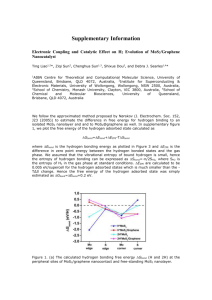
Tunable MoS2 bandgap in MoS2-graphene heterostructures
advertisement

Tunable MoS2 bandgap in MoS2-graphene heterostructures Abbas Ebnonnasir, Badri Narayanan, Suneel Kodambaka, and Cristian V. Ciobanu Citation: Applied Physics Letters 105, 031603 (2014); doi: 10.1063/1.4891430 View online: http://dx.doi.org/10.1063/1.4891430 View Table of Contents: http://scitation.aip.org/content/aip/journal/apl/105/3?ver=pdfcov Published by the AIP Publishing Articles you may be interested in Toward epitaxially grown two-dimensional crystal hetero-structures: Single and double MoS2/graphene heterostructures by chemical vapor depositions Appl. Phys. Lett. 105, 073501 (2014); 10.1063/1.4893448 Mechanical properties of MoS2/graphene heterostructures Appl. Phys. Lett. 105, 033108 (2014); 10.1063/1.4891342 Density functional theory study of chemical sensing on surfaces of single-layer MoS2 and graphene J. Appl. Phys. 115, 164302 (2014); 10.1063/1.4871687 Strain-induced magnetism in MoS2 monolayer with defects J. Appl. Phys. 115, 054305 (2014); 10.1063/1.4864015 Theoretical study on strain induced variations in electronic properties of 2H-MoS2 bilayer sheets Appl. Phys. Lett. 104, 053107 (2014); 10.1063/1.4863827 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 138.67.128.106 On: Tue, 10 Mar 2015 23:42:42 APPLIED PHYSICS LETTERS 105, 031603 (2014) Tunable MoS2 bandgap in MoS2-graphene heterostructures Abbas Ebnonnasir,1,2 Badri Narayanan,1 Suneel Kodambaka,2,a) and Cristian V. Ciobanu1,a) 1 Department of Mechanical Engineering and Materials Science Program, Colorado School of Mines, Golden, Colorado 80401, USA 2 Department of Materials Science and Engineering, University of California Los Angeles, Los Angeles, California 90095, USA (Received 10 April 2014; accepted 15 July 2014; published online 25 July 2014) Using density functional theory calculations with van der Waals corrections, we investigated how the interlayer orientation affects the structure and electronic properties of MoS2-graphene bilayer heterostructures. Changing the orientation of graphene with respect to MoS2 strongly influences the type and the value of the electronic bandgap in MoS2, while not significantly altering the binding energy between the layers or the interlayer spacing. We show that the physical origin of this tunable bandgap arises from variations in the S–S interplanar distance (MoS2 thickness) with the interlayer orientation, variations which are caused by electron transfer away from the Mo–S bonds. C 2014 AIP Publishing LLC. [http://dx.doi.org/10.1063/1.4891430] V Two-dimensional (2-D) materials can vary in terms of their electronic properties (e.g., graphene is metallic, hexagonal-BN (hBN) is insulating, and MoS2 is semiconducting) and, hence, are attractive, often in combinations, for technological applications. Among this class of materials, many studies have focused on molybdenum disulfide, which exhibits a number of interesting properties such as bandgap variation with the number of layers,1,2 high carrier mobility,3,4 high Seebeck coefficient,5 photoconductivity,6,7 large excitonic effects,8 environmental sensitivity,9,10 and high mechanical strength,11—properties which make this material promising for next-generation optoelectronic and nanoelectronic devices.12,13 Recent efforts have focused on taking advantage of the individual properties of different 2-D materials by fabricating heterostructures,14 which are vertical stacks of 2-D layers of dissimilar materials held together by van der Waals (vdW) forces. Such heterostructures not only can lead to opening of a bandgap in graphene (e.g., in graphene-hBN bilayers)15 without impairing its electronic mobility but can also develop direct bandgaps in multilayer heterostructures16 or improve the on/off current ratios drastically (104 in graphene-MoS2).12,17,18 Motivation for the present study stems from the fact that interlayer orientation can affect the electronic properties of monolayers on their growth substrate,19,20 as well in multilayered structures of the same material.21 