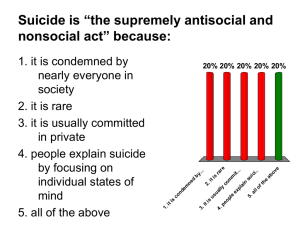
An interpretation of the polarized crystal spectra of mixed metal... salts
advertisement

An interpretation of the polarized crystal spectra of mixed metal trimethylammoniumchlorometallate salts by Martha Jean Lindbeck A thesis submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Chemistry Montana State University © Copyright by Martha Jean Lindbeck (1983) Abstract: Crystals fitting the formula (formula not captured in OCR)where M and M' represent the divalent ions of the first row transition metals Mn, Fe, Co, Ni and Cu have been found to be striking examples of pleochroism. Because these highly colored crystals possess orthorhombic symmetry, their unit cell axes are easily oriented with respect to the electric vector in polarized light. Through observations of the polarized crystal spectra coupled with group theoretical calculations, energy level diagrams have been constructed for the various ions in (formula not captured in OCR) symmetry. In order to explain all the observed peaks transition moment integrals for both electric and magnetic dipole transitions had to be considered. In addition to the spectral work cell parameters for the previously unpublished pure Fe and Ni analogs have been determined using a Supper precession camera. Analyses of the mixed crystals for individual metal content established the relative ease with which these ions were incorporated into the lattice site. AN INTERPRETATION OF THE POLARIZED CRYSTAL SPECTRA OF MIXED METAL TRlMETHYLAMMONIUMCHLOROMETALLATE SALTS by MARTHA JEAN LINDBECK A thesis submitted in p a r tia l fu lfillm e n t o f the requirements fo r the degree Of DOCTOR OF PHILOSOPHY in Chemistry MONTANA STATE UNIVERSITY . Bozeman, Montana March, 1983 D 37g L M _ C o p .^ 11 APPROVAL o f a thesis submitted by Martha Jean Lindbeck This thesis has been read by each member o f the thesis committee and has been found to be s a tis fa c to ry regarding content, English usage, form at, c ita tio n s , b ibliographic s ty le , and consistency, and is ready fo r submission to the College of Graduate Studies. Date Chairperson, Graduate Committee Approved fo r the Major Department IAdr. ^8 __ IW CgA7 // Head, Major Department Date Approved fo r the College o f Graduate Studies "3 Date / - f 2 y y * Graduate Dean iii STATEMENT OF PERMISSION TO USE In presenting th is thesis in p a r tia l fu lfillm e n t o f the req u ire­ ments fo r a doctoral degree a t Montana S tate U n iv e rs ity , I agree th at the Library shall make i t a v a ila b le to borrowers under the rules o f the L ib rary . I fu rth e r agree th a t copying o f th is thesis is allowable on­ ly fo r scholarly purposes, consistent w ith " f a i r use" as prescribed in the U.S. Copyright Law. Requests fo r extensive copying or reproduc­ tio n o f th is thesis should be re fe rre d to U niversity M icrofilm s In te r ­ n a tio n a l, 300 North Zeeb Road, Ann Arbor, Michigan 48106, to whom I have granted "the exclusive rig h t to reproduce and d is trib u te copies of the d is s e rta tio n in and from m icrofilm and the r ig h t to reproduce and d is trib u te by abstract in any fo rm a t." Signature Date a3 CtIanUia. J. _____ H y s ____________ V TABLE OF CONTENTS Page L is t o f Tables.......................... v ii L is t o f Figures........................ v iii A bstract.......................................................................... x INTRODUCTION................................. I EXPERIMENTAL METHODS.............................. 11 Syntheses...................'........................................ .— ................................. 11 Analyses....... .......................................................... 14 Morphology and S tru c tu re ....................................................................... 22 Spectra.................. 28 DISCUSSION.............................................................................. 43 Manganese Spectra..................................................................................... 86 Cobalt Spectra................................................... 94 Iron Spectra............................................................................................... 105 Nickel Spectra.................. 109 Copper Spectra................ 113 CONCLUSION................................................................ 122 LITERATURE CITED.................................... 124 vi LIST OF TABLES TABLE I. II. Page Cell Dimensions and Space Group fo r TMAMCl3 -EH2OS a lt s .. . 6 Inform ation on Metals o f In te re s t fo r the Series TMAMxM ^ xC l 3 -EH2O..................................... 9 Chloride and Metal Content o f the Series TMAMCi 3 =EH2O------ 15 Mole Fractions in Cu Containing S a lts ................ EO V. Mole Fractions in Mn Containing S a lt s .......................... ............ EO V I. Cell Dimensions fo r Members o f the Series TMAMCl3 =EH2O ... E6 III. IV . V II. Theoretical Values o f b. in Square Planar Arrangement fo r TMAMCl3 =EH2O in Pnma............................. .......................... .................. 11 V III. C ell Dimensions o f Some C rystals in the Mn-Cu S ub series.. 53 IX . Cell Dimensions o f Some Crystals in the Mn-Co S ubseries.. 53 X. Colors Exhibited by TMACoxM ^ xC l3=EH2O C ry s ta ls .................... 66 X I. X II. Colors Exhibited by TMAMnxMj xC l 3 =EH2O C ry s ta ls .................. . 70 Russell-Saunders Terms fo r Free Io n s .............................. 76 X III. S p littin g o f R-S Terms in Various Symmetries.......................... 78 XIV. Ground States in Various Symmetries................ '......... ................ 79 XV. Representations o f the Dipole Moment Operator................... 80 XVI. Normal V ib ratio n M odes................................................................. 83 Location and P o la riza tio n o f M n (II) Peaks............ ................... 9E X V II. vi i LIST OF FIGURES FIGURE Page 1. The Two Metal S ites in TMAgMgCl^................................ 4 2. U nit Cell o f T M A M n C l g i n the b-c Plane........................... 7 3. Some Habits Displayed by the Series TMAMxM ^ xClgeEHgO----- 23 4. Location o f C rystallographic Axes and P o la riza tio n D ire c tio n s ............................................................... 28 5. Polarized Crystal Spectra o f TMAMnClgeEHgO........................ 31 6. Crystal Spectrum o f TMACoClgeEHgO in x- or y;-P o la r ized L ig h t........................................... 7. Crystal Spectrum o f TMACoClgeEHgO in z-P o larized L ig h t ... 8. Polarized Crystal Spectra o f TMAMnxC o ^ xClgeEHgO (V is ib le Region).............................................................. 9. 32 33 34 P olarized Crystal Spectra o f TMAMnxC o ^ xC lg»EHgO or TMACoClgeEHgO (IR Region)................................................................ 35 10. P olarized Crystal Spectra of TMAMnxF e ^ xClg-EHgO ( I R ) ------ 36 11. P olarized Crystal Spectra o f TMAMnxN i ^ xClgeEHgO (V is ib le Region)................ 37 12. Polarized Crystal Spectra o f TMAMnxN i ^ xClg-EHgO( I R ) ; . . . 38 13. Polarized Crystal Spectra o f TMAMnxC u ^ xC lg*EHgO (V is ib le Region)........................ 14. 39 Polarized Crystal Spectra o f TMAMnxC u ^ xC lg»EHgO (IR . Region)................................... - ............................................................... 40 15. Unpolarized Crystal Spectrum o f TMAMnxFe^ xClg-EHgO........... 4.1 16. Unpolarized Crystal Spectrum o f TMACoClg-EHgO...................... 42 17. Mole Fraction Plots o f TMAMnM1ClgeEHgO...................................... 45 vi I i FIGURE , ' Page 18. Mole Fraction Plots o f TMACoM'C l 3 » 19. M o le F ra c tio n p io ts o fT M A C u M 1C l 3 ^ H 2O . . . . ........ .................... 47 20. Packing Hole Formed by Four C hlorides.................... 50 21. T e tra g o n a l, In d ic a tric e s ....................................... 22. Double Refraction o f Iceland Spar................ 57 23. Ray D irections and Wave Normals.................................................... 59 24. B ia x ia l In d ic a t r ix ................................................... 61 25. U nit Cell Axes and Bonding D ire c tio n s ....................................... 64 26. Colors Observed in Co-Ni C rystals in Polarized L ig h t........ 68 27. S p littin g o f the d O rb itals in Various Symmetries............... 73 28. C2 Rotation in Normal Coordinates................................................ 82 29. M n (II) Spectral T ra n s itio n s ................... 89 30. Possible Energy Level Diagram fo r C o ( I I ) ................................. 98 31. P referred Energy Level Diagram fo r C o ( I I ) ............................... 101 32. Possible Energy Level Diagrams fo r F e ( I I ) ................ 106 33. Energy Level Diagram fo r N i ( I I ) ................ HO 34. S p littin g o f the d O rb itals in D2h............................. ........... . . 116 35. Energy Level Diagram fo r A2CuCl^ in D2h............................ 117 36. Energy Level Diagram fo r TMACuC13 *2H20 in D2h. ..................... 117 ...................... 46 .56 ix ABSTRACT C rystals f i t t i n g the formula [(CH 3 ) 3NH]MxMj_xC l 3 «2H20 where M and M1 represent the d iv a le n t ions o f the f i r s t row tra n s itio n metals Mn, Fe, Co, Ni and Cu have been found to be s trik in g examples o f pleochroism. Because these highly colored crystals possess orthorhombic symmetry, t h e ir u n it c e ll axes are e a s ily oriented w ith respect to the e le c tr ic vector in po larized lig h t . Through observations of the pola­ rize d crystal spectra coupled w ith group th e o re tic a l c a lc u la tio n s , energy level diagrams have been constructed fo r the various ions in D_. symmetry. In order to explain a ll the observed peaks tra n s itio n moment in te g ra ls fo r both e le c tr ic and magnetic dipole tra n s itio n s had to be considered. In addition to the spectral work c e ll parameters fo r the previously unpublished pure Fe and Ni analogs have been deter­ mined using a Supper precession camera. Analyses o f the mixed crys­ ta ls fo r in d ivid u al metal content established the r e la tiv e ease with which these ions were incorporated in to the la t t ic e s it e . I INTRODUCTION The h is to ry o f a lk y lammonium chlo ro m etallates, as i t appears in the s c ie n tific lit e r a t u r e , dates as fa r back as 1908 a t which time Groth ( I ) published his work on the c o p p e r(II) s a lt [(CH 3 ) 3NH]CuC13 -2H20. This is one member, although a hydrated one, of a series o f compounds represented by the general formula: AmnMXn t2 where Am = an ammonium s a lt M = a metal X = a halogen n = an in teg er such th a t I < n & 4 In 1933 Reniy and Laves (2 ) prepared a large number o f these s a lts using various a lk y l amine hydrochlorides and c o p p e r(II) chloride as s ta rtin g m a te ria ls . For the most part t h e ir work, lik e Groth's , was prepara­ tiv e and d e s c rip tiv e . Except fo r the preceding papers, there is a dearth o f lit e r a t u r e concerning th is series u n til the 1960's. Research in te re s t in these s a lts was renewed a t th is time fo r a number o f rea­ sons. Instrumental technology had caught up with theory in the areas o f magnetics and spectroscopy, and the structures o f these compounds be­ came the focus o f increased in te re s t. Good crys ta ls o f these salts were e a s ily prepared, and thus the series provided a convenient source o f m aterial fo r te s tin g e x is tin g th eo ries. In recent years there has been considerable in te re s t in compounds which contain metals with unpaired d electrons and which are composed . o f lin e a r chains and layers held together p rim a rily by hydrogen bonding and van der Waals forces. Some o f these serve as e x c e lle n t experimental 2 models o f one- and two-dimensional magnetic systems; low temperature heat capacity and s u s c e p tib ility data have been obtained on such com­ pounds and f i t t e d to various th e o re tic a l models. As experimental data accumulated on th is s e rie s , i t was discovered th a t some members displayed a property known as thermochromism. In 1968 Day (3 ) published a paper in which he defined thermochromism as "the re v e rs ib le change in color o f a compound when i t is heated or cooled" and went on to review the e x is tin g lit e r a t u r e on such compounds. Thermochromism can be c la s s ifie d as e ith e r continuous or discontinuous. In the former the color change is gradual and is due to the temperature dependence o f the lin e widths of the absorption bands. In discontin­ uous thermochromism, which is more in te re s tin g chem ically, there is a sharp color change corresponding to some c h a ra c te ris tic temperature. This is due to a change in coordination geometry or ligand conn ectivity which re su lts in an energy s h if t o f the v is ib le absorption bands. Remy and Laves (3) had noted th a t some o f t h e ir c o p p e r(II) s a lts were th e rmochromic,. but beginning in 1964, W ille t t was among the f i r s t to explore the underlying causes o f these observed color changes. . In 1974 he pub­ lish ed a paper (4 ) in which he described two members o f the follow ing series which were e x c e lle n t examples o f discontinuous thermochromism: Am2CuCl4 where Am = (CH3 ) 2CHNHg+ or IPA = (C2H5 ) 2NH2 1- or DEA Crystals o f both o f these s a lts are green a t room temperature and upon heating undergo an abrupt color change to yellow ; a t 43°C and the IPA2CuCl4 changes a t 56°C. . the DEA2CuCl4 changes D iffe re n tia l thermal 3 analysis data and near IR spectra above and below the tra n s itio n temper­ ature suggest a change in coordination geometry o f the CuCl4- species. Comparison o f these spectra with those o f known structures indicated a square planar geometry fo r the low temperature stru ctu re and a fla tte n e d tetrah ed ral geometry fo r the high temperature s tru c tu re . This work has recently been extended to the IPACuClg s a lt (5) which changes from brown to orange a t S l 0C. The corresponding s tru c tu ra l s h if t is from bibridged lin e a r chains o f the dimer CUgClg- to trib rid g e d chains of (CuClg)JI". In conjunction with the in te re s t in the magnetics o f these systems, EPR and s u s c e p tib ility data were collected along w ith the s tru c tu ra l inform ation. In te re s t in the geometry, stru ctu re and re la te d properties o f the s a lts in th is p a rtic u la r series led to the synthesis o f a wide v a rie ty o f c ry s ta ls . In the course o f these in vestig atio n s some s im ila r com­ pounds were discovered which held a s lig h tly d iffe r e n t stoichiom etry than the one previously c ite d . One such class o f compounds which received a tte n tio n is characterized by the formula: TMA3M2C l 7 where TMA = t r i methyl ammonium, (CH3 ) 3NH+ Members o f th is series fo r which M = Mn and Cu have been prepared and studied ( 6 ,7 ) . In th is stru ctu re the d iv a le n t metal ions e x is t in two d iffe r e n t environments; one is characterized by chains o f face-sharing MClg octahedra, the other by discrete MCl4- tetrahed ra. tu ra l arrangement is illu s t r a te d in Figure I . This struc­ 4 Figure I The Two Metal S ites in TMA3M2Cly The existence o f two d iffe r e n t s ite s in such a system leads quite lo g ic a lly to an attempt to grow s a lts with a d iffe r e n t metal in each o f the s ite s . M n (II) and C u (II) w ill re a d ily adopt e ith e r coordination, but other f i r s t row tra n s itio n metals are not so f le x ib le . For example, C o (II) and Z n ( II) p re fe r the tetrahed ral geometry in MCl4" whereas cations such as N i ( I I ) and C d (II) form the octahedral chains more read­ ily . Clay, e t. al_., (7) attempted to grow the mixed metal s a lt with the formula TMA3CoCuCly where C o (II) was expected to occupy the te t r a ­ hedral s ite . Continuous s o lid solutions were obtained ra th e r than a single product with the desired stoichiom etry. Other than th is paper, nothing fu rth e r appears in the lit e r a t u r e in reference to mixed metal 5 s a lts o f th is p a rtic u la r series. The existence o f two d iffe r e n tly coordinated metal s ite s w ithin a sing le stru ctu re is a ra th e r unusual occurrence. The question arises as to whether, under the appropriate conditions, various metals could be forced to assume t h e ir preferred geometries in such a system. This research began as an e f f o r t to prepare such c rystals with the stoichiom­ e try TMAgMM1Cl^ where trim ethyl amine hydrochloride and the s ix possible pairs from the d iv a le n t chlorides of Mn, Co, Ni and Cu were used as s ta rtin g m a te ria l. When attempts to grow these mixed metal s a lts from stoichiom etric amounts in anhydrous solutions o f methanol and ethanol proved unsuccessful, the syntheses were performed in aqueous solution. Q u a lita tiv e analyses of the crys ta ls obtained from the l a t t e r procedure indicated th a t both metals were present in each p a ir. However, quanti­ ta tiv e analyses along with precession photographs of the Mn-Co analog subsequently showed i t to be isomorphic with the compound TMAMnClg-EHgO. A mixed metal product had indeed been prepared, but not one with the desired tw o -s ite .s tru c tu re . The crys ta ls which had been grown had the same stoichiom etry as the C u (II) s a lt studied by Groth in 1908 ( I ) ; these are the hydrated mem* bers o f the series f i r s t prepared and studied by Remy and Laves (2 ) . A lit e r a t u r e search revealed th a t fo r th is hydrated series crystals con­ ta in in g Mn, Co and Cu had been studied in d e t a il, again in connection with th e ir s tru c tu ra l arid magnetic p ro p erties. s a lt was prepared f i r s t ; H is to r ic a lly , the Cu i t s crystal stru ctu re was the f i r s t to be e lu ­ cidated and there is the most inform ation on th is compound in the l i t ­ eratu re ( 8 ) . Heat capacity and s u s c e p tib ility measurements have also 6 been made on both the Cu and Co analogs ( 8 ,9 ) ; only s tru c tu ra l work has been done on the Mn member o f th is series (1 0 ). Of these three s a lts , the Mn and Co analogs are isomorphic; to the Pnma space group. they are orthorhombic and belong The Cu analog is monoclinic as Groth o rig in ­ a l l y reported, and it s space group is PZ1Zc. Cell dimensions and the space group fo r these compounds are reported in Table I . Table I Cell Dimensions and Space Group fo r TMAMClg-ZHgO S alts O . a.(A) O b (A) O c (A) TMAMnCl3 -ZH2O 16.779(3) 7.434(1) 8.Z27(1) Pnma TMACoCl3-ZHgO 16.671(3) 7.Z73(1) 8 . 113(Z) . Pnma 7.864(11) 16.730(Z3) TMACuCl3-ZHgO 7.479(10) Group PZ1Zc The names o f these s a lts are in d ic a tiv e o f th e ir s tru c tu re ; fo r example, the manganese analog is named trim eth yl ammonium ca ten a-d i-p Chlorodiaquomanganese(II) c h lo rid e. It s stru ctu re is composed o f in ­ f i n i t e chains o f edge-sharing MnCl4 (HgO) 2 octahedra which run p a ra lle l to the b^ axis with bridging Cl atoms and O atoms in the trans positions. The chains are hydrogen bonded along the .c axis through the fre e Cl™ anions; the trimethylammonium cations l i e between the planes o f hydro­ gen bonded chains. th is ; The cobalt stru ctu re is completely analogous to the only differences occur in the c e ll dimensions due to the s lig h tly sm aller crystal io n ic radius o f the d iv a le n t cobalt. Figure Z on the follow ing page is a view along the a_ axis o f the u n it c e ll o f the TMAMnCl3-ZHgO s a lt . The chains l i e along the 7 Z Figure 2 N V- U nit Cell o f TMAMnCl3 -ZH2O in the b-c Plane 8 c rys ta llo g rap h ic axis and are hydrogen bonded in the £ d ire c tio n . In the c o p p e r(II) s a lt d is to rtio n o f the CuCl4 (H2O) 2 octahedra re ­ sults in a s lig h tly d iffe r e n t packing arrangement which is s u ffic ie n t to force th is analog o f the series into a d iffe r e n t space group. By con­ ventional choice o f axes in the monoclinic P2^/c group* the chains now run p a ra lle l to a ra th e r than jb as in the orthorhombic c e ll. the bonding patterns remain the same. However, Two o f the coordinated Cl atoms are found a t a s lig h tly g reater distance from the central Cu atom than are the remaining four atoms o f the coordination sphere. Although one angle is now d isto rte d from 9 0 °, the chains remain edge-shared with hydrogen bonding and van der. Waals forces s t i l l in e ffe c t. A ll o f the work done on the trim e th y lammoniurn chlorom etallate ser­ ies has been w ith these three pure metal compounds. A look a t the per­ io d ic ta b le raises the question, can the pattern be completed? That is to say, can the F e ( I I ) , N i ( I I ) and Z n ( II) analogs be prepared, and w ill they adopt one of the above structures? Table I I li s t s the metal atoms o f in te re s t along with t h e ir electron configurations and crystal io nic r a d ii. The values o f the ra d ii are those reported by Shannon and P re w itt (1 1 ). Synthesis o f the series TMAMxM ^ xC l 3 -ZH2O, although inadvertent, yield ed some in trig u in g observations on the chemical and o p tical proper­ tie s o f the pairs as compared to the pure metal end members. Since the mixed metal c rystals in th is series were e a s ily grown, i t was deemed worthwhile to pursue the hydrated s in g le -s ite metal p a ir system in some d e ta il. There are several aspects o f th is series which m erit a tte n tio n . 