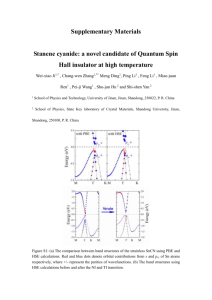
Cation-Disordered Oxides for Rechargeable Lithium Battery Cathodes Jinhyuk Lee
advertisement

Cation-Disordered Oxides for Rechargeable Lithium Battery Cathodes by Jinhyuk Lee B.S. Materials Science and Engineering Seoul National University (2010) Submitted to the Department of Materials Science and Engineering in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Materials Science and Engineering at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY September 2015 © Massachusetts Institute of Technology, 2015. All rights reserved. Signature of author .................................................................................................... Department of Materials Science and Engineering July 21, 2015 Certified by ............................................................................................................... Gerbrand Ceder R. P. Simmons Professor of Materials Science and Engineering Thesis Supervisor Accepted by............................................................................................................... Donald R. Sadoway Chair, Departmental Committee on Graduate Students 2 Cation-Disordered Oxides for Rechargeable Lithium Battery Cathodes by Jinhyuk Lee Submitted to the Department of Materials Science and Engineering On July 21, 2015, in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Materials Science and Engineering Abstract The demands for high-energy density cathode materials for rechargeable lithium batteries are ever increasing. This is because such cathode materials will enable smaller and lighter rechargeable lithium batteries for complex applications such as in electric vehicles or in grid energy storage. Nearly all high-energy density cathode materials for rechargeable lithium batteries have been sought from well-ordered close-packed oxides in which lithium and transition metal ions occupy distinct sites. In contrast, cation-disordered oxides, whose cation distribution is fully or partially random, have received only a limited attention, because lithium diffusion tends to be limited by their cation-disordered structure. In the first part of this thesis, from the study of Li1.211Mo0.467Cr0.3O2, it is demonstrated that cation-disordered oxides can be promising cathode materials if they contain enough lithium excess (x >1.09 in LixTM2-xO2). Li1.211Mo0.467Cr0.3O2 forms into a layered structure, but transforms almost completely to a cation-disordered structure during cycling. While common wisdoms would expect poor cyclability of this material due to limited lithium diffusion in its structure, the reversible capacity of this material is remarkably high (~265 mAh/g), which demonstrates that lithium diffusion can be facile in the cation-disordered structure. Using ab initio calculations, we show that this counterintuitive behavior is due to percolation of a certain type of active diffusion channels (0-TM channels) in disordered Li-excess materials, which becomes more extensive as the lithium-excess level increases. In the second part of this thesis, a new class of high capacity cation-disordered oxides (Li-Ni-Ti-Mo oxides) is designed based on the 0-TM percolation theory. As the theory predicts, the reversible capacity and rate capability in these materials considerably improve with lithium excess. In particular, Li1.2Ni1/3Ti1/3Mo2/15O2 delivers high capacity and energy density up to 250 mAh/g and 750 Wh/kg at 10 mA/g. Combining in situ X-ray diffraction, electron energy loss spectroscopy, and X-ray absorption near edge spectroscopy, we investigate the redox mechanism of the new materials and discuss how oxygen loss with lattice densification can affect lithium diffusion in the materials by decreasing the lithium-excess level. From these understandings, strategies for further improvements are proposed, setting new guidelines for the design of high performance cation-disordered oxides for rechargeable lithium batteries. Thesis Supervisor: Gerbrand Ceder Title: R. P. Simmons Professor of Materials Science and Engineering 3 Acknowledgements First, I would like to thank my thesis advisor, Professor Gerbrand Ceder, for his intellectual and mental supports. I was very excited and honored to work with him who is not only insightful but also fearless in conducting research. Thanks to him, I could finally become an independent researcher who can pursue ideas that can greatly contribute to the materials research society. I would also like to thank my thesis committee members, Professor Donald Sadoway and Professor Jeffrey Grossman, for their valuable comments and supports on my research. Thanks to their generosity and supports, I could successfully complete my doctoral research. I also thank my collaborators in the CEDER group (Dr. Alexander Urban, Dr. Xin Li, and Dr. Dong-Hwa Seo) for their continuous support on my research. Without them, none of my work could have been realized. The entire CEDER group members (Aziz Abdellahi, Rahul Malik, Stephen Dacek, Sai Gautam, Eric Wang, Rui Wang, Lei Liu, Wenxuan Huang, Daniil Kitchaev, Ian Matts, William Richards, Ziqin Rong, Wenhao Sun, Alexandra Toumar, Piere Manuele Canepa, Shinyoung Kang, Jae Chul Kim, Dong-Hwa Seo, Nancy Twu, Alexander Urban and Kathy Simons) are greatly acknowledged. The greatest thing about the CEDER group is that you can work with the nicest and the smartest people in the world whom you can be greatly inspired by. In particular, I thank the Korean members in the CEDER group (Dr. Jae Chul Kim, Dr. Shinyoung Kang, Dr. Dong-Hwa Seo, and Prof. Byoungwoo Kang) for their support. Thanks to them, my life in the CEDER group could be more lively and vivid. I always felt joys when spending time with them. Moreover, Nancy Twu and Wenhao Sun are greatly acknowledged. I had to go through many different moments during the graduate life, and I was so happy to share happy and harsh moments with them as the same-year CEDER group members. 4 Samsung Scholarship is greatly acknowledged for supporting me both financially and mentally. I will not forget happy moments that I shared with the Samsung Scholarship community. I also thank the Robert Bosch Corporation and the Umicore Specialty Oxides and Chemicals for their financial support on my research. All the Korean graduated students in the DMSE are greatly acknowledged. They have been a social family to me, guiding me toward a right direction for my graduate life. In particular, I thank all the Korean students who joined DMSE MIT in the same year of 2010 (Donghun Kim, Mansoo Park, Jiyoun Chang, Gyehyun Kim, and Sangtae Kim). I spent more time with them than any other people in MIT, and shared all the fun and happy moments with them during the graduate life. I hope to keep in touch with them and wish their future stays as bright as now. I cannot thank enough to my parents and my brother. I could not have grown up strong and confident without their endless support and love. Finally, I thank my beloved wife, Lina Dahye Song, whose patience, support and love made this possible. 5 6 Contents List of Figures and Tables ................................................................................................ 11 Part I. Introduction .......................................................................................................20 1.1 Motivation.....................................................................................................................21 1.2 Overview of rechargeable lithium batteries .........................................................22 1.3 Layered lithium transition metal oxides for Li-ion battery cathodes ........... 26 1.4 Ordering paradigm for the oxide cathodes ...........................................................30 1.5 Overview of the thesis ...............................................................................................35 Part II. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries ................................................................................36 2.1 Introduction ..................................................................................................................37 2.2 Methodology ................................................................................................................39 2.2.1 Experimental methodology ........................................................................................ 39 2.2.1.1 Synthesis .............................................................................................................39 2.2.1.2 Electrochemistry..................................................................................................45 2.2.1.3 Characterization ..................................................................................................45 2.2.2 Computational methodology...................................................................................... 46 2.2.2.1 Density-functional theory calculations ............................................................... 46 2.2.2.2 Structural model ..................................................................................................47 2.2.2.3 Percolation simulation ........................................................................................ 48 7 2.3 Experimental results ...................................................................................................49 2.3.1 Characterization of the as-synthesized Li1.211Mo0.467Cr0.3O2 .....................................49 2.3.2 The electrochemical properties of Li1.211Mo0.467Cr0.3O2 ............................................51 2.3.3 The structural change in Li1.211Mo0.467Cr0.3O2 during cycling ...................................55 2.3.4 Redox mechanism of Li1.211Mo0.467Cr0.3O2 ................................................................ 59 2.4 Computational results ................................................................................................61 2.4.1 Li diffusion channels in cation-disordered oxides ..................................................... 61 2.4.2 Li diffusion barriers in cation-disordered oxides: active 0-TM channels ..................62 2.4.3 Percolation of 0-TM diffusion channels ....................................................................65 2.5 Discussions ...................................................................................................................67 2.6 Conclusion ....................................................................................................................69 Part III. A new class of high capacity cation-disordered oxides for rechargeable lithium batteries: Li-Ni-Ti-Mo oxides .................................71 3.1 Introduction ..................................................................................................................72 3.2 Methodology ................................................................................................................74 3.2.1 Experimental methodology ........................................................................................ 74 3.2.1.1 Synthesis .............................................................................................................74 3.2.1.2 Electrochemistry .................................................................................................74 3.2.1.3 X-ray diffraction (XRD), scanning electron microscopy (SEM), and electron energy loss spectroscopy (EELS) .......................................................... 75 3.2.1.4 In situ X-ray diffraction ...................................................................................... 76 3.2.1.5 Ex situ X-ray near edge spectroscopy (XANES) ................................................76 8 3.2.2 Computational methodology...................................................................................... 77 3.3 Experimental results ..................................................................................................79 3.3.1 Design strategy form percolation theory....................................................................79 3.3.2 Characterization of Li-Ni-Ti-Mo oxides ....................................................................81 3.3.3 The electrochemical properties of Li-Ni-Ti-Mo oxides .............................................86 3.3.4 The structural evolution of Li1.2Ni1/3Ti1/3Mo2/15O2 during cycling ............................ 92 3.3.5 Investigation on the redox mechanism ......................................................................93 3.4 Computational results ................................................................................................99 3.4.1 Oxygen loss mechanisms ........................................................................................... 99 3.4.2 Calculated voltage profiles from possible redox mechanisms .................................101 3.4.3 Reduction of Mo and Ti after oxygen loss ............................................................... 