Monte Carlo Simulations for Proteins: Binding Affinities for Trypsin Benzamidine -

J. Phys. Chem. B 1997, 101, 9663
-
9669 9663
Monte Carlo Simulations for Proteins: Binding Affinities for Trypsin
-
Benzamidine
Complexes via Free-Energy Perturbations
Jonathan W. Essex,*
,†
Daniel L. Severance,
‡
Julian Tirado-Rives,
§
and William L. Jorgensen*
,
|
Department of Chemistry, Uni
V ersity of Southampton, Highfield, Southampton SO17 1BJ, U.K.,
Roche Bioscience, 3401 Hill
V iew A
V enue, Palo Alto, California 94303, and Department of Chemistry,
Yale Uni
V ersity, New Ha
V en, Connecticut 06520-8107
Recei
V ed: June 16, 1997; In Final Form: August 21, 1997 X
Monte Carlo computer simulations have been performed in conjunction with free-energy perturbation calculations to determine the relative binding constants of four benzamidine inhibitors with trypsin. The protein backbone was constrained in the simulations, but sampling of the side chains was allowed. The calculated free energies are very precise and are shown to yield closed thermodynamic cycles. The calculations correctly predict p-aminobenzamidine to be the strongest inhibitor and give relative free energies of binding for p-methyl- and p-chlorobenzamidine in excellent agreement with experiment. The predicted overly weak binding of the parent benzamidine is most likely due to a deficiency in the partial charges. The relative binding affinities are justified in terms of bulk-solvation arguments whereby the more polar inhibitors are preferentially stabilized in water. The calculations demonstrate that Monte Carlo computer simulations can be used to determine accurate and precise relative binding constants for protein systems.
Introduction
In this paper the application of Monte Carlo computer simulations to the calculation of relative binding constants in protein/inhibitor complexes is described. It is approximately
10 years since free-energy perturbation calculations were attempted on such systems.
1 The initial optimism that this procedure would allow relative binding free energies to be calculated quickly and accurately, thereby providing an important counterpart for experiments, has been diminished. Most practitioners now recognize that considerable care must be taken in deriving parameters and ensuring that the simulations are of sufficient length for the conformational space of the system to be sampled adequately.
2,3 To date, only the molecular dynamics simulation procedure has been used in these free-energy calculations because of concerns regarding the sampling efficiency of the Metropolis Monte Carlo method. The trial move of a backbone dihedral angle in the center of a protein will yield large displacements at remote sites and will consequently have a very low acceptance probability.
4 The Monte Carlo method does, however, offer a number of intrinsic advantages over molecular dynamics simulations in that constraints are easily applied, the statistical mechanical ensemble is rigorously enforced without having to resort to extra coupling algorithms, and thermal artifacts associated with the abrupt truncation of nonbonded interactions are not introduced.
5 The latter may be an important source of noise in free-energy calculations performed using molecular dynamics simulations. Furthermore, recent studies on liquid hexane suggest that the Monte Carlo procedure is more efficient than molecular dynamics methods in equilibrating the conformational populations.
4 Monte Carlo sampling of the internal coordinates of protein side chains is therefore likely to be as efficient as molecular dynamics but with none of the disadvantages outlined above. This, together
* To whom correspondence should be addressed.
†
University of Southampton.
‡ Roche Bioscience.
|
X
Yale University.
Abstract published in Ad
V
ance ACS Abstracts, October 1, 1997.
with the considerable success of Monte Carlo free-energy calculations on host
guest systems, 6,7 has encouraged the authors to develop and apply this methodology to protein/ inhibitor complexes. As a first approximation, the internal coordinates of the protein backbone were not sampled. However, the extension of this methodology to include the sampling of the backbone through the implementation of a crankshaft algorithm is envisaged.
8
The benzamidine series of inhibitors complexed with trypsin was chosen to test this approach for a number of reasons. First, reliable binding constants are available in the literature.
9
Second, this system has been the target of previous free-energy calculations using molecular dynamics simulations, allowing some comparisons to be made with other simulation results.
