182. Hu Y, Bock G, Wick G, Xu Q. Activation of PDGF receptor alpha in vascular smooth muscle cells by mechanical stress. FASEB J 12: 1135–1142, 1998. 202. Jin L, Ying Z, Hilgers RH, Yin J, Zhao X, Imig JD, Webb RC. Increased RhoA/Rhokinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. J Pharmacol Exp Ther 318: 288 –295, 2006. 183. Huang M, Satchell L, Duhadaway JB, Prendergast GC, Laury-Kleintop LD. RhoB links PDGF signaling to cell migration by coordinating activation and localization of Cdc42 and Rac. J Cell Biochem 112: 1572–1584, 2011. 203. Jones SV. Role of the small GTPase Rho in modulation of the inwardly rectifying potassium channel Kir2.1. Mol Pharmacol 64: 987–993, 2003. 184. Hunter JJ, Tanaka N, Rockman HA, Ross J Jr, Chien KR. Ventricular expression of a MLC-2v-ras fusion gene induces cardiac hypertrophy and selective diastolic dysfunction in transgenic mice. J Biol Chem 270: 23173–23178, 1995. 204. Kaarbo M, Crane DI, Murrell WG. RhoA is highly up-regulated in the process of early heart development of the chick and important for normal embryogenesis. Dev Dyn 227: 35– 47, 2003. 185. Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev 91: 119 –149, 2011. 205. Kai H, Muraishi A, Sugiu Y, Nishi H, Seki Y, Kuwahara F, Kimura A, Kato H, Imaizumi T. Expression of proto-oncogenes and gene mutation of sarcomeric proteins in patients with hypertrophic cardiomyopathy. Circ Res 83: 594 – 601, 1998. 186. Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, King MR, Schaffer CB, Reinhart-King CA. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci Transl Med 3: 112–122, 2011. 187. Ichiba T, Kuraishi Y, Sakai O, Nagata S, Groffen J, Kurata T, Hattori S, Matsuda M. Enhancement of guanine-nucleotide exchange activity of C3G for Rap1 by the expression of Crk, CrkL, and Grb2. J Biol Chem 272: 22215–22220, 1997. 188. Indolfi C, Avvedimento EV, Rapacciuolo A, Di Lorenzo E, Esposito G, Stabile E, Feliciello A, Mele E, Giuliano P, Condorelli G. Inhibition of cellular ras prevents smooth muscle cell proliferation after vascular injury in vivo. Nat Med 1: 541–545, 1995. 189. Ishimaru K, Ueno H, Kagitani S, Takabayashi D, Takata M, Inoue H. Fasudil attenuates myocardial fibrosis in association with inhibition of monocyte/macrophage infiltration in the heart of DOCA/salt hypertensive rats. J Cardiovasc Pharmacol 50: 187–194, 2007. 190. Ismat FA, Xu J, Lu MM, Epstein JA. The neurofibromin GAP-related domain rescues endothelial but not neural crest development in Nf1 mice. J Clin Invest 116: 2378 – 2384, 2006. 191. Ito K, Hirooka Y, Sakai K, Kishi T, Kaibuchi K, Shimokawa H, Takeshita A. Rho/Rhokinase pathway in brain stem contributes to blood pressure regulation via sympathetic nervous system: possible involvement in neural mechanisms of hypertension. Circ Res 92: 1337–1343, 2003. 192. Ito M, Adachi T, Pimentel DR, Ido Y, Colucci WS. Statins inhibit beta-adrenergic receptor-stimulated apoptosis in adult rat ventricular myocytes via a Rac1-dependent mechanism. Circulation 110: 412– 418, 2004. 193. Ito T, Ikeda U, Shimpo M, Ohki R, Takahashi M, Yamamoto K, Shimada K. HMG-CoA reductase inhibitors reduce interleukin-6 synthesis in human vascular smooth muscle cells. Cardiovasc Drugs Ther 16: 121–126, 2002. 194. Itoh S, Ding B, Bains CP, Wang N, Takeishi Y, Jalili T, King GL, Walsh RA, Yan C, Abe J. Role of p90 ribosomal S6 kinase (p90RSK) in reactive oxygen species and protein kinase C beta (PKC-beta)-mediated cardiac troponin I phosphorylation. J Biol Chem 280: 24135–24142, 2005. 195. Itzen A, Goody RS. GTPases involved in vesicular trafficking: structures and mechanisms. Semin Cell Dev Biol 22: 48 –56, 2011. 196. Iwai M, Chen R, Li Z, Shiuchi T, Suzuki J, Ide A, Tsuda M, Okumura M, Min LJ, Mogi M, Horiuchi M. Deletion of angiotensin II type 2 receptor exaggerated atherosclerosis in apolipoprotein E-null mice. Circulation 112: 1636 –1643, 2005. 197. Iwashita S, Kobayashi M, Kubo Y, Hinohara Y, Sezaki M, Nakamura K, Suzuki-Migishima R, Yokoyama M, Sato S, Fukuda M, Ohba M, Kato C, Adachi E, Song SY. Versatile roles of R-Ras GAP in neurite formation of PC12 cells and embryonic vascular development. J Biol Chem 282: 3413–3417, 2007. 198. Izumo S, Nadal-Ginard B, Mahdavi V. Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc Natl Acad Sci USA 85: 339 –343, 1988. 199. Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet 7: 353– 361, 1994. 1710 206. Kajimoto K, Shao D, Takagi H, Maceri G, Zablocki D, Mukai H, Ono Y, Sadoshima J. Hypotonic swelling-induced activation of PKN1 mediates cell survival in cardiac myocytes. Am J Physiol Heart Circ Physiol 300: H191–H200, 2011. 207. Kamei M, Saunders WB, Bayless KJ, Dye L, Davis GE, Weinstein BM. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature 442: 453– 456, 2006. 208. Kamijo H, Matsumura Y, Thumkeo D, Koike S, Masu M, Shimizu Y, Ishizaki T, Narumiya S. Impaired vascular remodeling in the yolk sac of embryos deficient in ROCK-I and ROCK-II. Genes Cells 16: 1012–1021, 2011. 209. Kamiyama M, Utsunomiya K, Taniguchi K, Yokota T, Kurata H, Tajima N, Kondo K. Contribution of Rho A and Rho kinase to platelet-derived growth factor-BB-induced proliferation of vascular smooth muscle cells. J Atheroscler Thromb 10: 117–123, 2003. 210. Kanda T, Hayashi K, Wakino S, Homma K, Yoshioka K, Hasegawa K, Sugano N, Tatematsu S, Takamatsu I, Mitsuhashi T, Saruta T. Role of Rho-kinase and p27 in angiotensin II-induced vascular injury. Hypertension 45: 724 –729, 2005. 211. Kandabashi T, Shimokawa H, Miyata K, Kunihiro I, Kawano Y, Fukata Y, Higo T, Egashira K, Takahashi S, Kaibuchi K, Takeshita A. Inhibition of myosin phosphatase by upregulated rho-kinase plays a key role for coronary artery spasm in a porcine model with interleukin-1beta. Circulation 101: 1319 –1323, 2000. 212. Kandabashi T, Shimokawa H, Mukai Y, Matoba T, Kunihiro I, Morikawa K, Ito M, Takahashi S, Kaibuchi K, Takeshita A. Involvement of rho-kinase in agonists-induced contractions of arteriosclerotic human arteries. Arterioscler Thromb Vasc Biol 22: 243– 248, 2002. 213. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 9: 517–531, 2008. 214. Karpushev AV, Ilatovskaya DV, Pavlov TS, Negulyaev YA, Staruschenko A. Intact cytoskeleton is required for small G protein dependent activation of the epithelial Na⫹ channel. PLoS One 5: e8827, 2010. 215. Karpushev AV, Levchenko V, Ilatovskaya DV, Pavlov TS, Staruschenko A. Novel role of Rac1/WAVE signaling mechanism in regulation of the epithelial Na⫹ channel. Hypertension 57: 996 –1002, 2011. 216. Kataoka C, Egashira K, Inoue S, Takemoto M, Ni W, Koyanagi M, Kitamoto S, Usui M, Kaibuchi K, Shimokawa H, Takeshita A. Important role of Rho-kinase in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide synthesis in rats. Hypertension 39: 245–250, 2002. 217. Kavamura MI, Pomponi MG, Zollino M, Lecce R, Murdolo M, Brunoni D, Alchorne MM, Opitz JM, Neri G. PTPN11 mutations are not responsible for the Cardiofaciocutaneous (CFC) syndrome. Eur J Hum Genet 11: 64 – 68, 2003. 218. Kawai M, Kawashima S, Sakoda T, Toh R, Kikuchi A, Yamauchi-Takihara K, Kunisada K, Yokoyama M. Ral GDP dissociation stimulator and Ral GTPase are involved in myocardial hypertrophy. Hypertension 41: 956 –962, 2003. 219. Ke Y, Lei M, Collins TP, Rakovic S, Mattick PA, Yamasaki M, Brodie MS, Terrar DA, Solaro RJ. Regulation of L-type calcium channel and delayed rectifier potassium channel activity by p21-activated kinase-1 in guinea pig sinoatrial node pacemaker cells. Circ Res 100: 1317–1327, 2007. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 220. Ke Y, Lei M, Solaro RJ. Regulation of cardiac excitation and contraction by p21 activated kinase-1. Prog Biophys Mol Biol 98: 238 –250, 2008. 240. Kuersten S, Ohno M, Mattaj IW. Nucleocytoplasmic transport: Ran, beta and beyond. Trends Cell Biol 11: 497–503, 2001. 221. Ke Y, Wang L, Pyle WG, de Tombe PP, Solaro RJ. Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circ Res 94: 194 – 200, 2004. 241. Kumai T, Takeba Y, Matsumoto N, Nakaya S, Tsuzuki Y, Yanagida Y, Hayashi M, Kobayashi S. Fasudil attenuates sympathetic nervous activity in the adrenal medulla of spontaneously hypertensive rats. Life Sci 81: 1193–1198, 2007. 222. Keating M, Atkinson D, Dunn C, Timothy K, Vincent GM, Leppert M. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science 252: 704 –706, 1991. 242. Kuroda S, Fukata M, Kobayashi K, Nakafuku M, Nomura N, Iwamatsu A, Kaibuchi K. Identification of IQGAP as a putative target for the small GTPases, Cdc42 and Rac1. J Biol Chem 271: 23363–23367, 1996. 223. Kharlap MS, Goriunova LE, Timofeeva AV, Smolianova GG, Khaspekov GL, Raskin VA, Dzemeshkevich SL, Akchurin RS, Golitsyn SP, Bibilashvili R. Gene expression analysis in myocytes of right atrial appendages in patients with atrial fibrillation using cDNA microarray technique. Kardiologiia 48: 34 – 42, 2008. 243. Kuster GM, Pimentel DR, Adachi T, Ido Y, Brenner DA, Cohen RA, Liao R, Siwik DA, Colucci WS. Alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin-1-sensitive oxidative modification of thiols on Ras. Circulation 111: 1192–1198, 2005. 224. Kim KS, Takeda K, Sethi R, Pracyk JB, Tanaka K, Zhou YF, Yu ZX, Ferrans VJ, Bruder JT, Kovesdi I, Irani K, Goldschmidt-Clermont P, Finkel T. Protection from reoxygenation injury by inhibition of rac1. J Clin Invest 101: 1821–1826, 1998. 225. Kirchhoff S, Nelles E, Hagendorff A, Kruger O, Traub O, Willecke K. Reduced cardiac conduction velocity and predisposition to arrhythmias in connexin40-deficient mice. Curr Biol 8: 299 –302, 1998. 226. Kishi T, Hirooka Y, Masumoto A, Ito K, Kimura Y, Inokuchi K, Tagawa T, Shimokawa H, Takeshita A, Sunagawa K. Rho-kinase inhibitor improves increased vascular resistance and impaired vasodilation of the forearm in patients with heart failure. Circulation 111: 2741–2747, 2005. 227. Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol 137: 481– 492, 1997. 228. Kobayashi N, Horinaka S, Mita S, Nakano S, Honda T, Yoshida K, Kobayashi T, Matsuoka H. Critical role of Rho-kinase pathway for cardiac performance and remodeling in failing rat hearts. Cardiovasc Res 55: 757–767, 2002. 229. Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol 67: 39 – 67, 2005. 