103 3.5 Discussions .................................................................................................................105 3.5.1 Redox mechanism ....................................................................................................105 3.5.2 Electrochemical performance .................................................................................. 107 3.5.3 Strategies for improvements .................................................................................... 109 3.6 Conclusion .................................................................................................................. 111 Part IV. Conclusions .................................................................................................... 112 References .......................................................................................................................... 117 9 10 List of Figures and Tables Figure 1-1 Gravimetric power and energy densities for different rechargeable batteries, which are currently being investigated for grid storage applications. Figure 1-2 Schematic description of Li-ion batteries: reversible shuttling of Li+ ions between the cathode and the anode enables Li-ion batteries. Figure 1-3 Illustrations of the crystal structure of (a) a layered Li-TM oxide, (b) a spinel Li-TM oxide, and an olivine LiMPO4 (M = Fe, Mn etc.) Figure 1-4 The first-cycle voltage profile of bare and coated LiCoO2 when cycled between 2.75−4.4 V at a rate of 70 mA/g. Figure 1-5 The first-cycle voltage profile of Li/LiNi1/3Mn1/3Co1/3O2 with a current density of 0.2 mA/cm2. Figure 1-6 The first-cycle voltage profiles of 0.3Li2MnO3·0.7LiMn0.5Ni0.5O2. Figure 1-7 The discharge profile at various C rates for the ion-exchanged (IE)-LiNi0.5Mn0.5O2 (above) and sold state (SS)-LiNi0.5Mn0.5O2 (below). In the test, the cell was charged at C/20 to 4.6 V, then held at 4.6 V for 5 hours and discharged at different rates. 1C corresponds to 280 mA/g. Figure 1-8 (a) The octahedral to tetrahedral to octahedral Li diffusion in general rocksalt-type Li-TM oxides (b) The 1-TM channel in stoichiometric Li-TM oxides Figure 1-9 (a) The decrease in the Li slab distance (tetrahedron height) by cation mixing: the layered structure has much larger Li slab spacing than the cation-disordered structure. (b) The calculated activation barrier for Li migration in LiNiO2 with cation mixing (stars) and without cation mixing (squares). The Li slab distances with and without cation mixing are indicated 11 with thick and thin dashes at different Li contents, respectively. The shift on the left is from fully lithiated state and the one on the right is from partially delithiated state. Figure 1-10 (a) The X-ray powder diffraction patterns of LiCoO2 powders prepared at high temperature after subsequent exposure to different milling times. (b) Voltage profiles of cathodes prepared from LiCoO2 samples exposed to different milling times. Figure 1-11 (a) Charge-discharge curves of high-temperature α-LiFeO2 and low-temperature ɑLiFeO2 (b) Capacity retention of low-temperature α-LiFeO2. Figure 2-1 Illustration of the layered rocksalt structure and disordered rocksalt structure: cation mixing between Li and TM layers lead to a disordered rocksalt structure, which tends to result in poor cyclability of Li-TM oxides by slowing down Li diffusion. Figure 2-2 The voltage profile of Li2VO3. Figure 2-3 High resolution transmission electrons microscopy images in two directions of the carbon-coated Li1.211Mo0.467Cr0.3O2 particle (top), and uncoated Li1.211Mo0.467Cr0.3O2 particle (bottom) Figure 2-4 The two upper left images show a Li metal foil and a separator from a Li1.211Mo0.467Cr0.3O2 half-cell that was charged/discharged for 100 cycles between 1.5−4.3 V at 164 mA/g at room temperature. The upper right SEM image shows spherical particles on the colored area on the separator (dotted circle). The lower image shows an energy dispersive Xray spectroscopy (EDS) spectrum of a particle attached to the separator, indicating that the particles from the colored area are composed of Mo and Cr (Mo:Cr = 8.875:1). Figure 2-5 Li1.211Mo0.467Cr0.3O2 half-cells were disassembled at points A, B, and C on the first charge voltage profile of Li1.211Mo0.467Cr0.3O2 at 3.2 mA/g. The three inset images with red boundaries show Li metal anode foils (attached to a current collector rod) from Li1.211Mo0.467Cr0.3O2 half cells that were disassembled after four days of resting at the first charge state of A) 200 mAh/g, B) 250 mAh/g, and C) 300 mAh/g. The inset image with a blue boundary shows a Li metal anode foil that was disassembled from a carbon-coated 12 Li1.211Mo0.467Cr0.3O2 half-cell after four days of resting at the same first charge state of 300 mAh/g as point C. Figure 2-6 The discharge-capacity retention of carbon-coated and uncoated Li1.211Mo0.467Cr0.3O2 during cycling between 1.5−4.3 V at 16.4 mA/g at room temperature. The two inset images show Li metal anode foils from uncoated and carbon-coated Li1.211Mo0.467Cr0.3O2 half-cells after ten cycles. Carbon coating retards transition metal dissolution, improving the capacity retention of carbon-coated Li1.211Mo0.467Cr0.3O2. Figure 2-7 The first-cycle voltage profile of the carbon-coated and uncoated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at 32.7 mA/g at room temperature. Figure 2-8 Structural model of disordered-Li2MoO3: The simulation cell contains 12 formula units (72 atoms). In the figure, large red balls represent oxygen atoms, and small green and violet balls represent the octahedral sites of lithium and molybdenum, respectively. Figure 2-9 0-TM wrapping probability (a) and accessible lithium fraction (b) as a function of the Li-excess level for different FCC supercell sizes. Results for fully disordered structures are shown. The legends refer to supercells of the primitive FCC unit cell. Figure 2-10 Rietveld refinement on the XRD pattern of pristine Li1.211Mo0.467Cr0.3O2 powder. The atomic ratio between elements was first set to the ICP result, and then Mo and Cr occupancy in Li and TM layers were refined. The refinement suggests no transition metals in Li layers at all. Li occupancy was refined after Mo and Cr occupancy had been determined. Figure 2-11 (a) The (003) peak in the XRD pattern on Li1.211Mo0.467Cr0.3O2 [black] and that of LiCrO2 [blue] (b) The electron diffraction pattern along ZA[010] of Li1.211Mo0.467Cr0.3O2. Figure 2-12 The broader XRD peaks of the carbon-coated Li1.211Mo0.467Cr0.3O2 powder (lower left) compared to the sharper XRD peaks of pristine Li1.211Mo0.467Cr0.3O2 powder (upper left) indicate that the carbon-coated Li1.211Mo0.467Cr0.3O2 particles are smaller than the uncoated particles. Furthermore, SEM images (right) confirm that the particles of the carbon-coated Li1.211Mo0.467Cr0.3O2 are smaller than those of uncoated one. During the carbon coating process, 13 sucrose (carbon precursor) and the Li1.211Mo0.467Cr0.3O2 powder were mixed in a planetary ballmill for six hours at 300 rpm, resulting in smaller particles after carbon coating. Figure 2-13 The voltage profile of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at the rate of 16.4 mA/g at room temperature Figure 2-14 The 10-cycle voltage profiles of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at the rate of (a) 4.1 mA/g, (b) 8.2 mA/g, and (c) 16.4 mA/g at room temperature (d) Corresponding specific capacity vs. cycle number plot Figure 2-15 The voltage profile of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at a rate of C/10, C/2, 1C, 2C, 4C and 10C (1C = 327 mA/g) Figure 2-16 Capacity retention of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5-4.3 V at a rate of C/10, C/2, 1C, 2C, 4C and 10C (1C = 327 mA/g) Figure 2-17 The ex situ XRD pattern of the carbon-coated Li1.211Mo0.467Cr0.3O2 electrode before cycle, after one cycle, after two cycles, and after 10 cycles between 1.5−4.3V at 32.7 mA/g. Figure 2-18 Rietveld refinement on the XRD pattern of a carbon-coated Li1.211Mo0.467Cr0.3O2 electrode after ten cycles between 1.5−4.3 V at 32.7 mA/g. Kapton film was applied to prevent oxidation, resulting in humps below 30°. From XRD refinement, it is difficult to precisely quantify and distinguish the level of Mo migration and Cr migration to Li layers. Therefore, we refined the XRD pattern with two different methods: (a) Refine Mo occupancy first then Cr occupancy. Here, refinement suggests that only Mo migrates (0.2634 Mo in Li layers). (b) Assume all Cr are in Li layers then refine Mo occupancy. Refinement is much improved by assuming 0.097988 Mo in addition to 0.3 Cr in Li layers. In both methods, Li occupancy was later refined by distributing 0.92 Li (based on the cumulative charge-discharge capacity after ten cycles) to Li and TM layers. However, the values are more subject to error. The two ways of refining give two extremes of the transition metal (TM) migration level. Refinement based 14 on the method A suggests a TM migration level of ~34.3% (i.e. ~34.3% of overall TM ions are in Li layers), and refinement based on the method B suggests a TM migration level of ~51.88%. From the two values, we can expect that at least ~34% to ~52% of TM ions have migrated to Li layers after ten cycles, indicating substantial cation mixing in Li1.211Mo0.467Cr0.3O2. Note that the TM migration level can slightly vary with the rate of cycling which changes the delithiation level at the end of charge. To determine the average tetrahedron height of the disorderedLi1.211Mo0.467Cr0.3O2, eight tetrahedron heights (four from the tetrahedron in Li layers, and four from that in TM layers) were averaged out, as Li1.211Mo0.467Cr0.3O2 is not yet completely disordered. Method A yields 2.39701 Å (× 3), 2.38362 Å (× 3), 2.41831 Å (× 1), and 2.37791 Å (× 1), resulting in the average tetrahedron height of 2.39226 Å . Method B yields 2.39816 Å (× 3), 2.38775 Å (× 3), 2.40875 Å (× 1), and 2.37756 Å (× 1), resulting in the average tetrahedron height of 2.393 Å . Therefore, both methods suggest the average tetrahedron height of disordered- Li1.211Mo0.467Cr0.3O2 to be ~2.39 Å . Figure 2-19 Left: the scanning transmission electron microscopy images along the [010] zone axis in a carbon-coated Li1.211Mo0.467Cr0.3O2 particle before cycling and after 1 and 10 cycles between 1.5−4.3 V at 16.4 mA/g. Right: Corresponding line profiles of the Z-contrast information with the measured spacing of Li-Mo-Cr layers. Figure 2-20 (a) The cyclic voltammetry profile of the carbon-coated Li1.211Mo0.467Cr0.3O2, (b) Cr EELS L3/L2 ratio in Li1.211Mo0.467Cr0.3O2 before cycling, after the first charge, and after the first cycle. (c) Integrated spin density of Mo and Cr in Li1.211Mo0.467Cr0.3O2 as a function of the delithiation level, obtained by DFT calculations. Figure 2-21 (a) o-t-o diffusion: Two tetrahedral paths connect each pair of neighboring octahedral sites (b-d) The activated Li+ ion can face-share with no octahedral transition metals (0-TM channel) (b), one transition metal (1-TM channel) (c), or two transition metals (2-TM channel) (d): note that the 2-TM channels do not support divacancy diffusion mechanism, and thus diffusion barrier through the 2-TM channels are extremely high. Figure 2-22 Calculated Li migration barriers along 1-TM (Mo4+) channels (red squares), 1-TM (Cr3+) channels (blue triangles), and 0-TM (Li+) channels (black circles) as a function of the 15 average tetrahedron height of model disordered structure (disordered Li2MoO3, disordered LiCrO2). Error bars denotes standard deviation. The shaded area highlights typical tetrahedron heights of disordered materials Figure 2-23 (a) Computed probability to find a percolating network of 0-TM channels (color) vs. Li content (x in LixTM2-xO2) and cation mixing (TMLi layers/TMTM layers ×100%) (b) The accessible Li contents by a percolating 0-TM network (color) vs. Li content and cation mixing. In the simulation, cations were randomly distributed at each cation mixing level Table 2-1 The average tetrahedron height of disordered rocksalt-type Li-TM-oxides, deduced from the literatures. The tetrahedron height of a disordered Li-TM-oxide is equivalent to its (111) plane distance, which can be derived by dividing the a-lattice parameter of each material by √3. Figure 2-24 (a) The XRD patterns of the carbon-coated Li1.211Mo0.467Cr0.3O2 after ten cycles, after ten cycles then charged to Li0.6165Mo0.467Cr0.3O2, and after ten cycles then charged to Li0.3082Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at 32.7 mA/g. (b) The c- and a-lattice parameter in disordered Li1.211Mo0.467Cr0.3O2 (based on the space group of R-3m) at different delithiation states (x = 0.291, 0.5945, 0.90275 in Li1.211-xMo0.467Cr0.3O2). The change in the lattice parameters is very small, leading to negligible volume change (<~0.12 %) upon delithiation Figure 3-1 (a) The crystal structure of disordered rocksalt-type solid solution compounds between LiNi0.5Ti0.5O2 and Li1+xM1-xO2 (M = Ti4+, Nb5+, Mo6+) (b-d) Theoretical capacities are given when M is (b) Ti4+ (c) Nb5+ and (d) Mo6+. Each figure plots three different capacities: Li capacity that assumes full extraction of available Li-ions, Ni2+/Ni4+ redox