Evaluating silica gel supported titanium dioxide for use as a... by Michael Owen Hardiman
advertisement

Evaluating silica gel supported titanium dioxide for use as a photocatalyst
by Michael Owen Hardiman
A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in
Chemical Engineering
Montana State University
© Copyright by Michael Owen Hardiman (2001)
Abstract:
With increasing attention given to the clean up of polluted waters, new technologies need to be
developed to offer safer, cheaper methods than those currently employed. Most current systems employ
oxidizing agents which are costly, consumed in the process and often hazardous to work with. The use
of titanium dioxide as a photocatalyst offers a safe alternative to current technologies and has been
proven to degrade a wide number of contaminants. The majority of recent work in photocatalysis uses
titanium dioxide slurry type reactors and separation is required to reclaim the catalyst. This will limit
the applicability of this method due to the added separation expense. Supporting titanium dioxide for
use in a packed bed configuration represents a viable alternative. Experiments were developed for the
evaluation of supported titanium dioxide as a photocatalyst. Silica gel was chosen as a support material
due to its unique properties regarding UV transmission, surface area and adsorptive capability for water
and organics. The support materials were optimized for light transmission properties with respect to
manufacturer, grade and size fraction. Coating methods were developed, and four continuous flow,
annular reactors were designed to evaluate the supported photocatalysts. Through light measurements
and kinetic experiments, four supported catalysts were evaluated in the four reactors for their ability to
degrade formic acid. The degradation rates observed with the supported catalysts were found to have a
strong dependence on catalyst loading and support material, but nonetheless outperformed slurry
catalysts under identical conditions. EVALUATING SILICA GEL SUPPORTED TITANIUM
DIOXIDE FOR USE AS A PHOTOCATALYST
by
.
.
Michael Owen Hardiman
A thesis submitted in partial fulfillment
o f the requirements for the degree
of
. Master o f Science
■
in
Chemical Engineering
'
MONTANA STATE UNIVERITY
Bozeman, Montana
December 2001
APPROVAL
o f a thesis submitted by
Michael Owen Hardiman
This thesis has been read by each member o f the thesis committee and has been
found to be satisfactory regarding content, English usage, format, citations, bibliographic
style, and consistency, and is ready for submission to the College o f Graduate Studies.
S ) / ' JO'O J
(Date)
Approved for the Department o f Chemical Engineering
0/ - /O " 0O~
Dr. John Sears
^(Signature)
(Date)
Approved for the College o f Graduate Studies
Dr. Bruce Mcj
(Signature)
(Date)
iii
STATEMENT OF PERMISSION TO USE
In presenting this thesis in partial fulfillment o f the requirements for a master’s,
degree at Montana State University, I agree that the Library shall make it available to
borrowers under the rules o f the library.
If I have indicated my intentions to copyright this thesis by including a copyright
notice page, copying is allowable only for scholarly purposes, consistent with “fair use”
as prescribed in the U.S. Copyright Law. Requests for permission for extended quotation
from or reproduction o f this thesis in whole or in part may be granted only by the
copyright holder.
Signature
iv
TABLE OF CONTENTS
1. INTRODUCTION AND BACKGROUND........................ ......................................I
Photocatalysis and Photocatalysts............................................. ............................3
Titanium Dioxide..................... ............................... ................. .......... ....................5
Titania Surface.............................................................................. ............. .............. 5
Mechanisms for Heterogeneous Photocatalysis using TiOz..........;.................. 8
Catalyst Supports.................... ............................................ ........:.......................... 11
Silica Gel.................................... . . . . : .......:...................................... ......... ........ .
15
Photoreactofs........................................... .............................. ........ ................... . 16
Radiant Flux............................. ...........!................. :—
.................................. 18
Statement o f Objectives........................................;............................. ..................20
2. EXPERIMENTAL METHODS AND MATERIALS....... .................................... 22
Catalyst Supports..................................'................................................................... 22
Light Penetration Tests................................................................: ................... .
23
P25 TiO2 Catalysts.......................................: .......................................................... 25
TiO2 Assay................................ ........................... .............. — :................ ......... . 26
Annular Reactors.......................... ......................................................................... 26
Reaction Runs........................................................................................................... 29
Slurry Runs.......... ...........................................................
30
Sample A nalysis........
31
3. RESULTS AND DISCUSSION................................................................. .............32
Choosing the Support M aterial.............
..32
Light Penetration Through Annular Spaces....... . ........................................ — . 35
Coatings............. .......................................— .........................................................r 36Formic Acid R uns.................... ................. ................................... : . . . . .......'............ 37
Packed Bed Annular Reactor Runs...................
41
Slurry R uns.....,....................:.................................................................................. 46
Data Analysis.................................
49
Volume Correction...............
49
Normalization to Rate o f Energy Absorbed................... — ... I.............:............52
SEM ...............................'............................... ............................................... ........ 58
C onclusion....................
60
Future W ork....................................... :..................................................................... 62
V
TABLE OF CONTENTS - CONTINUED
REFERENCES CITED......................................... !.................................................... '.... 63
A PPEN D IC ES........................................................
65
APPENDIX A .............................................................;................................................... 66
Formic Acid Runs Data..........................................................
67
APPENDIX B ....................................................................................
74
Spectrophotometric Method for the Determination
o f TiOz Loading........................................:............................................ ;...................75
LIST OF TABLES
Table
Page
1. Support Materials Used According to Size Fraction and M anufacturer............... 22
2. BET Data o f the Silica Gels Used.............................................................................. 23
3.
Annular Reactor Dimensions......................................................................................27
4. Five Consecutive Runs in 2.0” Reactor with
0.4 W eight Percent P25 on Grade 58............... ...................................................... 40
5. Summary o f Supported and Suspended Observed Rates........................................ 48
6. Volume Corrected Rate Constants for the Various Catalysts................................51
7. System Specific Extinction Coefficients.
......................... ■................................. 54
8. Summary o f Intensity Normalized Rate
Constants for the Various Catalysts................ .........................................................55
9. Comparison o f Spectrophotometric and ICP-AS M ethods..........
...76
Vll
LIST OF FIGURES
Figure
Page
1. Charge Separation in a Semiconductor
Particle Upon Excitation with Light........................................................................ 4
2. Surface Hydroxyl Groups on Titania......................................................................... 6
3. Photodegradation Rate Dependence on Radiant Flux..............................................19
4. Acrylic Box Used in Support Material Light Penetration Tests.......... ............. .
24
5. Light Intensity Measurements in Packed Annular Reactors...................................25
6. Annular Photoreactor.......................................................................
28
7. Schematic o f Experimental Photoreactor Setup.................................................. ;. .29
8. Light Transmission Through Grade 58 Silica Gel and
Borosilicate Glass Beads................................................. !'...................— ............... 32.
J
9. Light Penetration Through Different Brands o f 12-18 Mesh Silica..................... 33
10. Light Penetration Through Various Size Fraction o f Grade 58 Silica Gel...........34
11. Comparison o f Light Penetration Through Coated and Uncoated Silica.............. 35
12. Light Penetration Through Annular Reactors Packed with
Coated and Uncoated Silica......................................................................
36
13. 2.00" Reactor Packed with 0.4 Weight Percent P25 on Grade 58 Silica.............. 39
14. 2.0” Reactor Packed with 0.4 Weight Percent P25 on Grade 58
Silica at Varying Flow Rates.................................................................................... 40
15. Observed Rate Versus Mass TiO2 in Annular Reactors Packed with
0.4 W eight Percent P25 on Grade 58 Silica............................................. :............41
viii
LIST OF FIGURES - CONTINUED
Figure
Page
16. Observed Rate Versus Mass TiOz in Annular Reactors Packed with
0.04 W eight Percent P25 on Grade 58 Silica.......................................... i............ 42
17. Comparison o f Grade 58 and Grade 55 Silica Gels................................................. 43
18. Light Penetration Tests with Uncoated 12-18 Mesh Silica G el.................... ........ 44
i
'
19. Comparison o f Observed Rates Versus Mass o f Titania for the
Various Catalysts.......................................
45
20. Comparison o f Observed Rates with Supported and Suspended
P25 T itania.............................
47
21. Experimental Photoreactor System.......................
50
22. Light Measurements for Intensity Balance on Photoreactors................................ .53
23. Linearization o f Equation 32 with Experimental Data to Give b Term s................54
24. Radial Dependence ofk ic-............................................................................................ 58
25. SEM o f Silica Gel Particles.. , ..................................................................................... 59
ABSTRACT
W ith increasing attention given to the clean up o f polluted waters, new technologies need
to be developed to offer safer, cheaper methods than those currently employed. Most
current systems employ oxidizing agents which are costly, consumed in the process and
often hazardous to work with. The use o f titanium dioxide as a photocatalyst offers a safe
alternative to current technologies and has been proven to degrade a wide number o f
contaminants. The majority o f recent work in photocatalysis uses titanium dioxide slurry
type reactors and separation is required to reclaim the catalyst. This will limit the
applicability o f this method due to the added separation expense. Supporting titanium
dioxide for use in a packed bed configuration represents a viable alternative. Experiments
were developed for the evaluation o f supported titanium dioxide as a photocatalyst. Silica
gel was chosen as a support material due to its unique properties regarding UV
transmission, surface area and adsorptive capability for water and organics. The support
materials were optimized for light transmission properties with respect to manufacturer,
grade and size fraction. Coating methods were developed, and four continuous flow,
annular reactors were designed to evaluate the supported photocatalysts. Through light
measurements and kinetic experiments, four supported catalysts were evaluated in the
four reactors for their ability to degrade formic acid. The degradation rates observed with
the supported catalysts were found to have a strong dependence on catalyst loading and
support material, but nonetheless outperformed slurry catalysts under identical
conditions.
I
CHAPTER I
INTRODUCTION AND BACKGROUND
This past century has seen a rapid increase in pollutants in our environment.
Toxic pollutants in the soil, air and water are widespread. Continual human contact with
toxins and the steady degradation o f the environment is a growing concern on national
and international agendas. The sources o f these contaminants are extensive, ranging from
(but not limited to) the chemical/petroleum industries, textiles, pulp and paper milling to
increases in modem agriculture practices.
A considerable amount o f research is currently being directed towards improving
methods o f contaminant treatment. Current, processes in use such as air stripping or
activated carbon adsorption offer only a phase change o f the contaminant, likely
requiring further treatment. Other methods, such as treatment with a strong oxidant (e.g.
ozone or chlorine), generally offer less than complete removal o f organic contaminants
while consuming large quantities oxidants. Relatively new to the arena are methods
collectively termed advanced oxidation processes (AOP’s). These methods may be
cheaper and offer better degradation than some o f the above-mentioned methods.
Examples o f A O P’s include direct UV photolysis o f the contaminant, UV/ozone or
'
UVZH2O2, and heterogeneous photocatalysis. The latter three o f the these processes are o f
particular interest in that they have shown potential to kill bacteria as well as degrade
organic compounds (I). Direct photolysis o f an organic contaminant requires that it
absorb the incident light and suffer degradation as a result. This is difficult to achieve
2
with m ost contaminants, especially at the low concentrations generally encountered in
waste treatment. UV/ozone and UV/H 2 O 2 have shown promise as effective treatment
methods. W hen UV is used in conjunction with ozone or hydrogen peroxide, highly
reactive hydroxyl ( OH) radicals are generated, which will non-selectively attack most
organic compounds. While complete degradation is possible with these methods, large
amounts o f oxidant are consumed in the process. On the contrary, in heterogeneous
photocatalysis reactive radicals (thought to be primarily 'OH radicals) are generated in
situ from air, oxygen or water (2) without the need for costlier and more hazardous
oxidizers. In heterogeneous photocatalysis, a solid photocatalyst (usually an n-type
semiconductor such as Ti02, ZnO, or CdS) is illuminated with UV-visible light creating a
reduction-oxidation (redox) environment (to be described in more detail later) that has
been shown to be capable (3) o f degrading a wide variety o f organic contaminants. Other
features that make heterogeneous photocatalysis attractive is that it generally uses low
cost, non-toxic catalysts, can often be operated at ambient conditions and is capable o f
absorbing a wider portion o f the UV spectrum efficiently as compared to other UV
methods (2). In addition to air, water and wastewater treatment, heterogeneous
photocatalysis has been used for a wide range o f reactions including: m ild organic
oxidations, dehydrogenation, hydrogen transfer,
02
18- 0 216 and deuterium-alkane isotopic
exchange, and metal deposition (4). Heterogeneous photocatalysis has been confirmed by
numerous researchers to be effective for the total oxidation o f organic water contaminants
(5).
