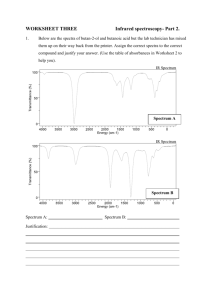
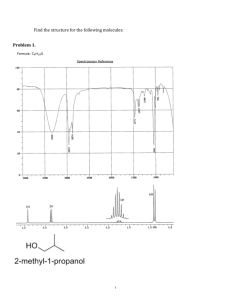
Structure Determination
advertisement

Structure Determination There are more than 5 million organic compounds, the great majority of which are colourless liquids or white solids. Identifying or at least characterising – determining some of its properties and features – organic compounds is an important technique in the pharmaceutical industry, and in research laboratories. Until the last forty years, chemical methods dominated this area, but they are now to a great extent, supplanted by instrumental methods, particularly spectroscopic techniques. This chapter concentrates on the application of spectroscopic techniques in the structure determination of organic compounds. The instrumental aspects of these techniques are covered in other chapters or subjects. 1. Chemical Tests Solubility Tests The principle of these tests is that a substance is most soluble in that solvent to which it is most closely related in structure. Compounds are divided on the basis of water solubility into two main groups (soluble and insoluble), which are then subdivided into other groups according to their solubility in other solvents (e.g. acids and bases, ethanol and hexane). All solubility measurements are done at room temperature with two drops of liquid or 50mg of solid in 1.5 mL of solvent. Functional group tests These have been extensively covered in Chemistry 3, and you should refresh your memory of the following tests. Test Functional Groups Giving +ve Observation For +ve Test bromine NaHCO3 DNP Fehlings Physical Parameters This includes such things as melting point, boiling point, refractive index, density etc. These are determined and compared to literature values. A close match to a complete set of literature values is strong but not definite evidence for identification of a substance. Structure Determination 2. Spectroscopic Analysis In Structure Determination Spectroscopic techniques have now become the main method of determining the structure of organic substances. Sophisticated instruments are now available which greatly simplify organic structure determination. The main techniques involved are listed below, along with the information which they provide. Each instrument will be examined individually, but generally data from all techniques is used and combined when determining the structure of a substance. TABLE 1 Spectroscopic techniques Spectroscopic Technique Information provided by the Technique UV-visible spectroscopy The degree of conjugation of the molecule, and some evidence of identity through spectral matching. Not particularly useful. Infra-red spectroscopy The functional groups present in the molecule and a means of absolute confirmation of identity through spectral fingerprint matching NMR spectroscopy Details of the carbon-hydrogen framework of the molecule Mass spectrometry The molecular mass of the substance (in many cases), the presence of certain species (Cl, Br, and N) through isotope peaks, spectral identification through fragmentation pattern matching. 3. Ultraviolet-Visible Spectroscopy UV-visible spectrophotometry is not generally useful in qualitative analysis, as quite different compounds can have a similar spectrum. It can still be useful however, to distinguish between substances as only conjugated substances, and those containing unpaired electrons (those containing N, O, and halogens) absorb in the UV-visible region above 220nm. CLASS EXERCISE 1 What does the term conjugation mean? UV-visible spectrophotometry is used in organic analysis to assess the degree of conjugation of a molecule. What this means is that the molecule will absorb energy at longer wavelengths if it has more conjugated double bonds. Normally at least two or more conjugated double (or triple) bonds to produce a UV-visible absorption peak above 220 nm. For example 1,3-pentadiene exhibits an absorption peak above 220 nm whilst the isomeric 1,4 pentadiene does not. Hence these two can be differentiated by UV-visible spectroscopy. Each extra conjugated double (or triple) bond adds about 15-20 nm to the wavelength of maximum absorption. Aromatic rings – with 3 conjugated double bonds – typically absorb around 250-260 nm. The groups with unpaired electrons, when attached to conjugated systems, significantly increase the intensity of absorption, and slightly increase the wavelength. 