capacity, and 0-TM capacity that is the Li capacity accessible by the percolating 0-TM network Figure 3-2 The X-ray diffraction patterns of LiNi0.5Ti0.5O2 (LNTO), Li1.05Ni11/24Ti11/24Mo1/30O2 (LNTMO5), Li1.1Ni5/12Ti5/12Mo1/15O2 (LNTMO10), Li1.15Ni3/8Ti3/8Mo1/10O2 (LNTMO15), and Li1.2Ni1/3Ti1/3Mo2/15O2 (LNTMO20): insets are the a-lattice parameters of the compounds Table 3-2 Target vs. actual Li:Ni:Ti:Mo atomic ratio as determined by direct current plasma 16 emission spectroscopy. Figure 3-3 Rietveld refinements on the XRD patterns of (a) LiNi0.5Ti0.5O2 [LNTO], (b) Li1.05Ni11/24Ti11/24Mo1/30O2 [LNTMO5] (c) Li1.1Ni5/12Ti5/12Mo1/15O2 [LNTMO10], (d) Li1.15Ni3/8Ti3/8Mo1/10O2 [LNTMO15], and (d) Li1.2Ni1/3Ti1/3Mo2/15O2 [LNTMO20]: structural parameters from the refinements are listed in Table 3-2. Table 3-2 Structural parameters from the Rietveld refinements in Figure 3-3: crystallographic information file of Fm-3m LiFeO2 was first used as an input file. The atomic occupancies were initially set to the atomic ratio obtained from the elemental analysis by direct current plasma emission spectroscopy, based on which the lattice parameters were initially refined. Then, we further refined the lattice parameters and the atomic occupancies simultaneously: TM occupancies were first refined, and then Li occupancy was refined. O occupancy did not change after the refinement. Figure 3-4 Scanning electron microscope (SEM) images of LNTO, LNTMO5, LNTMO10, LNTMO15, LNTMO20, and high-energy ball-milled LNTMO20 (HB-LNTMO20) Figure 3-5 (a) The first-cycle voltage profiles of LNTO, LNTMO5, LNTMO10, LNTMO15 and LNTMO20 [1.5−4.5 V, 20 mA/g], and (b) 20-cycle capacity retention of the compounds. Figure 3-6 The voltage profiles for 10 cycles of (a) LNTO, and (b) LNTMO20 [1.5−4.5 V, 20 mA/g, room temperature]. Figure 3-7 The voltage profiles of (a) LNTO and (b) LNTMO20 when charged and discharged once at 10 mA/g, and then at 20, 40, 100, 200, and 400 mA/g for the subsequent cycles. Figure 3-8 (a) The first discharge voltage profile of LNTMO20 from a galvanostatic intermittent titration test after charging to 270 mAh/g: the inset zooms in the time range between 240 h and 270 h. (b) The voltage profile of LNTMO20 when charged-discharged three times between 2.0−4.1 V (black) then between 1.5−4.5 V (blue) at 20 mA/g. 17 Figure 3-9 (a) The In situ XRD patterns of LNTMO20 upon two galvanostatic chargedischarge cycles between 1.5−4.8 V at 10 mA/g (b) The corresponding voltage profile (c) and the a-lattice parameter from single phase XRD refinements are shown. (d) The (002) peak is zoomed in for the intensity comparison. Figure 3-10 The X-ray absorption near-edge structures of (a) Ni, (b) Ti, and (c) Mo in LNTMO20 before cycle [black], after the first charge to 4.8 V [blue, ~300 mAh/g charged], and after the first discharge to 1.5 V [red, ~250 mAh/g discharged] at 20 mA/g. Figure 3-11 Electron energy loss spectra of Ti L-edge and O K-edge in LNTMO20 before cycling [black] and after 20 cycles between 1.5−4.5 V [red] at 20 mA/g: the inset focuses on the Ti L-edges. Figure 3-12 The first-cycle cyclic voltammetry profiles of LNTMO20 when voltage-swept between 1.5−4.5 V [black solid] and 1.5−4.1 V [red dash] at 0.1 mV/s. Figure 3-13 Back-of-the-envelope calculation: Average Mo oxidation state expected after the first charge to 300 mAh/g then discharge to 250 mAh/g as a function of the oxygen-loss capacity during the first charge. It was assumed that Ni, Ti, and O stay as Ni2+, Ti4+, and O2after the first discharge, and that there is no loss in the TM content during the first cycle. Figure 3-14 The calculated energies of densified Li0.52Ni0.37Ti0.37Mo0.15O2 structures and Li0.47Ni0.33Ti0.33Mo0.13O1.8 structures with oxygen vacancies: From the plot, it is seen that the energies of the densified Li0.52Ni0.37Ti0.37Mo0.15O2 structures are lower than those of Li0.47Ni0.33Ti0.33Mo0.13O1.8 structures with oxygen vacancies. This indicates that oxygen loss with densification is thermodynamically more favorable than that with oxygen vacancies in the lattice. Figure 3-15 (a) The voltage profiles of Li1.2-xNi0.33Ti0.33Mo0.13O2: The black curve is an experimental profile from the galvanostatic intermittent titration test (GITT) during the first charge, and the red line is calculated voltage profile of Li1.2-xNi0.33Ti0.33Mo0.13O2 that assumes 18 no oxygen loss during the first charge: the dotted red arrows specify the region of Ni oxidation and O oxidation. Finally, the dashed blue line indicates the oxygen-loss potential of Li1.2xNi0.33Ti0.33Mo0.13O2-y. (b) The calculated contribution of Ni [black] and oxygen [red] oxidation upon delithiation in Li1.2-xNi0.33Ti0.33Mo0.13O2 when no oxygen loss is assumed (the red curve in Figure 3-15a) Figure 3-16 (a) The voltage profiles of Li1.11-xNi0.37Ti0.37Mo0.15O2. (b) The average net moments of Ni, Ti, Mo and oxygen ions in Li1.11-xNi0.37Ti0.37Mo0.15O2 (x = 0, 0.074, 0.148, 0.222, 0.296, 0.370, 0.444, 0.519, 0.593, 0.667, 0.741, 0.815, 0.889, 0.963 and 1.037). Figure 3-17 Proposed first charge mechanism of LNTMO20: Ni2+/Ni~3+ oxiation, oxygen loss, and oxygen oxidation largely in sequence. The voltage profile of LNTMO20 from the in situ XRD test and the corresponding a-lattice parameters from single-phase XRD refinements are overlayed. Figure 3-18 Illustrations of a LNTMO20 particle before and after oxygen loss with densification near the surface: oxygen loss with densification reduces the Li-excess level, and thus decreases the 0-TM capacity. 19 -------------Part I------------- Introduction 20 1.1 Motivation Sustainable growth of our modern society requires clean energy production, storage, and transport. For the energy storage, rechargeable lithium batteries (i.e. Li-ion batteries) have played an irreplaceable role as they outperform the other types of technologies in terms of energy densities and power densities (Fig. 1-1).1,2 Due to their great performance, the Li-ion batteries have enabled many of the convenience of modern life, powering increasingly capable electronics. However, as the batteries are requested for more complex technologies where higher power consumptions are required, such as for electric vehicles and grid energy storage, the demand for higher performance Li-ion batteries has been ever increasing.2–6 Figure 1-1 Gravimetric power and energy densities for different rechargeable batteries, which are currently being investigated for grid storage applications.2 The development of better Li-ion batteries requires improvements in the electrode materials.1,5 In particular, current cathode materials do not satisfy all desired characteristics for 21 high-performance Li-ion batteries, including energy density, rate capability, safety, cycle life, cost, and toxicity.5 Hence, the development of novel cathode materials is critical for the design of high-performance Li-ion batteries for future applications such as in grid storage and longrange electric vehicles, thereby our society can grow sustainably and continuously.2,3,6 1.2 Overview of rechargeable lithium batteries Figure 1-2 Schematic description of Li-ion batteries: reversible shuttling of Li+ ions between the cathode and the anode enables Li-ion batteries. The Li-ion batteries are electrochemical energy storage devices that operate by shuttling Li+ ions and electrons between the cathode and the anode both of which serve as a reservoir for Li (Fig. 1-2).5,7–9 They are composed of three main components: the cathode, the anode, and the electrolyte. The cathode and anode are the positive and negative electrodes, respectively, where oxidation and reduction occur during charge and discharge.5,7 The electrolyte is an ionic conductor but electronic insulator, allowing Li+ ions to migrate between the cathode and the 22 anode. Upon charge, an external power supply is applied to convert electrical energy into chemical energy, transferring Li+ ions (through the electrolyte) and electrons (through an external circuit) from the cathode to the anode, for example, with the following reactions:4,5,9 (1) Cathode: (2) Anode: LixMOy (cathode) σ Li+ (electrolyte) + σ e- + Lix-σMOy (cathode) σ Li+ (electrolyte) + σ e- + σ C6 (anode) σ LiC6 (anode) During charge, oxidation and reduction occurs at the cathode and anode, respectively. Upon discharge, the reverse reactions occur, thus Li+ ions and electrons migrate from the anode to the cathode. Concurrently, reduction and oxidation occurs at the cathode and anode, respectively, releasing chemical energy into electrical energy. In commercial Li-ion batteries, graphite is typically used as the anode, which allows intercalation of Li+ ions between its sheets to form LiC6.4 The cathode usually is composed of a cathode material from inorganic compounds with transition metal ions as redox active species, carbon black to enhance electronic conductivity, and a polymeric binder such as poly tetrafluorethylene (PTFE). The electrolyte typically consists of Li salts, such as LiPF6, dissolved in organic solvents, such as ethylene carbonate (EC) with dimethyl carbonate (DMC).7,10,11 One of the greatest aims in the Li-ion battery research is to develop very high energy density Li-ion batteries. The energy density of a battery is defined as the energy per unit weight or volume, which is equal to the product of the cell voltage and specific capacity.5,12 Voltage of a Li-ion battery whose cathode and anode are both intercalation compounds for Li is determined by the difference in the chemical potential of Li in the cathode and the anode:5,12 V=− µLi(𝑐𝑎𝑡ℎ𝑜𝑑𝑒) − µLi(𝑎𝑛𝑜𝑑𝑒) 𝐹 In most commercial Li-ion batteries, LiCoO2 and graphite are used as the cathode and the anode, respectively, which gives an average voltage of ~3.6 V.1,8 23 The specific capacity of Li-ion batteries is determined by the cyclable Li content per unit weight or volume.5 As both cathode and anode are intercalation compounds for Li ions, the specific capacity of a battery depends on those of both electrodes. The specific capacity of an intercalation electrode can be calculated using the following equation: Specific capacity = 𝛥𝑥·𝐹 𝑀 × 1000 3600 [mAh/g] where Δx is the number of moles of Li that can reversibly inserted/extracted in and out of the electrode structure within a designated voltage window. M is the molar mass of the electrode compound. Finally, F is the Faraday constant. As can be inferred from the above formula, a high specific capacity of an electrode requires a light electrode material (small M) that can reversibly store and release a large amount of Li+ ions (high Δx). High power density (or high rate capability) is also a highly desired property of Li-ion batteries, particularly for high-power applications such as for electric vehicles or power tools.13,14 The power density of a battery is defined as the battery power per unit weight, which is the product of the current and the cell voltage.13 The power density is determined by the cell resistances from various factors such as mass transfer resistance of electrodes (related to Li diffusion), charge-transfer resistance at the electrode/electrolyte interface (related to solvation and desolvation of Li salts), and electric resistances (related to ionic conduction) in the electrolyte.8,13,14 A battery with large resistances will have a poor rate capability, and a large portion of the chemically stored energy will dissipate into heat upon discharge. Cycle life, cost, and safety are the other desired properties of Li-ion battery.8 Cycle life is defined as the number of charge-discharge cycles that can be performed before the specific capacity decreases below a certain cut-off limit. Using Li-ion batteries for a while, many kinds of side reactions can occur, which can build up internal resistances on the batteries. Also, the crystal structures of electrode materials can change during cycling, adding resistances as well. 24 Such increase in the resistances can lead to poor cycle life.8 Low cost is also very critical for successful Li-ion batteries, because the cost determines the economic viability of a battery.8 The deployment of electric vehicles has been slow because of the high cost of Li-ion batteries, resulting in no advantages of buying the electric vehicles instead of conventional gasoline vehicles.15 The cost of a battery will decrease as processing cost and materials’ cost decrease. The performance of the Li-ion batteries is largely determined by the properties of electrode materials. Thus, the search for better cathode/anode materials has been intense.4,5,8 Electrode materials for high performance Li-ion batteries should have the following characteristics. First, the materials should have a stable and sturdy crystal structure with many available Li sites to reversibly store and release a large amount of Li+ ions. Second, the material’s (molar) mass should be light such that the charge-storage capacity per unit weight can be high. Third, the Li-intercalation potential of the electrode material should be within a voltage window of an electrolyte for safety.5,8 Within an acceptable voltage window, high (low) intercalation