ELECTRONIC CONNECTION BETWEEN THE C AND ITS ROLE IN REGULATION OF
advertisement

Physiol Rev 95: 219 –243, 2015 doi:10.1152/physrev.00006.2014 ELECTRONIC CONNECTION BETWEEN THE QUINONE AND CYTOCHROME C REDOX POOLS AND ITS ROLE IN REGULATION OF MITOCHONDRIAL ELECTRON TRANSPORT AND REDOX SIGNALING Marcin Sarewicz and Artur Osyczka Department of Molecular Biophysics, Faculty of Biochemistry, Biophysics, and Biotechnology, Jagiellonian University, Kraków, Poland Sarewicz M, Osyczka A. Electronic Connection Between the Quinone and Cytochrome c Redox Pools and Its Role in Regulation of Mitochondrial Electron Transport and Redox Signaling. Physiol Rev 95: 219 –243, 2015; doi:10.1152/physrev.00006.2014.—Mitochondrial respiration, an important bioenergetic process, relies on operation of four membranous enzymatic complexes linked functionally by mobile, freely diffusible elements: quinone molecules in the membrane and water-soluble cytochromes c in the intermembrane space. One of the mitochondrial complexes, complex III (cytochrome bc1 or ubiquinol: cytochrome c oxidoreductase), provides an electronic connection between these two diffusible redox pools linking in a fully reversible manner two-electron quinone oxidation/reduction with one-electron cytochrome c reduction/oxidation. Several features of this homodimeric enzyme implicate that in addition to its well-defined function of contributing to generation of proton-motive force, cytochrome bc1 may be a physiologically important point of regulation of electron flow acting as a sensor of the redox state of mitochondria that actively responds to changes in bioenergetic conditions. These features include the following: the opposing redox reactions at quinone catalytic sites located on the opposite sides of the membrane, the inter-monomer electronic connection that functionally links four quinone binding sites of a dimer into an H-shaped electron transfer system, as well as the potential to generate superoxide and release it to the intermembrane space where it can be engaged in redox signaling pathways. Here we highlight recent advances in understanding how cytochrome bc1 may accomplish this regulatory physiological function, what is known and remains unknown about catalytic and side reactions within the quinone binding sites and electron transfers through the cofactor chains connecting those sites with the substrate redox pools. We also discuss the developed molecular mechanisms in the context of physiology of mitochondria. L I. II. III. IV. V. VI. VII. VIII. IX. X. XI. INTRODUCTION PRINCIPLES OF ELECTRON TRANSFER... QUINONES AND THE Q POOL CYTOCHROME c AND THE C POOL REDOX CONNECTION OF THE Q POOL... COFACTOR CHAINS OF CYTOCHROME... INTERACTIONS OF QUINONE AND... GENERATION OF REACTIVE OXYGEN... H-SHAPED ELECTRON TRANSFER... COMPOUNDS THAT INFLUENCE THE... CONCLUSIONS 219 221 222 224 224 226 228 231 233 234 235 I. INTRODUCTION Energy conversion is one of the fundamental processes supporting life of all organisms. It gives power to maintain homeostasis inside each cell and among cells. To achieve this, the process of energy conversion itself must be efficient, safe, and appropriately regulated. As the universal source of energy directly available for the cells to do useful work is stored in the concentrations of ATP, ADP, and Pi that are kept far from their equilibrium concentrations, the bioenergetic processes evolved to use the energy released from various chemical or physical processes to power the synthesis of ATP from ADP and Pi. While some amounts of ATP can be synthesized by water-soluble enzymes, the major fraction of ATP synthesis is associated with the operation of enzymes that are assembled to form specific bioenergetic paths (194). These enzymes are embedded in biological membranes involved in energy conversion (bioenergetic membranes). The factor that unifies a diversity of substrates used by these enzymes and a diversity of molecular mechanisms of conversion of these substrates is a proton-motive force (PMF), a central element of Peter Mitchell’s chemiosmotic theory (176, 179). PMF depends on the difference of pH values over a bioenergetic membrane (⌬pH) and the membrane potential (⌬⌿; difference in electrical po- 0031-9333/15 Copyright © 2015 the American Physiological Society 219 MARCIN SAREWICZ AND ARTUR OSYCZKA tentials between two aqueous compartments separated by a membrane). PMF, generated by the enzymes that use external source of energy to transfer protons between compartments separated by a bioenergetic membrane, forces ATP-synthase to convert ADP and Pi into ATP. Generation of PMF commonly relies on operation of several enzymes that are functionally linked together. In mitochondria, such assembly is located in the mitochondrial inner membrane forming the mitochondrial respiratory chain (FIGURE 1). The chain usually consists of four complexes, termed complex I to IV, which catalyze the transfer of electrons from substrates, such as NADH or succinate, to the final electron acceptor O2. Three of these complexes (complex I, III, and IV) couple the electron transfer with proton translocation across the membrane. These enzymes catalyze step-by-step electron transfer from the molecules of lower redox midpoint potential1 to the molecules of higher midpoint redox potential, and the stepwise release of energy in these reactions is used to translocate protons across the membrane. Within the protein, electrons travel through the redox cofactors (like hemes or iron-sulfur clusters) that form chains connecting various catalytic sites (4, 183). The 1 The term redox midpoint potential (Em) of a redox couple commonly used in biology is a derivative of “standard redox potential” (E0) and refers to the “actual redox potential” (Eh) at pH other than 0 (typically pH 7) for which concentration of oxidized and reduced forms are equal. E0 is equal to Eh at pH 0, under which concentrations of oxidized and reduced forms are equal and their activities are maintained at unity. cofactor chains and catalytic sites of respiratory complexes are linked together by small electron carriers that move between complexes. These carriers form two redox pools, each confined to the environment of different polarity. One pool, formed by quinone molecules (the Q pool), is confined to the lipid phase of inner mitochondrial membrane while the second pool, formed by cytochrome c (the C pool), is confined to the water phase of mitochondrial intermembrane space. Such a separation prevents energy-wasting direct electron exchange between the pools despite a significant difference in redox midpoint potential of quinone and cytochrome c (FIGURE 2). This is critically important for the efficiency of mitochondrial respiration. It is equally important that electrons do exchange between these two pools in the energy-conserving process. Such connection is secured by the operation of one of the complexes of mitochondrial respiratory chain. This complex, named cytochrome bc1 (EC 1.10.2.2; quinol-cytochrome c-reductase, complex III), catalyzes a net reaction of oxidation of ubihydroquinone (UQH2)2 2 Abbreviations for quinone nomenclature used in the text: USQ⫺, semiquinone anion; USQH, protonated semiquinone; USQ, general description of semiquinone radical regardless of its protonation state (in some cases it is used when protonation state is unknown); USQo, semiquinone in the Qo site; USQi, semiquinone in the Qi site; UQ, ubiquinone (fully deprotonated); UQH2, ubihydroquinone (ubiquinol); Q or quinone, general reference to quinone irrespective of the redox and protonation state of the molecule. FIGURE 1. Components of mitochondrial electron transport chain: membranous complexes I–IV linked by diffusible quinone (yellow sticks) in the membrane (forming the Q pool) and cytochrome c in the intermembrane space (forming the C pool). Cytochrome bc1 (complex III) connects electronically the Q pool with the C pool. 220 Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA 400 cytochrome c2 cytochrome c Midpoint redox potential [mV] 300 200 100 ΔG qu ino ne FIGURE 2. Dependence of average Em of the Q pool (green) and Em of mitochondrial C pool (red) on pH. The energy associated with cytochrome bc1-catalyzed electron transfer between the pools (⌬G) increases upon rising pH. Bacterial cytochromes c2 (blue) have generally higher Em than mitochondrial counterparts. 0 -100 -200 -300 5 6 7 8 9 10 11 pH and reduction of cytochrome c (20, 56, 65). The energy released during this electron transfer powers the reaction of oxidation of UQH2 and reduction of ubiquinone (UQ), each taking place at the distinct catalytic site located on the opposite side of the membrane. At one site (Qo site) UQH2 is oxidized and protons are released, while at the other site (Qi site) UQ is reduced and protons are uptaken. The joint action of these two catalytic sites, as originally proposed by Peter Mitchell in his Q cycle (177), leads to a net translocation of protons across the membrane contributing to PMF. This defines a basic bioenergetic role of this enzyme. An unusual feature of this enzyme, described on the basis of the Q cycle, is that the product of one catalytic site becomes the substrate for the second site, and vice versa (i.e., UQ produced in the Qo site is the substrate for reaction in the Qi site, while UQH2 produced in the Qi site is the substrate for reaction in the Qo site). This, together with the fact that these sites are connected with the Q and C pools, makes cytochrome bc1 a potential point of the regulation of the mitochondrial respiration at molecular level. The other potential role of this enzyme is related to the observation that under some conditions cytochrome bc1 can generate reactive oxygen species (ROS) in the form of superoxide (26, 83). In fact, cytochrome bc1 is considered a second, after complex I, source of ROS delivered from components of mitochondrial respiratory chain (33, 47). ROS, according to current views, cannot only be damaging to the cells, but also, in certain instances, were postulated to be important in cellular signaling (97, 122, 191, 240). Indeed, the superoxide generated by cytochrome bc1 can be related to both types of effects associated with ROS generation. Understanding of the energetic and regulatory roles of cytochrome bc1 requires the unification of knowledge on how physiological and energy-conserving reactions are supported while the energy-wasting and often thermodynamically favorable side reactions are avoided. The following text discusses current view on efficiency, safety, and regulation of the physiological connection between the Q pool and the C pool secured by cytochrome bc1 in the context of mitochondrial function. It begins with a brief outline of basic principles of electron transfer and the organization of electron transfer chains followed by a summary of selected properties of quinone and cytochrome c. It then discusses the molecular and physiological aspects of the mechanisms of cytochrome bc1 interactions with its substrates, of quinone redox reactions within the catalytic sites, and of ROS production by cytochrome bc1 to end with an emerging new physiological perspective afforded by recent studies that exposed a dimeric function of the enzyme. II. PRINCIPLES OF ELECTRON TRANSFER BETWEEN COFACTORS IN PROTEINS It is generally accepted that biological electron transfer is a process of tunneling, the quantum mechanical phenomenon in which an electron can cross a barrier of insulating protein medium between the redox active donor and acceptor. The rate of the tunneling is the product of two terms. The first is an electronic term arising from the strength of the coupling of electron donor/acceptor wave functions that decreases exponentially upon increasing the distance between donor and acceptor. The second term depends on both the driving force (⌬G, proportional to the difference in redox midpoint potentials between donor and acceptor) and the reorgani- Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 221 MARCIN SAREWICZ AND ARTUR OSYCZKA zation energy required to change the nuclear coordinates upon electron transfer (14, 170, 184, 185, 203). Within the protein matrix the dominant factor that influences the electron transfer rate is the distance between redox centers. The physiologically relevant electron transfer rate (usually millisecond or less to support the turnover of the enzymes) requires that the edge-to-edge distance between donor and acceptor should not exceed ⬃14 Å (203). Indeed, the geometric arrangement of cofactors that form chains in known proteins follows this rule, and the interacting redox centers are usually within 14 Å (182, 183). The reactions that are unproductive and adverse from physiological point of view usually face distances larger than 14 Å, which makes the rates slow enough to be insignificant. However, there are cases where such long-distance suppression of unproductive reactions is not possible and other mechanisms must be involved. One of the intriguing examples includes the Qo site of cytochrome bc1 (199, 200). The second important factor influencing electron transfer rate is the driving force, which in general should be considered as the free energy released during a spontaneous reaction of electron transfer from the low- to high-potential cofactor. Thus, to push electron along the chain in specific direction, the consecutive cofactors exhibit the increasing midpoint redox potentials. Interestingly, many cofactor chains possess energetically unfavorable “uphill” steps related to the presence of cofactors of lower potentials flanked by the two cofactors of higher potentials. Although the physiological significance of such arrangement is not clear, it appears that strikingly uphill electron transfer can occur at functionally relevant times provided that there is an adequate proximity between the centers and an overall driving force along the chain (5, 158, 203). and this observation was adopted by Peter Michell to propose the mechanism of protonmotive Q cycle (177, 178). Nowadays, apart from the bioenergetic function, quinones are considered to be engaged in a variety of cellular functions that are not only associated with their redox properties but also with the capability to change the fluidity of the lipid bilayers (48, 96, 256). The later point is supported by the fact that occurrence of quinones is not restricted to mitochondrial membrane (91), and the changes in their content is also correlated with age and with several diseases (10, 86, 87, 138, 156, 167, 263). While eukaryotic cells use two major types of quinones as elements of energy conversion systems: ubiquinones (or coenzyme Q) in respiration and plastoquinone (present in membranes of chloroplasts) in photosynthesis, prokaryotic cells can also use other types of quinones for those purposes. One example includes a variety of menaquinonerelated compounds (198) that are evolutionary more ancient than ubiquinones and plastoquinones and, by virtue of their low redox midpoint potential, are proposed to be engaged in bioenergetics of cells living in reducing atmosphere of preoxygenic eon (227). On the other hand, in humans, some menaquinones (identified as vitamin K) are also present but play other roles (24, 28). A. Redox Properties Benzoquinone ring of quinone may exist in nine different redox states (212) that result from three possible electronic and three protonation states (FIGURE 3). However, only six UQ USQ- UQ2- UQH+ USQH UQH- UQH22+ USQH2+ UQH2 III. QUINONES AND THE Q POOL Quinone derivatives are ubiquitous constituents of almost all lipid membranes of all organisms. Only rare exceptions are recognized in some prokaryotic cells of obligatory fermentative bacteria and methanogenic archae (198). Quinones are redox active compounds with capability to uptake/release two protons and two electrons (213, 214). The transitions between reduced and oxidized form involve formation of an intermediate semiquinone (USQ) which is an unstable oxygen-centered free radical (161). The role of isoprenoid quinones in the bioenergetics of cells was first proposed in the late 1950s (58). The observation that extraction of quinones from bioenergetic membranes inhibited the activity of mitochondrial complexes led to the conclusion that quinones are responsible for shuttling electrons between respiratory complexes (92, 146, 196). Further experiments revealed that semiquinone intermediate is formed during the catalytic cycle of cytochrome bc1 (265), 222 Em(UQ/UQH2) = [Em(USQ/UQH2) + Em(UQ/USQ)]/2 Kstab(USQ) = [USQ]2 [UQ] [UQH2] FIGURE 3. Possible redox and protonation states of quinone molecules and definition of Em of UQ/UQH2 couple and Kstab of semiquinone. The states that can be detected experimentally in solution or in catalytic sites of proteins are shown in black. Gray represents the states not present in biological environment. Vertical and horizontal arrows denote steps of protonation and reduction of the molecules, respectively. See References 115, 212. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA states were detected experimentally. Among these six states, only two are found to be relatively stable in bulk lipid phase (115, 281). However, in catalytic sites, the number of stable states can be larger. The redox midpoint potential of ubiquinone/ubiquinol (UQ/UQH2) couple [Em(UQ/UQH2) in FIGURE 3] is usually described as an average redox potential of two redox couples: ubiquinone/semiquinone (UQ/USQ) and semiquinone/ubihydroquinone (USQ/UQH2) (217). The difference between the redox midpoint potentials between these two couples defines the stability constant for USQ (Kstab in FIGURE 3): the larger the difference, the lower equilibrium concentration of USQ (79, 200). Kstab of USQ in bulk lipid phase is very low; therefore, in membranes only, UQ and UQH2 forms are present, at least in physiological pH. Nevertheless, the quinone binding sites within proteins often stabilize USQ as intermediate state of the catalytic cycles of the enzymes (115, 161). The degree of the stabilization of USQ may differ significantly from site to site. B. The Q Pool The size of the Q pool (160) may vary depending on type of membrane, organism, and tissue (1). In bioenergetic membrane, quinones are in excess in relation to the membranous complexes of electron transport chain. In mitochondria of mammals and plants, the estimated amount of quinone molecules per cytochrome oxidase is in the range of 6 – 8, while in yeast it is larger (⬃36) (124). In humans, the size of the Q pool has been found to increase within the first 20 years of life, then it decreases to eventually reach the size that is smaller compared with the size after birth (138). The size of the Q pool may be modified by certain diseases and even changed upon the progress of a disease (256). In general, these types of changes are expected to influence the bioenergetic function of mitochondria and perhaps also vary the contribution of the Q pool in the overall antioxidative activity of mitochondria. One of the most important parameters of the Q pool is its redox state defined as the ratio of UQ to total quinone content in membrane. The redox state of the Q pool is directly related to the bioenergetic state of the cells and is considered as one of the markers of oxidative stress (246, 271). The redox state of the Q pool is expected to change depending on the accessibility of the cells to oxygen, as for example observed in hypoxia where level of UQ increases in some tissues (105). Interestingly, hypoxia also induces a decrease in a size of the Q pool. The redox state of the Q pool may also change in diseases, as reported for cardiovascular diseases or cancer (256). Early experiments on the kinetics of electron transfer revealed that the Q pool creates rather homogeneous ensemble of molecules rapidly diffusing within lipid bilayer, being in equilibrium with binding sites of proteins (105, 146). An alternative view assumes the compartmentalization of quinone molecules into separate pools within the membrane (107). The estimation of the amount of quinone molecules that are bound to the proteins in mammals is in the range of 10 –32% of the total quinone (106, 159). Mobile molecules of quinones in membrane can collide with each other, which theoretically can lead to self-exchange of electrons between the colliding molecules. This reaction, however, in lipid environment seems unlikely, or occurs at a very low rate, as it requires deprotonation of UQH2 (213, 214). C. Other Functions of Quinones Quinones, besides playing a crucial role in energy conversion, are one of the most important antioxidants in the cells. It has been shown that UQH2 is effective in inhibiting both the initiation and propagation of lipid peroxidation chain reaction (18). In these reactions, UQH2 undergoes one-electron oxidation to USQ by reducing perferryl radicals or lipid peroxyl radicals. As USQ is a quite reactive electron donor for molecular oxygen, it must be neutralized effectively. In humans, USQ is reduced to UQH2 by ␣-tocopherol (vitamin E). However, there are evidences that UQH2 is still an efficient antioxidant compound even in the absence of vitamin E (104). This requires the existence of other mechanisms of neutralization of semiquinone radicals without engagement of vitamin E, possibly via dismutation (25). The interest in antioxidant properties of quinones became inspiration for synthesis of water-soluble derivatives such as MitoQ or SkQ (7, 8, 134, 235). These compounds penetrate into mitochondrial intermembrane space where they undergo reduction to hydroquinone form by the enzymes of respiratory chain. At low concentrations, SkQ was shown to prevent hydroxyl-radical-mediated cardiolipin peroxidation and inhibited ROS-induced apoptosis and necrosis of human fibroblasts (7, 235). It is now usually considered that the antioxidant properties of quinones may have a beneficiary effect on health and the lifespan. This has drawn the attention of the pharmacological industry to produce dietary supplements containing ubiquinone (coenzyme Q). However, understanding of the complex effect of quinone on an organism and its relation to the lifespan is still incomplete, especially in the view of reports of the increased lifespan of the nematode Caenorhabditis elegans under ubiquinone deficiency proposed to affect metabolism of the nematode cells (155, 180). Another possible function of quinone is related to the action of the uncoupling proteins, which in the inner mitochondrial membrane translocate H⫹ down the electrochemical proton gradient. This transport, not coupled to oxidative phosphorylation, leads to the dissipation of energy stored in PMF and generates heat. It was reported that quinone is an obligatory cofactor for uncoupling protein function (85), Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 223 MARCIN SAREWICZ AND ARTUR OSYCZKA and the regulation by quinone depends on its redox state (243). Indeed, UQH2 but not UQ functions as a negative regulator of uncoupling proteins inhibition by purine nucleotides (267). In addition to the functions discussed above, this molecule appears to be engaged in other molecular reactions of the cell, as for example in modulating the action of mitochondrial permeability pore (101). The detailed description of all postulated functions of quinones is beyond the scope of this review. IV. CYTOCHROME c AND THE C POOL Mitochondrial cytochrome c, since its description by Keilin in 1925 (139), became one of the best-characterized proteins (181, 229). It is a small, water-soluble, globular protein containing one high-potential heme that undergoes one electron oxidation/reduction. In mitochondrial respiration, cytochrome c works as a mobile one-electron carrier connecting cytochrome bc1 with complex IV (119). In living cells, the localization of cytochrome c is restricted to the intermembrane space of mitochondria. Any release of this protein to cytoplasm triggers an apoptotic pathway in which cytochrome c becomes a part of apoptosome which activates capsase-9 (3, 166, 211). Cytochrome c also appears to act as a cardiolipin oxygenase during early stages of apoptosis (137). Cytochrome c in solution can undergo an electron selfexchange reaction that refers to an electron transfer between cytochromes c without involvement of enzymes (81, 116, 170, 181). The physiological meaning of this reaction is not clear. In the textbook schemes of mitochondrial electron transport chain, this reaction is usually not considered; as for mitochondrial cytochrome c, it is relatively slow. Consequently, the connection between cytochrome bc1 and complex IV is described as a process in which a single molecule of cytochrome c takes electron from cytochrome bc1, then this molecule diffuses to complex IV where it undergoes oxidation. However, taking into account the effect of macromolecular crowding (89) and high concentration of cytochrome c in the intermembrane space (0.1–5 mM) (102), the self-exchange of electrons between cytochrome c molecules in the pool might also take place. In this context, the spatial distribution of electrons within the C pool, involving both the diffusion of molecules and the self-exchange reaction, would differ from the spatial distribution of electrons within the Q pool involving just the diffusion of quinone molecules that do not undergo self-exchange at significant rates. Interestingly, the measured rates of selfexchange reaction between bacterial cytochromes c, which have a lower net charge comparing to mitochondrial cytochrome c, is much faster than that measured for horse heart cytochrome c (181). 224 The alternative view of the transfer of electron between cytochrome bc1 and complex IV evokes the idea of supercomplexes (27, 154), which when formed create different conditions of electronic connection. According to this concept, quinone and cytochrome c molecules are confined to form small subpools operating just within boundaries of the supercomplexes. However, several studies, including recent time-resolved kinetics on intact cells, suggest that diffusion of cytochrome c is not restricted and the protein is not trapped within supercomplexes (119, 252). The relatively high redox potential of the heme c and the positively charged surface near the heme crevice make cytochrome c an effective reactant for superoxide anion (41, 172). This reaction seems advantageous over the dismutation reaction as the superoxide anion is converted back to oxygen instead of hydrogen peroxide. Thus high concentration of cytochrome c in the intermembrane space provides a very efficient and safe way of neutralizing potentially deleterious superoxide that is released into the intermembrane space by cytochrome bc1. For this reason, cytochrome c is considered as an ideal antioxidant (102, 205). V. REDOX CONNECTION OF THE Q POOL WITH THE C POOL To effectively drive the electron transport through the mitochondrial respiratory chain, the value of the midpoint redox potential (Em) of the Q pool is set in the middle between the Em values of the reducing equivalents that come from the tricarboxylic acid cycle and the Em of C pool that delivers electron for the terminal electron acceptor (161, 183). Cytochrome bc1 as a point of connection between the Q and C pools uses energy stored in the difference in redox potentials of quinone and cytochrome c and their physical separation by confinements to chemically unmixable phases. The Em of the Q pool is ⬃90 mV at pH 7 and, because oxidation/reduction of quinone involves deprotonation/ protonation, decreases by ⬃60 mV when pH increases by 1 (245, 258). The Em of the C pool is ⬃260 mV at pH 7 (171) and does not change significantly with pH. Because the Em of the Q pool changes with pH while the Em of the C pool does not, the energy associated with electron transfer (⌬G) from the Q pool to the C pool should in principle increase as pH rises (FIGURE 2). This could in part account for the observed pH dependence of rate of cytochrome bc1 catalysis (35, 125, 199, 201). Because the redox state of the Q pool and the C pool can dynamically change in the cell, the real reducing power of the Q pool and oxidizing power of the C pool depends not only on Em but also on the actual ratio of oxidized to reduced components of the pool that establishes the Eh of the pool according to Nernst equation (194). For example, Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA in healthy humans, the Q pool is generally above half-reduced, but in some tissues such as pancreas, it is almost fully reduced which makes Eh much lower than Em (249). The electron exchange between the Q pool and C pool through cofactor chains of cytochrome bc1 will depend on the actual conditions that encompass the redox states of the pools and the membrane potential. The later factor is particularly important in operation of cytochrome bc1 as, unlike other mitochondrial complexes, during the catalytic Q cycle half of the electrons that are transferred from UQH2 to cytochrome c must be “pushed” against the existing transmembrane electric field (this phenomenon is a result of bifurcation of electron transfer in the Qo site upon oxidation of UQH2 and spatial arrangement of the heme cofactors in relation to the vector of membrane potential as described in sect. VI). In mitochondria, under most physiologic conditions, the energy available to cytochrome bc1 released upon transfer of electrons from UQH2 to cytochrome c is used to do work A ADP H+ H+ on proton transport across the membrane (this will be referred in the remaining text as the “forward mode” of operation of cytochrome bc1). This occurs when oxygen and reducing equivalents from the tricarboxylic acid cycle are continuously supplied while ATP is continuously used up by the cellular processes (FIGURE 4A). However, as the membrane potential increases, the energy may become insufficient and the electron flow from the Q pool to the C pool slows down. At conditions of high proton electrochemical gradient and high degree of reduction of the C pool, the electron flow through cytochrome bc1 can be reversed (“reverse mode” of operation) (FIGURE 4B). This was shown by classic experiments in which artificially added ATP stimulated reverse electron flow in mitochondria (46). This phenomenon was also observed in experiments with cytochrome bc1 reconstituted into lipid bilayer (174). In addition, the reverse electron transfer within cytochrome bc1 induced by electric field gradient generated by photosynthetic reaction center was observed in membranes of purple bacteria (233). In fact, the growth of some acidophilic chemolitotrophic bacteria at low pH on ferrous iron ATP H+ H+ – UQH2 UQ Q pool UQH2 Δψ UQ UQH2 UQH2 UQH2 + Qi Reduction Qo Oxidation cyt. c Reduction H+ H+ H+ H+ H+ C pool B ADP ATP H+ UQ UQ H+ UQ Q pool UQ Qi Oxidation Qo Reduction cyt. c Oxidation Δψ UQH2 UQ – FIGURE 4. Two possible net directions of electron and proton transport catalyzed by cytochrome bc1. A: forward mode: in the presence of low or moderate membrane potential and low ATP/ADP ratio, cytochrome bc1 transports protons from matrix to intermembrane space while electrons are transferred from the Q pool to the C pool. The flow of protons back to matrix through ATPase results in ATP synthesis. B: reverse mode: under high membrane potential and high ATP/ADP ratio, the enzymes work in the opposite direction. Cytochrome bc1 transfers electrons from the C pool to the Q pool, while ATPase hydrolyzes ATP to ADP. UQ H+ H+ H+ C pool + H+ H+ H+ H+ H+ H+ Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 225 MARCIN SAREWICZ AND ARTUR OSYCZKA as a source of electrons depends on cytochrome bc1 that catalyzes the oxidation of cytochrome c from the C pool and reduction of UQ from the Q pool (38, 88). The UQH2 is then used to reduce NAD, which is required for CO2 fixation. This peculiar reverse electron transfer from cytochrome c is a consequence of mediocre reducing power of Fe3⫹/Fe2⫹ couple and the fact that Em of UQ/UQH2 at low pH at the periplasmic site is much more positive than at the cytoplasmic site where pH is close to neutral. The electron recirculation part works on the basis of action of the two quinone binding sites that together form a redox loop (161, 178) that plays a role of a “machine” translocating protons across the membrane. The UQ reduction site (Qi or Qn) is located at the matrix side of the inner mitochondrial membrane, while the UQH2 oxidation site (Qo or Qp) is located towards the intermembrane space (in this case the terms reduction and oxidation in the names of the sites refer to the forward mode of operation of cytochrome bc1). The Qo and Qi sites are connected electronically by the b-type hemes located in between. The Qo site is also connected electronically with the C pool by two cofactors: Rieske type iron-sulfur [2Fe-2S] cluster (FeS) and c-type heme (heme c1) (FIGURE 5B). This connection constitutes the second functional part of the enzyme. VI. COFACTOR CHAINS OF CYTOCHROME bc1 AS A FRAME FOR THE Q CYCLE Understanding of the catalytic Q cycle of cytochrome bc1 may appear difficult to a nonspecialist at first contact. In our opinion, it is a consequence of the fact that a complete enzymatic cycle of cytochrome bc1 involves two cycles in which two UQH2 are oxidized and two cytochrome c molecules are reduced while one UQ is reduced (64). This gives net oxidation of one UQH2 per reduction of two cytochrome c molecules. Such reaction must involve a recirculation of half of the electrons that derive from the Q pool back to the Q pool (36). The catalytic cycle can thus be understood as a product of a cooperation of two functional parts. The first part recirculates electrons from and to the Q pool, while the second part steers the electrons to the C pool powering the electron recirculation (FIGURE 