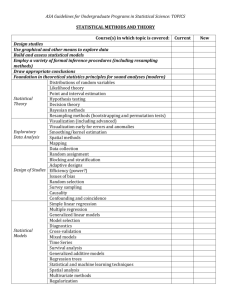
Sequential importance sampling and resampling in population genetic inference Paul Jenkins
advertisement

Sequential importance sampling
and resampling in population
genetic inference
Paul Jenkins
25 October 2012
CRiSM seminar
1
2
Single nucleotide polymorphisms (SNPs)
1
2
3
4
5
6
7
8
9
10
AACGAGTACTGGCTAAAGCTCGACTCGCTTACGTCAGTCTCTTT!
AACGAGTACTGGCTAAAGCTCGACTCGCTTACGTCAGTCTCTTT!
AACGGGTACTGGCTAAAGCTCGACTCGCTTACGTCAGTCTCTTT!
AACGGGTACTGGCTAAAGCTCGACTCGCTTACGTCAGTCTCTTT!
AACGGGTACTGGCTAAAGCTCGACTCGCTTACGTCAGTCTCTTT!
AACGGGTACTGGCTAAAGCTCGACTCGCCTACGTCAGTCTCTTT!
AACGGGTACTGGCTAAAGCTCGACTCGCCTACGTCAGTCTCTTT!
AACGGGTACTGGCTAAAGCTCGACTCGCCTACGTCAGTCTCCTT!
AACGAGTACTGGCTAAAGCTCGACTCGCTTACGTCAGTCTCTTT!
AACGGGTACTGGCTAAAGCTCGACTCGCCTACGTCAGTCTCCTT!
3
Inference using population genetic data
D=
Model-based approach
Main question: “What is the likelihood associated with the
observed configuration of�data under the assumed model?”
L(Θ) = P(D | Θ) =
P(D | H, Θ)P(H | Θ)dH
Coalescent model
Genealogies are hidden random binary trees with a given
distribution (known from the principles of coalescent theory)
4
The coalescent
� �
θ
Exp
2
θ/2
Time
Exp(1)
The “Infinite sites” model
Coalescence
Mutation
Θ = {θ}
5
Recombination
Exp
�ρ�
2
Recombination
Image: Laird & Lange (2011)
6
The coalescent with recombination
Coalescence
Mutation
Exp
�ρ�
2
Time
Exp(1)
� �
θ
Exp
2
Recombination
Θ = {θ, ρ}
7
Ancestral recombination graphs (ARGs)
P(H | Θ)
8
Likelihood-based inference
L(Θ) = P(D | Θ) =
�
P(D | H, Θ)P(H | Θ)dH
N
�
1
�MC (D | Θ) =
P
I{D} (D(i) )
N i=1
H(i) ∼ P(·|Θ)
N
(i)
(i)
�
P(H
|Θ)I
(D
)
1
{D}
IS
�
P (D | Θ) =
N i=1
Q(H(i) |Θ)
H(i) ∼ Q(·|Θ)
N
N
(i)
�
�
1
P(H |Θ)
1
(i)
=
=:
w
,
(i)
N i=1 Q(H |Θ)
N i=1
when P(·|D, Θ) � Q(·|Θ) � P(·|D, Θ)
9
Sequential Importance Sampling
H = (D = H0 , H−1 , . . . , H−m )
P(H0 |H−1 )
w=
Q(H−1 |H0 )
P(H0 |H−1 ) P(H−1 |H−2 ) P(H−2 |H−3 ) P(H−3 |H−4 ) P(H−4 |H−5 ) P(H−5 |H−6 ) P(H−6 |H−7 )
P(H−7 )
Q(H−1 |H0 ) Q(H−2 |H−1 ) Q(H−3 |H−2 ) Q(H−4 |H−3 ) Q(H−5 |H−4 ) Q(H−6 |H−5 ) Q(H−7 |H−6 )
10
Designing proposal distributions
Griffiths & Tavaré (1994):
QGT (H−(k+1) |H−k ) ∝ P(H−k |H−(k+1) )
Easy to implement
Greedy and inefficient
Stephens & Donnelly (2000), De Iorio & Griffiths (2004):
Describe an approximation to the coalescent and infer a
proposal distribution from that
Example – The “infinite-sites” model of mutation (without
nj
recombination):
QSD (H−(k+1) |H−k ) =
n◦
where, of the n◦ sequences that could be involved in the next
coalescence or mutation event, type j appears nj times and is
that which is chosen to modify H−k �→ H−(k+1)
11
Example
H = (D = H0 , H−1 , . . . , H−m )
..