9 Table I I Information on Metals of. In te re s t fo r the Series TMAMxM|_x C13 -2H20 column a Metal a_ Jb M n (II) 3d5 0.82 Pnma F e ( II) 3d6 0.78 Pnma* C o (II) 3d7 0.74 Pnma N i(II) 3d8 0.70 Pnma* C u (II) 3d9 0.73 P21/c Z n ( II) 3d1 0 ‘ 0.75 ? C d (II) 4d 10 0.95 ? C column b outer shell configuration O crystal io n ic r a d ii (A) fo r high spin six coordinate complex column c space group, a s te ris k s ig n ifie s compound not reported in the lit e r a t u r e Composition and stru ctu re are two o f the areas which were investigated in th is work. These properties are o f p a rtic u la r in te re s t in the Cu containing c rystals since, the pure Cu analog has a d iffe r e n t structure than the other members o f the s e rie s . Examination o f the Cu subseries to ascertain whether these compounds c r y s ta lliz e as the monoclinic P 2 j/c , orthorhombic Pnma or perhaps some new stru ctu re was one o f the goals o f th is research. C rystals o f the Fe and Ni members o f th is series were also synthesized and t h e ir structures determined to see i f they followed the e x is tin g trend indicated by the Mn and Co s a lts . 10 These compounds also e x h ib it a phenomenon known as dichroism, a property o f c rystals which is discussed a t length in a la t e r section. B r ie fly , they display two d iffe r e n t colors depending on th e ir spatial o rie n ta tio n . This dichroism is not to be confused with the discontin­ uous thermochromism mentioned e a r lie r . The color change observed in the la t t e r usually involves a s tru c tu ra l s h if t and is temperature depen­ dent while the colors in the former are a re s u lt o f the o p tic al proper­ tie s o f the crystal and are both v is ib le a t room temperature. The presence o f two metals seems to enhance th is e f f e c t , as the mixed metal s a lts are much more strongly dichroic than are the pure metal end mem­ bers o f th is s e ries . For dichroism to be observable in th is chlorom etallate s e rie s , absorption bands must be present in the v is ib le region o f the e le c tro ­ magnetic spectrum. In conjunction with the o p tical properties manifes­ ted by these c ry s ta ls , spectral data were collected in both polarized and unpblarized lig h t in order to gain a greater understanding o f what governing facto rs control the colors displayed by the various mixed metal s a lts . The question as to whether metal-metal in teractio n s could occur in the mixed metal compounds, thus giving ris e to the s trik in g d i­ chroism o f th e s e .c ry s ta ls , was addressed and a careful comparison o f the r e la tiv e in te n s itie s and locations o f absorption bands in various mem­ bers o f the series was made. 11 EXPERIMENTAL METHODS SYNTHESES C rystals o f s a lts f i t t i n g the formula TMAMXMJ_XC13 - ZH2O were grown fo r eigh t o f the ten possible combinations o f the f i r s t row tra n s itio n elements from Mn to Cu, in c lu s iv e . The pure end members, x = 0 , were prepared by dissolving equimolar q u a n titie s o f the d iv a le n t metal ch lo r­ ides and trim ethyl amineihydrochloride in a minimal amount o f water and allowing the aqueous solution to slowly evaporate. The c rystals which contained metal pairs were prepared by mixing varying mole fractio n s from x = 0 .2 to % = 0 .8 o f the m etal( I I ) chlorides with a stoichiom etric amount of the TMA solution and again merely le t t in g the solution evap­ orate slow ly. I t is c h a ra c te ris tic o f t;he mixed metal series th a t the crystals do not grow with the same stoichiom etry as th a t o f the o rig in a l solu tio n . I f a crystal such as TMAMng 5CUq ^Cl3-ZH2O is desired, the s ta rtin g solution must have a x^n soi n = 0.65 in order to produce a s o lid with XMn x ta l " 0 -5 0. The mole fra c tio n found in the crystal vs^ th a t found in the s ta rtin g solution w ill be tabulated fo r the individual series and discussed in a la t e r section. P e tri dishes o f various sizes and surface areas were u t iliz e d ; practice with the ra te o f evaporation even tu ally led to the optimum methods o f obtaining crystals o f the desired size and q u a lity . Even a f t e r much p ra c tic e , though, there oftentim es seemed to be as much luck as s k ill involved in the process. As these s a lts are extremely soluble in w ater, the solutions were sometimes q u ite viscous before crystals began to grow. This s o lu b ility 12 made i t impossible to obtain crystals o f th is series containing only Zn which has a f i l l e d 3d subshell or calcium, with an empty 3d subshell. Attempts to grow the pure Cd analog y ie ld ed the anhydrous s a lt TMACdCl3. Addition o f excess chloride in the form o f concentrated HCl to the solutions o f the pure Co and Co-Ni mixtures resulted in the formation o f la rg e r crystals in these systems in a shorter period o f tim e. This same e ffe c t was noted as a general ru le fo r the other s a lts as w e ll. No special precautions were taken in the syntheses o f the Mn, Co, Ni or Cu containing s a lts , but in the case o f the F e ( II) s a lt and any mixture in the subseries containing th is m e ta l, a ir oxidation proved to be a problem. Solutions o f i r o n ( I I ) chloride were prepared from the hydrate, the anhydrous s a lt and from iron w ire dissolved in concentra­ ted HCl. Because i r o n ( I I ) is not very stable in a i r , there is the p o s s ib ility th a t some i r o n ( I I I ) is present in solutions made from the i r o n ( I I ) s a lts . For th is reason the most r e lia b le syntheses o f the Fe subseries were those done using the solutions o f F e ( II) prepared from the iron w ire . In order to obtain good q u a lity crystals o f the pure Fe analog and it s mixed metal subseries, the s ta rtin g solutions were placed in a vacuum desiccator containing concentrated HgSO^ as the desiccant and an in e r t nitrogen atmosphere. A successful synthesis of the Fe-Cu mixed s a lt w ith the proper stoichiom etry was never accom­ plished, presumably because o f the high p ro b a b ility o f a redox reaction occurring between these two metals when both are present in the +2 ox­ id atio n s ta te . The Fe-Co analog was also not prepared. The crystals containing iron were collected and stored under a nitrogen atmosphere. 13 Upon exposure to a i r the pure Fe end member began to show signs o f de te rio ra tio n in about a week, undergoing a color change from very pale yellow /green to amber and beginning to look wet. 14 . ANALYSES Once s u ita b le c rystals o f varying mole fra c tio n had been prepared, the next step undertaken involved determination o f the actual s to ic h i­ ometry. Q u a lita tiv e analyses o f each batch indicated th a t both metals were present; q u a n tita tiv e measurements were then necessary to deter­ mine whether the mole fra c tio n s found in the crystal showed any r e la ­ tionship to th a t in the solution from which i t was grown. Because only a lim ite d amount o f these crys ta ls was a v a ila b le , under fiv e grams in most cases, a ll samples were weighed on an M5 microbalance and a ll tit r a t io n s were performed with a 1 . 0 - or 2 .5 -ml microburet. The chloride content was determined by Fajan's method (1 2 ), i . e . t it r a t io n o f Cl" w ith AgNO3 using d ich io ro fIu o re sc e in , DCF", as the in d ic a to r. The AgCl p re c ip ita te ex is ts as a white c o llo id and adsorp­ tio n o f the in d ic a to r anion, DCF", a t the endpoint causes a color change in the p re c ip ita te from white to pink. In d ilu te solutions the AgCl w ill coagulate as w ell as change c o lo r. The DCF" in solution is yellow-green and th is may somewhat mask the white c o llo id a l AgCl p rio r to the endpoint. In order to keep the suspension highly dispersed, and thus the color change more v is ib le , d extrin is sometimes added. I f the analysis is done a t the micro le v e l, th is work in dicated th a t the AgNO3 t i t r a n t should be standardized with and without the dextrin present to make c e rta in th a t th is reagent does not in te r fe r e . The chloride analyses were used to characterize the pure metal s a lts and to estab lish th a t th e ir stoichiom etry and th a t o f the mixed metal s a lts as well were indeed c o rre c t. Table I I I lis t s ! t h e %C1 found and compares these values to those calculated fo r the pure 15 compounds o f the series TMAMxM ^ xC l 3-ZH2O. In the e a rly stages o f th is work c h a rac te riza tio n o f the mixed metal compounds by th e ir chlo r­ ide content was considered, since these analyses can be performed with a r e la tiv e precision o f two or three parts per thousand, ppt. However, i t was subsequently determined th a t th is system is a c tu a lly a s o lid so­ lu tio n whose composition can be contro lled only w ith in a c e rta in range. The growth o f any one batch o f c rystals with a given stoichiom etry has an inherent e rro r of as much as 50 ppt r e la tiv e in it s metal composition The magnitude o f th is v a ria tio n w ith in the c rystals grown from one syn­ thesis makes i t impossible to u t i l i z e the small changes which occur in the %C1 determination to c a lcu late an accurate weighted average o f the two metals present. Since the mole fra c tio n of the metals present in these crystals was o f p a r tic u la r in te r e s t, other a n a ly tic a l techniques had to be considered. Table I I I Chloride and Metal Content o f the Series TMAMCK-2Ho0 M %C1 Found %C1 Calculated %M Found %M Calculated Molar Ratio Mn 41.50 41.32 Z1.46 Z1.34 2.997 Fe ' 41.17 ----- ' Z1.6Z —- Z2.54 40.68 Co Ni 40. Zl 4 0 .7Z ZlJl 22.48 3.059 Cu 40.53 39.98 Z4.41 23.89 2.976 16 - Included in Table I I I are the molar ra tio s o f chlo ride to metal fo r each o f the pure compounds analyzed. These values are q u ite close to the expected value o f 3.000 which indicates th a t the c rys ta ls were ac­ tu a lly the desired stru ctu re and stoichiom etry. The chloride content was not determined fo r the Fe and Co members o f th is series since X-ray data showed these c rystals to also be the orthorhombic Pnma stru ctu re. Because o f the wide range o f mole frac tio n s involved in.any one metal p a ir subseries, methods o f analyses had to be developed not only to determine the metals in question, but also to determine them in the pres­ ence o f one another in a single sampleJ I t is possible to determine a ll o f the metals used in th is s e rie s , Mn, Fe, Co, Ni and Zn, as well as Cu, . complexom etricalIy u t iliz in g . EDTA as the t i t r a h t (1 3 ). EDTA is an ex­ tremely v e rs a tile reagent, forming s ta b le , soluble stoichiom etric com­ plexes with most metal ions, including a ll o f those lis te d above. This very v e r s a t il it y , which makes EDTA such a valuable t i t r a n t , becomes a problem when s e le c tiv ity is desired; in a ll pairs in question the metals w ill in te r fe r e with one another and conditions and reagents must be very c a re fu lly selected in order to obtain r e lia b le data on in divid ual metal content. By judicious u t iliz a t io n o f masking agents, back titr a tio n s and redox reactio n s, various techniques were patched together making i t possible to t i t r a t e both metals in one sample fo r any o f the pairs which did not contain F e ( I I ) . Before describing the procedures used in the mixed metal analyses, the simpler methods o f t it r a t in g a sing le metal ion as in the pure s a lts w ill be discussed. Cu may be determined by forming the ammine complex and t it r a t in g w ith EDTA using murexide as the in d ic a to r (1 3 -1 6 ). The EDTA was 17 standardized against both Cu and Zn solutions prepared by dissolving the respective metal in acid. Welcher (13) recommends a saturated solution o f the murexide, but Elbeih (14) uses a powdered m ixture o f the in d ic a to r and NaCl. Previous work in th is lab (15) suggests th a t the la t t e r method produces a more s a tis fa c to ry endpoint. Elbeih reports good re ­ s u lts when t it r a t in g 25-100 pg o f m e ta l 9 but found th a t fo r samples containing less than 25 pg the endpoint became in d is tin c t. The copper content can also be determined io dom etrically using Na2S2O3 as the t i t r a n t (1 7 ). - Standardizations done using e ith e r Cu ribbon or K2Cr2O7 did not give r e lia b le re su lts a t the micro le v e l; th e re fo re , KIOg was u ltim a te ly chosen as the primary standard to d e te r­ mine the concentration o f the t i t r a n t . The Cu containing crystals were dissolved in doubly d i s t i lle d w ater, buffered with NH^HF2 and t it r a t e d to the starch endpoint. To prevent the endpoint from d r if t in g due to the adsorption o f fre e I 2 on the Cu2I 2 p re c ip ita te , KSCN was added a f t e r the starch. T itra tio n s of the pure Cu s a lt without KSCN gave the same re s u lts , but required more tim e. In comparative t it r a t io n s both the complexometric and the iodometr ic methods y ield ed the same re s u lts ; to avoid in terfe ren ce problems the l a t t e r method was chosen fo r the Cu analyses o f the mixed metal s a lts , since i t is s p e c ific fo r the C u (H ) ion. Samples containing only Ni were t it r a t e d with EDTA by the same procedure described above fo r Cu (1 3 ,1 4 ). The Co t it r a t io n s performed with EDTA also u t iliz e d the murexide in d ic a to r prepared in s o lid form as described above. Welcher (13) provides a f a i r l y d e ta ile d procedure fo r th is t it r a t io n ; additional 18 inform ation appears in Houk1s thesis previously c ite d (1 5 ). The pH must be c o n tro lled very c a re fu lly in th is t it r a t io n in order to obtain the correct color change a t the endpoint. The e asiest way to do th is was to add a c e tic acid or ammonium hydroxide as needed to maintain a yellow color p rio r to the endpoint; a f t e r the endpoint the solution remains purple regardless o f the amount o f acid or base added. The Co t it r a t io n may also be performed as a back t it r a t io n using Eriochrome Black T (EBT) as the in d ic a to r a t a pH of 10 (1 8 ). The Mn determination in the pure s a lt was done follow ing the pro­ cedure ou tlin ed by Welcher (13) except th a t the ZnO and KCN were not added as there were no other metals present to in te r fe r e . Ascorbic acid or hydroxyI amine hydrochloride must be added to prevent the oxidation of M n ( II) . The system is buffered a t pH = 10; tr a tio n is again EBT in the s o lid form. the in d ic a to r in th is t i ­ The M n (II) ion may thus be t it r a t e d d ir e c tly or an excess o f EDTA may be added and a back t it r a t io n performed w ith a standard Zn s o lu tio n . The same basic methods used to t i t r a t e the pure metal compounds . were u tiliz e d in the mixed s a lt analyses w ith some m odifications to pre­ vent any in terfe ren ce s . In the Cu containing subseries the Mn-Cu1 Co-Cu and Ni-Cu pairs were f i r s t analyzed fo r Cu io d o m e trica lly . A fte r the Cu content was determined, the Co-Cu and Ni-Cu pairs were t it r a t e d with EDTA fo r to ta l metal content follow ing the procedure described fo r the pure Cu s a lt . In the Mn-Cu subseries the Mn was subsequently t t - ' tra te d as in the pure s a lt except th a t KCN was added to form the Cu(CN)^ complex, thus preventing the Cu ion from in te r fe r in g . In the Mn-Co and Mn-Ni. subseries to ta l metal was found f i r s t , KCN 19 was added to mask the Co and Ni re s p e c tiv e ly , and the lib e ra te d EDTA was t it r a t e d w ith a standard metal s o lu tio n . The method described by Welcher (13) fo r determination o f both Co and Ni simultaneously proved impossible to d u p licate. A spectrophoto- m etric method described by Kratochvil and H arris was u t iliz e d to fin d the mole fra c tio n s o f these two metals in the Co-Ni subseries (2 0 ). From the t it r a t io n data the r e la tiv e s o lu b ilitie s o f the metals in th is series can be determined. For example, in the Cu containing sub­ s e rie s , the Cu ion occupies the metal s ite o f the crystal most re a d ily . In th is p a rtic u la r subseries the Cu may be regarded as the solvent; Co and Ni are doped in to the Cu s a lt . Mn, Mole fra c tio n data in dicate th a t the M n (II) ion is the most soluble and the N i ( I I ) is the le a s t s o l­ uble o f the three solute ions. The Ni analog was the most d i f f i c u l t of the pure compounds in the e n tire series fo r which to grow crystals and th is trend is re fle c te d in the low s o lu b ility o f the N i ( I I ) ion in the mixed metal compounds as w e ll. In a. solu tion .contain ing equimolar amounts o f the two metals such th a t xCu so-jn = 0 .5 0 , the c rystals which form w ill have Xqu x ta -j = 0.74 fo r the Mn-Cu s a lt , 0.96 fo r the Co-Cu s a lt and 0.99 fo r the Ni-Cu s a lt . The re la tio n s h ip between Xs0] n and xXtal fo r the Cu containing subseries is presented in Table IV ; a graphic presentation w ill be u t iliz e d in the discussion o f these re ­ s u lts in a .la t e r section. Table V l i s t s the corresponding mole fra c tio n data fo r the Mn con­ ta in in g subseries. The Cu is more re a d ily incorporated in to the crys­ ta l stru ctu re than Mn as was observed in the preceding system; displays a g re a ter s o lu b ility than Ni in th is series as w e ll. Co 20 Table IV Mole Fractions in Cu Containing S alts Cu-Mn Subseries Cu-Co Subseries Cu-Ni Subseries XCu, soln XCu, x ta l XCu, x ta l XCu, x ta l 0.2000 0.1887(25) 0.4057(50) 0.6814(14) 0.2500 0.2825(19) , 0.6398(12) 0.8489(12) 0.3333 0.5188(44) 0.8330(14) I 0.5000 0.7500(14) 0.9574(11) I 0.6667 0.9198(17) 0.9640(24) I 0.7500 0.9830 (2) 0.9825(24) I 0.8000 0.9785 (2) Table V I •1 Mole Fractions in Mn Containing S alts Mn-Cu Subseries Mn-Co Subseries Mh-Ni. Subseries XMn, soln XMn, x ta l XMn, xtal XMn, x ta l 0.2000 0.0215(89) 0.3973(29) 0.7567(14) 0.2500 0.0170(138) 0.5181(10) 0.7745 (2) . 0.0802(190) 0.6310 (3) 0.8704 ( 6 ) 0.3333 . . 0.5000 0.2500(43) 0,7694 (2) . 0.9440 (3) 0.6667 0.4812(48) 0.8713 (5) 0.9746 ( I ) 0.7500 0.7175 ( 8 ) 0.8321 (4) 0.9608 (4) 0.8000 0.8112 ( 6 ) 0.9386 (5) 0.9757 ( I ) 21 . There is also a change in stru ctu re in the Cu containing subseries which serves to complicate th is system. The remaining pairs o f a ll the subseries re ta in the orthorhombic s tru c tu re regardless o f metal compo­ s itio n and in a ll cases Nt displays the le a s t tendency to be incorpor­ ated in to the crystal s it e . 