potential is preferred for a cathode (anode) material, because it will result in high voltage thus high energy densities. Fourth, Li diffusion should be facile in the crystal structure, allowing for fast Li (de)intercalation reaction with negligible resistances.13,14 Fast Li diffusion is critical to achieve high energy densities and high rate capability from an electrode material and thus from a battery. Finally, the electrode materials should be thermally safe (stable) such that they do not release oxygen and heat at a high temperature. Oxygen evolution is an exothermic reaction, and thus can substantially increase temperature inside a battery. With oxygen gas, high temperature, and a flammable organic electrolyte, a Li-ion battery can catch fire, which must be prevented for safety.16,17 25 1.3 Layered lithium transition metal oxides for Li-ion battery cathodes While all components are important in Li-ion batteries, the cathode has been the limiting component on the current stage of Li-ion battery technology. This is because the cathode is the major factor that determines the energy density, rate capability, safety, and cost of Li-ion batteries.5,8 As the cathode materials, intercalation compounds have been most widely used due to their peculiar ability to accommodate Li+ ions over large concentration intervals. Figure 1-3 Illustrations of the crystal structure of (a) a layered Li-TM oxide, (b) a spinel Li-TM oxide, and an olivine LiMPO4 (M = Fe, Mn etc.) Today, three types of intercalation cathodes are being intensively studied, so called layered (rocksalt-type) lithium transition metal oxides (Li-TM oxides),18–24 spinel-type Li-TM oxides,25–30 and olivine LiMPO4 (M = Fe, Mn etc.) (Fig. 1-3).14,31–37 While each class has its own strength, none of them are perfect. For example, Li-excess layered oxides offer the highest capacity (> 220 mAh/g) and energy density (~900 Wh/kg), but are less safe than other types of cathode materials.38–45 On the other hand, spinel-type oxides have excellent rate capability but 26 with low specific capacity (~100 mAh/g), low energy density, and poor cycle life at high temperature.25,27,30,46–49 Finally, olivines such as LiFePO4 are cheap, safe and have excellent cycle life and rate capability, but with low gravimetric and volumetric energy densities (590 Wh/kg, 2000 Wh/l) due to their low density (~3.4 kg/l) and low average voltage (~3.5 V vs. Li/Li+).14,16,32,33 Among the cathode materials, the layered Li-TM oxides have been the most successful class, delivering high capacities, high volumetric, and gravimetric energy densities.40,41,50 The layered Li-TM oxide consists of close-packed oxygen planes that are stacked with the ABC stacking. Between the oxygen planes, layers of TM ions are alternating with layers of Li ions.13,51 TM ions in the TM layers can release or take electrons by redox reactions. Li layers are where Li ions are stored and Li diffusion takes place. Many commercial Li-ion batteries adopt cathode materials from the layered oxides. Figure 1-4 The first-cycle voltage profile of bare and coated LiCoO2 when cycled between 2.75−4.4 V at a rate of 70 mA/g.52 27 LiCoO2 and LiNiO2 were the first to be investigated as cathode materials.19,21,52–57 In particular, LiCoO2 was the cathode material in one of the first commercially successful Li-ion batteries, and has been used most widely until now. LiCoO2 delivers a high gravimetric (~600 Wh/kg) and volumetric (~3000 Wh/l) energy density due to its high voltage (~3.9 V) and high density (5 kg/l) (Fig. 1-4). Nevertheless, high cost of cobalt, a limited specific capacity from LiCoO2 (~160 mAh/g) and poor thermal safety have encouraged researchers to search for the other types of layered Li-TM oxides to replace LiCoO2.20,43 To develop layered cathode materials that outperform LiCoO2 both in performance and cost, Li(Ni/Mn/Co/Al)O2 have been intensively studied, in which TM layers are occupied mixed TM ions of Mn, Ni, Co and Al. In particular, LiNi1/3Mn1/3Co1/3O2, LiNi0.5Mn0.5O2, and LiNi0.8Co0.15Al0.05O2 have been most intensively studied (Fig. 1-5).13,22,58 They deliver high energy densities (~700 Wh/kg) and specific capacities (~180 mAh/g). Furthermore, they are cheaper than LiCoO2 as well, because they contain less cobalt which is expensive. Thus, the second generation Li-ion batteries use one of the materials as the cathode material. Figure 1-5 The first-cycle voltage profile of LiNi1/3Mn1/3Co1/3O2 with a current density of 0.2 mA/cm2.22 28 While most of the traditional layered materials are stoichiometric materials, there have been great efforts to achieve very high capacities and energy densities from Li-excess layered materials.40,45,59,60 Here, Li-excess refers to a composition in which the number of Li sites is greater than that of TM sites (x > 1 in LixTM2-xO2). In particular, many high capacity materials have been designed out of solid-solution compounds (or nano-composite compounds) between Li(Li1/3Mn2/3)O2 and Li(Ni/Mn/Co)O2.40,41,59–61 These materials can cycle more than 250 mAh/g at an average voltage of ~3.6 V, delivering a high energy density (~ 900 Wh/kg) (Fig. 1-6). Nevertheless, their crystal structures evolve during cycling, leading to voltage fading.41,62–64 Such evolution of the voltage profile is undesirable for Li-ion batteries, because the state of charge cannot be determined if a voltage profile evolves during cycling. Therefore, substantial efforts have been made to stabilize the crystal structure of the Li-excess materials.62 Moreover, the rate capability (i.e. power density) of the materials is moderate, which needs improvement.65 Figure 1-6 The first-cycle voltage profiles of 0.3Li2MnO3·0.7LiMn0.5Ni0.5O2.61 29 1.4 Ordering paradigm for the oxide cathodes As we have discussed, nearly all high energy density cathode materials have been sought from well-ordered close-packed oxides, such as layered Li-TM oxides or spinel-type Li-TM oxides. In these ordered materials, Li sites and pathways (a 2D slab in the layered oxides and a 3D network of tetrahedral sites in the spinels) are separated from the TM sublattice, which provides stability and electron storage capacity.13,30 Having well-ordered structures where there is little or no intermixing between the Li and the TM sublattice is generally considered critical to obtain high-capacity cathode materials with good cycle life.7,8 In some cases, improvements in cation ordering have notably improved the power or energy density.13,66–68 Figure 1-7 shows an example of the effect of cation (dis)ordering to the performance of the ordered oxides.13 In general, LiNi0.5Mn0.5O2 forms into a slightly cation-mixed layered structure in which ~ 9 % of Li sites are occupied by TM ions (accordingly ~9 % TM sites are occupied by Li ions) when standard high-temperature solid state methods are applied for synthesis. In 2006, Kang et al. demonstrated that removing the cation mixing by ion-exchange dramatically improves the performance of the layered LiNi0.5Mn0.5O2.13 The ion-exchanged layered LiNi0.5Mn0.5O2 has almost a perfectly layered structure with negligible cation mixing, and shows much higher capacity and better rate capability than a standard LiNi0.5Mn0.5O2 with a partially cation-mixed layered structure.13 Such improvement largely originates from that Li diffusion tends to be faster in a cationordered structure than in a (partially or fully) cation-disordered structure. In general rocksalt-type oxides, both Li and TM ions occupy a cubic close-packed lattice of octahedral sites, and Li diffusion proceeds by hopping from one octahedral site to 30 Figure 1-7 The discharge profile at various C rates for the ion-exchanged (IE)-LiNi0.5Mn0.5O2 (above) and sold state (SS)-LiNi0.5Mn0.5O2 (below). In the test, the cell was charged at C/20 to 4.6 V, then held at 4.6 V for 5 hours and discharged at different rates. 1C corresponds to 280 mA/g.13 another octahedral site via an intermediate tetrahedral site.51,69 Li in the intermediate tetrahedral site is the activated state in Li diffusion. The activated tetrahedral Li ion shares faces with four octahedral sites: the site previously occupied by the ion itself; the vacancy it will move into; and two sites that can be occupied by Li, TM, or a vacancy. The energy in this site, which reflects the Li migration barrier, is largely determined by electrostatic repulsion between the activated Li+ ion and its face-sharing species, and thus depends on (i) the valence of the face-sharing species and (ii) the available space for relaxation between the activated Li+ ion and the face-sharing species.13,51,68 This space is measured as the Li slab distance in layered structures or more generally as the height of the tetrahedron along which the relaxation occurs.13,51,70 31 Figure 1-8 (a) The octahedral to tetrahedral to octahedral Li diffusion in general rocksalt-type Li-TM oxides (b) The 1-TM channel in stoichiometric Li-TM oxides.70 As electrostatic repulsion on an activated Li+ ion is too strong when there are two facesharing cations, Li dominantly diffuses with the divacancy mechanism, involving a second vacancy beside the vacancy the migrating Li will move into.13,51,69,70 In rocksalt-type Li-TM oxides, two kinds of diffusion channels support this mechanism: 0-TM channels, involving no face-sharing TM ion, and 1-TM channels, involving one face-sharing TM ion.70–72 Note that in the traditional stoichiometric layered Li-TM oxides such as LiCoO2, Li diffusion takes place through the 1-TM channels only. In general, cation disorder (i.e. cation mixing) limits Li diffusion in the layered (rocksalt-type) oxides, inhibiting Li cycling from the layered materials.13,66,68,73 The negative effect of cation mixing on Li diffusion has been understood to originate from the following reasons. First, cation mixing greatly decreases the relaxation space (i.e. Li slab distance or tetrahedron height) for an activated Li+ ion against a face-sharing species upon Li migration, increasing the electrostatic energy in the activated state and thus the Li diffusion barrier (Fig. 1-9).13,70 Moreover, some people have argued that TM ions in Li layers can physically block the Li diffusion paths as well.74 32 Figure 1-9 (a) The decrease in the Li slab distance (tetrahedron height) by cation mixing: the layered structure has much larger Li slab spacing than the cation-disordered structure.70 (b) The calculated activation barrier for Li migration in LiNiO2 with cation mixing (stars) and without cation mixing (squares).51 The Li slab distances with and without cation mixing are indicated with thick and thin dashes at different Li contents, respectively. The shift on the left is from fully lithiated state and the one on the right is from partially delithiated state.51 The negative effects of cation disorder have been experimentally demonstrated as well. For example in 1998, Obravac et al. demonstrated that mechanochemical synthesis of cationdisordered LiMO2 (M = Ti, Mn, Co, Ni) is possible with ball milling (Fig. 1-10).75 In their experiments, increasing the ball-milling time resulted in more severe cation mixing in layered materials such as LiCoO2 and LiNiO2. Then, they demonstrated that the reversible capacity of layered materials rapidly decreases as their crystal structure changes from a layered rocksalt to disordered rocksalt structure.75 Furthermore, most disordered rocksalt-type Li-TM oxides such as α-LiFeO2 are electrochemically inactive, delivering a limited capacity (Fig. 1-11).75–77 Thus, only a negligible attention has been given to cation-disordered oxides as cathode materials, based on the experimental observations and the understanding of the negative effects of cation disorder to Li diffusion. 33 Figure 1-10 (a) The X-ray powder diffraction patterns of LiCoO2 powders prepared at high temperature after subsequent exposure to different milling times. (b) Voltage profiles of cathodes prepared from LiCoO2 samples exposed to different milling times.75 Figure 1-11 (a) Charge-discharge curves of high-temperature α-LiFeO2 and low-temperature ɑLiFeO2 (b) Capacity retention of low-temperature α-LiFeO2.76 34 1.5 Overview of the thesis This thesis explores the potential of cation-disordered Li-TM oxides as promising electrode materials for rechargeable Li batteries, enlarging the search space of high-energy density cathode materials to cation-disordered materials. In the first part, we demonstrate that contrary to the conventional wisdom, cationdisordered materials can be promising electrode materials from the study of novel Li1.211Mo0.467Cr0.3O2.70 Li1.211Mo0.467Cr0.3O2 forms as a layered rocksalt structure but transforms almost completely to a disordered rocksalt structure after several cycles. Nevertheless, the reversible capacity of this material is remarkably high, indicating that Li diffusion can be facile in the cation-disordered structure.70 Using ab inito computations, we show that this unexpected behavior is due to percolation of a certain type of active Li diffusion channels (0-TM channels) that are available in disordered Li-excess materials.70,72 In the second part, we apply 0-TM percolation to design a new class of high capacity cation-disordered oxides, Li-excess Ni-Ti-Mo oxides, which deliver both high capacities and high voltage. Combining experiments with ab initio calculations, we investigate their electrochemical performance, structural changes, and redox mechanism. From the understandings, strategies to further improve the new materials are presented, providing new guidelines for the design of high-energy density cation-disordered cathode materials for the rechargeable Li batteries. 