3
Photocatalysis and Photocatalvsts
Though there is currently some dispute as to a proper definition o f photocatalysis,
a general definition is the “acceleration o f a photoreaction by the presence o f a catalyst”
(3). For the purpose o f water treatment, heterogeneous photocatalysis can be loosely
defined as the illumination o f a particulate with UV-visible light o f suitable energy to
initiate redox chemistries (I) while the catalyst itself undergoes no overall change.
Semiconductors have largely been employed for use as photocatalysts because o f their
electronic structure, light absorption properties, charge transport characteristics, and
excited state lifetimes (3). A detailed description o f semiconductor materials is beyond
the scope o f this thesis and only some o f the properties applicable to photocatalysis will
be addressed. For a more complete treatment see Reference 6.
A semiconductor is characterized by a filled valence band and an empty
conduction band. For a semiconductor material to be used as a photocatalyst, an excited
state must be induced in the material by the absorption o f light. Upon excitation by the
absorption o f a photon o f sufficient energy, charge separation takes place as an electron
(e") is promoted from the valence band to the conduction band. The absence o f an
electron in the valance band (or a positive charge) is known as a hole (h+). The act o f
excitation by light is determined by the band gap, or the energy gap, between the valance
and the conduction band energy levels. For promotion o f an electron to occur, the photon
energy m ust exceed the energy o f the band gap.
4
Figure I . Charge separation in a semiconductor particle upon excitation with light
(hv > Ebg). Ox and Red are oxidant (electron acceptor) and reductant (electron
donor) respectively.
The existence o f the band gap prevents rapid recombination (or deactivation o f
the excited state) o f the electron-hole pairs (3). In the excited state, the electron and hole
pairs will either recombine or diffuse to the surface o f the semiconductor where they can
participate in charge transfer reactions, though certain conditions must exist at the surface
for this to occur. From Gratzel (3), “For semiconductor particles dispersed in a gaseous,
liquid, or solid medium, the charge transfer can take place only in the presence o f an
electroactive species acting as electron donor or acceptor. Thus an interfacial redox
reaction is required to produce the electric field within the particle”. Species on the
surface o f semiconductors act as traps for the electrons and holes and prevent their
recombination, allowing charge transfer to take place. For heterogeneous photocatalysis
in the treatment o f water, the electron donors and acceptors are mainly in the form o f
adsorbed oxygen and water molecules or surface bound hydroxyls (this to be treated in
greater detail later).
5
Titanium Dioxide
In the area o f water and wastewater treatment, the principal focus has been on the use
o f titanium dioxide or titania (TiOz) as a photocatalyst. Mills and Le Hunte (7) proposed
the following list o f criteria in choosing a semiconductor for use as a photocatalyst:
1. Photoactive.
2. Able to utilize visible and/or near UV light.
3. Biologically and chemically inert.
4. Photostable (i.e. not liable to photoanodic corrosion for example).
5. Inexpensive.
Titania meets these criteria well and has been shown to exhibit a high photoactivity
towards a wide range o f compounds (7, 8). TiOz requires light less than 390 nm to be
photoactive and has been shown to be Capable o f using sunlight to degrade organic
pollutants (3). Approximately 4-6 % of the available sunlight (the amount o f sunlight in
the 300-390 nm wavelength region) has been demonstrated to initiate the photoactivity o f
titania (6).
Titania surface
In an aqueous environment, the surface o f titania will be covered with hydroxyl
groups. The extent o f this coverage will depend on crystal phase and preparation
conditions. The point o f zero charge (PZC) o f TiOz used in photocatalysis is in the
pH range o f 6 - 7 (3), with the surface positively charged in acidic environments. Titania
is known to exist in three different crystal phases: anatase, rutile, and brookite.
6
(a)
O Lattice oxygen
Q Oxygen
(b)
(c)
* Proton
@ Titanium ion
Figure 2. Surface hydroxyl groups on titania (17). (a) hydroxyl free surface; (b) physical
adsorption o f water; (c) dissociation o f water, giving two distinct OH" groups.
The annealing conditions will determine the crystal structure. Amorphous TiOa has been
shown to exhibit low photoactivity (9), possibly due to decreased charge carrier mobility
more easily attainable in definite crystal structures (10). Brookite is o f no commercial
significance and not used in photocatalysis. O f the remaining two forms, anatase has
demonstrated the most photoactivity towards the degradation o f organic compounds (3).
Fox et al. (I I) proposed that the lower photoactivity o f rutile TiOa was due to its higher
electron-hole recombination rate and its lowered capacity to adsorb oxygen, though
Sclafani (11) found the activity o f rutile to be influenced by its preparation conditions.
Despite recent findings indicating that rutile TiO2 may exhibit greater photoactivity
towards certain compounds, particularly for the oxidation o f CN" (8), anatase is still the
more practical form for widespread environmental applications. Anatase is
thermodynamically less stable than rutile, though it is kinetically favored at temperatures
< 600 0C (3). Optimizing the performance o f titania with regards to annealing
temperature has been the subject o f several recent papers (8). Aside from the crystal
7
structure, the annealing conditions can control the morphology, specific surface area and
surface, density o f OH groups (7). Hoffmann et al. (8) found the rates o f 4-chlorophenol
degradation to increase as the anatase form was calcined progressively from 100 to 400
0C and to decrease with TiOz calcined at 500 0C and above, Tanaka et al. (6) reported the
degradation o f several compounds to be dependent upon annealing conditions while
independent for others. They also reported that TCE degradation rates increased for
annealing temperatures up to 500 0C, and in some cases up to 700 0C, but decreased
above those temperatures. Tsai et al. (11) found the photoactivity o f laboratory produced
rutile TiOz to decrease with calcination temperature and correlated it to the amount o f OH
groups on the surface. The general consensus seems to indicate that the photoactivity o f
titania decreases as the annealing temperature approaches the temperature o f the rutile
phase change (~600 0C). Moreover the higher annealing temperatures relieve the TiOz
surface o f OH groups essential to photdactivity. The increased photoactivity o f anatase
therefore is thought to arise from a larger surface area and a higher surface density o f
i.
■
1
1.
•
active sites for adsorption and photocatalysis.
The majority o f titania used in photocatalysis comes in three forms: a pre-made
TiOz powder, TiOz particles precipitated from a sol-gel, and TiOz produced by chemical
vapor deposition (CVD) o f titanium chlorides (TiCl3 and TiCU) on some support. The
pre-made powders come in a controlled crystal structure and particle size (generally
anatase on the nm tq pm scale). Starting from a titanium salt, sol-gel techniques can
.
produce titania from a sequential process o f hydrolysis, polycondensation and drying o f a
colloidal suspension (12). Sol-gels can generally produce small (~10nm) amorphous and
8
crystalline TiOa particles, where crystal structure and particle size is a function o f
preparation conditions (pre-cursor sol, calcining conditions, pH, etc.). Titania from CVD
is obtained by exposing a surface to titanium chlorides and water vapor at high
temperatures (~200 -500 0C), where the water vapor will substitute for the chlorides.
Ultrafine thin films o f titania on silicon and quartz substrates have been reported using
CVD techniques (13). Obviously, there are numerous variations on sol-gel and CVD
techniques that can affect particle growth and crystal structure. The chemistry involved is
beyond the scope o f this paper. A good treatment o f colloidal semiconductors can be
found in Reference 3.
Mechanism for heterogeneous photocatalvsis using TiO?
Certain features o f photoinduced reactions involving semiconductor catalysts
have previously been mentioned. Some will be repeated here for clarity in the specific
case o f heterogeneous photocatalysis involving TiOz and the degradation o f aqueous
organic contaminants. The overall process o f the photodegradation o f organics with TiOz
can be summarized as follows
Organic Pollutant + O 2 —
C O2 + H 2O + mineral acids (I)
Though the exact mechanism for the heterogeneous photodegradation o f aqueous
organics is not completely understood, it is known that the primary process involves
electron-hole pair formation followed b y interfacial electron transfer to form surface
bound oxidants (7). Based on laser flash photolysis measurements, Hoffinan et al. (8)
proposed the following mechanisms, with their characteristic times, for surface reactions
on illuminated TiOz-
9
' Chargecarriergeneration
TiO2
>e^b +h^b
(6 )
(2)
Chargecarrier happing
h ;b + > T iivOH -> {> TiivOH-J+
(IOns) (3)
e;b + > TiivOH *-> {> TimOHJ-
(IOOps) (4)
Chargecarrier recombinaion
e" + h + -> heat
(ns)
(5)
Interfacii charge transfer
^ T i ivOH-J+ + R e d —> > TirvO H + R ed-+ (IOOns) (6)
{> TimOHJ- + Ox - » TiivO H +O x-"
(ms)
(7)
The overall success o f a photocatalytic reaction is dependent on the competition between
charge carrier trapping and recombination (Equations 3-4 & 5). The above mechanism
assumes all oxidation o f organics occurs via surface bound hydroxyl J^TiivO H J+ attack,
though oxidation may occur with between free radicals and liquid phase organics as well
(14). Also not mentioned in this scheme is the formation o f hydrogen peroxide from
dioxygen reduction with conduction band electrons and possibly from valance band hole
oxidation o f water (15). These two pathways can be represented as follows (14,15):
T iiv - O 2- + e"b + 2H +
(Tiiv)H 2O 2
2H 20 + 2h+b —» H 2O 2 + 2H +
( 8)
( 9)
It has been noted that H2Oa may contribute to the degradation o f pollutants by
acting as a direct electron acceptor or as a further source o f hydroxyl radicals due to
homolytic splitting (15).
Though it is assumed that hydroxyl radicals are the primary oxidant in TiO2 .
systems, it is not known if oxidation principally occurs via surface bound or free radicals.
10
Turchi and Ollis (14), in an effort to determine the role o f hydroxyl radicals in TiOa
photocatalysis, proposed four possible mechanisms for hydroxyl radical attack.
1. Reaction occurring while both organic and hydroxyl are adsorbed.
2. A non-bound radical reacts with a bound organic.
3. An adsorbed radical reacts with a free organic.
4. Reaction occurs between free radical and organic.
Four kinetic models were derived for the above, relating degradation o f a single
component by way o f hydroxyl attack. All resembled Langmuir-Hinshelwood (L-H) rate
forms.
r _ k QbsKCi
‘ I + KCi
( 10)
It was found that kobs was the same for each o f the four cases in the degradation o f 14
organic compounds, suggesting that it is a function only o f catalyst properties and
reaction conditions. Further differentiating between reactions o f adsorbed and free radical
to suggest a dominant form was not possible. Estimates o f diffusion/reaction rates with
free OH radicals and liquid phase organics suggest that even at low organic
concentration, free radicals would not diffuse very far into solution before reacting (~10"8
- IO"6 m). Based on these estimates, they found it plausible that OH is present as a
mobile radical. It is likely that some combination Of these mechanisms is present during
heterogeneous photocatalysis.
In m ost photocatalytic systems, oxygen acts as an electron acceptor. The current
view is than electrons are consumed by dioxygen to produce the superoxide radical anion,
11
O2 ' (I), with subsequent electron transfer reactions leading to the formation o f H 2 O 2 .
Supporting this view is work by Serpone et al. that noted no degradation occurs in the
absence o f oxygen (3). Control batch experiments by our group in which aqueous phenol
suspensions o f TiC^ were sparged with nitrogen also found no degradation to take place.
Serpone has observed superoxide radicals on the surface o f illuminated TiC>2 in the
presence o f oxygen (I). Hoffinann et al. (15), using 18O isotopic labeling with a ZnO
photocatalyst noted that all o f the phofochemically produced H 2 O 2 is from dioxygen.
They also reported that hydrogen peroxide was not detected in the absence o f oxygen.
Catalyst Supports
M uch o f the work in liquid phase photocatalysis using Ti02 has been conducted
in slurry type reactors using very fine titania particles (e.g. Degussa P25 Ti02: -3 Onm in
diameter). These types o f reactors have several advantages. In perfectly mixed reactors,
there will be no segregation o f phases and with fine particles (nm scale), the entire
external surface can be illuminated during the reaction (12). Also these reactors have a
large ratio o f surface area o f TiC^ per unit volume o f the reactor, limiting mass transfer
effects (16). In modeling these systems, they can be (at least to a first approximation)
treated as “pseudo-homogenous” and amenable to simple laws such as the Beer-Lambert
law (12). Overall though, the utility o f these reactors is limited, as recovery o f the catalyst
downstream o f the reactor is difficult.