230. Koera K, Nakamura K, Nakao K, Miyoshi J, Toyoshima K, Hatta T, Otani H, Aiba A, Katsuki M. K-ras is essential for the development of the mouse embryo. Oncogene 15: 1151–1159, 1997. 231. Komatsu M, Ruoslahti E. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat Med 11: 1346 –1350, 2005. 232. Komuro I, Kurabayashi M, Takaku F, Yazaki Y. Expression of cellular oncogenes in the myocardium during the developmental stage and pressure-overloaded hypertrophy of the rat heart. Circ Res 62: 1075–1079, 1988. 233. Kontaridis MI, Yang W, Bence KK, Cullen D, Wang B, Bodyak N, Ke Q, Hinek A, Kang PM, Liao R, Neel BG. Deletion of Ptpn11 (Shp2) in cardiomyocytes causes dilated cardiomyopathy via effects on the extracellular signal-regulated kinase/mitogen-activated protein kinase and RhoA signaling pathways. Circulation 117: 1423–1435, 2008. 234. Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett 579: 4966 – 4972, 2005. 235. Kouchi H, Nakamura K, Fushimi K, Sakaguchi M, Miyazaki M, Ohe T, Namba M. Manumycin A, inhibitor of ras farnesyltransferase, inhibits proliferation and migration of rat vascular smooth muscle cells. Biochem Biophys Res Commun 264: 915–920, 1999. 236. Kouklis P, Konstantoulaki M, Vogel S, Broman M, Malik AB. Cdc42 regulates the restoration of endothelial barrier function. Circ Res 94: 159 –166, 2004. 237. Kovacs K, Hanto K, Bognar Z, Tapodi A, Bognar E, Kiss GN, Szabo A, Rappai G, Kiss T, Sumegi B, Gallyas F Jr. Prevalent role of Akt and ERK activation in cardioprotective effect of Ca2⫹ channel- and beta-adrenergic receptor blockers. Mol Cell Biochem 321: 155–164, 2009. 244. Kuwahara K, Kinoshita H, Kuwabara Y, Nakagawa Y, Usami S, Minami T, Yamada Y, Fujiwara M, Nakao K. Myocardin-related transcription factor A is a common mediator of mechanical stress- and neurohumoral stimulation-induced cardiac hypertrophic signaling leading to activation of brain natriuretic peptide gene expression. Mol Cell Biol 30: 4134 – 4148, 2010. 245. Kuwahara K, Saito Y, Nakagawa O, Kishimoto I, Harada M, Ogawa E, Miyamoto Y, Hamanaka I, Kajiyama N, Takahashi N, Izumi T, Kawakami R, Tamura N, Ogawa Y, Nakao K. The effects of the selective ROCK inhibitor, Y27632, on ET-1-induced hypertrophic response in neonatal rat cardiac myocytes–possible involvement of Rho/ ROCK pathway in cardiac muscle cell hypertrophy. FEBS Lett 452: 314 –318, 1999. 246. Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, and Tournier-Lasserve E. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet 23: 189 – 193, 1999. 247. Lakkis MM, Epstein JA. Neurofibromin modulation of ras activity is required for normal endocardial-mesenchymal transformation in the developing heart. Development 125: 4359 – 4367, 1998. 248. Lakshmikanthan S, Sobczak M, Chun C, Henschel A, Dargatz J, Ramchandran R, Chrzanowska-Wodnicka M. Rap1 promotes VEGFR2 activation and angiogenesis by a mechanism involving integrin avb3. Blood 118: 2015–2026, 2011. 249. Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res 100: 782–794, 2007. 250. Lampugnani MG, Zanetti A, Breviario F, Balconi G, Orsenigo F, Corada M, Spagnuolo R, Betson M, Braga V, Dejana E. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol Biol Cell 13: 1175–1189, 2002. 251. Langdon Y, Tandon P, Paden E, Duddy J, Taylor JM, Conlon FL. SHP-2 acts via ROCK to regulate the cardiac actin cytoskeleton. Development 139: 948 –957, 2012. 252. Lasater EA, Bessler WK, Mead LE, Horn WE, Clapp DW, Conway SJ, Ingram DA, Li F. Nf1⫹/⫺ mice have increased neointima formation via hyperactivation of a Gleevec sensitive molecular pathway. Hum Mol Genet 17: 2336 –2344, 2008. 253. Lasater EA, Li F, Bessler WK, Estes ML, Vemula S, Hingtgen CM, Dinauer MC, Kapur R, Conway SJ, Ingram DA Jr. Genetic and cellular evidence of vascular inflammation in neurofibromin-deficient mice and humans. J Clin Invest 120: 859 – 870, 2010. 254. Lauffenburger DA. ROCK in a stiff place. Sci Transl Med 3: 112fs112, 2011. 255. Laufs U, Adam O, Strehlow K, Wassmann S, Konkol C, Laufs K, Schmidt W, Bohm M, Nickenig G. Down-regulation of Rac-1 GTPase by Estrogen. J Biol Chem 278: 5956 – 5962, 2003. 256. Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 273: 24266 –24271, 1998. 257. Laufs U, Marra D, Node K, Liao JK. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPaseinduced down-regulation of p27(Kip1). J Biol Chem 274: 21926 –21931, 1999. 238. Krijnen PA, Sipkens JA, Molling JW, Rauwerda JA, Stehouwer CD, Muller A, Paulus WJ, van Nieuw Amerongen GP, Hack CE, Verhoeven AJ, van Hinsbergh VW, Niessen HW. Inhibition of Rho-ROCK signaling induces apoptotic and non-apoptotic PS exposure in cardiomyocytes via inhibition of flippase. J Mol Cell Cardiol 49: 781–790, 2010. 258. Lee BH, Kim JM, Jin HY, Kim GH, Choi JH, Yoo HW. Spectrum of mutations in Noonan syndrome and their correlation with phenotypes. J Pediatr 159: 1029 –1035, 2011. 239. Krugmann S, Williams R, Stephens L, Hawkins PT. ARAP3 is a PI3K- and rap-regulated GAP for RhoA. Curr Biol 14: 1380 –1384, 2004. 259. Lee JE, Bokoch G, Liang BT. A novel cardioprotective role of RhoA: new signaling mechanism for adenosine. FASEB J 15: 1886 –1894, 2001. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1711 LOIRAND, SAUZEAU, AND PACAUD 260. Legius E, Marchuk DA, Collins FS, Glover TW. Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nat Genet 3: 122–126, 1993. 279. Liu W, Zi M, Naumann R, Ulm S, Jin J, Taglieri DM, Prehar S, Gui J, Tsui H, Xiao RP, Neyses L, Solaro RJ, Ke Y, Cartwright EJ, Lei M, Wang X. Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation 124: 2702–2715, 2011. 261. Lengfeld J, Wang Q, Zohlman A, Salvarezza S, Morgan S, Ren J, Kato K, RodriguezBoulan E, Liu B. Protein Kinase C delta regulates the release of collagen type I from vascular smooth muscle cells via regulation of Cdc42. Mol Biol Cell 23: 1955–1963, 2012. 280. Liu Y, Suzuki YJ, Day RM, Fanburg BL. Rho kinase-induced nuclear translocation of ERK1/ERK2 in smooth muscle cell mitogenesis caused by serotonin. Circ Res 95: 579 –586, 2004. 262. Levay M, Settleman J, Ligeti E. Regulation of the substrate preference of p190RhoGAP by protein kinase C-mediated phosphorylation of a phospholipid binding site. Biochemistry 48: 8615– 8623, 2009. 263. Li Y, Kelly WG, Logsdon JM Jr, Schurko AM, Harfe BD, Hill-Harfe KL, Kahn RA. Functional genomic analysis of the ADP-ribosylation factor family of GTPases: phylogeny among diverse eukaryotes and function in C. elegans. FASEB J 18: 1834 –1850, 2004. 264. Liang X, Butterworth MB, Peters KW, Frizzell RA. AS160 modulates aldosteronestimulated epithelial sodium channel forward trafficking. Mol Biol Cell 21: 2024 –2033, 2010. 265. Ligeti E, Dagher MC, Hernandez SE, Koleske AJ, Settleman J. Phospholipids can switch the GTPase substrate preference of a GTPase-activating protein. J Biol Chem 279: 5055–5058, 2004. 266. Ligeti E, Welti S, Scheffzek K. Inhibition and termination of physiological responses by GTPase activating proteins. Physiol Rev 92: 237–272, 2012. 267. Lim YS, Chua CE, Tang BL. Rabs and other small GTPases in ciliary transport. Biol Cell 103: 209 –221, 2011. 268. Linseman DA, Benjamin CW, Jones DA. Convergence of angiotensin II and plateletderived growth factor receptor signaling cascades in vascular smooth muscle cells. J Biol Chem 270: 12563–12568, 1995. 269. Lips DJ, Bueno OF, Wilkins BJ, Purcell NH, Kaiser RA, Lorenz JN, Voisin L, Saba-ElLeil MK, Meloche S, Pouyssegur J, Pages G, De Windt LJ, Doevendans PA, Molkentin JD. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation 109: 1938 –1941, 2004. 270. Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, Verlaan D, Balogun F, Hughes L, Leedom TP, Plummer NW, Cannella M, Maglione V, Squitieri F, Johnson EW, Rouleau GA, Ptacek L, Marchuk DA. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet 73: 1459 –1464, 2003. 281. Lohn M, Plettenburg O, Ivashchenko Y, Kannt A, Hofmeister A, Kadereit D, Schaefer M, Linz W, Kohlmann M, Herbert JM, Janiak P, O’Connor SE, Ruetten H. Pharmacological characterization of SAR407899, a novel rho-kinase inhibitor. Hypertension 54: 676 – 683, 2009. 282. Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res 98: 322–334, 2006. 283. Loirand G, Pacaud P. The role of Rho protein signaling in hypertension. Nat Rev Cardiol 7: 637– 647, 2010. 284. Loirand G, Scalbert E, Bril A, Pacaud P. Rho exchange factors in the cardiovascular system. Curr Opin Pharmacol 8: 174 –180, 2008. 285. Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology 21: 269 –280, 2006. 286. Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M, Laufs U. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation 108: 1567– 1574, 2003. 287. Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem 276: 341–347, 2001. 288. Maillet M, Lynch JM, Sanna B, York AJ, Zheng Y, Molkentin JD. Cdc42 is an antihypertrophic molecular switch in the mouse heart. J Clin Invest 119: 3079 –3088, 2009. 289. Mallat Z, Gojova A, Sauzeau V, Brun V, Silvestre JS, Esposito B, Merval R, Groux H, Loirand G, Tedgui A. Rho-associated protein kinase contributes to early atherosclerotic lesion formation in mice. Circ Res 93: 884 – 888, 2003. 290. Mangoni ME, Nargeot J. Genesis and regulation of the heart automaticity. Physiol Rev 88: 919 –982, 2008. 291. Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr 135: 703–706, 1999. 271. Liu F, Schaphorst KL, Verin AD, Jacobs K, Birukova A, Day RM, Bogatcheva N, Bottaro DP, Garcia JG. Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3beta. FASEB J 16: 950 –962, 2002. 292. Marshall AK, Barrett OP, Cullingford TE, Shanmugasundram A, Sugden PH, Clerk A. ERK1/2 signaling dominates over RhoA signaling in regulating early changes in RNA expression induced by endothelin-1 in neonatal rat cardiomyocytes. PLoS One 5: e10027, 2010. 272. Liu HW, Halayko AJ, Fernandes DJ, Harmon GS, McCauley JA, Kocieniewski P, McConville J, Fu Y, Forsythe SM, Kogut P, Bellam S, Dowell M, Churchill J, Lesso H, Kassiri K, Mitchell RW, Hershenson MB, Camoretti-Mercado B, Solway J. The RhoA/ Rho kinase pathway regulates nuclear localization of serum response factor. Am J Respir Cell Mol Biol 29: 39 – 47, 2003. 293. Martin G, Duez H, Blanquart C, Berezowski V, Poulain P, Fruchart JC, Najib-Fruchart J, Glineur C, Staels B. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J Clin Invest 107: 1423–1432, 2001. 273. Liu J, Fraser SD, Faloon PW, Rollins EL, Vom Berg J, Starovic-Subota O, Laliberte AL, Chen JN, Serluca FC, Childs SJ. A betaPix Pak2a signaling pathway regulates cerebral vascular stability in zebrafish. Proc Natl Acad Sci USA 104: 13990 –13995, 2007. 274. Liu JJ, Stockton RA, Gingras AR, Ablooglu AJ, Han J, Bobkov AA, Ginsberg MH. A mechanism of Rap1-induced stabilization of endothelial cell-cell junctions. Mol Biol Cell 22: 2509 –2519, 2011. 275. Liu L, Zhu S, Gong Z, Low BC. K-ras/PI3K-Akt signaling is essential for zebrafish hematopoiesis and angiogenesis. PLoS One 3: e2850, 2008. 276. Liu PY, Chen JH, Lin LJ, Liao JK. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J Am Coll Cardiol 49: 1619 –1624, 2007. 277. Liu PY, Liao JK. A method for measuring Rho kinase activity in tissues and cells. Methods Enzymol 439: 181–189, 2008. 278. Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK. Evidence for statin pleiotropy in humans: differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation 119: 131– 138, 2009. 1712 294. Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O’Connell P, Cawthon RM. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63: 843– 849, 1990. 295. Martin GA, Yatani A, Clark R, Conroy L, Polakis P, Brown AM, McCormick F. GAP domains responsible for ras p21-dependent inhibition of muscarinic atrial K⫹ channel currents. Science 255: 192–194, 1992. 296. Maruyama Y, Nishida M, Sugimoto Y, Tanabe S, Turner JH, Kozasa T, Wada T, Nagao T, Kurose H. Galpha(12/13) mediates alpha(1)-adrenergic receptor-induced cardiac hypertrophy. Circ Res 91: 961–969, 2002. 297. Marzesco AM, Galli T, Louvard D, Zahraoui A. The rod cGMP phosphodiesterase delta subunit dissociates the small GTPase Rab13 from membranes. J Biol Chem 273: 22340 –22345, 1998. 298. Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 38: 1307–1310, 2001. 299. Matsubara K, Kishida S, Matsuura Y, Kitayama H, Noda M, Kikuchi A. Plasma membrane recruitment of RalGDS is critical for Ras-dependent Ral activation. Oncogene 18: 1303–1312, 1999. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 300. Matsuda T, Fujio Y, Nariai T, Ito T, Yamane M, Takatani T, Takahashi K, Azuma J. N-cadherin signals through Rac1 determine the localization of connexin 43 in cardiac myocytes. J Mol Cell Cardiol 40: 495–502, 2006. 301. Matsumoto Y, Uwatoku T, Oi K, Abe K, Hattori T, Morishige K, Eto Y, Fukumoto Y, Nakamura K, Shibata Y, Matsuda T, Takeshita A, Shimokawa H. Long-term inhibition of Rho-kinase suppresses neointimal formation after stent implantation in porcine coronary arteries: involvement of multiple mechanisms. Arterioscler Thromb Vasc Biol 24: 181–186, 2004. 302. Mayers CM, Wadell J, McLean K, Venere M, Malik M, Shibata T, Driggers PH, Kino T, Guo XC, Koide H, Gorivodsky M, Grinberg A, Mukhopadhyay M, Abu-Asab M, Westphal H, Segars JH. The Rho guanine nucleotide exchange factor AKAP13 (BRX) is essential for cardiac development in mice. J Biol Chem 285: 12344 –12354, 2010. 303. Medlin MD, Staus DP, Dubash AD, Taylor JM, Mack CP. Sphingosine 1-phosphate receptor 2 signals through leukemia-associated RhoGEF (LARG), to promote smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol 30: 1779 –1786, 2010. 304. Mehta D, Rahman A, Malik AB. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem 276: 22614 –22620, 2001. 305. Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc’h F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ Res 102: 959 –965, 2008. 306. Mettouchi A, Klein S, Guo W, Lopez-Lago M, Lemichez E, Westwick JK, Giancotti FG. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol Cell 8: 115–127, 2001. 307. Miao L, Calvert JW, Tang J, Parent AD, Zhang JH. Age-related RhoA expression in blood vessels of rats. Mech Ageing Dev 122: 1757–1770, 2001. 308. Migeotte I, Grego-Bessa J, Anderson KV. Rac1 mediates morphogenetic responses to intercellular signals in the gastrulating mouse embryo. Development 138: 3011–3020, 2011. 309. Min LJ, Mogi M, Iwai M, Horiuchi M. Signaling mechanisms of angiotensin II in regulating vascular senescence. Ageing Res Rev 8: 113–121, 2009. 310. Min LJ, Mogi M, Iwanami J, Li JM, Sakata A, Fujita T, Tsukuda K, Iwai M, Horiuchi M. Cross-talk between aldosterone and angiotensin II in vascular smooth muscle cell senescence. Cardiovasc Res 76: 506 –516, 2007. 311. Min LJ, Mogi M, Li JM, Iwanami J, Iwai M, Horiuchi M. Aldosterone and angiotensin II synergistically induce mitogenic response in vascular smooth muscle cells. Circ Res 97: 434 – 442, 2005. 312. Minamino T, Yoshida T, Tateno K, Miyauchi H, Zou Y, Toko H, Komuro I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 108: 2264 –2269, 2003. 313. Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, Hayoz D, Ruffieux J, Rusconi S, Montani JP, Yang Z. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation 110: 3708 –3714, 2004. 314. Mita S, Kobayashi N, Yoshida K, Nakano S, Matsuoka H. Cardioprotective mechanisms of Rho-kinase inhibition associated with eNOS and oxidative stress-LOX-1 pathway in Dahl salt-sensitive hypertensive rats. J Hypertens 23: 87–96, 2005. 315. Mohri M, Shimokawa H, Hirakawa Y, Masumoto A, Takeshita A. Rho-kinase inhibition with intracoronary fasudil prevents myocardial ischemia in patients with coronary microvascular spasm. J Am Coll Cardiol 41: 15–19, 2003. 316. Momotani K, Artamonov MV, Utepbergenov D, Derewenda U, Derewenda ZS, Somlyo AV. p63RhoGEF couples Galpha(q/11)-mediated signaling to Ca2⫹ sensitization of vascular smooth muscle contractility. Circ Res 109: 993–1002, 2011. 317. Montanez E, Wickstrom SA, Altstatter J, Chu H, Fassler R. Alpha-parvin controls vascular mural cell recruitment to vessel wall by regulating RhoA/ROCK signalling. EMBO J 28: 3132–3144, 2009. 318. Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol 13: 13–22, 2003. 319. Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc’h F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ Res 97: 1296 –1304, 2005. 320. Morishige K, Shimokawa H, Eto Y, Kandabashi T, Miyata K, Matsumoto Y, Hoshijima M, Kaibuchi K, Takeshita A. Adenovirus-mediated transfer of dominant-negative rhokinase induces a regression of coronary arteriosclerosis in pigs in vivo. Arterioscler Thromb Vasc Biol 21: 548 –554, 2001. 321. Morissette MR, Sah VP, Glembotski CC, Brown JH. The Rho effector, PKN, regulates ANF gene transcription in cardiomyocytes through a serum response element. Am J Physiol Heart Circ Physiol 278: H1769 –H1774, 2000. 322. Mozzicato S, Joshi BV, Jacobson KA, Liang BT. Role of direct RhoA-phospholipase D1 interaction in mediating adenosine-induced protection from cardiac ischemia. FASEB J 18: 406 – 408, 2004. 323. Mukai Y, Shimokawa H, Matoba T, Kandabashi T, Satoh S, Hiroki J, Kaibuchi K, Takeshita A. Involvement of Rho-kinase in hypertensive vascular disease: a novel therapeutic target in hypertension. FASEB J 15: 1062–1064, 2001. 324. Murata M, Cingolani E, McDonald AD, Donahue JK, Marban E. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ Res 95: 398 – 405, 2004. 325. Muslin AJ. Role of raf proteins in cardiac hypertrophy and cardiomyocyte survival. Trends Cardiovasc Med 15: 225–229, 2005. 326. Muthalif MM, Karzoun NA, Gaber L, Khandekar Z, Benter IF, Saeed AE, Parmentier JH, Estes A, Malik KU. Angiotensin II-induced hypertension: contribution of Ras GTPase/Mitogen-activated protein kinase and cytochrome P450 metabolites. Hypertension 36: 604 – 609, 2000. 327. Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, BeckerHapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med 4: 1449 –1452, 1998. 328. Nagai T, Tanaka-Ishikawa M, Aikawa R, Ishihara H, Zhu W, Yazaki Y, Nagai R, Komuro I. Cdc42 plays a critical role in assembly of sarcomere units in series of cardiac myocytes. Biochem Biophys Res Commun 305: 806 – 810, 2003. 329. Nakamura K, Ichise H, Nakao K, Hatta T, Otani H, Sakagami H, Kondo H, Katsuki M. Partial functional overlap of the three ras genes in mouse embryonic development. Oncogene 27: 2961–2968, 2008. 330. Nancy V, Callebaut I, El Marjou A, de Gunzburg J. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J Biol Chem 277: 15076 –15084, 2002. 331. Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente-Manzanares M, Horwitz AR. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J Cell Biol 173: 587–589, 2006. 332. Negoro N, Hoshiga M, Seto M, Kohbayashi E, Ii M, Fukui R, Shibata N, Nakakoji T, Nishiguchi F, Sasaki Y, Ishihara T, Ohsawa N. The kinase inhibitor fasudil (HA-1077) reduces intimal hyperplasia through inhibiting migration and enhancing cell loss of vascular smooth muscle cells. Biochem Biophys Res Commun 262: 211–215, 1999. 333. Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S, Papadakis K, Voight BF, Scott LJ, Zhang F, Farrall M, Tanaka T, Wallace C, Chambers JC, Khaw KT, Nilsson P, van der Harst P, Polidoro S, Grobbee DE, Onland-Moret NC, Bots ML, Wain LV, Elliott KS, Teumer A, Luan J, Lucas G, Kuusisto J, Burton PR, Hadley D, McArdle WL, Brown M, Dominiczak A, Newhouse SJ, Samani NJ, Webster J, Zeggini E, Beckmann JS, Bergmann S, Lim N, Song K, Vollenweider P, Waeber G, Waterworth DM, Yuan X, Groop L, OrhoMelander M, Allione A, Di Gregorio A, Guarrera S, Panico S, Ricceri F, Romanazzi V, Sacerdote C, Vineis P, Barroso I, Sandhu MS, Luben RN, Crawford GJ, Jousilahti P, Perola M, Boehnke M, Bonnycastle LL, Collins FS, Jackson AU, Mohlke KL, Stringham HM, Valle TT, Willer CJ, Bergman RN, Morken MA, Doring A, Gieger C, Illig T, Meitinger T, Org E, Pfeufer A, Wichmann HE, Kathiresan S, Marrugat J, O’Donnell CJ, Schwartz SM, Siscovick DS, Subirana I, Freimer NB, Hartikainen AL, McCarthy MI, O’Reilly PF, Peltonen L, Pouta A, de Jong PE, Snieder H, van Gilst WH, Clarke R, Goel A, Hamsten A, Peden JF, Seedorf U, Syvanen AC, Tognoni G, Lakatta EG, Sanna S, Scheet P, Schlessinger D, Scuteri A, Dorr M, Ernst F, Felix SB, Homuth G, Lorbeer R, Reffelmann T, Rettig R, Volker U, Galan P, Gut IG, Hercberg S, Lathrop GM, Zelenika D, Deloukas P, Soranzo N, Williams FM, Zhai G, Salomaa V, Laakso M, Elosua R, Forouhi NG, Volzke H, Uiterwaal CS, van der Schouw YT, Numans ME, Matullo G, Navis G, Berglund G, Bingham SA, Kooner JS, Connell JM, Bandinelli S, Ferrucci L, Watkins H, Spector TD, Tuomilehto J, Altshuler D, Strachan DP, Laan M, Meneton P, Wareham NJ, Uda M, Jarvelin MR, Mooser V, Melander O, Loos RJ, Elliott P, Abecasis Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1713 LOIRAND, SAUZEAU, AND PACAUD GR, Caulfield M, Munroe PB. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet 41: 666 – 676, 2009. 334. Ni W, Egashira K, Kataoka C, Kitamoto S, Koyanagi M, Inoue S, Takeshita A. Antiinflammatory and antiarteriosclerotic actions of HMG-CoA reductase inhibitors in a rat model of chronic inhibition of nitric oxide synthesis. Circ Res 89: 415– 421, 2001. 351. Ohkawara H, Ishibashi T, Shiomi M, Sugimoto K, Uekita H, Kamioka M, Takuwa Y, Teramoto T, Maruyama Y, Takeishi Y. RhoA and Rac1 changes in the atherosclerotic lesions of WHHLMI rabbits. J Atheroscler Thromb 16: 846 – 856, 2009. 352. Olson EN, Srivastava D. Molecular pathways controlling heart development. Science 272: 671– 676, 1996. 335. Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, Kavamura MI, Kurosawa K, Ohashi H, Wilson L, Heron D, Bonneau D, Corona G, Kaname T, Naritomi K, Baumann C, Matsumoto N, Kato K, Kure S, Matsubara Y. Germline KRAS and BRAF mutations in cardio-faciocutaneous syndrome. Nat Genet 38: 294 –296, 2006. 353. Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269: 1270 –1272, 1995. 336. Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol 5: 236 –241, 2003. 355. Pai EF, Kabsch W, Krengel U, Holmes KC, John J, Wittinghofer A. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature 341: 209 –214, 1989. 337. Nishida M, Tanabe S, Maruyama Y, Mangmool S, Urayama K, Nagamatsu Y, Takagahara S, Turner JH, Kozasa T, Kobayashi H, Sato Y, Kawanishi T, Inoue R, Nagao T, Kurose H. G alpha 12/13- and reactive oxygen species-dependent activation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase by angiotensin receptor stimulation in rat neonatal cardiomyocytes. J Biol Chem 280: 18434 –18441, 2005. 338. Nishiki K, Tsuruoka S, Kawaguchi A, Sugimoto K, Schwartz GJ, Suzuki M, Imai M, Fujimura A. Inhibition of Rho-kinase reduces renal Na-H exchanger activity and causes natriuresis in rat. J Pharmacol Exp Ther 304: 723–728, 2003. 339. Nofer JR, Feuerborn R, Levkau B, Sokoll A, Seedorf U, Assmann G. Involvement of Cdc42 signaling in apoA-I-induced cholesterol efflux. J Biol Chem 278: 53055–53062, 2003. 340. Noma K, Goto C, Nishioka K, Jitsuiki D, Umemura T, Ueda K, Kimura M, Nakagawa K, Oshima T, Chayama K, Yoshizumi M, Liao JK, Higashi Y. Roles of rho-associated kinase and oxidative stress in the pathogenesis of aortic stiffness. J Am Coll Cardiol 49: 698 –705, 2007. 354. Pagler TA, Wang M, Mondal M, Murphy AJ, Westerterp M, Moore KJ, Maxfield FR, Tall AR. Deletion of ABCA1 and ABCG1 impairs macrophage migration because of increased Rac1 signaling. Circ Res 108: 194 –200, 2011. 356. Pan J, Singh US, Takahashi T, Oka Y, Palm-Leis A, Herbelin BS, Baker KM. PKC mediates cyclic stretch-induced cardiac hypertrophy through Rho family GTPases and mitogen-activated protein kinases in cardiomyocytes. J Cell Physiol 202: 536–553, 2005. 357. Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, Bos JM, Ommen SR, Esposito G, Lepri F, Faul C, Mundel P, Lopez Siguero JP, Tenconi R, Selicorni A, Rossi C, Mazzanti L, Torrente I, Marino B, Digilio MC, Zampino G, Ackerman MJ, Dallapiccola B, Tartaglia M, Gelb BD. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 39: 1007–1012, 2007. 358. Pando R, Cheporko Y, Haklai R, Maysel-Auslender S, Keren G, George J, Porat E, Sagie A, Kloog Y, Hochhauser E. Ras inhibition attenuates myocardial ischemia-reperfusion injury. Biochem Pharmacol 77: 1593–1601, 2009. 359. Pannekoek WJ, Kooistra MR, Zwartkruis FJ, Bos JL. Cell-cell junction formation: the role of Rap1 and Rap1 guanine nucleotide exchange factors. Biochim Biophys Acta 1788: 790 –796, 2009. 341. Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, Wang H, Ahl D, Sawada N, Okamoto R, Hiroi Y, Shimizu K, Luscinskas FW, Sun J, Liao JK. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest 118: 1632–1644, 2008. 360. Pavlov TS, Chahdi A, Ilatovskaya DV, Levchenko V, Vandewalle A, Pochynyuk O, Sorokin A, Staruschenko A. Endothelin-1 inhibits the epithelial Na⫹ channel through betaPix/14 –3-3/Nedd4 –2. J Am Soc Nephrol 21: 833– 843, 2010. 342. Noonan JA. Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am J Dis Child 116: 373–380, 1968. 361. Peng J, He F, Zhang C, Deng X, Yin F. Protein kinase C-alpha signals P115RhoGEF phosphorylation and RhoA activation in TNF-alpha-induced mouse brain microvascular endothelial cell barrier dysfunction. J Neuroinflammation 8: 28, 2011. 343. Noren NK, Niessen CM, Gumbiner BM, Burridge K. Cadherin engagement regulates Rho family GTPases. J Biol Chem 276: 33305–33308, 2001. 344. Nozoe M, Hirooka Y, Koga Y, Sagara Y, Kishi T, Engelhardt JF, Sunagawa K. Inhibition of Rac1-derived reactive oxygen species in nucleus tractus solitarius decreases blood pressure and heart rate in stroke-prone spontaneously hypertensive rats. Hypertension 50: 62– 68, 2007. 345. Numaguchi K, Eguchi S, Yamakawa T, Motley ED, Inagami T. Mechanotransduction of rat aortic vascular smooth muscle cells requires RhoA and intact actin filaments. Circ Res 85: 5–11, 1999. 346. Oceandy D, Pickard A, Prehar S, Zi M, Mohamed TM, Stanley PJ, Baudoin-Stanley F, Nadif R, Tommasi S, Pfeifer GP, Armesilla AL, Cartwright EJ, Neyses L. Tumor suppressor Ras-association domain family 1 isoform A is a novel regulator of cardiac hypertrophy. Circulation 120: 607– 616, 2009. 347. Odashima M, Usui S, Takagi H, Hong C, Liu J, Yokota M, Sadoshima J. Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ Res 100: 1344 –1352, 2007. 348. Oderich GS, Sullivan TM, Bower TC, Gloviczki P, Miller DV, Babovic-Vuksanovic D, Macedo TA, Stanson A. Vascular abnormalities in patients with neurofibromatosis syndrome type I: clinical spectrum, management, and results. J Vasc Surg 46: 475– 484, 2007. 349. Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and phospholipase Cepsilon regulate Ca2⫹ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J Biol Chem 284: 1514–1522, 2009. 350. Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac-mediated activation of phospholipase C(epsilon) plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2⫹ mobilization in cardiac myocytes. J Biol Chem 282: 5488 –5495, 2007. 1714 362. Pereira L, Metrich M, Fernandez-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Benitah JP, Lezoualc’h F, Gomez AM. The cAMP binding protein Epac modulates Ca2⫹ sparks by a Ca2⫹/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol 583: 685– 694, 2007. 363. Pfleiderer P, Sumandea MP, Rybin VO, Wang C, Steinberg SF. Raf-1: a novel cardiac troponin T kinase. J Muscle Res Cell Motil 30: 67–72, 2009. 364. Phrommintikul A, Tran L, Kompa A, Wang B, Adrahtas A, Cantwell D, Kelly DJ, Krum H. Effects of a Rho kinase inhibitor on pressure overload induced cardiac hypertrophy and associated diastolic dysfunction. Am J Physiol Heart Circ Physiol 294: H1804 – H1814, 2008. 365. Pilowsky PM, Goodchild AK. Baroreceptor reflex pathways and neurotransmitters: 10 years on. J Hypertens 20: 1675–1688, 2002. 366. Pochynyuk O, Medina J, Gamper N, Genth H, Stockand JD, Staruschenko A. Rapid translocation and insertion of the epithelial Na⫹ channel in response to RhoA signaling. J Biol Chem 281: 26520 –26527, 2006. 367. Pochynyuk O, Stockand JD, Staruschenko A. Ion channel regulation by Ras, Rho, and Rab small GTPases. Exp Biol Med 232: 1258 –1265, 2007. 368. Poling J, Szibor M, Schimanski S, Ingelmann ME, Rees W, Gajawada P, Kochfar Z, Lorchner H, Salwig I, Shin JY, Wiebe K, Kubin T, Warnecke H, Braun T. Induction of smooth muscle cell migration during arteriogenesis is mediated by Rap2. Arterioscler Thromb Vasc Biol 31: 2297–2305, 2011. 369. Popoff MR, Geny B. Multifaceted role of Rho, Rac, Cdc42 and Ras in intercellular junctions, lessons from toxins. Biochim Biophys Acta 1788: 797– 812, 2009. 370. Post GR, Swiderski C, Waldrop BA, Salty L, Glembotski CC, Wolthuis RM, Mochizuki N. Guanine nucleotide exchange factor-like factor (Rlf) induces gene expression and potentiates alpha 1-adrenergic receptor-induced transcriptional responses in neonatal rat ventricular myocytes. J Biol Chem 277: 15286 –15292, 2002. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 371. Potenza N, Vecchione C, Notte A, De Rienzo A, Rosica A, Bauer L, Affuso A, De Felice M, Russo T, Poulet R, Cifelli G, De Vita G, Lembo G, Di Lauro R. Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep 6: 432– 437, 2005. 372. Pracyk JB, Tanaka K, Hegland DD, Kim KS, Sethi R, Rovira II, Blazina DR, Lee L, Bruder JT, Kovesdi I, Goldshmidt-Clermont PJ, Irani K, Finkel T. A requirement for the rac1 GTPase in the signal transduction pathway leading to cardiac myocyte hypertrophy. J Clin Invest 102: 929 –937, 1998. 373. Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiology 24: 342–356, 2009. 374. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11: 761–774, 2011. 375. Qi Y, Liu J, Wu X, Brakebusch C, Leitges M, Han Y, Corbett SA, Lowry SF, Graham AM, Li S. Cdc42 controls vascular network assembly through protein kinase Ciota during embryonic vasculogenesis. Arterioscler Thromb Vasc Biol 31: 1861–1870, 2011. 376. Qian L, Wythe JD, Liu J, Cartry J, Vogler G, Mohapatra B, Otway RT, Huang Y, King IN, Maillet M, Zheng Y, Crawley T, Taghli-Lamallem O, Semsarian C, Dunwoodie S, Winlaw D, Harvey RP, Fatkin D, Towbin JA, Molkentin JD, Srivastava D, Ocorr K, Bruneau BG, Bodmer R. Tinman/Nkx2–5 acts via miR-1 and upstream of Cdc42 to regulate heart function across species. J Cell Biol 193: 1181–1196, 2011. 377. Qu J, Li X, Novitch BG, Zheng Y, Kohn M, Xie JM, Kozinn S, Bronson R, Beg AA, Minden A. PAK4 kinase is essential for embryonic viability and for proper neuronal development. Mol Cell Biol 23: 7122–7133, 2003. 378. Quarck R, Berrou E, Magnier C, Bobe R, Bredoux R, Tobelem G, Enouf J, Bryckaert M. Differential up-regulation of Rap1a and Rap1b proteins during smooth muscle cell cycle. Eur J Cell Biol 70: 269 –277, 1996. 