5A). A The cofactors that build the first and the second part are commonly referred as a low-potential chain (or b-chain) and a high-potential chain (or c-chain), respectively. These terms reflect the difference in Em of the cofactors in the chains: Em of hemes b span from ⫺160 to ⫹100 mV, while Em of FeS and heme c1 in high-potential chain is in the range between ⫹200 to ⫹400 mV (69). The redox properties of cofactors appear to fit properties of the substrate. This can be inferred from the observations that some bacteria operating on lowpotential menaquinones or living in alkaline environment B Qi site 7Å 12.2Å Q pool 14Å Heme bL e– b chain Heme bH Qo site C pool 23Å b-state c-state 9.5Å >23Å Heme c1 C D Supernumerary subunits H+ UQH2 UQ Q pool e– UQH2 UQ c chain Rieske cluster e– FIGURE 5. Major functional principles and structural elements of cytochrome bc1. A: when the enzyme steers electrons from the Q pool to the C pool, half of the electrons are recycled back to the Q pool. B: spatial arrangement and distances between cofactors and catalytic sites in the dimer. C: subunit composition of mitochondrial cytochrome bc1 with colormarked three catalytic subunits of one monomer. D: scheme of electron and proton transfers within the monomer in the forward mode. Dotted lines in B and D denote large-scale movement of the FeS head domain harboring Rieske cluster between b- and c-states. e– 2H+ Iron-sulfur protein 226 Cytochrome b Cytochrome c1 C pool Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA have downshifted redox potential of FeS below ⫹200 mV (162, 227). through the c-chain (FeS and heme c1 to reach heme c of diffusible cytochrome c), while the other passes through the b-chain (heme bL and heme bH across the membrane to reach the occupant of the Qi site) (FIGURE 5D). This reaction is often referred to as a bifurcation of electron paths and is a central feature of the catalytic cycle critical for the energetic efficiency of the enzyme. The enzyme must somehow ensure that the two electrons deriving from UQH2 do not end up traveling in the same chain nor the energetically favorable electron transfer from the low-potential b-chain components to the high-potential c-chain components takes place (37, 189, 199, 200). Those types of reactions dissipate energy and are often referred as short-circuits (FIGURE 6). All redox cofactors and the catalytic sites are embedded within the three subunits that are part of the catalytic core of cytochrome bc1: transmembrane cytochrome b harbors two hemes b (bL and bH) and provides environment for quinone binding Qo and Qi sites, membrane-anchored cytochrome c1 harbors heme c1, the membrane-anchored FeS subunit contains FeS (FIGURE 5C) (128, 132, 269, 277). Some bacterial cytochromes bc1 (for example in Rhodobacter capsulatus and Paracoccus denitrificans) can just be composed of these three subunits (21, 142, 216, 272). Other cytochromes bc1 may have accessory subunits, which number varies from one in bacterial cytochrome bc1 (for example, in Rhodobacter sphaeroides) (6, 273) to eight in mitochondrial cytochrome bc1 (19, 268). These subunits do not contain redox cofactors and are not involved in energy conversion. It is suggested that some accessory subunits may serve as processing peptidase for subunits of complex III, at least for the FeS subunit. They may also play a role in positioning the FeS subunit within the complex (19). The Qo-site-mediated bifurcation delivers one electron to the Qi site at the time. As full reduction of UQ requires two electrons, the compound that is created in the Qi site after delivery of one electron from the Qo site depends on whether UQ or USQ⫺ occupies the Qi site. If UQ occupies the Qi site, it is half-reduced to USQ⫺, but if USQ⫺ is present, it is reduced to UQH2. This way two cycles of the Qo site are needed to fully reduce UQ to UQH2 at the Qi site, explaining the overall stoichiometry of one UQH2 molecule per two UQ molecules formed (64). The arrangement of cofactors in the c- and b-chains creates a unique set of conditions for redox reactions at the Qo site: upon two-electron UQH2 oxidation, one electron passes A forward heme bH C D heme bL Rieske cluster UQH2 1 1 2 2 UQH2 1 3 3' O2- O2 B E reverse UQH2 2 2' O2 O2- F 2 UQ 2 UQ 1 2 2' O2 1 1 UQ 3 O2- 3' O2 O2- FIGURE 6. Energy-conservation, short-circuiting, and ROS generation in the Qo site. A: forward mode: complete oxidation of UQH2. B: reverse mode: complete reduction of UQ. Blocking reaction 2 in A or B leads to semiforward or semireverse reaction, respectively, resulting in the formation of USQo. C–F: possible engagement of short-circuit reactions or leaks of electrons on oxygen when USQo is formed in semiforward or semireverse reaction (CD or EF, respectively). Numbers in A–F denote the order of electron transfer. Reactions 2= and 3= are alternatives to 2 and 3, respectively, denoting possible competition between superoxide generation and short-circuits. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 227 MARCIN SAREWICZ AND ARTUR OSYCZKA X-ray crystal structures of known cytochrome bc1 complexes showed close positioning of heme bL and bH in the b-chain that promote fast electron transfer (21, 93, 128, 132, 142, 268, 277). At the same time, the crystals revealed that the position of the head domain of the FeS subunit varied. This cofactor was found very close to the Qo site (in “b-state,” where it can exchange electrons with quinone in the Qo site), close to heme c1 (in “c-state,” where it can exchange electrons with heme c1) (FIGURE 5B) or at various positions between these two states (19, 60, 277). This implicated that the change in the position of FeS head domain between these two states was needed to transfer electrons from the Qo site to cytochrome c1. Such a large-scale movement of the FeS head domain is now supported by many kinetic and biochemical studies (40, 70 –73, 222, 250, 251). These studies allowed the description of several conformational states where the movement is affected by mutations and, unlike in the native system, becomes rate-limiting for electron transfer. There is an ongoing debate (22) as to whether the movement of the FeS head domain is controlled by specific conformational changes in the region of substrate binding (94, 268) or by allostery (50, 52, 55, 187) or whether the changes between the b- and c-state are rather stochastic (49, 60, 199). Crystal structures also revealed that the FeS center, despite the motion, never approaches closer than 23 Å from heme bL (FIGURE 5B). This provides a simple way of separating the high- and low-potential chains by slowing direct electron transfer from heme bL to the FeS center beyond the catalytic timescale. The exact position of quinone within this 23 Å gap is unknown because quinone has never been resolved in the Qo site. Crystal structures including the Qo site inhibitors show two overlapping loci of binding at which quinone redox catalysis may occur: one inhibitor, stigmatellin, approaches within ⬃7 Å of FeS (22, 128) (FIGURE 7), while the other, myxothiazol, approaches within ⬃6 Å of heme bL (95). Regardless of the exact position of quinone in the Qo site, its presence as a redox-active cofactor bridges the 23 Å gap between heme bL and FeS with two connections, FeS-quinone and quinone-heme bL, each of less than 14 Å, suitable for fast electron transfer (FIGURE 7). This creates a potential way for short-circuiting the b-chain with the c-chain (FIGURE 6). While it is clear that the shortcircuits must be avoided, the molecular mechanism of how this is accomplished remains unclear (43, 63, 199, 200). The crystal structures revealed that cytochrome bc1, unlike other complexes of mitochondrial respiratory chain, is a homodimer. The two monomeric units assemble so that the FeS head domain interacting with cytochrome b and cytochrome c1 of one monomer has its membranous anchor associated with the other monomer. Dimeric nature of cytochrome bc1 was further confirmed in biochemical studies (270). Interestingly, two hemes bL in the dimer are in a close distance (14 Å), implicating that electronic connection between monomers via heme bL-bL electron transfer is possible (FIGURE 5B). All other distances between cofactors of the two monomers are large enough to prevent catalytically relevant electron transfer between them. VII. INTERACTIONS OF QUINONE AND CYTOCHROME c WITH CYTOCHROME bc1 A. Quinone Binding Qo Site Despite the fact that the general idea of electronic bifurcation in the Qo site is widely accepted and supported by numerous biochemical studies, the consequences of this phenomenon make the understanding of the principles of the Qo site engineering difficult. Furthermore, some of the key structural and kinetic elements of catalysis remain obscure. First of all, the lack of crystal structures containing quinone bound, at any redox state, in the Qo site leaves a FIGURE 7. Structural elements of the Qo site with bound inhibitor, stigmatellin. Three major regions in the pocket probed by mutagenic studies are shown in blue, green, and magenta. Green rings denote histidines coordinating Rieske cluster. Residues E272 (on cytochrome b subunit shown in gray) and H161 (on FeS subunit shown in amber) form hydrogen bonds with the inhibitor. Dashed lines show distances between chroman ring and cofactors: heme bL and Rieske cluster. 228 Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA high degree of freedom in constructing models of the binding, electron transfers, and proton release into the intermembrane space. The second important aspect is associated with the experimental difficulty in isolating intermediate states of the catalysis, which also leaves a high degree of freedom for mechanistic considerations. The first report of detection of semiquinone intermediate associated with the Qo site (75a) was questioned (136), and until recently (44, 223, 261, 276), no other intermediates have been reported. This general difficulty in detecting semiquinone in the Qo site (USQo) has been considered as confirmatory to Mitchell’s original idea that this species must be highly unstable (177). This became the basis for constructing thermodynamic properties of USQo (43, 79, 275). Comparative analysis of various crystal structures containing different inhibitors of the Qo site identified the region of quinone binding consistent in amino acid composition with earlier mapping by site-directed mutagenesis (39, 59). In addition, it showed displacements of polypeptide backbone in the region expected for the quinone binding (94, 268). The most recognized representatives of the inhibitors of the Qo site (262) are stigmatellin and myxothiazol. The former inhibitor was found to form a hydrogen bond between hydroxyl group of the chroman ring and one of the histidines coordinating Rieske cluster (128, 164). In this way, stigmatellin creates conditions in which the FeS head domain is in the closest position to the Qo site. On the basis of this observation, it is assumed that quinone binds to the Qo site in virtually the same conformation as stigmatellin (62, 164, 165). On the other hand, myxothiazol binds closer to the heme bL not in direct contact with the FeS head domain. The two different niches for inhibitor binding suggest that there is enough degree of freedom for quinone to move within the site (62), or for two quinones to bind simultaneously but with different affinity as proposed in the double occupancy model (79, 80). In fact, the idea of dynamics of quinone within the site was exploited by some models of UQH2 oxidation and also can be considered as one of the explanations for lack of the recognized electron density of quinone in the Qo site in crystal structures. In addition, several biochemical studies reported that stoichiometry of the binding can be larger than 1 quinone per 1 Qo site, which would seem consistent with relatively spacious binding pocket (16, 36, 79, 80, 232). On the other hand, recent molecular dynamic simulations indicated that there is not enough room in the cavity to accommodate more than one molecule of substrate (207). Extensive site-directed mutagenesis systematically explored structural and functional elements of the Qo site. The mutations examined so far appear to come mainly from three regions of cytochrome b subunit (FIGURE 7): helix C, helix cd, and proximity to the conserved PEWY motif on the ef loop (23, 39, 111). These mutations can have various effects on phenotype, assembly of the whole complex, measured rates of electron transfer, enzymatic activity, quinone binding, and occupancy of the Qo site (11, 78, 79, 175, 225, 226). The mutations that affect assembly of the complex almost inevitably lead to the loss of function (226). On the other hand, within the group of the existing Qo site mutations that allow a proper assembly of the complex, only a few mutations fully abolish the functionality of cytochrome bc1 (23, 39, 111). A number of positions can be mutated with little or no effects, while the energetic efficiency of cytochrome bc1 is retained at levels that support growth of the cells under conditions that require the presence of functional cytochrome bc1. Although in some cases sensitivity to the type of the introduced by mutation amino acid side chain can be observed, many positions appear to tolerate many types of side chain change. In general, this indicates that the Qo site pocket may have a rather significant structural flexibility and a lot of engineering tolerances for a mutational change. Interestingly, this flexibility is in part explained by recent molecular simulations indicating that benzoquinone moiety of quinone does not directly interact with the side chains of amino acids of cytochrome b that create the binding pocket. Instead, it may interact with atoms of the polypeptide main chain through the molecules of water (207). This would explain why defining specific quinone binding motif is difficult (100). Because quinone at the Qo site is flanked by two redox centers, each coming from a different chain of cofactors, there are two possible general reaction schemes of twoelectron UQH2 oxidation or UQ reduction that can be considered. The “sequential” scheme considers two separate one-electron reactions occurring sequentially one after the other (37, 62, 165, 189, 199). If UQH2 is to be oxidized (forward mode), the first reaction is a reduction of FeS by UQH2 leading to generation of USQo intermediate while the second reaction is a reduction of heme bL by USQo leading to formation of UQ (completing the oxidation of UQH2) (FIGURE 6A). If UQ is to be reduced (reverse mode), the first reaction is reduction of UQ to USQo by heme bL followed by the reduction of USQo to UQH2 by FeS (FIGURE 6B). The “concerted” scheme assumes that the two oneelectron reactions involving FeS and heme bL required for oxidation of UQH2 or reduction of UQ occur simultaneously with no USQo intermediate formed during this reaction (129, 199, 236, 253, 280). The concerted scheme became a subject of considerations for two main reasons. It helped explain difficulty in detecting USQo intermediate. It also provided a built-in mechanism for prevention of unwanted short-circuit reactions (199, 200). This is because the concerted scheme assumes that one-electron reactions of the sequential scheme are not allowed, eliminating the short-circuits that are the result of these one-electron reactions occurring in “inappropriate” order (FIGURE 6, C–F). Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 229 MARCIN SAREWICZ AND ARTUR OSYCZKA There is still no consensus which scheme best describes reaction mechanism at the Qo site. Recent reports of detection of USQo seem to point towards the sequential scheme. In two cases, small amounts of USQo were identified by electron paramagnetic resonance (EPR) spectroscopy as typical radical signal of semiquinones (44, 276). The low levels of detected USQo were consistent with the concept of high instability of this intermediate. In another study, however, two populations of USQo were identified (223). The first population was represented by a radical signal similar to the other semiquinones but, unlike in previous reports (44, 75a, 276), was strongly affected by interaction with metal cofactors of the Qo site (reminiscent of its presence in the site). Intriguingly, the second population was recognized as a semiquinone spin-coupled to reduced Rieske cluster generating a new spectroscopic identity. The relatively large amount of the new signal suggested that USQo is not as highly unstable as commonly assumed, at least when USQo interacts with Rieske cluster. It was proposed that USQo coupled to Rieske cluster might represent an initial state of oxidation of UQH2 when oxidized FeS is withdrawing electron from UQH2 (223). Regardless of properties of all so far detected USQo, its presence in the site according to the sequential scheme requires additional level of control to prevent short-circuits. Mechanism of such control remains unknown and is subject of various proposals (43, 63, 186, 200, 215) that all introduce additional complexity to the system. In this context, the built-in mechanism for short-circuit prevention in a concerted scheme still remains an attractive alternative. When discussing sequential versus concerted scheme, one should bear in mind that a clear distinction between these two mechanisms is not as obvious as intuition might tell. The problem concerns relative timescales of processes. One could ask what is the minimum time separation between the first and the second electron transfer to consider the reaction as “virtually concerted.” Kinetically, the concerted term means that time separation between the two electron transfer steps is shorter than time required for significant atomic rearrangement (femtoseconds) (199). An important aspect of operation of the Qo site is rapid reversibility of electron transfers between the substrate and FeS and heme bL. At the same time, biochemical studies indicate that quinone bound at this site freely equilibrates with the Q pool (79). This, in the context of reversibility, may have important consequences on the mechanism of the Qo site catalysis. The molecule of quinone present in the site makes connection between the b- and c-chains allowing rapid exchange of electrons in both directions (199). This means that the net electron flow (forward or reverse) is a consequence of an evolution of the site into the new state that does not allow the reaction to be reversed, for example, 230 by replacing the substrate at the site (49). It follows that the redox state of the Q pool may actively influence the direction of electron flow through the site. Proton transfers at the Qo site remain largely unknown. Using the stigmatellin binding mode as a reflection of quinone binding in the catalytically active state, most of the proposed scenarios assume that upon UQH2 oxidation the first proton coming from UQH2 is used to protonate His161 (one of the ligand of the Rieske cluster), while the second proton is used to protonate Glu-272 (Glu-295 in bacteria) in PEWY motif (62, 129, 186, 204). Those two residues seem best suited as initial proton acceptors in crystal structures (FIGURE 7) given that they form hydrogen bonds with stigmatellin bound at the Qo site (130). However, until now, apart from the observation that histidine is the redox-linked protonation site of Rieske cluster (131), there is no direct experimental evidence for any of the proton reactions at the Qo site. Mutating His-161 (His-156 in bacteria) is not informative in this case as it disrupts assembly of the whole FeS subunit (74). On the other hand, mutating Glu-272 into nonprotonable amino acids does not abolish enzymatic activity of cytochrome bc1, indicating that if this residue is involved in proton transfers, it is not the only possible proton acceptor in the site (157, 201, 230, 264). As with other quinone oxidation/reduction sites, the pathways for proton appear relatively insensitive to perturbations (202). Another alternative, indicated by recent MD simulations, is that water molecules can act as direct acceptors of both protons from UQH2 (207). B. Quinone Binding Qi Site The Qi site is a terminal part of the low-potential chain that catalyzes two-electron reduction of UQ to UQH2 when cytochrome bc1 works in a forward mode. This reaction results from two sequential steps in which electrons are delivered from the same cofactor chain. Such reaction requires stabilization of USQ intermediate formed in the site after every second UQH2 oxidation cycle of the Qo site. Indeed, the stable semiquinone originating from cytochrome bc1, and as we now know being the semiquinone bound in the Qi site (USQi), was detected even before the formulation of the Q cycle (265). The possibility to detect USQi intermediate greatly facilitated structural and functional studies on the Qi site. Additionally, the crystal structures of cytochrome bc1 containing UQ molecule in this site provided direct and detailed information on the quinone binding (126, 128). The fact that UQ can be crystallized in the Qi site, but not in the Qo site, indicates that the binding mode at these two sites is different and the binding affinity for the Qi site is larger. Equilibrium redox titrations suggested that UQH2 binding to the Qi site is slightly stronger than UQ from the Q pool, while the binding constant for USQi must be several orders of magnitude larger than that for UQ (217). Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA Mutagenic studies and EPR spectroscopy identified conservative residues responsible for binding and stabilization of quinone and USQi (76, 112, 120, 143). One of them is His-201 (His-217 in bacteria) which appears to stabilize USQi through the H-bonding of ⬎C⫽O group to imidazole ring of histidine. Other important residues include Asp-228 (Asp-252 in bacteria) and Ser-205 (Asn-221). Quinone molecule binds very closely to heme bH with the edge-toedge distance between heme moiety and benzoquinone ring of ⬃6 Å. Such close distance allows for rapid electron transfer from reduced heme bH to UQ (or USQi). Intriguingly, in a closely related cytochrome b6f, the position of quinone is occupied by an additional heme ci (or cx) not present in cytochrome bc1, while quinone is shifted further away from heme bH (149, 242). As the complete reduction of UQ requires an uptake of two protons from matrix, the reaction scheme can be divided into following steps (143): UQ ⫹ bH2⫹ ↔ USQi⫺ ⫹ bH3⫹ (1) and USQi⫺ ⫹ bH2⫹ ⫹ 2H⫹ ↔ UQH2 ⫹ bH3⫹ (2). Despite the lack of experimental data on the electron-associated proton transfer, it can be envisaged that two protons are attached to the benzoquinone ring in response to the appearance of the second electron. This scheme is supported by the observation that USQi is in anionic form (USQi⫺) (76, 143). Crystal structures revealed two regions rich in protonable groups of amino acid side chains and water molecule paths for consideration as proton paths linking the catalytic site with the membrane surface (129, 153). Interestingly, the proton transfer is suggested to engage polar groups of cardiolipin as this lipid has been shown to bind in proximity to one of the putative proton channels (9, 129, 153, 208). Considering the reversibility of reactions, USQi⫺ can originate either from the reaction (1) or reversing the reaction (2). Nevertheless, USQi⫺ detected in experiments appear to come from reversing reaction (2), as it is present along with reduced heme bH. It is important to bear in mind that the reverse reaction (2) fills b-chain with electrons, which in consequence may slow down the rate of oxidation of UQH2 at the Qo site. This effect can be compared with the effect of the weak competitive inhibitor of the Qi site. As a result, at high range of UQH2 concentrations, the activity of cytochrome bc1 may decrease (29, 35). This “downregulation” may play an important role in controlling the electron flux through the respiratory chain. Although the crystal structures do not show any significant structural changes caused by binding of antimycin (126), a potent inhibitor of the Qi site, biochemical and spectroscopic studies have implicated that it might influence the average position of FeS head domain in the complex (51, 222, 260). While this long-range structural effect has been exploited by various models that propose allostery within the dimer of cytochrome bc1 (50, 52– 54), the origin and the time domain of this inhibitorinduced effect are unknown. C. Cytochrome c Binding Site Cytochrome bc1 interacts with soluble cytochrome c at the solvent-exposed surface of cytochrome c1 subunit. The interaction regions of both cytochrome c and c1 surround the area where hemes are exposed. Consequently, the association of the two proteins allows two hemes to attain distances in the range of 10 Å (152), which is short enough that fast electron exchange between them becomes possible. It is generally accepted that interactions between proteins involve long-range electrostatic interactions that facilitate formation of an encounter complex, while short-range interactions foster the stabilization of a tight complex in a proper spatial configuration (197, 228, 231, 257). Electrostatic forces are considered to be a dominant factor that contributes to binding of cytochrome c to cytochrome bc1, which is inferred from a significant salt dependence of electron transfer between these proteins (90, 114, 121, 135, 249). An importance of short-range hydrophobic interactions between surfaces of cytochrome c1 subunit and cytochrome c was proposed on the basis of X-ray structures of cytochrome bc1 co-crystallized with its redox partner (152, 238). Extensive chemical modifications and mutagenic studies identified crucial amino acid residues that stabilize the complex of the proteins through interactions of negatively and positively charged residues on the surfaces of cytochrome c1 and c, respectively (30, 163, 241, 249). The transient binding of cytochrome c to cytochrome bc1 in solution was inferred from the analysis of electron transfer between these proteins (175) and from the measurements based on the techniques that were independent of electron transfer, such as plasmon wave-guide resonance spectroscopy (75) or EPR (206, 221, 224). The complexes are relatively long-lived at low ionic strength, but their lifetime decreases rapidly with an increase of salt concentration. At ionic strength typical of physiological conditions, the complexes are very short-lived and the stationary concentration of bound cytochrome c is low. This implicates a mechanism in which under physiological conditions cytochrome c does not form stable, longlived complexes but rather constantly collides with the surface of cytochrome bc1 and electron transfer takes place coincidentally with one of such collision (117, 206, 221). VIII. GENERATION OF REACTIVE OXYGEN SPECIES Discussion on generation of ROS by cytochrome bc1 started in the mid 1970s when submitochondrial particles inhibited Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 231 MARCIN SAREWICZ AND ARTUR OSYCZKA with antimycin and fueled with succinate were able to produce a significant amount of superoxide anion (31, 32, 195). The superoxide production was subsequently linked to redox reactions of the Qo site (42, 123, 147, 190, 255). Consistent with that notion, current, widely shared view considers highly unstable USQo as a direct one-electron donor for oxygen (43). This is envisaged on the basis of very low potential of USQo/UQ couple (less than ⫺400 mV; Ref. 200) and relatively high redox potential of O2⫺/O2 couple (approximately ⫺160 mV in aqueous solution at pH 7; Ref. 266). While there are some suggestions that USQi may also contribute to ROS production (210), this much more stable semiquinone is not generally considered as a source of electrons that can leak on oxygen. Exclusion of the Qi site from ROS generation activity was supported by the fact that ROS from cytochrome bc1 are observed when inhibitor antimycin is bound to the Qi site. Furthermore, a complete inhibition of the Qo site by stigmatellin, which still allows formation of USQi (through reverse reaction), leads to a complete loss of superoxide generation activity (189). It is likely that stabilization of USQi through the H-bonding of ⬎C⫽O group to imidazole ring of histidine can protect USQi against the reaction with oxygen (259). In reversibly operating Qo site, USQo can be formed in two ways (29, 82, 145, 189, 209, 220): as a part of forward reaction, when FeS withdraws electron from UQH2 and the second electron transfer from USQo to heme bL does not take place, or as a part of reverse reaction when heme bL donates an electron to UQ and the second electron transfer from FeS to USQo does not take place. These two reactions can be referred to as “semiforward” and “semireverse,” respectively (FIGURE 6). Currently both semiforward and semireverse schemes are considered as possible reactions leading to USQo that can interact with oxygen. Both reaction schemes require as an initiation a reduced state of heme bL, which is consistent with the observation that ROS are observed when the electron transfer between heme bL and heme bH is impeded by blocking the Qi site with antimycin or by increase in membrane potential (144, 218). In the semiforward scheme, reduced heme bL is unable to take electron from USQo and thus cannot convert it to UQ (103, 145, 189, 209). In the semireverse scheme, the reduced heme bL initiates a formation of USQo (FIGURE 6) (29, 82, 220). As the semireverse reaction, unlike the semiforward reaction, uses UQ as a substrate, the recognition that the semireverse reaction may lead to ROS production came from analysis of a dependence of the rate of ROS production in submitochondrial particles and in isolated complexes on the redox state of the Q pool. The maximum rate of superoxide generation was detected when the Q pool was partly oxidized, which is consistent with the semireverse rather than semiforward mechanism (82), for which the rate of superoxide generation should increase monotonically with reduction level of 232 the Q pool. An impact of the Q pool redox state is also inferred from the stimulating effect of complex II inhibitors on ROS generation by cytochrome bc1 (82, 84, 168, 240). Further support for the semireverse scheme came from the analysis of various cofactor knockout mutants of cytochrome bc1 (29). With these studies the model was further developed to describe an important constraint associated with the kinetic effects of the motion of the FeS head domain. The model proposes that probability of ROS generation increases as the distance, separating the FeS head domain from the Qo site at the exact time of USQo formation, increases (29, 220). This is because the positions of FeS remote from the Qo site diminish a chance that FeS engages in electron transfer to interact with USQo (either by reducing USQo to UQH2 which would complete the reverse reaction, or oxidizing USQo to UQ which would complete the short-circuit reaction) before USQo reacts with oxygen. From the kinetic point of view, this can be ascribed as a competition taking place on timescale of seconds (i.e., timescale beyond the catalytic timescale) between the leaks of electrons on oxygen and the short-circuit reactions (FIGURE 6). When the short-circuits become effective (FeS is in or close to the Qo site), the leaks are diminished, and vice versa. This way, the short-circuits emerge as not only unwanted reactions that should be avoided at all costs as they dissipate energy, but in some cases they may be beneficial in helping to diminish or modulate the leaks of single electrons and the associated ROS levels. Interestingly, the notion about a “beneficial” role of short-circuits develops also for other bioenergetics enzymes (219). In closely related cytochrome b6f (this enzyme is part of photosynthetic chains in plants and catalyzes electron transfer from plastoquinol to plastocyanin) (57), the short-circuits were proposed to be important as an “emergency exit” pathway to bypass the Q-cycle in the enzyme with disabled Qi site to sustain the function of the entire photosynthetic chain (169). It should be noted that although the reaction schemes based on unstable USQo assume its high reactivity with oxygen, this reaction has never been directly confirmed experimentally. In this context, the possibility that other cofactors, such as low-potential heme bL, directly reduce oxygen cannot be ruled out. Interestingly, recent studies indicate that there might be states of USQo that are not highly reactive with oxygen (223). These include a state in which USQo is spin-coupled to reduced Rieske cluster. This state is observed in the presence of oxygen in particular in the FeS motion mutants that eliminate superoxide activity of cytochrome bc1 (29, 220). In general, the level of ROS production by mitochondria appears to strongly depend on respiration state (2, 192). It is rather a common misconception that with faster respiration rate, the conversion of oxygen to superoxide should increase. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA However, it was observed that factors limiting the rate of oxygen consumption (respiration rate) favor superoxide production. Typically, this can occur under high membrane potential, nearly inactive ATPase, high NADH:NAD⫹ ratio, or hypoxia (2, 118, 144, 192, 237). Under those conditions, the reduction levels of individual cofactors in respiratory complexes increase. This, in view of the kinetic constraints inherent to the proposed mechanisms of superoxide generation by the Qo site, explains why complex III can be one of the contributors to ROS generation under those types of conditions. return back to respiratory chain via reaction of superoxide anion with oxidized cytochrome c (205, 279). Because of this reaction, cytochrome c is considered an important element of antioxidative system of mitochondria. IX. H-SHAPED ELECTRON TRANSFER SYSTEM IN DIMER AND ITS PHYSIOLOGICAL IMPLICATIONS In most textbooks, the principles of Q cycle are described considering cytochrome bc1 monomer, which has its justification in the fact that each monomer is equipped with all structural elements necessary to complete the catalytic cycle. All of these were described above: quinone binding Qo, and Qi sites and cytochrome c binding site linked together by two cofactor chains. However, the dimeric structure of this enzyme invoked several intriguing mechanistic issues that became a subject of intense debate. From all components of mitochondrial electron transport chain that are considered as possible contributors to mitochondrial ROS (complex I, cytochrome bc1, and recently also complex II), only cytochrome bc1 is an enzyme that appears to release most of superoxide to the intermembrane space (34, 123, 239), as expected considering the localization of the Qo site (FIGURE 8). This topology of ROS release from the Qo site is proposed to have implications for redox signaling (26, 84). Generated superoxide after rapid dismutation to hydrogen peroxide may target components of the antioxidative system both in the intermembrane space and in cytosol (98). In this reaction scheme, electrons irreversibly leak from the respiratory chain due to dismutation of superoxide. In an alternative pathway, electrons on superoxide molecules present in the intermembrane space may Matrix The most fundamental question concerns the electronic connection between the monomers, which given the distances between the cofactors in dimeric structure, is possible at the level of hemes bL. While the existence of this connection has been considered since the first crystal structures of cytochrome bc1 were solved (50, 55, 61, 110, 199, 234, 269), the experimental Cytochrome bc1 – TQ Δψ 3 O2 e– 4 1 UQ TOC + Activation Regulation Signaling UQH2 UQH2 O2– H2O2 Reduced e– Cyt. c selfexchange superoxide dismutase 2 Oxidized O2 Intermembrane space H2O Complex IV FIGURE 8. Regulation and ROS-generating activity of cytochrome bc1. Factors influencing the electron transfer between the Q and C pools catalyzed by cytochrome bc1 include the following: 1 and 2, redox state of the Q and C pools, respectively; 3, magnitude of the membrane potential; 4, constituents of the membrane that can interact with quinone binding sites: tocopherol (TOC) and tocoquinone (TQ). Under some conditions, Qo site may become source of superoxide which is released to intermembrane space. Lines denote the following: dotted black, electron transfer paths; solid black, diffusion of O2 and H2O; solid red, diffusion of ⫺ and H2O2); dashed green, participation of ROS in activation, cytochrome c; solid green, diffusion of ROS (O2 regulation, and/or signaling; solid yellow, diffusion of TOC and TQ. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 233 MARCIN SAREWICZ AND ARTUR OSYCZKA evidence for it was not available until recently. It came from mutagenic studies demonstrating that the derivatives of the enzyme with inactivated Qo site in one monomer and Qi site in the other (cross-inactivated forms) are able to support enzymatic turnover using effectively the heme bL-bL electron transfer as the only available connection linking the functional Qo and Qi sites (67, 68, 150, 244). The electron transfer through this two-heme bridge occurs in a millisecond timescale, which falls within the timescale of the catalytic turnover of native enzyme. It was also observed that these cross-inactivated forms of cytochrome bc1 are able to sustain growth of the bacterial cells under conditions that obligatorily require presence of functional cytochrome bc1 (87a, 150, 151). This indicates that enzyme relying just on the paths involving heme bL-bL electron transfer performs the bioenergetic function in vivo. The at least millisecond timescale of electron transfer between two hemes bL converts the cofactor chains in the dimer into an H-shaped electron transfer system connecting all four quinone catalytic sites (FIGURES 5B AND 9). A basic operational principle of this system is that it enables any connection between catalytic sites of the opposite sides of the membrane to be enzymatically competent. The physiological meaning of this design remains an open issue. With our current knowledge on the thermodynamics of the reactions in cytochrome bc1 taken into account, it can be anticipated that electron transfer between hemes bL and bH of each monomer is favored over heme bL-bL electron transfer as long as there are no factors that influence the electron transfer route (234). The factor inherently present and dynamically changing in respiring mitochondria is membrane potential, which when increases, impedes the electron transfer from heme bL to heme bH, but at the same time it may increase the fraction of electrons that travel through the hemes bL-bL bridge (234). Thus the membrane potential comes as one of the prominent factors that can modulate the inter- versus intramonomer electron transfer. The presence of the intermonomer connection creates a possibility of dismutation of two semiquinones that may occupy quinone binding sites. This way it might be possible to fully reduce one UQ molecule in one of the Qi sites using two electrons, each delivered from a different Qo site. This could provide physiological advantage for the cells as it may enhance efficiency of the Q cycle under shortage of UQH2 in the Q pool. Another possible physiological advantage of the H-shaped electron transfer system is related to the effects of accumulation of mitochondrial damage. This process is related to a relatively high frequency of mutations within mitochondrial DNA (mtDNA) and a possibility that the mutations occurring in genes coding for subunits of respiratory enzymes affect operation of the enzymes expressed from these genes (13, 17, 77, 99, 173, 254). It is generally believed that in the process of mitochondrial damage, mutations in 234 mtDNA progressively accumulate in time, which may be inherent to aging and/or progress of development of several mitochondria-related diseases (148). For cytochrome bc1, this concerns cytochrome b subunit which is coded by a gene located in mtDNA. Given that this gene does not differentiate between monomers and that mtDNA is present in several copies in mitochondria, the presence of mutations in just a part of the copies of the gene is likely to result in expression of both symmetrically and asymmetrically mutated dimers of cytochrome bc1 (FIGURE 9) as observed in bacterial two-plasmid model systems (45, 141, 151). Thus it can be anticipated that the amount of the asymmetrically damaged complexes increases with age. In this context, the H-shaped electron transfer provides a clear advantage as it builds in redundancy to allow physiological function of the enzyme even after operational damage of one part. Yet another possible physiological advantage of the Hshaped electron transfer system is related to the ROS generation by cytochrome bc1. All current models of ROS generation consider the prolonged state of reduced heme bL as condition increasing probability of superoxide generation by the Qo site (29, 43, 82, 83, 145, 189, 209, 220). Such a state becomes more frequent when the electron outflow from the Qi site is blocked or suppressed by inhibition or mutation. If such damage or inhibition occurs asymmetrically in a dimer (is present only in one monomer), the connection between monomers helps remove electrons from hemes in the b-chain, decreasing the chance of superoxide formation by the enzyme. Considering that cytochrome bc1 evolved in an anoxygenic environment, it is not clear whether possible protective role of geometric proximity of two hemes bL against ROS was a coincidental effect or an evolutionarilydriven design. Nevertheless, one cannot exclude that some tuning of energy levels might have occurred during evolution (219), for example, to minimize the probability of direct reaction of reduced heme bL with oxygen. X. COMPOUNDS THAT INFLUENCE THE ACTIVITY OF CYTOCHROME BC1 So far, many chemical compounds have been recognized as efficient modulators of cytochrome bc1 activity. These are not only naturally occurring inhibitors and synthetic drugs or pesticides but also some substances that are an inherent part of the cellular environment like vitamin E and its metabolites or certain lipids. Typically, these compounds target the catalytic Qo or Qi sites by competing for binding with natural substrates. The very strong inhibitors that tightly bind to the catalytic sites are considered as toxic for cells as they significantly restrict or even prevent the electron transfer through complex III leading to disruption of mitochondrial respiration. Some of these compounds (such as myxothiazol, stigmatellin, or antimycin) (262, 247, 248) are naturally produced Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA Mutated mtDNA Crossinactivated Mutated mtDNA Asymmetricallymutated functional Unmutated mtDNA ROS Qi FIGURE 9. H-shaped electron transfer system of cytochrome bc1 and consequences of symmetric and asymmetric mutation patterns in the context of mitochondrial damage. The system connects electronically all four catalytic sites of the dimer, and only symmetrically inactivated enzymes with interrupted connection of both Qo sites with both Qi sites are nonfunctional. Qi ROS Qo Qo Symmetricallymutated nonfunctional Unmutated cytochrome bc1 by certain bacteria. Many Qo site inhibitors (such as strobilurin derivatives or famoxadone) are widely used in agriculture as pesticides because they are efficient fungicides but are, at the same time, relatively less toxic for mammals. Other applications include medicine. For example, the synthetic inhibitor of the Qo site atovaquone is used in combination with other drugs to treat parasite diseases, in particular malaria, babesia, and toxoplasmosis (12, 140, 193). The potential use of cytochrome bc1 inhibitors as drugs stimulates research to seek new compounds that would effectively target cells of protozoa parasites (15, 127, 278). The weak inhibitors that reversibly bind to the catalytic sites are potential regulators of cytochrome bc1 activity. One such compound is vitamin E (tocopherol) and the products of its degradation (tocoquinones). It appears that vitamin E has not only antioxidant properties but can also modify the activity of cytochrome bc1 (108, 109, 113, 188, 274). There are also observations that vitamin E can influence ROS production by mitochondria, and it is proposed that this effect results from the interference of vitamin E with electron transfer at the level of the Qo site (66, 188) (FIGURE 8). The concept of binding of vitamin E to the Qo site is supported by the fact that this compound shares the same chroman ring as stigmatellin. Interestingly, ␣-tocoquinone appears to act as an alternative substrate for the Qi site where it can be reduced to ␣-tocopheryl hydroquinone (109) (FIGURE 8). Because ␣-tocopheryl hydroquinone cannot be oxidized at the Qo site, this reaction partially uncouples the Q cycle. On the other hand, it is proposed that this reaction may be beneficial, especially under oxidative stress, as ␣-tocopheryl hydroquinone is a compound of antioxidative properties. It remains to be seen how the weakly bound external compounds can modulate the activity of cytochrome bc1 in the context of its dimeric function and its possible role in regulation of respiration on the molecular level. It also remains to be seen how and why they modulate ROS production. The regulatory function of these compounds may be of great importance for tissues under oxidative stress and aging. XI. CONCLUSIONS Cytochrome bc1 is one of the key enzymes of mitochondrial respiratory chain, where it plays a major role as proton translocating machinery utilizing the energy stored in the difference in redox potentials between the membrane pool of quinone (Q pool) and water-soluble pool of cytochrome c (C pool). Several features of this homodimeric enzyme Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 235 MARCIN SAREWICZ AND ARTUR OSYCZKA make it a potential point of regulation that dynamically modulates the electron flux through the mitochondrial respiratory chain in response to the actual physiological conditions. Our goal was to point out those features and discuss them in the context of the molecular mechanism of cytochrome bc1 operation and physiological functioning of mitochondria (FIGURE 8). First, each monomer builds in two quinone binding sites where two opposite redox reactions take place. This means that the substrate for one site is the product of the second site, and vice versa. These sites are located on the opposite sides of the membrane and are electronically connected through the chain of cofactors providing a path for reversible exchange of electrons between the sites. In this system the direction of electron flow depends on rates of all partial reversible reactions within the cofactor chains, including the exchange of UQ/UQH2 within the catalytic sites. Overall, this makes the activity of the whole complex sensitive to redox state of the Q pool. In addition, as the enzyme connects the Q pool with C pool, it may switch between the forward and the reverse mode of operation in response to the changes in bioenergetic state of the respiratory chain (FIGURE 4). Second, the electronic connection between monomers of the dimer links functionally all four quinone catalytic sites, forming the H-shaped electron transfer system which increases the number of available connections between the sites. In this system, any connection between the two sites on the opposite sides of the membrane is enzymatically competent (FIGURE 9). The H-shaped electron transfer system can provide physiological advantage for the cells as it may enhance the efficiency of the enzymatic cycle under shortage of UQH2 in the pool. It also builds in redundancy to allow physiological function of the enzyme even after operational damage of one part. This may be advantageous in the context of mitochondrial mutations that accumulate upon aging or under oxidative stress. Third, in addition to the energy-conserving reactions, cytochrome bc1 under certain conditions generates superoxide which, unlike in the case of other respiratory complexes, is released to the intermembrane space of mitochondria. This reaction results in electron leakage diminishing proton pumping efficiency of the enzyme. However, because of its location, superoxide generated by cytochrome bc1 can be considered as a potential factor engaged in the redox signaling (FIGURE 8). The role of this compound in sensing the redox state of mitochondria is conceivable given that the level of superoxide generation strongly depends on the reduction levels of cofactors in cytochrome bc1 which, in turn, change in response to various factors including the redox state of the Q pool and the C pool and the magnitude of membrane potential. In fact, the exact amounts of superoxide released by cytochrome bc1 may be under redox and 236 kinetic control given that this reaction appears to compete with other electron transfer reactions that take place within the enzyme. One type of those reactions includes the shortcircuits that, at cost of dissipating energy, retain single electrons within the enzyme preventing their leakage (FIGURE 6). The other type of reactions is inherent to a uniting action of the H-shaped electron transfer system which can collect and neutralize single electrons generated by cytochrome bc1 to diminish ROS. Fourth, some of the constituents naturally present in mitochondrial membrane may modulate the activity of cytochrome bc1 or ROS production by interfering with quinones in binding to the catalytic sites (FIGURE 8). Also, some metabolites of vitamin E can serve as an alternative substrate which undergoes reduction at quinone-reducing site of cytochrome bc1. On one hand, these types of reactions interfere with the catalytic cycle; on the other hand, they become a support for antioxidative system in membranes. At present, considering all the structural and functional details of the quinone binding catalytic sites and the cofactor chains of cytochrome bc1, understanding of the operation of cytochrome bc1 is at the advanced stage of molecular characterization. Nevertheless, several crucial aspects of operation of this enzyme still remain elusive. For example, at the molecular level, further progress in description of intermediates at the quinone oxidation site and reactions leading to ROS generation is needed. At the cellular level, the potential role of ROS generated by cytochrome bc1 in signaling requires further studies. Undoubtedly, as our thinking and methodology advance to better link physics/ chemistry with physiology, these masterpieces of bioenergetic machinery will excite us with new discoveries at all levels. ACKNOWLEDGMENTS Address for reprint requests and other correspondence: A. Osyczka, Dept. of Molecular Biophysics, Faculty of Biochemistry, Biophysics, and Biotechnology, Jagiellonian University, ul. Gronostajowa 7, 30-387 Kraków, Poland (e-mail: artur.osyczka@uj.edu.pl). GRANTS This work was supported by The Wellcome Trust International Senior Research Fellowship (to A. Osyczka). DISCLOSURES No conflicts of interest, financial or otherwise, are declared by the authors. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA REFERENCES 21. Berry EA, Huang LS, Saechao LK, Pon NG, Valkova-Valchanova M, Daldal F. X-Ray structure of Rhodobacter capsulatus cytochrome bc1: comparison with its mitochondrial and chloroplast counterparts. Photosynth Res 81: 251–275, 2004. 1. Aberg F, Appelkvist EL, Dallner G, Ernster L. Distribution and redox state of ubiquinones in rat and human tissues. Arch Biochem Biophys 295: 230 –234, 1992. 2. Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci 27: 639 – 645, 2006. 3. Adrain C, Martin SJ. The mitochondrial apoptosome: a killer unleashed by the cytochrome seas. Trends Biochem Sci 26: 390 –397, 2001. 4. Al-Attar S, de Vries S. Energy transduction by respiratory metallo-enzymes: from molecular mechanism to cell physiology. Coord Chem Rev 257: 64 – 80, 2013. 5. Alric J, Lavergne J, Rappaport F, Verméglio A, Matsuura K, Shimada K, Nagashima KVP. Kinetic performance and energy profile in a roller coaster electron transfer chain: a study of modified tetraheme-reaction center constructs. J Am Chem Soc 128: 4136 – 4145, 2006. 6. Andrews KM, Crofts AR, Gennis RB. Large-scale purification and characterization of a highly active four-subunit cytochrome bc1 complex from Rhodobacter sphaeroides. Biochemistry 29: 2645–2651, 1990. 22. Berry EA, Huang L. Observations concerning the quinol oxidation site of the cytochrome bc1 complex. FEBS Lett 555: 13–20, 2003. 23. Berry EA, Lee DW, Huang LS, Daldal F. Structural and mutational studies of the cytochrome bc1 complex. In: The Purple Phototrophic Bacteria, edited by Hunter CN, Daldal F, Thurnauer MC, Beatty JT. New York: Springer Science, 2009, p. 425– 450. 24. Beulens J, Booth SL, van den Heuvel EG, Stoecklin E, Baka A, Vermeer C. The role of menaquinones (vitamin K2) in human health. Br J Nutr 110: 1357–1368, 2013. 25. Beyer RE. The role of ascorbate in antioxidant protection of biomembranes: interaction with vitamin E and coenzyme Q. J Bioenerg Biomembr 26: 349 –358, 1994. 26. Bleier L, Dröse S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta 1827: 1320 –1331, 2013. 27. Boekema EJ, Braun HP. Supramolecular structure of the mitochondrial oxidative phosphorylation system. J Biol Chem 282: 1– 4, 2007. 28. Booth SL. Roles for vitamin K beyond coagulation. Annu Rev Nutr 29: 89 –110, 2009. 7. Antonenko YN, Avetisyan AV, Bakeeva LE, Chernyak BV, Chertkov VA, Domnina LV, Ivanova OY, Izyumov DS, Khailova LS, Klishin SS, Korshunova GA, Lyamzaev KG, Muntyan MS, Nepryakhina OK, Pashkovskaya AA, Pletjushkina OY, Pustovidko AV, Roginsky VA, Rokitskaya TI, Ruuge EK, Saprunova VB, Severina Simonyan RA 2nd, Skulachev IV, Skulachev MV, Sumbatyan NV, Sviryaeva IV, Tashlitsky VN, Vassiliev JM, Vyssokikh MY, Yaguzhinsky LS, Zamyatnin AA, Skulachev VP. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: synthesis and in vitro studies. Biochemistry Mosc 73: 1273–1287, 2008. 8. Antonenko YN, Roginsky VA, Pashkovskaya AA, Rokitskaya TI, Kotova EA, Zaspa AA, Chernyak BV, Skulachev VP. Protective effects of mitochondria-targeted antioxidant SkQ in aqueous and lipid membrane environments. J Membr Biol 222: 141–149, 2008. 9. Arnarez C, Mazat JP, Elezgaray J, Marrink SJ, Periole X. Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J Am Chem Soc 135: 3112–3120, 2013. 10. Artuch R, Vilaseca MA, Moreno J, Lambruschini N, Cambra FJ, Campistol J. Decreased serum ubiquinone-10 concentrations in phenylketonuria. Am J Clin Nutr 70: 892– 895, 1999. 11. Atta-Asafo-Adjei E, Daldal F. Size of the amino acid side chain at position 158 of cytochrome b is critical for an active cytochrome bc1 complex and for photosynthetic growth of Rhodobacter capsulatus. Proc Natl Acad Sci USA 88: 492– 496, 1991. 12. Baggish AL, Hill DR. Antiparasitic agent Atovaquone. Antimicrob Agents Chemother 46: 1163–1173, 2002. 13. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 120: 483– 495, 2005. 14. Barbara PF, Meyer TJ, Ratner MA. Contemporary issues in electron transfer research. J Phys Chem 100: 13148 –13168, 1996. 15. Barton V, Fisher N, Biagini GA, Ward SA, O’Neill PM. Inhibiting Plasmodium cytochrome bc1: a complex issue. Curr Opin Chem Biol 14: 440 – 446, 2010. 16. Bartoschek S, Johansson M, Geierstanger BH, Okun JG, Lancaster CR, Humpfer E, Yu L, Yu CA, Griesinger C, Brandt U. Three molecules of ubiquinone bind specifically to mitochondrial cytochrome bc1 complex. J Biol Chem 276: 35231– 35234, 2001. 17. Bénit P, Lebon S, Rustin P. Respiratory-chain diseases related to complex III deficiency. Biochim Biophys Acta 1793: 181–185, 2009. 18. Bentinger M, Brismar K, Dallner G. The antioxidant role of coenzyme Q. Mitochondrion 7 Suppl: S41–S50, 2007. 29. Borek A, Sarewicz M, Osyczka A. Movement of the iron-sulfur head domain of cytochrome bc1 transiently opens the catalytic Qo site for reaction with oxygen. Biochemistry 47: 12365–12370, 2008. 30. Bosshard HR, Wynn RM, Knaff DB. Binding site on Rhodospirillum rubrum cytochrome c2 for the Rhodospirillum rubrum cytochrome bc1 complex. Biochemistry 26: 7688 – 7693, 1987. 31. Boveris A, Cadenas E. Mitochondrial production of superoxide anions and its relationship to the antimycin insensitive respiration. FEBS Lett 54: 311–314, 1975. 32. Boveris A, Cadenas E, Stoppani AOM. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J 156: 435– 444, 1976. 33. Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 37: 755–767, 2004. 34. Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol 45: 466 – 472, 2010. 35. Brandt U, Okun JG. Role of deprotonation events in ubihydroquinone:cytochrome c oxidoreductase from bovine heart and yeast mitochondria. Biochemistry 36: 11234 – 11240, 1997. 36. Brandt U. Energy conservation by bifurcated electron-transfer in the cytochrome-bc1 complex. Biochim Biophys Acta 1275: 41– 46, 1996. 37. Brandt U. The chemistry and mechanics of ubihydroquinone oxidation at center P (Qo) of the cytochrome bc1 complex. Biochim Biophys Acta 1365: 261–268, 1998. 38. Brasseur G, Levican G, Bonnefoy V, Holmes D, Jedlicki E, Lemesle-Meunier D. Apparent redundancy of electron transfer pathways via bc1 complexes and terminal oxidases in the extremophilic chemolithoautotrophic Acidithiobacillus ferrooxidans. Biochim Biophys Acta 1656: 114 –126, 2004. 39. Brasseur G, Saribaş AS, Daldal F. A compilation of mutations located in the cytochrome b subunit of the bacterial and mitochondrial bc1 complex. Biochim Biophys Acta 1275: 61– 69, 1996. 40. Brugna M, Rodgers S, Schricker A, Montoya G, Kazmeier M, Nitschke W, Sinning I. A spectroscopic method for observing the domain movement of the Rieske iron-sulfur protein. Proc Natl Acad Sci USA 97: 2069 –2074, 2000. 41. Butler J, Jayson GG, Swallow AJ. The reaction between the superoxide anion radical and cytochrome c. Biochim Biophys Acta 408: 215–222, 1975. 19. Berry EA, De Bari H, Huang LS. Unanswered questions about the structure of cytochrome bc1 complexes. Biochim Biophys Acta 1827: 1258 –1277, 2013. 42. Cadenas E, Boveris A, Ragan CI, Stoppani AOM. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys 180: 248 –257, 1977. 20. Berry EA, Guergova-Kuras M, Huang L, Crofts AR. Structure and function of cytochrome bc complexes. Annu Rev Biochem 69: 1005–1075, 2000. 43. Cape JL, Bowman MK, Kramer DM. Understanding the cytochrome bc complexes by what they don’t do. The Q cycle at 30. Trends Plant Sci 11: 46 –55, 2006. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 237 MARCIN SAREWICZ AND ARTUR OSYCZKA 44. Cape JL, Bowman MK, Kramer DM. A semiquinone intermediate generated at the Qo site of the cytochrome bc1 complex: importance for the Q-cycle and superoxide production. Proc Natl Acad Sci USA 104: 7887–7892, 2007. 45. Castellani M, Covian R, Kleinschroth T, Anderka O, Ludwig B, Trumpower BL. Direct demonstration of half-of-the-sites reactivity in the dimeric cytochrome bc1 complex. J Biol Chem 285: 502–510, 2009. 46. Chance B, Hollunger G. The interaction of energy and electron transfer reactions in mitochondria. IV. The pathway of electron transfer. J Biol Chem 236: 1562–1568, 1961. 47. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol Chem 278: 36027– 36031, 2003. 48. Cheng B, Yang Z, Dong Y, Cheng K, Lin K. Effects of redox state for ubiquinone-10 on the membrane fluidity of cardiolipin-containing liposomes. In: Bioenergetics Molecular Biology, Biochemistry, and Pathology, edited by Kim CH, Ozawa T. New York: Plenum, 1990, p. 83– 88. 49. Cieluch E, Pietryga K, Sarewicz M, Osyczka A. Visualizing changes in electron distribution in coupled chains of cytochrome bc1 by modifying barrier for electron transfer between the FeS cluster and heme c1. Biochim Biophys Acta 1797: 296 –303, 2010. 50. Cooley JW, Lee DW, Daldal F. Across membrane communication between the Qo and Qi active sites of cytochrome bc1. Biochemistry 48: 1888 –1899, 2009. 51. Cooley JW, Ohnishi T, Daldal F. Binding dynamics at the quinone reduction (Qi) site influence the equilibrium interactions of the iron sulfur protein and hydroquinone oxidation (Qo) site of the cytochrome bc1 complex. Biochemistry 44: 10520 –10532, 2005. 52. Cooley JW. Protein conformational changes involved in the cytochrome bc1 complex catalytic cycle. Biochim Biophys Acta 1827: 1340 –1345, 2013. 53. Covian R, Gutierrez-Cirlos EB, Trumpower BL. Anti-cooperative oxidation of ubiquinol by the yeast cytochrome bc1 complex. J Biol Chem 279: 15040 –15049, 2004. 54. Covian R, Trumpower BL. Regulatory interactions between ubiquinol oxidation and ubiquinone reduction sites in the dimeric cytochrome bc1 complex. J Biol Chem 281: 30925–30932, 2006. 55. Covian R, Trumpower BL. Regulatory interactions in the dimeric cytochrome bc1 complex: the advantages of being a twin. Biochim Biophys Acta 1777: 1079 –1091, 2008. 56. Cramer WA, Hasan SS, Yamashita E. The Q cycle of cytochrome bc complexes: a structure perspective. Biochim Biophys Acta 1807: 788 – 802, 2011. 57. Cramer WA, Zhang H, Yan J, Kurisu G, Smith JL. Transmembrane traffic in the cytochrome b6f complex. Annu Rev Biochem 75: 769 –790, 2006. 58. Crane FL. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion 7S: S2–S7, 2007. 59. Crofts A, Hacker B, Barquera B, Yun CH, Gennis R. Structure and function of the bc-complex of Rhodobacter sphaeroides. Biochim Biophys Acta 1101: 162–165, 1992. 60. Crofts AR, Guergova-Kuras M, Huang L, Kuras R, Zhang Z, Berry EA. Mechanism of ubiquinol oxidation by the bc1 complex: role of the iron sulfur protein and its mobility. Biochemistry 38: 15791–15806, 1999. 61. Crofts AR, Holland JT, Victoria D, Kolling DRJ, Dikanov SA, Gilbreth R, Lhee S, Kuras R, Kuras MG. The Q-cycle reviewed: how well does a monomeric mechanism of the bc1 complex account for the function of a dimeric complex? Biochim Biophys Acta 1777: 1001–1019, 2008. 62. Crofts AR, Hong S, Ugulava N, Barquera B, Gennis R, Guergova-Kuras M, Berry EA. Pathways for proton release during ubihydroquinone oxidation by the bc1 complex. Proc Natl Acad Sci USA 96: 10021–10026, 1999. 65. Crofts AR. The cytochrome bc1 complex: function in the context of structure. Annu Rev Physiol 66: 689 –733, 2004. 66. Cuddihy SL, Ali SS, Musiek ES, Lucero J, Kopp SJ, Morrow JD, Dugan LL. Prolonged alpha-tocopherol deficiency decreases oxidative stress and unmasks alpha-tocopherol-dependent regulation of mitochondrial function in the brain. J Biol Chem 283: 6915– 6924, 2008. 67. Czapla M, Borek A, Sarewicz M, Osyczka A. Enzymatic activities of isolated cytochrome bc1-like complexes containing fused cytochrome bc1 subunits with asymmetrically inactivated segments of electron transfer chains. Biochemistry 51: 829 – 835, 2012. 68. Czapla M, Cieluch E, Borek A, Sarewicz M, Osyczka A. Catalytically-relevant electron transfer between two hemes bL in the hybrid cytochrome bc1-like complex containing a fusion of Rhodobacter sphaeroides and capsulatus cytochromes b. Biochim Biophys Acta 1827: 751–760, 2013. 69. Darrouzet E, Cooley JW, Daldal F. The cytochrome bc1 complex and its homologue the b6f complex: similarities and differences. Photosynth Res 79: 25– 44, 2004. 70. Darrouzet E, Daldal F. Movement of the iron-sulfur subunit beyond the EF loop of cytochrome b is required for multiple turnovers of the bc1 complex but not for single turnover Qo site catalysis. J Biol Chem 277: 3471–3476, 2002. 71. Darrouzet E, Daldal F. Protein-protein interactions between cytochrome b and the Fe-S protein subunits during QH2 oxidation and large-scale domain movement in the bc1 complex. Biochemistry 42: 1499 –1507, 2003. 72. Darrouzet E, Moser CC, Dutton PL, Daldal F. Large scale domain movement in cytochrome bc1: a new device for electron transfer in proteins. Trends Biochem Sci 26: 445– 451, 2001. 73. Darrouzet E, Valkova-Valchanova M, Moser CC, Dutton PL, Daldal F. Uncovering the [2Fe2S] domain movement in cytochrome bc1 and its implications for energy conversion. Proc Natl Acad Sci USA 97: 4567– 4572, 2000. 74. Davidson E, Ohnishi T, Atta-Asafo-Adjei E, Daldal F. Potential ligands to the Rieske cluster of the cytochrome bc1 complex of Rhodobacter capsulatus probed by sitedirected mutagenesis. Biochemistry 31: 3342–3351, 1992. 75. Devanathan S, Salamon Z, Tollin G, Fitch JC, Meyer TE, Berry EA, Cusanovich MA. Plasmon waveguide resonance spectroscopic evidence for differential binding of oxidized and reduced Rhodobacter capsulatus cytochrome c2 to the cytochrome bc1 complex mediated by the conformation of the Rieske iron-sulfur protein. Biochemistry 46: 7138 –7145, 2007. 75a.De Vries S, Albracht SPJ, Berden JA, Slater EC. A new species of bound ubisemiquinone anion in QH2:cytochrome c oxidoreductase. J Biol Chem 256: 11996 –11998, 1981. 76. Dikanov SA, Holland JT, Endeward B, Kolling DRJ, Samoilova RI, Prisner TF, Crofts AR. Hydrogen bonds between nitrogen donors and the semiquinone in the Qi-site of the bc1 complex. J Biol Chem 282: 25831–25841, 2007. 77. DiMauro S. Mitochondrial diseases. Biochim Biophys Acta 1658: 80 – 88, 2004. 78. Ding H, Daldal F, Dutton PL. Ion pair formation between basic residues at 144 of the Cyt b polypeptide and the ubiquinones at the Qo site of the Cyt bc1 complex. Biochemistry 34: 15997–16003, 1995. 79. Ding H, Moser CC, Robertson DE, Tokito MK, Daldal F, Dutton PL. Ubiquinone pair in the Qo site central to the primary energy conversion reactions of cytochrome bc1 complex. Biochemistry 34: 15979 –15996, 1995. 80. Ding H, Robertson DE, Daldal F, Dutton PL. Cytochrome bc1 complex [2Fe-2S] cluster and its interaction with ubiquinone and ubihydroquinone at the Qo site: a double-occupancy Qo site model. Biochemistry 31: 3144 –3158, 1992. 81. Dixon DW, Hong X, Woehler SE. Electrostatic and steric control of electron selfexchange in cytochromes c, c551, and b5. Biophys J 56: 339 –351, 1989. 63. Crofts AR, Lhee S, Crofts SB, Cheng J, Rose S. Proton pumping in the bc1 complex: A new gating mechanism that prevents short circuits. Biochim Biophys Acta 1757: 1019 – 1034, 2006. 82. Dröse S, Brandt U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J Biol Chem 283: 21649 –21654, 2008. 64. Crofts AR, Meinhardt SW, Jones KR, Snozzi M. The role of the quinone pool in the cyclic electron-transfer chain of Rhodopseudomonas sphareoides: A modified Q-cycle mechanism. Biochim Biophys Acta 723: 202–218, 1983. 83. Dröse S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. In: Mitochondrial Oxidative Phosphorylation, edited by Kadenbach B. New York: Springer, 2012, p. 145–169. 238 Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA 84. Dröse S, Hanley PJ, Brandt U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochim. Biophys Acta 1790: 558 – 565, 2009. 85. Echtay KS, Winkler E, Klingenberg M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 408: 609 – 613, 2000. 86. Edlund C, Söderberg M, Kristensson K, Dallner G. Ubiquinone, dolichol, and cholesterol metabolism in aging and Alzheimer’s disease. Biochem Cell Biol 70: 422– 428, 1992. 87. Edlund C, Söderberg M, Kristensson K. Isoprenoids in aging and neurodegeneration. Neurochem Int 25: 35–38, 1994. 87a.Ekiert R, Czapla M, Sarewicz M, Osyczka A. Hybrid fusions show that inner-monomer electron transfer robustly supports cytochrome bc1 function in vivo. Biochem Biophys Res Commun 451: 270 –275, 2014. 