Fixing the catalyst on a support o f some sort is the alternative. Certain tradeoffs
come with immobilizing the catalyst though. Degradatiori rates may be mass transfer
12
limited and the catalyst-photon interactions will be more limited. These limitations will
be dependent on specific reactor/support configurations.
Pozzo et al. (13), in a recent review o f research on supported TiOa for use in
environmental photocatalysis, suggested the following attributes o f a good photocatalyst
support:
1.
Transparent to UV radiation,
2.
Favor strong surface chemical-physical bonding with the TiOa particles
without negatively affecting their reactivity.
3.
Offer a high specific surface area.
4.
Have good adsorption capability for the organic compounds to be
degraded.
5.
Be in a physical configuration which favors the ultimate liquid-solid phase
separation.
6.
Allow reactor designs that facilitate the mass transfer processes.
7.
Be chemically inert.
Early work on immobilized photocatalysts involved coating the inner glass walls
in an annular type reactor (17) or the inner wall o f a glass coil wrapped around a circular
light source (18,19). Degradation rates for these flow-by type reactors were found to
suffer from boundary layer mass transfer limitations, as only a portion o f substrate will be
in contact with the catalyst at any given time (20).
In the last decade, much attention has been directed towards immobilizing TiOa
on various substrates including; particles (silica, alumina, zeolites, activated carbon, glass
13
beads etc.), quartz optical fibers, glass fiber or wool (12, 13). Various forms o f reactor
design have been employed, including packed, fluidized and floating bed reactors. Based
on the current interest here, the following discussion will be restricted to particle supports
applicable for packed bed applications.
Recent research has shown that supports can alter photoactivity in several ways.
For example, Xu and Langford (21) comparing two different zeolites, silica gel, and
alumina, related increased adsorption o f organics on the support to increased
photoactivity. Concerning zeolites, they proposed increased photoactivities could be
related to the rigid framework o f the surface, where reactive radicals, such as OH and
O 2"', could be stabilized on the surface where they can be efficiently reached by organic
molecules. Xu et al. (9), in a comparative study o f preparation methods for coating silica
gel with sol based TiOz, also related, increased photoactivity to increased adsorption o f
organics. Rates reported for supported catalysts were considerably higher than those
obtained with bare catalysts. In these experiments degradation rates were found to be
higher with smaller silica gel particles. They reported that smaller silica particles resulted
in the formation o f smaller TiOz particles on the surface. It was suggested that smaller
silica particles cause the TiOz to be more dispersed on the surface (possibly affecting
crystallinity) and more efficient as a photocatalyst.
Surface characterization o f supported catalysts is important in that the crystal
structure o f titania on the support can be determined and relate^ to its photocatalytic
behavior. Also, determining how TiOz is adhering to the support will allow predictions o f
the durability o f a specific catalyst under reaction conditions. Xu et al. (9) found that
14
supporting TiOa on silica restricts crystal formation even after calcining. They reported a
film o f amorphous TiOa to exist on the surface, limiting photoactivity. Xu and Langford
(21) found that at low coverage, TiOa on zeolite is amorphous, only forming well-defined
anatase crystals at higher coverage. Structure determination o f supported titania is made
difficult at lower loadings commonly encountered in environmental photocatalysis
(generally less than I wt. %). Differentiating between X-rays o f the catalyst/support
surface in X-ray diffraction (XRD) analysis is difficult at such low loadings, making the
results suspect. Similarly, scanning electron microscopy (SEM) o f supported surfaces
reveals little about the true nature o f the catalyst-support interactions . Islands o f what
appears to be titania have been reported to appear on supported Surfaces (12), suggesting
a random, highly dispersed coating. Xu and Langford (21) suggest that these particles
m ay form on a thin coat o f titania after monolayer capacity has been exceeded. Verifying
this would require slicing a coated particle and viewing it from the side. This would allow
determination o f the thickness o f a titania layer if it formed as a uniform coat. The small
size o f m ost support materials (pm - mm scale) makes this difficult and slicing would
likely damage the coating.
TiCb adherence to the support is an essential aspect in supported photocatalysts,
yet many papers on the subject neglect it entirely (9 ,1 6 ,1 8 , 21). If supports readily lose
their titania (or a substantial portion o f it) to the fluid phase, the reason for its use is
compromised. Adherence is generally evaluated at reaction conditions. Analysis o f the
catalyst after subjecting it to flows high enough to eliminate mass transfer concerns will
allow quantification o f adherence. Bideau et al. (12) in a comparison o f supports, titania,
15
and coating methods, found pre-made powder (P25 TiO2) coatings to adhere better to
silica gel than coatings using a sol-gel process (loss o f 52% for sol compared to 21% for
P25 powder). Zhang et al. (22), using powder (Aldrich TiO2) coated silica gels, reported
no noticeable attrition OfTiO2 after I hour under flow and no decrease in photoactivity
after a 20-day run. Though coating methods used by the Bideau arid Zhang groups were
similar for powder coatings, widely varying results were obtained. These results can be
somewhat misleading in that reactor configurations differed. The setup used by Zhang et
al. was a packed tubular reactor and that used by Bideau et al. was a CSTR with the
catalyst placed on the bottom. It would be natural to assume that supported catalyst in a
packed bed would be subjected to greater stress and yet they reported no loss o f catalyst.
Such results could be explained by the fact that different silica gels, titania powders, and
calcining conditions will produce different coatings, one performing well and one not.
This makes direct comparisons between different researchers difficult.
SilicaG el
In this study, silica gel (SiO2) was chosen as a photocatalyst support for its ability
to meet several o f the above mentioned criteria. It transmits UV light well, has high
specific surface area (~1 OO-1OOO m2/g) and is chemically inert. The surface o f silica gel is
covered by hydroxyl and siloxane groups. Hydroxyl groups can be arranged on the
surface in various ways with one or two hydroxyls bound to a single Si atom (23).
Siloxane groups are formed by a partially reversible dehydroxylation between hydroxyl
groups at about 500 K (3). This can be represented as follows
2 = Si - OH o = S i- O - S i = + H 2 O
(11)
16
In an aqueous environment, silica gel is capable o f taking up water and organics by
means o f physical adsorption (23), with the amount dependent on temperature, surface
density o f hydroxyl groups, pH, and specific surface area. The point o f zero charge (PZC)
for m ost silica gels is at a pH o f about 2 (3). Above this pH, the surface will be negatively
charged, limiting its adsorptive capabilities for negatively charged ions. Aside from the
above support criteria, silica gels also exhibit high thermal stability and high mechanical
strength (24).
Coating a support with sols or pre-made powders generally involves mechanical
mixing o f a liquid suspension o f titania and the support material followed by calcining
and washing. For the case o f silica gel, titania bonding likely occurs at surface hydroxyls
with the attraction being Van der Waals forces (24). The degree o f dispersion o f titania
on silica will therefore be dependent on the surface density o f hydroxyls on silica (and
also titania particle size) (24). This surface density o f hydroxyls will change with
calcining temperature. W ith other coating methods such as CVD there is evidence o f TiO-Si linkages at the surface, which will alter the electronic properties o f the catalyst (24).
Photoreactors
Photodegradation rates will be dependent on reactor design, as the geometry will
determine light distribution. Numerous reactor designs utilizing sunlight and artificial
light have been employed in photocatalytic work and are described elsewhere (4, 8,17,
22,25, 26). By far, the configuration most used in bench scale operations is the lamp in
tube annular reactor. This reactor represents a simple and very efficient configuration
utilizing artificial light (27) and is probably the best type for commercial applications.
17
Reliable design equations for annular photoreactors using a solid catalyst have yet to be
developed. These equations are complicated by the coupled mass and energy (photon)
balances that would have to be performed and the uncertainty o f certain parameters
(namely light absorption and scattering coefficients) (28). Theoretically, mass/energy
balances would have to be performed for each wavelength when using polychromatic
light (27). Pseudo-homogenous approximations for absorption and scattering coefficients
have been determined for some slurry reactors but are lacking for supported catalysts. In
theory, these parameters would be different for each catalyst/support combination and
have to be determined on case-by-case basis. Due to the nature o f these systems,
absorption and scattering coefficients for larger, supported catalysts are generally
unattainable and built into the few models that exist as assumptions (27, 28). This
subjects the coefficients to a high degree o f uncertainty.
Since lamps come in finite sizes, the majority o f bench reactors are scaled to
utilize a light o f certain dimensions, often with no other criteria. Obviously, reactor
materials should be chosen that transmit light o f the desired wavelengths. Parameters that
can be easily ascertained are the light intensity as a function o f distance and light
penetration through packed beds o f certain catalysts. Using this knowledge, the outer
diameter o f the reactor should be fixed such that a percentage o f the entering photons can
reach the outer m ost catalyst. Despite such measures, a photon gradient will exist with
radial distance from the lamp in an annular reactor. If it is substantial, a resultant
concentration gradient may develop where faster degradation occurs closer to the light.
18
Lamps should be chosen that emit light below the threshold wavelength (i.e. o f enough
energy to overcome the bandgap energy o f the catalyst) and ideally operate in the regime
where first order dependence on intensity holds (see below). It should also be verified
that the liquid phase reactants do not absorb light in order to conserve light for the
catalyst.
Radiant Flux
The rate o f a photocatalyzed reaction will be proportional to the flow o f photons
reaching the catalyst. Using titania for aqueous degradation o f organics, many researchers
have found the relationship to resemble Equation 13.
Rate oc (Intensity)11
(12)
The power n has been found to vary from a first order dependence at low intensities to a
square root dependence at high intensities (8). Hoffinann et al. (15) explained this on a
kinetic basis using ZnO (an n-type semiconductor like TiOz) in the photoproduction o f
hydrogen peroxide. Assuming that the reduction o f oxygen by surface trapped electrons
is the rate-limiting step, they proposed the following equation to calculate the
concentration o f photoexcited electrons.
= I - k a [ecb ] [h+b ] - k b [Zn11OH+ ][ecb 1+ k„b [ Z n ^ H ^ ]
(13)
W here I is the light intensity generating electron/hole pairs, k a[e~b][h^b] represents the
r
19
Radiant Flux
Figure 3. Photodegradation rate dependence on radiant flux.
rate o f electron/hole recombination, and the remaining terms represent the rate of
reversible surface trapping o f a conduction band electron. Using a steady state
approximation for the concentration o f electrons and assuming that [ecb"] = [hvb+], solving
for intensity gives
I = k b [Z nIIO H + ][e c b ]-k .b [Z n IOH-2 ] + k a [ecb ]2
(M )
At high light intensities (i.e. high electron concentrations) the third term in Equation 14 is
much larger than the other two and simplifies to
Z t V
[e Cbl =
(15)
Vk a )
Whereas, at low intensities the first two terms in Equation 14 will dominate, giving first
order dependence on light intensity.
20
I + k_b [Zn1O H ^]
( 16)
Ocb 1:
k b [Zn11OH2 ]
This assumes that the production rate o f electrons by the reverse o f the electron trapping
reaction is small compared to the rate o f production by photon absorption. Thus, at higher
light intensities second order recombination will dominate over first order trapping
mechanisms.
Furthering this, Hoffmann et al. (15) determined quantum yields for the rate o f
adsorbed oxygen reduction with surface trapped electrons (assuming this to be the rate
,
limiting step). Quantum yields can be defined as the rate o f reaction to the rate o f photon
absorption (15). It was found that the quantum yield will be proportional to intensity to
the negative one half power at high light intensity and independent o f intensity at low
light intensity.
Statement o f Objectives •
The work presented here represents a preliminary attempt in the development o f
supported TiOz catalysts for use in photoreactors. Through the use o f multiple reactors,
light measurements and kinetic studies it was desired to evaluate the effect o f catalyst
loading on light penetration and degradation rates o f formic acid. The focus o f this study
breaks down as follows:
o
Optimization o f support materials,
o
Light penetration through uncoated supports,
o
Light penetration through coated support materials.