35 -------------Part II------------- Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries [Reproduced from Ref. 70] Lee, J. et al., Science 343, 519-522 (2014). 36 2.1 Introduction Most high-energy density cathode materials for Li-ion batteries have been sought from the well-ordered close-packed oxides, such as layered LiCoO2 and spinel LiMn2O4.8,26,53 In these ordered materials, Li and TM ions occupy distinct sites such that Li sites and pathways are separated from the TM sublattice which provides stability and electron storage capacity. Having well-ordered structures with negligible mixing between the Li and the TM sublattice is generally considered necessary for obtaining high-capacity cathode materials with good cycle life.13,66 This is largely because Li diffusion tends to be facile in the well-ordered crystal structure, which is critical for achieving high energy and high power densities. As discussed in Part I: Introduction, cation mixing tends to limit Li diffusion because it decreases the Li slab spacing, increasing the Li diffusion barrier.51,68,70 Moreover, randomly distributed TM ions in the cation-disordered structure can physically block Li diffusion paths as well. Therefore, cation-disordered materials are generally disregarded as electrode materials, and have received only a limited attention (Fig. 2-1). Figure 2-1 Illustration of the layered rocksalt structure and disordered rocksalt structure: cation mixing between Li and TM layers lead to a disordered rocksalt structure, which tends to result in poor cyclability of Li-TM oxides by slowing down Li diffusion. 37 However, in fact there are a few cation-disordered Li-TM oxides with good cycling performance. For example, cation-disordered Li2VO3 can cycle up to 250 mAh/g (Fig. 2-2),78 and recently it has been demonstrated that cation-disordered Li1.3Mn0.4Nb0.3O2 can cycle up to 300 mAh/g as well.79 These results show that Li diffusion can be facile in the cation-disordered structure. By understanding the mechanism how these cation-disordered materials allow for facile Li diffusion, we can design a whole new class of high-capacity cation-disordered materials for rechargeable lithium battery electrodes. Figure 2-2 The voltage profile of Li2VO3.78 In this part of the thesis, from the study of Li1.211Mo0.467Cr0.3O2 we demonstrate that cation-disordered materials can be promising electrode materials if they contain enough Liexcess (x > 1.09 in LixTM2-xO2). Li1.211Mo0.467Cr0.3O2 forms into a layered structure, but transforms to a cation-disordered structure after several charge-discharge cycles. Nevertheless, its reversible capacity is remarkably high, delivering as high as ~265 mAh/g. In this work, we not only demonstrate the high capacity in this material, but also provide a fundamental understanding of how cation-disordered materials can be promising electrode materials, using 38 ab initio calculations. We believe this work unlocks the potential of cation-disordered oxides for rechargeable lithium batteries. 2.2. Methodology 2.2.1 Experimental methodology 2.2.1.1 Synthesis To synthesize Li1.211Mo0.467Cr0.3O2 [original target: Li1.233Mo0.467Cr0.3O2], Li2CO3 (Alfa Aesar, ACS, 99% min), MoO2 (Alfa Aesar, 99%), and Cr3(OH)2(OOCCH3)7 (Alfa Aesar, Cr 24%) were used as precursors. 5% excess Li2CO3 from the stoichiometric amount required to synthesize Li1.233Mo0.467Cr0.3O2 was used to compensate for possible Li loss during high temperature solid state reaction. The precursors were dispersed into acetone and ball-milled for 24 hours, and then dried overnight in an oven. The precursor mixture was pelletized and then fired at 1050°C for 15 hours under argon (Ar) gas, followed by furnace cooling to room temperature. After firing, the pellets were manually ground into fine powder. From inductively coupled plasma atomic emission spectroscopy (ICP-AES, bomb digestion method), slight Li deficiency (~1.8%) from the target composition was found in the final product (Li1.211Mo0.467Cr0.3O2). Carbon coating was applied to the Li1.211Mo0.467Cr0.3O2 powder (i) to prevent transition metal (TM) dissolution (mainly Mo) at high delithiation states (Fig. 2-8), and (ii) to improve the electrochemical properties of Li1.211Mo0.467Cr0.3O2 by decreasing its particle size, through planetary ball-milling in the carbon coating process (Fig. 2-7). Sucrose 39 (C12H22O11, EMD, GR ACS), the carbon precursor, was mixed with the Li1.211Mo0.467Cr0.3O2 powder in the weight ratio of 80 (Li1.211Mo0.467Cr0.3O2): 20 (Sucrose) by planetary ball-milling (Retsch, PM200) for six hours at 300 rpm in Ar-filled jars. Finally, the mixture was annealed at 700°C for three hours under Ar gas to obtain the carbon-coated Li1.211Mo0.467Cr0.3O2 powder. The CHN analysis (combustion method) detected 5.17 wt% of carbon from the carbon-coated Li1.211Mo0.467Cr0.3O2. Figure 2-3 shows the Li1.211Mo0.467Cr0.3O2 particle with and without carbon coating. As can be seen from the figures, thin layers of amorphous carbon are coated to the Li1.211Mo0.467Cr0.3O2 particle after carbon coating, which are not seen from the pristine Li1.211Mo0.467Cr0.3O2 particle. Figure 2-3 High resolution transmission electrons microscopy images in two directions of the carbon-coated Li1.211Mo0.467Cr0.3O2 particle (top), and uncoated Li1.211Mo0.467Cr0.3O2 particle (bottom).70 40 The carbon-coating layers prevent TM dissolution (mainly Mo) from Li1.211Mo0.467Cr0.3O2 upon cycle. It is known that Mo6+ dissolves into an electrolyte.80 As Mo4+ becomes oxidized to Mo6+ upon charging Li1.211Mo0.467Cr0.3O2, it is necessary to prevent surface from Mo dissolution for stable cycling performance. Figure 2-4 demonstrates how Mo dissolution can be significant if without coating. High energy ball-milled Li1.211Mo0.467Cr0.3O2 was cycled for 100 cycles between 1.5−4.3 V at 164 mA/g at room temperature. After 100 cycles, significant coloration is seen from the Li metal anode and the separators, which are confirmed to be from dissolved Mo compounds based on the energy dispersive X-ray spectroscopy. Figure 2-4 The two upper left images show a Li metal foil and a separator from a Li1.211Mo0.467Cr0.3O2 half-cell that was charged/discharged for 100 cycles between 1.5−4.3 V at 164 mA/g at room temperature. The upper right SEM image shows spherical particles on the colored area on the separator (dotted circle). The lower image shows an energy dispersive Xray spectroscopy (EDS) spectrum of a particle attached to the separator, indicating that the particles from the colored area are composed of Mo and Cr (Mo:Cr = 8.875:1).70 41 However, such Mo dissolution can be retarded with carbon coating. Figure 2-5 shows the Li metal anode of half-cells from uncoated and carboncoated Li1.211Mo0.467Cr0.3O2. Without carbon coating, charging to a higher capacity beyond 250 mAh/g leads to significant Mo dissolution as can be inferred from the coloration on the Li metal anode. With carbon coating, the coloration becomes much less, indicating that Mo dissolution is retarded. Figure 2-5 Li1.211Mo0.467Cr0.3O2 half-cells were disassembled at points A, B, and C on the first charge voltage profile of Li1.211Mo0.467Cr0.3O2 at 3.2 mA/g. The three inset images with red boundaries show Li metal anode foils (attached to a current collector rod) from Li1.211Mo0.467Cr0.3O2 half cells that were disassembled after four days of resting at the first charge state of A) 200 mAh/g, B) 250 mAh/g, and C) 300 mAh/g. The inset image with a blue boundary shows a Li metal anode foil that was disassembled from a carbon-coated Li1.211Mo0.467Cr0.3O2 half-cell after four days of resting at the same first charge state of 300 mAh/g as point C.70 42 Figure 2-6 The discharge-capacity retention of carbon-coated and uncoated Li1.211Mo0.467Cr0.3O2 during cycling between 1.5−4.3 V at 16.4 mA/g at room temperature. The two inset images show Li metal anode foils from uncoated and carbon-coated Li1.211Mo0.467Cr0.3O2 half-cells after ten cycles. Carbon coating retards transition metal dissolution, improving the capacity retention of carbon-coated Li1.211Mo0.467Cr0.3O2.70 Preventing Mo dissolution can lead to significant improvement in the cycle retention of Li1.211Mo0.467Cr0.3O2. Figure 2-6 compares the cycle retention of Li1.211Mo0.467Cr0.3O2 with and without carbon coating. As can be seen from the plot, the carbon-coated Li1.211Mo0.467Cr0.3O2 delivers a much higher capacity with better capacity retention than without carbon coating. The carbon coating process is further beneficial because during the process the particle size of Li1.211Mo0.467Cr0.3O2 becomes smaller, improving the reversible capacity by increasing the surface to volume ratio. Figure 2-7 compares the first cycle voltage 43 profile of the carbon-coated and uncoated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at 32.7 mA/g at room temperature. As can be seen from the profiles, the reversible capacity is higher for the carbon-coated Li1.211Mo0.467Cr0.3O2. This is due to the smaller particle size of the carbon-coated Li1.211Mo0.467Cr0.3O2 than the uncoated Li1.211Mo0.467Cr0.3O2 (Fig. 2-12) Figure 2-7 The first-cycle voltage profile of the carbon-coated and uncoated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at 32.7 mA/g at room temperature. 44 2.2.1.2 Electrochemistry The cathode film was composed of the carbon-coated Li1.211Mo0.467Cr0.3O2 powder (70 wt%), carbon black (20 wt%) as conductive agent, and polytetrafluoroethylene (PTFE) (10 wt%) as binder. The components were manually mixed for 40 minutes and rolled into a thin film inside an argon-filled glove box. 1M of LiPF6 in 1:1 volume ratio of ethylene carbonate: dimethyl carbonate (EC:DMC) solution was used as an electrolyte. Celgard 2500 polypropylene separator and Li metal foil were used as the separator and the counter electrode, respectively. The Swagelok cells were assembled inside an argon-filled glove box and tested on a Maccor 2200 at room temperature in galvanostatic mode unless otherwise specified. The loading density of the cathode film was approximately 3.6 mg/cm2. The current density at 1C (= 327.486 mAg-1) was based on the theoretical capacity of Li1.233Mo0.467Cr0.3O2 (= 327.486 mAhg-1). The specific capacity was calculated strictly on the amount of Li1.211Mo0.467Cr0.3O2 (66.318 wt%) in the cathode film. 2.2.1.3 Characterization The X-ray diffraction (XRD) patterns were collected on a PANalytical multipurpose diffractometer using Cu Kα radiation in the two-theta range of 5-85°. Kapton film was applied when running XRD on electrode films. Rietveld refinement was done using PANalytical X’pert HighScorePlus software. Scanning electron microscopy (SEM) images were collected on a FEI Phillips XL30 field-emission gun environmental SEM at an accelerating voltage of 15 and 25 kV, respectively. High Resolution Transmission Electron Microscopy (HRTEM) images were recorded with JEOL 2010F at MIT. High Angle Annular Dark Field (HAADF) 45 Scanning Transmission Electron Microscopy (STEM) images were recorded with Hitachi HD 2700C 200 kV using 1 Å probe of 28mrad semi-convergence angle and ADF detector with semi-collection angle of 50-28mrad. 2.2.2 Computational methodology 2.2.2.1 Density-functional theory calculations Structural energies and migration barriers were calculated based on density-functional theory (DFT)81,82 in the generalized-gradient approximation (GGA) using the PBE exchangecorrelation functional83 and projector-augmented wave (PAW) pseudopotentials,84,85 as implemented in the Vienna ab-initio Package (VASP).86,87 A plane-wave cutoff of 520 eV was employed to guarantee numerical convergence in variable-cell calculations. For chromium, a Hubbard-U correction was employed, using the U-value of reference 89 (U= 3.5 eV).88 Gamma centered k-point meshes for the integration of the Brillouin zone were selected to converge energies to 1 meV per formula unit and structural parameters to 10-3 Å . All geometries were fully optimized with atomic forces below 0.01 eV/Å . The climbing-image nudged elastic band (CI-NEB) method was used to compute migration barriers based on five images along the diffusion paths.89–91 All reported migration barriers were converged to 10 meV. The calculations were conducted for the well-established divacancy diffusion mechanism.51,69 All NEB calculations resulted in diffusion paths that traverse tetrahedral sites, in agreement with the mechanism found in layered oxides. 