1,10
Third, the inhibitors are small and have only a few rotatable bonds. Fourth, a number of crystal structures of trypsin are available in the literature that have been prepared under different conditions, with different inhibitors, and indeed crystallize with different space groups and unit cell dimensions.
11 These structures show remarkably little variation in backbone coordinates, suggesting that changing the nature of the smallmolecule inhibitor is likely to have a negligible effect on the protein, thereby justifying the assumption of backbone rigidity applied in the simulations. Finally, it should be emphasized that the calculation of relative binding free energies for protein/ inhibitor complexes requires two series of simulations to be performed. The first involves the simulation of single inhibitor molecules under aqueous conditions, and the second involves the simulation of the hydrated protein/inhibitor complexes.
Methodology
The X-ray crystal structure of trypsin complexed with the parent benzamidine was taken from the Brookhaven protein data bank (structure 3ptb).
12 The structure consists of 223 amino acid residues, the benzamidine inhibitor, a single calcium ion, and 62 water molecules. To simplify the calculation, the size of the simulation system was reduced by including only those residues with atoms within 20 Å of the inhibitor.
S1089-5647(97)01990-1 CCC: $14.00
© 1997 American Chemical Society
This
9664 J. Phys. Chem. B, Vol. 101, No. 46, 1997 Essex et al.
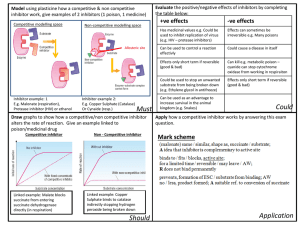
Figure 1. Division of the protein into sampled, constrained, and eliminated regions. Only the protein backbone is displayed. Residues outside the outer sphere were removed from the simulation. Residues inside the inner sphere were sampled in the course of the simulation.
simplification reduced the number of amino acids explicitly simulated to 153. Only those residues with atoms within 14 Å of the inhibitor were sampled in the course of the simulation, unless they also had side-chain atoms further than 17 Å from the inhibitor. All cystine, alanine, and glycine residues were constrained. Thus, side-chain motion in a total of 52 residues was included in the simulation.
This choice of distances represents a compromise between simulating as large a fragment of the protein as possible and the available computational resources. Since a nonbonded cutoff of 12 Å was applied in the simulations, the radius of the sampled region of the protein should ideally exceed this value.
Furthermore, including residues out to 20 Å will mean that the critical amino acids in the binding pocket will experience their full complement of nonbonded interactions. Finally, constraining residues with atoms within 3 Å of the simulation boundary will reduce artifacts arising from the incomplete solvation of charged amino acid side chains.
The four inhibitors, p-aminobenzamidine, pchlorobenzamidine, p-methylbenzamidine, and the parent benzamidine, were placed in the specificity pocket of the enzyme by fitting to the crystallographic coordinates of benzamidine.
The division of the protein system into sampled, constrained, and eliminated regions is illustrated in Figure 1. Since the experimental data were obtained at pH 8.0, all histidines were electrically neutral; experimental titration data on various trypsins and trypsinogens gave pK a
’s for the histidines of less than 8.
13 The appropriate tautomeric states were assigned manually to allow the maximum number of hydrogen bonds to be formed. Assignment of the conformation of the hydroxyl protons of serine, threonine, and tyrosine was performed with the assistance of the 1ntp structure obtained by neutron diffraction.
14 The system was hydrated by a sphere of TIP4P water 15 of radius 20 Å centered on the ipso-carbon of the inhibitor. If any water oxygen atom was within 2.5 Å of a protein/inhibitor heavy-atom, then that water molecule was discarded. Crystallographic water molecules identified in the
3ptb structure were explicitly added to the simulation system with random orientations. A total of 482 water molecules were included in the simulation. To prevent the evaporation of water molecules, a half-harmonic restraining potential of 0.5 kcal
Figure 2.