21
o
Reactor design for flow through packed bed system (titanium dioxide supported on
silica gel).
o
Design a kinetic experimental system.
o
Determine catalyst loading and reactor dimensions that are most effective in
degrading formic acid.
’
22
CHAPTER 2
EXPERIMENTAL METHODS AND MATERIALS
Catalysts Supports
Several silica gels were examined for their light transmission properties. Unless
noted, the silica gels were crashed and screened to the indicated size fractions. Listed
below in Table I are the various support materials used in this project. Glass beads were
used as a comparison.
Table I. Support materials used according to size fraction and manufacturer.
Support
Mesh (U.S.
____________ _____ Standard)
12-18
Silica Gel Grade 55
35- 60
Silica Gel Grade 646*
12-18
Silica Gel Grade 41
Silica Gel CS - 0470
12-18
4 -7
Silica Gel Grade 58
7-12
12-18
18 - 25
25 - 35
35 - 60
18 .
Borosilicate Glass Beads
18
Glasperlen Glass Beads
Size (mm)
Manufacturer
________________________
1.68-1.00
Grace Davison
0.50-0.25
Grace Davison
1.68-1.00
Grace Davison
1.68-1.00
PQ Corporation
4.76-2.83
Grace Davison
2.83-1.68
1.68-1.00
1.00-0.71
0.71 - 0.50
0,50 - 0.25
Chem Glass
1.00
Braun
1.00
* Used as purchased
Following crashing and screening, the gels were washed several times with MilliQ water to remove fines and dried at I IO0C. BET-Nitrogen adsorption (Micromeritics
Model ASAP 2000) was used to characterize surface area and pore size distribution o f the
23
coated and uncoated silica gels. Subsequent to size fractionation, support materials to be
coated were fired in a muffle furnace with a ramp rate o f 30CZmin to 3 SO0C and soaked
for seven hours at that temperature. The reason for the heat treatment was to prepare the
materials for firing following coating. If a material was to experience surface alterations
during firing it would be preferable if it occurred prior to coating. During the heat
treatment, the surface area o f some silica gels would decrease up to 25%, probably the
result o f pore widening. The decrease in surface area o f coated silica gels is likely due to
both pore widening and pore blocking by the titania.. Scanning electron microscopy o f
some coated and uncoated Grade SB silica gel was performed with a JEOL Model 6100
SEM.
Table 2. BET data o f the silica gels used.
B ET surface
area
Avg. Pore
Diam .
m 2/g
A0
Gd.SB-Not fired
Gd. SB-Fired
270
236
161'
195
Gd.58-0.4%TiO2-P25
232
192
Gd.58-0.04%TiO2-P25
224 '
197
Gd.41-Fired
Gd.41-0.4%TiO2-P25
707
655
23
22
Sam ple
Light Penetration Tests
A box made o f cast clear acrylic was constructed for tests o f light penetration
through packed beds o f support materials. The acrylic transmitted nearly 100% o f the UV
light. The interior o f the box was partitioned to allow for varying bed depths. On the back
24
o f the adjustable portion o f the box an International Light Super-Slim (model SSLOO I)
UV probe was attached and used in conjunction with an International Light photometer
(model 1400A).
XXXXXXXXXXXX Support Materials XXXXXXXXXXX
UV Probe - To Photometer
Figure 4. Acrylic box used in support material light penetration tests (top view).
The probe was factory calibrated to detect UV in the 250-400 nm range with a maximum
at 375 nm. A 15-Watt black light was fixed in place and centered on the front o f the box.
Bed depths o f 0.5, 1.0, 1.5, 2.5 and 3.5 cm were examined. Radiant flux readings, in
mW /cm2, for the various packed beds were normalized with empty bed values.
In the annular photoreactors, light intensity readings were taken with the UV
probe fixed at azimuth angle o f ninety-degrees and in a direct line to the center o f the
light. Measurements were taken with the probe butting the surface o f the inner and outer
reactor tubes. As there was some variability in intensity readings with the packed reactors
(particularly with the two smaller reactors as the annular space was just a few particle
diameters), four readings were taken by rotating the reactor ninety-degrees around the
axis. These four values were averaged. A slight correction was made to outer intensity
25
readings to account for the change in radial distance due to the tubing wall thickness as
this tubing varied for the four reactors. Intensity readings taken on the outer surface o f the
inner tube (r;) were denoted initial intensity, I,, for each reactor.
Catalyst
photometer
UV Probe
Figure 5. Light intensity measurements in packed annular reactors. Four readings taken at
ninety-degree intervals around outer tube (end view).
P25 TiO? Catalysts
The P25 brand TiC^ powder was a gift from the Degussa-Huls Corporation. As
supplied in powder form, P25 TiOz particles are on the order o f 30-60 nm in diameter.
For coating silica gel, the desired amount o f P25 was added to enough Milli-Q water to
cover the silica. The suspension was then sonicated for 15 minutes using a Heat SystemsUltrasonics sonicator (model W-380) to break up any agglomerates. Following sonication
the suspension was added to the silica gel and mixed. Coated silica was dried at 60 0C for
several days until all free liquid was evaporated, and then dried at 110 0C for one day to
drive out any pore liquid. The coated silica was finally fired with the same conditions for
the uncoated silica. These conditions were chosen based on catalyst preparation work
performed at the University o f Wisconsin-Madison Water Chemistry Department. The
26
fired catalysts were then washed and dried at 110 0C. W ith this method 40-50% o f
desired adherence was possible, the remainder dropping out o f solution as a powder.
Further refinement o f P25 coatings was not attempted.
TiO? Assay
To determine the amount o f titania adhering to the support material, a
spectrophotometric method was adapted from Jackson et al. (29) (see Appendix B for
details o f this method). Briefly, the coated support was digested in a sulfuric
acid/ammonium sulfate solution and the T i(SC )^ content was assayed by addition o f
H 2 O2 , which forms a yellow colored complex with an absorption maximum at 410nm.
The absorbance was linear with titanium complex concentration over the range
employed. To determine the reliability o f this method, some standards and samples were
also analyzed with inductively coupled plasma atomic spectroscopy (Model AKL
Accuris). The two methods correlated well (See Appendix B for data).
Annular Reactors
Four annular reactors o f varying outer tube diameter were constructed. The top
priority in material selection for these photoreactors was light transmission o f the inner
reactor wall between the light source and the photocatalyst. This wall m ust transmit light
at the proper wavelength to irradiate the catalyst. For titania to be photoactive, the
wavelength must be less than 390nm. The lights used in these experiments emitted near
UV light between 300 - 400nm with a maximum at ~355nm. This emission spectrum
allows the use o f borosilicate glass, instead o f more expensive quartz which is required
27
for medium and short wave UV applications. For all reactors, borosilicate glass was
chosen as it transmits UV greater than 300 nm. The three criteria for material selection
for reactor end pieces were; strength (as considerable compression had to be exerted to
seal the reactor), workability, and inertness to the reacting fluid. Teflon (PTFE) fit these
criteria well and was the chosen material for reactor end pieces and tubing. Silicone Orings served as sealing gaskets between the glass tubes and Teflon end pieces.
TableS. Annular reactor dimensions.
R ea cto r
D esig n a tio n
2.00
2.25
2.50
2.75
O u terT u b e D iam eter
(mm)
50.80
57.15
63.50
69.85
Annular S p a c e
(mm)
3.20
6.35
9.53
12.70
B ed V olum e
(m l)
64.90
135.70
216.70
327.20
The annular reactors were designed to utilize a 15 W black light I inch in
diameter by 18 inches in length. The reactor was chosen to be 7 inches in length and use
the center portion o f the lamp, as light intensity decreases towards the ends o f the lamp.
The light intensity was approximately constant over this portion o f the lamp. All four
annular reactor configurations used an inner glass tube o f 1.5 inches in diameter and had
a bed length o f 6.5 inches. Three ports for in flow and out flow were spaced at 120° in the
annulus and the end pieces were staggered so as not to provide straight flow through the
reactors. To further support the catalyst in the reactor and prevent large inlet void spaces
due to flow, glass wool was placed at the top and bottom o f the reactors.
28
Ieactor
Figure 6. Annular photoreactor (end caps not shown).
The experimental setup was a continuous recycle system consisting o f a reservoir
bottle in line with a pump, sample valve and the reactor. Oxygen was supplied to the
reservoir bottle through a sparging line during reaction runs. Stirring was accomplished
with a magnetic stir plate and a Teflon coated stir bar. The lines to and from the reactor
were manifolded with a four way connector to supply the three inlet and outlet ports.
Once packed and assembled the reactor was vertically mounted to provide consistent
flow.
29
Vertically
Supported
Photoreactor
O2 Sparge Line
pi
Magnetically
Stirred
Reservoir
Sample
Valve
Figure 7. Schematic o f experimental photoreactor setup.
Reaction Runs
Unless noted, all experiments were performed at a concentration o f I OO-PPM
carbon (corresponding to 383-PPM formic acid). Prior to placing the reactor on recycle, 2
to 5 liters o f the formic acid solution (dependent on reactor volume) was run through in
single pass mode to establish adsorptive equilibrium. Following the single pass run, the
light was turned off, the reactor drained o f all free liquid and I L o f I OO-PPM C was set
to run on continuous recycle. When the reactor was full o f liquid, the lamp was turned
back on marking time zero for the run. Reservoirs containing the formic acid solution
were sparged with oxygen one hour prior to use and sparged continuously throughout the
run while being stirred magnetically. A valve installed in line between the pump and the
30
reactor was used for sample withdrawal. Samples (approximately I mL) were taken
starting at t = 60 min. and intermittently there after until run completion. Depending on
catalyst loading, runs would last from four to eight hours, usually until it was estimated
that at least 80% degradation had occurred. On average, eight to ten samples were taken
per run. After completion o f all experiments with a particular reactor, fluid was drained
and the reactor was air dried. Catalyst removed from the reactor was further dried at
HO 0C and weighed. Assumptions made for this setup were as follows:
o
Absorptive equilibrium was established after running solution
through single pass.
o
Solution was saturated with oxygen,
o
Perfect mixing was present in the resevoir.
o
Plug flow through reactors was present.
Slurry Runs
As a comparison, experiments using powdered TiOi slurries were also conducted
in each o f the four annular reactors. Procedures for the suspended runs were similar to
those for the packed beds. A measured amount o f P25 TiOi was added to the reservoir
containing the formic acid solution to be degraded. To finely disperse the titania particles,
the resultant slurry was sonicated for fifteen minutes. The P25 powder in the formic acid
solution remained finely dispersed throughout the runs (no significant agglomeration or
settling o f particles was observed). Aluminum foil was wrapped around the reservoir
bottle to prevent stray UV light from causing degradation in the reservoir. The bottle was
sparged with oxygen for I hour prior to pumping and sparged continuously throughout
31
the run while under magnetic stirring., To rule out the disappearance o f formic acid due to
adsorption on the titania, samples were taken prior to powder addition and after the
addition o f TiOa and I hour o f stirring and sparging. No statistically significant,
adsorption o f formic acid was noted. The slurry was run through the reactor on
continuous recycle using a peristaltic pump at flows corresponding to those o f the packed
bed runs. Sampling procedures were the same as mentioned above for the packed bed
runs. Prior to analysis, samples were centrifuged for 6 minutes at 14000 rpm using a
Fisher Scientific Micro 14 microcentrifuge.
Sample Analysis
Formic acid concentrations were determined using a Dorhman Carbon (TOC)
Analyzer (DC-80). Direct aqueous injections o f 100 pL were possible and no refinement
o f samples was necessary. Repeatable results were obtained with an average percent
standard deviation o f less than 2 percent. All samples were refrigerated immediately and
usually analyzed within 24 hours.
32
CHAPTER 3
RESULTS AND DISCUSSION
Choosing the Support Material
For preliminary evaluation o f catalyst support materials, light penetration studies
were conducted using the acrylic box described previously. As indicated in Figure 8,
silica gel does not strongly absorb UV light and in comparison with borosilicate glass
beads, it can be seen that the silica gel would be the better choice for a photocatalyst
support.
12-18
BoroBaII
Bed Depth (cm)
Figure 8. Light transmission through Grade 58 silica gel and borosilicate glass beads.
UV transmission through silica gel was found to be dependent on manufacturer and/or
grade and size fraction. Light penetration tests were conducted to find the optimum grade
and size fraction for the support material to be used in the photoreactors. Intensity values
were normalized with empty bed values. Regarding size fraction for the support
33
materials, consideration was given to minimizing pressure drop, reducing loss due to
entrainment under flow conditions, and working with reasonable bench scale annular
spaces.
