Enediyne Natural Products: Biosynthesis and Prospect Towards Engineering Novel Antitumor Agents ,
advertisement

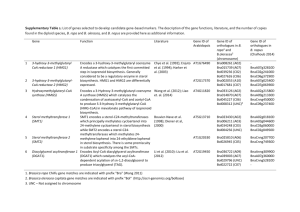
Current Medicinal Chemistry, 2003, 10, 2317-2325 2317 Enediyne Natural Products: Biosynthesis and Prospect Towards Engineering Novel Antitumor Agents Ben Shen1,2*, Wen Liu1 and Koichi Nonaka1 1 Division of Pharmaceutical Sciences and Wisconsin, Madison, WI 53705, USA 2 Department of Chemistry, University of A b s t r a c t : This review gives a brief account on the current status of enediyne biosynthesis and the prospective of applying combinatorial biosynthesis methods to the enediyne system for novel analog production. Methods for cloning enediyne biosynthetic gene clusters are first reviewed. A unified paradigm for enediyne biosynthesis, characterized with (a) the enediyne PKS, (b) the enediyne PKS accessory enzymes, and (c) tailoring enzymes, is then presented. Strategies and tools for novel enediyne analog production by combinatorial biosynthesis are finally discussed. The results set the stage to decipher the molecular mechanism for enediyne biosynthesis and lay the foundation to engineer novel enediynes by combinatorial biosynthesis for future endeavors. Keywords: Biosynthesis, C-1027, calicheamicin, combinatorial biosynthesis, enediyne, polyketide synthase. INTRODUCTION Neocarzinostatin (NCS), the first member of the enediyne family of antitumor antibiotics, was originally discovered as a macromolecular antitumor antibiotic from the culture filtrates of a Streptomyces carzinostaticus strain in 1965 [1]. Although it became clear shortly after its discovery that all biological activities of NCS resided in a nonprotein chromophore, the NCS chromophore structure (1) was not elucidated until 1985 [2], revealing an unprecedented bicyclo[7,3,0]dodecadiynene system, i.e., the 9-membered enediyne core, decorated with an aminosugar and a naphthoic acid moiety. However, this seminal work was underappreciated in the subsequent two years, and it was the discovery of the calicheamicins (2) from a Micromonospora echinospora strain [3,4] and the esperamicins (3) from an Actinomadura verrucosospora strain [5,6] in 1987 that grabbed the attention of chemists and biologists alike to the enediynes. In contrast to 1, structural elucidation of 2 and 3 unveiled a novel biocyclo[7,3,1]tridecadiynene system, i.e., the 10-membered enediyne core, decorated with several deoxysugar moieties. Since then, the enediyne family of natural products has been the focus of intense research activity in the fields of chemistry, biology, and medical sciences because of their highly unusual molecular architectures, biological activities, and modes of actions [713]. Over twenty enediyne natural products are currently known, and new members of the enediyne family are continuously to be discovered with the newest addition, the shishijimicins (4) from a marine ascidian D i d e m n u m proliferum species, reported in early 2003 [14]. The enediyne natural products could be classified into two sub*Address correspondence to this author at the Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, 777 Highland Ave., Madison, WI 53705, USA; Tel: +1-608-263-2673; Fax: +1-608-262-5345; E-mail: bshen@pharmacy.wisc.edu 0929-8673/03 $41.00+.00 categories according to the enediyne core structures. Members of the 9-membered enediyne core sub-category are chromoproteins consisting of an apo-protein and the enediyne chromophore, with N1999A2 (5 ) from Streptomyces sp. AJ9493 as the only exception that was isolated as a chromophore alone [15]. The apo-protein acts as a stabilizer and specific carrier for the otherwise unstable chromophore and its transport and interaction with target DNA. Due to their intrinsic reactivity, only three other chromoprotein chromophore structures, in additional to 1 and 5 , are currently known—kedarcidin (6 ) from Actinomycete L585-6 [16], C-1027 (7) from Streptomyces globisporus [17-20], maduropeptin (8) from Actinomadura madurae [21]. Members of the 10-membered enediyne core sub-category are in general more stable, all of which were isolated as discrete small molecules. In addition to 2, 3, 4, other members of this sub-category whose structures have been elucidated include dynemicin (9 ) from Micromonospora chersina sp. nov. No. M965-1 [22,23] and namenamicin (10) from a marine ascidian Polysyncraton lithostrotum species [24] (Figure 1). Members of both sub-categories of enediynes share a common mechanism of action, despite their structural difference. The enediyne core undergoes an electronic rearrangement (Bergman or Myers rearrangement) to form a transient benzenoid diradicals that damage DNA by abstracting hydrogen atoms from the deoxyribose moiety on both strands. Subsequent reactions of the resultant deoxyribose carbon-centered radicals with molecular oxygen initiate a process that leads in both single-strand and doublestrand DNA cleavage [7-13,25-27]. This novel mechanism of DNA damage has important implications for the development of the enediynes into clinical anticancer drugs [25-36]. Although the natural enediynes have seen limited use as clinical drugs mainly because of substantial toxicity, various polymer-based delivery systems or enediyneantibody conjugates