2 Structure Determination CLASS EXERCISE 2 How would the UV spectrum of phenylmethanal differ from that of phenylethanal? O H H O 4. Infrared (IR) Spectroscopy Infra red radiation is the region of the electromagnetic spectrum between microwave and visible light. It is the region of the electromagnetic spectrum which provides the correct amount of energy to make the covalent bonds in some organic molecules to vibrate, bend, stretch, wag and rock. Specific bonds absorb at specific frequencies, and by working backwards with correlation tables (comprehensive listings of functional groups and IR frequencies) we can deduce the types of bonds present in a molecule from its infra red spectrum. Full interpretation of an infra red spectrum is extremely difficult, so it is normal to only look for common absorptions in key areas. Interpreting IR spectra The IR spectrum is simply a graph of % transmittance (which tells us how much light is absorbed) versus frequency (usually given in wavenumbers, cm-1). Each functional group has its own characteristic absorption (frequency and strength) which change very little from one compound to another. This makes their identification very simple. For example the C=O group always absorbs in the region from 1670–1780 cm-1, while the O-H absorption of alkanols always occurs in the region from 3200–3600 cm-1, etc. All of these can be easily recognised in an infra red spectrum with a little practice. Additionally many absorptions occur with specific shapes and intensities which also aid their identification. The C=O group for example is generally a very strong sharp band (often the strongest in the spectrum), whilst the O-H peak is generally broad, often with a small sharp peak at the high wavenumber end. Infra red spectroscopy can also be used in the identification of a molecule. The spectrum is extremely complicated and often too difficult to interpret any meaningful information from, but it can be used however as a “molecular fingerprint” which can be matched with literature spectra. If the fingerprint region of a substance is identical to a literature spectrum then this can be taken as a form of unambiguous identification, as no two substances have identical infra red spectra. The infra red spectrum can be divided into two regions which provide the two different types of information (see Figure 1): the functional group region from 4000–1500cm-1 gives information about the functional groups present in a molecule and can be further sub-divided, the fingerprint region between 1500 - 650cm-1 provides a method of identification. 4000 Bonds to H C-H O-H N-H 2500 2000 1500 cm Triple bonds Double bonds C=C CC C=O CN C=N Functional group region -1 Fingerprint region FIGURE 1 The regions of the infra red spectrum. 3 Structure Determination A correlation table (see Table 1) lists the important peaks for each common functional group, allowing you to identify the presence of particular groups in a the spectrum of a compound. Figures 2-10 show the functional group region for selected examples of each type of functional group. CLASS EXERCISE 3 Examine Figures 2-10 in conjunction with Table 2 to see what the various functional group peaks really look like, and what some of the variations mean. TABLE 2 IR correlation chart Bond Frequency Region Relative Strength C-C-H 2800-2950 strong* C=C-H & Ar-H 3020-3070 weak CC-H 3100-3200 medium O-H 3400-3700 strong broad when H-bonding is pronounced, and sharp at higher end of range when OH free N-H 3300-3500 1590-1650* medium medium one peak for each N-H bond in higher region; peak marked * is for primary amines & amides only CC 2100-2200 medium CN 2200-2400 medium C=C 1620-1660 weak-medium 1600,1580,1500,14 50 medium one or more of these peaks may not be present in spectrum C=O 1660-1800 strong normally strongest peak in spectrum; position dependent on type of compound (acid halides highest, amides lowest); aromatic group attached lowers frequency COOH 3300-2600 strong very broad, due to OH group in acid, C=O peak as normal NO2 1500-1560 1300-1350 strong strong Aromatic ring Comments strength proportional to number of C-C-H bonds NOTE - Since C-O, C-N and C-X bonds do not yield distinctive peaks in the functional group region, ethers, haloalkanes and tertiary amines and amides do NOT give specific characteristic IR functional group peaks. 4 Structure Determination Figures 2-10 on pages 5-14 will be provided by your teacher in hardcopy form as the detail in the spectra cannot be satisfactorily gained by scanning. 5 Structure Determination CLASS EXERCISE 4 (a) Using Table 1, associate the spectra below with one of each of the following substances. It may help if you draw the structures of each compound first. 