Statistical power when testing for genetic differentiation
advertisement

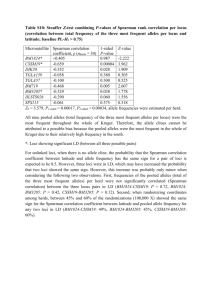
MEC1345.fm Page 2361 Friday, September 21, 2001 3:36 PM Molecular Ecology (2001) 10, 2361–2373 Statistical power when testing for genetic differentiation Blackwell Science, Ltd N . RY M A N * and P. E . J O R D E † *Division of Population Genetics, Stockholm University, S-106 91 Stockholm, Sweden, †Division of Zoology, Department of Biology, University of Oslo, PO Box 1050 Blindern, N-0316 Oslo, Norway Abstract A variety of statistical procedures are commonly employed when testing for genetic differentiation. In a typical situation two or more samples of individuals have been genotyped at several gene loci by molecular or biochemical means, and in a first step a statistical test for allele frequency homogeneity is performed at each locus separately, using, e.g. the contingency chi-square test, Fisher’s exact test, or some modification thereof. In a second step the results from the separate tests are combined for evaluation of the joint null hypothesis that there is no allele frequency difference at any locus, corresponding to the important case where the samples would be regarded as drawn from the same statistical and, hence, biological population. Presently, there are two conceptually different strategies in use for testing the joint null hypothesis of no difference at any locus. One approach is based on the summation of chi-square statistics over loci. Another method is employed by investigators applying the Bonferroni technique (adjusting the P-value required for rejection to account for the elevated alpha errors when performing multiple tests simultaneously) to test if the heterogeneity observed at any particular locus can be regarded significant when considered separately. Under this approach the joint null hypothesis is rejected if one or more of the component single locus tests is considered significant under the Bonferroni criterion. We used computer simulations to evaluate the statistical power and realized alpha errors of these strategies when evaluating the joint hypothesis after scoring multiple loci. We find that the ‘extended’ Bonferroni approach generally is associated with low statistical power and should not be applied in the current setting. Further, and contrary to what might be expected, we find that ‘exact’ tests typically behave poorly when combined in existing procedures for joint hypothesis testing. Thus, while exact tests are generally to be preferred over approximate ones when testing each particular locus, approximate tests such as the traditional chi-square seem preferable when addressing the joint hypothesis. Keywords: allele frequency, Bonferroni, chi-square, contingency table, Fisher’s exact test, statistical analysis Received 8 January 2001; revision received 9 April 2001; accepted 20 April 2001 Introduction An increasingly common question in conservation and evolutionary biology is whether a set of samples are likely to represent the same gene pool. Several statistical techniques are being applied when addressing this type of problem, but there has been little discussion about their relative merits for detection of genetic heterogeneity. This lack is particularly obvious for methods used to combine the information from multiple loci. Correspondence: Nils Ryman. Fax: +46 8 154041; E-mail: Nils.Ryman@popgen.su.se © 2001 Blackwell Science Ltd In a typical situation an investigator has collected tissue samples from two or more groups of individuals that are separated in space or time. Application of some biochemical or molecular techniques provides genotypes of the sampled individuals at one or more nuclear loci or at the mitochondrial genome, and each sample is described in terms of its size and allele (or haplotype) frequencies. The specific scientific questions may vary from study to study, but a very basic one, which frequently determines how to proceed with the analysis is the following one: Are the allele frequency differences observed among samples large enough to suggest that all the samples are not drawn from the same population (gene pool)? It appears that in most MEC1345.fm Page 2362 Friday, September 21, 2001 3:36 PM 2362 N . RY M A N and P. E . J O R D E cases the underlying evolutionary model is one of ‘selective neutrality — isolation — genetic drift’, which implies that all polymorphic loci examined are potentially informative with respect to the question of overall genetic heterogeneity. The general statistical approach most frequently used — and the one dealt with in this paper — is first to conduct a contingency test for allele frequency homogeneity for each locus separately, and in a second step to evaluate the simultaneous, or joint, information from all loci examined. The test procedure applied to each individual locus (contingency table) implies assessment of the probability of obtaining — if the null hypothesis (H0 ) of equal allele frequencies is true — an outcome that is as likely as, or less likely than, the observed one. This probability (P-value) can either be calculated exactly (Fisher 1950; Mehta & Patel 1983), iterated or simulated to a desired degree of precision (Roff & Bentzen 1989; Raymond & Rousset 1995a,b), or approximated by means of some test statistic expected to follow a chi-square distribution (Fisher 1950; Everitt 1977; Sokal & Rohlf 1981). In the second step the results from the separate tests are combined for evaluation of the joint null hypothesis (H0,J ) that there is no allele frequency difference at any locus (i.e. H0,J is true when all the separate H0s are true). Presently there appears to be two conceptually different strategies in use for testing H0,J. One technique is based on the summation of chi-square statistics and utilizes the fact that the sum of a series of chi-square distributed variables also follows a chi-square distribution (e.g. Everitt 1977; Sokal & Rohlf 1981). Another approach is used by investigators applying the Bonferroni technique to test if the heterogeneity observed at any particular locus can be regarded significant when considered separately. The general idea behind the Bonferroni method is to account for the increased probability of obtaining, by pure chance when the null hypothesis is true, a significant result at one or more loci when performing multiple tests (e.g. Rice 1989). Under this approach the joint null hypothesis (H0,J ) is rejected if one or more of the component contingency tests is considered significant, i.e. at least one ‘Bonferroni significance’ is required for rejecting the joint null hypothesis of equal allele frequencies at