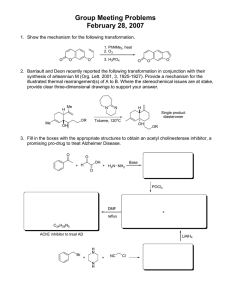
Protein simulation system. The protein is displayed schematically by a ribbon representation using the program Molscript.
33
R
-Helixes are displayed in blue and β -sheets in purple.
mol
-
1 Å
-
2 was applied if the distance from a water oxygen atom to the sphere center exceeded 20 Å. Use of a spherical hydration scheme rather than conventional periodic boundary conditions has been demonstrated to affect calculated free energies of hydration in simple systems.
16 To reduce the influence of this approximation on free energies of binding, an identical approximation should be adopted for the perturbations conducted in the aqueous phase.
3 The aqueous-phase simulation system was therefore constructed by placing a sphere of TIP4P water of radius 20 Å on the ipso-carbon atom of each inhibitor.
Water molecules were discarded from the simulation using identical distance criteria to that described in the protein case, leaving a total of 1120 water molecules to be sampled. An identical half-harmonic boundary restraint was also applied. The protein simulation system is illustrated in Figure 2.
The OPLS united-atom force field was adopted in this study for the protein.
17 However, the basic force field was extended through the explicit inclusion of hydrogen atoms on aromatic rings. These hydrogen atoms are required if aromatic
aromatic and aromatic
charge interactions are to be modeled correctly.
18
The Lennard-Jones parameters for the inhibitors were derived by comparison with related molecules for which OPLS parameters have been reported.
18,19 To derive the electrostatic parameters for the inhibitors, ab initio calculations at the RHF/
6-31G(d) level were performed using the Gaussian 92 program.
20
The CHELPG procedure 21 was used to fit atom-centered point charges to the quantum mechanical electrostatic potentials of each inhibitor optimized with C
2 symmetry. This optimized geometry was also used in the Monte Carlo simulations. This method of electrostatic parameter generation has been demonstrated to give reliable free energies of hydration for a range of organic molecules.
22 The nonbonded parameters assigned to each inhibitor are given in Figure 3. Bond angles and dihedrals of the amino acid side chains were sampled during the Monte
Carlo simulations, and the size of the attempted moves was adjusted to give an acceptance probability of approximately
40%. In the complex, the inhibitors were allowed to undergo rigid-body translation and rotation, and sampling of the important dihedral angles was also performed. Identical sampling was performed in the aqueous phase, except that rigid-body
Trypsin
-
Benzamidine Complexes J. Phys. Chem. B, Vol. 101, No. 46, 1997 9665
Figure 3. Nonbonded parameters of the benzamidine inhibitors (q, σ , ).
translations were not performed. The size of the attempted moves was again adjusted to yield an acceptance probability of approximately 40%. The parameters for the dihedral angles of the inhibitors were derived by fitting to the ab initio barriers calculated at the RHF/6-31G(d) level using a combination of energy minimization and transition-state optimization.
A number of the stationary points were characterized by frequency calculations to confirm that the appropriate dihedral-angle transition states had been found. The force field parameters adopted for each dihedral angle are presented in Table 1, where the functional form is given in eq 1:
V( φ )
)
V
0 +
1
/
2
V
2
[1
cos(2 φ )]
+
1
/
2
V
4
[1
cos(4 φ )] (1)
Relative free energies of binding ( ∆∆ G b
) for the benzamidine inhibitors were calculated using the thermodynamic cycle given in Scheme 1. Free energy is a state function, and consequently, the relationship given in eq 2 holds:
∆ G
1
-
∆ G
2
)
∆ G u
-
∆ G b
(2)
TABLE 1: Dihedral Energy Parameters for the
Benzamidine Inhibitors torsion amidine NH
2 in all inhibitors
p-NH
2 in p-aminobenzamidine amidine in p-aminobenzamidine amidine in p-chlorobenzamidine amidine in benzamidine amidine in p-methylbenzamidine
V
0 kcal/mol
0.000
0.000
2.006
3.272
3.315
2.913
V
2 kcal/mol
V
4 kcal/mol
20.700
10.500
3.500
-
0.493
-
-
0.000
0.000
3.540
3.020
-
0.534
-
3.040
0.376
-
3.100
∆ G
1
-
∆ G
2 corresponds to the difference in the free energy of association between the two inhibitors, and ∆ G b and ∆ G u are the differences in free energy between the two inhibitors while bound to the enzyme and free in solution, respectively.