1-hexene 1-hexanol 2-hexanone phenylhexane (i) (ii) (iii) 15 hexanoic acid 1-hexanamine Structure Determination (iv) (v) (vi) 16 Structure Determination (b) (i) How would you distinguish between the following pairs of compounds by their IR spectra, assuming you couldn’t compare them to standard spectra for fingerprint matching? methylbenzene and cyclohexane (ii) chlorobenzene and 2-methylchlorobenzene (iii) 1-hexanamine and N-methyl-1-hexanamine (iv) phenylethene and phenylethyne (v) benzoyl chloride and benzamide (vi) 4-methylphenol and phenylmethanol 17 Structure Determination (c) What structural information can be obtained from the following IR spectra? It is not necessary to identify what groups are not present. (i) (ii) 18 Structure Determination (iii) (iv) 19 Structure Determination 5. Nuclear Magnetic Resonance (NMR) Spectroscopy This form of spectroscopy involves the interaction between electromagnetic radiation in the radio frequency region (60-600 x 106 Hz) and the nuclei of some of the atoms in organic molecules. It is an absorption technique, measuring the intensity of absorption at different radio frequency energies. In this chapter, we will only examine the use of NMR spectroscopy for the purposes of organic structure determination. Only a very basic coverage of how the instrument works will be provided. What causes the absorption? Atomic nuclei spin on their axis (like the earth) , and for some elements this creates a magnetic field. The magnetic fields of the spinning nuclei are oriented randomly (see Figure 11 (a)). When this small magnetic field is placed in a much stronger magnetic field it will align with the applied field – the ground state (see Figure 11(b)). If energy is supplied in the form of radiowaves of exactly the correct frequency, radiation is absorbed and the spinning nuclear magnet flips and becomes aligned against the magnetic field in a higher energy – the excited state (see Figure 11(c)). (a) random orientation of nuclei magnetic fields (b) alignment of nuclei magnetic fields with external field (ground state) (c) alignment of nuclei magnetic fields against external field (excited state) FIGURE 11 Behaviour of atomic nuclei in NMR spectroscopy (arrow indicates N to S poles of magnetic field) 20 Structure Determination Not all nuclei behave in this manner, and of the three isotopes most common in organic compounds (1H, 12C and 16O) only the hydrogen atom behaves in this manner. We can however, obtain NMR spectra on the C skeleton as 13C is NMR active. Other important NMR active isotopes include 19F and 31P. Different nuclei and nuclei in different atomic groupings and environments require different amounts of rf energy to be excited. This is the basis for NMR spectroscopy. Information provided by the NMR spectrum Figure 12 shows a typical NMR spectrum (for bromoethane). It will look quite different to the spectra of other types you are used to. FIGURE 12 A typical NMR spectrum The NMR spectrum provides four main pieces of information about the structure of the compound: the number of “different” types of hydrogen atoms the functional groups surrounding each different hydrogen the number of atoms of each type of hydrogen atom the number of non-equivalent hydrogen atoms on adjacent atoms CLASS EXERCISE 5 You will be assigned a simple organic compound, for which you will build up a representation of its NMR spectrum. This will occur in four stages, corresponding to the four pieces of information obtained from the spectrum. Compound name Compound structure 21 Structure Determination The number of different hydrogens in the compound What are different hydrogens? First, let’s introduce the jargon used in NMR spectroscopy: non-equivalent hydrogens. Non-equivalent hydrogens are those attached to different carbons (or other atoms), where the surrounding groups are different. Rules for identifying equivalent hydrogens hydrogens attached to the same atom are equivalent, as long as that is NOT an alkene carbon hydrogens attached to different atoms, where the molecule has some symmetry, making the two groups the same EXAMPLE 1A How many types of non-equivalent hydrogens are in (a) 3-pentanol (b) butanone? (a) – four CH 3CH 2CHCH 2CH 3 OH 1. The H on the O is obviously different to all the others. 2. The H on the C attached to the O is different to the others. 