have shown great clinical success and/or promise in anticancer chemotherapy [8,10,12,13,37,38]. For © 2003 Bentham Science Publishers Ltd. 2318 Current Medicinal Chemistry, 2003, Vol. 10, No. 21 Shen et al. example, the poly(styrene-co-maleic acid)-conjugated NCS was approved in Japan in 1993 and has been marketed since 1994 for use against hepatoma [10]. A CD33 monoclonal antibody (mAB)-calicheamicin conjugate was approved in US in 2000 and has been marked under the trade name of Mylotarg to treat acute myeloid leukemia [37]. Several antiheptoma mAB-C-1027 conjugates have also been prepared and showed to display high tumor specificity and to exert a strong inhibitory effect on the growth of established tumor xenografts [38]. These examples clearly demonstrate that the enediynes can be developed into powerful drugs when their extremely potent cytotoxicity is harnessed and delivered onto the target tumor cells. A great challenge will be to develop ways to make new enediynes for mechanistic and clinical studies. Access to complex natural products such as the enediynes and their analogs by total synthesis poses a monumental challenge to synthetic chemists, and yet tour de force effort by synthetic chemists has led to the total syntheses of almost every member of the enediyne family of natural products as well as a myriad of analogs. Significant progress in developing the enediynes into clinical agents has been made towards (a) improving cancer cell specificity, (b) developing efficient delivery systems to the tumor targets, O A O O OCH3 H2C O O N OCH3 O O O H3C O H3C O (CH3 )2 N O OH O O CH3 OH OH CH3HN O R O O 22 HO O HO Cl CH3 N R = OH (7) R = H (11) (1) NH2 O Cl O HO O H OH O OH H3 CO OH O NH HO CH3 CH3 (8) H3C i-PrO OH HO O H3CO OCH3 Cl NH O N O H3C OCH3 N(CH3) 2 Cl OH O O O O O HOH2C O OH (5) O H3C HO H3C OH O O (6) OH Biosynthesis and Prospect Towards Engineering Novel Antitumor Agents Current Medicinal Chemistry, 2003, Vol. 10, No. 21 2319 (Fig. 1). contd..... B SSSCH3 O NHCO2CH3 OH HO O O O CH3 N O OH H CH3 S O OCH3 NHEt O H3C OCH3 I OCH3 H3C HO CH3O (2) O O OH SSSCH3 O NHCO2CH3 O O HO O O O H 3C HO OH CH3 N O OH H CH3 SCH3 O OCH3 NHPr-i O O H3CO O H3CO (3) NH OCH3 O SSSCH3 CH3 H OH O O NHCO2CH3 CO2H HN HO O O O O OCH3 HO O OH O OH CH3 OH O O CH3 SCH3 SCH3 OH OCH3 NHPr-i (10) (9) OH SSSCH3 O HO N NHCO2CH3 O O HO O N H CH3 O SCH3 OCH3 NHPr-i O (4) Fig. (1). The chromophore structures of naturally occurring 9-membered enediyne chromoproteins: neocarzinostatin (1), C-1027 (7), maduropetin (8), kedarcidin (6), and N1999A2 (5) and as well as the engineered deshydroxy-C-1027 (11). (B) Structural representation of naturally occurring 10-membered enediynes: calicheamicin r1 I (2), esperamicin A1 (3), dynemicin A (9), namenamicin (10), and shishijimicin A (4). and (c) designing triggers and trapping the enediynes as prodrugs [11-13]. However chemical total synthesis has very limited practical value for complex natural products such as the enediynes, and analog generation by chemical modification of the natural products can only access to limited functional groups, often requiring multiple protection and deprotection steps. 2320 Current Medicinal Chemistry, 2003, Vol. 10, No. 21 Complementary to organic synthesis, genetic manipulations of genes governing secondary metabolism— an emerging technology also known as combinatorial biosynthesis—offer a promising alternative to preparing complex natural products and their analogs biosynthetically [39-46]. Specific structural alteration in the presence of other functional groups can often be achieved, and the target molecules will be produced by a recombinant organism that is amenable for large-scale fermentation, thereby lowering the production cost and reducing the environment concern associated with conventional chemical synthesis. The success of this approach depends on (a) the cloning and genetic and biochemical characterization of the biosynthetic pathways of the target metabolites and (b) the development of strategies, methods, and expedient tools for combinatorial manipulation of natural product biosynthetic gene clusters. The enediynes offer an outstanding opportunity to investigate the genetic and biochemical basis for the biosynthesis of structurally complex natural products and to apply the combinatorial biosynthesis principle and methods to enediyne biosynthesis for generating novel enediyne analogs, some of which could potentially be developed into anticancer drugs. In this review, we provide a brief account on the current status of enediyne biosynthesis and the prospective of applying combinatorial biosynthesis methods to the enediyne system for novel analog production. We first review the strategies available for cloning enediyne biosynthetic gene clusters from various organisms. We then examine the genetic and biochemical data for the biosynthesis of 7 and 2, as examples for the 9- and 10membered enediynes, respectively, to illustrate a common polyketide pathway for the enediyne family of natural products, characterized by an iterative type I polyketide synthase (PKS). We finally discuss challenges and possible solutions for combinatorial biosynthesis of enediyne natural products with the production of deshydroxy-C-1027 (11) to demonstrate the