∆ G b and ∆ G u were calculated using statistical perturbation theory with well-established procedures.
7,23 In the aqueous-phase calculations, simulations were performed at coupling parameter values of 0.0, 0.1, 0.2, ..., 0.9, 1.0, giving a total of 11 separate windows. Double-wide sampling 24 was adopted such that the change in coupling parameter used to generate the perturbed
9666 J. Phys. Chem. B, Vol. 101, No. 46, 1997
SCHEME 1
Essex et al.
state was
(
0.05. In the bound calculations, simulations were performed at coupling parameter values of 0.0, 0.05, 0.1, ...,
0.9, 0.95, 1.0, giving 21 separate windows. A change in coupling parameter of
(
0.05 was used to generate the perturbed states, with the result that two values for the overall free-energy change were obtained from each complete set of simulations.
In practice, the simulations at each window were prepared and equilibrated independently, thereby minimizing the effect of trajectory propagation. The free-energy results for each window are therefore as statistically independent as possible.
The Metropolis Monte Carlo method 25 was used for the simulations, and a temperature of 15
°
C was applied for consistency with the experimental data. The solute
solute nonbonded interactions were truncated at a range of 12 Å using a residue-based cutoff. Thus, if two atoms of different residues were within 12 Å, then all the interactions between the residues were evaluated. An identical cutoff was applied to the solute
solvent and solvent
solvent interactions, but for the latter this was based solely on the oxygen atoms of the water molecules.
For the aqueous-phase calculations, a solute move was attempted every 200 configurations. At each window, the simulation system was equilibrated for a total of 8 million (M) configurations, although only water moves were attempted in the first
2M configurations. Three batches of data collection, each of
6M configurations, were then performed.
For the protein calculations, a single amino acid was moved every 11 configurations and an inhibitor move was attempted every 115 configurations. At each window, the system was equilibrated for 5000 configurations with full sampling followed by 2M configurations of solvent moves and then 4M configurations of full sampling. Three batches of data collection, each of 4M configurations, were then performed. Coordinate sets were saved for analysis every 0.1M Monte Carlo configurations. All calculations were performed using the MCPRO program 26 on
Silicon Graphics computers.
Results and Discussion
The aqueous-phase free-energy results are given in Figure 4.
Each value is the average of three consecutive batches of data collection, and the quoted statistical uncertainty corresponds to the standard error on this average. A more reliable discriminator of the precision of the simulation results is the determination of the extent to which the calculations yield closed thermodynamic cycles.
27 To this end, the free-energy change for the closed cycle p-aminobenzamidine f
p-chlorobenzamidine f benzamidine f
p-aminobenzamidine was computed and found to be 0.23
(
0.25 kcal mol
-
1 , very close to the ideal result of zero. Thus, the precision of the aqueous-phase calculations is excellent, and moreover, the calculated hysteresis is consistent with the reported standard errors, giving support to this method of error estimation. The free-energy results obtained from the protein/inhibitor complexes are also presented in Figure 4. Each value is the average of six results obtained from three consecutive batches of data collection. The hysteresis calculated over the closed thermodynamic cycle was 0.41
(
0.13 kcal mol
-
1 , again a value close to zero.
Figure 4. Aqueous (aq) and bound (prot) free-energy changes for the benzamidine inhibitors (kcal mol
-
1 ). Standard errors are given in parentheses.