all loci. This strategy for testing H0,J is conceptually adequate in the present context, although it has been noted that it may be quite conservative resulting in too few rejections (Legendre & Legendre 1998). We are aware of no study, however, that evaluates the two approaches with respect to their ability to detect true heterogeneity. This paper compares the power of the above statistical methods — summation of chi-square vs. application of the Bonferroni method to determine if any one of the separate locus tests can be considered significant — for detecting genetic heterogeneity when multiple loci have been scored. The results show that the efficiency may differ dramatically between the two approaches and, contrary to what might be expected, this difference may become enhanced as the number of loci increases. Exemplifying the problem As an example of the statistical test options, Table 1 presents sample allele frequency data for 12 codominant and di-allelic allozyme loci from two consecutive yearclasses of brown trout (Salmo trutta) collected from Lake Blanktjärnen in central Sweden (see Jorde & Ryman 1996 for details). Are the observed allele frequency differences large enough to suggest that there are true genetic differences between year-classes? The allele frequency difference at each individual locus was tested using Fisher’s exact test, the conventional chisquare 2 × 2 contingency statistic ( X2, degrees of freedom, d.f. = 1), and the chi-square statistic with Yates’ continuity correction (X C2, d.f. = 1; chi-square test statistics are denoted by X2 to distinguish them from values of a theoretical χ2 distribution). Both chi-square approximations provide Pvalues that are reasonably similar to the exact ones, but those from the conventional X2 are generally smaller (less conservative) whereas X C2 tends to produce larger ones. All methods yield significant results (P < 0.05) for the same two loci (sAAT-4 and bGALA-2). When testing the joint null hypothesis (H0,J) that there are no allele frequency differences at any locus, we note that the sum of a set of χ2 distributed variables also follows a χ2 distribution with d.f. equal to the sum of d.f. of the contributing variables. This summation is straightforward for X2 and X C2, which are both expected to follow approximately a χ2 distribution under the null hypothesis. With respect to the P-values obtained in the exact tests, Fisher (1950) has shown that when the null hypothesis is true the quantity –2ln(P) is expected to follow asymptotically a χ2 distribution with d.f. = 2. Thus, summing the negative of twice the natural logarithm of the 12 P-values results in a X2 statistic that is to be evaluated against a χ2 distribution with d.f. = 24 (Table 1; this technique of summing –2ln(P) is sometimes referred to as ‘Fisher’s method’, but it should not be confused with Fisher’s exact test). Under each of the three approaches the summed chi-square statistic is significant (P < 0.05) and results in rejection of the joint null hypothesis (H0,J ) although the level of significance differs among them (X2 and X C2 yielding the smallest and largest summation P-value, respectively). In contrast to the above ‘summation’ approaches, application of the Bonferroni method results in a different conclusion. The Bonferroni logic implies that no individual test in a series of k-tests is to be judged significant unless the Pvalue is smaller than α/k, where α is the preassigned significance level for rejecting the null hypothesis (Rice 1989 and references therein; see below). In the present case with k = 12 (12 loci) and α = 0.05 a P-value less than 0.05/12 = © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361– 2373 MEC1345.fm Page 2363 Friday, September 21, 2001 3:36 PM D E T E C T I N G G E N E T I C D I F F E R E N T I AT I O N 2363 Table 1 Allele frequencies (the 100 allele) and 2 × 2 contingency test statistics for 12 di-allelic allozyme loci in two cohorts (1992 and 1993) of brown trout from Lake Blanktjärnen, Sweden. The number of fish is 43 and 27 in 1992 and 1993, respectively Allele frequency (100 allele) Cohort Fisher’s exact test Chi-square test X2 Locus 1992 1993 P sAAT-4 DIA bGALA-2 bGLUA G3PDH-2 sIDHP-1 LDH-5 aMAN sMDH-2 ME MPI PEPLT 0.6395 0.7791 0.7791 0.8256 0.7558 0.4302 0.7209 0.9884 0.9767 0.8953 0.1047 0.6395 0.4074 0.8148 0.9444 0.7407 0.6296 0.5741 0.6852 0.9259 0.9815 0.8704 0.2222 0.6667 0.009 0.673 0.009 0.285 0.129 0.119 0.705 0.073 1.000 0.786 0.087 0.856 Sum Sum of d.f. P 0.0042 is thus required to be regarded significant. No individual (single locus) P-value in Table 1 is that small, and consequently no locus can be considered displaying significant heterogeneity when applying the Bonferroni technique. The joint null hypothesis (H0,J ) is therefore also accepted, regardless of whether the basic contingency P-values were obtained by means of chi-square approximation or exact calculation. Thus, in this example the ‘summation’ method indicates that the joint null hypothesis should be rejected for each of the tests statistics applied, whereas application of the Bonferroni technique consistently suggests acceptance when using the same set of individual P-values. The difference is crucial, but on the basis of the data available an investigator cannot determine what decision is most appropriate. It appears that many scientific journals would accept either approach without requiring additional data analysis. In the remainder of this paper we present results of computer simulations aimed at evaluating the probability of detecting a true genetic difference (statistical power) and the probability of falsely rejecting a true null hypothesis (α or type I error) when addressing the joint H0,J (Bonferroni vs. ‘summation’). We recognize that the present problem can also be addressed by means of exact binomial or multinomial calculations, but we chose the simulation approach for practical reasons. Simulations Random sampling of 2n genes (n diploid individuals) from populations with known allele frequencies was simulated © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361–2373 –2ln(P) 9.421 0.793 9.489 2.512 4.097 4.261 0.700 5.240 0.000 0.482 4.881 0.311 7.222 0.258 6.849 1.454 2.550 2.748 0.205 3.756 0.036 0.205 3.596 0.107 42.19 24 0.012 28.98 12 0.004 Chi-square test Yates’ correction P X C2 P 0.007 0.611 0.009 0.228 0.110 0.097 0.651 0.053 0.851 0.651 0.058 0.743 6.331 0.087 6.298 0.969 1.954 2.207 0.068 2.099 0.165 0.032 2.662 0.021 0.012 0.768 0.012 0.325 0.162 0.137 0.794 0.147 0.685 0.858 0.103 0.885 22.89 12 0.029 by means of pseudo random number generation. At each locus the hypothesis of equal