3. The three Hs on the end carbon are the same as each other and the same as the three on the other end C, but they are different to the others 4. The two sets of hydrogens (the CH2 groups) are the same as each other, but different to the other 3 types (b) – three CH 3CH 2CCH 3 O 1. The hydrogens on the CH2 are different to the others, because they are next to the carbonyl, and there are only two of them 2. The three hydrogens on C-1 are not equivalent to the 3 Hs on C-4, because they are attached to different groups (CH2 vs C=O). The number of types of non-equivalent hydrogens determines the number of peaks in the spectrum. EXAMPLE 1B How many peaks are in the spectrum of (a) 3-pentanol and (b) butanone? (a) four (b) three CLASS EXERCISE 5A How many peaks are in the spectrum of your compound? 22 Structure Determination EXAMPLE 2A Look back at the real spectrum in Figure 12. How many peaks are present? How many different types of hydrogens are present in this compound? It seems as though there are seven other peaks – does this mean there are seven types of hydrogen? Actually there are only two true peaks – the group of three and the group of four. This is known as splitting, and will be explained later. CLASS EXERCISE 6A How many types of hydrogens are present in the spectra in Figures 13 and 14? FIGURE 13 NMR spectrum for Exercise 6 FIGURE 14 NMR spectrum for Exercise 6 23 Structure Determination The functional groups surrounding each different hydrogen As with most forms of spectroscopy, the molecular environment around the absorbing species – in this case, the hydrogen nuclei – affects its absorption frequency. Electrons in the immediate environment of a proton shield it very slightly from the external magnetic field, and hence they reduce the energy required to cause absorption. All NMR spectra are displayed on charts that show the absorption energy increasing from left to right. Thus the left part of the chart is called the downfield (deshielded) side (because protons with less electrons around them will absorb in this region as it requires less energy to bring the nucleus into resonance), and the right part of the chart is called the upfield (shielded) side. The scale units in NMR spectroscopy are either rf frequency (Hz) or more commonly, the chemical shift () (in ppm units, which are unfortunately named, and nothing to do with parts per million). The chemical shift has values of 0-12 for proton NMR, and will be what we refer to here. For reasons we will look at in the NMR instrumentation chapter, it is necessary to calibrate the NMR spectrum with an internal standard: this is usually the compound known as TMS (tetramethylsilane, Si(CH3)4). The chemical shift of this is assigned as 0. It is used as a reference peak for several reasons including: it absorbs upfield from almost all other peaks it is volatile and can easily be removed from the sample after use by heating it gives a very strong signal (as it has 12 equivalent protons) it is inert and does not react with the compounds being analysed. As with infrared spectroscopy, NMR absorptions for similar functional groups tend to occur in similar regions, as shown in Figure 15. A more extensive correlation table is provided in Table 3. O=C-O-H, O=C-H alkanoic acids & alkanals aromatic compounds 12 - 10 9 – 6.5 = C-H -C- X = C-C-H -C-C-H H attached to double bond where X = O, N or halides H adjacent to double bond saturated hydrocarbons 6.5 - 4.5 4.5 - 3 3-2 1.5 – 0.5 (ppm) FIGURE 15 Proton NMR absorption regions EXAMPLE 1B What would the chemical shifts for peaks in the spectra of (a) 3-pentanol (b) butanone be? (a) It helps if you label the different protons (a, b , c etc), so it makes discussion of the spectrum easier. a b c b a CH 3CH 2CHCH 2CH 3 OH d Proton a b c d Bonding H-C-C H-C-C H-C-O H-O Chemical Shift 1.0 – 1.8 1.0 – 1.8 3.3 – 4.0 2-5 Note that for proton c, the path could have been H-C-C also. Always look for the highest chemical shift alternative. 24 Structure Determination TABLE 3 NMR correlation chart Grouping Chemical Shift (ppm) H-C-C 1.0 - 1.8 H-C=C 4.5 - 6.0 H-C-C=(O, C or N) 2.0 - 3.0 H-CC 1.8 - 3.0 H-C-O 3.3 - 4.4 H-C-N 2.3 - 3.0 H-C-X 3.0 - 4.5 H-Ar 6-9 H-O-C 2–5 H-O-Ar 4-7 H-N-C 1- 5 H-N-Ar 3-6 H-C=O 8 - 10 H-O-C=O 10 - 12 NOTES 1. These chemical values are for hydrogens attached to single functional groups. If a hydrogen is attached to a carbon bonded to two functional groups (e.g. Ar-CH2-CO-), it will be shifted to higher values compared to that for a single functional group (e.g. ArCH2-C- or -CH2-CO). 2. In general, CH3 (methyl) protons occur at the lower of the chemical range for a particular grouping, CH2 (methylene) in the middle and CH (methine) at the higher end. EXAMPLE 1B (CONT’D) (b) a b c CH 3CH 2CCH 3 O Proton a b c Bonding H-C-C H-C-C= H-C-C= CLASS EXERCISE 5B What would be the chemical shifts for peaks in your compound? 