379. Raaijmakers R, Noordam C, Noonan JA, Croonen EA, van der Burgt CJ, Draaisma JM. Are ECG abnormalities in Noonan syndrome characteristic for the syndrome? Eur J Pediatr 167: 1363–1367, 2008. 380. Rahaman SO, Swat W, Febbraio M, Silverstein RL. Vav family Rho guanine nucleotide exchange factors regulate CD36-mediated macrophage foam cell formation. J Biol Chem 286: 7010 –7017, 2011. 381. Rajashree R, Blunt BC, Hofmann PA. Modulation of myosin phosphatase targeting subunit and protein phosphatase 1 in the heart. Am J Physiol Heart Circ Physiol 289: H1736 –H1743, 2005. 382. Ramchandran R, Mehta D, Vogel SM, Mirza MK, Kouklis P, Malik AB. Critical role of Cdc42 in mediating endothelial barrier protection in vivo. Am J Physiol Lung Cell Mol Physiol 295: L363–L369, 2008. 383. Ramirez MT, Sah VP, Zhao XL, Hunter JJ, Chien KR, Brown JH. The MEKK-JNK pathway is stimulated by alpha1-adrenergic receptor and ras activation and is associated with in vitro and in vivo cardiac hypertrophy. J Biol Chem 272: 14057–14061, 1997. 384. Rankinen T, Church T, Rice T, Markward N, Blair SN, Bouchard C. A major haplotype block at the rho-associated kinase 2 locus is associated with a lower risk of hypertension in a recessive manner: the HYPGENE study. Hypertens Res 31: 1651–1657, 2008. 385. Rashid M, Tawara S, Fukumoto Y, Seto M, Yano K, Shimokawa H. Importance of Rac1 signaling pathway inhibition in the pleiotropic effects of HMG-CoA reductase inhibitors. Circ J 73: 361–370, 2009. 386. Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol 151: 33– 40, 2000. 387. Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U. S. death certificates. Am J Hum Genet 68: 1110 –1118, 2001. 388. Resh MD. Targeting protein lipidation in disease. Trends Mol Med 18: 206 –214, 2012. 389. Reynolds JF, Neri G, Herrmann JP, Blumberg B, Coldwell JG, Miles PV, Opitz JM. New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement–the CFC syndrome. Am J Med Genet 25: 413– 427, 1986. 390. Ribbeck K, Lipowsky G, Kent HM, Stewart M, Gorlich D. NTF2 mediates nuclear import of Ran. EMBO J 17: 6587– 6598, 1998. 391. Ridley AJ. Life at the leading edge. Cell 145: 1012–1022, 2011. 392. Ridley AJ. Rho proteins, PI 3-kinases, and monocyte/macrophage motility. FEBS Lett 498: 168 –171, 2001. 393. Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, Liao JK. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1⫹/⫺ haploinsufficient mice. Circulation 112: 2959 –2965, 2005. 394. Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, Young T, Neri G. The cardiofaciocutaneous syndrome. J Med Genet 43: 833– 842, 2006. 395. Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, Kucherlapati RS. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet 39: 70 –74, 2007. 396. Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311: 1287–1290, 2006. 397. Rojas AM, Fuentes G, Rausell A, Valencia A. The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J Cell Biol 196: 189 –201, 2012. 398. Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME, Roberts AE, Robinson W, Takemoto CM, Noonan JA. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 126: 746 –759, 2010. 399. Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev 90: 1507–1546, 2010. 400. Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 6: 167–180, 2005. 401. Roy N, Kahlem P, Dausse E, Bennaceur M, Faure S, Weissenbach J, Komajda M, Denjoy I, Coumel P, Schwartz K. Exclusion of HRAS from long QT locus. Nat Genet 8: 113–114, 1994. 402. Ruan H, Mitchell S, Vainoriene M, Lou Q, Xie LH, Ren S, Goldhaber JI, Wang Y. Gi alpha 1-mediated cardiac electrophysiological remodeling and arrhythmia in hypertrophic cardiomyopathy. Circulation 116: 596 – 605, 2007. 403. Sadhu DN, Ramos KS. Cyclic AMP inhibits c-Ha-ras protooncogene expression and DNA synthesis in rat aortic smooth muscle cells. Experientia 49: 567–570, 1993. 404. Sadoshima J, Izumo S. The heterotrimeric G q protein-coupled angiotensin II receptor activates p21 ras via the tyrosine kinase-Shc-Grb2-Sos pathway in cardiac myocytes. EMBO J 15: 775–787, 1996. 405. Sagara Y, Hirooka Y, Nozoe M, Ito K, Kimura Y, Sunagawa K. Pressor response induced by central angiotensin II is mediated by activation of Rho/Rho-kinase pathway via AT1 receptors. J Hypertens 25: 399 – 406, 2007. 406. Sah VP, Hoshijima M, Chien KR, Brown JH. Rho is required for Galphaq and alpha1adrenergic receptor signaling in cardiomyocytes. Dissociation of Ras and Rho pathways. J Biol Chem 271: 31185–31190, 1996. 407. Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW, 2nd Ross J Jr, Chien KR, Brown JH. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest 103: 1627–1634, 1999. 408. Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, Touchman JW, Gallione CJ, Lee-Lin SQ, Kosofsky B, Kurth JH, Louis DN, Mettler G, Morrison L, Gil-Nagel A, Rich SS, Zabramski JM, Boguski MS, Green ED, Marchuk DA. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet 8: 2325–2333, 1999. 409. Sakamoto K, Nakahara T, Ishii K. Rho-Rho kinase pathway is involved in the protective effect of early ischemic preconditioning in the rat heart. Biol Pharm Bull 34: 156 –159, 2011. 410. Sakata H, Sakabe M, Matsui H, Kawada N, Nakatani K, Ikeda K, Yamagishi T, Nakajima Y. Rho kinase inhibitor Y27632 affects initial heart myofibrillogenesis in cultured chick blastoderm. Dev Dyn 236: 461– 472, 2007. 411. Sakurai A, Fukuhara S, Yamagishi A, Sako K, Kamioka Y, Masuda M, Nakaoka Y, Mochizuki N. MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol Biol Cell 17: 966 –976, 2006. 412. Sanders LC, Matsumura F, Bokoch GM, de Lanerolle P. Inhibition of myosin light chain kinase by p21-activated kinase. Science 283: 2083–2085, 1999. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1715 LOIRAND, SAUZEAU, AND PACAUD 413. Sanematsu F, Hirashima M, Laurin M, Takii R, Nishikimi A, Kitajima K, Ding G, Noda M, Murata Y, Tanaka Y, Masuko S, Suda T, Meno C, Cote JF, Nagasawa T, Fukui Y. DOCK180 is a Rac activator that regulates cardiovascular development by acting downstream of CXCR4. Circ Res 107: 1102–1105, 2010. 433. Sedding DG, Trobs M, Reich F, Walker G, Fink L, Haberbosch W, Rau W, Tillmanns H, Preissner KT, Bohle RM, Langheinrich AC. 3-Deazaadenosine prevents smooth muscle cell proliferation and neointima formation by interfering with Ras signaling. Circ Res 104: 1192–1200, 2009. 414. Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis 3: 13, 2008. 434. Seizinger BR, Rouleau GA, Lane AH, Farmer G, Ozelius LJ, Haines JL, Parry DM, Korf BR, Pericak-Vance MA, Faryniarz AG. Linkage analysis in von Recklinghausen neurofibromatosis (NF1) with DNA markers for chromosome 17. Genomics 1: 346 –348, 1987. 415. Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci USA 103: 7432– 7437, 2006. 416. Satoh S, Rensland H, Pfitzer G. Ras proteins increase Ca muscle contraction. FEBS Lett 324: 211–215, 1993. 2⫹ -responsiveness of smooth 417. Sauzeau V, Le Jeune H, Cario-Toumaniantz C, Smolenski A, Lohmann SM, Bertoglio J, Chardin P, Pacaud P, Loirand G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2⫹ sensitization of contraction in vascular smooth muscle. J Biol Chem 275: 21722–21729, 2000. 418. Sauzeau V, Sevilla MA, Montero MJ, Bustelo XR. The Rho/Rac exchange factor Vav2 controls nitric oxide-dependent responses in mouse vascular smooth muscle cells. J Clin Invest 120: 315–330, 2010. 419. Sawada J, Urakami T, Li F, Urakami A, Zhu W, Fukuda M, Li DeanÂY, Ruoslahti E, Komatsu M. Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell 22: 235–249, 2012. 420. Sawada N, Itoh H, Ueyama K, Yamashita J, Doi K, Chun TH, Inoue M, Masatsugu K, Saito T, Fukunaga Y, Sakaguchi S, Arai H, Ohno N, Komeda M, Nakao K. Inhibition of rho-associated kinase results in suppression of neointimal formation of balloon-injured arteries. Circulation 101: 2030 –2033, 2000. 421. Sawada N, Li Y, Liao JK. Novel aspects of the roles of Rac1 GTPase in the cardiovascular system. Curr Opin Pharmacol 10: 116 –121, 2010. 422. Sawada N, Salomone S, Kim HH, Kwiatkowski DJ, Liao JK. Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circ Res 103: 360 –368, 2008. 423. Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 100: 416 – 424, 2007. 424. Scheidig AJ, Burmester C, Goody RS. The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure 7: 1311–1324, 1999. 425. Schmidt A, Brixius K, Bloch W. Endothelial precursor cell migration during vasculogenesis. Circ Res 101: 125–136, 2007. 426. Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, Nguyen H, West B, Zhang KY, Sistermans E, Rauch A, Niemeyer CM, Shannon K, Kratz CP. Germline KRAS mutations cause Noonan syndrome. Nat Genet 38: 331–336, 2006. 427. Schuhmacher AJ, Guerra C, Sauzeau V, Canamero M, Bustelo XR, Barbacid M. A mouse model for Costello syndrome reveals an ANG II-mediated hypertensive condition. J Clin Invest 118: 2169 –2179, 2008. 428. Scruggs SB, Solaro RJ. The significance of regulatory light chain phosphorylation in cardiac physiology. Arch Biochem Biophys 510: 129 –134, 2011. 429. Seasholtz TM, Majumdar M, Kaplan DD, Brown JH. Rho and Rho kinase mediate thrombin-stimulated vascular smooth muscle cell DNA synthesis and migration. Circ Res 84: 1186 –1193, 1999. 430. Seasholtz TM, Wessel J, Rao F, Rana BK, Khandrika S, Kennedy BP, Lillie EO, Ziegler MG, Smith DW, Schork NJ, Brown JH, O’Connor DT. Rho kinase polymorphism influences blood pressure and systemic vascular resistance in human twins: role of heredity. Hypertension 47: 937–947, 2006. 431. Seasholtz TM, Zhang T, Morissette MR, Howes AL, Yang AH, Brown JH. Increased expression and activity of RhoA are associated with increased DNA synthesis and reduced p27(Kip1) expression in the vasculature of hypertensive rats. Circ Res 89: 488 – 495, 2001. 432. Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol 3: 346 –352, 2001. 1716 435. Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, Nakano T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res 92: 411– 418, 2003. 436. Selvakumar B, Hess DT, Goldschmidt-Clermont PJ, Stamler JS. Co-regulation of constitutive nitric oxide synthases and NADPH oxidase by the small GTPase Rac. FEBS Lett 582: 2195–2202, 2008. 