88. Elbehti A, Brasseur G, Lemesle-Meunier D. First evidence for existence of an uphill electron transfer through the bc1 and NADH-Q oxidoreductase complexes of the acidophilic obligate chemolithotrophic ferrous ion-oxidizing bacterium Thiobacillus ferrooxidans. J Bacteriol 182: 3602–3606, 2000. 89. Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci 26: 597– 604, 2001. 90. Engstrom G, Rajagukguk R, Saunders AJ, Patel CN, Rajagukguk S, Merbitz-Zahradnik T, Xiao K, Pielak GJ, Trumpower B, Yu CA, Yu L, Durham B, Millett F. Design of a ruthenium-labeled cytochrome c derivative to study electron transfer with the cytochrome bc1 complex. Biochemistry 42: 2816 –2824, 2003. 91. Ericsson J, Dallner G. Distribution, biosynthesis, and function of mevalonate pathway lipids. In: Sub-cellular Biochemistry, edited by Borgese N, Harris JR. New York: Plenum, 1993, p. 229 –272. 92. Ernster L, Lee IY, Norling B, Persson B. Studies with ubiquinone-depleted submitochondrial particles. Essentiality of ubiquinone for the interaction of succinate dehydrogenase, NADH dehydrogenase, and cytochrome b. Eur J Biochem 9: 299 –310, 1969. 105. Galinier A, Carrière A, Fernandez Y, Bessac AM, Caspar-Bauguil S, Periquet B, Comtat M, Thouvenot JP, Casteilla L. Biological validation of coenzyme Q redox state by HPLC-EC measurement: relationship between coenzyme Q redox state and coenzyme Q content in rat tissues. FEBS Lett 578: 53–57, 2004. 106. Genova ML, Lenaz G. New developments on the functions of coenzyme Q in mitochondria. Biofactors 37: 330 –354, 2011. 107. Genova ML, Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta 1837: 427– 443, 2014. 108. Gille L, Gregor W, Staniek K, Nohl H. Redox-interaction of alpha-tocopheryl quinone with isolated mitochondrial cytochrome bc1 complex. Biochem Pharmacol 68: 373– 381, 2004. 109. Gille L, Staniek K, Rosenau T, Duvigneau JC, Kozlov AV. Tocopheryl quinones and mitochondria. Mol Nutr Food Res 54: 601– 615, 2010. 110. Gong X, Yu L, Xia D, Yu CA. Evidence for electron equilibrium between the two hemes bL in the dimeric cytochrome bc1 complex. J Biol Chem 280: 9251–9257, 2005. 111. Gray KA, Daldal F. Mutational studies of the cytochrome bc1 complexes. In: Anoxygenic Photosynthetic Bacteria, edited by Blankenship RE, Madigan MT, Bauer CE. New York: Kluwer Academic, 1995, p. 747–774. 112. Gray KA, Dutton PL, Daldal F. Requirement of histidine 217 for ubiquinone reductase activity (Qi site) in the cytochrome bc1 complex. Biochemistry 33: 723–733, 1994. 113. Gregor W, Staniek K, Nohl H, Gille L. Distribution of tocopheryl quinone in mitochondrial membranes and interference with ubiquinone-mediated electron transfer. Biochem Pharmacol 71: 1589 –1601, 2006. 114. Gu¨ner S, Willie A, Millett F, Caffrey MS, Cusanovich MA, Robertson DE, Knaff DB. The interaction between cytochrome c2 and the cytochrome bc1 complex in the photosynthetic purple bacteria Rhodobacter capsulatus and Rhodopseudomonas viridis. Biochemistry 32: 4793– 4800, 1993. 115. Gunner MR, Madeo J, Zhu Z. Modification of quinone electrochemistry by the proteins in the biological electron transfer chains: examples from photosynthetic reaction centers. J Bioenerg Biomembr 40: 509 –519, 2008. 93. Esser L, Elberry M, Zhou F, Yu CA, Yu L, Xia D. Inhibitor-complexed structures of the cytochrome bc1 from the photosynthetic bacterium Rhodobacter sphaeroides. J Biol Chem 283: 2846 –2857, 2008. 116. Gupta RK. Electron transfer in cytochrome c, role of the polypeptide chain. Biochim Biophys Acta 292: 291–295, 1973. 94. Esser L, Gong X, Yang S, Yu L, Yu CA, Xia D. Surface-modulated motion switch: capture and release of iron-sulfur protein in the cytochrome bc1 complex. Proc Natl Acad Sci USA 103: 13045–13050, 2006. 117. Gupte S, Wu ES, Hoechli L, Hoechli M, Jacobson K, Sowers AE, Hackenbrock CR. Relationship between lateral diffusion, collision frequency, and electron transfer of mitochondrial inner membrane oxidation-reduction components. Proc Natl Acad Sci USA 81: 2606 –2610, 1984. 95. Esser L, Quinn B, Li YF, Zhang M, Elberry M, Yu L, Yu CA, Xia D. Crystallographic studies of quinol oxidation site inhibitors: a modified classification of inhibitors for the cytochrome bc1 complex. J Mol Biol 341: 281–302, 2004. 96. Fato R, Bertoli E, Parenti CG, Lenaz G. Fluidizing effect of endogenous ubiquinone in bovine heart mitochondrial membranes. FEBS Lett 172: 6 –10, 1984. 97. Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem 287: 4434 – 4440, 2012. 98. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol 194: 7–15, 2011. 99. Fisher N, Meunier B. Effects of mutations in mitochondrial cytochrome b in yeast and man. Deficiency, compensation and disease. Eur J Biochem 268: 1155–1162, 2001. 100. Fisher N, Rich PR. A motif for quinone binding sites in respiratory and photosynthetic systems. J Mol Biol 296: 1153–1162, 2000. 101. Fontaine E, Ichas F, Bernardi P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J Biol Chem 273: 25734 –25740, 1998. 118. Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol 91: 807– 819, 2006. 119. Hackenbrock CR, Chazotte B, Gupte SS. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J Bioenerg Biomembr 18: 331–368, 1986. 120. Hacker B, Barquera B, Crofts AR, Gennis RB. Characterization of mutations in the cytochrome b subunit of the bc1 complex of Rhodobacter sphaeroides that affect the quinone reductase site (Qc). Biochemistry 32: 4403– 4410, 1993. 121. Hall J, Kriaucionas A, Knaff D, Millett F. The reaction domain on Rhodospirillum rubrum cytochrome c2 and horse cytochrome c for the Rhodospirillum rubrum cytochrome bc1 complex. J Biol Chem 262: 14005–14009, 1987. 122. Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 35: 505–513, 2010. 102. Forman H, Azzi A. On the virtual existence of superoxide anions in mitochondria: thoughts regarding its role in pathophysiology. FASEB J 11: 374 –375, 1997. 123. Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J 353: 411– 416, 2001. 103. Forquer I, Covian R, Bowman MK, Trumpower BL, Kramer DM. Similar transition states mediate the Q-cycle and superoxide production by the cytochrome bc1 complex. J Biol Chem 281: 38459 –38465, 2006. 124. Hauska G, Hurt E. Pool function behavior and mobility of isoprenoid quinones. In: Function of Quinones in Energy Conserving Systems, edited by Trumpower B. London: Academic, 1982, p. 87–110. 104. Forsmark P, Aberg F, Norling B, Nordenbrand K, Dallner G, Ernster L. Inhibition of lipid peroxidation by ubiquinol in submitochondrial particles in the absence of vitamin E. FEBS Lett 285: 39 – 43, 1991. 125. Hong S, Ugulava N, Guergova-Kuras M, Crofts AR. The energy landscape for ubihydroquinone oxidation at the Qo site of the bc1 complex in Rhodobacter sphaeroides. J Biol Chem 274: 33931–33944, 1999. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 239 MARCIN SAREWICZ AND ARTUR OSYCZKA 126. Huang LS, Cobessi D, Tung EY, Berry EA. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J Mol Biol 351: 573–597, 2005. 127. Hughes LM, Lanteri CA, O’Neil MT, Johnson JD, Gribble GW, Trumpower BL. Design of anti-parasitic and anti-fungal hydroxy-naphthoquinones that are less susceptible to drug resistance. Mol Biochem Parasitol 177: 12–19, 2011. 128. Hunte C, Koepke J, Lange C, Rossmanith T, Michel H. Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure 8: 669 – 684, 2000. 129. Hunte C, Palsdottir H, Trumpower B. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett 545: 39 – 46, 2003. 130. Hunte C. Insights from the structure of the yeast cytochrome bc1 complex: crystallization of membrane proteins with antibody fragments. FEBS Lett 504: 126 –132, 2001. 131. Iwaki M, Yakovlev G, Hirst J, Osyczka A, Dutton PL, Marshall D, Rich PR. Direct observation of redox-linked histidine protonation changes in the iron-sulfur protein of the cytochrome bc1 complex by ATR-FTIR spectroscopy. Biochemistry 44: 4230 – 4237, 2005. 132. Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 281: 64 –71, 1998. 134. James AM, Sharpley MS, Manas ARB, Frerman FE, Hirst J, Smith RAJ, Murphy MP. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem 282: 14708 –14718, 2007. 135. Janzon J, Eichhorn AC, Ludwig B, Malatesta F. Electron transfer kinetics between soluble modes of Paracoccus denitrificans cytochrome c1 and its physiological redox partners. Biochim Biophys Acta 1777: 250 –259, 2008. 136. Ju¨nemann S, Heathcote P, Rich PR. On the mechanism of quinol oxidation in the bc1 complex. J Biol Chem 273: 21603–21607, 1998. 137. Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1: 223–232, 2005. 138. Kalén A, Appelkvist EL, Dallner G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 24: 579 –584, 1989. 139. Keilin D. On cytochrome, a respiratory pigment, common to animals, yeast, and higher plants. Proc R Soc London Ser B 98: 312–339, 1925. 140. Kessl JJ, Lange BB, Merbitz-Zahradnik T, Zwicker K, Hill P, Meunier B, Palsdottir H, Hunte C, Meshnick S, Trumpower BL. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J Biol Chem 278: 31312–31318, 2003. 148. Kujoth GC, Bradshaw PC, Haroon S, Prolla TA. The role of mitochondrial DNA mutations in mammalian aging. PLoS Genet 3: 161–173, 2007. 149. Kurisu G, Zhang H, Smith JL, Cramer WA. Structure of the cytochrome b6f complex of oxygenic photosynthesis: tuning the cavity. Science 302: 1009 –1014, 2003. 150. Lanciano P, Khalfaoui-Hassani B, Selamoglu N, Daldal F. Intermonomer electron transfer between the b hemes of heterodimeric cytochrome bc1. Biochemistry 52: 7196 –7206, 2013. 151. Lanciano P, Lee DW, Yang H, Darrouzet E, Daldal F. Intermonomer electron transfer between the low-potential b hemes of cytochrome bc1. Biochemistry 50: 1651–1663, 2011. 152. Lange C, Hunte C. Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. Proc Natl Acad Sci USA 99: 2800 –2805, 2002. 153. Lange C, Nett JH, Trumpower BL, Hunte C. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J 20: 6591– 6600, 2001. 154. Lapuente-Brun E, Moreno-Loshuertos R, Acín-Pérez R, Latorre-Pellicer A, Colás C, Balsa E, Perales-Clemente E, Quirós PM, Calvo E, Rodríguez-Hernández MA, Navas P, Cruz R, Carracedo Á, López-Otín C, Pérez-Martos A, Fernández-Silva P, Fernández-Vizarra E, Enríquez JA. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340: 1567–1570, 2013. 155. Larsen PL, Clarke CF. Extension of life-span in Caenorhabditis elegans by a diet lacking coenzyme Q. Science 295: 120 –123, 2002. 156. Lass A, Kwong L, Sohal RS. Mitochondrial coenzyme Q content and aging. Biofactors 9: 199 –205, 1999. 157. Lee DW, El Khoury Y, Francia F, Zambelli B, Ciurli S, Venturoli G, Hellwig P, Daldal F. Zinc inhibition of bacterial cytochrome bc1 reveals the role of cytochrome b E295 in proton release at the Qo site. Biochemistry 50: 4263– 4272, 2011. 158. Léger C, Lederer F, Guigliarelli B, Bertrand P. Electron flow in multicenter enzymes: theory, applications, and consequences on the natural design of redox chains. J Am Chem Soc 128: 180 –187, 2006. 159. Lenaz G, Fato R, Formiggini G, Genova ML. The role of Coenzyme Q in mitochondrial electron transport. Mitochondrion 7 Suppl: S8 –33, 2007. 160. Lenaz G, Genova ML. Mobility and function of coenzyme Q (ubiquinone) in the mitochondrial respiratory chain. Biochim Biophys Acta 1787: 563–573, 2009. 161. Leonard MA, Sharp RE, Darrouzet E, Moser CC, Ohnishi T, Gibney BR, Daldal F, Dutton PL. Coenzyme Q oxidation reduction reactions in mitochondrial electron transport. In: Coenzyme Q: Molecular Mechanisms in Health and Disease, edited by Kagan VE, Quinn, PJ. Boca Raton, FL: CRC, 2000, p. 65– 82. 141. Khalfaoui-Hassani B, Lanciano P, Daldal F. A robust genetic system for producing heterodimeric native and mutant cytochrome bc1. Biochemistry 52: 7184 –7195, 2013. 162. Lewis RJ, Prince RC, Dutton PL, Knaff DB, Krulwich TA. The respiratory chain of Bacillus alcalophilus and its nonalkalophilic mutant derivative. J Biol Chem 256: 10543– 10549, 1981. 142. Kleinschroth T, Castellani M, Trinh CH, Morgner N, Brutschy B, Ludwig B, Hunte C. X-ray structure of the dimeric cytochrome bc1 complex from the soil bacterium Paracoccus denitrificans at 2.7-Å resolution. Biochim Biophys Acta 1807: 1606 –1615, 2011. 163. Li J, Osyczka A, Conover RC, Johnson MK, Qin H, Daldal F, Knaff DB. Role of acidic and aromatic amino acids in Rhodobacter capsulatus cytochrome c1. A site-directed mutagenesis study. Biochemistry 42: 8818 – 8830, 2003. 143. Kolling DRJ, Samoilova RI, Holland JT, Berry EA, Dikanov SA, Crofts AR. Exploration of ligands to the Qi site semiquinone in the bc1 complex using high-resolution EPR. J Biol Chem 278: 39747–39754, 2003. 164. Link TA, Iwata S. Functional implications of the structure of the “Rieske” iron-sulfur protein of bovine heart mitochondrial cytochrome bc1 complex. Biochim Biophys Acta 1275: 54 – 60, 1996. 144. Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 416: 15–18, 1997. 165. Link TA. The role of the “Rieske” iron sulfur protein in the hydroquinone oxidation (QP) site of the cytochrome bc1 complex. FEBS Lett 412: 257–264, 1997. 145. Kramer DM, Roberts AG, Muller F, Cape J, Bowman MK. Q-cycle bypass reactions at the Qo site of the cytochrome bc1 (and related) complexes. Methods Enzymol 382: 21– 45, 2004. 146. Kröger A, Klingenberg M. The kinetics of the redox reactions of ubiquinone related to the electron-transport activity in the respiratory chain. Eur J Biochem 34: 358 –368, 1973. 147. Ksenzenko M, Konstantinov AA, Khomutov GB, Tikhonov AN, Ruuge EK. Effect of electron transfer inhibitors on superoxide generation in the cytochrome bc1 site of the mitochondrial respiratory chain. FEBS Lett 155: 19 –24, 1983. 240 166. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86: 147–157, 1996. 167. Maes M, Mihaylova I, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E. Lower plasma Coenzyme Q10 in depression: a marker for treatment resistance and chronic fatigue in depression and a risk factor to cardiovascular disorder in that illness. Neuro Endocrinol Lett 30: 462– 469, 2009. 168. Malinska D, Kulawiak B, Kudin AP, Kovacs R, Huchzermeyer C, Kann O, Szewczyk A, Kunz WS. Complex III-dependent superoxide production of brain mitochondria contributes to seizure-related ROS formation. Biochim Biophys Acta 1797: 1163–1170, 2010. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA 169. Malnoë A, Wollman FA, de Vitry C, Rappaport F. Photosynthetic growth despite a broken Q-cycle. Nat Commun 2: 1– 6, 2011. 192. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. 170. Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta 811: 265–322, 1985. 193. Nayak SK, Mallik SB, Kanaujia SP, Sekar K, Ranganathan KR, Ananthalakshmi V, Jeyaraman G, Saralaya SS, Rao KS, Shridhara K, Nagarajan K, Row TNG. Crystal structures and binding studies of atovaquone and its derivatives with cytochrome bc1: a molecular basis for drug design. Cryst Eng Comm 15: 4871– 4884, 2013. 171. Margalit R, Schejter A. Cytochrome c: a thermodynamic study of the relationships among oxidation state, ion-binding and structural parameters. 1. The effects of temperature, pH and electrostatic media on the standard redox potential of cytochrome c. Eur J Biochem 32: 492– 499, 1973. 172. McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244: 6049 – 6055, 1969. 194. Nicholls DG, Ferguson SJ. Bioenergetics (4th ed.). Amsterdam: Academic, 2013. 195. Nohl H, Hegner D. Do mitochondria produce oxygen radicals in vivo? Eur J Biochem 82: 563–567, 1978. 173. Meunier B, Fisher N, Ransac S, Mazat JP, Brasseur G. Respiratory complex III dysfunction in humans and the use of yeast as a model organism to study mitochondrial myopathy and associated diseases. Biochim Biophys Acta 1827: 1346 –1361, 2013. 196. Norling B, Glazek E, Nelson BD, Ernster L. Studies with ubiquinone-depleted submitochondrial particles. Quantitative incorporation of small amounts of ubiquinone and its effects on the NADH and succinate oxidase activities. Eur J Biochem 47: 475– 482, 1974. 174. Miki T, Miki M, Orii Y. Membrane potential-linked reversed electron transfer in the beef heart cytochrome bc1 complex reconstituted into potassium-loaded phospholipid vesicles. J Biol Chem 269: 1827–1833, 1994. 197. Northrup SH, Erickson HP. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc Natl Acad Sci USA 89: 3338 –3342, 1992. 175. Millett F, Havens J, Rajagukguk S, Durham B. Design and use of photoactive ruthenium complexes to study electron transfer within cytochrome bc1 and from cytochrome bc to cytochrome c. Biochim Biophys Acta 1827: 1309 –1319, 2013. 198. Nowicka B, Kruk J. Occurrence, biosynthesis and function of isoprenoid quinones. Biochim Biophys Acta 1797: 1587–1605, 2010. 176. Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemiosmotic type of mechanism. Nature 191: 144 –148, 1961. 177. Mitchell P. The protonmotive Q cycle: a general formulation. FEBS Lett 59: 137–139, 1975. 178. Mitchell P. Protonmotive redox mechanism of the cytochrome b-c1 complex in the respiratory chain: Protonmotive ubiquinone cycle. FEBS Lett 56: 1– 6, 1975. 