feasibility of this approach. Early studies on enediyne biosynthesis by isotope feeding experiments and on cloning and characterization of the apo-proteins can be seen in a previous review [47]. METHODS FOR CLONING OF BIOSYNTHETIC GENE CLUSTERS ENEDIYNE Antibiotic production genes are often clustered in one region of the microbial chromosome, consisting of structural, resistance, and regulatory genes. This clustering phenomenon has become the cornerstone from which strategies for cloning microbial natural product biosynthetic gene clusters were developed [39,48-51]. Practically, once one gene is cloned and confirmed to encode the production of the target molecule, chromosomal walking from this locus is virtually certain to lead to the identification of the entire biosynthetic gene cluster (ranging in size from 20-200kb [48]). Recent technological advancement in DNA sequencing has dramatically reduced the time and effort in determining the DNA sequence once the cluster is cloned. However, it remains to be a great challenge to unambiguously identify, localize, and confirm the target gene cluster from a genome of 9 Mbp (the approximate size Shen et al. of genome of the antibiotic-producing actinomycetes), a daunting task that is further complicated by the fact that most of these organisms contain multiple secondary metabolite biosynthetic gene clusters [49-51]. dNDP-Glucose 4,6-Dehydratase as A Probe This strategy was exemplified by the cloning of the biosynthetic gene cluster for 7 from S. globisporus [52,53] and should be applicable to gene clusters of both the 9- and 10-membered enediynes. Except for 5 and 9, enediynes with known structures all contain at least one deoxyhexose moiety. The biosynthesis of various deoxyhexoses shares a common key intermediate, 4-keto-6-deoxyglucose nucleoside diphosphate, the formation of which from glucose nucleoside diphosphate is catalyzed by the dNDP-glucose 4,6-dehydratase [54,55]. Taking advantage of the high amino acid sequence homology observed among dNDP-glucose 4,6-dehydratases from actinomycetes, Bechthold and coworkers developed a general approach for cloning dNDPglucose 4,6-dehydratase genes by PCR with primers designed according to the conserved regions [56]. We adopted this PCR method to clone the biosynthetic gene cluster for 7 from S. globisporus [52,53]. We first cloned a putative dNDP-glucose 4,6-dehydratase gene, sgcA, from S. globisporus by PCR and confirmed its involvement in 7 biosynthesis by gene disruption and complementation [52]. We then screened the S. globisporus genomic library using sgcA as a probe and localized the entire biosynthetic gene cluster for 7 to an 85-kb contiguous region of the S . globisporus chromosome [53]. We finally sequenced and genetically characterized the cloned sgc cluster, revealing that sgcA is indeed clustered with the other structural genes, the previously cloned cagA gene that encodes the C-1027 apoprotein [57], as well as resistance and regulatory genes for 7 production [53]. Apo-Protein as A Probe This strategy depends on the availability of the apoprotein, many of which have been previously cloned and characterized [47], and its applicability, therefore, is limited only to the chromoprotein enediynes as exemplified by the cloning of the biosynthetic gene cluster for 1 from S . carzinostaticus [48]. We initially attempted to localize the 1 biosynthetic gene cluster by the PCR method [56] to clone the dNDP-glucose-4,6-dehydratase as a probe, following the same strategy as we did for 7 [52]. Although we were successful in amplifying a putative dNDP-glucose-4,6dehydratase gene from S. carzinostaticus, inactivation of it failed to abolish 1 production, auguring against its involvement in 1 biosynthesis. We then reasoned that the apo-protein gene could be used as a probe to clone the chromoprotein enediyne gene cluster, viewing the apoprotein as a resistance mechanism via drug sequestering. This hypothesis gained further support after we confirmed that the cagA gene, encoding the CagA aproprotein for 7, was indeed clustered with the rest of its biosynthesis, resistance, and regulatory genes [52,53]. We then took advantage of the previously cloned ncsA gene, which encodes the NcsA apoprotein for 1 [58,59], as a probe to screen the S. carzinostaticus genomic library and localized Biosynthesis and Prospect Towards Engineering Novel Antitumor Agents the biosynthetic gene cluster for 1 to a 95-kb contiguous region of the S. carzinostaticus chromosome. Our confidence that the cloned gene cluster encoded 1 biosynthesis was further increased after identifying another copy of the dNDP-glucose-4,6-dehydratase gene, different from the one amplified by PCR, within this locus. The colocalization of ncsA and a dNDP-glucose-4,6-dehydratase gene, reminiscent to that for the 7 biosynthetic gene cluster [53], was viewed as an evidence of clustering between biosynthesis and resistance genes. DNA sequence of the 95kb region finally unveiled the ncs cluster, consisting of the ncsA gene, a dNDP-glucose-4,6-dehydratase gene, as well as other biosynthesis, resistance, and regulatory genes, a genetic organization that is highly homologous to that of 7 [48,53]. Resistance Gene as A Probe This strategy was exemplified by the cloning of the