- A - G d 41
- X - G d 58
Bed Depth (cm)
Figure 9. Light penetration through different brands o f 12-18 mesh silica.
Based on the data shown in Figure 9, Grade 58 silica gel was chosen for further
study. Though Grade 41 showed similar light transmittance to the Grade 58, it was found
to burst on contact with water. BET analysis revealed it to be microporous with a surface
area greater than 700 m2/g. It is possible that a high heat o f adsorption with water is the
cause. This obviously makes it ill suited as a support in an aqueous environment.
To determine the most effective size fraction for use, Grade 58 (obtained in 4-7
mesh size from the distributor) was crushed and screened to five smaller fractions. As
indicated in the Figure 10, approximately 50 to 75 percent o f the light is absorbed in the
first IOmm o f packed bed with the chosen silica gels. It should be noted that light
intensity readings at the 5 mm bed depth for the larger size fractions are highly dependent
34
on packing as this space is only 1-3 particle diameters. Based on the size fraction tests it
was concluded that 12-18 mesh grade 58 silica gel would perform best as a photocatalyst
support material.
0.90
0.80
0.70
0.60
0.50
0.40
0.30
-
0.20
0.10
-
• — 7-12
± — 12-18
»3— 18-25
• — 25-35
•-3 5 -6 0
Bed Depth (cm)
Figure 10. Light transmission through various size fraction o f Grade 58 silica gel.
Referring to Figure 11, the effect o f loading the silica gel with titania can be seen
to be substantial. Light penetration tests through a 0.02 weight percent TiC^ loading
shows a marked decrease in light transmission within the first few millimeters o f bed.
Approximately a 70 percent difference in light transmission was observed between the
unloaded and loaded catalyst at a bed depth o f 1.0 cm.
These measurements served as a basis for the design o f the photoreactors. Four
annular reactors were designed with annular spaces ranging from 3.2 to 12.7mm.
These were practical sizes for the reactors, considering the catalyst size, drop in light
transmission and the available materials.
35
0 .9 0 -
Gd 58 Uncoated
0 .8 0 0 .7 0 -
H B - Gd 58 - 0.02% TiO2
0.60
0.50
0 .4 0 0.30
0.20
0.10
-
0.00
0 .5 0
1.00
1.50
2.0 0
2.50
3.00
3.50
Bed Depth (cm)
Figure 11. Comparison o f light transmission through coated and uncoated silica.
Light Penetration Through Annular Spaces
Light penetration data taken in the acrylic box gave a general idea o f the UV
transmission properties o f silica gels. It should be noted that the light penetration tests
conducted in the acrylic box may be subject to some error due to the geometry o f the box.
There exists the possibility that light can exit the box in several different directions other
than the direct line between the light and the probe. UV transmission through a
cylindrical geometry is expected to differ from linear bed measurements in that the bed
area increases with radial distance. In this configuration, light can be assumed to only exit
through the outer glass of the reactor (the Teflon end caps will transmit light but this was
determined to be negligible). After packing each reactor with coated and uncoated
supports, intensity readings were taken at the outer surface o f the four outer tubes.
Referring to Figure 12, the first data points represent light readings on the outer surface
36
o f the inner glass and the successive data points represent light readings on the outer
surface o f each o f the four reactors.
■
Gd 58 Uncoated
0.04wt.% Ti0 2
_
3.00
♦
<
2.50 -
A 0.4wt.% Ti02
^
2.00
E 1.50 -
1.00
-
0.50 -
Figure 12. Light penetration through annular reactors packed with coated and uncoated
silica.
Similar to linear bed measurements, the effect o f loading silica gel with TiC^
greatly affects light distribution through the annular reactors. Approximately 95 % o f the
incoming light is absorbed in the first three millimeters (the smallest reactor) o f reactor
space with a 0.4 weight percent T 1O 2 loading.
Coatings
From the light penetration tests, a 0.4 weight percent loading was determined to
be the practical upper limit. At higher loadings, light would likely be undetectable outside
even the smallest reactor. To test the adherence o f titania to the silica, the 2.0” reactor
was packed with a 0.4 percent loading and reaction conditions simulated for several
hours. Following the run, the catalyst from the reactor was assayed at a 0.35 weight
37
percent loading. As this is within the variability o f the analysis method, it is difficult t o '
determine if there was an actual loss o f titania. It is possible that unattached fines not
removed during catalyst washing, were fully removed under flow. Fines in this system
■:
would contribute little to the overall degradation rate, as they would not be under
.
constant illumination.
Formic Acid Runs
Formic acid was chosen as the compound to test the photoactivity o f the catalysts
because it has a simple molecular structure, is easily photodegraded, is highly soluble in
water, and there is limited possibility for intermediate radical formation (30). The
photooxidation o f formic acid can be represented as follows
CH2 O2 + - O 2
Tl° 2,hv >C O 2 + H 2 O
(17)
2
Two blank runs were performed to confirm that the disappearance o f formic acid
occurred only in the presence o f illuminated titania.. To rule out photolysis o f formic acid
due to absorption o f UV light and volatilization from the reservoir during sparging, a
blank run was performed in an empty, illuminated annular reactor. Another run was
conducted in an illuminated annular reactor packed with uncoated silica gel. This
experiment was performed to rule out degradation o f formic acid due to an interaction
with the silica gel. There was no loss o f formic acid in either blank run after several hours
o f illumination. It was also noted that formic acid, at the concentrations used, did not
absorb UV light.
Numerous researchers involved in heterogeneous photocatalysis have found some
form o f the Langmuir-Hihshelwood rate expression to fit degradation data well (4,22,26).
38
^o b sicCj
I + KCi
( 18)
W here k0bs is the reaction rate constant, k is the adsorption constant, and Q is the '
concentration o f substrate. In cases o f high and low substrate concentration, apparent zero
order and first order rates, respectively, have been observed. If the LangmuirHinshelwood rate expression accurately describes the kinetics, the second term in the
denominator o f Equation 18 would dominate at high substrate concentrations and cancel
with the numerator giving:
rL = k Obs
,
(19)
For the case o f low substrate concentration, the I in the denominator o f Equation 18
would dominate giving apparent first order rates. .
, ri = k obsKCi = k appc i
(20)
As indicated in Figure 13, initial runs using a 0.4 weight percent P25 loading on
Grade 58 silica gel show that the degradation o f formic acid follows apparent zero order
kinetics at the higher concentrations used (100 PPM C). The linearity o f concentration
versus time data is seen to decline at concentrations below about 20 PPM C. This was
confirmed in a run with a starting concentration o f 15 PPM C where apparent first order
degradation was observed. The observed kinetics are consistent with data presented by
Kim et al. (35), which shows a transition between zero order and first order kinetics
between 17.5 and 35 PPM formic acid. All subsequent runs used an initial concentration
o f 100 PPM C.
39
100.00
_
'Cr
£
I
d
c
O
°
-|
I
80.00 60.00
40.00 -
20.00
-
0.00
-
♦
0
100
200
Time (min)
300
*
400
Figure 13. 2.00" Reactor packed with 0.4 weight percent P25 on Grade 58 silica.
It is known that reaction rates in packed bed configurations will have a
dependence on flow as mass transfer limitations may exist below a threshold flow rate.
To determine if there were mass transfer limitations in the annular systems, four runs
were conducted at different flow rates to see if a drop in rate was observed. Starting at a
flow o f 150 mL/min (the highest possible with the pump and setup used) four runs were
conducted, incrementally reducing the flow to 25 mL/min.
The observed rate remained constant for flows o f 50, 100 and 150 mL/min with
approximately a ten-percent drop for a flow o f 25 mL/min (Figure 14). All subsequent
runs were performed at flows between 100 and 150 mL/min.
As the assay test to determine the adherence o f TiO] to the silica gel was
inconclusive, further tests were conducted. The 2.0” reactor was packed with fresh
40
W 18.0
<d 16.0 ■o 14.0
100
Flow rate (mL/min)
Figure 14. 2.0” reactor packed with 0.4 weight percent P25 on Grade 58 silica at varying
flow rates.
catalyst and subjected to five consecutive runs at a flow rate o f 100 mL/min to determine
if the degradation rate decreased as a result o f catalyst loss. After the fourth run, the
reactor remained under flow for 90 hours before the fifth run. W ith approximately a
three-percent variability in reaction rates, there does not appear to be a change in reaction
rates due to erosion o f titania.
Table 4. Five consecutive runs in 2.0” reactor with 0.4 weight percent P25 on Grade 58.
Run
I
2
3
4
5 (After 90 Hrs. under
Flow)
Rate (PPM /hr)
16.8
17.4
18.2
17.2
17.4
41
Packed Bed Annular Reactor Runs
Several o f the experiments with the supported catalysts in the four reactors were
randomly performed twice to demonstrate repeatability with the setup and methods used.
Rate data generated from the repeated runs was used to calculate an average standard
deviation in rates o f approximately 5 percent. Error bars in all subsequent figures
showing rate data represent plus and minus one standard deviation.
As stated above, a 0.4 weight percent loading was taken as the upper limit o f
catalyst loading in the evaluation o f the annular photoreactors. Using a 0.4 weight percent
P25 loading on Grade 58 silica gel, identical runs were conducted in the four reactors.
Figure 15 depicts a run in each o f the four annular reactors with increasing mass
o f TiOi corresponding to the increasing volume o f the reactors. It can be seen that
increasing the mass o f TiOi had little or no effect. This was not unexpected as 95 % o f
the light was absorbed in the first 3.2mm o f annular space at this loading and therefore,
titania in the reactor past this point is not activated.
T
20 t----------------------------------------------------------------------------
ICL 1 8 ®
t
t
T
Io
----------------------------
I
I
^
16 -
5
-CS
o
14
-
Z
$
12-
o
10
(A
Si
4-—— — ,--------- 1---------- 1----------T---------0
100
200
300
400
500
Mass TiO 2 (mg)
Figure 15. Observed rate versus mass TiOi in annular reactors packed with 0.4 weight
percent P25 on Grade 58 silica.
42
In the next set o f experiments, the TiC^ loading was decreased an order o f
magnitude to 0.04 weight percent P25. It was desired to see more o f a pronounced effect
on rates with increasing reactor volume. The absorption o f light was less severe at this
loading and light was able to penetrate the packing in all reactors (i.e. light was detected
at the outer surface o f each reactor).
As indicated in Figure 16, the observed rate is seen to increase with mass o f TiC>2
but appears to be leveling o ff with the largest two reactors. At this loading the light
detected outside the largest reactor corresponded to absorption o f 99 % o f the incident
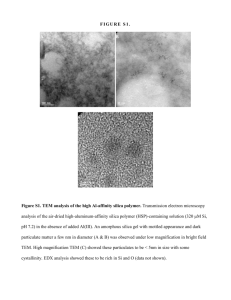
light entering the reactor. Increasing the bed volume past this point would have little
effect at this loading and the observed rates would likely have a maximum around 13-14
PPM/hr.
Based on the data presented, experiments were planned for titania loadings
between 0.04 and 0.4 weight percent to better examine the effect o f light distribution on
rates. However, new silica gel had to be obtained at this point as the previous loadings
S
M12
-
3
10
-
O
4
Mass TiO 2 (mg)
Figure 16. Observed rate versus mass TiO2 in annular reactors packed with 0.04 weight
percent P25 on Grade 58 silica.
43
used the remainder o f the stock. Upon consulting the supplier (Aldrich Chemicals), it was
discovered that the manufacturer had discontinued Grade 58 but was supplying the same
gel in different size fractions as Grades 55 and 59 (both > 8 mesh). As indicated in Figure
17, a 0.1 weight percent P25 loading prepared with Grade 55 silica (12-18 mesh) did not
give the results expected. A small increase in degradation rates was observed in the two
smallest reactors with 0.1 weight percent P25 on Grade 55, but quickly leveled off to
perform worse than the 0.04 weight percent P25 Grade 58 catalyst in the larger two
reactors.
■a
6
E
4-
O
0
Gd 55-0.1 wt.% P25
Gd 58 0.04wt.% P25
Mass TiO 2 (mg)
Figure 17. Comparison o f Grade 58 and Grade 55 silica gels.
