Document 11743579
advertisement

10/2/14 The Neutral Theory of Molecular Evolution
v Kimura (1968)
² initially viewed as a challenge to Darwinian
evolution
² e.g., King & Jukes 1969 Science 164:788-798.
v many genetic polymorphisms have no
effect on fitness and are therefore
selectively neutral
v neutral polymorphisms are maintained by
the combined effects of mutation and drift
² mutations introduce new alleles as others are
lost through drift
Origins of the “Selectionist-Neutralist”
Debate
v the only “mutations” early biologists saw were ones that
had phenotypic effects
v 1953 - structure of DNA (Watson & Crick / Franklin)
v 1960-70’s - protein electrophoresis
² revealed allelic diversity for many genes
v 1968 - Motoo Kimura - the neutral theory
² motivated by allozyme (amino acid) variation
v discovery of “junk DNA”
² 98.5% of human genome is non-coding
v DNA sequencing has revealed substantial silent and noncoding variation, suggesting that much genetic variation
is selectively neutral (or nearly so!)
1 10/2/14 Infinite Alleles/Sites Model
v what is the expected level of genetic
diversity (heterozygosity) given mutation
and drift in a finite population?
v suppose a gene is 900 base pairs long,
coding for 300 amino acids
² there are 4900 = 10542 possible sequences
(sorta…)
v thus, we can reasonably assume that each
new mutation generates a unique allele…
Infinite Alleles/Sites Model
v it follows that alleles with the same
sequence are identical by descent
v autozygous - a genotype with two alleles
that are identical by descent
v allozygous - a genotype with alleles that are
not identical by descent (is this possible?)
² arbitrarily declare all alleles unique at t = 0
v autozygous = homozygous under the infinite
alleles model
² thus, the level of heterozygosity can be
predicted from the expected level of
autozygosity
2 10/2/14 Infinite Alleles/Sites Model
v Ft = probability that two randomly chosen
alleles are IBD
² same as autozygosity if we randomly choose
alleles to form genotypes
" 1 %
"
1 %
2
2
Ft = $ '(1− µ) + $1−
'(1− µ) Ft−1
# 2N &
# 2N &
² in this model, mutations generate new alleles
and “erase” IBD
€
Infinite Alleles Model
v in a random-breeding population of
constant size, an equilibrium is reached
where the increase in autozygosity (~IBD)
due to loss of alleles by drift is exactly
countered by the increase in heterozygosity
produced by new mutations
v solving for Ft = Ft-1 yields:
Fˆ =
€
1
1+ 4Nµ
3 10/2/14 Infinite Alleles Model
Fˆ =
1
1
1
=
=
1+ 4Nµ 1+ 4N e µ 1+ θ
v given the assumption of infinite alleles, any
genotype that is not autozygous is
heterozygous, so
€
Ĥ = 1− F̂ =
4N eµ
θ
=
1+ 4N eµ 1+ θ
Neutral Expectations for Genetic Diversity
4 10/2/14 F̂ =
1
1
=
1+ 4N µ 1+ θ
v although an ideal population is expected to
reach an “equilibrium” value of F, the
population is not really at equilibrium, but
rather in a “dynamic steady state” because
there is a continual turnover of alleles
² the most common allele is periodically
replaced by another, other alleles are lost,
and new alleles are produced by mutation
v DNA sequencing
revealed
surprisingly high
levels of neutral
genetic variation
5 10/2/14 DNA Sequence-based Measures of
Genetic Variation
v S = number of segregating sites
v ∏ = average number of pairwise
differences between sequences
v ∏ analogous to heterozygosity
v can derive theoretical expectations for
both measures for an idealized, random
breeding population (and also assuming
an “infinite sites” model)…
Segregating sites
v expected number of segregating sites:
" 1 1 1
1 %
E ( S ) = θ $1+ + + +... +
'
# 2 3 4
k −1 &
v where θ = 4Nµ and k = the number of
sequences in the sample