179. Mitchell P. Possible molecular mechanisms of the protonmotive function of cytochrome systems. J Theor Biol 62: 327–367, 1976. 199. Osyczka A, Moser CC, Daldal F, Dutton PL. Reversible redox energy coupling in electron transfer chains. Nature 427: 607– 612, 2004. 200. Osyczka A, Moser CC, Dutton PL. Fixing the Q cycle. Trends Biochem Sci 30: 176 – 182, 2005. 201. Osyczka A, Zhang H, Mathé C, Rich PR, Moser CC, Dutton PL. Role of the PEWY glutamate in hydroquinone-quinone oxidation-reduction catalysis in the Qo site of cytochrome bc1. Biochemistry 45: 10492–10503, 2006. 202. Paddock ML, Feher G, Okamura MY. Proton transfer pathways and mechanism in bacterial reaction centers. FEBS Lett 555: 45–50, 2003. 180. Miyadera H, Amino H, Hiraishi A, Taka H, Murayama K, Miyoshi H, Sakamoto K, Ishii N, Hekimi S, Kita K. Altered quinone biosynthesis in the long-lived clk-1 mutants of Caenorhabditis elegans. J Biol Chem 276: 7713–7716, 2001. 203. Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402: 47–52, 1999. 181. Moore GR, Pettigrew GW. Cytochromes c, Evolutionary, Structural and Physicochemical Aspects. Heidelberg: Springer-Verlag, 1990. 204. Palsdottir H, Lojero CG, Trumpower BL, Hunte C. Structure of the yeast cytochrome bc1 complex with a hydroxyquinone anion Qo site inhibitor bound. J Biol Chem 278: 31303–31311, 2003. 182. Moser CC, Anderson JLR, Dutton PL. Guidelines for tunneling in enzymes. Biochim Biophys Acta 1797: 1573–1586, 2010. 183. Moser CC, Farid TA, Chobot SE, Dutton PL. Electron tunneling chains of mitochondria. Biochim Biophys Acta 1757: 1096 –1109, 2006. 184. Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. Nature of biological electron transfer. Nature 355: 796 – 802, 1992. 185. Moser CC, Page CC, Chen X, Dutton PL. Electron transfer in natural proteins theory and design. In: Enzyme-Catalyzed Electron and Radical Transfer, edited by Holzenburg A, Scrutton NA. New York: Kluwer Academic Press, 2002. 205. Pereverzev MO, Vygodina TV, Konstantinov AA, Skulachev VP. Cytochrome c, an ideal antioxidant. Biochem Soc Trans 31: 1312–1315, 2003. 206. Pietras R, Sarewicz M, Osyczka A. Molecular organization of cytochrome c2 near the binding domain of cytochrome bc1 studied by electron spin-lattice relaxation enhancement. J Phys Chem B 118: 6634 – 6643, 2014. 207. Postila PA, Kaszuba K, Sarewicz M, Osyczka A, Vattulainen I, Róg T. Key role of water in proton transfer at the Qo-site of the cytochrome bc1 complex predicted by atomistic molecular dynamics simulations. Biochim Biophys Acta 1827: 761–768, 2013. 186. Mulkidjanian AY. Ubiquinol oxidation in the cytochrome bc1 complex: reaction mechanism and prevention of short-circuiting. Biochim Biophys Acta 1709: 5–34, 2005. 208. Pöyry S, Cramariuc O, Postila PA, Kaszuba K, Sarewicz M, Osyczka A, Vattulainen I, Róg T. Atomistic simulations indicate cardiolipin to have an integral role in the structure of the cytochrome bc1 complex. Biochim Biophys Acta 1827: 769 –778, 2013. 187. Mulkidjanian AY. Proton translocation by the cytochrome bc1 complexes of phototrophic bacteria: introducing the activated Q-cycle. Photochem Photobiol Sci 6: 19 – 34, 2007. 209. Quinlan CL, Gerencser AA, Treberg JR, Brand MD. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J Biol Chem 286: 31361– 31372, 2011. 188. Müllebner A, Patel A, Stamberg W, Staniek K, Rosenau T, Netscher T, Gille L. Modulation of the mitochondrial cytochrome bc1 complex activity by chromanols and related compounds. Chem Res Toxicol 23: 193–202, 2010. 210. Raha S, McEachern GE, Myint AT, Robinson BH. Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic Biol Med 29: 170 –180, 2000. 189. Muller F, Crofts AR, Kramer DM. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc1 complex. Biochemistry 41: 7866 –7874, 2002. 211. Reed JC. Cytochrome c: can’t live with it– can’t live without it. Cell 91: 559 –562, 1997. 190. Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry 42: 6493– 6499, 2003. 191. Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, Rhee SG, Thornalley PJ, Partridge L, Gems D, Nyström T, Belousov V, Schumacker PT, Winterbourn CC. Unraveling the biological roles of reactive oxygen species. Cell Metab 13: 361–366, 2011. 212. Rich PR, Bendall DS. A mechanism for the reduction of cytochromes by quinols in solution and its relevance to biological electron transfer reactions. FEBS Lett 105: 189 –194, 1979. 213. Rich PR. Electron transfer reactions between quinols and quinones in aqueous and aprotic media. Biochim Biophys Acta 637: 28 –33, 1981. 214. Rich PR. Electron and proton transfers in chemical and biological quinone systems. Faraday Discuss Chem Soc 74: 349 –364, 1982. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 241 MARCIN SAREWICZ AND ARTUR OSYCZKA 215. Rich PR. The quinone chemistry of bc complexes. Biochim Biophys Acta 1658: 165– 171, 2004. 216. Robertson DE, Ding H, Chelminski PR, Slaughter C, Hsu J, Moomaw C, Tokito M, Daldal F, Dutton PL. Hydroubiquinone-cytochrome c2 oxidoreductase from Rhodobacter capsulatus: definition of a minimal, functional isolated preparation. Biochemistry 32: 1310 –1317, 1993. 217. Robertson DE, Prince RC, Bowyers JR, Matsuura K, Dutton PL, Ohnishi T. Thermodynamic properties of the semiquinone and its binding site in the ubiquinol-cytochrome c (c2) oxidoreductase of respiratory and photosynthetic systems. J Biol Chem 259: 1758 –1763, 1984. 218. Rottenberg H, Covian R, Trumpower BL. Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. J Biol Chem 284: 19203–19210, 2009. 219. Rutherford AW, Osyczka A, Rappaport F. Back-reactions, short-circuits, leaks and other energy wasteful reactions in biological electron transfer: redox tuning to survive life in O2. FEBS Lett 586: 603– 616, 2012. 220. Sarewicz M, Borek A, Cieluch E, S´wierczek M, Osyczka A. Discrimination between two possible reaction sequences that create potential risk of generation of deleterious radicals by cytochrome bc1. Implications for the mechanism of superoxide production. Biochim Biophys Acta 1797: 1820 –1827, 2010. 221. Sarewicz M, Borek A, Daldal F, Froncisz W, Osyczka A. Demonstration of short-lived complexes of cytochrome c with cytochrome bc1 by EPR spectroscopy: implications for the mechanism of interprotein electron transfer. J Biol Chem 283: 24826 –24836, 2008. 222. Sarewicz M, Dutka M, Froncisz W, Osyczka A. Magnetic interactions sense changes in distance between heme bL and the iron-sulfur cluster in cytochrome bc1. Biochemistry 48: 5708 –5720, 2009. 223. Sarewicz M, Dutka M, Pintscher S, Osyczka A. Triplet state of the semiquinoneRieske cluster as an intermediate of electronic bifurcation catalyzed by cytochrome bc1. Biochemistry 52: 6388 – 6395, 2013. 224. Sarewicz M, Pietras R, Froncisz W, Osyczka A. Reorientation of cytochrome c2 upon interaction with oppositely charged macromolecules probed by SR EPR: implications for the role of dipole moment to facilitate collisions in proper configuration for electron transfer. Metallomics 3: 404 – 409, 2011. 225. Saribas¸ AS, Ding H, Dutton PL, Daldal F. Tyrosine 147 of cytochrome b is required for efficient electron transfer at the ubihydroquinone oxidase site (Qo) of the cytochrome bc1 complex. Biochemistry 34: 16004 –16012, 1995. 226. Saribas¸ SA, Valkova-Valchanova M, Tokito MK, Zhang Z, Berry EA, Daldal F. Interactions between the cytochrome b, cytochrome C1, and Fe-S protein subunits at the ubihydroquinone oxidation site of the bc1 complex of Rhodobacter capsulatus. Biochemistry 37: 8105– 8114, 1998. 227. Schoepp-Cothenet B, Lieutaud C, Baymann F, Verméglio A, Friedrich T, Kramer DM, Nitschke W. Menaquinone as pool quinone in a purple bacterium. Proc Natl Acad Sci USA 106: 8549 – 8554, 2009. 228. Schreiber G. Kinetic studies of protein-protein interactions. Curr Opin Struct Biol 12: 41– 47, 2002. 229. Scott RA, Mauk AG. (Editors). Cytochrome c, a Mutidisciplinary Approach. Sausalito, CA: University Science Books, 1996. 230. Seddiki N, Meunier B, Lemesle-Meunier D, Brasseur G. Is cytochrome b glutamic acid 272 a quinol binding residue in the bc1 complex of Saccharomyces cerevisiae? Biochemistry 47: 2357–2368, 2008. 231. Selzer T, Schreiber G. New insight into the mechanism of protein-protein association. Proteins 45: 190 –198, 2001. 232. Sharp RE, Gibney BR, Palmitessa A, White JL, Dixon JA, Moser CC, Daldal F, Dutton PL. Ubiquinone binding capacity of the Rhodobacter capsulatus cytochrome bc1 complex: effect of diphenylamine, a weak binding Qo site inhibitor. Biochemistry 38: 3440 – 3446, 1999. 233. Shinkarev VP, Crofts AR, Wraight CA. The electric field generated by photosynthetic reaction center induces rapid reversed electron transfer in the bc1 complex. Biochemistry 40: 12584 –12590, 2001. 242 234. Shinkarev VP, Wraight CA. Intermonomer electron transfer in the bc1 complex dimer is controlled by the energized state and by impaired electron transfer between low and high potential hemes. FEBS Lett 581: 1535–1541, 2007. 235. Skulachev VP, Antonenko YN, Cherepanov DA, Chernyak BV, Izyumov DS, Khailova LS, Klishin SS, Korshunova GA, Lyamzaev KG, Pletjushkina OY, Roginsky VA, Rokitskaya TI, Severin FF, Severina II, Simonyan RA, Skulachev MV, Sumbatyan NV, Sukhanova EI, Tashlitsky VN, Trendeleva TA, Vyssokikh MY, Zvyagilskaya RA. Prevention of cardiolipin oxidation and fatty acid cycling as two antioxidant mechanisms of cationic derivatives of plastoquinone (SkQs). Biochim Biophys Acta 1797: 878 – 889, 2010. 236. Snyder CH, Gutierrez-Cirlos EB, Trumpower BL. Evidence for a concerted mechanism of ubiquinol oxidation by the cytochrome bc1 complex. J Biol Chem 275: 13535– 13541, 2000. 237. Solaini G, Baracca A, Lenaz G, Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta 1797: 1171–1177, 2010. 238. Solmaz S, Hunte C. Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer. J Biol Chem 283: 17542–17549, 2008. 239. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277: 44784 – 44790, 2002. 240. Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann NY Acad Sci 1147: 37–52, 2008. 241. Stonehuerners J, O’Brien P, Gerens L, Millett F, Steidl J, Yu L, Yu CA. Identification of the binding site on cytochrome c1 for cytochrome c. J Biol Chem 260: 5392–5398, 1985. 242. Stroebel D, Choquet Y, Popot JL, Picot D. An atypical haem in the cytochrome b6f complex. Nature 426: 413– 418, 2003. 243. Swida-Barteczka A, Woyda-Ploszczyca A, Sluse FE, Jarmuszkiewicz W. Uncoupling protein 1 inhibition by purine nucleotides is under the control of the endogenous ubiquinone redox state. Biochem J 424: 297–306, 2009. 244. Świerczek M, Cieluch E, Sarewicz M, Borek A, Moser CC, Dutton PL, Osyczka A. An electronic bus bar lies in the core of cytochrome bc1. Science 329: 451– 454, 2010. 245. Takamiya KI, Dutton PL. Ubiquinone in Rhodopseudomonas sphaeroides some thermodynamic properties. Biochim Biophys Acta 546: 1–16, 1979. 246. Tang PH, Miles MV, DeGrauw A, Hershey A, Pesce A. HPLC analysis of reduced and oxidized coenzyme Q10 in human plasma. Clin Chem 47: 256 –265, 2001. 247. Thierbach G, Kunze B, Reichenbach H, Höfle G. The mode of action of stigmatellin, a new inhibitor of the cytochrome bc1 segment of the respiratory chain. Biochim Biophys Acta 765: 227–235, 1984. 248. Thierbach G, Michaelis G. Mitochondrial and nuclear myxothiazol resistance in Saccharomyces cerevisiae. Mol Gen Genet 186: 501–506, 1982. 249. Tian H, Sadoski R, Zhang L, Yu CA, Yu L, Durham B, Millett F. Definition of the interaction domain for cytochrome c on the cytochrome bc1 complex. J Biol Chem 275: 9587–9595, 2000. 250. Tian H, White S, Yu L, Yu CA. Evidence for the head domain movement of the Rieske iron-sulfur protein in electron transfer reaction of the cytochrome bc1 complex. J Biol Chem 274: 7146 –7152, 1999. 251. Tian H, Yu L, Mather MW, Yun CA. Flexibility of the neck region of the Rieske iron-sulfur protein is functionally important in the cytochrome bc1 complex. J Biol Chem 273: 27953–27959, 1998. 252. Trouillard M, Meunier B, Rappaport F. Questioning the functional relevance of mitochondrial supercomplexes by time-resolved analysis of the respiratory chain. Proc Natl Acad Sci USA 108: E1027–E1034, 2011. 253. Trumpower BL. A concerted, alternating sites mechanism of ubiquinol oxidation by the dimeric cytochrome bc1 complex. Biochim Biophys Acta 1555: 166 –173, 2002. 254. Tuppen HAL, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 1797: 113–128, 2010. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org REGULATION AND FUNCTION OF CYTOCHROME bc1 IN MITOCHONDRIA 255. Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237: 408–414, 1985. 256. Turunen M, Olsson J, Dallner G. Metabolism and function of coenzyme Q. Biochim Biophys Acta 1660: 171–199, 2004. 257. Ubbink M. The courtship of proteins: understanding the encounter complex. FEBS Lett 583: 1060 –1066, 2009. 258. Urban PF, Klingenberg M. Redox potentials of ubiquinone and cytochrome b in the respiratory chain. Eur J Biochem 9: 519 –525, 1969. 259. Valgimigli L, Amorati R, Fumo MG, DiLabio GA, Pedulli GF, Ingold KU, Pratt DA. The unusual reaction of semiquinone radicals with molecular oxygen. J Org Chem 73: 1830 –1841, 2008. 260. Valkova-Valchanova M, Darrouzet E, Moomaw CR, Slaughter CA, Daldal F. Proteolytic cleavage of the Fe-S subunit hinge region of Rhodobacter capsulatus bc1 complex: effects of inhibitors and mutations. Biochemistry 39: 15484 –15492, 2000. 269. Xia D, Yu CA, Kim H, Xia JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science 277: 60 – 66, 1997. 270. Xiao K, Chandrasekaran A, Yu L, Yu CA. Evidence for the intertwined dimer of the cytochrome bc1 complex in solution. J Biol Chem 276: 46125– 46131, 2001. 271. Yamamoto Y, Yamashita S. Plasma ratio of ubiquinol and ubiquinone as a marker of oxidative stress. Mol Aspects Med 18: S79 –S84, 1997. 272. Yang XH, Trumpower BL. Purification of a three-subunit ubiquinol-cytochrome c oxidoreductase complex from Paracoccus denitrificans. J Biol Chem 261: 12282–12289, 1986. 273. Yu L, Tso SC, Shenoy SK, Quinn BN, Xia D. The role of the supernumerary subunit of Rhodobacter sphaeroides cytochrome bc1 complex. J Bioenerg Biomembr 31: 251–257, 1999. 274. Yu L, Yu CA. Inhibitory effect of alpha-tocopherol and its derivatives on bovine heart succinate-cytochrome c reductase. Biochim Biophys Acta 723: 139 –149, 1983. 261. Vennam PR, Fisher N, Krzyaniak MD, Kramer DM, Bowman MK. A caged, destabilized, free radical intermediate in the Q-cycle. Chem Bio Chem 14: 1745–1753, 2013. 275. Zhang H, Chobot SE, Osyczka A, Wraight CA, Dutton PL, Moser CC. Quinone and non-quinone redox couples in complex III. J Bioenerg Biomembr 40: 493– 499, 2008. 262. Von Jagow G, Link TA. Use of specific inhibitors on the mitochondrial bc1 complex. Methods Enzymol 126: 253–271, 1986. 276. Zhang H, Osyczka A, Dutton PL, Moser CC. Exposing the complex III Qo semiquinone radical. Biochim Biophys Acta 1767: 883– 887, 2007. 263. Weant KA, Smith KM. The role of coenzyme Q10 in heart failure. Ann Pharmacother 39: 1522–1526, 2005. 277. Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, Crofts AR, Berry EA, Kim SH. Electron transfer by domain movement in cytochrome bc1. Nature 392: 677– 684, 1998. 264. Wenz T, Hellwig P, MacMillan F, Meunier B, Hunte C. Probing the role of E272 in quinol oxidation of mitochondrial complex III. Biochemistry 45: 9042–9052, 2006. 265. Wikström MKF, Berden JA. Oxidoreduction of cytochrome b in the presence of antimycin. Biochim Biophys Acta 283: 403– 420, 1972. 278. Zhao PL, Wang L, Zhu XL, Huang X, Zhan CG, Wu JW, Yang GF. Subnanomolar inhibitor of cytochrome bc1 complex designed by optimizing interaction with conformationally flexible residues. J Am Chem Soc 132: 185–194, 2010. 266. Wood PM. The potential diagram for oxygen at pH 7. Biochem J 253: 287–289, 1988. 279. Zhao Y, Wang ZB, Xu JX. Effect of cytochrome c on the generation and elimination of O2·⫺ and H2O2 in mitochondria. J Biol Chem 278: 2356 –2360, 2003. 267. Woyda-Ploszczyca A, Jarmuszkiewicz W. Ubiquinol (QH2) functions as a negative regulator of purine nucleotide inhibition of Acanthamoeba castellanii mitochondrial uncoupling protein. Biochim Biophys Acta 1807: 42–52, 2011. 280. Zhu J, Egawa T, Yeh SR, Yu L, Yu CA. Simultaneous reduction of iron-sulfur protein and cytochrome bL during ubiquinol oxidation in cytochrome bc1 complex. Proc Natl Acad Sci USA 104: 4864 – 4869, 2007. 268. Xia D, Esser L, Tang WK, Zhou F, Zhou Y, Yu L, Yu CA. Structural analysis of cytochrome bc1 complexes: implications to the mechanism of function. Biochim Biophys Acta 1827: 1278 –1294, 2013. 281. Zhu Z, Gunner MR. Energetics of quinone-dependent electron and proton transfers in Rhodobacter sphaeroides photosynthetic reaction centers. Biochemistry 44: 82–96, 2005. Physiol Rev • VOL 95 • JANUARY 2015 • www.prv.org 243



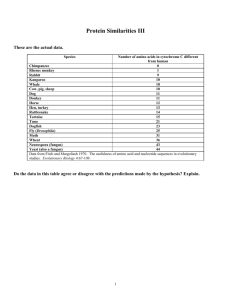


![Anti-Cytochrome C antibody [EP1326-80-5] ab76107 Product datasheet 2 Abreviews 2 Images](http://s2.studylib.net/store/data/012919405_1-aca2b1f1969a664ccaaf17570998f1d3-300x300.png)