437. Serban D, Leng J, Cheresh D. H-ras regulates angiogenesis and vascular permeability by activation of distinct downstream effectors. Circ Res 102: 1350 –1358, 2008. 438. Sethi T, Ginsberg MH, Downward J, Hughes PE. The small GTP-binding protein R-Ras can influence integrin activation by antagonizing a Ras/Raf-initiated integrin suppression pathway. Mol Biol Cell 10: 1799 –1809, 1999. 439. Shan L, Li J, Wei M, Ma J, Wan L, Zhu W, Li Y, Zhu H, Arnold JM, Peng T. Disruption of Rac1 signaling reduces ischemia-reperfusion injury in the diabetic heart by inhibiting calpain. Free Radic Biol Med 49: 1804 –1814, 2010. 440. Sheehan KA, Ke Y, Solaro RJ. p21-Activated kinase-1 and its role in integrated regulation of cardiac contractility. Am J Physiol Regul Integr Comp Physiol 293: R963–R973, 2007. 441. Sheehan KA, Ke Y, Wolska BM, Solaro RJ. Expression of active p21-activated kinase-1 induces Ca2⫹ flux modification with altered regulatory protein phosphorylation in cardiac myocytes. Am J Physiol Cell Physiol 296: C47–C58, 2009. 442. Shi J, Zhang YW, Summers LJ, Dorn GW 2nd, Wei L. Disruption of ROCK1 gene attenuates cardiac dilation and improves contractile function in pathological cardiac hypertrophy. J Mol Cell Cardiol 44: 551–560, 2008. 443. Shi J, Zhang YW, Yang Y, Zhang L, Wei L. ROCK1 plays an essential role in the transition from cardiac hypertrophy to failure in mice. J Mol Cell Cardiol 49: 819 – 828, 2010. 444. Shibata R, Kai H, Seki Y, Kato S, Morimatsu M, Kaibuchi K, Imaizumi T. Role of Rho-associated kinase in neointima formation after vascular injury. Circulation 103: 284 –289, 2001. 445. Shibata R, Kai H, Seki Y, Kusaba K, Takemiya K, Koga M, Jalalidin A, Tokuda K, Tahara N, Niiyama H, Nagata T, Kuwahara F, Imaizumi T. Rho-kinase inhibition reduces neointima formation after vascular injury by enhancing Bax expression and apoptosis. J Cardiovasc Pharmacol 42 Suppl 1: S43– 47, 2003. 446. Shibata S, Mu S, Kawarazaki H, Muraoka K, Ishizawa K, Yoshida S, Kawarazaki W, Takeuchi M, Ayuzawa N, Miyoshi J, Takai Y, Ishikawa A, Shimosawa T, Ando K, Nagase M, Fujita T. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest 121: 3233–3243, 2011. 447. Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, Miyoshi J, Takai Y, Fujita T. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med 14: 1370 –1376, 2008. 448. Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, Nagata I, Kikuchi H, Takemae T, Hidaka H. Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage Results of a prospective placebo-controlled double-blind trial. J Neurosurg 76: 571–577, 1992. 449. Shikata Y, Rios A, Kawkitinarong K, DePaola N, Garcia JG, Birukov KG. Differential effects of shear stress and cyclic stretch on focal adhesion remodeling, site-specific FAK phosphorylation, and small GTPases in human lung endothelial cells. Exp Cell Res 304: 40 – 49, 2005. 450. Shimokawa H, Hiramori K, Iinuma H, Hosoda S, Kishida H, Osada H, Katagiri T, Yamauchi K, Yui Y, Minamino T, Nakashima M, Kato K. Anti-anginal effect of fasudil, a Rho-kinase inhibitor, in patients with stable effort angina: a multicenter study. J Cardiovasc Pharmacol 40: 751–761, 2002. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 451. Shimokawa H, Rashid M. Development of Rho-kinase inhibitors for cardiovascular medicine. Trends Pharmacol Sci 28: 296 –302, 2007. 452. Shirai H, Autieri M, Eguchi S. Small GTP-binding proteins and mitogen-activated protein kinases as promising therapeutic targets of vascular remodeling. Curr Opin Nephrol Hypertens 16: 111–115, 2007. 453. Smith A, Brownawell A, Macara IG. Nuclear import of Ran is mediated by the transport factor NTF2. Curr Biol 8: 1403–1406, 1998. 454. Smith SD, Jaffer ZM, Chernoff J, Ridley AJ. PAK1-mediated activation of ERK1/2 regulates lamellipodial dynamics. J Cell Sci 121: 3729 –3736, 2008. 455. Solaro RJ. Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. J Biol Chem 283: 26829 –26833, 2008. 456. Somekawa S, Fukuhara S, Nakaoka Y, Fujita H, Saito Y, Mochizuki N. Enhanced functional gap junction neoformation by protein kinase A-dependent and Epac-dependent signals downstream of cAMP in cardiac myocytes. Circ Res 97: 655– 662, 2005. 472. Sun Z, Zhang J, Chen C, Du Q, Chang L, Cao C, Zheng M, Garcia-Barrio MT, Chen YE, Xiao RP, Mao J, Zhu X. Rad GTPase induces cardiomyocyte apoptosis through the activation of p38 mitogen-activated protein kinase. Biochem Biophys Res Commun 409: 52–57, 2011. 473. Sussman MA, Welch S, Walker A, Klevitsky R, Hewett TE, Price RL, Schaefer E, Yager K. Altered focal adhesion regulation correlates with cardiomyopathy in mice expressing constitutively active rac1. J Clin Invest 105: 875– 886, 2000. 474. Szaszi K, Kurashima K, Kapus A, Paulsen A, Kaibuchi K, Grinstein S, Orlowski J. RhoA and rho kinase regulate the epithelial Na⫹/H⫹ exchanger NHE3. Role of myosin light chain phosphorylation. J Biol Chem 275: 28599 –28606, 2000. 475. Takagi H, Hsu CP, Kajimoto K, Shao D, Yang Y, Maejima Y, Zhai P, Yehia G, Yamada C, Zablocki D, Sadoshima J. Activation of PKN mediates survival of cardiac myocytes in the heart during ischemia/reperfusion. Circ Res 107: 642– 649, 2010. 476. Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxiainduced downregulation of endothelial nitric oxide synthase. Circulation 106: 57– 62, 2002. 457. Somlyo AP, Somlyo AV. Ca2⫹ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83: 1325– 1358, 2003. 477. Tan W, Palmby TR, Gavard J, Amornphimoltham P, Zheng Y, Gutkind JS. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J 22: 1829 –1838, 2008. 458. Srinivasan R, Zabuawala T, Huang H, Zhang J, Gulati P, Fernandez S, Karlo JC, Landreth GE, Leone G, Ostrowski MC. Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS One 4: e8283, 2009. 478. Taparowsky E, Shimizu K, Goldfarb M, Wigler M. Structure and activation of the human N-ras gene. Cell 34: 581–586, 1983. 459. Srivastava D. Building a heart: implications for congenital heart disease. J Nucl Cardiol 10: 63–70, 2003. 460. Staruschenko A, Nichols A, Medina JL, Camacho P, Zheleznova NN, Stockand JD. Rho small GTPases activate the epithelial Na⫹ channel. J Biol Chem 279: 49989 – 49994, 2004. 461. Staruschenko A, Patel P, Tong Q, Medina JL, Stockand JD. Ras activates the epithelial Na⫹ channel through phosphoinositide 3-OH kinase signaling. J Biol Chem 279: 37771–37778, 2004. 462. Staruschenko A, Pochynyuk OM, Tong Q, Stockand JD. Ras couples phosphoinositide 3-OH kinase to the epithelial Na⫹ channel. Biochim Biophys Acta 1669: 108 –115, 2005. 463. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10: 513–525, 2009. 464. Stepanova OV, Chadin AV, Masiutin AG, Kulikova TG, Gurin Ia V, Sergeeva IA, Shirinskii VP. Rho-associated protein kinase is involved in establishing the contractile phenotype of cardiomyocytes. Biofizika 55: 880 – 885, 2010. 465. Stockand JD, Spier BJ, Worrell RT, Yue G, Al-Baldawi N, Eaton DC. Regulation of Na⫹ reabsorption by the aldosterone-induced small G protein K-Ras2A. J Biol Chem 274: 35449 –35454, 1999. 466. Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med 207: 881– 896, 2010. 467. Storey NM, O’Bryan JP, Armstrong DL. Rac and Rho mediate opposing hormonal regulation of the ether-a-go-go-related potassium channel. Curr Biol 12: 27–33, 2002. 468. Su L, Hattori M, Moriyama M, Murata N, Harazaki M, Kaibuchi K, Minato N. AF-6 controls integrin-mediated cell adhesion by regulating Rap1 activation through the specific recruitment of Rap1GTP and SPA-1. J Biol Chem 278: 15232–15238, 2003. 469. Sugden PH, Clerk A. Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid Redox Signal 8: 2111–2124, 2006. 470. Sugimoto K, Ishibashi T, Sawamura T, Inoue N, Kamioka M, Uekita H, Ohkawara H, Sakamoto T, Sakamoto N, Okamoto Y, Takuwa Y, Kakino A, Fujita Y, Tanaka T, Teramoto T, Maruyama Y, Takeishi Y. LOX-1-MT1-MMP axis is crucial for RhoA and Rac1 activation induced by oxidized low-density lipoprotein in endothelial cells. Cardiovasc Res 84: 127–136, 2009. 471. Sugita M, Sugita H, Kaneki M. Farnesyltransferase inhibitor, manumycin a, prevents atherosclerosis development and reduces oxidative stress in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 27: 1390 –1395, 2007. 479. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29: 465– 468, 2001. 480. Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, Ustaszewska A, Martin J, Bristow J, Carta C, Lepri F, Neri C, Vasta I, Gibson K, Curry CJ, Siguero JP, Digilio MC, Zampino G, Dallapiccola B, Bar-Sagi D, Gelb BD. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet 39: 75–79, 2007. 481. Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol 1: 2–26, 2010. 482. Thompson-Torgerson CS, Holowatz LA, Flavahan NA, Kenney WL. Rho kinasemediated local cold-induced cutaneous vasoconstriction is augmented in aged human skin. Am J Physiol Heart Circ Physiol 293: H30 –H36, 2007. 483. Thorburn A, Thorburn J, Chen SY, Powers S, Shubeita HE, Feramisco JR, Chien KR. HRas-dependent pathways can activate morphological and genetic markers of cardiac muscle cell hypertrophy. J Biol Chem 268: 2244 –2249, 1993. 484. Thorburn J, Xu S, Thorburn A. MAP kinase- and Rho-dependent signals interact to regulate gene expression but not actin morphology in cardiac muscle cells. EMBO J 16: 1888 –1900, 1997. 485. Tian Y, Autieri MV. Cytokine expression and AIF-1-mediated activation of Rac2 in vascular smooth muscle cells: a role for Rac2 in VSMC activation. Am J Physiol Cell Physiol 292: C841–C849, 2007. 486. Tian Y, Lei L, Cammarano M, Nekrasova T, Minden A. Essential role for the Pak4 protein kinase in extraembryonic tissue development and vessel formation. Mech Dev 126: 710 –720, 2009. 487. Tolle M, Pawlak A, Schuchardt M, Kawamura A, Tietge UJ, Lorkowski S, Keul P, Assmann G, Chun J, Levkau B, van der Giet M, Nofer JR. HDL-associated lysosphingolipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production. Arterioscler Thromb Vasc Biol 28: 1542–1548, 2008. 488. Torsoni AS, Fonseca PM, Crosara-Alberto DP, Franchini KG. Early activation of p160ROCK by pressure overload in rat heart. Am J Physiol Cell Physiol 284: C1411– C1419, 2003. 489. Tsimberidou AM, Chandhasin C, Kurzrock R. Farnesyltransferase inhibitors: where are we now? Expert Opin Investig Drugs 19: 1569 –1580, 2010. 490. Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389: 990 –994, 1997. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1717 LOIRAND, SAUZEAU, AND PACAUD 491. Ueno H, Yamamoto H, Ito S, Li JJ, Takeshita A. Adenovirus-mediated transfer of a dominant-negative H-ras suppresses neointimal formation in balloon-injured arteries in vivo. Arterioscler Thromb Vasc Biol 17: 898 –904, 1997. 509. Wang G, Zhu X, Xie W, Han P, Li K, Sun Z, Wang Y, Chen C, Song R, Cao C, Zhang J, Wu C, Liu J, Cheng H. Rad as a novel regulator of excitation-contraction coupling and beta-adrenergic signaling in heart. Circ Res 106: 317–327, 2010. 492. Urosevic J, Sauzeau V, Soto-Montenegro ML, Reig S, Desco M, Wright EM, Canamero M, Mulero F, Ortega S, Bustelo XR, Barbacid M. Constitutive activation of B-Raf in the mouse germ line provides a model for human cardio-facio-cutaneous syndrome. Proc Natl Acad Sci USA 108: 5015–5020, 2011. 510. Wang HW, Liu PY, Oyama N, Rikitake Y, Kitamoto S, Gitlin J, Liao JK, Boisvert WA. Deficiency of ROCK1 in bone marrow-derived cells protects against atherosclerosis in LDLR⫺/⫺ mice. FASEB J 22: 3561–3570, 2008. 493. Vahebi S, Kobayashi T, Warren CM, de Tombe PP, Solaro RJ. Functional effects of rho-kinase-dependent phosphorylation of specific sites on cardiac troponin. Circ Res 96: 740 –747, 2005. 494. Van Nieuw Amerongen GP, Beckers CM, Achekar ID, Zeeman S, Musters RJ, van Hinsbergh VW. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler Thromb Vasc Biol 27: 2332–2339, 2007. 495. Van Nieuw Amerongen GP, Musters RJ, Eringa EC, Sipkema P, van Hinsbergh VW. Thrombin-induced endothelial barrier disruption in intact microvessels: role of RhoA/ Rho kinase-myosin phosphatase axis. Am J Physiol Cell Physiol 294: C1234 –C1241, 2008. 496. Van Nieuw Amerongen GP, Vermeer MA, van Hinsbergh VW. Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol 20: E127–133, 2000. 497. Van Wetering S, van Buul JD, Quik S, Mul FP, Anthony EC, ten Klooster JP, Collard JG, Hordijk PL. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J Cell Sci 115: 1837–1846, 2002. 498. Vecchione C, Aretini A, Marino G, Bettarini U, Poulet R, Maffei A, Sbroggio M, Pastore L, Gentile MT, Notte A, Iorio L, Hirsch E, Tarone G, Lembo G. Selective Rac-1 inhibition protects from diabetes-induced vascular injury. Circ Res 98: 218 – 225, 2006. 499. Vecchione C, Carnevale D, Di Pardo A, Gentile MT, Damato A, Cocozza G, Antenucci G, Mascio G, Bettarini U, Landolfi A, Iorio L, Maffei A, Lembo G. Pressure-induced vascular oxidative stress is mediated through activation of integrin-linked kinase 1/betaPIX/Rac-1 pathway. Hypertension 54: 1028 –1034, 2009. 500. Vetter IR, Nowak C, Nishimoto T, Kuhlmann J, Wittinghofer A. Structure of a Ranbinding domain complexed with Ran bound to a GTP analogue: implications for nuclear transport. Nature 398: 39 – 46, 1999. 501. Vicente-Steijn R, Kolditz DP, Mahtab EA, Askar SF, Bax NA, VDG LM, Wisse LJ, Passier R, Pijnappels DA, Schalij MJ, Poelmann RE, Gittenberger DEGAC, Jongbloed MR. Electrical activation of sinus venosus myocardium and expression patterns of RhoA and Isl-1 in the chick embryo. J Cardiovasc Electrophysiol 21: 1284 –1292, 2010. 502. Vlasblom R, Muller A, Beckers CM, van Nieuw Amerongen GP, Zuidwijk MJ, van Hardeveld C, Paulus WJ, Simonides WS. RhoA-ROCK signaling is involved in contraction-mediated inhibition of SERCA2a expression in cardiomyocytes. Pflügers Arch 458: 785–793, 2009. 503. Voss AK, Gruss P, Thomas T. The guanine nucleotide exchange factor C3G is necessary for the formation of focal adhesions and vascular maturation. Development 130: 355–367, 2003. 504. Voss K, Stahl S, Schleider E, Ullrich S, Nickel J, Mueller TD, Felbor U. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 8: 249 –256, 2007. 505. Vouret-Craviari V, Boquet P, Pouyssegur J, Van Obberghen-Schilling E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol Biol Cell 9: 2639 –2653, 1998. 506. Vouret-Craviari V, Bourcier C, Boulter E, van Obberghen-Schilling E. Distinct signals via Rho GTPases and Src drive shape changes by thrombin and sphingosine-1-phosphate in endothelial cells. J Cell Sci 115: 2475–2484, 2002. 507. Vouret-Craviari V, Grall D, Van Obberghen-Schilling E. Modulation of Rho GTPase activity in endothelial cells by selective proteinase-activated receptor (PAR) agonists. J Thromb Haemost 1: 1103–1111, 2003. 508. Wang DZ, Hammond VE, Abud HE, Bertoncello I, McAvoy JW, Bowtell DD. Mutation in Sos1 dominantly enhances a weak allele of the EGFR, demonstrating a requirement for Sos1 in EGFR signaling and development. Genes Dev 11: 309 –320, 1997. 1718 511. Wang L, Hauser ER, Shah SH, Pericak-Vance MA, Haynes C, Crosslin D, Harris M, Nelson S, Hale AB, Granger CB, Haines JL, Jones CJ, Crossman D, Seo D, Gregory SG, Kraus WE, Goldschmidt-Clermont PJ, Vance JM. Peakwide mapping on chromosome 3q13 identifies the kalirin gene as a novel candidate gene for coronary artery disease. Am J Hum Genet 80: 650 – 663, 2007. 512. Wang L, Proud CG. Ras/Erk signaling is essential for activation of protein synthesis by Gq protein-coupled receptor agonists in adult cardiomyocytes. Circ Res 91: 821– 829, 2002. 513. Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, Surks HK. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res 104: 531–540, 2009. 514. Wang YX, da Cunha V, Martin-McNulty B, Vincelette J, Li W, Choy DF, Halks-Miller M, Mahmoudi M, Schroeder M, Johns A, Light DR, Dole WP. Inhibition of Rho-kinase by fasudil attenuated angiotensin II-induced cardiac hypertrophy in apolipoprotein E deficient mice. Eur J Pharmacol 512: 215–222, 2005. 515. Wang Z, Zhang Y, Gao M, Wang J, Wang Q, Wang X, Su L, Hou Y. Statin therapy for the prevention of atrial fibrillation: a meta-analysis of randomized controlled trials. Pharmacotherapy 31: 1051–1062, 2011. 516. Wassmann S, Laufs U, Baumer AT, Muller K, Konkol C, Sauer H, Bohm M, Nickenig G. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol 59: 646 – 654, 2001. 517. Wei BR, Martin PL, Hoover SB, Spehalski E, Kumar M, Hoenerhoff MJ, Rozenberg J, Vinson C, Simpson RM. Capacity for resolution of Ras-MAPK-initiated early pathogenic myocardial hypertrophy modeled in mice. Comp Med 61: 109 –118, 2011. 518. Wei BR, Simpson RM, Johann DJ, Dwyer JE, Prieto DA, Kumar M, Ye X, Luke B, Shive HR, Webster JD, Hoover SB, Veenstra TD, Blonder J. Proteomic profiling of H-RasG12V induced hypertrophic cardiomyopathy in transgenic mice using comparative LC-MS analysis of thin fresh-frozen tissue sections. J Proteome Res 2012. 519. Wei G, Srinivasan R, Cantemir-Stone CZ, Sharma SM, Santhanam R, Weinstein M, Muthusamy N, Man AK, Oshima RG, Leone G, Ostrowski MC. Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood 114: 1123– 1130, 2009. 520. Wei L, Imanaka-Yoshida K, Wang L, Zhan S, Schneider MD, DeMayo FJ, Schwartz RJ. Inhibition of Rho family GTPases by Rho GDP dissociation inhibitor disrupts cardiac morphogenesis and inhibits cardiomyocyte proliferation. Development 129: 1705– 1714, 2002. 521. Wei L, Roberts W, Wang L, Yamada M, Zhang S, Zhao Z, Rivkees SA, Schwartz RJ, Imanaka-Yoshida K. Rho kinases play an obligatory role in vertebrate embryonic organogenesis. Development 128: 2953–2962, 2001. 522. Wei L, Taffet GE, Khoury DS, Bo J, Li Y, Yatani A, Delaughter MC, Klevitsky R, Hewett TE, Robbins J, Michael LH, Schneider MD, Entman ML, Schwartz RJ. Disruption of Rho signaling results in progressive atrioventricular conduction defects while ventricular function remains preserved. FASEB J 18: 857– 859, 2004. 523. Wei Y, Zhang Y, Derewenda U, Liu X, Minor W, Nakamoto RK, Somlyo AV, Somlyo AP, Derewenda ZS. Crystal structure of RhoA-GDP and its functional implications. Nat Struct Biol 4: 699 –703, 1997. 524. Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol 3: 950 –957, 2001. 525. Westphal RS, Coffee RL Jr, Marotta A, Pelech SL, Wadzinski BE. Identification of kinase-phosphatase signaling modules composed of p70 S6 kinase-protein phosphatase 2A (PP2A) and p21-activated kinase-PP2A. J Biol Chem 274: 687– 692, 1999. 526. Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res 301: 43– 49, 2004. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org SMALL G PROTEINS IN THE CARDIOVASCULAR SYSTEM 527. Whitehead KJ, Chan AC, Navankasattusas S, Koh W, London NR, Ling J, Mayo AH, Drakos SG, Jones CA, Zhu W, Marchuk DA, Davis GE, Li DY. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med 15: 177–184, 2009. 528. Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development 131: 1437–1448, 2004. 529. Wilk-Blaszczak MA, Singer WD, Quill T, Miller B, Frost JA, Sternweis PC, Belardetti F. The monomeric G-proteins Rac1 and/or Cdc42 are required for the inhibition of voltage-dependent calcium current by bradykinin. J Neurosci 17: 4094 – 4100, 1997. 530. Wimmer R, Cseh B, Maier B, Scherrer K, Baccarini M. Angiogenic sprouting requires the fine tuning of endothelial cell cohesion by the Raf-1/Rok-alpha complex. Dev Cell 22: 158 –171, 2012. 531. Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12–G13-LARG-mediated signaling in vascular smooth muscle is required for saltinduced hypertension. Nat Med 14: 64 – 68, 2008. 532. Wirth A, Schroeter M, Kock-Hauser C, Manser E, Chalovich JM, De Lanerolle P, Pfitzer G. Inhibition of contraction and myosin light chain phosphorylation in guineapig smooth muscle by p21-activated kinase 1. J Physiol 549: 489 –500, 2003. 533. Wittchen ES. Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front Biosci 14: 2522–2545, 2009. 534. Wittchen ES, Aghajanian A, Burridge K. Isoform-specific differences between Rap1A and Rap1B GTPases in the formation of endothelial cell junctions. Small GTPases 2: 65–76, 2011. 535. Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci 114: 1343–1355, 2001. 536. Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vasc Pharmacol 39: 187–199, 2002. 537. Wojnowski L, Zimmer AM, Beck TW, Hahn H, Bernal R, Rapp UR, Zimmer A. Endothelial apoptosis in Braf-deficient mice. Nat Genet 16: 293–297, 1997. 538. Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, Dominiak P, Liao JK. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/ protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol 24: 1842–1847, 2004. 