.
2
QSD (H−1 |H0 ) =
3
P(H0 |H−1 ) P(H
1/(3
+−2
θ))
1/(3 + θ) 2θ/(6 + 3θ)
−1 |H
w=
=
=
Q(H−1 |H0 ) Q(H−22/3
|H−1 )
2/3
1/2
12
“Approximation to the coalescent”
Approximation: If
Bj := {Type j is involved in the previous event back in time}
nj
then assume
�
P(Bj |H−k ) = ◦
n
Write down a system of equations for genealogical histories
P(Hk ) =
�
{Hk−1 }
P(Hk |Hk−1 )P(Hk−1 )
Each path corresponds to a genealogical history
Solving the system is as hard as computing the likelihood
But the assumption above simplifies the system and defines a
distribution over backwards transitions
Use this as the proposal
De Iorio & Griffiths (2004)
13
“Approximation to the coalescent”
Combine Bayes’ rule:
P(Hk−1 )
P(Hk−1 |Hk ) = P(Hk |Hk−1 )
P(Hk )
with the asserted approximation:
P(Hk )P(Bj |Hk ) = P(Hk , Bj )
�
=
{Hk−1 reached by events
involving type j}
P(Hk |Hk−1 )P(Hk−1 )
This leaves a soluble system for backwards transitions
14
Incorporating recombination
The work described so far ignores recombination
Introducing recombination causes problems
Problem 1: These approximations no longer provide soluble
backwards transitions; the system is still nonlinear
Problem 2: These approximations no longer lead to an
efficient proposal distribution
Solution 1 [Griffiths, Jenkins & Song, 2008]
Exploit hidden structure in the recursion system to find a
solution
Solution 2 [Jenkins & Griffiths, 2011]
Assert different well-motivated approximations to the
coalescent
15
Incorporating recombination: Problem 2
Recombine sequence 1
1
2
Mutate sequence 2
Recombine sequence 2
1
2
16
Incorporating recombination: Solution 2
Solution: Decouple recombination in this approximation
nj
Recall the previous assumption:
�
P(Bj |H−k ) =
n◦
Bj := {Type j is involved in the previous event back in time}
We modify this to:
� j |H−k , R) ∝ nj
P(B
� j |H−k , R� ) ∝ nj
P(B
where R := {The previous event back was a recombination}
The proposal proceeds first by choosing whether or not a
recombination occurred, then proceeding as before
ρ
Q(R|Hk , Θ) =
ρ+θ+n−1
17
Results
Comparison of new proposal (Jenkins & Griffiths, 2011) versus
‘canonical’ choice (Griffiths & Marjoram, 1996):
QGM (H−(k+1) |H−k ) ∝ P(H−k |H−(k+1) )
l = 0.1
l=1
2
1
1
2
3
4
log10(ESS) (G&M)
ï1
ï1
Relative error (J&G)
Relative error (J&G)
1
0
1
2
3
4
log10(ESS) (G&M)
4
3
2
1
0
5
1
0.5
ï0.5
2
0
5
1
0
3
0
1
2
3
4
log10(ESS) (G&M)
5
2
Relative error (J&G)
0
4
10
3
5
log (ESS) (J&G)
4
0
l = 10
5
log10(ESS) (J&G)
log10(ESS) (J&G)
5
0.5
1
N
Effective sample size (ESS) =
2 /w̄ 2
1
+
s
ï0.5
ï1 w
ï0.5
0
0.5
Relative error (G&M)
1
0
ï1
ï1
ï0.5
0
0.5
Relative error (G&M)
1
0
ï2
ï2
ï1
0
1
Relative error (G&M)
2
Results
0.1
Probability
0.08
4
0.06
0.04
3
Likelihood (10
ï57
)
0.02
2
0
1
0
10
0
25
20
30
40
50
# Recombination events
60
5
20
4
15
3
10
2
5
l
Jeffreys et al. (2000)
1
0
0
e
19
Recursion relation
Sample complexity
(n + s – 1)
One locus: 27 ACs
Four loci with recombination:
900,000,000 ACs!