46 2.2.2.2 Structural model Structural energies and migration barriers were obtained from structural models containing 72 atoms in the fully lithiated state, i.e., 12 Li2MoO3 formula units and 18 LiCrO2 formula units, respectively (Fig. 2-8). Cation disorder was imposed by randomly distributing Li and TM ions over all cation sites. For the migration barrier calculations, a divacancy was created in the fully lithiated structure. Figure 2-8 Structural model of disordered-Li2MoO3: The simulation cell contains 12 formula units (72 atoms). In the figure, large red balls represent oxygen atoms, and small green and violet balls represent the octahedral sites of lithium and molybdenum, respectively.70 47 2.2.2.3 Percolation simulation Percolation simulations were conducted following the Monte-Carlo algorithm proposed by Newman and Ziff and employed periodic 8×8×8 supercells of the conventional (4 atom) FCC cell containing 2048 sites and 1000 MC steps per each combination of cation mixing and Li content.92 Percolation was approximated by periodic wrapping in one lattice dimension. Since the wrapping probability is sensitive with respect to the size of the simulation cell, we verified the convergence of the percolation threshold for the limit of fully disordered structures, corresponding to 100% cation mixing in Figure 2-9a. As evident from Figure 2-9A, the inflection point of the wrapping probability rapidly converges with the cell size and therefore is a good estimate for the percolation probability even in small simulation cells. Finite size effects are less pronounced for the accessible lithium contents above the percolation threshold, as shown in Figure 2-9b. Figure 2-9 0-TM wrapping probability (a) and accessible lithium fraction (b) as a function of the Li-excess level for different FCC supercell sizes. Results for fully disordered structures are shown. The legends refer to supercells of the primitive FCC unit cell.70 48 2.3 Experimental results 2.3.1 Characterization of the as-synthesized Li1.211Mo0.467Cr0.3O2 As-synthesized Li1.211Mo0.467Cr0.3O2 has a layered rocksalt structure, which can be inferred from its XRD pattern in Figure 2-10. In this material, layers of Li and of Li-Mo-Cr are alternating in the oxygen cubic close-packed framework. The XRD refinement on the assynthesized Li1.211Mo0.467Cr0.3O2 shows no cation mixing between the Li and the Li-Mo-Cr layers, indicating that this material forms into a well-layered structure. Figure 2-10 Rietveld refinement on the XRD pattern of pristine Li1.211Mo0.467Cr0.3O2 powder. The atomic ratio between elements was first set to the ICP result, and then Mo and Cr occupancy in Li and TM layers were refined. The refinement suggests no transition metals in Li layers at all. Li occupancy was refined after Mo and Cr occupancy had been determined.70 While Li1.211Mo0.467Cr0.3O2 is targeted to be a solid-solution compound between Li(Li1/3Mo2/3)O2 (i.e. Li2MoO3) and LiCrO2, one can suspect local phase-separation of Li1.211Mo0.467Cr0.3O2 into domains of Li(Li1/3Mo2/3)O2 and LiCrO2. However, neither in XRD or TEM is there any indication of the local domains. The c-lattice parameter of Li2MoO3 49 (~14.91 Å ) and LiCrO2 (~14.43 Å ) are quite different from each other. Thus, if there were such distinct domains, the XRD signature near 18.4o from LiCrO2 (in blue in Figure 2-11a) would be visible in the diffraction pattern of Li1.211Mo0.467Cr0.3O2. However, such feature is not observed in the XRD pattern of Li1.211Mo0.467Cr0.3O2 (black). In addition, the electron diffraction pattern along ZA [010] of pristine Li1.211Mo0.467Cr0.3O2 in Figure 2-11b shows a single phase, which further excludes the existence of small LiCrO2 domains (< 5 nm) beyond the detectability of XRD. Hence, all the evidence points towards a single solid solution. Figure 2-11 (a) The (003) peak in the XRD pattern on Li1.211Mo0.467Cr0.3O2 [black] and that of LiCrO2 [blue] (b) The electron diffraction pattern along ZA[010] of Li1.211Mo0.467Cr0.3O2. The scanning electron microscopy (SEM) images show that the primary particles around 200 nm in diameter are highly agglomerate in the as-synthesized Li1.211Mo0.467Cr0.3O2, and the primary particle size decreases to around 100 nm after the carbon-coating process (Fig. 2-12). The decrease in the particle size can also be inferred from the change in the XRD patterns in which peak broadening is observed after carbon coating. 50 Figure 2-12 The broader XRD peaks of the carbon-coated Li1.211Mo0.467Cr0.3O2 powder (lower left) compared to the sharper XRD peaks of pristine Li1.211Mo0.467Cr0.3O2 powder (upper left) indicate that the carbon-coated Li1.211Mo0.467Cr0.3O2 particles are smaller than the uncoated particles. Furthermore, SEM images (right) confirm that the particles of the carbon-coated Li1.211Mo0.467Cr0.3O2 are smaller than those of uncoated one. During the carbon coating process, sucrose (carbon precursor) and the Li1.211Mo0.467Cr0.3O2 powder were mixed in a planetary ballmill for six hours at 300 rpm, resulting in smaller particles after carbon coating.70 2.3.2 The electrochemical properties of Li1.211Mo0.467Cr0.3O2 Figure 2-13 shows the voltage profile of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at the rate of 16.4 mA/g at room temperature. From the profile, we find a very high capacity of ~265 mAh/g, corresponding to cycling ~1 Li per formula unit of Li1.211Mo0.467Cr0.3O2. Considering that conventional well-layered materials show a capacity of ~160 to ~190 mAh/g, we can see that the capacity from Li1.211Mo0.467Cr0.3O2 is remarkably 51 high. The gravimetric energy density of this material is also promising (~660 Wh/kg), which exceeds those of the conventional cathode materials such as LiCoO2 (~590 Wh/kg), LiMn2O4 (~480 Wh/kg), and LiFePO4 (~595 Wh/kg).14,26,57 From the voltage profile, we find that the first-charge profile is different from those of further cycles. This indicates that the crystal structure of Li1.211Mo0.467Cr0.3O2 changes during the first charge. Figure 2-13 The voltage profile of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at the rate of 16.4 mA/g at room temperature. 52 Figure 2-14 The 10-cycle voltage profiles of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at the rate of (a) 4.1 mA/g, (b) 8.2 mA/g, and (c) 16.4 mA/g at room temperature (d) Corresponding specific capacity vs. cycle number plot.70 The rate capability of the carbon-coated Li1.211Mo0.467Cr0.3O2 is also promising. Figure 2-14a, 14b, and 14c shows the 10-cycle voltage profiles of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at the rate of 4.1 mA/g, 8.2 mA/g and 16.2 mA/g, respectively. From the profiles, we find a gentle decrease in the reversible capacity with increasing rates. The carbon-coated Li1.211Mo0.467Cr0.3O2 still shows a high capacity above 200 mAh/g at 4.1 mA/g, indicating that the rate capability of this material is promising. The cycle retention of Li1.211Mo0.467Cr0.3O2 is also promising. During 10 cycles, the average loss of the discharge capacity per cycle is ~2 mAh/g, which is reasonably good for a new material without any significant engineering (Fig. 2-14d). As the rate further increases, the reversible 53 capacity further decreases (Figs. 2-15 and 2-16). Nevertheless, the carbon-coated Li1.211Mo0.467Cr0.3O2 can still deliver as much as 135 mAh/g at a very high rate of 1300 mA/g (4C rate). Figure 2-15 The voltage profile of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at a rate of C/10, C/2, 1C, 2C, 4C and 10C (1C = 327 mA/g) Figure 2-16 Capacity retention of the carbon-coated Li1.211Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at a rate of C/10, C/2, 1C, 2C, 4C and 10C (1C = 327 mA/g) 54 2.3.3 The structural change in Li1.211Mo0.467Cr0.3O2 during cycling The electrochemical performance of the Li1.211Mo0.467Cr0.3O2 is promising, but its voltage profile upon first charge is different from those upon later cycles (Fig. 2-13). Voltage is determined by the difference in the chemical potential of Li in a cathode and an anode.12 In our experiment, we used Li metal as the anode. Thus, the chemical potential of Li in the anode does not change. Thus, the change in the voltage profile must come from the change in the chemical potential of Li in the cathode during the first cycle. As the chemical potential of Li is largely determined by the crystal structure of the cathode compounds, the change in the voltage profile indicates that the crystal structure of the material changes substantially upon first charge. To analyze the change in the crystal structure during cycling, we performed ex situ XRD on the carbon-coated Li1.211Mo0.467Cr0.3O2 electrode before cycling, after one cycle, after two cycles, and after ten cycles between 1.5−4.3V at 32.7 mA/g (Fig. 2-17). From the XRD patterns, dramatic changes in the pattern are seen after the first cycle. Before cycling, the XRD pattern shows a pattern of a layered-rocksalt structure (space group: R-3m) in which the Li layers and the TM layers form alternating layers within an oxygen FCC framework (Fig. 2-1a). However, after the first cycle, the (003) peak which is an indicative of the layered structure is almost completely gone,73 and the XRD pattern resembles a pattern of a disordered-rocksalt structure (space group: Fm-3m). Based on the XRD refinements (Fig. 2-18), we estimate 34 to 52 % of the TM ions to be in Li layers after ten cycles. This indicates substantial cation-mixing between the Li and TM layers, known to be detrimental to the performance of layered materials. The structural evolution of Li1.211Mo0.467Cr0.3O2 to a disordered structure can also be confirmed in real space with scanning transmission electron microscopy (STEM) (Fig. 2-19). The bright and dark columns in the “before” image correspond to atomic columns of mixed Li-Mo-Cr 55 ions and Li ions, respectively. The Z-contrast decreases after one cycle and is very weak after 10 cycles, indicating increased cation mixing. As both XRD and STEM show, Li1.211Mo0.467Cr0.3O2 transforms to a disordered rocksalt-structure after several cycles. Nevertheless, the carbon-coated Li1.211Mo0.467Cr0.3O2 still delivers a high capacity up to 265 mAh/g after substantial cation mixing. It has been understood that cation-disordered structures do not allow for facile Li diffusion, largely because the Li slab spacing (i.e. tetrahedron height) becomes very small in the structures.13,66,68 Indeed, the Li slab spacing of Li1.211Mo0.467Cr0.3O2 decreases considerably from ~2.63 Å to ~2.39 Å after cation mixing. Thus, the high capacity after cation mixing is very counterintuitive and exciting, because the mechanism by which this material delivers a very high capacity will allow us to design promising cation-disordered materials as high-capacity lithium battery electrodes. Figure 2-17 The ex situ XRD pattern of the carbon-coated Li1.211Mo0.467Cr0.3O2 electrode before cycle, after one cycle, after two cycles, and after 10 cycles between 1.5−4.3V at 32.7 mA/g. 56 Figure 2-18 Rietveld refinement on the XRD pattern of a carbon-coated Li1.211Mo0.467Cr0.3O2 electrode after ten cycles between 1.5−4.3 V at 32.7 mA/g. Kapton film was applied to prevent oxidation, resulting in humps below 30°. From XRD refinement, it is difficult to precisely quantify and distinguish the level of Mo migration and Cr migration to Li layers. Therefore, we refined the XRD pattern with two different methods: (a) Refine Mo occupancy first then Cr occupancy. Here, refinement suggests that only Mo migrates (0.2634 Mo in Li layers). (b) Assume all Cr are in Li layers then refine Mo occupancy. Refinement is much improved by assuming 0.097988 Mo in addition to 0.3 Cr in Li layers. In both methods, Li occupancy was later refined by distributing 0.92 Li (based on the cumulative charge-discharge capacity after ten cycles) to Li and TM layers. However, the values are more subject to error. The two ways of refining give two extremes of the transition metal (TM) migration level. Refinement based on the method A suggests a TM migration level of ~34.3% (i.e. ~34.3% of overall TM ions are in Li layers), and refinement based on the method B suggests a TM migration level of ~51.88%. From the two values, we can expect that at least ~34% to ~52% of TM ions have migrated to Li layers after ten cycles, indicating substantial cation mixing in Li1.211Mo0.467Cr0.3O2. Note that the TM migration level can slightly vary with the rate of cycling which changes the delithiation level at the end of charge. To determine the average tetrahedron height of the disordered57 Li1.211Mo0.467Cr0.3O2, eight tetrahedron heights (four from the tetrahedron in Li layers, and four from that in TM layers) were averaged out, as Li1.211Mo0.467Cr0.3O2 is not yet completely disordered. Method A yields 2.39701 Å (× 3), 2.38362 Å (× 3), 2.41831 Å (× 1), and 2.37791 Å (× 1), resulting in the average tetrahedron height of 2.39226 Å . Method B yields 2.39816 Å (× 3), 2.38775 Å (× 3), 2.40875 Å (× 1), and 2.37756 Å (× 1), resulting in the average tetrahedron height of 2.393 Å . Therefore, both methods suggest the average tetrahedron height of disordered- Li1.211Mo0.467Cr0.3O2 to be ~2.39 Å .70 Figure 2-19 Left: the scanning transmission electron microscopy images along the [010] zone axis in a carbon-coated