539. Woolfolk EA, Eguchi S, Ohtsu H, Nakashima H, Ueno H, Gerthoffer WT, Motley ED. Angiotensin II-induced activation of p21-activated kinase 1 requires Ca2⫹ and protein kinase C␦ in vascular smooth muscle cells. Am J Physiol Cell Physiol 289: C1286 – C1294, 2005. 540. Work LM, McPhaden AR, Pyne NJ, Pyne S, Wadsworth RM, Wainwright CL. Shortterm local delivery of an inhibitor of Ras farnesyltransferase prevents neointima formation in vivo after porcine coronary balloon angioplasty. Circulation 104: 1538 – 1543, 2001. 541. Wu CH, Lin CS, Hung JS, Wu CJ, Lo PH, Jin G, Shyy YJ, Mao SJ, Chien S. Inhibition of neointimal formation in porcine coronary artery by a Ras mutant. J Surg Res 99: 100 –106, 2001. 542. Wu G, Yussman MG, Barrett TJ, Hahn HS, Osinska H, Hilliard GM, Wang X, Toyokawa T, Yatani A, Lynch RA, Robbins J, Dorn GW 2nd. Increased myocardial Rab GTPase expression: a consequence and cause of cardiomyopathy. Circ Res 89: 1130 – 1137, 2001. 543. Wuertz CM, Lorincz A, Vettel C, Thomas MA, Wieland T, Lutz S. p63RhoGEF–a key mediator of angiotensin II-dependent signaling and processes in vascular smooth muscle cells. FASEB J 24: 4865– 4876, 2010. 544. Xiang SY, Vanhoutte D, Del Re DP, Purcell NH, Ling H, Banerjee I, Bossuyt J, Lang RA, Zheng Y, Matkovich SJ, Miyamoto S, Molkentin JD, Dorn GW 2nd, Brown JH. RhoA protects the mouse heart against ischemia/reperfusion injury. J Clin Invest 121: 3269 – 3276, 2011. 545. Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, Sawyer DB. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol 282: C926 –C934, 2002. 546. Xiaolu D, Jing P, Fang H, Lifen Y, Liwen W, Ciliu Z, Fei Y. Role of p115RhoGEF in lipopolysaccharide-induced mouse brain microvascular endothelial barrier dysfunction. Brain Res 1387: 1–7, 2011. 547. Xu GF, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62: 599 – 608, 1990. 548. Xu J, Ismat FA, Wang T, Lu MM, Antonucci N, Epstein JA. Cardiomyocyte-specific loss of neurofibromin promotes cardiac hypertrophy and dysfunction. Circ Res 105: 304 – 311, 2009. 549. Xu J, Ismat FA, Wang T, Yang J, Epstein JA. NF1 regulates a Ras-dependent vascular smooth muscle proliferative injury response. Circulation 116: 2148 –2156, 2007. 550. Xu K, Chong DC, Rankin SA, Zorn AM, Cleaver O. Rasip1 is required for endothelial cell motility, angiogenesis and vessel formation. Dev Biol 329: 269 –279, 2009. 551. Xu X, Marx SO, Colecraft HM. Molecular mechanisms, and selective pharmacological rescue, of Rem-inhibited CaV1.2 channels in heart. Circ Res 107: 620 – 630, 2010. 552. Yada H, Murata M, Shimoda K, Yuasa S, Kawaguchi H, Ieda M, Adachi T, Murata M, Ogawa S, Fukuda K. Dominant negative suppression of Rad leads to QT prolongation and causes ventricular arrhythmias via modulation of L-type Ca2⫹ channels in the heart. Circ Res 101: 69 –77, 2007. 553. Yamada T, Sakisaka T, Hisata S, Baba T, Takai Y. RA-RhoGAP, Rap-activated Rho GTPase-activating protein implicated in neurite outgrowth through Rho. J Biol Chem 280: 33026 –33034, 2005. 554. Yamaguchi O, Watanabe T, Nishida K, Kashiwase K, Higuchi Y, Takeda T, Hikoso S, Hirotani S, Asahi M, Taniike M, Nakai A, Tsujimoto I, Matsumura Y, Miyazaki J, Chien KR, Matsuzawa A, Sadamitsu C, Ichijo H, Baccarini M, Hori M, Otsu K. Cardiacspecific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J Clin Invest 114: 937–943, 2004. 555. Yamakawa T, Tanaka S, Numaguchi K, Yamakawa Y, Motley ED, Ichihara S, Inagami T. Involvement of Rho-kinase in angiotensin II-induced hypertrophy of rat vascular smooth muscle cells. Hypertension 35: 313–318, 2000. 556. Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, Molina CA, Yatani A, Vatner DE, Vatner SF, Sadoshima J. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest 111: 1463–1474, 2003. 557. Yamazaki T, Komuro I, Shiojima I, Yazaki Y. The molecular mechanism of cardiac hypertrophy and failure. Ann NY Acad Sci 874: 38 – 48, 1999. 558. Yamazaki T, Komuro I, Yazaki Y. Signalling pathways for cardiac hypertrophy. Cell Signal 10: 693– 698, 1998. 559. Yan J, Li F, Ingram DA, Quilliam LA. Rap1a is a key regulator of fibroblast growth factor 2-induced angiogenesis and together with Rap1b controls human endothelial cell functions. Mol Cell Biol 28: 5803–5810, 2008. 560. Yanazume T, Hasegawa K, Wada H, Morimoto T, Abe M, Kawamura T, Sasayama S. Rho/ROCK pathway contributes to the activation of extracellular signal-regulated kinase/GATA-4 during myocardial cell hypertrophy. J Biol Chem 277: 8618 – 8625, 2002. 561. Yang T, Suhail Y, Dalton S, Kernan T, Colecraft HM. Genetically encoded molecules for inducibly inactivating CaV channels. Nat Chem Biol 3: 795– 804, 2007. 562. Yang T, Xu X, Kernan T, Wu V, Colecraft HM. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol 588: 1665–1681, 2010. 563. Yang X, Li Q, Lin X, Ma Y, Yue X, Tao Z, Wang F, McKeehan WL, Wei L, Schwartz RJ, Chang J. Mechanism of fibrotic cardiomyopathy in mice expressing truncated Rhoassociated coiled-coil protein kinase 1. FASEB J 26: 2105–2116, 2012. 564. Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, Motoshima H, Taguchi T, Sonoda K, Kukidome D, Takuwa Y, Kawada T, Brownlee M, Nishikawa T, Araki E. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinasedependent cyclooxygenase-2 expression in macrophages. Circ Res 100: 1442–1451, 2007. 565. Yatani A, Irie K, Otani T, Abdellatif M, Wei L. RhoA GTPase regulates L-type Ca2⫹ currents in cardiac myocytes. Am J Physiol Heart Circ Physiol 288: H650 –H659, 2005. Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org 1719 LOIRAND, SAUZEAU, AND PACAUD 566. Yatani A, Okabe K, Polakis P, Halenbeck R, McCormick F, Brown AM. ras p21 and GAP inhibit coupling of muscarinic receptors to atrial K⫹ channels. Cell 61: 769 –776, 1990. 579. Zhang J, Chang L, Chen C, Zhang M, Luo Y, Hamblin M, Villacorta L, Xiong JW, Chen YE, Zhang J, Zhu X. Rad GTPase inhibits cardiac fibrosis through connective tissue growth factor. Cardiovasc Res 91: 90 –98, 2011. 567. Yatani A, Quilliam LA, Brown AM, Bokoch GM. Rap1A antagonizes the ability of Ras and Ras-Gap to inhibit muscarinic K⫹ channels. J Biol Chem 266: 22222–22226, 1991. 580. Zhang J, Li XX, Bian HJ, Liu XB, Ji XP, Zhang Y. Inhibition of the activity of Rho-kinase reduces cardiomyocyte apoptosis in heart ischemia/reperfusion via suppressing JNKmediated AIF translocation. Clin Chim Acta 401: 76 – 80, 2009. 568. Ying Z, Giachini FR, Tostes RC, Webb RC. PYK2/PDZ-RhoGEF links Ca2⫹ signaling to RhoA. Arterioscler Thromb Vasc Biol 29: 1657–1663, 2009. 569. Ying Z, Jin L, Dorrance AM, Webb RC. Increased expression of mRNA for regulator of G protein signaling domain-containing Rho guanine nucleotide exchange factors in aorta from stroke-prone spontaneously hypertensive rats. Am J Hypertens 17: 981– 985, 2004. 570. Yoshikawa Y, Satoh T, Tamura T, Wei P, Bilasy SE, Edamatsu H, Aiba A, Katagiri K, Kinashi T, Nakao K, Kataoka T. The M-Ras-RA-GEF-2-Rap1 pathway mediates tumor necrosis factor-alpha dependent regulation of integrin activation in splenocytes. Mol Biol Cell 18: 2949 –2959, 2007. 571. Young A, Lyons J, Miller AL, Phan VT, Alarcon IR, McCormick F. Ras signaling and therapies. Adv Cancer Res 102: 1–17, 2009. 572. Yudin D, Fainzilber M. Ran on tracks– cytoplasmic roles for a nuclear regulator. J Cell Sci 122: 587–593, 2009. 573. Yue TL, Wang C, Gu JL, Ma XL, Kumar S, Lee JC, Feuerstein GZ, Thomas H, Maleeff B, Ohlstein EH. Inhibition of extracellular signal-regulated kinase enhances ischemia/ reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res 86: 692– 699, 2000. 574. Yutzey KE, Colbert M, Robbins J. Ras-related signaling pathways in valve development: ebb and flow. Physiology 20: 390 –397, 2005. 575. Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, Marchuk DA. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet 14: 2521–2531, 2005. 576. Zeidan A, Javadov S, Karmazyn M. Essential role of Rho/ROCK-dependent processes and actin dynamics in mediating leptin-induced hypertrophy in rat neonatal ventricular myocytes. Cardiovasc Res 72: 101–111, 2006. 581. Zhang S, Ren J, Khan MF, Cheng AM, Abendschein D, Muslin AJ. Grb2 is required for the development of neointima in response to vascular injury. Arterioscler Thromb Vasc Biol 23: 1788 –1793, 2003. 582. Zhang YM, Bo J, Taffet GE, Chang J, Shi J, Reddy AK, Michael LH, Schneider MD, Entman ML, Schwartz RJ, Wei L. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. FASEB J 20: 916 –925, 2006. 583. Zhao Z, Rivkees SA. Rho-associated kinases play a role in endocardial cell differentiation and migration. Dev Biol 275: 183–191, 2004. 584. Zhao Z, Rivkees SA. Tissue-specific expression of GTPas RalA and RalB during embryogenesis and regulation by epithelial-mesenchymal interaction. Mech Dev 97: 201– 204, 2000. 585. Zheng M, Dilly K, Dos Santos Cruz J, Li M, Gu Y, Ursitti JA, Chen J, Ross J, Jr, Chien KR, Lederer JW, Wang Y. Sarcoplasmic reticulum calcium defect in Ras-induced hypertrophic cardiomyopathy heart. Am J Physiol Heart Circ Physiol 286: H424 –H433, 2004. 586. Zhou Q, Liao JK. Pleiotropic effects of statins. Basic research and clinical perspectives. Circ J 74: 818 – 826, 2010. 587. Zhou Q, Liao JK. Rho kinase: an important mediator of atherosclerosis and vascular disease. Curr Pharm Des 15: 3108 –3115, 2009. 588. Zieba BJ, Artamonov MV, Jin L, Momotani K, Ho R, Franke AS, Neppl RL, Stevenson AS, Khromov AS, Chrzanowska-Wodnicka M, Somlyo AV. The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J Biol Chem 286: 16681–16692, 2011. 577. Zeisler E, Becker S. Generalized lentigo: its relation to systematic nonelevated nevi. Arch Dermat Syph 33: 109 –125, 1936. 589. Zimmerman MC, Dunlay RP, Lazartigues E, Zhang Y, Sharma RV, Engelhardt JF, Davisson RL. Requirement for Rac1-dependent NADPH oxidase in the cardiovascular and dipsogenic actions of angiotensin II in the brain. Circ Res 95: 532–539, 2004. 578. Zhang J, Bian HJ, Li XX, Liu XB, Sun JP, Li N, Zhang Y, Ji XP. ERK-MAPK signaling opposes rho-kinase to reduce cardiomyocyte apoptosis in heart ischemic preconditioning. Mol Med 16: 307–315, 2010. 590. Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res 95: 210 –216, 2004. 1720 Physiol Rev • VOL 93 • OCTOBER 2013 • www.prv.org