In this Letter, we investigate the use of interlayer orientation between two different 2-D materials as a tuning parameter for the electronic properties of the bilayer heterostructure. We present results from vdW-corrected density functional theory (DFT) calculations on the effect of interlayer orientation on the electronic structure of the MoS2-graphene bilayers. We find that the relative orientation of graphene with respect to the MoS2 layer strongly influences the value and type of the bandgap in MoS2, while having little effect on both the interlayer spacing and the binding energy. The physical origin of this strong orientation dependence stems from particular atomic a) Authors to whom correspondence should be addressed. Electronic addresses: cciobanu@mines.edu and kodambaka@ucla.edu 0003-6951/2014/105(3)/031603/5/$30.00 registries in which carbon atoms of graphene can effectively weaken a significant number the MoS2 bonds, thus changing both the thickness and the bandstructure of MoS2. Using a tight-binding approach to rationalize the results of DFT calculations, we show that it is valence band edge at the C point that is the most sensitive to the variations in MoS2 thickness, sensitivity which gives rise to the computed changes in MoS2 bandgap with graphene orientation. These results are relevant for heterostructures as potential photovoltaic devices in which exciton pairs could be created in MoS2 and collected by graphene electrodes,22 and furthermore, provide perspective on previous related works23–25 showing the tunability of the bandgap in single-layer MoS2 via tensile strain. To study the effect that graphene has on the electronic structure of the MoS2 monolayer, we constructed commensurate moire patterns of graphene on MoS2 for two interlayer orientations with large spatial periodicities (refer to Fig. 1). The DFT calculations were performed using the SIESTA Package26 with Troullier-Martins pseudopotentials,27 vdW exchange-correlation functional of Klimes et al. (optB88vdW version),28 and double-f basis set with polarization functions. This basis set is sufficient, since the use of larger sets did not lead to significant changes in the structural and electronic properties.29 We have validated our computational approach by reproducing the structural and electronic properties of MoS2 single-layers and bulk crystals; the validation for graphene was performed in our earlier works20,21,30 on epitaxial graphene. We have ensured commensurability of the graphene and MoS2 layers by applying small biaxial strains to graphene while keeping the lattice constant of MoS2 at the relaxed equilibrium value of 3.204 Å. This value for single-layer MoS2 is close to those reported from X-ray diffraction experiments,31,32 and consistent with values from other DFT approaches.33,34 Our use of the equilibrium value for the lattice constant of MoS2 ensures that we do not introduce spurious strains that might obfuscate the effects of interlayer orientation that we seek to reveal. For atomic relaxations and bandstructure calculations, we used Monkhorst-Pack C-centered 10 10 1 and 24 24 1 grids, respectively. The structural optimization was carried 105, 031603-1 C 2014 AIP Publishing LLC V This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 138.67.128.106 On: Tue, 10 Mar 2015 23:42:42 031603-2 Ebnonnasir et al. Appl. Phys. Lett. 105, 031603 (2014) FIG. 1. Surface unit cells of the MoS2-graphene bilayer heterostructures considered in this work: (a) 4:5z, denoting a 4 4 MoS2 cell and a 5 5 graphene cell, oriented so as the ½2 1 10 crystallographic directions of the two layers in the heterostructure are parallel pffiffiffi to one pffiffiffianother, and (b) 4:3a, denoting a 4 4 MoS2 cell matched to a 3 3 3 3 graphene unit cell, in which 1 10 axis. the armchair graphene direction is parallel to MoS2 ½2 out until the residual forces fell below 0.01 eV/Å. The electronic convergence tolerance was set to 106 eV. For all calculations, an energy cutoff of 250 Ry was chosen for the integration mesh. Our results on interlayer binding energies and structural