Subsequent light penetration tests conducted on the reactors packed with uncoated
Grades 55 and 58 silica (12-18 mesh) showed the two gels to have similar light
transmission properties. The difference in intensities between the two gels (Figure 18) is
insignificant as there was some variability in measurements due to packing arrangements
in the reactors.
44
^
3.00 -
♦ Grade 55
O 2.50 -
■ Grade 58
1-50
1.00
0.50 -
Radius (cm)
Figure 18. Light penetration tests with uncoated silica gel (12-18 mesh).
To better examine the apparent lower activity o f the new silica, a batch o f Grade
55 silica with 0.40 weight percent P25 was prepared. This would allow a more direct
comparison between the two support materials, weighing its performance against the 0.40
weight percent P25 on Grade 58 silica. With 98 percent o f the light absorbed in the
smallest reactor with this catalyst, it was expected that no significant increase in rates
would be observed in the larger reactors. Runs performed with this catalyst in the largest
and smallest reactors confirmed this with little difference in observed degradation rates
between the two reactors. However, similar to the 0 .1 weight percent loading, rates did
not approach the rates attainable with the Grade 58 silica. It is not likely that the
difference in activity (nearly 30 % difference in observed rates) can be attributed to a
slight increase in light absorption (Figure 18). The lower activity may be related to
impurities present in higher quantities in the Grade 55 gel that act to deactivate the
45
titania. Another possibility is that impurities are present in the Grade 58 silica that
enhance the photoactivity o f this supported catalyst. Verifying this would require
analysis o f the silica gels to determine the concentration o f any impurities that may be
present.
S
18 -
TJ
8 6 -
>
Gd 55-0.1 wt.% P25
Gd 58 0.04wt.% P25
Gd 55-0.4 wt.% P25
Gd 58-0.4 wt.% P25
50
100
150
200
250
300
350
400
450
500
Mass TiOa (mg)
Figure 19. Comparison o f observed rates versus mass o f titania for the various catalysts.
The rate data for the four catalysts are summarized in Figure 19. Upon comparing
the data for the two Grade 58 catalysts, it is apparent that reducing the TiC^ loading
results in decreased catalyst activity. This decrease in activity is likely due to increased
absorption o f the incident light by the silica gels. It is also apparent that different grades
o f silica, with similar light transmission properties, will behave quite differently as a
support material.
Based on this data, further refinements to the catalysts, with regards to P25
loading, was not attempted. For both grades o f silica gel, a 0.40 weight percent loading
would appear to be the maximum practical loading for these reactors. Higher loadings
46
may result in slightly higher rates but would result in unilluminated catalyst in even the
smallest reactor.
Slurry Runs
With much o f the current research in photocatalysis conducted using TiOz
slurries, it was desirable to compare the above degradation rates to those obtained with
slurries. In reviews o f current literature, it was noted that mixed results have been
obtained in experiments comparing suspended and supported photocatalysts. Several
different researchers (9, 21) have attributed the higher photoactivity o f supported titania
to increased surface area o f the substrate/catalyst and to increased water/organic/oxygen
adsorption at the surface. Others (12, 16) have reported better efficiencies using slurries,
indicating a higher catalyst surface availability for reactant and photon interactions to be
a contributing factor. W ith the number o f variables involved in photocatalysis and the
widely varying systems employed, this is not surprising. As much o f the work here is
preliminary, these experiments were intended to provide a quick comparison between the
supported and suspended catalysts in this system and an attempt was not made to extend
these findings to other systems.
Slurries were prepared to give total concentrations o f P25 in each o f the reactors
comparable to that for the 0.40 weight percent loaded catalyst. In other words, at any
instant during the slurry experiment, the amount o f TiOz within the annulus would
compare to the amount o f stationary TiOz in the reactor for a 0.40 weight percent loaded
silica. At this slurry concentration virtually all o f the incoming light was absorbed in the
47
four reactors and very little or no light was detected at the outer glass once the slurry was
introduced.
Runs were conducted in each reactor at the equivalent 0.40 weight percent
concentration and one in the largest reactor at double this concentration. The results are
presented below in Figure 20.
Slurry
0.4 wt%-Gd58
0.4 wt%-Gd55
500
7
Mass TiO2 (mg)
Figure 20. Comparison o f observed rates with supported and suspended P25 titania.
Noting that rate increases were observed with increasing reactor diameter, it is
probable that light was able to penetrate deeper into the reactors than it was with the
packed beds. Though an increase in the rate o f degradation was observed with increasing
reactor volume, it did not approach rates attainable with the Grade 58 supported catalysts.
As it was not feasible to further increase the outer diameter, it was decided to increase
slurry concentration in the largest reactor to determine if higher rates were possible.
Though changing the concentration o f slurry in the reactors changes the depth that light
can penetrate, it was reasoned that if rates were significantly higher at a higher slurry
48
concentration, it would be possible for rates to continue to increase with increasing
reactor diameter at the equivalent 0.40 weight percent concentration. More than twice the
amount o f powder in the largest reactor did little to improve the performance o f the
slurry. It is evident that sufficient light is not available to increase degradation rates much
beyond that attained with the 1210 PPM slurry in the largest reactor. This result
correlates well with results presented by Herrmann (4) who found the optimal slurry
concentration to be 2500 PPM. Beyond this concentration, rates were determined to be
independent o f mass o f titania.
The fact that higher rates were achieved with Grade 58 supported catalysts in this
system does indicate that there are beneficial aspects o f immobilizing titania. Whether
those benefits are associated with increased adsorption o f organics of oxygen remains to
be determined and requires further investigation. Another possibility is that the heat
treatment o f the supported catalysts enhances photoactivity. The powder for the slurries
was used as supplied and underwent no refinement. Rate data for the supported and
suspended catalyst experiments are summarized in Table 5 below.
Table 5. Summary o f supported and suspended observed rates.
Degradation Rates
R eactor OD 0.04% P25- 0.4% P25(in.)
G d58
Gd58
. (PPM /hr)
0.1% P25- 0.4% P25- Slurry 0.4 Equiv.*
Gd55
Gd55______________________
4.1
. 7.9
11.9
13.2
17.6
17.5
17.9
17.9
8.7
10
10.7
10.2
12.2
8.1 (780 PPM TiO2)
9.3 (1020 PPM T iO 2)
10.2(1060 PPM T iO 2)
12.2 (1210 PPM TiO2)
11.5
12.8 (2650 PPM TiO2)
*Plus one run in largest reactor at more than twice this concentration
2.00
2.25
2.50
2.75
—
49
Data Analysis
,
Due to the nature o f photocatalytic systems, rate constants will be a function, o f
several external parameters such as light intensity, catalyst loading, support material, and
catalyst preparation methods. W ith the variety o f experimental conditions used by current
research groups; a significant comparison o f data becomes difficult. Consideration was
given to factor out some parameters in this project. All runs were performed at the same
flow rate, initial substrate concentration, initial light intensity (i.e. same light used in each
reactor), and make o f titanium dioxide. The inadvertent use o f different support materials
in these experiments further complicated internal comparisons. To better correlate the
data, an attempt was made to compare rate constants based on the rate o f light absorption
in each reactor.
Volume Correction
It was noted above that formic acid degradation follows apparent zero order
kinetics at higher concentrations. Referring to the experimental setup used, rate constants
were calculated using the concentration-time history in the tank. The setup used is
essentially a constant volume batch system consisting of.a plug flow photoreactor (PFR)
in a recirculation loop and a well stirred, nonreacting mixing tank. The formic acid
solution is degraded only while under illumination in the reactor and is returned to the
tank where it dilutes the bulk solution. Small differential conversions were obtained with
each pass through these photoreactors (generally less than I %).
50
I*
PFR
C.
Pump
Sample
Valve
Figure 21. Experimental photoreactor system.
The observed rates were reported in units o f PPM/hr. All experiments were run
using I L o f 100 PPM carbon (from formic acid), making the units on rate constants
PPM-C/hr or mg-C/hr*L. The observed rate constants were thus expressed on a total
system volume basis. Several researchers (31,32,33,34) noted that rate constants
calculated this way would be in error if the reactor volume is not negligible compared to
the tank volume. In these experiments, the ratio o f reactor to tank volume ranged from
about 10 to 35 percent for the four reactors. For the above comparisons o f catalyst
performance and reaction conditions in our system, using the concentration-time history
in the tank is sufficient, but for deeper data analysis purposes these rate constants should
be corrected. Wolfrum and Turchi (32) suggested that the volume corrected rate constant
can be approximated using the concentration-time history in the mixing tank and
accounting for the ratio o f reactor to tank volumes.
(21)
51
In the case o f heterogeneous catalysis, the reactor volume in Equation 21 is the void
volume in the reactor. In a rigorous analysis Klausner et al. (31) determined this
approximation to give rate constants accurate to within five percent o f the actual rate
constants over a wide range o f conditions. Equation 21 can be simplified as follows:
^Vol-Corr —^obs
V Tank + V Reac.Void
(22)
ViRe ac.Void
For the four reactors used, the average void fraction was 0.3 with 12-18 mesh silica gel.
Considering the numerator in Equation 22 to be the total volume o f the system, this can
be expressed as:
kVoLCorr
^obs
^
v Total
^
(23)
, v ReacVoid y
The volume corrected rate constants can be further converted to an empty bed basis.
k EB = k VoLCorr
v ReacVoid
(24)
Vr
It was now assumed that kvoi.coir or ksB depended only on the light intensity, TiC^
loading, and the support material used (neglecting compound effects and pH).
Table 6. Volume corrected rate constants for the various catalysts.
kEB '
S ilica G rade
58
58
55
55
Slurry
T iO 2 Loading R eacto r 1 R eacto r 2
0.04
0.4
0.1
0.4
0.4 Equiv.
63.7
27 6 .8
136.2
190.9
132.8
6 0.8
134.6
76.5
—
77.8
(PPM /hr)
R eacto r 3
R eacto r 4
58.6
88
52.5
44 .4
60.1
34.2
—
57.3
38.7
4 9.5
■52
Normalization to Rate o f Energy Absorbed
Light intensity will remain one o f the more difficult parameters to evaluate in
photocatalysis, with degradation rates dependent on the amount o f light absorbed by TiOz
o f sufficient energy to induce charge-carrier separation. Analysis o f the rate data becomes
more challenging when the catalyst support absorbs some light and competes with the
titania. As stated earlier, silica gel will absorb some light (some grades more than others)
and differentiating between the light absorbed by the catalyst and support is difficult. The
light intensity at any point in the reactors will be a function o f the concentration o f
absorbing media, support material and distance from the source.
Normalization o f rate constants to the rate o f energy absorbed was performed
■using light measurements at the inner and outer glass o f the packed reactors. The
difference in light intensity between the inner and outer annular reactor tubes provided a
means to determine the amount o f light absorbed in the reactors.
A simple energy balance taken over the volume o f the reactors yielded th e .
following equation treating intensity as a function o f radius.
l|r (27tLr) - l|r+dr 27tL(r + dr) - (Abs.)(7t[(r + dr)2 - r 2 ]L) = O
(25)
The first term represents the light entering the reactors, the second term is the light
exiting the reactor, and the third term is the light energy absorbed as a function o f
position in the reactors.
53
Absorbed Light
(mW/cm3)
Figure 22. Light measurements for intensity balance on photoreactors.
The Abs. term in Equation 25 is the rate o f light energy (mW) absorbed per unit volume
(cm3) in the reactors. This balance ignores reflectance and scattering effects. Expanding
terms, simplifying, and solving for the Abs. term yields Equation 26.
(Abs.)rdr = l|r (r) - l|r+dr(r + dr)
(26)
In simplifying, the dr2 term was considered to be negligible compared with rdr. The Abs.
term can also be modeled as intensity (mW/cm2) as a function o f radius multiplied by a
system specific extinction coefficient b (c m 1).
Abs. = b lr
(27)
Solving for the b term would allow for a more qualitative comparison o f the catalysts by
considering b to be a function o f TiOz loading and the support used. Substituting
Equation 27 into Equation 26 and simplifying gives the following.
b I rdr = -d(Ir)
(28)
d @ L _ b .I T
(29)
Integrating Equation 29 and expanding gives intensity in the reactors as a function o f
position (Equation 31).
r i p / r o d(I-r)
(30)
54
Z
Ir
\
I r 2l • exp(-b • (r0 - r , ) )
<r0 y
(31)
Further manipulation allowed for the determination o f b with the experimental data.