biosynthetic gene cluster for 2 from M. echanospora [60,61] and should be applicable to gene clusters of both the 9- and 10-membered enediynes. To clone the 2 biosynthetic gene cluster, Thorson and co-workers first screened a M . echanospora genomic library with probes designed according to the conserved regions of both type I and type II PKSs on the assumption that the enediyene core is of polyketide origin. Since it is known that most actinomycetes contain multiple polyketide pathways [4951,62,63], the resultant clones were further screened with probes designed according to the conserved regions of both glucose-1-phosphate nucleotidyltransferase and dNDPglucose 4,6-dehydratase targeting the biosynthesis of the deoxyhexose moieties of 2. While these preliminary screenings were effective in identifying putative clones for the 2 biosynthetic locus, it was the finding that some of the clones, positive towards both the PKS and deoxyhexose probes, also conferred 2 resistance that ultimately convinced these researchers that they had localized the biosynthetic gene cluster for 2 [47]. They subsequently mapped the 2 resistance gene to calC and characterized CalC as a nonheme iron metalloprotein, although the precise mechanism of how CalC confers 2 resistance remains to be established [60]. DNA sequencing of these clones finally unveiled the entire c a l gene cluster, consisting of PKS genes, deoxyhexose biosynthesis genes, the calC resistance gene, as well as other biosynthesis, resistance, and regulatory genes. Inactivation of the calE8 enediyne PKS gene abolished 2 production, unambiguously confirming the involvement of the cloned cal gene cluster in 2 biosynthesis [61]. Genome Scanning Method This strategy was exemplified by the cloning of the biosynthetic gene clusters for 9 from M. chersina strain M956-1, macromomycin (the precise structure remains unknown) from Streptomyces macromyceticus strain M480M1, as well as several putative novel enediynes whose structures are yet to be determined from other actinomycete strains that previously were not known as enediyne producers. Since this method does not rely on specific biosynthetic machinery, it should be applicable to gene clusters of both the 9- and 10-membered enediynes as well Current Medicinal Chemistry, 2003, Vol. 10, No. 21 2321 as other natural products [48]. Farnet and co-workers developed this so-called “high-throughput genome scanning method to discover metabolic loci” on the basis that genes encoding secondary metabolite biosynthesis in microorganisms are clustered [48]. Similar to the so-called “perfect probes” approach to identify and clone type I PKS gene clusters in a genome [63], this method consists essentially three steps: (a) shotgun DNA sequencing to generate short (700 bp) random genome sequence tags (GSTs) from a genomic library, (b) sequence comparison of GSTs to a database of microbial gene clusters known to be involved in natural product biosynthesis to identify candidate genes, and (c) genomic library screening with selected GTSs as probes for clones containing genes of interest as well as neighboring ones that together may constitute a biosynthetic gene cluster. This method provides an efficient way to survey the biosynthetic potential for a given microorganism and to access specific biosynthetic gene cluster of interest. Using this method, Farnet and coworkers cloned and sequenced several enediyne biosynthetic loci from various microorganisms, including not only these that were known to be enediyne producers such as M . c h e r i s i n a strain M956-1 for 9 and S t r e p t o m y c e s macromyceticus strain M480-M1 for macromomycin but also ones that previously were not known as enediyne producers, revealing several novel enediyne biosynthetic gene clusters. It came as a surprise that the enediyne PKS loci were found at a very high frequency (~15%) among the organisms surveyed, suggesting that the ability of these organisms to produce enediynes or other bioactive natural products might have been greatly underestimated. Guided by these genetic data, enediyne production by these organisms, including those that were not known as enediyne producers previously, were demonstrated under selective growth conditions and detected by the enediyne-specific biochemical induction assays, supporting the involvement of these clusters in enediyne biosynthesis [48]. Enediyne PKS Gene as A Probe This strategy was exemplified by the cloning of the biosynthetic gene clusters for 3 from A. verrucosopora, 8 from A. madurea, and 9 from M. chersina and should be applicable to gene clusters of both 9- and 10-membered enediynes. The cloning and characterization of 7 and 2, as model systems for the 9- and 10-membered enediynes, respectively, revealed a common polyketide pathway for enediyne biosynthesis characterized by the enediyne PKS, iterative type I PKSs that exhibited head-to-tail sequence homology and have identical domain organization (Figure 2) [53,61,64]. Taking advantage of the remarkable similarity in both sequence and domain organization of the enediyne PKSs and their associated accessory enzymes, we developed a PCP-based approach to access the enediyne PKS loci using a set of primers designed according to various conserved region of these enzymes. The effectiveness of this approach was demonstrated by the cloning of the enediyne PKS and its accessory enzymes for 3, 8, and 9 biosynthesis from the corresponding producer, respectively [65]. The availability of the enediyne PKS loci has now set the stage to use them as probes to clone the entire biosynthetic gene clusters. 