Li1.211Mo0.467Cr0.3O2 particle before cycling and after 1 and 10 cycles between 1.5−4.3 V at 16.4 mA/g. Right: Corresponding line profiles of the Z-contrast information with the measured spacing of Li-Mo-Cr layers.70 58 2.3.4 Redox mechanism of Li1.211Mo0.467Cr0.3O2 (a) (b) (c) Figure 2-20 (a) The cyclic voltammetry profile of the carbon-coated Li1.211Mo0.467Cr0.3O2, (b) Cr EELS L3/L2 ratio in Li1.211Mo0.467Cr0.3O2 before cycling, after the first charge, and after the first cycle. (c) Integrated spin density of Mo and Cr in Li1.211Mo0.467Cr0.3O2 as a function of the delithiation level, obtained by DFT calculations. To understand the redox mechanism of Li1.211Mo0.467Cr0.3O2, we performed the cyclic voltammetry (CV) test. From the CV test (Fig. 2-20a), we see a redox peak at ~2.4 V, and a small peak at ~ 4 V after Li1.211Mo0.467Cr0.3O2 becomes cation-disordered. The L3/L2 intensity ratio in electron energy loss spectroscopy (EELS) on the carbon-coated Li1.211Mo0.467Cr0.3O2 shows that Cr oxidation state increases mainly after Li1.211Mo0.467Cr0.3O2 is being charged to above 4 V (~240 mAh/g, C/20), as shown in Figure 2-20b, which suggests that Mo 4+/Mo6+ redox (~248 mAh/g) is largely being used before Cr redox is being used (~80 mAh/g): the error 59 bar on each data point in Figure 2-20b gives the distribution of the results as obtained from ten different particles on the sample region with less than 0.3 inelastic mean free path. The blue and yellow bands in Figure 2-20b are the distributions of Cr EELS L3/L2 ratios for Cr3+ and Cr4+, respectively, from different Cr oxides standards in the literature.93 These results indicate that Cr is only being oxidized above 4.0 V and to some oxidation state between +3 and 4+. Therefore, we determine the redox peak at ~2.4 V to be of Mo4+/Mo6+ and the peak at ~4 V to be of Cr3+/Cr4+. Note that the ~2.4 V redox reaction is consistent with the Mo4+/Mo6+ redox voltage in Li2MoO3.80 Additionally, we computationally assessed the redox mechanism of the cationdisordered Li1.211Mo0.467Cr0.3O2. Our calculations show that Mo is mostly being oxidized before Cr oxidation, which is in agreement with our experimental observations. Only after more than ~75 % of Mo4+ is oxidized to Mo6+, does the oxidation of Cr3+ start together with the remainder of Mo oxidation. This can be seen from the change in the average integrated spin densities of Mo and Cr in Figure 2-20c. While the spin on Mo decreases continuously upon Li removal, the spin on Cr3+ only decreases slightly at the top of charge. Therefore, combining CV, EELS and DFT calculations, we propose that Mo4+/Mo6+ redox is mainly being utilized, and Cr3+/Cr4+ redox can be used as well after Mo4+ ions are fully oxidized to Mo6+. 60 2.4 Computational results Li1.211Mo0.467Cr0.3O2 delivers a high capacity and energy density even after substantial cation mixing, indicating that Li diffusion can be facile in the cation-disordered structure. This is in stark contrast to the conventional wisdom that an ordered crystal structure is necessary for facile Li diffusion. Then, how can we understand this counterintuitive behavior? To explain this, we need to understand the Li diffusion mechanism in general rocksalt-type oxides. 2.4.1 Li diffusion channels in cation-disordered oxides In general rocksalt-type oxides, both Li and TM ions occupy the octahedral sites, and Li diffusion takes place by traversing an intermediate tetrahedral site (Fig. 2-21a).13,51,69–71 Li in this tetrahedral site is the activated state in Li diffusion, whose electrostatic energy largely determines the Li diffusion barrier. Therefore, the species in the face-sharing octahedral sites and the tetrahedron height that serves as the relaxation space for the activated Li+ ion govern the Li mobility through each diffusion channel.13,51,66,68 As electrostatic repulsion on an activated Li ion is too strong when there are two facesharing cations, Li dominantly diffuses with the divacancy mechanism, involving a second vacancy beside the vacancy the migrating Li will migrate into.51,69,71 As discussed in the Part I: Introduction, two types of diffusion channels support this mechanism in rocksalt-type oxides thus in cation-disordered oxides: 0-TM channels, involving no face-sharing TM ion (Fig 221b), and 1-TM channels, involving one face-sharing TM ion (Fig. 2-21c).70–72 Note that the 1TM channels are responsible for Li diffusion in typical layered Li-TM oxides. 61 Figure 2-21 (a) o-t-o diffusion: Two tetrahedral paths connect each pair of neighboring octahedral sites (b-d) The activated Li+ ion can face-share with no octahedral transition metals (0-TM channel) (b), one transition metal (1-TM channel) (c), or two transition metals (2-TM channel) (d): note that the 2-TM channels do not support divacancy diffusion mechanism, and thus diffusion barrier through the 2-TM channels are extremely high.70 2.4.2 Li diffusion barriers in cation-disordered oxides: active 0-TM channels To investigate which channels in disordered-Li1.211Mo0.467Cr0.3O2 allow for reasonable hopping rates, Li migration barriers for 1- and 0-TM channels were calculated using density-functional theory, according to the divacancy mechanism. 62 Figure 2-22 Calculated Li migration barriers along 1-TM (Mo4+) channels (red squares), 1-TM (Cr3+) channels (blue triangles), and 0-TM (Li+) channels (black circles) as a function of the average tetrahedron height of model disordered structure (disordered Li2MoO3, disordered LiCrO2). Error bars denotes standard deviation. The shaded area highlights typical tetrahedron heights of disordered materials.70 The red and blue dashed lines in Figure 2-22 show the mean migration barriers along a 1-TM channel as a function of the average tetrahedron height of model disordered structures (disordered-Li2MoO3, disordered-LiCrO2) when the face-sharing octahedral species is Mo4+ and Cr3+, respectively. Note that migration barriers in disordered structures vary with the local atomic environment, which accounts for a distribution of migration barriers. The mean barrier increases as the tetrahedron height (h) decreases, and reaches ~510 meV along a 1-Mo4+ channel and ~490 meV along a 1-Cr3+ channel at h ~ 2.39 Å , the average tetrahedron height in disordered-Li1.211Mo0.467Cr0.3O2. Note that these barriers increases as the TM ion becomes oxidized in charge.13,51 Considering that typical 1-TM barriers in layered oxides are 63 ~300 meV,51 such high barriers in disordered-Li1.211Mo0.467Cr0.3O2 indicate limited Li diffusion along 1-TM channels. This is because the small tetrahedron height in disorderedLi1.211Mo0.467Cr0.3O2 confines the activated Li+ ion close to a face-sharing high-valent octahedral TM ion in 1-TM channels, resulting in strong electrostatic repulsion on the Li+ ion. The black line in Figure 2-22 shows the mean migration barriers along 0-TM channels. In contrast to the high barriers in 1-TM channels, the low ~290 meV barrier at h ~ 2.39 Å indicates that Li migration along 0-TM channels will still be facile in disorderedLi1.211Mo0.467Cr0.3O2, with a ~4400 times [exp(500 meV/kBT)/exp(290 meV/kBT)] higher hopping rate than along 1-TM channels at room temperature. The low valence of a face-sharing octahedral Li+ ion (vs. Mo4+ or Cr3+) results in much weaker electrostatic repulsion on the activated Li+ ion in 0-TM channels. At highly charged states, tetrahedral Li may form in some 0-TM channels since high delithiation should leave no face-sharing octahedral Li at all. However, the mean migration barrier between two 0-TM tetrahedral sites was calculated to be ~415 meV, indicating that Li can easily escape from these sites. . Herein lies the real issue of cation-disordered structures: 1-TM channels, which account for the great Li mobility in the layered Li-TM oxides which currently dominate the battery industry, become nearly inactive in the cation-disordered materials due to their small tetrahedron heights. In contrast, 0-TM channels are active in disordered rocksalts, but are much less frequent than 1-TM channels. Nevertheless, as we will show below, 0-TM channels start to enable facile macroscopic Li diffusion in disordered structures, once enough Li excess is introduced. 64 2.4.3 Percolation of 0-TM diffusion channels For 0-TM channels to allow for macroscopic Li diffusion, they must be continuously connected through the entire material, forming a percolating network uninterrupted by 1- and 2-TM channels. To establish a general understanding of 0-TM percolation, we investigated 1) when 0-TM channels percolate in a rocksalt-type Li-TM-oxide and 2) which fraction of the Li ions become part of a percolating network of 0-TM channels. Figure 2-23a shows the probability to find a percolating network of 0-TM channels (0TM network) in a rocksalt-type Li-TM-oxide as a function of Li content (x in LixTM2-xO2) and cation mixing (TMLi layers/TMTM layers × 100 %), as obtained by Monte-Carlo simulations. The probability (color-coded) steeply increases from zero (red) to one (blue) across the black line in Fig. 2-23a (percolation threshold), varying from x ~ 1.13 for layered oxides to x ~ 1.09 for fully disordered oxides. As 0-TM channels require a locally Li-rich environment, excess Li (x ≥ ~1.09) is crucial to open the percolating 0-TM network. To estimate the contribution of a percolating 0-TM network to macroscopic Li diffusion, we investigated how Li excess and cation mixing affect the Li content in the network (Fig. 223b), which we refer to as accessible Li.70,72 This Li can diffuse through the network without traversing 1- or 2-TM channels, while inaccessible Li must traverse 1- or 2-TM channels to reach the percolating 0-TM network. The three black lines in Fig. 2-12b are the contour lines where the accessible Li content is 0.8 Li, 1 Li, and 1.2 Li per LixTM2-xO2. For x ≤ 1, no percolating 0-TM network exists (Fig. 2-23a), thus there is no accessible Li content, explaining why stoichiometric Li-TM oxides have low capacity when cation disordered.73,75–77 However, the accessible Li content gradually increases as x exceeds ~1.09 (percolation threshold), and becomes as high as 1 Li as x exceeds ~1.22 regardless of cation mixing. Increasing Li excess 65 adds more 0-TM channels to a percolating 0-TM network, improving the network’s connectivity.70,72 Figure 2-23 (a) Computed probability to find a percolating network of 0-TM channels (color) vs. Li content (x in LixTM2-xO2) and cation mixing (TMLi layers/TMTM layers ×100%) (b) The accessible Li contents by a percolating 0-TM network (color) vs. Li content and cation mixing. In the simulation, cations were randomly distributed at each cation mixing level.70 66 The above results explain how Li diffusion can be facile in the cation-disordered Li1.211Mo0.467Cr0.3O2. Li1.211Mo0.467Cr0.3O2 is a Li-excess material with x = 1.233 in LixTM2-xO2. With this Li content, 0-TM channels will be percolating (Fig. 2-23a), accessing as high as ~1 Li per formula unit (Fig. 2-23b). Therefore, even as 1-TM channels become nearly inactive after cation mixing, a large fraction of Li in the material can still be cycled through the percolating active 0-TM network. 