properties of the heterostructures35 shown in Fig. 1 are summarized in Table I. The strains in graphene are sufficiently small that they do not unduly influence our conclusions regarding the electronic properties of the MoS2 layer. The binding energies and interlayer distance, measured between graphene and the closest S atomic plane, do not change significantly with the graphene orientation. The atomic-scale corrugations of MoS2 and graphene are practically inexistent. Large 2-D crystals undergo the formation of spontaneous long-range ripples, whose effect on the bandstructure is presently not fully understood;36–40 by studying virtually flat bilayers (i.e., without significant corrugation, Table I), we ensure that the effects reported here are strictly due to interlayer orientation. The S–S interplanar spacing—i.e., the thickness of the MoS2 layer—varies little with the orientation (Table I); however, these small variations in thickness largely control the electronic properties of the MoS2 layer. As seen in the table, the binding energies are small, since the two layers of the bilayer heterostructures are physically bonded. The binding energy is virtually the same for the two structures (Table I), and, by itself, it does not affect their electronic properties. The fact that there are no significant changes in the (weak) binding implies that many different graphene-MoS2 interlayer orientations would likely be encountered in experiments, a situation which is typical for weakly bonded 2-D systems;19,21,41 our models in Fig. 1 are representative of two orientations. The properties of the heterostructure, as we shall see, are controlled by the interlayer orientation. At a given interlayer orientation in a largeperiod moire pattern, the in-plane sliding of the layers does not change their binding or the electronic properties.42 Figure 2 shows the bandstructures and orbital-decomposed densities of states for the structures studied (Fig. 1). Graphene retains its semi-metallic character, as there is no distinguishable bandgap in the pz states. However, the bandgap in MoS2 varies: 1.674 eV for the 4:5z supercell, and 1.561 eV for 4:3a. As seen in Fig. 2, the nature of the bandgap is different for the two systems (Fig. 1): the gap is direct at the K point for the 4:5z structure, and indirect (from K to C) for 4:3a. Although the assignment of a bandgap type (direct versus indirect) when using supercells that consist of multiple primitive cells is affected by the well-known Brillouin zone folding effect,43 the variations in badgap values and type are physically significant for cells of the same size (4:5z and 4:3a) for which the folding across the Brillouin zone is identical. Next, we will focus on understanding why the orientation of graphene affects the bandstructure of MoS2. Figures 2(b) and 2(c) show that the MoS2 bandgap is direct for the 4:5z configuration, but indirect for 4:3a. Since both systems have the same 4 4 MoS2 cell, we focus on the Table I information pertaining to MoS2 that can be influenced by the proximity of graphene. This key information is the MoS2 thickness t, which changes from 3.227 Å to 3.174 Å. To understand how the thickness of MoS2 can affect its bandstructure, we first study the bandgap as a function of thickness using 1 1 unit cells—which ensures the removal of zone folding artifacts. Our results for MoS2 1 1 are shown in Figs. 3(a) and 3(b); in these calculations, the atomic positions are kept fixed, and the bandgap is computed for different values of t. At equilibrium (t ¼ 3.21 Å), the bandgap is direct, while a slight decrease in the thickness makes the bandgap indirect. As seen in Fig. 3(b), the bandgap can be tuned over a span of 0.6 eV by varying the MoS2 thickness over a span of only 0.2 Å—which is consistent with other reports.24,25 The reason for this high TABLE I. Calculated properties of the MoS2-graphene systems: binding energy, corrugation of graphene, average spacing between graphene and MoS2, strain imposed for commensurability, thickness, and corrugation of MoS2. Strain % System 4:5z 4:3a Binding Energy