In
I q ' ro
- b - (ro - r i )
(32)
XIo
The data plotted with Equation 32 is shown in Figure 23 with the system specific
extinction coefficients (slope o f the lines in Figure 23) given in Table 7.
-
1.00
♦ 0.40%-Gd58
■ 0.04%-Gd58
”o -3.00
A 0.10%-Gd55
u -5.00
* 0.40%-Gd55
rQ-H (cm)
(Outer radius-inner radius)
Figure 23. Linearization o f Equation 32 with experimental data to give b terms.
Table 7. System specific extinction coefficients.
C atalyst
0.40%-Gd55
0.40%-Gd58
Slurry 0.4 Equiv.
0.10%-Gd55
0.04%-Gd58
b (c m 1)
10.1
8.2
7.9
5.3
3.0
55
Using b, the above defined Ir (Equation 31) and integrating over the volume o f the
four reactors, a volumetric rate o f energy absorption (VREA) was determined. A
volumetric rate o f energy absorption (VREA) can be defined as the rate o f energy
absorbed (REA) per unit empty bed volume with units o f HiWZVeb or mJ/s*VEB.
V R E A = £ 5 ^ = _ ! — (1P (b-I(r))-2-ji-r-L -dr
Vbb Veb ^rI
(33)
W ith the VREA the rate constant could be normalized to the rate o f energy absorbed.
kREA = J eb_ = IcebVeb = W i r
. kj^a
VREA
REA
(34)
REA
After converting time, Erea will have units o f mg-C degraded per J o f energy absorbed
(mg-C/J).
Table 8. Summary o f intensity normalized rate constants for the various catalysts.
kREA(mg-C/J)
Silica Grade T iO i Loading Reactor I R eactor 2
58
58
55
55
Slurry
0.04
0.4
3.00E -03
4.33E -03
R eactor 4
R eactor 3
6.03E -03
6.67E -03
8.70E -03
8.22E -03
8.54E -03
8.79E -03
0.1
0.4
4.87E -03
4.82E -03
5.14E -03
5.03E -03
5.78E -03
'
0.4 Equiv.
4.20E -03
4.76E -03
5 .6 6 E -0 3 .
5.56E -03
,
7.27E -03.
By inspecting the data in Table 8, it can be seen that the rate o f reaction per rate
o f energy absorbed (Erea) is essentially constant for all catalysts except the 0.04 weight
percent P25 on Grade 58 silica and the slurry. W ith these catalysts, Erea increases with
increasing reactor diameter. SeeEing a quantitative explanation for this trend is difficult
as the functional relation between light intensity and VREA is complex. Recalling
56
Hoffinann ’s (15) explanation o f rate dependence on light intensity, it was demonstrated
that this relation can vary from a power o f 0.5 at high intensities to 1.0 at lower
intensities.
Rate oc (Intensity)11
(35)
Using this relationship, an expression can be written describing the local rate constant
(ki) as a function o f the local volumetric rate o f energy absorption (LVREA).
kL =km t(LV REA )n
n = 0 .5 -» 1 .0
(36)
W here Iqnt is the intrinsic rate constant. The intrinsic rate constant is assumed to be
constant at a given temperature for a given catalyst. With small differential conversions
per pass in these reactors, Iqj can be considered to vary only in the radial direction. The
change in kL is a result o f both LVREA and n varying as intensity decreases in the radial
direction. W ith these assumptions, the calculated empty bed rate constant
(L e b )
can
therefore be viewed as the local rate constant integrated over the volume o f the reactor.
kEB =
Jpj0
2 %-r L dr°
V
V EB
(37)
y
Further substitution and manipulation yields an expression relating the observed rate
constant with a varying dependence on light intensity (Equation 40).
Ti •r •L • dr
W
(39)
kobs=
(b'lto)” -Z-TtT-L.*
(40)
57
Extending this to the above defined intensity normalized rate constant (Equation 34) the
following is obtained:
J^0 (b • I(r))n - 2 -TC-T-L-dr
l REA
(41)
‘■int •
J!°(b -I(r))-2 -T c-r-L -d r
As light intensity decreases in the radial direction, n will approach unity and the
ratio o f integrals in Equation 41 will increase. Therefore, for the case o f the 0.04 weight
percent P25-Grade 58 catalyst, Erea will increase with increasing reactor diameter as
indicated in Table 8. Past the limiting radius for full light absorption, degradation rates
will be constant. Where the shift in n occurs in this system will depend on catalyst
loading and the support material used. These findings are similar to those made by
Hoffinann et al. (15) where the derived quantum yield showed a negative half order
dependence on intensity at high intensities and was found to be independent o f intensity
at lower intensities. For the other three supported catalysts (0.4% on Grade 58 and both
Grade 55 catalysts), essentially all light is absorbed in the smallest reactor and therefore
Icrea should be the same for each reactor as is indicated in Figure 24.
58
1. E-02
9.E-03
8.E-03
3 7.E-03
9 6.E-03
E 5.E-03
^ 4.E-03
J f 3.E-03
2. E-03
1.E-03
O.E+OO
2.15
0.04 wt%-Gd58
0.4 wt%-Gd58
0.1 wt%-Gd55
0.4 wt%-Gd55
2.65
3.15
3.65
Reactor Radius (cm)
Figure 24. Radial dependence o f kic-
SEM
An attempt was made to discern the nature o f the coatings through SEM. Images
o f some o f the coated and uncoated silica can be seen on the following page. Two
conclusions that can be drawn from the SEM photos are (I) the surface is covered by an
uneven coating o f TiCb that can be seen as small whitish islands, or (2) the surface has a
uniform coating o f TiCb, possibly with small islands o f titania built up on the exterior o f
it. Figure 25.a could be explained as a near uniform layer o f TiOi that has cracked due to
silica gel surface features and/or preparation methods. Finer resolution (Figure 25.b &
25.c) reveals islands o f what may be titania deposited in random groups. It is not possible
to discern as to whether these islands are deposited on top o f a uniform layer o f TiOi or
randomly agglomerated directly to the silica. SEM o f an uncoated bead (Figure 25.d)
does reveal a cleaner surface than similar resolution o f coated beads. A further possibility
59
Figure 25. SEM o f a silica gel particles (12-18 mesh), (a) 0.40 weight percent
loaded titania, 1700X resolution, (b) 0.40 weight percent loaded titania
2000X resolution, (c) 0.40 weight percent loaded titania 6000X
resolution, (d) Uncoated silica 2000X resolution.
Note: Samples were mistakenly labeled 0.5 % - actual specimens are 0.4 %.
60
is that the resolution is not good enough to distinguish the TiOi on the surface and the
images are revealing different surface features o f the silica gel. It was noted during
imaging that the surface o f the silica beads varied greatly in roughness depending on
where the beam was directed. This should be expected o f a crystal subjected to grinding
and screening.
The above degradation rate data suggests that at lower loadings, increased
absorption o f light by silica gel may affect the photoactivity o f the supported catalysts. If
the titania was deposited in small, randomly agglomerated islands on the surface, the
lower loadings would result in more o f the silica surface being exposed, thus enabling it
to absorb m ore light. Based on this assumption and the SEM images, it can be proposed
that a uniform coating is not present and the white groups in Figure 25.b and 25.c are
indeed random islands o f TiOz attached directly to the substrate. In order to confirm this
using SEM, methods would need to be developed to slice the silica beads. A comparison
o f sliced coated and uncoated beads would allow for the identification o f a uniform
coating. This would be a difficult task considering the size o f the beads used and that the
coating m ust remain intact and unaffected from any cutting.
Conclusion
Past work in environmental photocatalysis has largely involved slurries o f TiOz
which will require separation to reclaim the catalyst. This is one reason this technology
has not seen much use past bench scale applications. Supported photocatalysts in packed
bed reactors represents an industrially feasible alternative to slurries and possibly a viable
waste water treatment method in the near future.
61
The proven conclusions o f these experiments are summarized as follows:
o
UV light transmission through silica gel will be a function o f size'fraction,
manufacturer and/or grade.
o
UV light transmission through silica gel will decrease sharply when coated
with titanium dioxide.,
o
Good adherence o f the titania to the silica gel was obtained,
o
Mass transfer limitations were negligible at the flow rates used,
o
Apparent zero order kinetics were observed for the degradation o f formic acid
at the concentrations used.
o
Coating TiOa on silica gel can enhance photocatalytic rates as compared to
slurries run under identical conditions.
o
The optimum catalyst for the degradation o f formic acid in this system would
be 0.40 weight percent P25 on Grade 58 silica gel.
The assumed conclusions o f these experiments are summarized as follows:
o
The difference in rates between the 0.40 weight percent loadings can be
attributed to impurities present in one o f the grades o f silica,
o
The lower rates observed for catalysts with lower titania loadings can be
attributed to increased absorption o f light by the silica gel.
o
The trend in the intensity normalized rate constants is related to the varying
h alf order to first order dependence o f phofocatalytic degradation rates on
light intensity.
o
A uniform coating was not attained with the coating methods employed.
62
Future Work
W ithout a proper understanding o f the nature o f the coating it will be difficult to
gauge the extent to which a support material will influence degradation rates in a
photocatalytic system. It was proposed here that a uniform coating was not attained with
the coating methods used, but this was not confirmed. A better understanding o f the
nature o f the coatings is required. The relatively low loadings used in this work makes
conventional analysis difficult. A reliable method o f slicing the small beads employed
would facilitate better evaluation using SEM. This would allow determination o f both the
thickness and the nature o f the coating.
Experiments should be performed to determine if there is a certain impurity
present in silica gels that could affect photoactivity. Examples o f this could be the
digestion o f silica gel followed by spectrophotometric or ICP analysis.
A significant comparison o f data among different researchers in photocatalysis is
complicated by the number o f variables associated with this field. The introduction o f
various support materials offers yet another variable and it is clear that certain standard
methods need to be employed to further this line o f study. It can be proposed that with
the development o f some standard methods, the above approach for the determination o f
catalyst specific extinction coefficients could provide for useful comparisons between
different researchers working with supported photocatalysts.
63
REFERENCES CITED
1. Serpone5N. Solar Energy Mats. Solar Cells. 38 (1995) 369-379.
2. M elz5 G.R., Zepp5R.G., Crosby5D.G., Eds. Aquatic and Surface Photochemistry.
Lewis Publishers, Ann Arbor5Mi. 1994.
3. Serpone5N., Pelizzetti5E. Eds. Photocatalysis: Fundamentals, and Applications. John
W iley and Sons. New York. 1989.
4. Herrmann5J. Catalysis Todayi 53 (1999) 115-129.
5. Mills, A., Davies5R. J. Photochem. Photobiol. A: Chem. 85 (1995) 173-178. '
6. Ollis5D.F., Al-Ekabi5H. Eds. Photocatalytic Purification and Treatment o f Water and
Air. Elsevier. Amsterdam. 1993.
7. Mills, A., Le Hunte5 S. J. Photochem. Photobiol. A: Chem. 108 (1997) 1-35.
8. Hoffinann5M.R., M artin5 S.T., Choi5W., Bahnemann5D.W. Chem. Rev. 95 (1995)
69-96.
9. X u5Y., Zheng,. W., Liu5W. J. Photochem. Photobiol. A: Chem. 122 (1999) 57-60.
10. Shackelford, J.F. Introduction to Materials Science for Engineers 4th Ed. Prentice
Hall. Upper Saddle River5N.J. 1996.
11. Tsai5 S., Cheng5 S. Cat. Today. 33 (1997) 227-237.
12. Bideau5M., Claudel, B., Dubien5C., Faure5L., Kazouan5H. J Photochem. Photobiol.
A: Chem. 91 (1995) 137-144.
13. Pozzo5R.L., Baltanas5M.A., Cassano5A.E. Cat. Today. 39 (1997) 219-231.
14. Turchi5C.S., Ollis5D.F. J. Catal. 122 (1990) 178-192.
15. Hoffinann5A J., Carraway5E.R., Hoffinann5M.R. Environ. Sci. Technol 28 (1994)
776-785.
16. Kobayakawa5K., Sato5 C., Sato5Y., Fujishima5A., J. Photochem. Photobiol. A:
Chem. 118 (1998) 65-69.
64
17. Sebate5J., Anderson5M.A., Kikkawa5H., Edwards5M., Hill5C-Gr. J Catal. 127
(1991) 167-177.