22 MORPHOLOGY AND STRUCTURE Before discussing the c o lle c tio n o f X-ray data, the shape or habit th a t these crys ta ls adopt w ill be described. Taken in conjunction with the observed dichroism, the habit can provide inform ation as to the location o f the p rin cip a l axes in a c ry s ta l. This f a c ilit a t e s the mounting and o rie n ta tio n o f in divid ual crys ta ls p rio r to taking pre­ cession photographs. I f no e f f o r t is made to control t h e ir s iz e , stoichiom etric prepa­ rations o f the compounds in th is series y ie ld small a c ic u la r crystals w ith the needle axis p a ra lle l to the chain d ire c tio n . T y p ic a lly , these c rystals are two m illim e ters or less in length and about the thickness o f a h a ir. Slow growth w ill sometimes produce a la t h - lik e habit with dimensions roughly 0 .5 x 1.0 x 2-3 mm. This la th is an external ex­ pression of the u n it c e ll and is bounded by ( 100) and ( 001) ; most of the s tru c tu ra l work w ith the precession camera was done on crystals w ith th is h a b it. The needle axis o f the la th lie s along the b^ c ry s ta llo ­ graphic axis and corresponds to the chain d ire c tio n o f the MCl^(H2O) 2 octahedra. In terms o f chemical bonding and cleavage, th is external morphology is a lo g ic a l outgrowth of the u n it c e ll o f the c ry s ta l. The c c rystallo g rap h ic axis is shorter than the a_ axis and the growth pat­ tern of the laths w ill sometimes r e fle c t th is ; however, there is no p a rtic u la r preference fo r a crystal to develop along the £ axis rath er S than along a. The chain axis coincides w ith the needle axis o f a ll crystals except those in the stru ctu re tra n s itio n region. Very large crys ta ls 10-15 mm in length and several mm on a side 23 which are grown from s o lu tio n s containin g an excess o f C l" develop random faces; h a b its . even the needle axis may be d i f f i c u l t to locate in such Figure 3 shows some o f the ty p ic a l habits fo r th is s e rie s . Figure 3 Some Habits Displayed by the Series TMAMxM^_xC l3*2H20 24 Growth in odd habits was e s p e c ia lly common fo r mixed metal crys­ ta ls which contained Cu. In these subseries a s tru c tu ra l change from the orthorhombic Pnma to the monoclinic P2^/c must occur a t some par­ t ic u la r mole fr a c tio n . In the tra n s itio n region crys ta ls bounded by ( I O l ) 9 ( I O l ) , (H O ) and (H O ) are the ty p ic a l h a b it observed; shown in Figure 3c. th is is In such cases i t was extremely d i f f i c u l t to lo ­ cate the p rin cip a l axes and to o rie n t the crystal fo r X-ray w ork.solely by morphology. A fte r mounting, the p rin cip a l d ire c tio n in such crystals was found more or less by t r i a l and e rro r. Normally, the crystal was mounted with the needle axis coincident with the axis o f the goniometer head and a p rin cip al d ire c tio n located on the basis o f a re fle c te d face. Crystals with the orthorhombic stru ctu re were found to display two colors depending on o rie n ta tio n ; the a-b and b-c planes could be found by. color i f the h a b it o f the crys­ ta l did not happen to contain ( 100) or ( 001) planes which could be lo ­ cated e a s ily . This made i t possible to o rie n t crys ta ls o f any habit in the orthorhombic stru ctu re with a minimum o f e f f o r t . In the Mn-Cu, Co-Cu and Ni-Cu s a lts the habit in Figure 3c predom­ inates when Xqu x ta ] approaches 0 .7 5 -0 .8 0 . The longest dimension in these c rystals is along the a_ axis which corresponds to the chain d i­ rectio n in the monoclinic c e ll. orthorhombic s tru c tu re ; However, these c rystals re ta in the a f u l l set o f precession photographs was re ­ quired to demonstrate th a t these mixtures were indeed c r y s ta lliz in g in the Pnma space group. At xCu x ta i £ 0.80 the Cu containing s a lts w ill c r y s ta lliz e in the monoclinic s tru c tu re . The X-ray data were co llected on a Supper precession camera and 25 Mo Ka ra d ia tio n with X = 0.70926 A was u t iliz e d . The zero, f i r s t and second le v e ls o f the pure Mn and pure Cu s a lts were taken shooting in to the a-b and b-c planes o f lath-shaped c ry s ta ls . These c rys ta ls were subsequently remounted and the zero, f i r s t and second le v e ls of the a-c plane were obtained as w e ll. This provided a complete set o f preces­ sion photographs fo r both the orthorhombic Pnma and monoclinic P 2 j/c s tru c tu re s . As a f i r s t approximation o f s tru c tu re , the zero levels of any crystal o f the mixed metal s a lts in th is series could now be com­ pared to those o f the pure end members to determine whether or not the spot pattern matched one or the o th e r. Full data decks were collected on some, but not a l l , o f the mixed s a lts as a check to make certain th a t no anomalies occurred in the upper le v e ls . The zero and f i r s t le vels fo r two planes were co llected on a t le a s t three mole fractio n s o f each subseries. S alts in the series TMAMClg^HgO where M = Mn, Co and Cu have been reported in the lit e r a t u r e previously (8 -10 ) and t h e ir structures are well documented. Synthesis o f the Fe and Ni end members completes the 5 progression across the f i r s t row tra n s itio n metals from configuration d to d^ in c lu s iv e . Spot patterns on the precession photographs in dicate th a t these two new compounds fo llo w the trend set by the Mn and Co and adopt the orthorhombic stru ctu re with space group Pnma. U t iliz in g the formulas provided by Buerger (21) and Stout and Jensen (22) c e ll dimen­ sions were calculated fo r a ll of the pure compounds from precession data. These values are s lig h tly lower than those reported in the lit e r a t u r e fo r the three compounds on which data is a v a ila b le , but these v a riatio n s are ty p ic a l o f in te rla b comparisons. For the data co llected in th is 26 work, the Fe and Ni analogs f i t the trend developed w ith decreasing s ize o f the d iv a le n t metal cations q u ite n ic e ly . This c o rre la tio n be­ tween the io n ic r a d ii and c e ll dimensions is illu s tr a te d in Table V I. Table VI Cell Dimensions fo r Members of the Series TMAMClg'BHgO Metal Radius a b £ Mn 0.82 16.59 7.419 8.141 Fe 0.78 16.56 ■ 7.331 8.095 Co 0.74 16.52 7.234 8.046 Ni 0.70 16.52 , 7.110 8.031 Cu* 0.73 7.517 7.849 16.70 * axes change in Cu s a lt due to d iffe r e n t space group. One can also c a lc u la te a th e o re tic a l value fo r b^ based on a square planar arrangement o f the chloride anions around the central metal atom and a knowledge o f the ra d ii involved fo r the various ions. Such a O c a lc u la tio n using a value o f 1.81 A fo r the Cl radius y ie ld s c e ll d i­ mensions which are in remarkably good agreement w ith those observed. C Table V II contains these values fo r the d s e rie s . Q through d analogs o f th is 27 Table V II Theoretical Values o f j) in Square Planar Arrangement fo r TMAMCl3- 2H20 in Pnma Metal Radius ]d calc found Mn 0.82 7.467 7.419 Fe 0.78 7.326 7.331 Co 0.74 7.227 7.234 Ni 0.70 7.071 7.110 28 SPECTRA The fin a l phase o f the in ve s tig atio n o f the mixed metal series TMAM^Mj ^Clg'ZHgO e n ta ile d running x.-» and z-p o la rize d spectra as w ell as unpolarized spectra o f these crys ta ls on the Cary 14. When discussing these spectra, z -p o la rize d lig h t re fers to lig h t which has it s only allowed v ib ra tio n d ire c tio n p a ra lle l to the z a x is ; x., and z correspond to the a_, b and jc c rystallo g rap h ic axes in the u n it c e ll. These o rie n ta tio n s are depicted in Figure 4. z(c ) Figure .4 Location o f C rystallographic Axes and P o la riza tio n D irections Crystal spectra were taken a t room temperature in the near in fra re d ( IR ) , v is ib le and u lt r a v io le t (UV) regions. In the pure Mn compound three peaks were observed in the UV region, but in a ll other cases th is region y ie ld ed very l i t t l e useful inform ation as charge tra n s fe r bands appeared which were o f f scale. Most o f the in te re s tin g peaks, were ob­ served in the v is ib le and near IR regions. In order to run spectra on these c ry s ta ls , specimens a t le a s t a 29 m illim e te r wide had to be grown, but they could not be so th ick as to e ffe c tiv e ly block a ll transm itted lig h t . With th is p a rtic u la r series such dimensions proved d i f f i c u l t to a tta in ; a d d itio n a lly , the dark colors and intense absorption bands inherent in some o f the crystals reduced transmission. Of the pure s a lts the Mn analog w ith very weak bands is the most s u ita b le sample on which to take spectra. The pure Co and Cu end members must be cut and ground to get th in enough crystals to produce reasonable spectra. Pure Fe and Ni c rystals large enough to record spectra were never obtained. Among the mixed metal com­ pounds, the e n tire Mn subseries produced crys ta ls which allowed useful spectra to be recorded with a minimum o f e f f o r t . This translucence was due p a rtly to the h a b it, but was probably due in la rg e r p a rt to the d i­ lu tio n e ffe c t imparted to these s a lts by the presence o f the M n (II) ion. Special sample c e lls had to be improvised to hold the crystal in place w hile running so lid spectra on the Cary 14. Brass plates were cut to f i t in to the normal c e ll holder o f the instrument and holes a m illim e te r in diameter were then d r il le d , positioned in the center of the transm itted beam. The c rystals were taped over the hole o f the sample p la te , oriented w ith one o f the c rys tallo g rap h ic axes p a ra lle l to the long dimension o f the p la te . To obtaiin polarized spectra, s trip s o f Polaroid film were taped over the holes o f both reference and sample plates and the o rie n ta tio n and color o f the crys ta l were care­ f u l ly noted. Many blanks were run w ith ju s t the p la te s , the plates w ith polarized film in place and w ith d iffe r e n t pieces o f the film to determine the so rt o f background to expect. The film has weak bands in the v is ib le and near IR , but they are very small compared to the crystal 30 sc atte rin g and cannot be mistaken fo r c rys ta l absorption bands in these regions. The film does absorb in the UV region; t h is , along w ith the presence o f charge tra n s fe r bands, made i t impossible to get polarized UV spectra. i Log In te n s ity Wavelength (nm) Figure 5 Polarized Crystal Spectra of TMAMnCl3 -EH2O Log In te n s ity 32 Wavelength (nm) Figure 6 Crystal Spectrum o f TMACoClyZHgO in x- or y -P o la rize d Light Log In te n s ity 33 Wavelength (nm) Figure 7 Crystal Spectrum of TMACoC I3*ZH2O in Zr Polarized Light Log In te n s ity Wavelength (nm) Figure 8 Polarized Crystal Spectra of TMAMnxCol xCl3- 28^0 (V isible Region) Log In te n s ity 35 Wavelength (nm) Figure 9 Polarized Crystal Spectra of TMAMnxC o^xClg• ZH2O or TMACoClg 'ZHgO (IR Region) Log In te n s ity 36 Wavelength (nm) Figure 10 Polarized Crystal Spectra of TMAMnxF e ^ xCl3 eEH2O (IR) Log In te n s ity Wavelength (nm) Figure 11 Polarized Crystal Spectra of TMAMnxN i ^ xCI3- ZH2O (V isible Region) Log In te n s ity 38 Wavelength (nm) Figure 12 Polarized Crystal Spectra of TMAMnxN i ^ xCl3 -ZH2O (IR) Log In te n s ity 39 — ,--------------------------------------1--------------------------------------h 400 500 600 Wavelength (nm) Figure 13 Polarized Crystal Spectra of TMAMnxCu1^ C l 3* 2H20 (V is ib le Region) Log In te n s ity 40 Wavelength (nm) Figure 14 Polarized Crystal Spectra of TMAMnxCu^xCl3 -ZH2O (IR Region) Wavelength (ran) Figure 15 Unpolarized Crystal Spectrum TMAMnxF e ^ xCl3- ZH2O Log In te n s ity 42 Wavelength (nm) Figure 16 Unpolarized Crystal Spectrum TMACoClEHgO 43 DISCUSSION As mentioned e a r l ie r , th$ o rig in a l in te n t o f th is research was the synthesis of mixed metal crystals o f the tw o -s ite system TMA3M2C l 7 where M and M1 were expected to assume th e ir preferred geometries. Prelim inary in ve s tig atio n o f the c rystals grown in th is attempt in d ica­ ted th a ti although they were not the desired stoichiom etry, they were an in te re s tin g system in t h e ir own r ig h t. Crystals o f the mixed metal series a c tu a lly obtained, TMAMxM ^ xC l 3* 2H20 , whdn observed in the p e tri dishes in which they were grown, displayed two contrasting colo rs, making i t appear th a t two d iffe r e n t products had formed. Closer exam­ in atio n o f in divid ual c rystals revealed th a t both colors were v is ib le in each; subsequently, t it r a t io n data indicated th a t a mixed metal product o f f a i r l y uniform composition had indeed been prepared. The stru ctu re and composition of these mixed metal compounds are now dis­ cussed in d e t a il. Dichroism, the v i s i b i l i t y o f two d iffe r e n t colors w ith in a single c r y s ta l, is discussed in the follow ing section. I n i t i a l analyses o f the c rystals prepared from solutions containing equimolar amounts o f the two m etals, plus a stoichiom etric amount of TMA, suggested a 3:1 preference in moles fo r one metal over the other. The molar r a tio of chloride to metal was monitored and X-ray pictures were taken to v e rify th a t the mixtures were a c tu a lly the stoichiom etry and stru ctu re expected and not double s a lts or defect stru ctu res. Although there is only one metal s ite a v a ila b le in the hydrated s e rie s , the existence o f a reproducible 3:1 metal r a tio in the crystals grown from aqueous solutions with a 1:1 metal r a tio raised the question as to whether these mixed s a lts could in fa c t be s to ic h io m e tric. As 44 the range o f mole fra c tio n s in the s ta rtin g solutions was expanded, and the re s u ltin g products analyzed, i t became evident th a t th is system was a c tu a lly a s o lid solution with c e rta in re s tric tio n s on the mole fra c tio n o f a p a rtic u la r metal which could occupy the metal s ite o f the c r y s ta l. In the experimental section on analyses in Tables IV and V the % values found in the c rys ta ls were compared to those in the s ta rtin g solutions fo r various metal p a irs . Figures 17-19 are graphic representations o f the same data. These mixed metal compounds are described in terms o f s o lid solu­ tio n s . One o f the pure end members may be designated as the solvent . and the remaining tra n s itio n metal ions are solute p a r tic le s . I f a ll the metal cations were id e n tic a l in a ll o f th e ir p ro p e rtie s , and th e ir fre e energies were the same whether in solution or in the crystal s it e , such a system could be used to represent an ideal s o lu b ility curve. In th is s itu a tio n the x p lo t would be a s tra ig h t lin e w ith slope = 1 ; the crystals grown would have the same compositiion as the s ta rtin g solu­ tio n . The observed deviation from lin e a r it y re fle c ts the fa c t th a t the ions are not a c tu a lly id e n tic a l; p ro p erties. they do possess s lig h tly d iffe r e n t The nature and extent o f the deviation fo r d iffe r e n t metal pairs allows one to compare the r e la tiv e s o lu b ilitie s o f the metal ions in th is s e ries . . I f the Cu s a lt is chosen as the s o lven t, the mole fra c tio n o f the solute present in the crystal is represented by: _ v % M1, x ta l nM1 + nCu ' 45 x ta l Mn Subseries x Ml , soln Figure 17 Mole Fraction Plots of TMAMnM'Cl^*ZHgO 46 x ta l Co Subseries Figure 18 Mole Fraction Plots of TMACoM'Cl^'ZHgO 47 x ta l Cu Subseries xM ', soln Figure 19 Mole Fraction Plots of TmACuM1CI3- 48 where n re fers to the number o f moles o f the designated metal ion. Figure 1|9 indicates the re la tio n s h ip between Xjvp Vn the crys ta l and th a t in the solution fo r the Cu containing subseries. a ll three curves l i e below the ideal lin e ; p r e fe re n tia lly in the s o lid phase. I t is noted that in each case Cu is taken up I f one examines the Mn-Cu curve, i t is seen to possess one p o in t, th a t fo r the highest Mn content, which a c tu a lly does l i e on the ideal lin e . Because a ll o f the pure end mem­ bers, where Xm■ - I , do e x is t, a ll o f these curves must approach the ideal case as xM« ^ 1 -0 . Thus, even in the real s o lid s o lu tio n s, the two end points are fix e d ; what happens to these curves between the end points is useful in describing the chemistry o f th is system. The o rig in a l aqueous solutions in the Mn-Cu subseries with xMn=0.20 0.25 and 0.33 produce crys ta ls which e ffe c tiv e ly exclude the solute m etal, having mole frac tio n s xMn xi;a-] = 0 .0 2 , 0.02 and 0.07 resp ectively S tructural determinations u t iliz in g data collected with the precession camera in d icate th a t these mixed metal crys ta ls which are nearly a ll Cu adopt the monoclinic stru ctu re o f the pure Cu analog. The ions whose . pure s a lts grow in the orthorhombic stru ctu re show very l i t t l e to dope in to the monoclinic system o f lower symmetry; tendency only trace a- mounts o f Mn, Co or Ni are found in crys ta ls of the P2^/c space group. When Xjvji in the crystal reaches a value o f about 0 .2 0 , the crystal undergoes a s tru c tu ra l tra n s itio n from P2^/c to Pnma; c h a ra c te ris tic o f th is region is also e x h ib ite d . a d iffe r e n t habit The habit o f the o r­ thorhombic Mn-Cu crystal with the highest Cu content is a d isto rte d octahedron elongated along a_, the chain d ire c tio n in the monoclinic c e l l , but the d ire c tio n o f van der Waals forces in the orthorhombic c e ll 49 This habit was shown previously in Figure 3. In addition to the change in h a b it in th is tra n s itio n region, twinning commonly occurs in the Co-Cu and Ni-Cu crys ta ls as w e ll. I t is in te re s tin g to note how l i t t l e o f the solute metal is neces­ sary to allow the Pnma stru ctu re to predominate. Only 20% o f the metal s ite s in the crystal need be occupied by the solute metal ions to enable the orthorhombic stru c tu re to m anifest i t s e l f . When th is structure p re v a ils , the solute metals whose pure s a lts are orthorhombic enter the crys ta l s ite much more re a d ily than they did in the monoclinic case. T h e ir s o lu b ility increases sharply follow ing the s tru c tu ra l tra n s itio n and. ex h ib its the order: Cu > Mn > Co > N i. The most s trik in g pattern in the observed order is the decrease in the s ize o f the metal cation as the s o lu b ility decreases. Although the q C u (II) ion is not the la rg e s t c a tio n , i t has a d electron configuration and displays a Ja h n -T e lle r e ffe c t when i t occupies the metal s ite o f the c r y s ta l; i t thus tends to d is to rt the regular octahedral s ite and ap­ pears to have a la rg e r radius than the numbers would in d ic a te . This d is to rtio n is s u ffic ie n t to destroy the Pnma symmetry when more than 80% o f the s ite s are involved. Because o f the energy considerations w ith in the c ry s ta l, the M n (II) ion which is the la rg e s t o f the s e rie s , seems to pack b e tte r in th is environment than the other sm all6r cations, ex­ cluding Cu. C o (II) with a sm aller radius would not be expected to sub­ s t it u t e fo r the Cu ion in the la t t ic e s ite s quite as re a d ily as the Mn; th is is indeed what is observed in the Cu subseries. considered, N i( I I ) tic e s it e ; Of a l l the ions is the most d i f f i c u l t to force in to the crystal l a t ­ i t is also the sm allest. The pure Ni end member o f th is * 50 Series tends to form an amorphous powder ra th e r than a c ry s ta llin e soK id,. and th is e ffe c t is observed in the mixed metal s a lts as w e ll. Ni is by fa r the le a s t soluble in the s o lid phase o f the tra n s itio n metals studied in th is s e ries . This e ffe c t may be explained by comparing the s ize o f the metal ion to the hole formed by four chloride anions ju s t touching in a square planar arrangement as shown in Figure 20. Figure 20 Packing Hole Formed by Four Chlorides In order fo r the metal cation to touch these ch lo rid es, it s ionic radius must be 0.75 A or la rg e r; C o(IT) ju s t f i t s . N i ( I I ) with an O io nic radius o f only 0.70 A is too sm all. Since th is cation does not f i t in to the crystal s it e , growth o f the Ni crystal is very d i f f i c u l t to achieve. The la rg e r cations such as Fe and Mn, and Cu as w e ll, tend to push the chloride anions apart when they occupy the S it e 9 thus reducing the e le c tro s ta tic repulsions among the Cl" ions. Absolute values fo r the ra d ii o f the ions involved in th is exer­ cise are impossible to determine. How well the actual numbers in these 51 calculatio ns f i t the observed s o lu b ilitie s is therefore dependent on the choice o f r a d ii one makes. Those calculated by Shannon and P rew itt (11) take in to account the spin and coordination number o f the metals and were therefo re preferred over Pauling's values. As i t turned ou t, these values also in d ic a te qu ite w ell th a t N i ( I I ) is too small to occupy the crystal s ite in th is s e rie s . the trend remains the same. Regardless o f whose r a d ii are chosen, N i ( I I ) is always the sm allest and predic­ ta b ly the most d i f f i c u l t to force in to the crystal s it e . Based on the s o lu b ilitie s observed in the Cu containing subseries and the ra tio n a le proposed to account fo r these observations, one would p red ict th a t in the Mn subseries Cu w ill again be more soluble than Mn, and th a t Co w ill dissolve more re a d ily than Ni with the l a t t e r two both less soluble than Mn. s e rie s . This is borne out in the x plots of these sub­ In the Co subseries both Cu and Mn are more soluble than Co which is indicated by the p o sitiv e deviations from the id e a l.lin e fo r these curves; Ni has a negative deviatio n showing th a t i t once again is the le a s t soluble o f the tra n s itio n metal ions in th is s e ries . A spectrophotometric technique (20) was u tiliz e d to determine both Co and Ni simultaneously in a single sample; th is a n a ly tic a l method was not s e n s itiv e enough to detect very low concentrations o f N i, y ie ld in g xNi x ta l values o f zero fo r xN i^ oln values less than 0 .50. This method does in d ic a te a t what mole fra c tio n in the aqueous solution appreciable amounts o f Ni begin to appear in the re s u ltin g c ry s ta ls . The technique, although not as s e n sitive as the t it r a t io n methods, is s u ffic ie n t to corroborate the e x is tin g s o lu b ility p a tte rn . F a m ilia rity with th is system allows one to a c tu a lly rank a group of 52 crystals from the same subseries according to r e la tiv e metal content by visual examination alone. Although d i f f i c u l t to describe q u a n tita tiv e ­ ly , a gradual, subtle change in color is observed in a group o f crystals in the orthorhombic stru ctu re as mole fra c tio n changes. One must ex­ ercise care in selecting crys ta ls o f roughly the same s ize and q u a lity when doing such a comparison, however. C rystals from solutions which contain progressively more Ni e x h ib it a gradual, but d e f in it e , color change i f they are compared to one another, supplying fu rth e r evidence th a t more Ni is indeed being dissolved in the s o lid s o lu tio n . These q u a lita tiv e observations on the crys ta ls can be