eV/Å2 Graphene Corrugation Å Interlayer Distance Å MoS2 Gr MoS2 Thickness Å Corrugation Å 0.051 0.051 0.086 0.014 3.110 3.131 0.0 0.0 þ3.0 0.7 3.227 3.174 0.001 0.002 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 138.67.128.106 On: Tue, 10 Mar 2015 23:42:42 031603-3 Ebnonnasir et al. FIG. 2. Bandstructure and projected density of states for the bilayer heterostructures shown in Fig. 1. The pz, dxy =dx2 y2 , and dz2 characters of the bands are indicated in black, green, and blue, respectively. sensitivity of the bandgap is that the small variations in t (e.g., 0.05 Å) amount to changing the length of the Mo–S bonds by 1%: Since these bonds are strong and covalent, a bondlength variation of 1% will significantly affect the bandstructure.23,24 Fig. 3(a) shows that a small decrease in the thickness t results in a large increase in energy of the valence band at C, while the energy values of both the valence and conduction edges at the K point remain largely unchanged. This disparity in the thickness-dependence of the energies at the valence band maxima at C and K causes the direct-indirect band gap transition in the MoS2 layer [Fig. 3]. To understand the physical origin of such a disparity, we turn to a tight-binding (TB) model that describes the band structure of MoS2 in terms of electronic hopping processes associated with the 4d Mo and 3p S orbitals.44,45 In this description, the energy of the valence edge Ej (j ¼ C, K) at C or K points is expressed as:45 ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi j s 2 2 j h þ h hj1 hj2 j 1 2 þ E ¼ þ 2 hj3 ; (1) 2 2 where hji are hopping terms related to different types of atom-pairs. The terms hj1 and hj2 describe electronic Mo–Mo FIG. 3. Bandgap variation for 1 1 MoS2 layer with respect to its thickness. The direct bandgap is measured from the K point on valence band to the K point on conduction band. The indirect bandgap is from the C point on the valence band to the K point on conduction band. Appl. Phys. Lett. 105, 031603 (2014) hopping and S–S hopping, respectively, while hj3 is associated with Mo–S nearest-neighbor pairs. In general, hopping terms hji (i ¼ 1, 2, 3; j ¼ C, K) in Eq. (1) are influenced by the variation in the distances between relevant atom pairs.44 However, in the range of thickness values considered here (i.e., 3 Å–3.3 Å), the h1 and h2 parameters should not be affected by t. This is because they depend primarily only on the in-plane S–S and Mo–Mo distances, which do not change. Therefore, the variations in band edges at K and C (Eq. (1)) are mostly governed by changes in hK3 and hC3 , respectively, which do vary significantly with t because the Mo–S bonds are directly affected by thickness variations. Figures 4(a) and 4(b) show the parameters and the correthickness-dependence of the hK;C 3 sponding valence band edges at K and C. The calculations were performed35 using the parameters in Ref. 45. As seen in Fig. 4, the variations of hC3 and EC are more pronounced than their counterparts at the K point, which leads to significant thickness-dependence of the valence band energy at C and to the direct-to-indirect bandgap transition upon decreasing layer thickness below its equilibrium value. Last, we discuss the reason why the in-plane rotation of graphene affects the MoS2 thickness, which, as we have seen, leads to changes in the bandgap (Fig. 3). To this end, we have determined the electronic transfer that occurs when the two layers (graphene and MoS2) are brought together. For the larger supercells, 4:5z and 4:3a, this electron transfer is shown in Figs. 5(a) and 5(b). Focusing on Fig. 5(a), we note that nine out of the 48 Mo–S bonds on the graphene side (the three-lobed areas identified for clarity in Fig. 5(a)), lose electronic density, meaning that they become weaker and slightly longer (by 0.25%). In turn, the thickness t increases beyond the equilibrium value of 3.22 Å and the bandgap is direct according to Fig. 3. On the other hand, in Fig. 5(b) where graphene is rotated by 30 , the three-lobed clusters are not present because graphene has a different registry