allele frequencies was tested by various r × c contingency tests (r = number of samples and c = number of alleles) using both Fisher’s exact method and chi-square tests with d.f. = (r – 1) (c – 1). The number of replicates (runs) of each simulation was typically in the interval 5000–10 000, and the frequency of replicates resulting in rejection and acceptance of a null hypothesis provided estimates of the statistical power (probability of rejecting a false hypothesis) or the α (type I) error (probability of rejecting a true hypothesis), respectively. The intended α level was consistently kept at 0.05, rejecting the null hypothesis for P < 0.05. A replicate was discarded if the random sampling of genes resulted in less than c alleles being observed in the combined material from the r samples, and a new set of r samples was drawn in those cases. Population allele frequencies were generally chosen to result in a ‘realistic’ proportion of simulated contingency tables where at least one cell had a small expected value (expectancy less than 5 or 1) at low or moderate sample sizes. This was done in order not to provide an overly optimistic view of the fit of the various chi-square approximations to the expected χ2 distribution. When simulating situations where multiple loci (k > 1) are scored, all loci within a population were assumed to segregate at identical allele frequencies (for example, for di-allelic loci the allele under consideration may occur at the frequency 0.10 at all loci in population 1 and at 0.15 at all loci in population 2). Of course, in the real world MEC1345.fm Page 2364 Friday, September 21, 2001 3:36 PM 2364 N . RY M A N and P. E . J O R D E it is not very likely to encounter a situation where all the loci examined in a population segregate at exactly the same frequency (although the true allele frequency distribution is unknown in most cases). This model is appropriate for the present purpose of examining differences between test procedures, however. When dealing with relative frequencies both the power and the α error are dependent on the population frequencies, and varying those frequencies might obscure the comparison of test procedures. A ‘regular’ chi-square test statistic (X2) was calculated for each simulated contingency table. In addition, Yates’ continuity correction was applied to provide a corrected chi-square (X C2) for all 2 × 2 tables, and for larger tables the G-statistic with and without Williams’ correction was used (Sokal & Rohlf 1981; 737 – 38). Fisher’s exact test for 2 × 2 tables was performed as described by Sokal & Rohlf (1981; 740 – 42), and the algorithm of Mehta & Patel (1983) was applied for larger tables. Computational results from the statistical routines developed for the simulation programs were checked with outputs from softwares such as biom (Rohlf 1987 ), statistica (StatSoft Inc. 1998), StatXact-Turbo (CYTEL Software Corporation 1992), and genepop (Raymond & Rousset 1995b), and the simulated power estimates were checked against exact calculations and those obtained using the ‘standard’ normal approximation for power assessment (e.g. Zar 1984; 398). When combining the information from multiple loci for evaluation of the joint null hypothesis (H0,J ) of no difference at any locus the approaches of Bonferroni and summation of chi-square statistics were applied as exemplified in the preceding section. A considerable number of simulations have been conducted during the course of this study using different combinations of allele frequencies, sample sizes, number of alleles, loci, and populations. In order not to burden the presentation unnecessarily, however, we have tried to choose as simple combinations as possible for illustration of general trends and principles. Thus, most of the paper focuses on situations like that in Table 1 where the basic statistical tests refer to 2 × 2 tables (2 samples and 2 alleles per locus). 2 × 2 contingency tables Four examples of typical simulation results for 2 × 2 contingency tests are depicted in Fig. 1(a). In the most basic case we consider a single locus (k = 1) with two alleles (A and A′), and a random sample of 20 diploid individuals (40 genes) is drawn from each of two populations (1 and 2) where the A allele occurs in the true frequency Q1 and Q2, respectively. In the case of multiple loci (k > 1) all of them have the same allele frequency (Q1) in population 1, and within population 2 all frequencies are Q2. Every plate in Fig. 1(a) represents a specific combination of Q1 and Q2, and for each test procedure the proportion of replicates resulting in rejection of the joint null hypothesis (y-axis) is indicated for different number of loci examined (x-axis; k = 1, 2 … 5, 10, 20 … 50). As in the introductory example (Table 1) the joint null hypothesis (H0,J ) was rejected for P < 0.05 (summation of chi-squares), and when using the Bonferroni method rejection of H0,J required at least one single locus P-value smaller than 0.05 /k. For k = 1 the ‘summation’ is over a single value only, and the Bonferroni rejection criterion coincides with that of the basic test. When Q1 is different from Q2 (H0 is false; upper plates) the proportion of H0,J rejections estimates the power of the test, and when Q1 = Q2 (H0 is true; lower plates) it estimates the realized α error. In a first step we focus on the situations where H0 is false (Q1 ≠ Q2; Fig. 1a, upper plates). Considering a single locus only (k = 1) the power estimates for the Q1/ Q2 combination of 0.10/0.15 are 0.107, 0.041, and 0.052 when using X2, X C2, and Fisher’s exact test, respectively, and for the 0.50/0.60 combination the corresponding estimates are 0.150, 0.103, and 0.103. The difference between these simulated power estimates and the values expected theoretically is noticeable in the third decimal place only, indicating that the simulations provide reasonably accurate results. Also, Fisher ’s exact and the X C2 tests are both expected to be more conservative than the X2 test (e.g. Everitt 1977 ), and they yield accordingly lower power estimates within each combination of Q1/Q2. In contrast to the power observed for a single locus, the results for tests involving multiple loci are more surprising. Here, one might expect a reliable test to detect the true divergence between populations more frequently as additional loci are examined. It is only the ΣX2 approach, however, that behaves consistently in this way at both combinations of Q1 and Q2. In the case of Q1/ Q2 = 0.10/ 0.15 the probability of rejecting H0 actually tends to decrease when the number of loci included in the test increases for all methods except the ΣX2. When