q u an tifie d by a nonde-s tru c tiv e method, u t iliz in g the precession photographs. As one d is ­ solves more o f the solute metal ion in to a c r y s ta l, not only the color but also the c e ll dimensions should change gradually as a. re s u lt of the d iffe r e n t sizes o f the two ions. X-ray data in d ic a te th a t th is e ffe c t can indeed be seen. For a p a rtic u la r subseries o f the mixed metal c ry s ta ls , the values of a_» j) and £ should vary in a p red ictab le way with the molar, r a tio o f the component metals. A large d iffe re n c e in ra d ii occurs between the M n (II) and C u (II) ions; thus, th is e ffe c t should be noticeable in th is subseries. However, a s tru c tu ra l tra n s itio n also occurs in th is case which lowers the range o f Xqu which can be studied. Cell dimensions fo r orthorhom­ bic c rystals o f the Mn-Cu subseries w ith xCu = 0.20 and Xqu = 0.75 are presented in Table V I I I on the follow ing page. The Mn-Co subseries also displays a f a i r l y wide range o f mole fr a c ­ tio n s , Xq0 = 0.05 to Xq0 = 0 .6 0 , and there is no change in structure between end members to complicate m atters. Table IX compares the c e ll 53 dimensions o f the extreme x values o f the Mn-Co subseries to those of the pure end members. Table V I I I Cell Dimensions o f Some C rystals in the Mn-Cu Subseries XCu 0.00 Metal . Mn a_ b C 16.78 7.434 8.227 0.20 Mn^Cu 16.64 7.469 8.095 0.75 Mn-Cu 16.59 7.419 7.828 1.00 Cu* 16.73 7.479 ■ 7.864 * Cu axes redefined, a-*b, b->c, c-*a Table IX Dimensions o f Some Crystals in the Mn'-Co Subseries Metal a 0.00 Mn 16.78 7.434 8.227 0.05 Mn-Co 16.76 7.426 8.213 0.60 Mn-Co , 16.61 7.277 8.085 1.00 Co 16.52 7.234 8.046 xCo b C The Mn-Co c rystals seem to f i t the expected trend in c e ll dimen­ sions. The crystal which is nearly pure Mn has values which are very close to those fo r pure Mn, and the crystal with Xq0 = 0.60 has values 54 which are approaching those fo r the pure Co end member. In the case o f the Mn-Cu c ry s ta ls , th a t with a low mole fra c tio n o f Cu f i t s the trend, but the crystal which is predominantly Cu is anomalous. This mole fra c tio n also occurs in the region in which the habit is q u ite d iffe r e n t. The preference fo r th is new habit may be re la te d to some in te ra c tio n a t local s ite s between the Mn and th e .Cu cations which could also give ris e to the break in the pattern o f c e ll dimensions which is noted in th is p a rtic u la r region. An anomaly may well be.expected fo r these crystals in -this region o f s tru c tu ra l and habit tra n s itio n s . For many o f the observed patterns in th is work the C u (II) ion has proven anomalous. The dichroism exhibited by these crys ta ls has been mentioned ear­ l i e r , and i t is th is property above a ll else which f i r s t captures the a tte n tio n and in te re s t o f the observer. One o f the special a ttra c tio n s o f the mixed metal series is the s trik in g v i s i b i l i t y of two and some­ times three d iffe r e n t colors w ith in a sin g le c ry s ta l. This dichroism may be characterized by visual examination o f the c ry s ta ls , by observa­ tio n o f the c rystals w ith a p o la rizin g microscope, or by comparison of polarized crystal spectra in various o rie n ta tio n s . The wavelength at which an absorption maximum occurs has an e ffe c t on the value o f the r e fra c tiv e index, n, determined fo r a c ry s ta l. The index o f re fra c tio n of a substance, defined as the r a tio of the v e lo c ity o f Tight in a vacuum to th a t in the substance, is one o f the most basic physical properties measured by chemists. The substance of in te re s t is usually a liq u id , but may also be a gas or a s o lid . Meas­ urement o f the r e fra c tiv e index o f a c r y s ta llin e s o lid is o f p a rtic u la r in te re s t to the m ineralogist (2 3 -2 5 ); the physical method used to 55 determine n is necessarily d iffe r e n t fo r a s o lid than a f lu id . In addition to the problems associated with a more involved procedure, d iffe r e n t values may be obtained fo r n depending on the o rie n ta tio n i f the s o lid ex h ib its anisotropy. The v e lo c ity o f lig h t m ay.actually vary depending on what d ire c tio n i t is tra v e lin g through the c ry s ta l. Crystals are c la s s ifie d as is o tro p ic or aniso tro p ic. In an iso ­ tro p ic crystal the v e lo c ity o f li g h t , or the r e fra c tiv e index, is the same in a ll d ire c tio n s , as is the case fo r liq u id s and glasses. If a three-dimensional surface in d ic a tin g the value o f the re fr a c tiv e index, n, in any d ire c tio n w ith in the crystal were constructed fo r an is o tro p ic c r y s ta l, i t would be s p h e ric a l; a trix . such a surface is defined as an in d ic - Perhaps the most common example o f an is o tro p ic crystal is ta b le s a lt , NaCl. This is a cubic system, the highest possible symmet­ ry class, and is completely described i f one c e ll dimension is known; to use a chemist's expression, i t has one degree of freedom. On the basis of the s tru c tu ra l symmetry o f NaCl, i t should not be surprising th a t a cubic crystal has only one value o f n. In anisotropic c rystals the v e lo c ity o f lig h t varies depending on the o rie n ta tio n o f the c ry s ta l. Such crys ta ls can be described in terms of t h e ir in d ic a tric e s as e ith e r u n iaxial or b ia x ia l. In uniaxial c rystals there is one, and only one, axis defined as the o p tic axis which is perpendicular to a c irc u la r or is o tro p ic cross section o f the in d ic a tr ix ; in b ia x ia l c rystals there are two such axes. The uniaxial crystal has the higher symmetry and belongs to e ith e r the te tra g o n a l, hexagonal or rhombohedral class. Two degrees o f freedom must be spec­ if ie d to completely define these systems; two edges, a_ and £ , fo r the A 56 f i r s t two and one edge and one angle fo r the la s t one. In a tetragonal system j* = b; thus, symmetry requires a 4 -fo ld p rin cip al axis p a ra lle l to the £ axis o f the u n it c e ll which w ill coin­ cide with the o p tic axis o f the in d ic a t r ix .. longer than or shorter than the a^ and Jd axes. The £ axis may be e ith e r The e llip s o id which represents the in d ic a trix may also be p ro late (n£ > n^) or oblate (n£ > nQ) ; c ry s ta l. or m ineralogists use th is d is tin c tio n to define the sign o f a A tetragonal crystal may be e ith e r uniaxial p o s itiv e , U (+ ), negative, U( - .) , as shown in Figure 21. Figure 21 Tetragonal In d ic a tric e s ’ The sign may a c tu a lly be determined with a microscope w ithout knowledge o f the absolute values o f n and n . e o While the c e ll dimensions define the o p tic a x is , they y ie ld no information on the r e la tiv e values of n. The m in e ra lo g is ts 's most dramatic example o f a u n iaxial crystal is Iceland spar, more commonly c a lle d cal c it e . C alc ite is calcium carbon­ a te , CaCOg, which f a lls in the rhombohedral class. crystal which cleaves n a tu ra lly in to rhombs. I t is .a transparent I f one o f these rhombs 57 is placed over a mark on a piece o f paper, two marks are v is ib le through the c ry s ta l. This is shown in Figure 22. Figure 22 Double Refraction o f Iceland Spar One of the observed images is produced by the ordinary ray, the other by the extraordinary ray. They can be distinguished by ro ta tin g the rhomb as i t lie s on the paper; the ordinary image remains fix e d while the extraordinary image rotates with the c r y s ta l. A close examination o f the two images reveals th a t both appear higher than the paper which is to be expected due to the d iffe re n c e in r e fra c tiv e index between a ir and the c ry s ta l. appears a l i t t l e However, the image produced by the extraordinary ray lower than th a t from the ordinary ray. The d iffe r e n t apparent depth is evidence fo r two r e fra c tiv e indices w ith in a single crystal in th is o rie n ta tio n . This illu s t r a te s the property o f double re fra c tio n exhibited by a ll anisotropic c ry s ta ls . M ineralogists quan­ t i f y th is property by defining a term, b ire frin g e n c e , which is the d i f ­ ference between the maximum and minimum r e fra c tiv e indices. 58 The la te r a l lo cation and ro ta to ry motion o f the extraordinary image depends on the wave nature o f lig h t and the a b il it y o f a crystal to resolve lig h t in to two m utually perpendicular v ib ra tio n d ire c tio n s . Light is electromagnetic ra d ia tio n ; to r, ordinary lig h t has an e le c tr ic vec­ capable o f v ib ra tio n in any d ire c tio n perpendicular to the prop­ agation d ire c tio n . In the cal c ite crystal a ll o f these v ib ra tio n d i­ rections are resolved in to two orthogonal d ire c tio n s . The vib ratio n d ire c tio n which represents t normal to the o p tic axis is th a t o f the ordinary ray. The E vector o f the ordinary ray is in the plane o f the c irc u la r cross section o f the in d ic a tr ix ; th is means th a t the e le c tric f ie ld o f th is ray in te ra c ts with th a t o f the electrons in the crystal in the same fashion in any d ire c tio n w ith in the a-b plane. The ordin­ ary ray propagates through the crystal as i f i t were i is o tro p ic . The E vector o f the extraordinary ray is perpendicular to th a t o f the ordinary ray and in the same plane as the optic a x is . By symmetry the atomic arrangement along c is d iffe r e n t than th a t along a or Jd. The vibration s due to the electrons in th is asymmetrical environment in te rfe re with t o f the extraordinary ray and produce a re s u lta n t vector which is no longer exactly perpendicular to the fay d ire c tio n . . However, the propagation d ire c tio n must s t i l l be perpendicular to the E vector; th is so-called wave normal no longer coincides with the ray as i t does in the ordinary ray. dinary ray s lig h t ly ; rhomb. This in te ra c tio n bends the path o f the extraor­ thus, two images appear as lig h t e x its the cal c ite The paths of these two rays are shown in Figure 23. 59 ordinary ray . extraordinary ray Figure 23 Ray D irections and Wave Normals The re s o lu tio n , i . e . the distance between these two images, is d i­ re c tly proportional to the birefringence and to the thickness of the c ry s ta l. In cal c ite lig h t tra v e ls fa s te r along the op tic a x is ; th is case, n^ < nQ. in This means th a t n£ is closer to nai-r than nQ so th a t the extraordinary image should appear lower than the ordinary one which is indeed the case. A th ic k e r rhomb w ill cause the two images to appear fu rth e r apart due to the longer path length the two rays fo llo w . This same process occurs in a ll anisotropic c ry s ta ls . Cal- c ite is unusual in th a t i t is transparent and has an extremely high b irefrin g en ce. Quartz w ill also form two images, but a crystal 15 times as th ic k is required to observe the same separation o f images. I t is possible to observe anisotropic crystals with lig h t which . has only one allowed v ib ra tio n d ire c tio n ; such lig h t is " p o la riz e d ." Plane polarized lig h t may be obtained in a number of ways; some 60 crys ta ls such as b io t it e or tourmaline n a tu ra lIy p o la rize lig h t . A Nicol prism often used in p o la rizin g microscopes consists o f a cal c ite crystal which is oriented so th a t only the c ry s ta l's extraordinary ray is allowed to pass through.. Large sheets of P olaroid, the substance which was used to obtain the polarized spectra in th is work, are made by embedding oriented c rystals o f herapathite in a p la s tic sheet. Even lig h t re fle c te d from a b rig h t, nonmetal surface is polarized to some e x ten t. I f the two images produced by cal c ite are.observed through a po­ la r iz e r as the rhomb is ro ta te d , each one w ill disappear or extinguish a lte rn a te ly a t 90° in te rv a ls . The plane o f the v ib ra tio n d ire c tio n of the ordinary and extraordinary rays can thus be determined. I f there is no vector component o f the ordinary ra y , th a t image w ill not be seen; i t extinguishes or is a t an e x tin c tio n po sitio n . There are two such positions fo r the extraordinary ray as w e ll, 180° apart and orthog­ onal to those fo r the ordinary ray. A ll the properties th a t have been discussed fo r un iaxial crystals are also present in b ia x ia l c ry s ta ls , but the la t t e r show other e ffec ts as well due to t h e ir g reater complexity. Crystals which f a l l into the orthorhombic, monoclinic or t r i c l i n i c classes are b ia x ia l. crystals the in d ic a trix is a t r i a x ia l e llip s o id ; fe re n t value fo r n. For such each axis has a d i f ­ The two extremes are denoted as n^ fo r the lowest value and n^ fo r the highest value. The b ia x ia l in d ic a trix has two op tic axes which are normal to c irc u la r cross sections whose ra d ii rep­ resent n^, the interm ediate re fra c tiv e index fo r b ia x ia l c ry s ta ls . The location o f these indices w ithin the in d ic a trix is illu s tr a te d in 61 Figure 24. n Y n a radius of c ir c le = n Figure 24 B iaxial In d ic a trix In orthorhombic c rystals symmetry requires th a t the three axes of the in d ic a trix coincide w ith the three crystallographic axes, but no re ­ s tric tio n s are placed on the actual mapping, i . e . the shortest c ry s ta l­ lographic axis need not correspond to n . is fy th is requirement; found by experiment. There are s ix ways to s a t­ the correct one fo r a p a rtic u la r system is In the monoclinic system one o f the 2-fo ld axes o f the in d ic a trix w ill coincide with the unique axis of the c r y s ta l, In the t r i c l i n i c system any o rie n ta tio n is possible. The differences in symmetry along the various axes in anisotropic crys ta ls are b a s ic a lly a re s u lt of d iffe r e n t atomic arrangements. This resu lts in the v a ria b le re fra c tiv e indices observed in such c ry s ta ls . The in te ra c tio n o f the crystal with lig h t is also what determines the color o f a c r y s ta l. An anisotropic crystal is therefo re capable of displaying, d iffe r e n t colors depending on the o rie n ta tio n and d ire c tio n in which i t is observed. I f a ll three axes in a b ia x ia l crystal have 62 a d iffe r e n t associated c o lo r, the general term fo r the phenomenon is pleochroism. I f only two colors are involved, i t is ca lle d dichroism. The la t t e r term is often used fo r a ll cases, since in any one o rie n ta ­ tio n o f the crystal only two colors can be observed. Tourmaline which grows as elongated prisms is an example o f a d ichroic c ry s ta l; rized lig h t . i t has been mentioned previously as a source of pola­ This mineral allows lig h t v ib ra tin g p a ra lle l to the prism axis to pass through w hile completely blocking lig h t v ib ra tin g normal to the prism a x is . It s dichroism may be described as black and w hite. Two c rystals of tourmaline with th e ir prism axes orthogonal w ill not allow any lig h t to pass. Most anisotropic crystals are dichroic or pleochroic to some ex­ te n t, but cases among minerals strong enough to see, p a rtic u la r ly with contrasting c o lo rs, are very ra re . is one such example; Andalu s ite , an aluminum s ilic a t e , some crystals of th is mineral which contain a small amount o f im pu rities are y ello w , green and red along the p r in c i­ pal axes o f it s in d ic a tr ix . When viewed along any p rin cip a l a x is , the color observed is the sum o f the colors o f the other two axes. In colored substances such as the dichroic and pleochroic minerals discussed above, the re fra c tiv e indices have unusually high or low v a l­ ues due to the presence of absorption bands in the v is ib le region. Because the r e fra c tiv e index is a function of wavelength, X, values of n are reported with reference to some known wavelength o f lig h t used to make the measurement, fo r example the Na D lin e . The follow ing equa­ tio n can be used to describe the re la tio n s h ip between the re fra c tiv e index, n, and wavelength, X (26): 63 A1 X2 • n2 = I + -------------- ■ where A1 is a constant and X1 is an absorbing wavelength. (I) I f a sub­ stance has more than one absorption band, addition al terms involving X are added. quation. There are three cases which can be considered fo r th is eI f the wavelength used to determine n is much greater than the absorbing wavelength, X » X1 , Equation I reduces to: n2 = I + A1 = k where k is the d ie le c tr ic constant. ( 2) This equation generally holds fo r gases and dispersion is not usually observed. I f X > X1 , but the r a tio X1ZX is sm all, Cauchy's Theorem fo r d is ­ persion re s u lts : 2 A, * ? n - I + A, + ----- (3) 1 X^ Let A = I +A1 B = A1X1 2 B n =A + — ^ X^ which is Cauchy's Theorem. (4) I f n is p lo tted against X, a normal dispersion curve re su lts from Equa­ tio n 3 with n increasing gradually as X decreases. As the wavelength used to measure the re fra c tiv e index approaches the absorbing wavelength. Equation I requires th a t n become very la rg e , going to i n f in i t y a t X = X1 . Since th is cannot a c tu a lly occur, the expression does not adequately define n a t th is p o in t. A damping term must be added to the equation in order fo r i t to apply to a ll values of 64 A, including absorption points (2 7 ). The Mn, Fe, Co and Ni s a lts as well as th e ir mixed metal analogs prepared in th is research a ll c r y s ta lliz e in the orthorhombic class with space group Pnma, and are thus examples o f b ia x ia l c ry s ta ls . Be­ cause they are highly colored, they possess v is ib le absorption bands which w ill give ris e to anomalous dispersion curves. is t ic s , These character­ lower than cubic symmetry and absorption bands in the v is ib le region, are two requirements a crystal must meet in order to be dichroic . The th ird and cru c ia l requirement is th a t the local s ite symmetry o f the chromophore be oriented or aligned in the u n it cel I so th a t the dichroic e ffe c t is not cancelled in the same way as o p tic al a c t iv it y in a racemate. The MCl^(H2O) 2 octahedra in the Pnma stru ctu re and in the P2-|/c stru ctu re as well also meet th is th ird requirement; th e re fo re , the c rystals in the TMAM^Mj_^C ly2H 20 series are d ich ro ic . The stru ctu re and c e ll dimensions o f these compounds have been discussed e a r lie r . To review b r ie f ly , the axes and corresponding bonding d irectio n s are shown in Figure 25. z (c ) x(a) Figure 25 U nit Cell Axes and Bonding D irections 65 Of the pure orthorhombic s a lts , the Co analog displays the most re a d ily v is ib le dichroism without the aid o f a p o la riz in g microscope, although the c rystals must be selected ju d ic io u s ly fo r the colors to be observed. L ite ra tu re reports these c rystals to be blue in ^ -p o la rize d lig h t propagating along the a^ a x is ; c rystals appear r e d -v io le t. th is work. in a ll other o rien tatio n s these These observations have been confirmed in The dichroism o f the Mn and Cu analogs has not been re ­ ported in the lit e r a t u r e previously. Crystals in the mixed metal subseries containing Co are a ll s t r i k ­ in g ly d ich ro ic ; Table X summarizes th e ir behavior when observed in po-^ la riz e d and in unpolarized lig h t . Light designated as ^ -p o la rize d has it s allowed v ib ra tio n d ire c tio n , which corresponds to ? , p a ra lle l to the a_ c rys tallo g rap h ic a x is , s im ila rly fo r the jD- and ^ -p o la rize d lig h t . I f the Co containing compounds were observed only in unpolarized lig h t , these crys ta ls would be classic examples of dichroism and pleochroism. In a crystal o f the Mn-Co s a lt the shorter wavelengths of v is ib le lig h t polarized in the ^ or Ib d ire c tio n are absorbed, and the crystal appears r e d -v io le t. When c_-polarized lig h t is propagated through the b-c plane o f the c r y s ta l, the longer wavelengths are ab­ sorbed, and the crystal appears blue. The iso tro p ic behavior in the a-b plane can be ra tio n a liz e d on the basis o f s tru c tu re , since the metal ion giving r is e to the absorption band is in an e s s e n tia lly square p la ­ nar environment with the the surrounding Cl" ions. In the b-c plane, however, the metal is bound to HgO in one d ire c tio n and to Cl in the o th e r, qu ite a d iffe r e n t atomic environment and thus a d iffe r e n t color may be expected fo r c -p o la r i zed lig h t than fo r b -polarized lig h t . In 66 Table X Colors Exhibited by TMACoxMJ ^ ly Z H g O Crystals Crystal P o la riza tio n pure Co a-b Face b-c Face none re d -v io le t blue a re d -v io le t b re d -v io le t blue C Co-Mn none red a . red b red . none red a lig h t red b dark red none dark red a dark red b dark red c red blue dark red blue C Co-Cu blue blue C Co-Ni r e d -v io le t green dark red green 67 such a stru ctu re dichroism seems to be a lo g ic a l r e s u lt. With the aid o f an instrument pleochroism can be demonstrated