2322 Current Medicinal Chemistry, 2003, Vol. 10, No. 21 Shen et al. Fig. (2). (A) Domain organization and sequence comparison between representative enediyne PKSs for a 9-membered enediyne core such as 7 (SgcE) and a 10-membered enediyne core such as 2 (CalE8). aa, amino acid (B) A unified paradigm for enediyne biosynthesis featuring the enediyne PKSs to account for the linear unsaturated polyketide intermediates, enediyne PKS accessory enzymes to branch into the 9- or 10-membered enediyne cores, and tailoring enzymes to imbue additional structural diversity. Atoms that were incorporated intact from the acyl CoA precursors to the enediyne cores are shown in bold. A UNIFIED PARADIGM FOR ENEDIYNE BIOSYNTHESIS INVOLVING AN ITERATIVE TYPE I PKS Although feeding experiments with 1 3 C - l a b e l e d precursors clearly established that both the 9- and 10membered enediyne cores were derived (minimally) from eight head-to-tail acetate units [47,64-66], controversy remained until the recent cloning and characterization of the biosynthetic gene clusters of 7 [53] and 2 [61] whether the enediyne cores are assembled by de novo polyketide biosynthesis or degradation from a fatty acid precursor. Elucidation of the 7 and 2 biosynthetic gene clusters and pathways, together with the cloning and sequence analysis of the enediyne PKS loci for 1, 3, 8, and 9, now clearly suggest a common polyketide pathway for the biosynthesis of both 9- and 10-membered enediyne cores, despite the fact that their incorporation pattern by 13C-labeled acetate feeding experiments are distinct [47,64-66]. Common to all enediyne biosynthetic gene clusters known to date is the enediyne PKS—an unprecedented type I PKS consisting of a ketoacyl synthase (KS), an acyltransferase (AT), an acyl carrier protein (ACP), a ketoreductase (KR), a dehydratase (DH), and a C-terminal domain (TD) (also proposed to encode a phosphopantetheinyl transferase) (Figure 2). Flanking the enediyne PKS gene are a group of five to ten genes that are organizationally conserved among all characterized enediyne gene clusters. These genes encode the enediyne PKS accessory enzymes that are unusual oxidoreductases or proteins of unknown functions that are Biosynthesis and Prospect Towards Engineering Novel Antitumor Agents only associated with enediyne biosynthesis. Also identified within the enediyne clusters are genes that encode a myriad of pathway-specific tailoring enzymes for the biosynthesis of the other structural moieties and their attachment to the enediyne cores [48,53,61,64]. Although these findings fell short of revealing the precise mechanism for enediyne core biosynthesis, they have provided sufficient data to formulate a unified paradigm for enediyne biosynthesis. The enediyne PKSs could be envisaged to catalyze the assembly of a linear polyunsaturated intermediate from the acyl CoA precursors in an iterative process. The nascent intermediate is subsequently desaturated to furnish the two yne groups and cyclized to afford either a 9- or 10-membered enediyne core intermediate. The enediyne PKS accessory enzymes serve as the excellent candidates for these processing reactions. Preliminary phylogenetic analysis of the enediyne PKSs and their accessory enzymes showed that they could be grouped into two distinct sub-families according to the 9- or 10membered enediyne core structures. This finding suggested that it might be even possible to predict the enediyne core structure on the basis of sequence analysis of its biosynthetic gene cluster [65]. Further decoration of the enediyne cores with the other structural moieties affords the structural diversity of the enediyne family of natural products, and the pathway specific tailoring enzymes are responsible for these modifications. This emerging paradigm for enediyne biosynthesis, consisting of (a) enediyne PKS-catalyzed synthesis of a linear polyketide intermediate, (b) enediyne PKS accessory enzymes-mediated processing of the enediyne core, and (c) pathway specific tailoring enzymes-driven structural diversification of the enediyne natural products, has now laid the foundation to ask the fundamental mechanistic questions for enediyne biosynthesis and to explore the potential of engineering novel enediyne compounds by applying combinatorial biosynthesis methods to the enediyne biosynthetic machinery. COMBINATORIAL BIOSYNTHESIS OF NOVEL ENEDIYNES A prerequisite for applying combinatorial biosynthesis methods to engineer novel natural products is to have a genetic system in place for the producing organism. Four general methods are available for introducing plasmid DNA into a Streptomyces species: (a) polyethyleneglycol (PEG)mediated protoplast transformation, (b) electroporation, (c) E. coli-Streptomyces conjugation, and (d) phage infection [69]. However, the applicability of each method to a given organism, cannot be predicted a priori, and the effectiveness of these methods to a particular