Q1 / Q2 = 0.50/ 0.60 all the procedures provide at least some increase of power when more loci are considered, but the ΣX2 approach is consistently the most powerful one. It is an important observation that the remarkably better power obtained through summation of X2 is not associated with an unduly large α error when H0,J is true. Rather, this method is the only one that consistently appears to provide an α error that is reasonably close to the intended one of α = 0.05 (Fig. 1a, lower panels). Simulation results for the considerably larger sample sizes of 500 individuals (1000 genes) are shown in Fig. 1(b). Here, the population allele frequencies are the same as those in Fig. 1(a) when estimating the α error (lower plates), but for the sake of illustration the assessment of power (upper plates) is based on smaller differences © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361– 2373 MEC1345.fm Page 2365 Friday, September 21, 2001 3:36 PM D E T E C T I N G G E N E T I C D I F F E R E N T I AT I O N 2365 Fig. 1 Proportion of statistical significances when testing the joint null hypothesis (H0,J) of no difference at any locus after simulated drawing of a sample of size (n) from each of two populations where the true allele frequency at di-allelic loci are Q1 and Q2, respectively. The number of loci examined is 1– 50. A test for allele frequency homogeneity is conducted for each locus separately using X2, XC2, and Fisher’s exact test, and the resulting Pvalues are combined into a joint test by the ‘summation’ and Bonferroni approaches. The intended α is 0.05, and each data point is based on 10 000 (Fig. 1a) and 5000 (Fig. 1b) replicates, respectively. See text for details. Figure 1(a): n = 20 individuals (40 genes). Figure 1(b): n = 500 individuals (1000 genes). between Q1 and Q2 to account for generally higher power associated with larger samples. (When sample sizes as large as 500 individuals the power of all methods is close to unity for the combinations of Q1 and Q2 used in Fig. 1a). At sample sizes of 1000 genes all methods are reasonably successful in keeping the α error close to the intended one of 0.05, except for those representing summation of X C2 and –2ln(P). This latter observation most likely reflects a somewhat slower approach to the limiting χ2 distribution for these two test statistics as compared to X2 (see below). When H0,J is false (upper panels, Fig. 1b) summation of X2 is the technique that consistently provides the highest probability of rejecting H0,J. For all methods, however, the power increases as the number of loci grows, although the ‘summation’ approach in all cases appears more powerful regardless of the statistical procedure applied in the basic © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361–2373 contingency tests. This is true also when summing X C2 and –2ln(P), in spite of the lower realized α observed for these statistics. Reasons for low power For both the Bonferroni and the ‘summation’ method the low power is associated with the approximations involved when assessing P-values for the basic 2 × 2 tables. Both methods are expected to work satisfactorily in theory when samples are large and allele frequencies intermediate. In practice, however, the approximate nature of the test statistics and the P-values produced by the primary contingency tests may provide a realized α in the combined test that is far below the intended one, and the reduced α typically results in a small power. MEC1345.fm Page 2366 Friday, September 21, 2001 3:36 PM 2366 N . RY M A N and P. E . J O R D E Fig. 1 Continued Bonferroni. As noted above, the objective behind the Bonferroni technique is to avoid excessive numbers of false significances when performing multiple tests through adjusting the P-value at which a particular component test is to be judged significant. The probability of observing false significances increases rapidly as the number of tests grows. When performing k-tests of a true null hypothesis at α = 0.05 the expected probability of obtaining one or more significances by pure chance is 1 – (1 – 0.05) k; for k = 10, for example, this probability exceeds 40%. In order to maintain the intended α level (here 0.05) of a particular test when a series of k-tests have been performed, the idea behind the Bonferroni method is to adjust the P-value for rejection such that the probability of observing one or more significances among the k-tests remains at α. This goal is met approximately if rejecting any particular H0 at P < α/k (rather than at P < α). The rationale for this criterion is based on the relationship 1 – (1 – α/k) k ≈ α. The above arguments form the basis for the ‘extended’ application of the Bonferroni correction in the context of testing the joint null hypothesis H0,J that all the components H0s are true when multiple tests have been performed. That is, if all the component (single locus) H0s are true (i.e. H0,J is true), the probability is α to observe one or more ‘Bonferroni significances’ (at the α/k level). Thus, H0,J is rejected when obtaining a single locus P-value less than α/k, otherwise it is accepted. © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361– 2373 MEC1345.fm Page 2367 Friday, September 21, 2001 3:36 PM D E T E C T I N G G E N E T I C D I F F E R E N T I AT I O N 2367 If the Bonferroni technique is to work in practice, however, two basic conditions must be met which are not satisfied in many realistic situations. First, it is necessary that each single locus test can produce a P-value that is smaller than α/k. However, this is not possible for many combinations of sample size (n) and population allele frequencies (Q1/Q2) because of the restricted number of potential outcomes of the sampling process. If basing the evaluation of H0,J on a suite of such contingency tests there is a substantial risk that the Bonferroni method yields a realized α that is considerably smaller than the intended one, and thereby a correspondingly reduced power. To clarify the point we may consider the extreme example of sampling two individuals (four genes) from each of two populations that are fixed for different alleles. The counts of the two types of alleles will be 4; 0 and 0; 4 for the two samples, respectively, which represents the most extreme outcome possible under the null hypothesis of equal allele frequencies. Fisher’s exact test yields a (twosided) P-value of 0.029, which is significant at the 5% level, and this is the smallest P-value that can be obtained with the present sample sizes. Analysis of, say, five alternately fixed loci would result in five P-values of 0.029, and intuition would justifiably make