v µ (“mu”) is the per locus mutation rate =
mutation rate per site per generation x
length of sequence
6 10/2/14 Coalescent theory often provides “easy”
derivations of classical theory
v e.g., number of segregating sites in a sample
² is a function of the total length (in
generations) of the coalescent tree E(T) times
the mutation rate per locus per generation
k
k−1
# k & k
4N
1
E (T ) = E %∑ iTi ( = ∑ iE (Ti ) = ∑ i
= 4N ∑
$ i= 2 ' i= 2
i= 2 i(i −1)
i=1 i
k−1
k−1
1
1
E ( S ) = µE (T ) = 4Nµ∑ = θ ∑
i
i
i=1
i=1
€
€
Coalescent theory often provides “easy”
derivations of classical theory
v e.g., number of segregating sites in a sample
² is a function of the total length (in
generations) of the coalescent tree E(T) times
the mutation rate per locus per generation
n
n
n
n−1
2k
1
1
= 2∑
= 2∑
k=2 k ( k −1)
k=2 k −1
k=1 k
E [total _ treelength ] = ∑ kE [tk ] = ∑
k=2
n−1
# n−1 1 & θ
1
E ( S ) = % 2∑ ( = θ ∑
$ k=1 k ' 2
k=1 k
7 10/2/14 Average number of pairwise differences
Π=
total number of nucleotide mismatches
total number of pairwise comparisons
v in an idealized population, the expected value
of ∏ is θ:
E ( Π) = θ = 4N µ
€
v θ can also be estimated from S:
" 1 1 1
1 %
θ = E ( S ) $1+ + + +... +
'
# 2 3 4
k −1 &
! 1 1 1$
θ = 16 #1+ + + & = 7.68
" 2 3 4%
# ( 6 × 6 ) + ( 4 × 9 ) + ( 7 ×1) + ( 0 × 484) &
E(θ ) = Π = %
( = 7.90
10
$
'
8 10/2/14 Key point!
v differences in the values for number of
segregating sites and average pairwise
differences lead to the inference that the
gene(s) or the population departs in one or
more ways from the ideal “null model” (i.e.,
constant population size, no selection, etc..)
mtDNA haplotypes
for big brown bats
(Eptesicus fuscus)
east of the Rockies
v θ (∏) = 5.35
v θ (S) = 10.42
9 10/2/14 Data versus histories
v generally, the coalescent history of a
sample is unknowable
v but, can be crudely approximated by
building a gene tree based on DNA
sequence data
“hanging” mutations on the tree…
Rosenberg & Nordborg 2002 Nat Rev Gen
10 10/2/14 Data versus histories
v generally, the coalescent history of a
sample is unknowable
v but, can be crudely approximated by
building a gene tree based on DNA
sequence data
v genealogical histories estimated with
sequence data typically collapse to
poorly resolved “networks”
Ornithine decarboxylase
intron 6 sequences for
mallards, black ducks,
and mottled ducks
Harrigan et al. 2008 Mol. Ecol. Resources
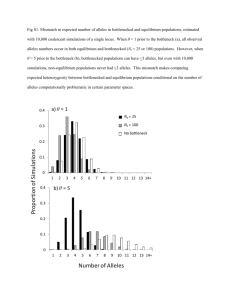
11 10/2/14 The Ewens Distribution
v beyond F, there is additional “information”
available in the number of alleles present
and the distribution of allele frequencies
² “allelic configuration”
² or “allele-frequency spectrum”
v Ewens (1972) - expected number of alleles k
in a sample of size n, depends on θ
E ( k ) = 1+
θ
θ
θ
+
+ ...+
θ +1 θ + 2
θ + n −1
€
Values of θ = 4Nµ
E ( k ) = 1+
Expected number of unique alleles
18
0.125
θ
θ
θ
+
+ ...+
θ +1 θ + 2
θ + n −1
0.25
16
0.5
14
1€
12
2
4
10
8
8
16
32
6
4
2
0
0
5
10
15
20
N (sample size)
12 10/2/14 The Ewens Sampling Formula
v but there’s more than just k (number of alleles)
v the Ewens Distribution specifies the probability
distribution on the set of all partitions of the
integer n
² a.k.a. the “Chinese Restaurant” problem
² broad applicability outside of population
genetics
Partitions of 8
v 8
v 7 + 1
v 6 + 2
v 6 + 1 + 1
v 5 + 3
v 5 + 2 + 1
v 5 + 1 + 1 + 1
v 4 + 4
v 4 + 3 + 1
v 4 + 2 + 2
v 4 + 2 + 1 + 1
v 4 + 1 + 1 + 1 + 1
v 3 + 3 + 2
v 3 + 3 + 1 + 1
v 3 + 2 + 2 + 1
v 3 + 2 + 1 + 1 + 1
v 3 + 1 + 1 + 1 + 1 + 1
v 2 + 2 + 2 + 2
v 2 + 2 + 2 + 1 + 1
v 2 + 2 + 1 + 1 + 1 + 1
v 2 + 1 + 1 + 1 + 1 + 1 + 1
v 1 + 1 + 1 + 1 + 1 + 1 + 1 + 1
13 10/2/14 Ewens’ Sampling Formula
(from Wikipedia!)