species has to be determined empirically. In spite of these difficulties, significant progress has been made in the past a few years for several enediyne producing organisms. We have developed efficient genetic systems for both the 7-producing S. globisporus and 1producing S. carzinostaticus strains by either PEG-mediated protoplast transformation or E. coli- Streptomyces conjugation. These methods were successfully applied to carry out in vivo genetic characterization of 7 or 1 biosynthesis by gene disruption, replacement, or complementation experiments, setting the stage to engineer Current Medicinal Chemistry, 2003, Vol. 10, No. 21 2323 the 9-membered enediyne biosynthetic pathways [48,52,53]. Thorson and co-workers have successfully developed a genetic system for M. echinospora ssp. calichensis by PEGmediated protopolast transformation and applied it to inactivate the calE8 enediyne PKS gene. The resultant calE8 mutant lost its ability to produce 2, demonstrating the feasibility of manipulating the biosynthesis of 10-membered enediynes such as 2 in vivo [61]. Complementary to manipulating natural product biosynthesis in their native producing organisms, progress has also been made to develop bacterial artificial chromosome (BAC) that can shuttle between E. coli and Streptomyces species for expression of the target gene cluster in a heterologous host. The entire biosynthetic gene cluster could be cloned directly from chromosomal DNA [70-73] or reconstructed from previously isolated overlapping cosmids [74-76]. The resultant BAC could be mobilized into genetically amenable host strains, such as Streptomyces coelicolor or Streptomyces lividans, allowing genetic analysis and manipulation of biosynthetic gene clusters that are otherwise intractable genetically in their native producers. While holding a great promise, this strategy has yet to demonstrate efficient expression of the entire gene cluster and production of the encoded natural product in the heterologous host. Attempts to clone the entire biosynthetic clusters of 1 or 7 into BAC for heterologous expression were not successful so far. In contrast, we have recently discovered that the biosynthetic gene clusters for 1 and 7 reside on giant linear plasmids. By protoplast fusion, we successfully mobilized the giant linear plasmid for 1 biosynthesis from S. carzinosaticus into S. lividans and confirmed 1 production in the heterologous host [Zhang, J.; Nonaka, K.; Shen, B. unpublished data]. These findings provided an exciting alternative to manipulate enediyne biosynthesis in one of the best-characterized Strteptomyces hosts. Although the engineering of novel enediynes with altered enediyne core has to wait for the fundamental understanding of how the enediyne PKS and its accessory enzymes catalyze the enediyne core biosynthesis, the proof-of-principle for combinatorial biosynthesis of novel enediynes has already been demonstrated by the production of deshydroxy-C-1027 (11) [53]. Characterization of the 7 biosynthetic gene cluster and elucidation of the 7 biosynthetic pathway established that the C22-OH group of the β-amino acid moiety of 7 is introduced by the SgcC hydroxylase. Inactivation of the s g c C gene resulted in the production of a novel chromoprotein complex, the chromophore of which was confirmed to be 11. It was noteworthy that 11 is not only biologically active as judged by bioassay with 7 as a positive control but also at least fivefold more stable than 7 at 25 °C in respect to undergoing the Bergman cyclization [53]. The latter property could be potentially explored in developing 7 into clinically useful enediyne drugs. Taking together, these results not only demonstrated the feasibility of generating novel enediynes by manipulating the genes governing their biosynthesis but also suggested that the tailoring enzymes could be as equally rewarding as the enediyne PKS and its accessory enzymes as the targets for engineering novel enediynes. Given the relatively conserved enediyne core structures yet distinct periphery moieties among the enediynes, it is tempting to speculate that some 2324 Current Medicinal Chemistry, 2003, Vol. 10, No. 21 Shen et al. of the tailoring enzymes could cross-talk among the various enediyne pathways, further expanding the enediyne structural diversity accessible by combinatorial biosynthesis methods. CA78747. B.S. is a recipient of National Science Foundation CAREER Award MCB9733938 and National Institutes of Health Independent Scientist Award AI51687. CONCLUSION AND PROSPECTIVE REFERENCES The studies of enediyne biosynthesis have enjoyed a renaissance in the past a few years. The enediyne family of natural products has seen a steady growth. Specific and general methods to access enediyne biosynthetic gene clusters have been developed. Biosynthetic gene clusters for three 9-membered enediynes (1, 7, 8) and three 10-membered enediynes (2, 3, 9) have been cloned, respectively. A unified paradigm for enediyne biosynthesis has emerged, featuring an unprecedented iterative type I PKS and its accessory enzymes as well as a myriad of tailoring enzymes. Genetic systems for representative 9- and 10-membered enediyne producing organisms have been developed, and genetic characterization of enediyne gene clusters in