the investigator suspect that the populations are genetically divergent. A Bonferroni evaluation would dismiss such an interpretation, however, because no P-value is smaller than 0.01 (0.05/5), a P-value which is impossible to obtain with the present sample sizes. Thus, in this situation the Bonferroni correction results in a realized α level of zero, and the power is thereby also reduced to zero. In the present simplified example it is easy to see how the Bonferroni method, as an effect of a discrete number of possible experimental outcomes, may result in a reduction of the realized α far below the intended one, and thereby in a corresponding decrease of power. The phenomenon is a general one, however, and the magnitude of the effect depends on the sample sizes, the number of loci (tests), and the true allele frequency differences (Fig. 1). The other requirement for the Bonferroni method to work satisfactorily is that the realized α error of each of the basic, single locus, contingency tests is reasonably close to the intended one. In other words, when the component H0 is true the probability of obtaining a P-value of 0.05 or less should be close to 0.05, that of obtaining P ≤ 0.01 should be close to 0.01, etc. It appears that this characteristic of the sampling distribution is frequently taken for granted, in spite of the fact that substantial deviations are quite common. To exemplify the difference between intended and realized α errors in basic contingency tests we may consider the occurrence of various P-values when drawing two independent samples of 20 individuals (40 genes) from a population with the allele frequency Q = 0.10. For illustration, all the 2 × 2 tables possible when drawing two samples of © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361–2373 40 items were created, P-values based on regular chisquare (X2) and Fisher’s test were computed, the exact frequency of occurrence of each table and its associated P-values was derived binomially, and the cumulative frequency of occurrence of P ≤ 0.05 was depicted graphically (Fig. 2, upper plate). As seen in Fig. 2 (upper plate) the realized α may be considerably smaller than the intended one, and particularly so for Fisher’s exact test. With Fisher’s exact test, for example, values of P ≤ 0.05 only occur in a frequency of 0.017 (less than half the ‘ideal’ rate), and the discrepancy is even more pronounced for smaller P-values. The corresponding cumulative distributions of P-values when sampling 50 and 500 individuals (100 and 1000 genes) are depicted in the central and lower plates of Fig. 2, respectively. For X2 the correspondence between intended and realized α is fairly good at sample sizes of 50 individuals or more. In contrast, at the present population allele frequency samples in the order of hundreds of individuals are required for this to occur with Fisher’s exact test. When realized α of the separate (single locus) contingency tests are smaller than intended, the Bonferroni approach to testing the joint H0,J of no difference at any locus may result in an overall realized α that is far below the anticipated one. To exemplify, Table 2 gives exact realized values of α for 1–50 loci when applying Fisher’s exact test to two independent samples of n = 10 (or n = 20) diploids from a population with the true allele frequency Q = 0.10. For n = 10 and k = 10 loci (tests), for instance, the Bonferroni method implies that H0,J should be rejected when observing at least one contingency P ≤ 0.005 (0.05/ 10). With two samples of n1 = n2 = 10 diploids the largest Fisher P-value meeting the criterion P ≤ 0.005 is 0.00385 (due to the restricted number of possible 2 × 2 tables at these sample sizes), and P-values this small or smaller occur at a frequency of 0.000107 (exact cumulative binomial probability). Thus, the realized α of the Bonferroni approach to testing H0,J corresponds to the probability of observing one or more single locus P-values of P ≤ 0.005, which is α = 1 – (1 – 0.000107)10 = 0.0011. Clearly, this is dramatically smaller than the intended α of 0.05, and with such a small realized α the chance of detecting anything but very large allele frequency differences is minor. Chi-square summation. As noted above, the logic of the summation approach is based on the fact that the sum of two or more χ2 distributed variables will also follow a χ2 distribution with d.f. equal to the summed d.f. of the component variables. Under the null hypothesis, the test statistic computed for each particular locus (contingency table) is expected to be asymptotically χ2 distributed with d.f. = (r – 1)(c – 1) where r is the number of samples (rows) and c is the number of alleles (columns). The fit may be quite poor, however, particularly for small sample sizes and skewed allele frequencies, and the sum of several such MEC1345.fm Page 2368 Friday, September 21, 2001 3:36 PM 2368 N . RY M A N and P. E . J O R D E Fig. 2 Exact cumulative frequency of occurrence of possible P-values when drawing two independent samples of n = 20, 50, or 100 diploids (40, 100 or 1000 genes) from a population where the true allele frequency at a di-allelic locus is 0.1. The null hypothesis of allele frequency homogeneity is tested using regular chi-square (X2) and Fisher’s exact test. Only the left-most part (P ≤ 0.05) of the distribution is shown. Table 2 Exact realized α error when applying the Bonferroni method to a series of k 2 × 2 tables. Each table represents two independent samples of n diploid individuals (2n genes) from a population where the true gene frequency at di-allelic loci is 0.10, and where the H0 of no gene frequency difference has been tested by Fisher’s exact method. Intended α = 0.05. Frequency of occurrence represents the cumulative binomial probability of obtaining the realized P-value or a smaller one. See text for details n = 10 n = 20 k = no. of tested loci Intended P-value for rejection Realized P-value for rejecting a separate 2 × 2 table Frequency of occurrence 1 2 5 10 20 50 0.05 0.025 0.01 0.005 0.0025 0.001 0.04837 0.02484 0.00953 0.00385 0.00220 0.00077 0.012002 0.003010 0.000622 0.000107 0.000015 0.000002 Realized α Realized P-value for rejecting a separate 2 × 2 table Frequency of occurrence Realized α 0.012002 0.006010 0.003107 0.001065 0.000304 0.000090 0.04817 0.02470 0.00931 0.00483 0.00240 0.00096 0.016770 0.005559 0.002022 0.000678 0.000514 0.000056 0.016770 0.011087 0.010067 0.006761 0.010225 0.002802 © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361– 2373 MEC1345.fm Page 2369 Friday, September 21, 2001 3:36 PM D E T E C T I N G G E N E T I C D I F F E R E N T I AT I O N 2369 ‘poorly