In Figure 5, the relative binding constants, ∆∆ G b
, calculated using eq 2, are presented. The hysteresis calculated over the closed thermodynamic cycle was 0.2
(
0.3 kcal mol
-
1 , showing that there was some cancellation of the discrepancies for the aqueous and bound cycles, yielding a smaller overall hysteresis for their difference. These are very propitious results for the further application of the methodology. The experimental values for the relative free energies of binding, 9 together with their estimated errors, are also presented in Figure 5. Although the differences in ∆∆ G b are not large, the calculations correctly find p-aminobenzamidine to be the strongest inhibitor. The
∆∆ G b values for p-methylbenzamidine and p-chlorobenzamidine calculated with respect to p-aminobenzamidine are in excellent agreement with experimental results. The ∆∆ G b values for benzamidine are not in agreement with experimental results.
The high precision of the simulation results suggests that the predicted overly weak binding of the parent benzamidine is not due to sampling problems. A more likely possibility is a deficiency in the partial charges derived from the RHF/6-31G-
(d) calculations. Calculations of free energies of hydration for a range of organic molecules have given an average error of
1.1 kcal mol
-
1 using this charge generation procedure, 22 and the discrepancy in the calculated ∆∆ G b observed here for the parent benzamidine is of this order of magnitude.
It is not possible to compare these results directly with other calculations described in the literature, since different inhibitors have been studied. Wong and McCammon 1 were able to calculate the difference in the free energy of binding between benzamidine and p-fluorobenzamidine using molecular dynamics simulations with an accuracy of approximately 0.5 kcal mol
-
1 , a value comparable to that observed here. Åqvist 10 has investigated benzamidine and p-carbethoxybenzamidine binding
Trypsin
-
Benzamidine Complexes J. Phys. Chem. B, Vol. 101, No. 46, 1997 9667
Figure 5. Calculated and experimental relative free energies of binding for the benzamidine inhibitors (kcal mol
-
1 ). Standard errors are given in parentheses.
using the linear interaction energy approximation. Excellent agreement with experimental results for the absolute binding free energies was obtained, but the relative free energy of binding between these inhibitors differed from experimental values by 0.4 kcal mol
-
1 . Furthermore, since this procedure relies on taking the difference between average energies that are typically large, the calculated ∆∆ G b statistical uncertainty,
(
0.7 kcal mol
-
1 suffers from significant in this case.
To determine the origin of the trend in binding free energies, the number of hydrogen bonds formed by each inhibitor was examined. Hydrogen bonds between the inhibitors and protein were assigned if the hydrogen-bonding distance was less than
2.5 Å. For solute
solvent hydrogen bonding, this distance criterion was applied, together with the requirement that the intermolecular interaction energy should be less than
-
2.25 kcal mol
-
1 . The analysis of the bound simulations was performed over the three batches of data collection, each consisting of 40 coordinate sets, whereas the aqueous phase results were analyzed over three batches each of 60 coordinate sets. In the aqueous phase, p-aminobenzamidine forms 5.3
(
0.1 hydrogen bonds,
p-methylbenzamidine forms 4.0
(
0.0, p-chlorobenzamidine 3.7
(
0.1, and benzamidine 3.7
(
0.1. Thus, on mutating the
p-amino group to any other substituent, between 1.3 and 1.6
hydrogen bonds are lost. In the protein/inhibitor complexes, removing the p-amino group loses a total of 1.6
(
0.2 hydrogen bonds. Thus, the change in the number of hydrogen bonds between the inhibitors was virtually identical in the aqueous and protein systems and hydrogen bonding cannot therefore be invoked to rationalize the trends in binding constant.
A wealth of structural information is available from the simulations. For example, in Figure 6 a single structure of the
p-aminobenzamidine complex, chosen at random, is presented.
Figure 6. Single structure of the trypsin active site taken from the simulation of the p-aminobenzamidine inhibitor. Hydrogen bonds are identified by dotted lines.