vivo has begun to unveil new mechanistic insights into enediyne biosynthesis. Judicial application of combinatorial biosynthesis methods to the enediyne biosynthetic machinery has demonstrated the production of novel enediynes. It is becoming increasingly clear that access to the genetic information for natural product biosynthesis is no longer the bottleneck for combinatorial biosynthesis. A great challenge in studying enediyne biosynthesis lies in the revelation of the fundamental mechanism of how the enediyne PKS and its accessory enzymes assemble the enediyne cores from the acyl CoA precursors. Although sequence analysis and functional comparison among the various enediyne gene clusters have enabled us to formulate a few rudimentary hypotheses, innovation in both biochemistry concepts and experimental designs are needed to make the breakthrough in deciphering the precise mechanism for the enediyne core biosynthesis. Outcomes from these studies will greatly accelerate the pace to realize the full potential of combinatorial biosynthesis of enediyne natural products. We remain optimistic that enediyne biosynthesis will continue to thrive in the coming years, particularly in mechanistic enzymology and in generation of novel enediynes by combinatorial biosynthesis methods. [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] ABBREVIATION [22] ACP AT DH KR KS Mab NCS PKS TD = = = = = = = = = Acyl carrier protein Acyl transferase Dehydratase Ketoreductase Ketoacyl synthase Monoclonal antibody Neocarzinostatin Polyketide synthase C-terminal domain ACKNOWLEDGEMENTS Enediyne biosynthesis described from our laboratory was supported in part by National Institutes of Health grant [23] [24] [25] [26] [27] [28] [29] [30] [31] Ishida, N.; Miyazaki, K.; Kumagai, K.; Rikimaru, M. J. Antibiot., 1965, 18, 68. Edo, K.; Mizugaki, M.; Koide, Y.; Seto, H.; Furihata, K.; Otake, N.; Ishida, N. Tetrahedron Lett., 1985, 26, 331. Lee, M.D.; Dunne, T.S.; Siegel, M.M.; Chang, C.C.; Morton, G.O.; Borders, D.B. J. Am. Chem. Soc., 1987, 109, 3464. Lee, M.D.; Dinne, T.S.; Chang, C.C.; Ellestad, G.A.; Siegel, M.M.; Morton, G.O.; McGahren, W.J.; Borders, D.B. J. Am. Chem. Soc., 1987, 109, 3466. Golik, J.; Clardy, J.; Dubay, G.; Groenewold, G.; Kawaguchi, H.; Konishi, M.; Krishnan, B.; Ohkuma, H.; Saitoh, K.; Doyle, T. W. J. Am. Chem. Soc., 1987, 109, 3461. Golik, J.; Dubay, G.; Groenewold, G.; Kawaguchi, H.; Konishi, M.; Krishnan, B.; Ohkuma, H.; Saitoh, K.; Doyle, T.W. J. Am. Chem. Soc., 1987, 109, 3462. Nicolaou, K.C.; Dai, W.-M. Angew. Chem. Int. Ed. Engl., 1991, 30, 1387. Doyle, T.W.; Border, D.B. Enediyne Entibiotics as Antitumor Agents. Marcel-Dekker: New York, 1995. Smith, A.L.; Nicolaou, K.C. J. Med. Chem., 1996, 39, 2103. Maeda, H.; Edo, K.; Ishida, N. Eds. Neocarzinostatin: The Past, Present, and Future of an Anticancer Drug. Springer-Verlag:New York, 1997. Xi, Z.; Goldberg, I.H. in Comprehensive Natural Products Chemistry, Barton, D.; Nakanish, K.; Meth-Cohn, O., Eds. Elesiver:New York, 1999, vol 7, p.553. Thrson, J.S.; Sievers, E.L.; Ahlert, J.; Shepard, E.; Whitwam, R.E.; Onwueme, K.C.; Ruppen, M. Curr. Pharm. Design., 2000, 6, 1841. Jones, G.B.; Fouad, F.S. Curr. Pharm. Des., 2002, 8, 2415. Oku, N.; Matsunaga, S.; Fusetani, N. J. Am. Chem. Soc., 2003, 125, 2044. Ando, T.; Ishii, M.; Kajiura, T.; Kameyama, T.; Miwa, K.; Sugiura, Y. Tetrahedron Lett., 1998, 39, 6495. Leet, J.E.; Schroeder, D.R.; Hofstead, S.J.; Golik, J.; Colson, K.L.; Huang, S.; Klohr, S.E.; Doyle, T.W; Matson, J.A. J. Am. Chem. Soc., 1992, 114, 7946. Minami, Y.; Yoshida, K.; Azuma, R.; Saeki, M.; Otani, T. Tetrahehron Lett., 1993, 34, 2633. Yoshida, K.; Minami, Y.; Azuma, R.; Saeki, M.; Otani, T. Tetrahehron Lett., 1993, 34, 2637. Iida, K.; Fukuda, S.; Tanaka, T.; Hirama, M.; Imajo, S.; Ishiguro, M.; Yoshida, K.; Otani, T. Tetrahedron Lett., 1996, 37, 4997. Otani, T.; Yoshida, K.; Sasaki, T.; Minami, Y. J. Antibiot., 1999, 52, 415. Schroeder, D.R.; Colson, K.L.; Klohr, S.E.; Zein, N.; Langley, D.R.; Lee, M.S.; Matson, J.A.; Dolye, T.W. J. Am. Chem. Soc., 1994, 116, 9351. Konishi, M.; Ohkuma, H.; Matsumoto, K.; Tsuno, T.; Kamei, H.; Miyaki, T.; Oki, T.; Kawaguchi, H. J. Antibiot., 1989, 42, 1449. Myers, A.G.; Fraley, M.E.; Tom, N.J.; Cohen, S.B.; Madar, D.J. Chem. Biol., 1995, 2, 33. McDonald, L.A.; Capson, T.L.; Krishnamurth, G.; Ding, W.-D.; Ellestad, G.A.; Bernan, V.S.; Maiese, W.M.; Lassota, P.; Discafani, C.; Kramer, R.A.; Ireland, C.M. J. Am. Chem. Soc., 1996, 118, 10898. Stassinopoulos, A.; Ji, J.; Gao, X.; Goldberg, I.H. Science, 1996, 272, 1943. Ikemoton, N.; Kumar, R.A.; Ling, T.T.; Elleastad, G.A. Proc. Natl. Acad. Sci. USA, 1995, 92, 10506. Dedon, P.C.; Goldberg, I.H. Chem. Res. Toxicol., 1992, 5, 311. SugiuraY.; Shiraki, T.; Konishi, M.; Oki, T. Proc. Natl. Acad. Sci. USA., 1990, 87, 3831. Zein, N.; Closon, K.L.; Leet, J.E.; Schroeder, D.R.; Solomon, W.; Dolye, T.W.; Casazza, A.M. Proc. Natl. Acad. Sci. USA., 1993, 90, 2822. Nicolaou, K.C.; Stabila, P.; Esmaeli-Azad, B.; Wrasidlo, W.; Hiatt, A. Proc. Natl. Acad. Sci. USA., 1993, 90, 3142. Kappen, L.S.; Goldberg, I.H. Science, 1993, 261, 1319. Biosynthesis and Prospect Towards Engineering Novel Antitumor Agents [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] [46] [47] [48] [49] [50] [51] [52] [53] [54] Yu, L.; Goldberg, I.H.; Dedon, P.C. J. Biol. Chem., 