fo r these s a lts which are a c tu a lly orthorhombic in symmetry. I f the p o la riz e r is removed, the color o f the b-c face should be r e d -v io le t, the re s u ltin g combination o f red and blue. the case. This is not The b-c planes o f the pure Co and the Mn-Co mixed s a lt are both blue when viewed e ith e r in ^ -p o la rize d lig h t or unpolarized lig h t . This is not normal dichroic behavior. The Cu-Co s a lt shows the same anomalous persistence o f the color green in it s b-c plane. In the section on the spectra o f these compounds which fo llo w s , i t is noted th a t the red absorption bands are indeed present in the unpolarized spectra. I t seems th a t the red colo r along the Jb axis is not intense enough to mask or even to mix appreciably with the blue color of the £ a x is . This problem is discussed in d e ta il below. The Co-Ni crys ta ls are v is ib ly pleochroic without the aid of an instrument. With polarized lig h t propagating perpendicular to the a-b plane, two shades of red are observed. This is re a d ily apparent i f two c rystals w ith th is o rie n ta tio n are placed a t r ig h t angles to one another as in Figure 26. The crystal with it s j) axis p a ra lle l to t is dark red whereas the crystal with it s £ axis p a ra lle l to t is a much lig h te r red. In a d d itio n , i f the p o la riz e r is ro ta te d , one red fades and the other darkens, u n til a t 45° the two shades o f red appear iden­ tic a l. This same e ffe c t is seen in the b-c face of a c rys ta l with the colors dark red and blue; a t 45° both crys ta ls appear purple. With these c rys ta ls i t is d i f f i c u l t to be c e rta in th at the red is ac­ tu a lly dark red due to o rie n ta tio n , because the c rystals tend to be 68 ■' c a t 0° same same < v io le t a f t e r ro ta tio n o f 45° a fte r ro ta tio n of 90° Figure 26 Colors Observed in Co-Ni Crystals in Polarized Light 69 th ic k e r along c than along _a which w ill also deepen the color observed. However, symmetry requires th is red to be the dark red observed in the a-b face. With three d iffe r e n t colors v is ib le depending on o rie n ta ­ tio n , th is system is a classic case o f pleochroism. In unpolarized lig h t the blue color again p e rs is ts . Spectral data in d ic a te th a t two d iffe r e n t reds ought to be d is tin g u is h a b le , but unpolarized spectra do not show only blue peaks fo r the b-c plane. The subseries containing Mn also ex h ib its these phenomena; colors are tabulated in Table X I. the Both the Mn and Mn-Ni analogs are pink in a-p o larize d lig h t and c le a r in depolarized lig h t . . In ^ -p o la ­ rize d lig h t the pure Mn crystal remains c le a r, but the Mn-Ni mixed metal crystal appears yello w . the Co-Ni analog. The l a t t e r is thus v is ib ly pleochroic as is However, i t is extremely d i f f i c u l t to distinguish between the yellow and c le a r o rie n ta tio n s unless the crys ta ls are about fiv e m illim e ters th ic k or there are c rys ta ls the same s ize with which to compare colors. The same technique used to separate the two reds in the Co-Ni case can be applied to th is system. The Mn-Cu analog is dichroic in the true sense o f the word as the m ineralogist uses the term. Two colors are v is ib le in polarized lig h t ; amber in ar or b^-polarized lig h t and green in ^ -p o la rize d lig h t . In unpolarized lig h t the color observed is a mixture o f the two seen in the two polarized d ire c tio n s . This may also be true of the Mn and Mn-Ni s a lts , but i t is impossible to t e l l v is u a lly because one o f the "colors" involved is transparent. The o p tical s p e c t r a ,p a r t ic u la r ly in the v is ib le region, o f spe­ cies w ith tra n s itio n metal ions are generally described in terms of d-d 70 Table XI xM i-xCV 2H2° Colors Exhibited by TMAMn Crystal P o la riza tio n pure Mn none a-b Face pink a pink b c le a r none pink a pink b c le a r none pink a pink b yellow none amber a : I t amber b dk amber C c lear c le a r c le a r y e l I ow yellow c le a r C Mn-Cu c le a r c le a r C Mn-Ni b-c Face c lear C Mn-Fe Crystals amber dk amber green 71 tra n s itio n s which have energies on the order o f IO^ cm""*'. The in ten ­ s itie s o f peaks due to d-d tra n s itio n s are weak as a consequence o f th e ir v io la tio n o f the selection ru le , A l = I l ; in complexes which contain high spin M n (II) these tra n s itio n s are spin forbidden as w e ll. In the high energy v is ib le and UV regions these d-d peaks may be hidden by the more intense chloride to metal charge tra n s fe r bands. Three d iffe r e n t approaches have been u t iliz e d to explain the ob­ served spectra o f tra n s itio n metal complexes; these are crystal f ie ld theory, CFT, ligand f ie ld theory, LFT, and molecular o rb ita l theory, MOT. The f i r s t assumes th a t the central metal ion and a ll surrounding ions are point charges which undergo purely e le c tro s ta tic in te ra c tio n s ; no overlap o f o rb ita ls is allowed to occur. LFT is a refinement on th is and allows some in te ra c tio n or overlapping of o rb ita ls to occur between the metal and neighboring atoms. In the la s t approach the atomic o rb ita ls o f the ligands and the metal in te ra c t to form molecular, o rb ita ls which may be constructed by group th e o re tic a l methods so as to a tta in maximum overlap. Because o f the nature o f the metal ions and ligands involved in th is work, CFT was selected as the best approach to explain the ob­ served spectra. Whatever approach is used, the in te rp re ta tio n o f the spectra requires some knowledge o f the i n i t i a l and fin a l s ta te s .o f the system. The to ta l energy o f a normalized wavefunction is expressed by Equation 5: E = J V f H ip d? = <^|H|^> The Hamiltonian op erator, H, fo r a many electron wavefunction which could be used to describe a tra n s itio n metal ion is a sum o f several (5) 72 terms: r Z Vt 2m z Z V, ( 6) .i> j r U Z V. (7) The f i r s t three terms are the k in e tic energy of the electrons', the coulombic a ttra c tio n between the nucleus and the electrons and the in te re le c tro n ic repulsions, re sp ec tiv e ly . Because these terms, are iden­ tic a l fo r ions with a 3d subshell, they may be collected and defined as H°. CFT defines V^. as the p o ten tial f ie ld introduced by a p a rtic u la r environment such as octahedral, Vo c t, or te tra g o n a l, Vt e t . In the metal complexes considered here, V. is small compared to the e lectro n electron repulsion term, arid may thus be treated as a perturbation of H°. To determine the r e la tiv e energy level s p littin g o f the d o rb ita ls in various complexes only the e ffe c t o f V. need be considered. The methods of group theory (28-30) provide a powerful tool in the analysis o f spectra without recourse to the tedious computations which solving Equation 5 e n ta ils . tonian; Symmetry operator commute with the Hamil­ i f the d ire c t product o f the characters of the terms in any in ­ tegral o f the form <ip|0 |^> where 0 is an operator does not contain the t o t a lly symmetric representation, the in te g ra l has a value o f zero. Use of the symmetry o f a complex and the character ta b le fo r it s assoc­ ia te d point group y ie ld s a great deal o f information about the possible tra n s itio n s which may be observed. The fiv e d o rb ita ls o f the tra n s itio n metal ions may be chosen as a basis s e t; metry. these are a ll degenerate in the free ion in spherical sym­ I f the metal ion is surrounded by ligands, the fiv e -fo ld 73 degeneracy is removed in a way which depends on the symmetry o f the com­ plex. When six id e n tic a l ligands are coordinated in octahedral sym­ metry, the point group fo r the complex is . In symmetry the fiv e degenerate d o rb ita ls are s p li t into two energy levels designated e and 9 2 2 2 tgg which contain the z , x -y and xy, xz, yz d o rb ita ls re sp ec tiv e ly . I f two axial ligands are removed or replaced, the symmetry of the com­ plex is lowered to D ^ . In th is environment a ll degeneracy is removed from the octahedral eg le v e l; (dx2_y2). i t s p lits in to an aig ( dz2) and a b ^ The tgg level becomes a b2g( dXy ) and a doubly degenerate eg(dxz and dy Z)* I f the symmetry is lowered fu rth e r to the point group, a ll degeneracy is removed so th a t each d o rb ita l has it s own as­ sociated energy level and is described by a one-dimensional representa­ tio n from th is group. . These s p littin g patterns are shown in Figure 27. Spherical . D, lig b ag dz 2 ag d*V Ig Zg b d b 3g d yz Zg Figure 27 d xy XZ S p littin g o f the d O rb ita ls in Various Symmetries 74 A ll of the above s p lit t in g assignments may be obtained d ir e c tly from the appropriate point group or can be derived by performing the operations o f the point group on the in divid ual d o rb ita ls and comparing the re s u l­ tin g representations with those o f the group. Group theory indicates the possible energy le v e ls , but does not enable one to estab lish the extent o f the s p littin g and thus the r e la ­ tiv e energies o f the re s u ltin g le v e ls . This s p lit t in g may be deter­ mined w ithout a c tu a lly solving Equation 5 in part by the location o f the ligands w ith respect to the lobes o f the d o r b it a ls , but is also depen- dent on the nature o f the ligands involved. Ligands are described as weak or strong f ie ld based on, the magnitude o f the s p lit t in g which they induce between energy le v e ls ; the octahedral s p littin g between eg and tgg is defined as A or IODq. Complexes containing weak f ie ld ligands generally possess high spin, because less energy is required to occupy the higher o rb ita l than to p a ir spins in the lower one. In the c rystals involved in th is work the metal ions are octahe­ dral Iy coordinated, M C l^ ^ O ^ , but with water molecules occupying the axial s ite s , the local metal ion symmetry is a c tu a lly D ^ . and the chloride anion are designated as weak fie ld , ligands. suggests th a t the s p littin g s which a ris e due to compared to IODq. Both water This symmetry are small I f th is is tru e , the p o ten tial term in the Hamilto­ nian due to the presence o f a tetragonal f i e l d , a perturbation o f the octahedral case. , may be treated as With th is assumption one can construct an energy level diagram fo r a complex in O^ symmetry and sub­ sequently apply group th e o re tic a l methods to determine the possible s p littin g patterns in lower symmetries. 75 Before tra n s itio n s can be assigned, the ground and excited states must be described in terms o f the possible states fo r the various sym­ m etries of in te r e s t. The ground s ta te fo r the free metal ions may be determined according to Hund1s Rule; in order to fin d a ll possible Russel I -Saunders(R-S) terms fo r the excited s ta te s , a ta b le o f states in d ic a tin g the number o f determinants fo r each Ml -Ms combination must be constructed. 7 As an example, fo r the d 8 or d ions in which two electrons or two holes, positrons, re s p e c tiv e ly , are placed in the fiv e o 101 d o rb ita ls with e ith e r o f two spins, there are C10 = = 45 possible s ta te s . With the aid o f an Ml -Ms ta b le these are resolved in to fiv e energy le v e ls , the R-S terms ^G, ^F, *D, terms Hund's Rule designates 3 and *S. F as the ground s ta te . Of these fiv e Table X II summar­ izes the number o f states and the term symbols fo r each configuration of the d o r b ita ls . The ground s ta te is lis te d f i r s t in each case. By applying the symmetry operations of any point group to an o r­ b i t a l , one can determine it s representations in th a t p a rtic u la r group. The d i f f i c u lt y of the ca lc u la tio n depends upon the o rb ita l involved; op­ e ratin g on the d o rb ita ls is a cumbersome task. Fo rtun ately, there is a method which allows the representation o f an o rb ita l to be w ritte n knowing the characters of only the ro ta tio n symmetry operators. Using the transform ation m atrix fo r ro ta tio n by an angle, a , the trace is given by Equation 8 : X(o) - I n=0 e( l- n ) ia = .sin (H%)o; (8) sin When a = 0 , which corresponds to the id e n tity operation, the trace is 21+1. The reducible representation can thus be found in any symmetry 76 Table X II Russell-Saunders Term fo r Free Ions Configuration QOO CL PO d1 (d9) Term Symbols Number of States 10 2D 45 3F, 1G, 1D, 3P, 1S d3 (d7 ) 4 F, 2F, 2H, 2G, 2D, 4 P, 2P 120 d4 <d6) 5D, 3D, 2x1D, 1I , 3H, 3G, 2x 1G, 210 2x 3F, 1F, 2x 3P, 2x 1S d5 6S, 2S, 2 I , 2H, 4 G5 2x 2G, 4 F 5 2x 2F, 252 4D, Sx2D, 4P, 2P fo r any o r b ita l by c a lc u la tin g the character of the various rotation s from Equation 8 . The states described by the irre d u c ib le representa­ tions are designated g i f they possess a center o f in ve rs io n , u i f they change sign upon inversion. The m atrix above is derived from eim^ , where the quantum number, m, may take on 21 + I values fo r the o r b ita l involved. S im ila rly , the wavefunction fo r a R-S term may be expressed as e "*^ , where M may have 2L + I values. This correspondence enables one to use the symmetry of the o rb ita ls to define the states in to which the analogous R-S terms are 77 s p lit in various environments. These representations are tabulated fo r Oh , D4h and D2h symmetries in Table X I I I . 2 8 3 In the d example^ which is equivalent to d , the. F ground state o f the fre e ion w ill s p li t in to A2g + B^g + B2g + 2Eg in a tetragonal f ie ld . Although the f o rb ita ls are antisym metric, the basis set o f d o rb ita ls is symmetric; th e re fo re , regardless o f the spin o r b it coupling introduced by F, the overall symmetry of the e le c tro n ic wavefunction must re ta in it s g character fo r any d configuration . I t is again pointed out th a t group th e o re tic a l calculatio ns only in d ic a te which en­ ergy le v e ls may be present in a given environment; these predictions reveal nothing about the r e la tiv e s p lit t in g . As a general ru le the R-S terms o f excited states are ranked in en­ ergy according to angular momentum, L, and spin m u lt ip lic it y , 2S + I ; the greater the value o f L or S, the lower in energy the term. ever, there are many exceptions to th is ru le . How­ The r e la tiv e energies o f the excited states of the fre e ions may be taken from the ordinate o f a Tanabe-Sugano (T-S) diagram. The energy states a ris in g from the s p littin g o f the R-S terms, previously obtained from the character ta ­ bles by group theory methods, may also be taken d ire c tly from T-S d ia ­ grams fo r ions in an octahedral, Oh, f i e l d . Since both D4h and D2h symmetries are treated as perturbations o f the octahedral environment of a metal io n , the weak f ie ld s p littin g o f Oh serves as a reasonable s ta rtin g point in the determination of the possible tra n s itio n s o f these lower symmetries. Reference to appropriate T-S diagrams provides a valuable piece o f addition al data, the ground sta te in Oh, a re s u lt unattainable from group theory alone. I f the ground s ta te in Oh is a T a b le X III S p littin g o f Russell Saunders Terms in Various Symmetries Term °h S P D F G H I. °4h Aig % T lu A2U + u + Tlu + T2u Aig + Eg + T ig + T2g Eu + 2T l u + Ag ■ Blu + b2u + 1B3u =U flIg + 8Ig + B2g + =g Eg + T2g A2 D2h T 2u Alg + A2g + =g + T lg + 2T2g A2u + Blu .+ B2 u + 2Eu 2A9 + Blg + B2g - a g Au + 28Iu + 2B2u 2B3u 2Alg + A2g + Blg + B2g + 2=g 3Ag Alu + 2A2 u + 6Iu + b2u + 3Eu 2Au + 36IU + 3B2u + 3B3u 2Alg + A2g + 28Ig + 2B2g+3Eg 4V 28Ig + 2B2g + 2B3g 36Ig + 3B2g 3B3g 79 one-dimensional representation , symmetry considerations allow the ground s ta te to be sp ecified in D4h and D2h also. Some ambiguity arises i f the octahedral ground s ta te is m ulti-dim ensional. For example, a s ta te s p lits in to an A2g + Eg in D4h, e ith e r o f which may be the new ground s ta te . Table XIV l i s t s the ground states possible fo r d^ - d^ configurations. Table XIV Ground States in Various Symmetries Term ?h_ °4h ° 2h Ai g Ai g Ag T 2g B2g ° r Eg Bl g ’ B2g ^ Tig A2g or Eg 6 Ig - B2g or 8Sg A2g Big B3g Ag Observed spectral bands fo r tra n s itio n metal species in the v i s i ­ ble region are generally produced by e le c t r ic dipole tra n s itio n s . Whether or not a given tra n s itio n is allowed in a free ion can be de­ termined by the p a r ity of the in te g ra l <^g|Ml^e> o f ij>e and where the symmetries are those o f the ground and excited e le c tro n ic s ta te s . The value of th is integral is defined as the transition moment, and M is the dipole moment operator. 80 M = ( 9) Z e.v% ( 10) where r = x t + y j" + The symmetry of the operator is th a t o f the ,x, and z coordinates; the representations fo r the components o f the operator w ill therefore always be odd. Table XV li s t s these representations as found in the character tables fo r 0h , D4h and D ^ . If and ^ are both even functions, as they must be a ris in g from d o r b ita ls , th is in te g ra l w ill be odd and w ill vanish. As mentioned e a r l ie r , d-d tra n s itio n s are p a rity forbidden. , , Table XV Representations o f the Dipole Moment Operator °h °4h Tlu (x , y , z) Eu ■ ° 2h (x , y ) A2u (z ) 6Iu <z > B2u (y) B3u M Magnetic dipole tra n s itio n s may also be observedj but are usually much less intense than e le c tr ic dipole tra n s itio n s . I f only the e lec­ tro n ic wavefunction is considered, an allowed e le c tr ic dipole tra n s itio n 3 is about 10 times as intense as an allowed magnetic dipole tra n s itio n . The magnetic dipole moment operator has the symmetry o f the cross product, r x p, and transforms as the ro ta tio n a l representations, Rx , Ry or Rz o f the point group in question. Because these representations possess g symmetry, magnetic dipole tra n s itio n s are p a rity allowed fo r 81 d-d tra n s itio n s . The in te g ra l which must be considered to determine whether the magnetic dipole tra n s itio n s are symmetry allowed is a IrI ^ ' The d ire c t product is found in the same way as fo r e le c ­ t r i c dipole tra n s itio n s and must s t i l l contain the t o t a lly symmetric representation fo r an allowed tra n s itio n to occur. U tiliz a t io n o f the symmetry o f the e le c tr ic dipole moment operator to determine whether or not a tra n s itio n is allowed assumes th a t the e le c tro n ic wayefunction, is completely independent o f the v ib ra ­ tio n a l wavefunction, ib . When the ion is in a c ry s ta l, v ib ra tio n of the atoms may momentarily destroy the g character o f s ite symmetry. coupling. Such in te ra c tio n between \pe and in the local is c a lle d vibronic An expanded in te g ra l can now be w ritte n , to describe the tra n s itio n moment. |M|^v^e> , I f th is in teg ra l is nonzero, the tra n s itio n is v ib ro n ic a lly allowed. The spectral bands which arise 3 from such tra n s itio n s are on the order o f 10 times weaker than those which are symmetry and p a r ity allowed fo r pure e le c tro n ic tra n s itio n s . The introduction o f vibronic coupling implies th a t the d ire c t product o f the characters o f fiv e representations must be calculated to determine whether or not the in te g ra l vanishes. fie d to some e x ten t. Since This can be sim p li­ is t o t a lly symmetric in the ground s ta te , m ultip lyin g by it s representation w ill not change anything, and i t need not be considered. Also, any representation m u ltip lie d by i t ­ s e lf must contain the t o t a lly symmetric representation. Therefore, i f one of the representations spanned by the d ire c t product o f ^ M \pe Is also one o f the odd noma I modes df v ib ra tio n o f the excited s ta te , i|/y , the in te g ra l w ill be nonzero. For a ll nonlinear complexes, SN - 6 82 v ib ra tio n a l modes e x is t , where N is the number o f atoms in the complex; in octahedral coordination N = 7, giving ris e to 15 modes. Their rep­ resentations are determined by assigning normal coordinates to each atom and observing how they transform under the symmetry operations of the point group in question. Each character is a sum o f 21 terms, most o f which fo rtu n a te ly turn out to be zero. the f i r s t operation other than E is C^iz) . For example, in D2h I f a MLg molecule is ro ta ­ ted 180° about the z_ a x is , four atoms change p o sitio n s, thus contribu­ tin g nothing to the tra c e . This ro ta tio n is shown in Figure 28 where is defined by the M-L^ bond. "I X ^ L3 L ■V Figure 28 Cg Rotation in Normal Coordinates The three atoms which l i e along the ro ta tio n axis do not move; coordinates transform according to the follow ing m atrix. indicates the coordinates a f t e r the ro ta tio n operation. Xl V1 ?! - 1 0 Zi 0 0 - 1 0 Z' 0 0 I th e ir The prime 83 The coordinate a x is , Z, which is also the ro ta tio n a x is , is unchanged by the op eration, but X and Y become -X and -Y . This occurs fo r three atoms so the trace is -3 fo r the C2 (z ) operation in This process is continued fo r each operation in the po int group. The re su ltin g rep­ resentation is reduced and the terms due to tra n s la tio n and ro ta tio n are removed. The remaining terms are the irre d u c ib le representations o f the normal modes o f v ib ra tio n o f the complex. fo r These are tabulated and D2^ symmetries in Table XVI. Table XVI Normal Modes o f V ib ratio n 'h '4h lZh % + Eg + 2Tlu + T 2g + T 2u 2Alg + 6Ig + B2g + Eg + 2A 2 u + 3Ag + Blg + B2g + B3g + 36Iu + B2 u 3B 2 u + + 3Eu 3B 3 u The tra n s itio n moment in teg ra l w ill also vanish i f the i n i t i a l and fin a l states do not possess the same spin; AS = 0. th is is the selection ru le The e le c tro n ic wavefunction may be w ritte n as the product of o rb ita l and spin components, ipe = • Since the dipole moment oper­ a to r does not involve spin , the in te g ra l may be w ritte n : <ifV |iK.