TABLE 2: Free-Energy Changes (kcal/mol) and Associated
Dipole Moments mutation
p-NH
2 f X
p-NH
2
p-H
p-CH
3
p-Cl
∆ G u calcd ∆ G b
∆∆ G b exptl a
∆∆ G b dipole moment
D
-
0.35
(
0.19
1.61
(
0.09 2.0
(
0.2 0.4
(
0.1
4.59
6.00
0.83
(
0.17
1.91
(
0.09 1.1
(
0.2 0.7
(
0.2
7.69
-
3.12
(
0.15
-
1.99
(
0.09 1.1
(
0.2 1.1
(
0.6
12.12
a Experimental data taken from ref 9.
As well as forming the salt bridge with Asp 189, hydrogen bonds are identified between the inhibitor and the oxygen of Gly 219, the side chain of Ser 195, and with two water molecules. On average, the side-chain oxygen atom of Ser 195 forms 0.4
(
0.2 hydrogen bonds with the p-amino inhibitor, but no hydrogen bonds with water. This atom cannot form any hydrogen bonds with the other inhibitors but instead recovers these interactions by forming an identical number of hydrogen bonds with water.
In general, the total number of hydrogen bonds involving protein atoms in the active site is independent of the inhibitor, to within statistical error, but the precise pattern of the hydrogen bonding varies.
In Table 2, the free-energy changes for the mutations are presented, together with the calculated dipole moments for each inhibitor. Since the inhibitors are charged, the dipole moments are dependent on the choice of coordinate origin; the data presented here were evaluated with respect to coordinate axes centered on each molecule’s center of mass.
On purely continuum electrostatic grounds, as the dipole moment of a molecule increases, its free energy of solvation should become more negative.
28 The higher the dielectric constant of the surrounding medium, the more pronounced this effect will be.
The dielectric constant of a protein lies somewhere between 2 and 30, 29 whereas that of bulk water is approximately 80.
30 In the complex, the inhibitor is partly exposed to water, so some solvent response to changes in the inhibitor dipole moment is expected, although this will be much less significant than that observed in the aqueous-phase simulations. On this basis, with all other effects being equal, the most polar molecule should bind the weakest, and the least polar the strongest. This bulk solvation effect might be expected to dominate, since it has already been shown that the change in the number of hydrogen bonds between the inhibitors was virtually identical in the aqueous and protein systems. Thus, the free energies of binding
9668 J. Phys. Chem. B, Vol. 101, No. 46, 1997 Essex et al.
Figure 7. Molecular surface of the trypsin binding-pocket. The surface is colored according to electrostatic potential.
of the inhibitors, on the basis of their dipole moments alone, should be p-aminobenzamidine
< benzamidine
<
p-methylbenzamidine
<
p-chlorobenzamidine. This is in exact accord with experimental results. In the experimental binding study, 9 a roughly linear relationship was found between pK i
Hammett σ p and the substituent constant for the para substituent with weaker binding for the more electron-withdrawing groups. This trend was rationalized with reference only to effects on the energetics for the bound structures. The explanation invoked a new dipole
dipole attraction in the complexes with an electron-donating para substituent like amino and a new dipole
dipole repulsion for an electron-withdrawing substituent like chloro. The present data indicate that this rationalization is false; the amino-to-chloro perturbation is favorable both in the aqueous phase and while bound to the protein. Electron-withdrawing groups serve only to make the molecule more polar, increasing the stability of the inhibitor in water over that in the complex.
Thus, the more polar an inhibitor is, the weaker its binding.
In Figure 7 the electrostatic potential of the protein in the vicinity of the binding pocket, as calculated using the linear
Poisson
-
Boltzmann equation, 31 is presented using the program
Grasp.