1994, 269, 4144. Hensens, O.D.; Chin, D.H.; Stassinopoulos, A.; Zink, D.L.; Kappen, L.S.; Goldberg, I.H. Proc. Natl. Acad. Sci. USA., 1994, 91, 4534. Ho, S.N.; Boyer, S.H.; Schreiber, S.L.; Danishefsky, S.J.; Crabtree, G.R. Proc. Natl. Acad. Sci. USA., 1994, 91, 9203. Schor, N.F.; Tyuina, Y.Y.; Fabisiak, J.P.; Tyurin, V.A.; Lazo, J.S.; Kagan, V.E. Brain Res., 1999, 831, 125. Dziegielewski, J.; Beerman, T.A. J. Biol. Chem., 2002, 277, 20549. Sielvers, E.L.; Appelbaum, F.R.; Spielberger, R.T.; Forman, S.J.; Flowers, D.; Smith, F.O.; Shannon-Dorcy, K.; Berger, M.S.; Bernstein, I.D. Blood, 1999, 93, 3678. Brukner, I. Curr. Opin. Oncol. Endocr. Met. Invest. Drugs., 2000, 2, 344. Hopwood, D.A. Chem. Rev., 1997, 97, 2465. Cane, D.E.; Walsh, C.T.; Khosla, C. Science, 1998, 6, 319. Shen, B. Curr. Top. Chem., 2000, 209, 1. Staunton, J.; Wessman, K.J. Nat. Prod. Rep., 2001, 18, 380. Strohl, W.R. Met. Engineer., 2001, 3, 4. Du, L.; Shen, B. Curr. Opinion Drug Discov. Dev., 2001, 4, 215. Rodriguez, E.; McDaniel, R. Curr. Opinion Microbiol., 2001, 4, 526. Walsh, C.T. ChemBioChem, 2002, 3, 124. Thorson, J.S.; Shen, B.; Whitwam, R.E.; Liu, W.; Li, Y.; Ahlert, J. Bioorg. Chem., 1999, 27, 172. Zazopoulos, E.; Huang K.; Staffa, A.; Liu, W.; Bachmann, B.O.; Nonaka, K.; Ahlert, J.; Thorson, J.S.; Shen, B.; Farnet, C.M. Nat. Biotechnol., 2003, 21, 187. Bentley, S.D.; Chater, K.F.; Cerdeno-Tarraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; Bateman, A.; Brown, S.; Chandra, G.; Chen, C.W.; Collins, M.; Cronin, A.; Fraser, A.; Goble, A.; Hidalgo, J.; Hornsby, T.; Howarth, S.; Huang, C.H.; Kieser, T.; Larke, L.; Murphy, L.; Oliver, K.; O'Neil, S.; Rabbinowitsch, E.; Rajandream, M.A.; Rutherford, K.; Rutter, S.; Seeger, K.; Saunders, D.; Sharp, S.; Squares, R.; Squares, S.; Taylor, K.; Warren, T.; Wietzorrek, A.; Woodward, J.; Barrell, B.G.; Parkhill, J.; Hopwood, D.A. Nature, 2002, 417, 141. Omura, S.; Ikeda, H.; Ishikawa, J.; Hanamoto, A.; Takahashi, C.; Shinose, M.; Takahashi, Y.; Horikawa, H.; Nakazawa, H.; Osonoe, T.; Kikuchi, H.; Shiba, T.; Sakaki, Y.; Hattori, M. Proc. Natl. Acad. Sci. USA, 2001, 98, 12215. Ikeda, H.; Ishikawa, J.; Hanamoto, A.; Shinose, M.; Kikuchi, H.; Shiba, T.; Sakaki, Y.; Hattori, M.; Omura, S. Nat. Biotechnol., 2003, 21, in press. Liu, W.; Shen, B. Antimicrob. Agents Chemother., 2000, 44, 382. Liu, W.; Christenson, S. D.; Standage, S.; Shen, B. Science, 2002, 297, 1170. Trefzer, A.; Salas, J.A.; Bechthold, A. Nat. Prod. Rep., 1999, 16, 283. Current Medicinal Chemistry, 2003, Vol. 10, No. 21 [55] [56] [57] [58] [59] [60] [61] [62] [63] [64] [65] [66] [67] [68] [69] [70] [71] [72] [73] [74] [75] [76] 2325 He, X.M.; Liu, H.-w. Annu. Rev. Biochem., 2002, 71, 701. Decker, H.; Gaisser, S.; Pelzer, S.; Schneider, P.; Westrich, L.; Wohlleben, W.; Bechthold, A. FEMS Microbiol. Lett., 1996, 141, 195. Sakata, N.; Ikeno, S.; Hori, M.; Hamada, M.; Otani, T. Biosci. Biotech. Biochem., 1992, 56, 1592. Maeda, H.; Glaser, C.B.; Kuromizu, K.; Meienhofer, J. Arch. Biochem. Biophys., 1974, 164, 379. Gibson, B.W.; Herlihy, W.C.; Samy, T.S.A.; Hahm, K.S.; Maeda, H.,; Meienhofer, J.; Biemann, K. J. Biol. Chem., 1984, 259, 10801. Whitwam, R. E.; Ahlert, J.; Holman, T.R.; Ruppen, M.; Thorson, J.S. J. Am. Chem. Soc., 2000, 122, 1556. Ahlert, J.; Shepard, E.; Lomovskaya, N.; Zazopoulos, E.; Staffa A.; Bachmann, B. O.; Huang, K.; Fonstein L.; Czisny, A.; Whitwam, R.E.; Farnet, C.M.; Thorson, J.S. Science, 2002, 297, 1173. Ruan, X.; Stassi, D.; Lax, S.A.; Katz, L. Gene, 1997, 203, 1. Santi, D.V.; Siani, M.A.; Julien, B.; Kupfer, D.; Roe, B. Gene, 2000, 247, 97. Shen. B. Curr. Opinion Chem. Biol., 2003, 7, 285. Liu, W.; Ahlert, J.; Gao, Q.; Wendt-Pienkowski, E.; Shen, B. Thorson, J.S.; submitted. Hensens, O.D.; Ginger, J.-L.; Goldberg, L.H. J. Am. Chem. Soc., 1989, 111, 3295. Tokiwa, Y.; Miyoshi-Saitosh, M.; Kobayashi, H.; Sunaga, R.; Konishi, M.; Oki, T.; Iwasaki, S. J. Am. Chem. Soc., 1992, 114, 4107. Lam, K.S.; Veitch, J.A.; Golik, J.; Krishnan, B. J. Am. Chem. Soc., 1993, 115, 12340. Kieser, T.; Bibb, M.J.; Buttner, M.J.; Chater, K.F.; Hopwood, D.A. Practical Streptomyces Genetics. The John Innes Foundation:Norwich, U.K., 2000. Gillespie, D.E.; Brady, S.F.; Bettermann, A.D.; Cianciotto, N.P.; Liles, M.R.; Rondon, M.R.; Clardy, J.; Goodman, R.M.; Handelsman, J. Appl. Environ. Microbiol., 2002, 68, 4301. Brady, S.F.; Chao, C.J.; Clardy, J. J. Am. Chem. Soc., 2002, 124, 9968. Zengler, K.; Toledo, G.; Rappe, M.; Elkins, J.; Mathur, E.J.; Short, J.M.; Keller, M. Proc. Natl. Acad. Sci., 2002, 99, 15681. Miao, V.; Cöeffet-LeGal, M.-F.; Penn, J.; Whiting, A.; Parr, I.; Bouchard, M.; Silva, C.; Doekel, S.; Alexander, D.; Martin, S.; Gibson, T.; Brost, R.; Sinneman, S., Henton-Jones, D.; Chapple, J.P.; Daavies, J.E.; Baltz, R.H.; Brian, P. Abstract S123, Society for Industrial Microbiology Annual Meeting, August 11-15, 2002, Philadelphia, PA. Sosio, M.; Giusino, F.; Cappellano, C.; Bossi, E.; Puglia, A.M.; Donadio, S. Nat. Biotechnol., 2000, 18, 343. Sosio, M.; Bossi, E.; Donadio, S. Nucleic Acids Res., 2001, 37, e37. Donadio, S.; Monciardini, P.; Aldunia, R.; Mazza, P.; Chiocchini, C.; Cavaletti, L.; Sosio, M.; Puglia, A.M. J. Biotechnol., 2002, 99, 187.