with respect to the S atoms in MoS2. Consequently, there is no electronic loss in the Mo–S bonds. Instead, we find a weak and directional increase in the charge associated with these bonds, which makes them slightly shorter; consequently, the S–S interlayer spacing t becomes smaller than its equilibrium value, and the bandgap becomes smaller and indirect. FIG. 4. Effect of thickness on the MoS2 valence band, calculated via the tight-binding model of Ref. 45. (a) Hopping terms hC3 and hK3 as functions of thickness. (b) Energies of the valence band edges EC and EK at C and K points, respectively, as functions of thickness. For clarity, we plot the variation of the quantities with respect to their corresponding equilibrium values (e.g., DhC3 ¼ hC3 hC3;eq and DEC ¼ EC ECeq ). The inset shows a part of the MoS2 structure (Mo: red, S: yellow) and defines the geometric parameters t, rMoS ; rSS , and h. This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 138.67.128.106 On: Tue, 10 Mar 2015 23:42:42 031603-4 Ebnonnasir et al. Appl. Phys. Lett. 105, 031603 (2014) 3 Y. Zhang, J. Ye, Y. Matsuhashi, and Y. Iwasa, Nano Lett. 12, 1136 (2012). B. Radislajevic, A. Radenovic, J. Brivio, V. Giacometti, and A. Kis, Nat. Nanotechnol. 6, 147 (2011). 5 M. Buscema, M. Barkelid, V. Zwiller, H. S. van der Zant, G. A. Steele, and A. Castellanos-Gomez, Nano Lett.13, 358 (2013). 6 H. S. Lee, S. W. Min, Y. G. Chang, M. K. Park, T. Nam, H. Kim, J. H. Kim, S. Ryu, and S. Im, Nano Lett. 12, 3695 (2012). 7 Z. Y. Yin, H. Li, L. Jiang, Y. M. Shi, Y. H. Sun, G. Lu, Q. Zhang, X. D. Chen, and H. Zhang, ACS Nano 6, 74 (2012). 8 A. Ramasubramaniam, Phys. Rev. B 86, 115409 (2012). 9 H. Qui, L. J. Pan, Z. N. Yao, J. J. Li, Y. Shi, and X. R. Wang, Appl. Phys. Lett. 100, 123104 (2012). 10 D. J. Late, B. Liu, H. S. S. R. Matte, V. P. David, and C. N. R. Rao, ACS Nano 6, 5635 (2012). 11 R. C. Cooper, C. Lee, C. A. Marianetti, X. D. Wei, J. Hone, and J. W. Kysar, Phys. Rev. B 87, 035423 (2013). 12 L. Britnell, R. V. Gorbachev, R. Jalil, B. D. Belle, F. Schedin, A. Mishchenko, T. Georgiou, M. I. Katsnelson, L. Eaves, S. V. Morozov, et al., Science 335, 947 (2012). 13 W. J. Yu, Z. Li, H. Zhou, Y. Chen, Y. Wang, Y. Huang, and X. Duan, Nat. Mater. 12, 246 (2012). 14 A. K. Geim and I. V. Grigorieva, Nature (London) 499, 419 (2013). 15 G. Giovannetti, P. A. Khomyakov, G. Brocks, P. J. Kelly, and J. van den Brink, Phys. Rev. B. 76, 073103 (2007). 16 H. Terrones, F. L opez-Urıas, and M. Terrones, Sci. Rep. 3, 1549 (2013). 17 N. Myoung, K. Seo, S. J. Lee, and G. Ihm, ACS Nano 7, 7021 (2013). 18 G.-H. Lee, Y.-J. Yu, X. Cui, N. Petrone, C.-H. Lee, M. S. Choi, D.-Y. Lee, C. Lee, W. J. Yoo, K. Watanabe et al., ACS Nano 7, 7931 (2013). 19 Y. Murata, E. Starodub, B. B. Kappes, C. V. Ciobanu, N. C. Bartelt, K. F. McCarty, and S. Kodambaka, Appl. Phys. Lett. 97, 143114 (2010). 20 B. B. Kappes, A. Ebnonnasir, S. Kodambaka, and C. V. Ciobanu, Appl. Phys. Lett. 102, 051606 (2013). 21 Y. Murata, S. Nie, A. Ebnonnasir, E. Starodub, B. B. Kappes, K. F. McCarty, C. V. Ciobanu, and S. Kodambaka, Phys. Rev. B 85, 205443 (2012). 22 K. Roy, M. Padmananabham, S. Goswami, T. P. Sai, S. Kaushal, and A. Ghosh, Solid State Commun. 175–176, 35 (2013). 23 E. Scalise, M. Houssa, G. Pourtois, V. Afanase, and A. Stesmans, Nano Res. 5, 43 (2011). 24 W. S. Yun, S. W. Han, S. C. Hong, I. G. Kim, and J. D. Lee, Phys. Rev. B 85, 033305 (2012). 25 M. Ghorbani-Asl, S. Borini, A. Kuc, and T. Heine, Phys. Rev. B 87, 235434 (2013). 26 J. M. Soler, E. Artacho, J. D. Gale, A. Garcia, J. Junquera, P. Ordej on, and D. S. Sanchez-Portal, J. Phys.: Condens. Matter 14, 2745 (2002). 27 N. Troullier and J. L. Martins, Phys. Rev. B 43, 8861 (1991). 28 J. Klimes, D. R. Bowler, and A. Michaelides, Phys. Rev. B 83, 195131 (2011). 29 A. Kumar and P. K. Ahluwalia, Modell. Simul. Mater. Sci. Eng. 21, 065015 (2013). 30 H. S. Mok, A. Ebnonnasir, Y. Murata, S. Nie, K. F. McCarty, C. V. Ciobanu, and S. Kodambaka, Appl. Phys. Lett. 104, 101606 (2014). 31 P. Joensen, E. D. Crozier, N. Alberding, and R. F. Frindt, J. Phys. C: Solid State Phys. 20, 4043 (1987). 