Ancestral
configurations (ACs)
0
1
1
1
2
4
3
5
4
5
5
5
6
3
7
2
8
Hein et al. (2005)
20
1
Sequential importance sampling algorithm
1. For i = 1, . . . , N :
• Draw H(i) from Q(·|H0 , Θ) by sequential construction:
(i)
(i)
(i)
(i)
(i)
Q(H(i) |H0 , Θ) = Q(H−1 |H0 )Q(H−2 |H−1 ) . . . Q(H−m |H−m+1 ).
• Compute the importance weight
(i)
w(i) =
P(H0 |H−1 )
(i)
Q(H−1 |H0 )
(i)
...
(i)
P(H−m+1 |H−m )
(i)
(i)
Q(H−m |H−m+1 )
.
2. Approximate the likelihood with
N
�
1
�IS (D|Θ) =
P
w(i) .
N i=1
21
Sequential importance resampling algorithm
1. Initialize each H(i) = (H0 ), w(i) = 1, i = 1, . . . , N .
2. Set j = 1. While not all genealogies have been completed:
• If ESS< B, resample:
(a) Multinomially draw N new samples from the existing collection
{H(i) : i = 1, . . . , N } of partial reconstructions according to the
probabilities {a(i) : i = 1, . . . , N }.
(b) If a slot is filled by resampling particle k, set its weight to be
w(k) /(N a(k) ).
(i)
(i)
• Extend each H(i) by appending H−j drawn from Q(·|H−j+1 ).
(i)
• Update each importance weight w(i) �→ w
• Set j �→ j + 1.
3. Approximate the likelihood with
(i)
P(H−j+1 |H−j )
(i)
(i)
(i)
Q(H−j |H−j+1 )
.
a(k) ∝ w(k)�.
a ∝ w(k) .
Typically, N
1 � (i)
IS
�
P (D|Θ)
=
w (k)
.
Alternatively,
N i=1
22
SIS with variable steps
Particles can take variable steps – this could be a problem.
Particle 1
Particle 2
Suppose resampling takes place at step j = 4
Resampling could punish particle 1 (e.g. if mutations have low
prior probability) even though it is close to completion
23
Stopping-time resampling
1. Initialize each H(i) = (H0 ), w(i) = 1, i = 1, . . . , N .
2. Set j = 1. While not all genealogies have been completed:
• If ESS< B, resample:
(a) Multinomially draw N new samples from the existing collection
{H(i) : i = 1, . . . , N } of partial reconstructions according to the
probabilities {a(i) : i = 1, . . . , N }.
(b) If a slot is filled by resampling particle k, set its weight to be
w(k) /(N a(k) ).
(i)
(i)
• Extend each H(i) by appending H−j drawn from Q(·|H−j+1 ).
(i)
• Update each importance weight w(i) �→ w
• Set j �→ j + 1.
(i)
(i)
Q(H−j |H−j+1 )
N
�
1
�IS (D|Θ) =
3. Approximate the likelihood with P
w(i) .
N i=1
(i)
(i)
P(H−j+1 |H−j )
(i)
(i)
.
(i)
• Extend each H(i) by appending (H−(Tj−1 +1) , H−(Tj−1 +2) , . . . , H−Tj ) drawn
(i)
from Q(·|H−Tj−1 ).
24(2005)
Chen et al.
Why does stopping-time resampling work?
For large N, at time T particle i survives approximately
(i)
independently of other particles, with probability CwT
The weight of each surviving particle is set to w̄ ≈ P(survival)/C
The final weight of a surviving particle is
(i)
(i)
(i)
(i)
P(H
|
H
)
P(H
|
H
P(survival)
−m )
−T
−T −1
−m+1
w� (H(i) ) =
·
·
·
·
P(H−m ),
(i)
(i)
(i)
(i)
C
Q(H−T −1 | H−T )
Q(H−m | H−m+1 )
Hence:
Eq [w� | survived]P(survived) = Eq [w� I{survived} ]
∞
�
=
Eq [w� CwT ]P(T = t)
=
t=1
∞ ��
�
t=1
�
wP(survived)q(H)dH P(T = t)
= P(survived)Eq (w) = P(survived)P(D).