fitted’ variables may deviate dramatically from the expected χ2 distribution. The mean and variance of a χ2 distribution is d.f. and 2d.f, respectively, and as an example of the fit to χ2 Table 3 gives the observed mean and variance of the test statistics obtained when simulating the drawing of two independent samples from a population with an allele frequency of 0.1 (Q1 = Q2 = 0.1; n = 3 – 500). Both of X2 and X C2 have d.f. = 1, and if the fit is perfect we expect these statistics to yield means and variances of 1 and 2, respectively. With respect to the P-value from Fisher’s exact test (exact P) the quantity –2ln(exact P) should be asymptotically χ2 distributed with d.f. = 2 (see above), and with a perfect fit we expect the mean and variance to be 2 and 4, respectively. With respect to the traditional chi-square statistic (X2), the fit of the observed sampling distribution to the theoretical χ2 is quite good, except for a reduced variance and a slightly inflated mean at the smallest sample sizes. In contrast, the approach to the limiting χ2 distribution is markedly slower for X C2 and –2ln(exact P). The observed sampling distributions tend to be located to the left of the Mean expected one, and this shift in location produces too few false significances (realized α < 0.05), and thereby a low power. The mean and variance of X2 are fairly close to their expected values when n ≥ 50 individuals, but X C2 and –2ln(exact P) both yield markedly smaller means and variances even at n = 500. The shift of location relative to χ2 may not seem alarming when plotting the test statistic sample distribution, but a variable representing the sum of several observations from such a distribution may deviate dramatically from the one expected for the sum. To exemplify, the simulated and expected distributions of the 2 × 2 contingency X C2 (d.f. = 1) for n = 20 diploids and Q1 = Q2 = 0.1 are shown in Fig. 3(a). The deviation from the χ2 distribution with d.f. = 1 may not appear overly large, but when summing 10 observations and comparing with χ2 with d.f. = 10 the difference is dramatic (Fig. 3b). Clearly, in a situation like this when X C2 for 10 loci is being summed, the realized α error is far below the expected one, and the probability of rejecting H0,J is reduced correspondingly. In the case of 2 × 2 contingency tables we have focused on the X2, X C2, and –2ln(exact P) test statistics, and it is Variance Test statistic n Observed Expected Observed Expected X2 X2 X2 X2 X2 X2 X C2 X C2 X C2 X C2 X C2 X C2 –2ln(exact P) –2ln(exact P) –2ln(exact P) –2ln(exact P) –2ln(exact P) –2ln(exact P) G G G G G G Williams’ G Williams’ G Williams’ G Williams’ G Williams’ G Williams’ G 3 10 20 50 100 500 3 10 20 50 100 500 3 10 20 50 100 500 3 10 20 50 100 500 3 10 20 50 100 500 1.09 1.04 1.01 1.00 1.02 1.00 0.25 0.44 0.54 0.67 0.78 0.88 0.34 0.84 1.13 1.41 1.59 1.80 1.43 1.24 1.10 1.04 1.03 1.01 1.05 1.07 1.02 1.02 1.01 1.00 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 1 1 1 1 1 1 1 1 1 1 1 1 0.75 1.52 1.77 1.95 2.04 1.98 0.15 0.49 0.83 1.28 1.53 1.75 0.59 1.74 2.47 3.15 3.49 3.70 1.30 2.58 2.44 2.20 2.14 1.95 0.86 2.01 2.12 2.09 2.08 1.94 2 2 2 2 2 2 2 2 2 2 2 2 4 4 4 4 4 4 2 2 2 2 2 2 2 2 2 2 2 2 © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361–2373 Table 3 Mean and variance of test statistics from 2 × 2 contingency tables when simulating the drawing of two independent samples of equal size (n = 3 – 500 diploids) from a population where the true allele frequency at a di-allelic locus is 0.1. The number of replicates (number of 2 × 2 tables) is 10 000 at each sample size. Expected values are those for a χ2 distribution with d.f. = 1 [or d.f. = 2 for –2ln(exactP) ]. See text for details MEC1345.fm Page 2370 Friday, September 21, 2001 3:36 PM 2370 N . RY M A N and P. E . J O R D E clear that the traditional chi-square test appears to provide the statistic (X2) that is to be preferred when combining information from multiple loci by summation (Fig. 1, Table 3). It is especially interesting to note that the fit of –2ln(exact P) is markedly poor when n is small, i.e. when the use of an exact test is typically regarded most warranted for comparisons of allele frequencies at each particular locus considered separately. As a comparison, Table 3 also gives simulated means and variances for G and Williams’ G for the case of Q1 = Q2 = 0.10. Here, Williams’ G performs as well as X2, and sometimes even better. In contrast to the other statistics, however, the sampling distribution of G seems to be shifted to the right of χ2 for many sample sizes (larger mean and variance than expected). The G-test therefore appears to produce an excess of false significances in the basic contingency tests (realized α > 0.05), and this tendency is expected to grow progressively stronger when testing H0,J through summation of G-values from multiple loci. two rows or columns (Everitt 1977), and when testing H0,J the realized α is therefore expected to be in better agreement with the intended one for both the Bonferroni and the ‘summation’ methods. As an example, Table 4 gives the results from simulated drawings of three samples (r = 3) from populations segregating for two and five alleles, respectively (c = 2 or 5), for the diploid sample sizes (n) of 10, 20, and 50. Under the conditions simulated the difference between the contingency test statistic sampling distribution and the expected χ2 is in most cases minimal for the largest sample size (n = 50). As a result, the ‘summation’ method for addressing H0,J generally provides a realized error rate that is reasonably close to the intended α at n = 50, although that of –2ln(exact P) is still a bit low in the 3 × 2 tests (Table 4). At all sample sizes the Bonferroni approach usually results in a realized α that is smaller than that obtained by summation. At the smaller sample sizes (10 and 20) the –2ln(exact P) statistic yields an α error that appears unduly small in the 3 × 2 tests, but in the 3 × 5 tables the error is close to the intended one. Most strikingly, however, summation of the G-statistic tends to produce unacceptably high rates of false significances (10 – 37%) at the smallest sample sizes, an effect of a markedly poor fit to the expected χ2 distribution. The traditional chi-square (X2) generally seems to behave in fairly good agreement with expectation, and the ‘summation’ method appears to perform better than Bonferroni over a wide range of sample sizes. Although the differences between test statistics and methods for evaluating H0,J are less pronounced than for the 2 × 2 tables, the tendencies are similar. Combining exact P-values by means of ‘Fisher’s method’ tends to result in an unduly small α error more frequently than when summing X2, particularly at small or moderate sample sizes, and it seems that ‘summation’ should be preferred before Bonferroni. Finally, it should be noted that the present results indicating generally better ‘summation properties’ of X2 relative to the other test statistics is not caused by choosing combinations of sample sizes (n) and allele frequencies (Q) that produce unduly few tables with low expectancy cells. Considering 2 × 2 contingency tables with n = 20 and Q = 0.1 (Table 3), for example, over 70% of the simulated tables have two (out of four) cells with an expectation less than five. Similarly, at n = 20 nearly 80% of the simulated 3 × 2 contingency tables (Table 4) have three cells with an expected value of less than five; almost all the 3 × 5 tables have 6–12 such cells and over 10% have 3 – 6 cells with an expectation of less than unity. General r × c contingency tables Concluding remarks and recommendations The fit of the test statistic sample distributions to χ2 is generally improved for contingency tables with more than It is obvious from the above that the choice of statistical method may be crucial for the probability of drawing Fig. 3 Simulated sampling distribution of the X2C test statistic (2 × 2 contingency chi-square with Yates’ correction) when drawing two independent samples of 20 diploids (40 genes) from a population where the true allele frequency at di-allelic loci is 0.1. The corresponding χ2 distributions are those expected asymptotically under large sample theory. (a) Examining a single locus; expected χ2 has d.f. = 1. (b) Testing 10 loci separately and summing the test statistic values; expected χ2 has d.f. = 10. © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361– 2373 MEC1345.fm Page 2371 Friday, September 21, 2001 3:36 PM D E T E C T I N G G E N E T I C D I F F E R E N T I AT I O N 2371 Table 4 Mean and variance of test statistics from 3 × c contingency tables when simulating the drawing of three samples of equal size (n = 10 – 50 diploids) from the same population. The number of alleles (c) is 2 and 5 occurring in the frequencies 0.1 and 0.9 (c = 2) and 0.7, 0.1, 0.1, 0.05, and 0.05 (c = 5). The number of replicates (number of 3 × c tables) at each sample size is 10 000 and 1000 for the 3 × 2 and 5 × 2 tables, respectively. Expected values are those for a chi-square distribution with d.f. = 2(c – 1) [d.f. = 2 for –2ln(exact P) ]. Realized α refers to a situation where the information for 10 loci is combined by means of the summation or the Bonferroni approach and the intended α is 0.05. See text for details Mean Realized α (10 loci) Variance c Test statistic d.f. n Obs. Exp. Obs. Exp. Summation Bonferroni 2 2 2 2 2 2 2 2 2 2 2 2 5 5 5 5 5 5 5 5 5 5 5 5 X2 X2 X2 –2ln(exact P) –2ln(exact P) –2ln(exact P) G G G Williams’ G Williams’ G Williams’ G X2 X2 X2 –2ln(exact P) –2ln(exact P) –2ln(exact P) G G G Williams’ G Williams’ G Williams’ G 2 2 2 2 2 2 2 2 2 2 2 2 8 8 8 2 2 2 8 8 8 8 8 8 10 20 50 10 20 50 10 20 50 10 20 50 10 20 50 10 20 50 10 20 50 10 20 50 2.01 2.02 2.00 1.26 1.62 1.83 2.38 2.17 2.04 2.09 2.05 1.99 8.19 8.14 7.78 1.96 2.04 1.86 9.82 9.21 8.07 8.11 8.38 7.79 2 2 2 2 2 2 2 2 2 2 2 2 8 8 8 2 2 2 8 8 8 8 8 8 3.25 3.71 3.94 2.70 3.60 3.79 4.77 4.86 4.26 3.82 4.30 4.07 10.96 14.33 13.90 3.65 4.21 3.26 15.06 19.86 15.66 10.65 16.53 14.58 4 4 4 4 4 4 4 4 4 4 4 4 16 16 16 4 4 4 16 16 16 16 16 16 0.043 0.049 0.052 0.001 0.019 0.029 0.150 0.100 0.068 0.072 0.066 0.057 0.030 0.060 0.040 0.040 0.070 0.050 0.370 0.240 0.060 0.030 0.070 0.060 0.027 0.035 0.047 0.013 0.025 0.037 0.052 0.079 0.064 0.018 0.061 0.056 0.000 0.049 0.020 0.020 0.068 0.020 0.049 0.086 0.020 0.000 0.058 0.020 the right conclusion when testing for genetic differentiation. To date, it appears that the primary statistical interest has been devoted to avoidance of false significances (α errors), whereas considerably less attention has been paid to the prospect of detecting true differences (power). Because the two quantities are interrelated, excessive focus on one of them may have undesirable effects on the other. It appears that the current trend in many fields of evolutionary biology is to score a steadily growing number of loci in a quite restricted number of individuals, frequently in the range, say, 5 – 30. The question of how to combine the information from multiple loci is therefore becoming increasingly significant. Our results indicate strongly that summation of chi-square (X2) tends to perform better than any of the alternatives examined, even at fairly small sample sizes. This approach should typically be the method of choice when testing the joint null hypothesis of no difference at any locus. The technique of summing twice the negative logarithm of P-values from Fisher’s exact test [Σ – 2ln(exact P) ] © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361–2373 appears to be used increasingly often. It should be stressed, though, that this approach (‘Fisher’s method’ applied to Fisher’s exact P) may be associated with a strikingly small power even when sample sizes are quite large, and particularly so for 2 × 2 tables. It may seem superficially appealing to base the joint test on probabilities that are computed ‘exactly’ for each contributing contingency table. Nevertheless, the poor fit to the asymptotically expected χ2 distribution frequently makes this method profusely conservative. Therefore, we suggest that results from summation of –2ln(exact P) should generally be accompanied by the results from chi-square summation. It should be stressed that we by no means suggest that exact contingency tests should be abandoned. Whenever possible, exact calculation of P is to be preferred before any approximation relying on large sample theory, and particularly so when sample sizes are modest or small. Although necessarily conservative, there is an obvious advantage with exact tests in that the investigator is guaranteed that the realized α will never exceed the MEC1345.fm Page 2372 Friday, September 21, 2001 3:36 PM 2372 N . RY M A N and P. E . J O R D E intended one. The problem we address arises when combining the information from several exact tests by means of an approximation such as ‘Fisher’s method’. There is no contingency test that is universally ‘best’ from every perspective and under all circumstances, and there are many situations where an investigator may have valid concern regarding the appropriateness of the chisquare statistic, which necessarily represents an approximation. With small expected values in one or more cells the risk of excessive