32 The electrostatic potential is predominantly negative, supporting the fact that positively charged ligands make good inhibitors of this enzyme. In Figure 8, the equivalent electrostatic potential maps are presented for the benzamidine inhibitors; the electrostatic potential surfaces are predominantly positive, showing good complementarity between the inhibitors and the active site. The molecular structures used are single representative configurations taken from the computer simulations. The electrostatic parameters are identical with those used in the simulations, and the molecular surface is determined using the default atomic radii provided with the program. The polarity of each molecule may also be estimated from these figures by observing the asymmetry in the electrostatic potential on the molecular surfaces. p-Aminobenzamidine has a positive electrostatic potential at both ends of the molecule and consequently the smallest dipole moment. The parent benzamidine also shows this effect and does indeed have the next smallest dipole moment. Inspection of the p-methylbenzamidine and p-chlorobenzamidine surfaces suggests that these molecules should be the most polar, as is observed.
Figure 8. Molecular surface of the benzamidine inhibitors. The surface is colored according to electrostatic potential.
Conclusion
Monte Carlo computer simulations have been performed in conjunction with free-energy perturbation calculations to determine the relative binding constants of a number of benzamidine inhibitors with trypsin. The simulation results are very precise; the standard errors calculated from batch averages are small and, more importantly, closed thermodynamic cycles are obtained. This conclusion supports the use of the Monte Carlo sampling procedure in the determination of association constants for protein/inhibitor complexes. The calculations correctly predict p-aminobenzamidine to be the strongest inhibitor and give ∆∆ G b values for p-methylbenzamidine and p-chlorobenzamidine that are in excellent agreement with experimental values.
The predicted overly weak binding of the parent benzamidine is most likely due to a deficiency in the partial charges. Analysis of the simulation trajectories reveals that changes in hydrogen bonding provide no assistance in rationalizing the calculated free energies. Instead, the relative binding affinities arise solely from bulk solvation effects whereby the more polar inhibitors are preferentially stabilized in water. This is in agreement with the conclusions of ref 1. The explanation for the trend in association constants given in the experimental binding study has been shown to be incorrect.
This study has demonstrated that Monte Carlo computer simulations of proteins can be used to calculate accurate and
Trypsin
-
Benzamidine Complexes precise free energies of binding. The wealth of structural information available from the simulations has been analyzed and used to rationalize the calculation results, enabling predictions to be made. These auspicious results bode well for the future application of this methodology to other protein/inhibitor complexes.
Acknowledgment. Gratitude is expressed to the National
Institutes of Health and NATO/EPSRC for support. J.W.E. is a
Royal Society University Research Fellow.
References and Notes
(1) Wong, C. F.; McCammon, J. A. J. Am. Chem. Soc. 1986, 108,
3830.
(2) Van Gunsteren, W. F.; King, P. M.; Mark, A. E. Q. Re
V
. Biophys.
1994, 27, 435.
947.
(3) Van Gunsteren, W. F.; Mark, A. E. Eur. J. Biochem. 1992, 204,
(4) Jorgensen, W. L.; Tirado-Rives, J. J. Phys. Chem. 1996, 100, 14508.
(5) Adams, D. J.; Adams, E. M.; Hills, G. J. Mol. Phys. 1979, 38,
387. Kitchen, D. B.; Hirata, F.; Westbrook, J. D.; Levy, R.; Kofke, D.;
Yarmush, M. J. Comput. Chem. 1990, 11, 1169. Oda, K.; Miyagawa, H.;
Kitamura, K. Mol. Simul. 1996, 16, 167.
(6) Jorgensen, W. L.; Nguyen, T. B. Proc. Natl. Acad. Sci. U.S.A. 1993,
90, 1194. Duffy, E. M.; Jorgensen, W. L. J. Am. Chem. Soc. 1994, 116,
6337.
(7) Jorgensen, W. L. Chemtracts: Org. Chem. 1991, 4, 91 and references therein.
(8) Dodd, L. R.; Boone, T. D.; Theodorou, D. N. Mol. Phys. 1993, 78,
961. Deem, M. W.; Bader, J. S. Mol. Phys. 1996, 87, 1245.
(9) Mares-Guia, M.; Nelson, D. L.; Rogana, E. J. Am. Chem. Soc. 1977,
99, 2331.
(10) Åqvist, J. J. Comput. Chem. 1996, 17, 1587.