32 D. Yang, S. J. Sandoval, W. M. R. Divigalpitiya, J. C. Irwin, and R. F. Frindt, Phys. Rev. B 43, 12053 (1991). 33 H.-P. Komsa and A. V. Krasheninnikov, Phys. Rev. B 88, 085318 (2013). 34 A. Molina-Sanchez and L. Wirtz, Phys. Rev. B 84, 155413 (2011). 35 See supplementary material at http://dx.doi.org/10.1063/1.4891430 for the atomic coordinates associated with each structure shown in Fig. 1, and for a detailed description of the tight-binding calculations. 36 J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth, and S. Roth, Nature (London) 446, 60 (2007). 37 J. Brivio, D. T. L. Alexander, and A. Kis, Nano Lett. 11, 5148 (2011). 38 S. Y. Zhou, G. H. Gweon, J. Graf, A. V. Fedorov, C. D. Spataru, R. D. Diehl, Y. Kopelevich, D. H. Lee, S. G. Louie, and A. Lanzara, Nat. Phys. 2, 595 (2006). 39 W. Jin, P. Yeh, N. Zaki, D. Zhang, J. T. Sadowski, A. Al-Mahboob, A. M. van der Zande, D. A. Chenet, J. I. Dadap, I. P. Herman, P. Sutter, J. Hone, and R. M. Osgood, Jr., Phys. Rev. Lett. 111, 106801 (2013). 40 P. Miro, M. Ghorbani-Asl, and T. Heine, Adv. Mater. 25, 5473 (2013). 41 Z. Yan, Y. Liu, L. Ju, Z. Peng, J. Lin, G. Wang, H. Zhou, C. Xiang, E. L. G. Samuel, C. Kittrell, V. I. Artyukhov, F. Wang, B. I. Yakobson, and J. M. Tour, Angew. Chem. Int. Ed. 53, 1565 (2014). 4 FIG. 5. Electronic charge transfer between the MoS2 and graphene layers for two different relative orientations present in (a) the 4:5z and (b) the 4:3a structures. The insets are side views for the normal cross-sections shown as dashed lines in the main (top) views. The three-lobed contours in panel (a) show the specific local registries that cause the weakening of Mo–S bonds. In conclusion, we have shown that the orientation of graphene in MoS2 heterostructures affects the bandgap of MoS2, and have traced the physical origin of this result to the highly sensitive dependence of the bandgap value on the thickness of the MoS2 layer. Changes in registry between graphene and MoS2 are accompanied by interfacial electronic transfer, which affects the Mo–S bondlengths and hence the bandstructure. This suggests that graphene-MoS2 heterostructures are suitable for photovoltaic devices, in which the exciton pair could be created in MoS2 and collected at graphene layers situated on either side of the MoS2 layer. Furthermore, our results provide another perspective on previous works23,24 which showed tunability of the bandgap via tensile strain in the MoS2 layer, and contributes to an increasing body of work that exploits different stacking registries for discovering useful electronic properties (e.g., Ref. 46). The authors thank Dr. E. Cappelluti for discussions regarding the tight-binding parametrization published in Ref. 45. The authors gratefully acknowledge funding from National Science Foundation (A.E., B.N., and C.V.C.) through Grant No. CMMI-0846858, and from the Office of Naval Research (A.E. and S.K.), ONR Award No. N0001412-1-0518 (Dr. Chagaan Baatar). Supercomputer time for this project was provided by the Golden Energy Computing Organization at Colorado School of Mines. 1 K. F. Mak, C. Lee, J. Hone, J. Shan, and T. F. Heinz, Phys. Rev. Lett. 105, 136805 (2010). 2 C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone, and S. Ryu, ACS Nano 4, 2695 (2010). This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 138.67.128.106 On: Tue, 10 Mar 2015 23:42:42 031603-5 42 Ebnonnasir et al. Y. Ma, Y. Dai, M. Guo, C. Niu, and B. Huang, Nanoscale 3, 3883 (2011). C. Hamaguchi, Basic Semiconductor Physics, 2nd ed. (Springer, 2010), p. 388. 44 A. Castellanos-Gomez, R. Roldan, E. Cappelluti, M. Buscema, F. Guinea, H. S. J. van der Zant, and G. A. Steele, Nano Lett. 13, 5361 (2013). Appl. Phys. Lett. 105, 031603 (2014) 45 43 46 E. Cappelluti, R. Roldan, J. A. Silva-Guillen, P. Ordej on, and F. Guinea, Phys. Rev. B 88, 075409 (2013). A. Shmeliov, M. Shannon, P. Wang, J. S. Kim, E. Okunishi, P. D. Nellist, K. Dolui, S. Sanvito, and V. Nicolosi, ACS Nano 8, 3690 (2014). This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 138.67.128.106 On: Tue, 10 Mar 2015 23:42:42