> (11) Orthogonality requires the second in te g ra l to be zero, i f the spin wavefunctions o f the excited and ground states d if f e r . With the symmetry and s p lit t in g inform ation contained in Tables 84 X II through XVI, one can determine whether a p a rtic u la r tra n s itio n is allowed or forbidden in the geometry described by a given point group. Ligand coordination fo r the complexes o f the series investigated in th is work suggests th a t local s ite s . is the highest possible symmetry fo r the Pleochroism and p o la riz a tio n data in d ic a te th a t an even lower symmetry, D2h, best describes the metal environment. T-S d ia ­ grams provide the r e la tiv e positions o f energy le v e ls fo r regular octa­ hedral complexes only. To construct an energy level diagram fo r D2h complexes, one may e ith e r perform the tedious numerical calculations required to fin d the sign and magnitude o f the s p lit t in g , or one may as sume th a t the fu rth e r s p lit t in g o f the octahedral le v e ls in lower sym­ metry is small compared to th a t which occurs going from spherical to octahedral local s ite symmetry. Since SCF computations o f open shell configurations are beyond the scope o f th is in v e s tig a tio n , the la t t e r assumption is made. In the complexes in question the tra n s itio n moment operator, T ^ in Oh, indicates th a t a ll the, tra n s itio n s are allowed. In D4h a ll of the tra n s itio n s are allowed in x- or ^ -p o la rize d li g h t , w hile some are forbidden in z-p o la rize d lig h t . but not pleochroism. This pattern allows fo r dichroism, Only in D2h symmetry do the three components of o f the tra n s itio n moment operator transform as three d iffe r e n t represen ta tio n s , and thus permit three d iffe r e n t spectra. A large number of workers have published crystal spectra of the di valen t ions o f Mn, Fe, Co, Ni and Cu in various environments, but very little inform ation c o rre la tin g p o la riz a tio n and pleochroic e ffe c ts ap­ pears in the lit e r a t u r e . In addition most o f the spectral 85 assignments are made u t iliz in g octahedral or square planar symmetry, an assumption which does not allow fo r the pleochroism so s trik in g ly e v i­ dent in the mixed metal c rystals o f the TMAiyixM ^ xC l 3 * 2H20 s e ries . Even when polarized spectra were taken in many cases a sing le crystal fa c e , th a t produced by the growth h a b it, was u t iliz e d . C rystals which grow with faces aligned along e ith e r a v ib ra tio n d ire c tio n or a u n it c e ll axis are not cpmmon; spectra remain unresolved. th e re fo re , many of these polarized crystal No system reported in the lit e r a t u r e o f­ fe rs the s im p lic ity of orthorhombic symmetry coupled with the wide v a ria tio n in color demonstrated by the metal ions in th is s e ries . Energy level diagrams fo r the follow ing spectral discussions are based on lit e r a t u r e data wherever possible; in most cases the symmetry has been lowered and s p littin g assignments have been made to best f i t the observed polarized spectra. lowing order: (4 2 -4 6 ), N i ( I I ) The spectra are discussed in the f o l ­ M n (II) ion (3 1 -3 6 ), C o (II) ion (3 4 -4 1 ), F e ( II ) ion ion (47-51) and C u (II) ion (5 2 -6 1 ). 86 MANGANESE SPECTRA The f i r s t spectra to be examined in d e ta il are those o f the pure . Mn. crystal TMAMnCl3* 2 ^ 0 . For a complex containing high spin M n (II) a ll tra n s itio n s are spin forbidden as w ell as p a rity forbidden, i f only the e le c tro n ic wavefunction is considered. The p a rity re s tr ic tio n may be removed by invoking vibronic coupling, but because these tra n ­ s itio n s are s t i l l spin forbidden, the observed peaks should be narrow and r e la tiv e ly weak in in te n s ity . The ground s ta te , ^S in R-S nota­ tio n , is a h a l f - f i l l e d subshell which transforms as the t o t a lly symmet­ r ic representation in any point group. The tra n s itio n s to excited states require the pairin g o f two electro ns; these excited states are the qu artet terms G, D, P and F lis te d in order of increasing energy. This ordering is obtained from the T-S diagram.which was constructed by u t iliz in g SCF methods. With reference to a T-S diagram and the in fo r ­ mation in Table X I I I an energy level diagram fo r the v is ib le spectra of weak f ie ld M n (II) complexes in symmetry may now be constructed. This is shown in Figure 29. For high spin M n (II) complexes the ground sta te is Ag in D2^, so only the characters o f the operator and the excited e le c tro n ic state need be considered to determine whether or not a given tra n s itio n is . v ib ro n ic a lly allowed. gy level fo r M n (II) in T-S diagrams show th a t the lowest excited ener­ symmetry is T^g which s p lits in to B^g + B2g + Bgg in D2^1 as shown in Table X I I I . I f x -p o larize d lig h t is used to e x cite the complex, the tra n s itio n moment in te g ra ls are <Bxg.lB3UlAg> » <B2g IB3u lAg> and <B3g lB3u lAg> fo r e le c tr ic dipole tra n s itio n s to these excited s ta te s . The d ire c t products are B2u, B^u and Ay resp ec tiv e ly . 87 Because Ay is not one o f the normal modes o f v ib ra tio n in D2h, the l a t ­ t e r tra n s itio n is v ib ro n ic a lly forbidden. tra n s itio n and v is ib le in polarized lig h t . A peak corresponding to th is or ^ -p o la riz e d lig h t would disappear in x- Calculations s im ila r to those above have been per­ formed fo r the possible tra n s itio n s in a ll p o la riza tio n s which would give ris e to peaks in the near IR and v is ib le regions o f the spectra fo r M n (II) complexes o f D2h symmetry. The re su lts in d icate one forbidden tra n s itio n fo r each p o la riz a tio n d ire c tio n : Ag ■+ Bgg in x-polarized lig h t Ag -> B2g in y - polarized lig h t Ag -> Blg in z^polarized lig h t A ll other tra n s itio n s are v ib ro n ic a lly allowed and should show no po­ la r iz a tio n e ffe c ts . The problem o f r e la tiv e ordering o f.th e energy le v e ls giving ris e to these peaks now becomes im portant. Group theory provides no in fo r ­ mation on the magnitude o f the s p lit t in g and SCF methods have not been applied in D2h symmetry fo r these complexes. The energy le vels were therefo re ranked a r b it r a r i ly so as to y ie ld the best c o rre la tio n of the observed peaks and th e ir p o la riza tio n s with the predictions of group theory. For the spectra of the TMA s a lts which contain M n (II) i t was necessary to invoke ra th e r la rg e s p littin g s in two cases to account fo r the experimental observations. With th is assumption, however, the M n (II) spectra can be s a tis fa c to r ily explained with the exception of a very weak band a t 460 nm. n e tic dipole tr a n s itio n . This band was u ltim a te ly assigned as a mag­ The tra n s itio n assignments fo r the observed peaks in order of increasing energy are discussed in. d e ta il below. 88 Figures 5-14 display the polarized spectra o f the Mn containing c ry s ta ls ; Figure 5 s p e c ific a lly contains the polarized spectra from crystals o f the pure s a lt , TMAMnCl3* . The follow ing discussion is i' lim ite d to those peaks due to the M n (II) ion. The lowest energy peak seen consistently in a ll c rystals occurs as a broad band with a maximum a t 530 nm; evidence exists in some crys ta ls o f another sm aller peak a t about 580 nm. The crys ta ls in which th is peak appears are o f very high q u a lity and thus i t is trea te d here as a real peak. In lig h t of v a r i­ ous p o la riza tio n s the band a t 530 nm shows d e fin ite changes in shape, the most dramatic o f which is a very large decrease in in te n s ity in y polarized lig h t . In x-p o larize d lig h t th is band is most intense while the band a t 580 nm has t o t a lly disappeared. both y - and ^ -p o la riz e d lig h t ; Two peaks are observed in th e ir in te n s itie s are greater in ^-po­ la riz e d li g h t , although not as large as the 530 nm peak in ^ -p o la rize d lig h t . Inspection o f Figure 5 suggests th a t the r e la tiv e ordering of these energy le v e ls a ris in g from the octahedral T ^ level should be Bgg < < Bgg; th is best f i t s the observed p o la riz a tio n e ffe c ts as is shown in the energy level diagram fo r M n (II) in Figure 29 on the f o l ­ lowing page. I t is lo g ic a l to assign the Ag -* B3g tra n s itio n to the peak a t 580 nm which disappears in ^ -p o la rize d li g h t , as i t should based on group th e o re tic a l pred ictio n s. The band centered around 530 nm can reasonably be B^g + Bgg as i t has maximum in te n s ity in ^ -p o la ­ rize d lig h t where both tra n s itio n s are allowed. The Ag Bgg tra n s i­ tio n seems to be stronger than the Ag -> B^g tr a n s itio n , because th is band, is more intense in ^ -p o la rize d lig h t than in y -p o la riz e d lig h t . However, due to v a ria tio n s in crystal thickness and sc atte rin g as the O h D2h Forbidden Observed P o la riza tio n Peaks Z T X ' I ---A n ------T 2g - " k l y none Z 350 nm y 367 nm X X '---A S : 330. nm none 412 nm 418 nm Z y 460 nm y 530 nm X ,580 nm Z CO CO CQ Z / Z A ig Figure 29 A g M n (II) Spectral Transitions 90 crystal is rotated to obtain a ll three p o la riz a tio n d ire c tio n s , in te n ­ s ity is not very conclusive evidence fo r the la t t e r statement. The peak a t 460 nm cannot be assigned s o le ly as an e le c tr ic dipole tr a n s itio n . I t is v is ib le in y - polarized lig h t , disappears in ^ -p o la ­ rize d lig h t and is barely v is ib le in ^ -p o la riz e d lig h t . There are two tra n s itio n s which should occur in th is region, Ag -> Blg and Ag -+ B^g ; the former is forbidden in ^z-polarized fo r an e le c tr ic dipole tra n s i­ tio n , allowed fo r a magnetic dipole tr a n s itio n . Since no peak appears in z - polarized li g h t , the weak peak a t 460 nm v is ib le in x-p o larized lig h t is assigned as the Ag -> Blg e le c tr ic dipole tr a n s itio n . This extremely weak tra n s itio n is lo s t under the la rg e r peak present in y polarized lig h t . The behavior of th is la rg e r peak suggests th at Ag -+ Bgg may be a magnetic dipole tr a n s itio n . The operator symmetry fo r these tra n s itio n s is even and magnetic dipole tra n s itio n s are a l ­ lowed in ju s t those p o la riza tio n s in which e le c tr ic dipole tran s itio n s are forbidden. This im plies th a t Ag -> Bgg is allowed only in y -p o la - rize d lig h t which f i t s the observed spectra. . Two d is tin c t peaks which behave q u ite d iffe r e n tly in polarized lig h t occur a t 412 nm and 418 nm. In ^ -p o la rize d lig h t the very sharp peak a t 412 nm disappears completely; the broader, less intense peak a t 418 nm displays no d e fin itiv e change in any p o la riz a tio n . If a large s p lit t in g of the Tgg(^G) energy level occurs, the tra n s itio n Ag -* Bgg could be the source o f the peak a t 412 nm; th is leaves the Ag -> Bgg tra n s itio n a t lower energy which corresponds with the peak a t 460 nm. The three Ag le vels a ris in g from Eg and Al g (^G) could y ie ld the unpolarized peak a t 418 nm, and th is peak is broad enough to 91 suggest th a t there may be more than one tra n s itio n under the envelope. A s im ila r large s p lit t in g o f the Tgg(^D) is required to account fo r the p o la riz a tio n o f the peaks a t 367 nm and 350 nm. Again the A 9 le v e ls from Eg(^D) should give ris e to an unpolarized peak9 but super­ imposed on th is peak a t 350 nm is the Ag -*■ tra n s itio n . . This peak is d r a s tic a lly reduced in in te n s ity in z-p o la rize d li g h t , but does not completely disappear, presumably because o f the presence o f the unpola­ rize d Ag -*■ Ag tra n s itio n s . At about 330 nm the highest energy peak in the spectral region is recorded. The s l i t on the Cary 14 reaches maximum aperture fo r useful spectra in th is region; strument a r t i f a c t . thus., th is peak could conceivably be an in ­ However, although not v is ib le in a ll c ry s ta ls , the peak is consistently a t 330 nm when i t is seen; ^ -p o la rize d lig h t . i t also disappears in I f i t is indeed a v ia b le tr a n s itio n , i t may be as- signed as the Ag -> B^g tra n s itio n a ris in g from the octahedral Tlg ( P ) . Foster and G ill (31) have published unpolarized crystal spectra fo r several M n (II) s a lts based on octahedral geometry. Although none of the c rystals studied by these authors possess the same chromophore as TMAMnCl2»ZHgO, the energies reported fo r the observed peaks in the v is ib le region agree q u ite well w ith those found in th is work. D ingle, e t. al_., (33) have examined the po larized crystal spectra a t low tem­ perature of [ (CH3)^N]MnClg; th is hexagonal s a lt e x h ib its .tw o d iffe r e n t spectra as expected fo r a un iaxial c ry s ta l. crystal spectra of MnClg«ZHgO. same chromophore as the TMA s a lt . Lawson (35) has reported This monoclinic compound contains the Because the tra n s itio n s fo r the M n (II) ion are spin forbidden, varying the ligand and geometry does not 92 re s u lt in a very large energy s h if t fo r the observed peaks. makes c o rre la tio n o f the spectra a straightforw ard task. This Table XVII li s t s the MnClg^HgO peaks along with those obtained in th is work and also assigns p o la riza tio n s to the tra n s itio n s . Table XVII Location and P o la riza tio n o f M n (II) Peaks MnCl2- 2H20 TMAMnCl3- 2H20 Forbidden Peaks (nm) Peaks (nm) P o la riza tio n 325 330 y? 348 350 z 367 373 412 367 X, y 412 x 416 418 none 466 460 x, z 525 530 y. z 580 X To account fo r the observed p o la riz a tio n of peaks in the TMA s a lt , a s p littin g o f the T2g(^G) energy level large enough to cause overlap w ith the Eg(^G) s ta te had to be invoked. be in accordante w ith the lit e r a t u r e data. This assumption appears to The major peaks o f the spectra observed in th is work are in e x c e lle n t agreement w ith those of Lawson recorded fo r the same chromophore. This close agreement 93 im plies th a t the o rig in a l assumption o f a small perturbation o f the energy le vels in octahedral symmetry to account fo r the s p littin g in D2J1 symmetry is a v a lid one. Although there are more possible tra n ­ s itio n s due to the decrease in symmetry, the s p lit t in g is small and broader peaks are usually seen ra th e r than more peaks. In one in ­ stance a sharp new peak is introduced (th a t a t 412 nm) which is a sin ­ g le , nondegenerate polarized tra n s itio n ; G ill has no evidence of th is p a rtic u la r tra n s itio n fo r the unpolarized spectra in octahedral sym­ metry. On the whole the predictions o f group theory are borne out q u ite well by the observed polarized crystal spectra o f the M n (II) ion. 94 COBALT SPECTRA Of the f i r s t row tra n s itio n metals u t iliz e d in th is work, M n (II) is the only ion fo r which the tra n s itio n s are spin forbidden as well as p a rity forbidden; in the spectra o f the Fe, Co, Ni and Cu s a lts the observed tra n s itio n s are spin allowed. The peaks fo r these ions are thus broader and much more intense than those fo r M n ( II ) . In addition the near IR region must be scanned fo r these ions, because some o f the lower energy peaks are seen there ra th e r than in the v is ib le region. In order to keep the peaks on scale fo r spectra o f c rystals containing the d® through d^ ions, e ith e r very th in crystals or crys ta ls with a very low mole fra c tio n o f these ions had to be selected. By grinding an extremely th in cross section o f a crystal o f TMACoC I3* 2H20 , p o larized spectra were obtained fo r th is pure s a lt in the same manner as fo r the Mn analog. The Mn-Co mixed crystals provided a much less intense spectrum fo r the C o (II) io n , although i t was complicated by the pres­ ence of the M n (II) ion. The higher energy peaks o f the Mn spectrum are s t i l l v is ib le and v ir t u a lly unaffected, but the two lowest energy tra n s itio n s are lo s t beneath the much la rg e r peaks due to the Co ion. Comparison of the spectra of the Mn, Mn-Co and Co analogs in d i­ cates th a t there is no in te ra c tio n between the metal ions in these c ry s ta ls ; the Mn and Co individual spectra are simply a d d itiv e . There is a marked loss o f in te n s ity in Mn and Co in divid ual peaks in the spectra o f the mixed metal c ry s ta ls , but th is is to be expected due to lower metal concentrations fo r both ions. The habit fo r the. mixed metal c rystals also made thinner crys ta ls a v a ila b le fo r these spectra. N evertheless, there is a good mapping o f the Co peaks in the pure s a lt 95 to those found in the mixed c ry s ta l, i f one ignores in te n s ity d iffe r e n C ces which cannot be avoided. The peaks due to the d ion also map, except fo r the two which are lo s t beneath the intense tra n s itio n s of the Co ion. Because o f the in te n s ity differences between a spin a l ­ lowed and spin forbidden tr a n s itio n , i t is assumed th a t any p o la riz a ­ tio n e ffe c ts fo r the peaks in th is region are due s o le ly to the C o (II) ion. The R-S ground s ta te fo r C o ( I I ) 9 a d io n , is F; in an octahe­ dral f ie ld th is s p lits in to A2g +■ T2g + T1 , the la t t e r o f which is the ground s ta te as seen in T-S diagrams. When the degeneracy of the Tlg level is removed by lowering the symmetry to D ^ , . three new energy le vels a r is e , Blg + B2g + Bgg , any o f which could be the new ground s ta te . Without access to SCF methods the actual ground s ta te may be assigned by c o rre la tin g the predicted tra n s itio n s with the observed p o la riza tio n data; fo r each ground s ta te nine spin allowed tra n s itio n s which may y ie ld peaks in the v is ib le or near IR regions must be con­ sidered. Group th e o re tic a l evaluation o f the tra n s itio n moment in te ­ grals and comparison with the polarized spectra in Figures 6-9 e lim in ­ ate B2g as the ground s ta te . Both Blg and Bgg f i t portions of the C o (II) spectra, but n e ith e r is s a tis fa c to ry fo r the e n tire IR -v is ib le range. Because id e n tific a tio n o f the ground s ta te is cru c ia l to the discussion o f the TMACoClg'2H20 spectra, previously published spectra o f other C o (II) crys ta ls were examined to establish a v a lid framework upon which to base tra n s itio n assignments. Several a r tic le s appear in the lit e r a t u r e (34-41) which contain polarized crystal spectra o f the C o (II) ion in an environment s im ila r to 96 th a t found in the TMA s a lt . Ferguson and Wood have published low temperature data fo r CoCl2-SH2O (38) and CoCl2-EH2O (3 9 ). The d ihy­ drate is o f p a rtic u la r in te r e s t, because the chromophore in th is com­ pound, CoCl^(H2O)2 , is the same as th a t found in the TMA s a lt . energy level diagram has been constructed fo r An symmetry based on the data fo r the hexahydrate which contains Co(H2O)^Cl2 octahedra. How­ ever, n e ith e r the spectra o f CoCl2-EH2O nor TMACoClg-EHgO allows a rea­ sonable in te rp re ta tio n o f the p o la riz a tio n patterns in tetragonal sym­ metry. Ferguson and Wood have attempted to explain the IR spectra of the d ihydrate as magnetic dipole tra n s itio n s in C2h symmetry. Choice o f ground s ta te is again ambiguous, and regardless o f the choice made, a ll o f the observed peaks cannot be assigned. For the TMA s a lts the e le c tr ic dipole moment operator was o rig in ­ a lly chosen to describe the spectra because most peaks persisted in two p o la riza tio n s ; such behavior is ty p ic a l o f d-d vibronic tra n s itio n s . In the Mn s a lt only one peak appeared which could not be assigned as an e le c tr ic dipole tra n s itio n . In the Co analog many o f the peaks de­ crease in in te n s ity in two p o la riz a tio n s , behavior in d ic a tiv e of mag­ n e tic dipole tra n s itio n s . Another problem encountered in dealing with the C o (II) spectra is th a t not a ll o f the possible tra n s itio n s are seen in the TMA s a lt , making assignment o f peaks a te n ta tiv e process. Because n e ith e r e le c tr ic nor magnetic dipole tra n s itio n s alone can account fo r the C o (II) spectra of these d