25
Choice of stopping-times
(i)
Chen et al. (2005): Choose TlC := inf{j ∈ N : Cj ≥ l},
(i)
where Cj is the number of coalescence events encountered by
particle i after k steps
(1)
T2
(1)
T1
(2)
T2
(2)
T1
Particle 1
Particle 2
“Finite-alleles model”
26
Problem with stopping-time resampling
Several authors were unsuccessful in applying Chen et al.’s
stopping-times to other variants of the coalescent model
This is because current and final weight may be negatively
correlated
27
Stopping-times ignore mutations
Particle 1
Particle 2
28
Intrinsic vs. extrinsic contribution to IS
weight
Extrinsic contribution: events which increase the correlation
between current and final weight (e.g. early optional, wasteful
decisions)
Intrinsic contribution: events which decrease the correlation
between current and final weight, (e.g. early but necessary
decisions overcoming probability ‘hurdles’)
Observations:
Resampling is really based on the extrinsic component
Stopping-time resampling (should) adjust path lengths so that
intrinsic components are approximately equal across particles
Solution:
Define a metric which makes this notion of progress explicit
29
Stopping times and (pseudo-)metrics
Chen et al. (2005)
(i)
(j)
(i)
(j)
d[H−ki , H−kj ] := |Cki − Ckj |
(i)
Tl := inf{k ∈ N : d[H−k , H−m ] ≤ d[H0 , H−m ] − l}.
30
Stopping times and (pseudo-)metrics
More generally:
(i)
(j)
�
(i)
(j)
(i)
(j)
�
d[H−ki , H−kj ] := ν |Cki − Ckj | + µ|Mki − Mkj | ,
(i)
Tl := inf{k ∈ N : d[H−k , H−m ] ≤ d[H0 , H−m ] − l}.
ν = 1, µ = 0
ν = 1, µ = 1
n−2
ν=
,
n + µs
n−1
µ = �n−1 1
θ r=1 r
31
Incorporating recombination
This approach is easy to
extend to more
complicated models,
e.g. a two-locus model
(i)
(j)
�
(i)
(j)
(j)
(i)A(i)
�
(j) A(j)
B(i)
d[H−ki , H−kj ] := ν |C
µ|M
| , | + µB |Mki
|Lkkii − C
L kj | + µ
−kM
A |M
kiki− M
j kj
(i)
B(j)
− Mkj
�
| ,
where Lj is the total length of ancestral material in the
remaining sample
Extensions in other directions are also possible, e.g. models for
microsatellite loci
32
Simulation study
Resampling events
Unsigned relative error
Scheme C
Scheme SCM
1
1
1
0.8
0.8
0.8
0.6
0.6
0.6
0.4
0.4
0.4
0.2
0.2
0.2
0
0
0
40
40
40
20
20
20
0
ï4
0
4
8 � 12
�
N
−
B
log2log2B
B
Scheme CM
0
ï4
0
8� 12
�4
N
−
B
log2log2B
B
0
ï4
0
8�
�4
N
−
B
log2 log2B
B
12
1 simulated dataset, 19 B values x 25 independent experiments:
n = 20, θ = 3.5, ρ = 5, N = 10,000; ‘truth’ estimated from N = 10,000,000
Unsigned relative error
Unsigned relative error
Simulation study
l = 0.1
1
0.5
0
ï4
ï2
0
2
4� 6 � 8
N −B
log2log2B
10
12
14
B
1
C
CM
SCM
0.5
0
0
10
20
30
Resampling events
40
50
1 simulated dataset, 19 B values x 25 independent experiments:
n = 20, θ = 3.5, ρ = 0.1, N = 10,000; ‘truth’ estimated from N = 10,000,000
Unsigned relative error
Unsigned relative error
Simulation study
l = 10
1
0.5
0
ï4
ï2
0
2
4� 6 � 8
N −B
log2log2B
10
12
14
B
1
C
CM
SCM
0.5
0
0
10
20
30
Resampling events
40
50
1 simulated dataset, 19 B values x 25 independent experiments:
n = 20, θ = 3.5, ρ = 10, N = 10,000; ‘truth’ estimated from N = 10,000,000
Unsigned relative error
Unsigned relative error
Simulation study
l = 20
1
0.5
0
ï4
ï2
0
2
4� 6 � 8
N −B
log2log2B
10
12
14
B
1
C
CM
SCM
0.5
0
0
10
20
30
Resampling events
40
50
1 simulated dataset, 19 B values x 25 independent experiments:
n = 20, θ = 3.5, ρ = 20, N = 10,000; ‘truth’ estimated from N = 10,000,000
Unsigned relative error
Unsigned relative error
Simulation study
l = 100
1
0.5
0
ï4
ï2
0
2
4� 6 � 8
N −B
log2log2B
10
12
14
B
1
C
CM
SCM
0.5
0
0
10
20
30
Resampling events
40
50
1 simulated dataset, 19 B values x 25 independent experiments:
n = 20, θ = 3.5, ρ = 100, N = 10,000; ‘truth’ estimated from N = 10,000,000
Remarks
Sequential importance sampling (SIS) is a potent tool for
analyzing DNA sequence data
Nonetheless, recombination inhibits its applicability to wholegenome data
Two of our approaches to getting the most out of SIS:
Careful design of proposal distribution based on wellmotivated coalescent approximations
Resampling mechanism with carefully chosen stopping times
based on metric space approximation
These contributions broaden the scope of SIS, enabling both
larger datasets and more elaborate models
Further extensions
Scalability: Combine SIS with other approximations to make it
applicable to whole chromosomes, e.g. composite likelihood,
sequentially Markovian coalescent
Better stopping times, e.g. incorporating the signals of
38
recombination apparent from the data (the ‘four-gamete test’)
Wider implications
Some properties of this statistical problem:
Variable particle path lengths
Weak/negative correlation between current and final IS weight
Final target distribution is ‘special’ compared to intermediate
targets
Efficient proposal distributions must be highly circumspect
…are shared with some other statistical problems. Here are three
more examples.
39
1. (Chen et al., 2005): The Dirichlet problem
2
2
Solve u(x, y) defined on (x, y) ∈ [0, 1] × [0, 1] satisfying ∂∂xu2 + ∂∂yu2 = 0 on the
interior, and with boundary condition
�
1 on top of the square
u(x, y) =
0 on the sides and bottom of the square.
Monte Carlo approach to finding u(a,b):
Discretize the square to an n by n grid
Set off N simple random walks from an interior point u(a,b)
and use a Monte Carlo approximation to:
u(a, b) = E[u(X, Y )]
where (X,Y) is the random point on the boundary the walk hits
Here, u(a,b) is approximated by the fraction of walks hitting
the top side of the square
A SIS algorithm would bias walks upwards
40
2. (Zhang & Liu, 2002): Self-avoiding walks
“In the 2D hydrophobic-hydrophilic model, a protein is abstracted
as a sequence of hydrophobic (H) and hydrophilic (P) residues.
The sequence occupies a string of adjacent sites on a twodimensional square lattice. Only the self-avoiding conformations
are valid with a simple interacting energy function:
e(HH) = -1, e(HP) = e(PP) = 0,
for contacts between noncovalently bounded neighbors. The
native structure of the sequence is defined as the conformation
with the minimum energy.”
41
3. (Lin et al., 2010): Diffusion bridges
1
dXt = Xt dt + dWt
5
Standard Monte Carlo approach to drawing sample paths:
Discretize time
Draw a skeleton path by SIS (e.g. proposing according to the
Euler approximation to the diffusion, with some modification to
push the path towards the fixed endpoint)
42
Wider implications
Some properties of this statistical problem:
Variable particle path lengths
Weak/negative correlation between current and final IS weight
Final target distribution is ‘special’ compared to intermediate
targets
Efficient proposal distributions must be highly circumspect
…are shared with some other statistical problems. Here are three
more examples…
How can we decide when a standard resampling will be
problematic? How can we characterize such problems?
What remedies are optimal in dealing with this?
Stopping-time resampling
Exact lookahead weighting, exact lookahead sampling,
forward pilot-exploration, backward pilot-exploration
Are there more general strategies for choosing a good
combination of proposal, auxiliary targets, and stopping times?
43
Acknowledgements
Bob Griffiths
Yun Song
Jotun Hein
Chris Holmes
Rune Lyngsø
Carsten Wiuf
EPSRC
NIH
44
Appendix
Jenkins (2012)
45