(11) Marquart, M.; Walter, J.; Deisenhofer, J.; Bode, W.; Huber, R. Acta
Crystallogr. 1983, B39, 480.
(12) Bode, W.; Schwager, P. J. Mol. Biol. 1975, 98, 693.
(13) Markley, J. L.; Poruscan, M. A. J. Mol. Biol. 1976, 102, 487.
Porubscan, M. A.; Neves, D. E.; Rausch, S. K.; Markley, J. L. Biochemistry
J. Phys. Chem. B, Vol. 101, No. 46, 1997 9669
1978, 17, 4640. Sakiyama, F.; Kawata, Y. J. Biochem. 1983, 94, 1661.
(14) Kossiakoff, A. A. Basic Life Sci. 1984, 27, 281.
(15) Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.;
Klein, M. L. J. Chem. Phys. 1983, 79, 926.
(16) Essex, J. W.; Jorgensen, W. L. J. Comput. Chem. 1995, 16, 951.
(17) Jorgensen, W. L.; Tirado-Rives, J. J. Am. Chem. Soc. 1988, 110,
1657.
(18) Jorgensen, W. L.; Severance, D. L. J. Am. Chem. Soc. 1990, 112,
4768.
(19) Duffy, E. M.; Severance, D. L.; Jorgensen, W. L. Isr. J. Chem.
1993, 33, 323. Jorgensen, W. L.; Laird, E. R.; Nguyen, T. B.; Tirado-Rives,
J. J. Comput. Chem. 1993, 14, 206.
(20) Frisch, M. J.; Trucks, G. W.; Head-Gordon, M.; Gill, P. M. W.;
Wong, M. W.; Foresman, J. B.; Johnson, B. G.; Schlegel, H. B.; Robb, M.
A.; Replogle, E. S.; Gomperts, R.; Andres, J. L.; Raghavachari, K.; Binkley,
J. S.; Gonzalez, C.; Martin, R. L.; Fox, D. J.; Defrees, D. J.; Baker, J.;
Stewart, J. J. P.; Pople, J. A. GAUSSIAN 92, Revision E.2; Gaussian, Inc.:
Pittsburgh, PA, 1992.
(21) Breneman, C. M.; Wiberg, K. B. J. Comput. Chem. 1990, 11, 361.
(22) Carlson, H. A.; Nguyen, T. B.; Orozco, M.; Jorgensen, W. L. J.
Comput. Chem. 1993, 14, 1240.
(23) Zwanzig, R. W. J. Chem. Phys. 1954, 22, 1420.
(24) Jorgensen, W. L.; Ravimohan, C. J. J. Chem. Phys. 1985, 83, 3050.
(25) Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A.
H.; Teller, E. J. Chem. Phys. 1953, 21, 1087.
(26) Jorgensen, W. L. MCPRO, Version 1.2; Yale University: New
Haven, CT, 1994.
(27) Mark, A. E.; van Helden, S. P.; Smith, P. E.; Janssen, L. H. M.; van Gunsteren, W. F. J. Am. Chem. Soc. 1994, 116, 6293.
(28) Onsager, L. J. Am. Chem. Soc. 1936, 58, 1486.
(29) Warshel, A.; Åqvist, J. Annu. Re
V
. Biophys. Biophys. Chem. 1991,
20, 267. Smith, P. E.; Brunne, R. M.; Mark, A. E.; van Gunsteren, W. F.
J. Phys. Chem. 1993, 97, 2009. Simonson, T.; Brooks, C. L., III. J. Am.
Chem. Soc. 1996, 118, 8452.
(30) Weast, R. C. Handbook of Chemistry and Physics; CRC Press:
Boca Raton, FL, 1987.
(31) Nicholls, A.; Honig, B. J. Comput. Chem. 1991, 12, 435.
(32) Nicholls, A.; Sharp, K. A.; Honig, B. Proteins 1991, 11, 281.
(33) Kraulis, P. J. J. Appl. Crystallogr. 1991, 24, 946.