25 Chemical Shift 1.0 – 1.8 2.0 – 3.0 2.0 – 3.0 Structure Determination EXAMPLE 2B Look back at the spectrum in Figure 12. What bond groups are suggested by the chemical shifts of its peaks? The two peaks have chemical shifts of 1.5-1.8 and 3.2-3.6. The former could be H-C-X or H-C-O. Since we know the compound contains Br and not O, we can eliminate the latter. The latter is certainly H-C-C. CLASS EXERCISE 6B What bond groups are suggested by the spectra? The number of atoms of each type of hydrogen atom The area of a NMR absorption peak is related to the number of protons causing that peak. However, there is no easy way of linking the area to an exact number, so all that can be done is a relative measure between peaks in the same spectrum. For example, if the peak area of peak A is double that of peak B, we can say that there are twice as many absorbing protons causing peak A as causing peak B. However, this could mean that there are 2:1 or 4:2 or 6:3 etc. EXAMPLE 1C What are the relative peak areas for (a) 3-pentanol (b) butanone? (a) Using the abc labels as in Example 1B, this gives a ratio of 6:4:1:1. (b) 3:2:3. CLASS EXERCISE 5C What are the relative peak areas for your compound? 26 Structure Determination EXAMPLE 3 The peak areas for three peaks in an NMR spectrum are recorded as 25, 49 and 38. What are the relative number of hydrogens? Divide the peak areas by the smallest number and round to 1 decimal place: 1: 2: 1.5. If necessary, multiply the ratio to get whole numbers: in this case x 2 to give 2: 4: 3. EXERCISE 6C Given the peak areas for the spectra in Figures 13 and 14 are as follows, determine the ratio of hydrogens of each type. The areas are from left to right (high shift to low: 13: 78, 161, 153, 235 14: 867, 348, 358, 172 The number of non-equivalent hydrogen atoms on adjacent atoms Finally, we come to the explanation of why the peaks in the spectra you have seen are not simple, single peaks, but split into multiple symmetrical sub-peaks. Any non-equivalent hydrogen atoms within three bonds lengths of the hydrogen causing a peak will “interfere” with the absorption energy required to flip the magnetic field of the absorbing proton. The absorption band is split, with each neighbouring non-equivalent hydrogen adding one subpeak, as shown in Figure 16. 2 extra excited states excited state splitting by 2 adjacent hydrogens CH-CH2 ground state ground state hydrogen causing peak hydrogens causing splitting FIGURE 16 Splitting of absorption by adjacent hydrogen atoms Splitting pattern rules 1. When a proton has n non- equivalent protons on a neighbouring C atom, then its NMR signal will be split into n + 1 peaks. 2. Chemically equivalent protons do not split each other. 3. Hydrogens on non-carbon atoms are not involved in splitting. Split peaks are given names for easy reference: singlet (one peak), doublet (two), triplet (three), quartet (four) and multiplet (many and non-symmetric). 27 Structure Determination EXAMPLE 1D What splitting patterns would be expected for (a) 3-pentanol (b) butanone? (a) Remember that the splitting is caused by hydrogens other than those causing the peak! Hydrogen group a: there are two hydrogens on the neighbouring carbon (within three bonds), so the peak for “a” types will be a triplet. b: there are 3 hydrogens on one adjacent C and 1 on the other, for a total of four. This means the peak will be split into five sub-peaks. c: Two hydrogens on each side means splitting into 5 peaks. The hydrogen on the O is not involved. d: the hydrogen is on the O, so it is not split. (b) a: triplet b: quartet c. singlet CLASS EXERCISE 5D What are the splitting patterns for your compound? Now draw the spectrum for your compound. 10 8 6 4 Chemical Shift (ppm) 28 2 0 Structure Determination EXAMPLE 2D Look back at the spectrum in Figure 12. How does the splitting pattern correlate with the structure? A quartet means that three hydrogens are on the adjacent C, ie a CH 3 group. A triplet means that two hydrogens are on the adjacent C, ie a CH 2 group. CLASS EXERCISE 6D What do the splitting patterns in Figures 13 and 14 indicate about the hydrogen groupings? Now try to combine all that you know about the compounds giving the spectra in Figures 13 and 14. 29 Structure Determination EXERCISE 7 What information can you obtain from the following NMR spectra? Peak areas 97 151 Peak areas 531 347 Peak areas 237 234 202 352 309 1049 371 30 Structure Determination CLASS EXERCISE 8 Why is impossible to have a spectrum that has one doublet only? What is the explanation for such a spectrum? 