rates of false significances cannot be ignored, although several reports suggest (as do the present simulation results) that the severity of this continuity problem may be overrated (e.g. Cochran 1954; Lewontin & Felsenstein 1965; Everitt 1977). Exact tests, on the other hand, may be overly conservative and fail to detect true differences more often than anticipated. When evaluating a single contingency table, however, exact calculation of the P-value should be the primary method of choice. It must be noted, though, that the observation of a nonsignificant P-value (P > 0.05) may be quite uninformative in the absence of minimal information on realized α and power of the test at the sample sizes at hand. When combining the information from several contingency tables, however, we recommend that the decision on overall genetic divergence is made on the basis of summation of chi-square (X2). The Bonferroni correction is also being applied more commonly as a tool for evaluating the joint null hypothesis of no difference at any locus, and its frequently poor performance in the present context may be perceived a bit surprising. It should be noted, though, that the Bonferroni correction was primarily designed to reduce the probability of obtaining false significances when performing several independent tests. It was not aimed at combining the information from multiple tests that address the same null hypothesis. The Bonferroni method focuses exclusively on the occurrence of (very) small P-values and largely ignores, for example, tendencies of weak significances to be overrepresented. When testing for genetic heterogeneity under a ‘selective neutrality — genetic drift’ model, however, any indication of allele frequency differences at any polymorphic locus (regardless of direction) should ideally contribute information that makes the joint null hypothesis less likely. The ‘summation’ method is directly aimed at picking up such tendencies, and the difference between the two approaches becomes particularly obvious when the underlying test statistic distributions are characterized by marked discontinuities. As with exact tests, we do not suggest that the Bonferroni method should be avoided in general. The Bonferroni (or the sequential Bonferroni) approach represents a most valuable tool for controlling the α error when an investigator, after conducting multiple tests, focuses on a particular null hypothesis (Rice 1989). Rather, because of the markedly low power we recommend against its ‘extended’ use when testing the joint null hypothesis (H0,J) that all the component H0s are true. This paper is focused on contingency tests for allele frequency heterogeneity, but problems with statistical power similar to those discussed here may also occur in other testing situations. The Bonferroni approach, for example, is often applied for evaluation of multiple P-values obtained when testing for Hardy–Weinberg proportions or linkage equilibrium between pairs of loci. For the Bonferroni method to work properly in such cases, it is also necessary that the contributing tests can produce P-values as small as α/k and that those P-values occur at frequency reasonably close to α/k when the null hypothesis is true. For instance, if it is impossible in practice to obtain P < α/k in many or most of the contributing tests, then the power of the joint test (the Bonferroni evaluation) may be very close to zero. In such a situation, any attempt to interpret an observed lack of significance in biological terms would typically be meaningless and potentially erroneous. Acknowledgements We thank Linda Laikre, Ole Christian Lingjærde, Stefan Palm, Associate Editor Laurent Excoffier, and two anonymous reviewers for comments on earlier versions of this paper. The study was supported by grants to N.R. from the Swedish Natural Science Research Council and from the Swedish research program on Sustainable Costal Zone Management, SUCOZOMA, funded by the Foundation for Strategic Environmental Research, MISTRA. P.E.J. was supported by a grant from the National Research Council of Norway. References Cochran WG (1954) Some methods for strengthening the common χ2 test. Biometrics, 10, 417–451. CYTEL Software Corporation (1992) StatXact-Turbo; statistical software for exact nonparametric inference. CYTEL Software Corporation, Cambridge, MA. Everitt BS (1977) The Analysis of Contingency Tables. Chapman & Hall, London. Fisher RA (1950) Statistical Methods for Research Workers. 11th edn. Oliver and Boy, London. Jorde PE, Ryman N (1996) Demographic genetics of brown trout (Salmo trutta) and estimation of effective population size from temporal change of allele frequencies. Genetics, 143, 1369–1381. Legendre P, Legendre L (1998) Numerical Ecology. 2nd edn. Elsevier, Amsterdam. Lewontin RC, Felsenstein J (1965) The robustness of homogeneity tests in 2xN tables. Biometrika, 36, 117 – 129. Mehta CR, Patel NR (1983) A network algorithm for performing Fisher’s exact test in r × c contingency tables. Journal of the American Statistical Association, 78, 427–434. Raymond M, Rousset F (1995a) An exact test for population differentiation. Evolution, 49, 1280–1283. © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361– 2373 MEC1345.fm Page 2373 Friday, September 21, 2001 3:36 PM D E T E C T I N G G E N E T I C D I F F E R E N T I AT I O N 2373 Raymond M, Rousset F (1995b) genepop (version 1.2): a population genetics software for exact tests and ecumenicism. Journal of Heredity, 86, 248 – 249. Rice WR (1989) Analyzing tables of statistical tests. Evolution, 43, 223 – 225. Roff DA, Bentzen P (1989) The statistical analysis of mitochondrial DNA polymorphisms: X2 and the problem of small samples. Molecular Biology and Evolution, 6, 539–545. Rohlf FJ (1987) BIOM. A Package of Statistical Programs to Accompany the Text of Biometry. Applied Biostatistics, Inc., New York. Sokal RR, Rohlf FJ (1981) Biometry. 2nd edn. W.H. Freeman, San Francisco, CA. StatSoft Inc. (1998) STATISTICA for Windows. StatSoft, Inc., Tulsa, OK. © 2001 Blackwell Science Ltd, Molecular Ecology, 10, 2361–2373 Zar JH (1984) Biostatistical analysis. 2nd edn. Prentice Hall, Inc., Englewood Cliffs, New Jersey. Nils Ryman is a professor of genetics and heads the Division of Population Genetics at the Stockholm University. His research has focused primarily on the genetic structure of natural populations, the genetic effects of human exploitation of such populations, and related conservation genetics issues. Per Erik Jorde graduated in genetics in Stockholm and is presently a postdoctoral fellow at the University of Oslo, working on microsatellite DNA analyses of fishes.