ihydrates, the question as to whether both could be present must be raised . The in te n s ity of a sym­ metry allowed magnetic dipole tra n s itio n is much weaker than th a t o f a symmetry allowed e le c tr ic dipole tr a n s itio n . However, in the solid 97 s ta te where thermal vib ratio n s are much weaker, one,can imagine th a t an e le c tr ic dipole tra n s itio n which is p a rity forbidden and can be ex­ plained only by vibronic coupling, might a c tu a lly be of the same order o f in te n s ity as the p a rity and symmetry allowed magnetic dipole tr a n s i­ tio n . Thus, in these crystals one might reasonably expect a magnetic dipole tra n s itio n to appear a t room temperature and an e le c tr ic dipole tra n s itio n to p e rs is t a t very low temperature. Work done in th is lab a t room temperature on several c rystals containing C o (II) has yielded consistent spectra with w e ll-d e fin e d peaks in the v is ib le region, but good IR spectra have proven d i f f i c u l t to a tta in . Lacking low tempera­ tu re data fo r the TMA s a lts , there is no evidence to distinguish be­ tween the two types o f spectra in th is work. The argument presented above can be tested on spectra taken from ■ the lit e r a t u r e such as those reported by Ferguson and Wood (3 8 ,3 9 ). The energy level diagram fo r C o (II) in symmetry is presented in Figure 30 along with possible s p littin g patterns i f the symmetry is lowered to D2^. Also included in th is fig u re are the locations of peaks in the CoC12*2H20 spectra and the p o la riza tio n s calculated assum­ ing th a t e le c t r ic dipole tra n s itio n s are involved. The r e la tiv e o r­ dering o f the Dz^1 energy le v e ls , which establishes A2g as the ground s ta te , is based on the spectral data o f Ferguson and Wood fo r the hexahydrate, CoCl2- SH2O. Explanation o f the spectra of the d ihydrate, CoCl2eEH2O5 requires a lower symmetry. The spectral measurements fo r the l a t t e r s a lt were made on c rystals which were o rie n te d .w ith orthog­ onal axes, a / , b_ and £ where a' and j) were defined by e x tin c tio n d ire c ­ tio n s . These axes correspond to £ , £ and Ib re sp ectively in the 98 Forbidden P o la riza tio n Calculated R-S - -B1 ' - * 29- C o C l 2H?0 Peaks^ ( R e f .39 ) Forbidden P o larizatio n Observed 395 nm CT y .z T --------- ' Tig< --B , ' - Eg < -B, z 505 nm z x 610 nm x,y y 830 nm y,x? none 1200 nm none 1630 nm z none 1 1 1 f 1 1 1 1 \ \ \ \ \ \ z \ \ \ \ \ Figure 30 Possible Energy Level Diagram fo r C o (II) 99 orthorhombic TMA s a lt . Careful comparison o f actual p o la riz a tio n d i­ rections with respect to the Co octahedra indicates th a t, although CoClg'ZHgO is m onoclinic, the v is ib le spectra in a ll p o la riza tio n s a t IO S are very s im ila r to those in TMACoClg'ZHgO a t room temperature; these compounds possess the same chromophore. The highest energy peak in the near IR also ex h ib its the same behavior in both s a lts . Such agreement demonstrates the v a lid it y of the assumption th a t the spectra o f these two hydrates a ris e from the same energy level diagram. The near IR spectra obtained by Ferguson and Wood cannot be s a tis fa c to r ily explained u t iliz in g magnetic dipole tra n s itio n s in Cg^ symmetry; in x- and y - polarized lig h t two peaks are always v is ib le in the observed spectra whereas only one is allowed based on group th e o re tic a l predic­ tio n s . In the IR spectra a ll o f the peaks can be explained i f they are assigned as e le c tr ic dipole tra n s itio n s u t iliz in g s ta te in symmetry. as the ground U nfortunately, th is requires a ra th e r a rb i­ tr a r y s p lit t in g of the Eg energy level which arises from the T2g( 4F) term as can be seen in Figure 30. The tra n s itio n s from B0 to B1 and B2g which a ris e from the same T^g term occur a t such low energy th a t they are not seen in the near IR region. The lowest energy peak ap­ pears a t 1630 nrh in Ferguson's spectra and may be assigned as the tra n ­ s itio n Bgg -> B2g which disappears in z-p o la rize d lig h t . The peak a t 1200 nm which shows no p o la riz a tio n e ffe c ts is assigned as the Bgg ->Bgg tr a n s itio n . The remaining peak a t 830 nm corresponds to the Bgg -» Blg tra n s itio n which is forbidden in y -p o la riz e d lig h t , but th is is an ex­ tremely weak tra n s itio n in the CoCl2-EH2O spectra. In addition to the 100 un satisfactory s p lit t in g o f the Eg and B2g levels from D4h$ th is model suffers due to the to ta l in a p p lic a b ility o f Bgg as the ground sta te fo r any of the peaks in the v is ib le region. Selection o f B^g as the ground s ta te in y ie ld s a more reason­ able s p lit t in g arrangement as shown in Figure 31, but leaves the peak a t 1630 nm which should be the B^g -> B2g tra n s itio n unexplained e ith e r as a magnetic or e le c tr ic dipole tr a n s itio n . The B^g -> B^g tra n s itio n a t 1200 nm is allowed in a ll p o la riza tio n s as an e le c tr ic dipole tra n ­ s itio n ; i t is forbidden in a ll p o la riza tio n s as a magnetic dipole tr a n s itio n . The peak a t 830 nm can be assigned as the, Blg Bgg e le c ­ t r i c dipole tra n s itio n which is forbidden in ^ -p o la rize d lig h t . It can also be assigned as a magnetic dipole tr a n s itio n , i f one assumes th a t th is peak has disappeared in the x-p o larize d spectrum. There is some ju s t if ic a tio n fo r both o f these arguments. This second model has another advantage in th a t the Blg ground state.can be used to in te rp re t the p o la riz a tio n patterns of the peaks in the v is ib le region as w e ll. I f s p lit t in g is small as symmetry is lowered from D4^ to D2^, one would expect Blg as the new ground s ta te , because A2g transforms as Bl g . Although th is is conjecture since the Eg and A2g energy le v e ls from Tlg (^F) cannot be lo cated , i t lends some addition al c r e d ib ilit y to the assignment o f Blg as the ground s ta te . The disadvantage o f the energy level diagram presented in Figure 31 is th a t the peaks in the spectra o f the TMA s a lt must be described as magnetic dipole tra n s itio n s . Explanation o f the v is ib le spectra also requires a tr a n s itio n , a broad band between 500 and 550 nm, which is very intense, in x - and ^ -p o la rize d l i g h t , but weak in z-p o la rized 101 °4h 4P— CO h-i /'V / D2h CD °h I I R"S Forbidden TMACoCly ZH9O Forbidden P o la riza tio n Peaks'^ P o larizatio n Calculated Observed none 425 nm ? X 515 nm y sz y 545 nm X sZ Z 610 nm x,y y 830 nm X 5Z - T1 9 V X CO I / CD CO ; - A2g— / g / X-E < - ' ' - B2S "Big " " "Ag I I Z I CD CO CO I I x---vcr-i, \ x \ \ \ \ Figure 31 Preferred Energy Level Diagram fo r C o (II) 102 lig h t . band; Ferguson and Wood have designated th is peak as an "anomalous" i t w ill be discussed separately below. On top o f th is broad band are two peaks which are assigned as mag­ n e tic dipole tra n s itio n s ; the Blg -> B2g and Blg these two peaks a t 515 and 545 nm represent Bgg tr a n s itio n s , re sp ec tiv e ly . 515 nm is allowed only in ^ -p o la riz e d lig h t ; The peak a t th a t a t 545 nm is allowed only in y -p o la riz e d lig h t . 1 The peak a t 610 nm is the lowest energy tra n s itio n in the v is ib le region; i t is assigned as the Blg -» Ag tra n s itio n allowed only in z- polarized lig h t . This peak is located a t the same wavelength and is polarized in the same way in both TMACoClg-ZHgO and CoCl2- 2H20 ; the la t t e r displays b e tte r reso lutio n due to lower temperature. The highest energy peak in the near IR occurs a t 830 nm fo r both of these complexes as well and also behaves s im ila r ly in polarized lig h t . lig h t ; s itio n . In the TMACoClg-ZHgO th is peak is seen only in ^ p o la r iz e d th is corresponds to the Blg Bgg magnetic dipole moment tra n ­ The remaining two IR peaks were never seen in the spectra of the TMA s a lt which were taken a t room temperature. The highest energy spin allowed magnetic dipole tr a n s itio n , Blg -> Blg (^P) is forbidden in a ll p o la riz a tio n s ; appear in any o f the C o (II) spectra. such a peak does not Ferguson and Wood report a charge tra n s fe r band a t 395 nm which increases in in te n s ity as tempera­ ture decreases; i t is polarized perpendicular to the chain d ire c tio n . This p a rtic u la r peak is not seen in the spectra of the TMA s a lt , but absorption is increasing as the s l i t opens beyond useful aperture in th is region. A very weak peak appears a t 425 nm in the TMACoClg-ZHgO 103 spectra which is not seen in the CoClg-ZHgO spectra. Although i t is most intense in ^ -p o la rize d lig h t , i t is v is ib le in x - and ^ -p o la rize d lig h t as w e ll. This is the only peak which is not c le a rly v is ib le in both the pure Co and the mixed Mn-Co spectra. Because o f it s low in ­ te n s ity in the pure s a lt , it s absence in the mixed Mn-Co crystal would not be s u rp risin g . Nevertheless, i f one examines the Mn-Co spectra c a r e fu lly , there is some evidence o f a shoulder on the M n (II) 418 nm peak which may be th is tra n s itio n . ways: This peak may be explained in two as the -> B ^ e le c tr ic dipole tra n s itio n or as a spin fo rb id 2 den tra n s itio n to one o f the energy le v e ls a ris in g from the G term which f a l l s in th is range as seen on the T-S diagrams. is very low fo r an e le c tr ic dipole tr a n s itio n ; The in te n s ity thus, the la t t e r in t e r ­ p re ta tio n is p re fe rre d . Description o f the C o (II) spectra in the TMA s a lt as magnetic d i­ pole tra n s itio n s leaves only one peak unexplained. This broad, in ­ tense band present in x- and ^ -p o la rize d lig h t almost completely disap­ pears in ^ -p o la rize d lig h t . Although one would expect i t to be an e le c tr ic dipole tr a n s itio n , since i t is present in two p o la riz a tio n s , no choice of ground s ta te in e ith e r Dg^ or it s p o la riz a tio n . symmetry can account fo r Ferguson and Wood have described th is peak as an "anomalous" crystal f ie ld band u t iliz in g water vib ratio n s to induce in ­ te n s ity fo r some o f the doublet terms which are spin forbidden. Be­ cause o f it s in te n s ity , i t is d i f f i c u l t to accept th is peak as a spin forbidden tr a n s itio n . The peak is w ell documented, appearing in a ll C o (II) spectra, but only in symmetry as low as Dgh does it s in te rp re ta tio n become a problem. I t has always been described as the T ^ ( F) to 104 4 T ig( P) tra n s itio n in octahedral geometry or the A2g to Eg e le c tr ic d i­ pole tra n s itio n in tetragonal geometry; without p o la riz a tio n data no c o n flic t arises in terms o f the energy level diagram. More spectral work needs to be done u t iliz in g polarized lig h t in complexes of lower symmetry in order to f u l ly explain the o rig in of th is peak. 105 IRON SPECTRA Crystals of the pure Fe and the pure Ni s a lts were never grown large enough fo r spectra to be recorded from them. However, preces­ sion photographs o f these needle-1ike crystals in d ic a te th a t they are also orthorhombic, isomorphic with the Mn analog. This inform ation, coupled with the fa c t th a t the Mn-Co mixed metal spectra are simply the sum o f the spectra of the in divid ual pure s a lts , leads one to assume th a t the F e ( II) and N i ( I I ) spectra may also be extracted from the spectra o f the corresponding mixed metal analogs. Crystals o f the mixed Mn-Fe and Mn-Ni s a lts which are s u ita b le fo r c o lle c tio n of spec­ t r a l data are obtained f a i r l y re a d ily . Even though spin allowed tra n ­ s itio n s e x is t fo r these two ions, t h e ir spectra should be r e la tiv e ly simple, because there are fewer possible tra n s itio n s . The energy level diagram fo r F e ( II) is presented in Figure 32. 5 The R-S ground s ta te , D, s p lits in to Eg + T2g in octahedral symmetry; the T2g level is the ground s ta te . As in the case o f the C o (II) io n , there is some ambiguity in designation o f the ground s ta te when the symmetry is lowered to D2^; any of the three le v e ls which a rise from T2g may be the D2^ ground s ta te . The number of peaks observed in the polarized spectra of the F e ( II) s a lt depends a great deal upon the magnitude of the s p lit t in g o f the energy le v e ls in orthorhombic symmet­ ry ! I f the s p lit t in g is sm all, only one peak may be v is ib le . I f the s p littin g o f the energy le v e ls from Eg is la rg e , one would expect two peaks, both ground s ta te to Ag tra n s itio n s . I f the s p lit t in g of the T2g level is also la rg e , one could expect a maximum o f four spin a l ­ lowed tra n s itio n s in the F e ( II) spectra. As in the C o (II) spectra. D2h (a) R-3 Oh / D4h (b) (c ) T ransitio n T ran sitio n Transition z-forbidden y - forbidden x-forbidden , - E V - aI S - - - - / " B1 ---------- ' / ig Ag \ Ag Ag Ag Ag \ 639 5D V \ x-E - ' C '-Vv 9 "B2g Figure 32 '-'V 8Ig ... B3g B2g Bi g 8Ig - B2g B3g Possible Energy Level Diagrams fo r F e ( II) Observed peaks 1060 nm 107 the peaks due to tra n s itio n s among the energy levels from s p littin g of the Tgg term are not considered, as they are so low in energy th at they are u n lik e ly to be observed. considered: Bgg and Bgg . Three possible ground states must be One or perhaps two peaks ought to occur in the near IR. A cleavage fragment, a c le a r crystal containing the b-c cross sec­ tio n , was u t iliz e d to c o lle c t the observed polarized spectra of the Mn-Fe s a lt . No peaks appear in the v is ib le region other than those ascribed to the M n (II) ion. One large peak appears in the near IR a t 1060 nm and is id e n tic a l in shape and in in te n s ity in both y - and polarized lig h t . Zr U nfortunately, due to the habit o f the Mn-Fe c r y s ta l, the spectrum in ^ -p o la rize d lig h t could not be obtained. Based on the evidence a v a ila b le , the assignment fo r th is peak im plies a Bgg ground s ta te fo r the F e ( II ) ion in Dg^ symmetry. However, lacking the x-po­ la riz e d spectrum, th is assumption cannot be v e rifie d . The o p tic a l and near IR spectrum fo r Fe(C^O4 ) (HgO)g in unpolarized lig h t has been determined by Long (4 2 ); no peaks show in the v is ib le region and two broad bands appear a t 930 and 1180 nm. Long assumes tetragonal symmetry and assigns these as tra n s itio n s from an undeter­ mined ground s ta te to A^g and B^g re s p e c tiv e ly . tween these two peaks is not good; The resolution be­ i f they were not resolved, the max­ imum would occur a t 1055 nm which is very close to the value observed fo r the sing le peak in the TMA s a lt . I t may be th a t the two energy levels are not s p li t very much in the TMA s a lt , and thus remain unre­ solved. Putnik, eix al_., (43) have examined polarized crystal spectra of 108 RbFeClg and CsFeClg a t 4 .2 K. These compounds c r y s ta lliz e in the P6g/mmc space group and e x h ib it two colors in polarized li g h t , red and yellow . However, the broad band a t about 1330 nm which these authors id e n tify as the T2g ^ Eg tra n s itio n remains unchanged in a- or ^ -p o la ­ rize d lig h t . The colpr change in th is compound appears to be due to p o la riza tio n o f the spin forbidden tra n s itio n s to t r i p l e t s ta te s . It is in te re s tin g to note th a t th is compound has hexagonal symmetry as did the [(CHg)^NjMnClg discussed e a r lie r (33) and is therefo re u n ia x ia l; two d iffe r e n t spectra are again predicted. Polarized spectra o f minerals which contain F e ( II) such as g ille s p it e , fa y a lit e and almandine also show a peak in the near IR a t about 1000 nm (4 4 ). Other workers have found s im ila r peaks (4 5 ,4 6 ). Thus the observed spectra o f the F e ( II) TMA s a lt corresponds to those found in the lit e r a t u r e fo r F e ( II) compounds. U nfortunately, without the x-p o larize d spectrum, the ground s ta te cannot be proven to be Bgg. 109 NICKEL SPECTRA The tra n s itio n s involved fo r the N i ( I I ) ion are illu s tr a te d in Figure 33. The ground s ta te in symmetry, A2g, is already a one­ dimensional representation so in th is case there is no ing the ground s ta te in symmetry; problem assign­ A2g transforms as Ag . The two t r i p l e t s , T^g and J 2 , which a ris e from the 3F term are each s p lit in to Blg + B2g + B3g in any order; the lower energy term, T2g, probably involves a tra n s itio n low enough in energy to be located in the near IR. There is also a T1 in octahedral symmetry which arises from a 3 P term; th is y ie ld s a th ird band composed o f Blg + B2g + B3g. The l a t t e r is the highest energy spin allowed tra n s itio n and should appear in the v is ib le region. The Mn-Ni c rystals large enough to be u t iliz e d fo r spectral meas­ urements contained very low mole frac tio n s o f N i ( I I ) ; le a s t soluble in the M n (II) la t t ic e s it e . th is ion is the The z-p o la rize d spectrum taken on a th in crystal y ie ld s peaks o f extremely low in te n s ity . Attempts to increase the in te n s ity o f the observed peaks by using l a r ­ ger crystals resulted in so much s c a tte r th a t peaks could no longer be id e n tifie d . In the Mn-Ni polarized spectra a broad band appears in the near IR a t 825 nm which is v is ib le in ^ -p o la rize d lig h t . I f i t is a c tu a lly present in x-p o larize d li g h t , i t is a very weak tr a n s itio n . I t does not show in ^ -p o la rize d lig h t , but th is may be due to the thinness o f the crystal and low N i ( I I ) concentration ra th e r than to p o la riza tio n e ffe c ts . In the v is ib le region there is a peak a t 440 nm; th is is superimposed on the M n (II) 460 nm peak which makes sorting out the HO Forbidden R-S °h D2h , , ' ' - S T ---------- --- " " - f Igt" - P o larizatio n Peaks y "Blg Z ' " B3g X 440 nm I C7> CO CO > V \ X \ > / - V c - - S 2g y 825 nm Z / '" - S / / / S y Z \ \ \ X X X Figure 33 I > CO X I . CD CO CO X X - v X Energy Level Diagram fo r N i ( I I ) I Ill p o la riza tio n e ffe c ts d i f f i c u l t . As the p o la riz a tio n d ire c tio n is changed, th is peak does indeed s h if t location and change in shape s lig h tly . This indicates th a t i t could be the high energy v is ib le tra n s itio n predicted by group theory, the e le c tr ic dipole tra n s itio n from Ag to the three closely spaced le v e ls , Bjg + B2g + B^g , a ris in g from T j ( P ) . Examination o f the p o la riz a tio n e ffe c ts suggests the s p littin g B3g lower than B^g lower than B2g. . The lit e r a t u r e on N i ( I I ) s a lts deals p rim a rily w ith low tempera­ ture in vestig atio n s o f magnetic chains o f the type AMX3 . The N i ( I I ) compounds RbNiCl3 , CsNiCl3 and [(CH 3 )^N lN iC l 3 (47,49) belong to the same general category as the F e ( II) compounds (43) which were discussed in the preceding section. The N i( I I ) s a lts KMgCl3 (48) and CsMgCl3 (4 9 ). ion can also be doped into the A ll o f the above compounds belong to the PG3Zmmc space group except [(CH 3 )^N lN iC l 3 which c r y s ta lliz e s in PG3Zm. Such crystals are uniaxial with the p o s s ib ility o f dichroism. These workers have obtained polarized spectra; however, they again de­ scribe the tra n s itio n s in terms o f octahedral symmetry. Many peaks are present in the spectra o f these compounds which are not seen in the TMA s a lt; spin forbidden as well as spin allowed tra n s itio n s appear in the low temperature spectra. The spin allowed tra n s itio n s from A2g to T jg( 3F) and T j g( 3P) are located at. 1430 nm, 850 nm and 460 nm, re sp ec tiv e ly . The lo catio n o f the two higher energy peaks correlates q u ite well with those observed in the Mn-Ni TMA s a lt a t 825 nm and 440 . nm. Resolution of the 440 nm peak allows assignment of the r e la tiv e order of the B jg + B2g + B3g le v e ls and it s location is supported by the lit e r a t u r e data. However, the peak in the near IR a t 825 nm cannot 112 be s p li t based on the p o la riz a tio n data with any degree o f c e rta in ty . The lowest energy spin allowed tra n s itio n is not seen a t a ll in the spectra o f the TMA s a lt taken a t room temperature. 