6. Mass Spectrometry (MS) This is a technique that allows us to measure the molecular weight of the molecule. Additionally we can also gain valuable information about unknown substances by measuring the masses of the fragments produced when high energy molecules fly apart. You will notice the different term used in the title: spectrometry, not spectroscopy. This is because there is no absorption or emission of radiation involved. The spectrum is a graph of particle mass (horizontal axis) against number of particles (vertical axis). How does mass spectrometry work? A very small amount of sample is introduced into the mass spectrometer and bombarded by a beam of high energy, most often in the form of rapidly moving electrons electrons. When this high energy beam strikes the organic molecule it dislodges a valence electron and produces a positively charged species, as shown in Equation 1. This ionised molecule is known as the molecular ion, and has the same mass as the neutral compound. M M+ + e Eqn 1 In addition to causing ionization, electron bombardment transfers such a large amount of energy to the target molecules that they often fly apart into a number of smaller pieces, some retaining the positive charge and some remaining neutral. All the positive ions of the various masses are separated from other particles and then by their different masses to produce the spectrum. The peaks corresponding to the break up of the molecule are called the fragmentation pattern. A typical mass spectrum is shown in Figure 17. base peak fragmentation pattern 135 77 intensity (%) 104 165 180 molecular ion m/e (mass to charge ratio) FIGURE 17 A typical mass spectrum 31 Structure Determination Interpreting mass spectra 1. Molecular weight – in most, but not all cases, some of the molecular ion remains unfragmented and reaches the detection system. Therefore, it produces a peak on the spectrum. Therefore, the highest mass peak in the spectrum is most likely to be that of the molecular ion, and therefore, equal to the molecular weight of the compound. For example, the highest mass peak in the spectrum in Figure 17 is 180, and could therefore be the molecular weight of the compound. How can you be sure that it is the molecular ion? This is not simply answered, but when we look at the instrumentation side of mass spectrometry, some clues will be gained. One point to make – in most spectra, you will notice a very small peak one higher than what is described as the molecular ion peak. This is due to the a small natural proportion of carbon-13 atoms in the carbon population. 1 in 100 carbon atoms weighs 13, not 12, so any molecule with this atom in it will weigh one more than the majority. It does not alter the molecular ion assignment. 2. Molecular fingerprint – each molecule will always fragment in the same way, so compounds can be positively identified by a comparison of their mass spectra in the same fashion as we do with infra red. However, the mass spectra of very similar compounds are very similar, and not as good a “fingerprint” as the infrared spectra. This is shown in Figure 18. 3. Isotope peaks – as mass spectrometry measures individual ions, it shows a peak for each isotope of a mixture. Hence in chlorine for example, we see the peaks due to both the 37Cl and 35Cl and these always occur in the natural 3:1 ratio. These are referred to as isotope peaks. Bromine shows peaks for 79Br and 81Br, and these are often observed as repeating peaks in their 1:1 isotopic ratios throughout the mass spectrum. These are shown in Figure 19. 4. The “nitrogen rule” – a molecule of even number molecular weight must contain either no nitrogen or an even number of nitrogen; likewise a molecular weight which is odd signifies an odd number of nitrogen. It doesn’t matter what other elements are in the compound. It isn’t easy to prove this, but you will not be able to find an example that disproves it! 5. Characteristic fragmentation losses – some functional groups fragment in a particular and consistent way; for example, ethyl esters always lose the OCH 2CH3 group, thus showing a strong peak which has a mass loss of 45 less than the molecular ion. This is shown in Figure 20. What You Need To Be Able To Do list non-spectroscopic test procedures for identifying organic compounds explain why UV spectra are not general useful for structure determination interpret IR spectra interpret NMR spectra interpret mass spectra 32 Structure Determination FIGURE 18 Mass spectra of (a) 1,3-dimethylbenzene and (b) 1,4-dimethylbenzene FIGURE 19 Mass spectra of (a) 2-chlorobenzoic acid and (b) 2-bromobenzoic acid FIGURE 20 Mass spectra of (a) ethyl benzoate and (b) ethyl hexanoate 33 Structure Determination CLASS EXERCISE 10 What information can you obtain from the mass spectra below? 34