v Ewens’ result provided the basis for a formula
(Karlin & McGregor, 1972) giving the probability of a
given allele frequency configuration (note: this
is just one formulation)…
a
n
n!
θ j
Pr{a1,...,an } =
∏
θ (θ + 1) ... (θ + n −1) j=1 j a j a j !
where a1,...,an are counts of the number of
alleles represented one, two,..., n times in
the sample. a1,...,an are nonnegative integers
that satisfy : a1 + 2a2 + 3a3 + ...+ nan = n
€
Karlin & McGregor 1972
v this equation works too…
r!
1
θk
Pr(θ ; n1, n 2 ,..., n k ) =
n1n 2 ... n k α1!α 2!...α p ! Lr (θ )
where Lr (θ ) = θ (θ + 1)(θ + 1) ... (θ + r −1)
€
14 10/2/14 15 10/2/14 Ewens’ Sampling Formula
v key point: the Ewens distribution provides
a basis for testing observed data against
the neutral model
Ewens’ Sampling Formula
v key point: the Ewens distribution provides
a basis for testing observed data against
the neutral model
v and if the data fit neutral expectations,
they can be used to estimate
demographic and historical parameters
16 10/2/14 Site Frequency Spectrum
v applicable to DNA sequence data
v what is the distribution of frequencies for
individual mutations (SNPs)
v number of derived “singletons” = θ
v why?
² length of “external branches” = 4N generations
² (note: t = 2 in Nielsen & Slatkin)
v frequency distribution for derived mutations
1/ j
E !" f j #$ = n−1 , for j = 1, 2, ... , n-1
1
∑k
k−1
Neutral Expectations with…
v ...constant population size and mutation
nucleotide diversity
(infinite sites)
# segregating sites
(infinite sites)
heterozygosity
(infinite alleles)
# unique alleles
(infinite alleles)
E ( Π) = θ = 4N µ
" 1 1 1
1 %
E ( S ) = θ $1+ + + +... +
'
# 2 3 4
k −1 &
θ
Ĥ =
1+ θ
θ
θ
θ
+
+ ...+
θ +1 θ + 2
θ + n −1
E ( k ) = 1+
allele frequency distribution
(infinite alleles)
Pr {a ,..., a
1
site frequency€
distribution
(infinite sites)
n} =
a
n
n!
θ j
∏
θ (θ +1) ... (θ + n −1) j=1 j a j a j !
1/ j
E !" f j #$ = n−1 , for j = 1, 2, ... , n-1
1
∑k
k−1
17 10/2/14 The coalescent with population growth
v coalescent trees are expected to be sparse
(few lineages) near the root for populations
of constant size
v in a growing or shrinking population, the
distribution of coalescence times differs
from expectations for the ideal population
v expanding populations have more nodes
closer to the root of the tree
² takes longer for alleles to “find each other” in
a growing population
constant
18 10/2/14 growing
declining
19 10/2/14 constant
growing
declining
So, what’s the point?
v coalescent modeling
² simulate genealogies under a given set of
population parameters
² “hang” random mutations on those trees in
equal number (or at the same rate) as in the
observed data
² evaluate whether the observed data could
have been produced by a random
coalescent process (the null hypothesis)
20