113 COPPER SPECTRA Several serious problems were encountered try in g to obtain crystal spectra fo r the C u (II) ion. Due to the Jah n -T eller d is to rtio n exhib­ ite d by the d^ io n , the pure Cu s a lt c r y s ta lliz e s in a monoclinic ra th e r than an orthorhombic space group. This makes o rie n ta tio n of the p rin cip a l v ib ra tio n directions, a more d i f f i c u l t problem than fo r the orthorhombic c ry s ta ls . As has been mentioned previously, the u n it c e ll axes in the pure Cu s a lt are redefined as compared to the ortho­ rhombic u n it c e ll; by c rystallo g rap h ic convention the unique axis in the monoclinic c e ll is defined as lb. In the Cu s a lt th is j) axis hap­ pens to correspond to the d ire c tio n o f hydrogen bonding; axis is along ja. the chain The a-b cross section o f the pure Cu u n it c e ll th erefo re corresponds to the orthorhombic b-c cross section. In the 9 2 2 2 d ion the x -y and z o rb ita ls s p li t so th a t the lone electron occu­ pies the higher energy o r b it a l. T h is , the J a h n -T eller e f f e c t , causes d is to rtio n o f the square planar CuCl^- arrangement with two o f the Cu-Cl bonds lengthening, two shortening. The overall re s u lt is th at the £ axis of the u n it c e ll is no longer perpendicular to the needle a x is , £ ; the c e ll is now monoclinic. However, even though the u n it c e ll axes are no longer orthogonal, the three p rin cip a l v ib ra tio n d i­ rections remain so. The unique axis may define one v ib ra tio n a l d ire c ­ tio n w ithin the monoclinic in d ic a tr ix , but there are no re s tric tio n s on the lo catio n o f the remaining two. Location of these v ib ra tio n d i­ rections w ith respect to the u n it c e ll axes is necessary i f unambiguous spectra are desired. Examination o f a TMACuClg-EHgO crys ta l mounted and oriented on a goniometer head fix e d to a graduated stage and 114 between crossed p o la rize rs indicated p a ra lle l e x tin c tio n in the a-b plane and an e x tin c tio n angle of approximately 30° from the a_ axis in the a-c plane. With respect to the chromophore these e x tin c tio n d i­ rections are id e n tic a l to those found by Ferguson and Wood in the dihydrate CoClg-ZHgO (3 9 ). Extremely th in plates of the pure Cu s a lt are e a s ily grown, y ie ld ­ ing samples fo r spectra o f the a-b cross section. Large c rystals are also re a d ily obtained which may be s lic e d in the proper o rie n ta tio n to give access to the remaining d ire c tio n necessary fo r a complete set of polarized spectra. Thus, i t is th e o r e tic a lly possible to solve the problem presented by the monoclinic stru c tu re of the pure Cu s a lt . However, decomposition o f the pure Cu crystal when exposed to the IR source made i t impossible to obtain the desired p o la riz a tio n data. In the Cary 14 the IR source, a high in te n s ity tungsten lamp, is located in the compartment adjacent to the sample c e ll. The extreme s e n s itiv ity o f the Cu TMA crystals to the IR source may be explained by th is proxim ity, coupled with the fa c t th a t the sample is irra d ia te d by the e n tire output o f the source throughout the scan. There are several possible explanations fo r th is decomposition: one is loss o f water which seems to be a problem fo r th is hydrated s a lt even a t room temperature. another explanation: Previous work done in th is lab (52) o ffe rs a photochemical redox reaction may take place be­ tween the C u (II) and the chloride ions. E ith e r or both o f the above may occur during a scan, rendering the re s u ltin g spectrum unusable. Mixed metal c rystals containing Cu can be grown in e ith e r the o r­ thorhombic or monoclinic space group; thus, the stru ctu re problem can 115 be circumvented by selecting a mixed metal crystal with a low mole fra c ­ tio n o f Cu fo r spectral wqrk. The Mn-Cu crystal with the le a s t Cu was chosen both because o f it s la t h - lik e habit fo r easy o rie n ta tio n and it s la c k .o f in te rfe rin g peaks due to the presence of the M n (II) ion. The mixed metal crys ta ls proved more durable than the pure Cu s a lt , but they were also susceptible to damage by the IR source. Despite a ll the precautions taken, inform ation on the C u (II) polarized spectra gained from the Mn-Cu crys ta ls is not as complete as th a t fo r some o f the other ions studied. Even though the crystal remained in ta c t during a scan, the in te n s ity of tra n s itio n s due to the d ion drove the peaks o f f scale. As the crys ta ls were ground th in n e r, decomposition once again occurred. Low temperature spectra would have been h e lp fu l, but th is c a p a b ility was unavailable on the Cary 14. This problem with in te n s ity of the chlorocuprate spectra has been observed by other workers as w e ll. There is a great deal of lit e r a t u r e data concerning C u (II) spectra, but few workers have looked a t th is ion in symmetry (5 3 -5 5 ). Hitchman and Cassidy have studied two s a lts in which the planar CuCl u n it is present; these are (methylphenethylammoniurn),,CuCl4 (54) and (creatiniuiri^CuCl^ (55) h e re a fte r designated as A2CuCl4 and A 12CuCl4 . There is no axial lig a tio n in these compounds, the nearest axial atom O ly in g more than 3 .0 A d is ta n t from the central Cu atom; there are two 0 O s lig h tly d iffe r e n t Cu-Cl bond lengths, 2.23 A and 2.27 A in A 12CuCl4 O O and 2.25 A and 2.28 A in A2CuCl4 , making the local s ite symmetry fo r these complexes D2^. Hitchman and Cassidy have assigned tra n s itio n s 9 fo r the d ion from which the r e la tiv e s p lit t in g of the d o rb ita ls can be determined. This is presented in Figure 34; they noted th a t the 116 dz2 o r b ita l was lowered in energy much more than was expected. The energy level diagram based on symmetry representations along with pola­ riz a tio n data is shown in Figure 3.5. A2CuCl4 TMACuC I3- 2H20 S p littin g Arrangement S p littin g Arrangement x2-y 2 ----- Z - - - *9 < xy t ~ < i x ------- ---2g Figure 34 x V xz yz yz XZ Z2 xy S p littin g o f the d O rbitals in D2^ Although the o rb ita l s p lit t in g pattern proposed by Hitchman and Cassidy (54) f i t s the spectra fo r the two planar complexes, i t cannot explain th a t o f the TMA s a lt . Only low temperature data were reported fo r A2CuCl4 , but the room temperature spectra of A 12CuCl4 in one po­ la r iz a tio n is v ir t u a lly id e n tic a l to one o f the polarized spectra of the TMA s a lt . However, the p o la riz a tio n e ffe c ts predicted by group theory do not match even when the c e ll axes are compared w ith respect to the chromophore o rie n ta tio n . Therefore, one must assume th a t a d iffe r e n t s p lit t in g arrangement is responsible fo r the C u (II) spectra 117 Representation m Oh Representation in D2h / . / / / / A Eg <'x I I I / T2g CO I / Forbidden P o la riza tio n Calculated Observed Peaks none 588 none X 692 X y 712 y Z 800 . Z Forbidden P o larizatio n Observed ' \ \ S v_Big \ 1 ■N X x A Energy Level Diagram fo r AgCuCl4 in D 2h Figure 35 Representation - m Oh Representation ^n D2h Forbidden P o la riza tio n Calculated Observed . Peaks Forbidden P o larizatio n Observed Z 805 Z y 860 y X 895 y? 960 none PO IO I / Z V I Co I ^2g I I I / S X E - r " Figure 36 -3 g Ag none Energy Level Diagram fo r TMACuCl 3 'ZHgO in Dgh 118 in the TMA s a lt . There is a x ia l lig a tio n in TMACuCly 2H20; the Cu-O bond length is about 2.0 A and the two Cu-Cl bond lengths are 2.3 A and 2.8 A; An­ other d iffe re n c e between the d ihydrate and Hitchman's s a lts is the ex­ te n t o f the d is to rtio n w ith in the CuCl4- plane. The Cu-Cl distances O d i f f e r by only 0.04 A in AgCuCl4 whereas in the TMA s a lt they d if f e r by O 0 .5 A. These d ifferences in geometry seem to favor a d iffe r e n t o r b it ­ al s p litt in g pattern in TMACuClg-2HgO. To compound the problem the C u (II) ion has been doped in to an orthorhombic s ite dominated by the , ' O M n (II) ion. In TMAMnClg -2Hg0 the Mn-O bond distance is 2.2 A; the ° 0 q Mn-Cl bond distances are 2.5 A and 2.6 A; This suggests th a t the d ion is forced in to an octahedral s tru c tu re where the a x ia l bond lengths are shorter than the equatorial bond lengths, although the actual en­ vironment is probably somewhere between th a t of the two pure end mem­ bers, o f the TMA s e ries . Several possible s p lit t in g arrangements e x is t fo r the d o r b ita ls ; the one proposed in Figure 34 can be used to construct an energy level diagram s u ita b le fo r the explanation o f the observed po larized spectra o f the Mn-Cu TMA s a lt . Symmetry representations based on th is s p li t ­ tin g arrangement are shown in Figure 36. There are only fiv e possible electro n c o n fig u ra tio n s .fo r the d® ion. Placing nine electrons in the d o rb ita ls resu lts in the same symmetry as placing one hole in these fiv e o r b ita ls . Therefore, the symmetry o f the o rb ita l occupied by the unpaired electron w ill be the symmetry o f the s ta te . The d o rb ita l s p lit t in g pattern proposed fo r the TMA s a lt in Dg^ is reasonable in th a t the Cu-O bond which defines 119 the z_ axis is the shortest bond; making the z 2 the most repulsion w ill be along z.9 o r b ita l the highest energy le v e l. w ith in the CuCl^- plane; The x and ^ axes l i e the Cu-Cl bonds are longer than the Cu-O bond, so there is le a s t repulsion along x a n d Presence of the C u (II) ion d is to rts the nearly square planar CuCl^" arrangement; w ill remove the degeneracy o f the xz and yz o r b ita ls . th is The tgg t r i p l e t is s p li t so th a t the xy o rb ita l has the lowest energy, the xz o rb ita l is s lig h tly higher and the yz o r b ita l is the highest in energy of these th ree. I f nine electrons are to occupy the d o rb ita ls as s p li t in Figure 34, the ground s ta te has the symmetry o f the d^2 o r b i t a l, Ag in The lowest energy excited s ta te is found by promoting an electron from 2 2 2 the x -y . o rb ita l to the z o r b i t a l; s ta te is also A . 9 the symmetry o f th is excited Transferral o f an electron from the y z , xz or xy o rb ita ls y ie ld s excited states with representations B3g, Bgg and Bl g , re sp ec tiv e ly . This gives ris e to the energy le v e l diagram presented in Figure 36. Spectra were taken of the b-c cross section o f the Mn-Cu c ry s ta ls ; in y_-polarized lig h t these c rystals are dark amber, in z-p o la rized lig h t they are lig h t green. Four peaks can be id e n tifie d in the near IR fo r the C u ( II) spectra obtained, but only one remains on scale. Due to the h a b it o f the Mn-Cu c rystals only the y - and z-p o la rize d d i­ rections were accessible. A large peak, the top of which is o ff s c ale, appears in the near IR in ^ -p o la riz e d lig h t ; dicates th a t the top o f th is peak lie s a t 805 nm. lig h t a broader double peak appears; in te rp o la tio n in ­ In ^ -p o la rize d the tops o f both are o f f scale. 120 but in te rp o la tio n suggests peak maxima a t 860 and 895 nm. In both of i these po larized spectra a 'w e ll-d e fin e d shoulder is evident a t 960 nm. The peak a t 960 nm is assigned as the Ari unpolarized. A„ tra n s itio n which is The peak a t 895 nm is the only one which does not f i t as an e le c tr ic dipole tr a n s itio n ; th is IR peak seems to disappear in %- po larized lig h t whereas i t should only disappear in x -p o larize d lig h t . Because the IR region was scanned ra p id ly to preserve the c r y s ta l, th is peak may be hidden under the large peak seen a t 805 nm. There is some s lig h t evidence o f a shoulder in the proper region, i f one exam­ ines the spectrum c lo s e ly . Ag Bgg tra n s itio n ; in y -p o la riz e d lig h t . The peak a t 860 nm. is assigned as the i t is v is ib le in ^ -p o la rize d lig h t and vanishes The highest energy peak in the IR region occurs a t 805 nm and is assigned as the Ag -*• B^g tra n s itio n which is forbidden in z-p o la rize d lig h t . The x-p o larize d spectrum would have provided valuable addition al d a ta , but could not be obtained due to the habit of these c ry s ta ls . In the v is ib le portion o f the Mn-Cu spectrum a peak appears a t 548 nm in ^ -p o la rize d lig h t ; i t is not present in ^ -p o la riz e d lig h t . This peak is te n ta tiv e ly assigned as a charge tra n s fe r band occurring a t r e la tiv e ly low energy; th is band is presumably the o rig in of the amber color o f the crystal in th is o rie n ta tio n . The peaks due to the M n (II) ion can be seen in the ^ -p o la rize d spectrum, but the in te n s ity goes o f f scale soon a f t e r the observed peak in the ^ -p o la riz e d spectrum. Crystals o f (CHgNHgJgMnCl^ and (enHg)MnCl^ when doped with small amounts o f C u (II) are deep red; W ille t t has reported a polarized peak a t 480 nm in these crys ta ls which is re sp o n sib le.fo r the color (5 8 ). 121 The chromophore fo r th is peak is CuCl5 whereas the chromophore fo r the peak seen in the TMA s a lt is CuCl4 (H2O)2 . The Cu-Cl bonds are longer in the l a t t e r , and two chlorides have been replaced by w ater, which should weaken the crystal f ie ld in te ra c tio n s . Thus, these two peaks may have s im ila r o rig in s . One other system has been reported in the lit e r a t u r e which con­ tains a Mn-Cu m ixture; have been studied (5 9 ). dimers of Cu(C5H5NO)Cl2- H2O and Cu(C5H5NO)2C l 2 The doped c rystals also e x h ib it a color change from the pure compounds (6 0 ). W i lle t t , e t. al_., (58) worked with a very small range o f mole fra c tio n s , claiming th a t the C u (II) ion cannot be forced to occupy the M n (II) s it e , even though the space groups are, id e n tic a l fo r both end members. Work done by Campana a t th is campus (61) indicates th a t the e n tire range is possible fo r (CH3NH3 ) 2MnxC u ^ xCl4 . There also seems to be no problem g e ttin g any mole fra c tio n in the TMA series although a s tru c tu ra l change occurs as Xqu -*■ I . The existence o f a d iffe r e n t color in mixed metal substances con­ ta in in g Mn and Cu than is present in e ith e r pure end member is well documented. The source fo r the peak in the v is ib le region which causes th is color change has been postulated to be an electron tra n s fe r of some s o r t, e ith e r -n-^ to Cu or Mn to Cu. I f an electron tra n s fe r oc curs between Mn and Cu, th is peak should be seen in a ll Mn-Cu systems; th is has not been obs ved. Therefore, the charge tra n s fe r from Cl to Cu induced by local s ite in tera c tio n s seems more probable. 122 CONCLUSION Orthorhombic c rystals of T IW y ij^ xC l 3 * 2H20 where M and M1 repre­ sent the d iv a le n t tra n s itio n metal ions Mn, Fe, Co, Ni and Cu were found to e x is t as s o lid solutions. The ease with which these ions are incorporated in to the la t t ic e s ite s was established: Ni < Co < Mn < Cu. S o lu b ility increases as the size of the ion increases; Cu is unusually soluble because J a h n -T e lle r d is to rtio n increases the e ffe c tiv e size of th is aon. Cell parameters were determined fo r the mixed metal s a lts and also fo r the previously unpublished pure Fe and Ni analogs. These values f ji t the trend expected, i . e . longer c e ll dimensions fo r the la rg e r cations. Because these highly colored crys ta ls are orthorhombic, th e ir u n it c e ll axes are e a s ily oriented with respect to the e le c tr ic vecto r, t , of polarized lig h t . U t iliz in g D2^ symmetry to account fo r the pleochro- ism exhibited by these c ry s ta ls , energy level diagrams were constructed to f i t the observed po larized spectra. In the M n (II) spectra one peak appears which must be assigned as a magnetic dipole tra n s itio n ; remainder were assigned as e le c tr ic dipole tra n s itio n s . the In the C o (II) spectra a ll o f the observed peaks must be assigned as magnetic dipole tra n s itio n s . One peak remained unexplained as e ith e r a magnetic or an e le c tr ic dipole tra n s itio n ; th is "anomalous" band needs fu rth e r study before it s o rig in can be f u l ly understood. In the F e ( II ) and N i ( I I ) spectra in s u ffic ie n t p o la riz a tio n data was obtained to estab lish and adequately support the r e la tiv e s p lit t in g pattern in a diagram. energy level In the C u (II) spectra the peaks were assigned s o le ly as e le c tr ic dipole tra n s itio n s with a charge tra n s fe r band appearing in 123 the v is ib le region. In a ll cases the observed spectral data corre­ la te well w ith those in the lit e r a t u r e fo r ions in s im ila r environ­ ments. Crystals with lower C u (II) concentrations and higher N i ( I I ) con­ centrations are necessary to v e rify the exact s p littin g pattern in symmetry fo r these ions. Low temperature spectra o f these crystals would provide a means to distinguish between magnetic and e le c tr ic d i­ pole tra n s itio n s experim entally ra th e r than so le ly by group th e o retical pred ictio n s. 124 LITERATURE CITED 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. P. Groth, Chemische K ris t a l l ographie, (Wilhelm Engleman, Leip zig, 1908), V o l. I , p. 375. H. Remy and G. Laves, Ber. , 66, 401 (1933). J. H. Day, Chem. R ev., 68, 649 (1968). ' R. D. W i lle t t , e t. al_., Inorg.- Chem., 1J3, 2510 (1974). Sv A. Roberts, e t. al_., J. Am. Chem. Soc., 103, 2603 (1981). R. E. Caputo, e t . al_., In o rg . Chem., .15^, 820 (1976). . R. M. C lay, J. Murray-Rust and P. Murray-Rust, J. Chem. Soc., D alton, 595 (1973). D. B. Losee, e t. al_., Phys. Rev. B, 4342 (1972). D. B. Losee5 e t . al_., Phys. Rev. B. 2185 (1973). R. E. Caputo, R. D. W ille t t and J. A. M uir, Acta. C ry s t., B32, ! 2639 (1976). R. D. Shannon and C. T. P re w itt, Acta. C ry s t., B26, 1046 (1970). I . M. K o lth o ff and E. B. SandelI , Textbook of Q u an titative In or­ ganic Analyses, (Third E d ., Macmillan C o., N. Y . , 1952), p. 543 F. J. Welcher5 The A n a ly tic a l Uses o f Ethylenediam inetetraacetic A cid , (D. van Nostrand C o., I n c . , Princeton, N. J . , 1958). I . I . M. E lb eih , Chem. A n a l., 45, 75 (1956). C. C. Houk, Doctor's Thesis, (Montana State U n iv e rs ity , 1966). D. N. G rin d ley, An Advanced Course in P ractical Inorganic Chem­ i s t r y , ( Butterw orths, London5 1964), p. 24. E. H. S w ift, A System o f Chemical Analysis fo r the Common Ele­ ments, (P re n tic e -H a ll, I n c . , 1939). R. P r ib il and Z. Roubal, Chem. L is ty 5 48, 818 (1954); C. A. 48, 13524 (1954). H. Flaschka, Chem. A n a l., 42, 56 (1953). W. E. H arris and B. K ra to c h v il, Chemical Separations and Measure­ ments , (W. B. Saunders C o., P h ila d e lp h ia , 1974), p. 141. M. J. Buerger5 Crystal Structure A nalysis, (John Wiley and Sons, I n c . , N. Y ., 1960)1 G. H. Stout and L. H. Jensen5 X-Ray Structure Determ ination5 (Mac­ m illan C o., N.' Y ., 1968). E. A. Wood5 C rystals and L ig h t5 (D. van Nostrand C o ., I n c . , Princeton, N. J . , 1964). C. Bunn5 C rystals: Th eir Role in Nature and Science, (Academic Press, N. Y ., 1964). P. F. K err5 Optical Mineralogy, (Third E d ., McGraw-Hill Book C o., I n c . , N. Y ., 1959). J. K, Robertson, Introduction to Physical O ptics, (D. van Nos­ trand C o ., I n c . , N. Y. , 1937), p. 352. R. W. Wood, Physical O ptics, (Macmillan C o., N. Y ., 1934), p. 487. F. A. Cotton, Chemical A pplications o f Group Theory, ( In t e r ­ science Publishers, N,. Y ., 1967). C. J. Ball hausen. Introduction to Ligand Field Theory, (McGrawH ill C o., N. Y ., 1962). D. S. McClure, E lectronic Spectra o f Molecules and Ions i n , C ry s ta ls , (Academic Press, N. Y ., 1959). 125 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. J. J. Foster and N. S. G i l l 5 J. Chem. Soc. (A )5 2625 (1968). L. L. Lohr and D. S. McClure, J. Chem. Phys., 49, 3516 (1968). R. D in g le 5 M. E. Lines and S. L. H o lt, Phys. R ev., 187, 643 (1969). 0. G. Holmes and D. S. McClure, J. Chem. Phys., 26, 1686 (1957). K. E. Lawson, J. Chem. Phys., 44, 4159 (1966). D. R. Rosseinsky and I . A. D o rrit y , Coord. Chem. R ev., 2JL 31 (1978). J.. Ferguson, D. L. Wood and K. . Knox, J. Chem. Phys., 3£, 881 (1963). J. Ferguson and I . E. Wood, In o rg . Chem., 1A, 184 (1975). J. Ferguson and I . E. Wood, In o rg . Chem., 14_, 190 (1975). B. Morosin and E. J. Graeber, Acta. C ry s t., 16, 1176 (1963). L. G. Vanquickenborne and E. Verdonck, In org. Chem., 15, 454 (1976). G. J. Long, In o rg . Chem., 27, 2702 (1978). C. F. Putnik , e t . aJL , In o rg . Chem., 15_, 826 (1976). R. G. Burns, M ineralogical A pplications o f Crystal F ie ld Theory. (Cambridge U niversity Press, 1970). G. D. Jones, Phys. R ev., 155, 259 (1967). F. A. Cotton and M. D. Myers, J . Am. Chem. S oc., 82^ 5023 (1960) G. L. McPherson and G. D. Stucky, J. Chem. Phys., 57^, 3781 (1972). J. Brynestad, H. L. Yakel and G. P. Smith, J. Chem. Phys., 45_, 4652 (1966). J. Ackerman, E. M. H olt and S. L. H o lt, J. Solid S tate Chem., 9_, 279 (1974). J. D. Lebedda and R. A. Palmer, Inorg. Chem., JU, 484 (1972). M. Habenschuss and B. C. G erstein, J. Chem. Phys., 61_, 852 (1974). K. D. Mannil a . M aster's Thesis, (Montana State U n iv e rs ity , 1969) D. W. Smith, Coord. Chem. R ev., 21_, 93 (1976). M. A. Hitchman and P. Cassidy, Inorg. Chem., 18, 745 (1979). M. A. Hitchman, J. Chem. S o c., Chem. Comm., 973 (1979). M. R. Udopa and B. Krebs, Inorg. Chim. Acta, 33, 241 (1979). R. L. Harlow, et> a]_., Inorg. Chem., _13i, 2106~Tl974). U. Schmid, H. Gudel and R. W i lle t t , Inorg. Chem., 21, 2977 (1982). D. A. Krost and G. L. McPherson, J . Am. Chem, S od., 100, 987 (1978). K. Emerson, Personal communication. K. Emerson, Personal communication. MONTANA STATE UNIVERSITY LIBRARIES stks D378.L64@Theses An interpretation o f the polarized cryst RL 3 1762 00178198 6 D 378 L64 c o p .2 L in d b e c k , M a r th a J e a n A n in te r p r e ta tio n o f th e p o la r iz e d c r y s ta l s p e c t r a o f m ix e d m e t a l . . . DATE 0) 3 7 % L6>4 IS S U E D TO