18. Abdullah, M., Low5 G., Matthews5R.W. J. Phys. Chem. 94 (1990) 6820-6825.
19. Al-Ekabi5H., Serpone5N. J. Phys. Chem. 92 (1988) 5726-5731.
20. Turchi5 C.S., Ollis5D.F. J Phys. Chem. 92 (1988) 6852-6853.
21. Xu5Y., Langford5 C.H. J Phys. Chem. 99 (1995) 11501-11507.
22. Zhang5Y., Crittenden5J.C., Hand5D.W., Perram5D.L. Environ. Sci. Technol. 28
(1994) . 435-442.
23. Her, R.K. The Chemistry o f Silica. John W iley and Sons. New York. 1979.
24. Gao5X., W achs51.E. Cat. Today. 51 (1999) 233-254.
25. Matthews5R.W. Solar Energy. 38 (1987) 405-413.
26. Raupp5 G.B., N ico5J.A., Annangi5 S., Changrani5R., Annapragada5R. AIChE
Journal. 43 (1997) 792-801.
27. Cassano5A.E., Martin5 C.A., Brandi5R J :, Alfano5O.M. Ind. Eng. Chem. Res. 34
(1 9 9 5 ) 2155-2201.
28. Romero5R.L., Alfano5 OJVL5 Cassano5A.E. Ind. Eng. Chem. Res. 36 (1997) 30943109.
29. Jackson, N.B., W ang5 C.M., Luo5Z., Schwitzgebel5J., Ekerdt5J.G., Brock, J 1R.,
Heller, A. Electrochem. Soc. 138 (1991) 3660-3664.
30. Aguado5M.A., Anderson5M.A., Hill5C.G. J. Molec. Cat. 89 (1994) 165-178.
31. Goswami5D.Y. J. SolarEnergyEngin. 119 (1997) 101-107.
32. W olffum5E.J., Turchi5 C .S .. J. Catal. 136 (1992) 626-628.
33. Davis5A.P., Hao5 O.J. J. Catal. 131 (1991) 285-288.
34. Davis5A.P., Hao5 O.J. J. Catal. 136 (1992) 629-630.
35. Kim5D.H., Anderson5M.A. J. Photochem. Photobiol. A: Chem. 94 (1996) 221-229.
65
APPENDICES
-i
APPENDIX A
FORMIC ACID RUNS DATA
67
0.40 Weight percent Degussa oh Grade 58 Silica Gel. Flowrate 150 mL/min
2.00" R eactor
Tim e (min) ,
0.00
60.00
90.00
120.00
180.00
240.00
270.00
300.00
330.00
2.25" Reactor
TOC
Cone, (ppm)
98.11
83.79
74.37
64.99
47.59
30.07
21.63
13.91
7.66
2.75" Reactor
2.50" R eactor
Tim e (min)
0.00
60.00
90.00
120.00
150.00
180.00 '
260.00
300.00
330.00
Tim e (min)
0.00 ,
. 60.00 .
90.00
120.00
150.00
180.00
210.00
■
245.00
300.00
TOC
Cone, (ppm)
95.70 ..
82.42
74.50
65.65
57.53
47.75
39.26
28.55
12.45
TOC
Cone, (ppm)
99.85
87.76
78.99
70.57
‘6 1.74
52.18
27.33
16.62
1 7.37
T im e (min)
0.00
60.00 ■
120.00
• 150.00 ,
180.00
240.00
462.00
TO C
Cone, (ppm )
99.36
88.78
, 71.27
63.00
52.88
35.21
2.23
68
0.04 Weight percent Degussa on Grade 58 Silica Gel. Flowrate 150 mL/min.
2.00" R eactor
Tim e (min)
0.00
66.00
90.00
120.00
226.00
256.00
285.00
335.00
2.25" Reactor
TOC
Cone, (ppm)
' 101.30
99.26
97.35 '
95.66
88.42
86.99
84.01
80.13
TOC
Cone, (ppm)
99.43
95.43
92.55
88.13
85.47
82.89
78.40
68,93
65.24
61,25
55.17
52.18
2.75" Reactor
2.50" R eactor
T im e (min)
0.00
62.00
91.00
120.00
150.00
183.00
211.00
240.00
270.00
300.00
Tim e (min)
0.00
60.00
92.00
127.00
150.00
180.00
210.00
270.00 .
300.00
332.00
374.00
394.00
TOC
Cone, (ppm)
9 4.16'
86.32
80.94
76.23
70.98
64.43
58.50
53.01
44.39
39.54
Tim e (min)
0.00
60.00
90.00
120.00
150.00
210.00
,
273.00
300.00
TOC
Cone, (ppm )
104.00
94.04
88.14
80.94
76.15
62.06
48.13
40.75
69
0.10 Weight percent Degussa on Grade 55 Silica Gel. Flowrate 150 mL/min.
2.00" R eactor
Tim e (min)
0.00
65.00
95.00
120.00
273.00
391.00
427.00
2.25" R eactor
TO C
Cone, (ppi
98.64
87.65
84.09
79.87
58.04
' 40.58
35.77
2.75" R eactor
2.50" R eactor
Tim e (min)
0.00
61.00
90.00
120.00
213.00
244.00
274.00
353.00
Tim e (min)
0.00
77.00 ,
120.00
152.00
180.00
210,00
276.00
330.00
375.00
400.00 .
TO C
Cone, (ppm )
94.61 ,
83.68
77.21
72.58
67.59
63.11
51.42
42.74
35.00
30.19
TO C
Cone, (ppm)
94.85
86.68
82.08
76.99
60.95
55.19
49.25
34.64
Tim e (min)
. .0.00
60.00
120.00
181.00
240.00
TO C
Cone, (ppm )
98.97
88.61
78.61
68.68
58.14
70
0.40 Weight percent Degussa on Grade 55 Silica Gel. Flowrate 150 mL/min.
2 . 00 "
R eactor
Tim e (min)
0.00
60.00
92.00
136.00
170.00
203.00
241.00
270.00
306.00
396.00
2.75"
Reactor
TOC
Cone, (ppm)
99.30
90.02
84.63 .
76.26
69.28
63.74
56.11
49.50
41.47
21.76
Tim e (min)
0.00
60.00
90.00
122.00
154.00
244.00
374.00
504.00
TOC
Cone, (ppm)
97.76
88.62
83.07
77.41
72.16
53.85 .
27.56
4.26
71
Flow rate D ependence Runs
2.00" R eactor
Flow rate 25 m L/m in
Tim e (min)
0.00
60.00
90.00
120.00
180.00
210.00
270.00
300.00
330.00
TOC
Cone, (ppm)
99.73
87.35
79.30
72.20
55.42
46.69
31.01
23.81
18.51
Flow rate 100 m L/m in
Tim e (min)
0.00
60.00
90.00
120.00
150.00
230.00
290.00
350.00
395.00
TOC
Cone, (ppm)
101.30
90.05
80.19
72.13
62.90
39.55
23.02
8.08
4.70
Flowrate 50 m L/m in
Tim e (min)
0.00
60.00
90.00
120.00
150.00
180.00
210.00
270.00
330.00
430.00
TOC
Cone, (ppm )
107.73
92.88
83.39 .
75.39
65.65
56.79
46.70
31.55
15.21
1.88
Flowrate 150 m L/m in
Tim e (min)
0.00
60.00
90.00
120.00
150.00
180.00
210.00
255.00
370.00
TOC
Cone, (ppm )
98.86
83.06
74.87
66.20
56.75
47.73
38.23
25.50
3.90
72
P25 Slurry Runs
2.0" reactor w /780 ppm P25 slurry.
TOC
Tim e
Cone.
(min)
(ppm)
0.00
95.39
47.00 ■
90.82
94.00
84.04
145.00
. 76.82.. .
182.00
71.05
225.00
66.12
261.00
62.08
324.00
, 53.63
500.00 .
29.19
Tim e (min)
0.00
60.00
126.00
154.00
186.00
217.00
248.00
278.00
393.00
2.50" reactor w /1060 pp m P25 slurry.
TOC
Tim e
Cone.
(min)
(ppm)
0.00
94.26
83.45
61.00
105.00
'7 5 .8 7
137.00
.71.06
66.76
167.00
194.00
61.91
382.00
28.84
515.00 ■
. 5.88
2.75" reactor w /1210 pp m P25 slurry,
TOC
Cone.
Tim e (min)
(ppm)
0.00.
98.01
75.00
83.32
110.00
. 76.58
150.00
68.97
180.00
62.06
240.00
49.74
270.00
44.07
331.00
31.53
2.75" reactor w /2650 ppm P25 slurry.
TOC
Cone.
Tim e
(ppm)
(min)
0.00
. 100.70
98.76
6.50
78.49
106.00
71.28
136.00
60.75
189.00
52.54
228.00
25.40
,
358.00 1
424.00 .
8.67
'
2.25" reactor w /1020 pp m P25 slurry.
.
•’
‘
TOC
Cone.
(ppm)
98.53
87.18
76.40
72.04
67.60
63.20
56.89
53.21
35.36
73
Flow R uns to D eterm ine A ttrition o f Titania
R un I
Tim e (min)
0.00
30.00
60.00
90.00
120.00
300.00
R un 2
TOC
Cone, (ppm)
113.95
108.10
99.93
92.72
83.75
33.28
R un 3
Tim e (min)
. 0.00
60.00
90.00
120.00
240.00
270.00
375.00
Tim e (min)
0.00
30.00'
60.00
150.00
180.00
330.00
TOC
Cone, (ppm)
106.00'
102.00
96.19
71.10
62.74,
' 18.07
R un 4
TOC
Cone, (ppm)
102.90
88.33
79.94
71.32
34.19
24.91
3.63.
R un 5 (A fter 90 Hours o f Flow)
T im e (min)
0.00
60.00
90.00
120.00
150.00
230.00
290.00
350.00
395.00
2.0” Reactor
TOC
Cone, (ppm)
101.30
90.05
80.19
72.13
62.90
39.55.
23:02
8.08
4.70
Tim e (min)
o.oo
60.00
90.00
150.00
180.00.
210.00
' 240.00
' 270.00
395.00 •
TOC
Cone, (ppm)
101:50
88.51 .
81.09 ,
66.33
56.60
47.84
'■ 37.78 ■
28.26
. 2.15
",
APPENDIX B
SPECTROPHOTOMETRIC METHOD FOR THE
DETERMINATION OF TiO? LOADING
75
The amount o f TiOa attached to the silica beads was determined with a
spectrophotometric method adapted from Reference 29. A sample o f coated silica beads
with an estimated 1 - 3 mg o f titania was weighted and added to 8.5 m L o f ammonium
sulfate/sulfuric acid solution (0.4 g o f (NH 4 )aS0 4 per mL H 2 SO 4) and heated at 320 0C
for one hour. This was found to be sufficient to dissolve the titanium present. No
dissolution o f the silica was noted. The liquid and silica beads were allowed to cool and
then quantitatively transferred to a 50 mL volumetric flask (this was done while canting
to leave the silica in the beaker). The flask was filled to exactly 50 mL with a 5/95
solution OfH 2 OZH2 SO4. Six drops OfH 2 O2 was added to a 10 mL sample o f this solution
(sufficient amount for rinsing a cuvette and two runs in the spectrophotometer). The flask
w ith the 10 mL o f solution was weighted before and after the addition o f peroxide to
account for the volume change. Upon addition OfH 2O 2 a yellow complex was formed and
allowed spectrophotometric determination (at 410 nm) o f titanium content against a
standard curve.
To determine the effectiveness o f this method some samples and standards were
send to the Soils Laboratory at Montana State University for inductively coupled plasma
atomic spectroscopy (ICP-AS) analysis. Digested and diluted samples (the 50 mL
dilutions) and standards had to be diluted to approximately 5 percent acid content to be
directly analyzed. Table 9 on the following page details the results from the two methods.
76
Table 9. Comparison o f spectrophotometric and ICP-AS determination o f TiOz loading
on silica gel.
A nalysis
M ethod
Spectrophotom etric
ICP-AS
W e ig h t P ercent Loading on S ilica
Sam ple
1
2
0 .47
0 .4 2
0.40
0 .42
' '
3
0.1 3
0.11
4
' 0 .09
0 .09
MONTANA STATE UNIVERSITY - BOZEMAN
Iiiiii Iii i i i i ni i
762 10366656 4