2.5 Discussions The principle of creating a percolating 0-TM network can be applied to design other high performing disordered Li-TM-oxides for two reasons. First, the 0-TM activated state is only surrounded by Li sites, making the effect of the TM species on the activation energy less pronounced. Secondly, as shown in Table 2-1, the tetrahedron height of most disordered rocksalts is such that 0-TM channels are calculated to be active (Fig. 2-22). Therefore, a percolating 0-TM network will likely enable facile Li diffusion in other disordered materials, assuming no other kinetic barriers become limiting. Note that the few cation-disordered materials in the literature that were electrochemically active are indeed Li excess materials, whereas stoichiometric disordered materials are usually not electrochemically active, which is consistent with our understanding.73,75–78,94 Disordered Li-excess rocksalts have considerable advantages over layered materials. First, we find that the changes in lattice parameters and volume, as function of Li concentration, are very small in disordered materials (< 1 % in Li1.211Mo0.467Cr0.3O2), which will lead to less mechanical stresses and capacity loss in an electrode (Fig. 2-24). Furthermore, as their cation distribution is more homogenous, they tend to have less significant changes of local 67 environment of the Li ions as a function of state of charge. This change in environment is particularly problematic in layered structures where the Li slab spacing decreases significantly when large amounts of Li are removed, leading to a substantial reduction of Li mobility.69,95,96 However, in the cation-disordered structure, homogenously distributed cations should lead to a Li diffusivity that is more independent of the Li concentration, as is the case for electrode materials with the spinel and olivine type structures. One issue that requires more investigation is whether cation disordering will lead to a more sloped voltage profile than for well-ordered materials, as one would expect from the wider distribution of Li-site energies in a cationdisordered material. However, this variance in the Li-site energy may be counteracted by a less effective Li-Li interaction, which is responsible for the slope of the voltage curve in layered materials.56 Therefore, careful tailoring of the TM-Li to Li-Li ion interaction may mitigate this effect. Given the insights presented in this work, it may not be surprising that the highest capacity layered materials are highly Li-excess materials45,61,97 which become more disordered in the first few cycles due to a particular overcharge mechanism.60,98,99 Materials 75 LiTiO2 LiMnO275 α-LiFeO2100 Li0.542Co1.458O275 Li0.8388Ni1.1612O275 LiNi0.5Ti0.5O2101 LiCo0.5Ti0.5O2102 a (Å ) Tetrahedron height (Å ) Ref. 4.149(1) 4.179(4) 4.157(1) 4.143(2) 4.074(1) 4.1453(6) 4.14 2.395(4) 2.412(9) 2.400(1) 2.392(0) 2.352(1) 2.3933(2) 2.39 75 75 100 75 75 101 102 Table 2-1 The average tetrahedron height of disordered rocksalt-type Li-TM-oxides, deduced from the literatures. The tetrahedron height of a disordered Li-TM-oxide is equivalent to its (111) plane distance, which can be derived by dividing the a-lattice parameter of each material by √3.70 68 Figure 2-24 (a) The XRD patterns of the carbon-coated Li1.211Mo0.467Cr0.3O2 after ten cycles, after ten cycles then charged to Li0.6165Mo0.467Cr0.3O2, and after ten cycles then charged to Li0.3082Mo0.467Cr0.3O2 when cycled between 1.5−4.3 V at 32.7 mA/g. (b) The c- and a-lattice parameter in disordered Li1.211Mo0.467Cr0.3O2 (based on the space group of R-3m) at different delithiation states (x=0.291, 0.5945, 0.90275 in Li1.211-xMo0.467Cr0.3O2). The change in the lattice parameters is very small, leading to negligible volume change (< ~0.12 %) upon delithiation.70 2.6 Conclusion In conclusion, through the high capacity of carbon-coated Li1.211Mo0.467Cr0.3O2, which transforms from a layered to a cation-disordered oxide during electrochemical cycling, we have shown that Li diffusion can be facile in cation-disordered materials. Using DFT calculations and percolation theory, we have explained this by a percolating network of transition states with no transition metals around them (0-TM channels), which provides macroscopic diffusion. The 0-TM percolation threshold is found to be x ~ 1.09 in LixTM1-xO2, but accessing 1 Li per formula unit requires x ≥ ~1.22, in great agreement with our experimental results. Our results may explain why disorder has not been pursued as a strategy before: Most materials 69 synthesized are near stoichiometry (LiTMO2), which is well below the percolation threshold for 0-TM diffusion. Thus, these materials quickly lose their capacity upon disorder as it renders typical 1-TM channels inactive, while 0-TM channels are not percolating.73,75–77 As a result, cation disorder may have appeared as a counterintuitive strategy. In contrast, our analysis points to cation-disordered materials as an exciting new class of materials that can deliver high capacity and high energy density, and therefore offers one of the best hopes to substantially improve the performance of rechargeable Li batteries. 70 -------------Part III------------- A new class of high capacity cation-disordered oxides for rechargeable lithium batteries: Li-Ni-Ti-Mo oxides 71 3.1 Introduction From the study of Li1.211Mo0.467Cr0.3O2, important progress has been made in the oxide space, which enlarges the search space of high energy density cathode materials to cation-disordered lithium transition metal oxides (Li-TM oxides). While cation-disordered materials were widely are disregarded as electrode materials,73,75–77 they can be promising cathode materials if containing enough Li excess (x > 1.09 in LixTM2-xO2).70,72,79,103–105 To briefly summarize our understanding, the Li-excess content in a close-packed oxide is important as it can strongly affect Li diffusion. In the close-packed oxides, Li diffusion occurs between two connected octahedral sites through an intermediate tetrahedral site.13,51,70–72 A Li ion in this tetrahedral site is the activated state in Li diffusion, whose electrostatic energy largely determines the Li diffusion barrier. 13,51,70–72 As the activated Li ion feels weaker electrostatic repulsion when it avoids face-sharing high valent TM ions, the diffusion barrier through a channel with no face-sharing TM ions around the activated state (0-TM channels) is lower than through other types of channels.70–72 In the cation-disordered oxides, Li diffusion can be facile only through these 0-TM channels. Nevertheless, for the channels to support macroscopic Li diffusion, they must be percolating in the disordered structure, which requires a Li-excess composition. As the Li-excess level increases, the percolating network of 0-TM channels becomes more extensive, enabling a higher fraction of Li ions in a disordered structure to cycle through the network.70,72 While this understanding is very exciting, the energy density of Li1.211Mo0.467Cr0.3O2 (from which the understanding is developed) is in fact not particularly high even with a very high capacity of ~ 265 mAh/g.70 This originates from a low redox potential (~ 2.6 V) of Mo4+/Mo6+ couple in the material. Therefore, in the second part of this thesis, we try to search 72 for high voltage cation-disordered materials to achieve higher energy densities. For the design of such materials, we directly apply the percolation theory, demonstrating the validity of percolation theory. In this work, we present a new class of high capacity and high voltage cation-disordered oxides: lithium nickel titanium molybdenum oxides (Li-Ni-Ti-Mo oxides). Combining electrochemistry with in situ X-ray diffraction, electron energy loss spectroscopy, and X-ray absorption near edge spectroscopy, we investigate their electrochemical properties, redox mechanism and structural changes upon cycling. From this understanding, strategies to improve the new materials are presented, setting new guidelines for the design of high energy density cation-disordered cathode materials for rechargeable lithium batteries. 73 3.2 Methodology 3.2.1 Experimental methodology 3.2.1.1 Synthesis To synthesize Li1+x/100Ni1/2-x/120Ti1/2-x/120Mox/150O2 (x = 0, 5, 10, 15, 20), Li2CO3 (Alfa Aesar, ACS, 99% min), NiCO3 (Alfa Aesar, 99 %), TiO2 (Alfa Aesar, 99.9 %), and MoO2 (Alfa Aesar, 99%) were used as precursors. Other than for LiNi0.5Ti0.5O2, a stoichiometric amount of precursors were used. For LiNi0.5Ti0.5O2, 5 % excess Li precursor and 4 % excess Ni precursor were used, because it resulted in the purest disordered rocksalt phase with a composition close to the desired composition (Fig. 2, Table 1). The precursors were dispersed into acetone and ball-milled for 15 hours, and then dried overnight in an oven. The mixture of the precursors was pelletized and then sintered at 750°C for two hours in air, followed by furnace cooling to room temperature. After the sintering, the pellets were manually ground into fine powder. 3.2.1.2 Electrochemistry To prepare a cathode film, the powder of the Li-Ni-Ti-Mo oxides and carbon black (Timcal, Super P) were first mixed by a planetary ball mill (Retsch PM200) in the weight ratio of 70:20 for two hours at 300 rpm. Then, polytetrafluoroethylene (PTFE, DuPont, Teflon 8C) was added to the mixture as a binder, such that the cathode film consists of the Li-Ni-Ti-Mo oxide powder, carbon black, and PTFE in the weight ratio of 70:20:10. The components were manually mixed 74 for 30 minutes and rolled into a thin film inside an argon-filled glove box. To assemble a cell for all cycling tests, except for in situ X-ray diffraction, 1 M of LiPF6 in ethylene carbonate (EC) and dimethyl carbonate (DMC) solution (1:1, Techno Semichem), Celgard 2500 polypropylene separator, and Li metal foil (FMC) were used as the electrolyte, the separator, and the counter electrode, respectively. Swagelok-type cells were assembled inside an argonfilled glove box and tested on a Maccor 2200 at room temperature in the galvanostatic mode otherwise specified. Cyclic voltammetry tests were performed on a Solartron electrochemical potentiostat (1470E) between 1.5−4.1 V (or 1.5−4.5 V) at 0.1 mV/s. The loading density of the cathode film was ~5 mg/cm2. The specific capacity was calculated on the amount of the Li-NiTi-Mo oxides (70 wt %) in the cathode film. 3.2.1.3 X-ray diffraction (XRD), scanning electron microscopy (SEM), and electron energy loss spectroscopy (EELS) The X-ray diffraction (XRD) patterns for the as-prepared compounds were collected on a PANalytical multipurpose diffractometer (Cu source) in the 2ϴ range of 5−85°. Rietveld refinement was completed using PANalytical X’pert HighScore Plus software. Scanning electron microscopy (SEM) images were collected on a Zeiss Merlin High-resolution SEM. Elemental analysis on the compounds was performed with direct current plasma emission spectroscopy (ASTM E 1097-12). Electron energy loss spectroscopy (EELS) spectra were obtained from thin specimens on a JEOL 2010F equipped with a Gatan spectrometer, using parallel incident electron beam and semi-collection angle of 8 mrad in TEM diffraction mode. EELS quantification was performed by using a signal integration window of 50 eV, HartreeSlater model of partial ionization cross section, and power law background subtraction. 75 3.2.1.4 In situ X-ray diffraction For in situ XRD, an in situ cell was designed with a Be window for X-ray penetration. The cell was configured with a Li1.2Ni1/3Ti1/3Mo2/15O2 electrode film as the working electrode, Li metal foil as the counter electrode, 1M of LiPF6 in EC:DMC (1:1) solution as the electrolyte, and glass fiber as the separator. Galvanostatic charge-discharge of the in situ cell was performed on a Solartron electrochemical potentiostat (SI12287) between 1.5−4.8 V at 10 mA/g. The in situ XRD patterns were obtained in one hour intervals from a Bruker D8 Advanced Da Vinci Mosource diffractometer (Mo source) in the 2Ө range of 7−36°. Rietveld refinement on the in situ XRD patterns was performed using PANalytical X’pert HighScore Plus software for every other scan. 3.2.1.5 Ex situ X-ray absorption near edge spectroscopy (XANES) Ni, Ti and Mo K-edge XANES measurements were performed in transmission made using beamline 20BM at the Advanced Photon Source. The incident energy was selected using a Si (111) monochromator. The energy calibration was performed by simultaneously measuring the spectra of the appropriate metal foil. Harmonic rejection was accomplished using a Rh-coated mirror. The samples for the measurements were prepared with the Li1.2Ni1/3Ti1/3Mo2/15O2 electrode films (a) before cycling, (b) after the first charge to 4.8 V at 20 mA/g, and (c) after the first charge to 4.8 V then discharge to 1.5 V at 20 mA/g. The loading density of the films was ~5 mg/cm2. Additionally, spectra of some reference standards were measured in transmission mode, to facilitate interpretation of the XANES data. Data reduction was carried out using the Athena software.106 76 3.2.2 Computational methodology First principles calculations were carried out with density functional theory (DFT) using the spin polarized generalized gradient approximation (GGA).83 Hubbard U parameters (GGA + U) were used to correct the self-interaction of GGA,107 using U values of 6.0 eV for Ni and U values of 4.4 eV for Mo.108 The projector-augmented wave pseudopotentials were used for all energy calculations as implemented to the Vienna Ab initio Simulation Package (VASP).86 To determine the cation-disordered Li1.2Ni0.33Ti0.33Mo0.13O2 structure, a large number of Li/Ni/Ti/Mo cation orderings were generated by using the genetic algorithm method109,110 within a 5 × 3 × 2 supercell containing thirty formula units of monoclinic LiMO2 primitive cell (space group: C2/m). To model the cation-disordered structure, the large supercell was selected and the compositions of cation were fixed to similar composition of Li1.2Ni0.33Ti0.33Mo0.13O2 for every layer along the c direction. One hundreds of Li/Ni/Ti/Mo orderings with the lowest electrostatic energy were calculated with GGA+U. Among them, the most stable configuration was selected as the cation-disordered Li1.2Ni0.33Ti0.33Mo0.13O2 structure. The cation-disordered Li0.467Ni0.37Ti0.37Mo0.15O2-α (α = 0.2) structure was also determined by same technique within a 3 × 3 × 3 supercell of the monoclinic LiMO2 primitive cell. The various oxygen/oxygenvacancy orderings were considered within Li0.467Ni0.33Ti0.33Mo0.13O2-α (α = 0.2) to determine the structure of Li0.467Ni0.33Ti0.33Mo0.13O1.8 with same technique. The Li/Li-vacancy orderings were also generated for Li1.2-xNi0.33Ti0.33Mo0.13O2 and Li1.11-xNi0.37Ti0.37Mo0.15O2 with same methods. Thirty Li/Li-vacancy orderings with the lowest electrostatic energy were calculated within GGA+U at each composition. The DFT energies of the most stable configurations at each composition were used to calculate the voltage profile with following equation; ⟨𝑉⟩ = − E[Lix1 MO2 ]−E[Lix2 MO2 ]−(x1 −x2 )E[Li] (x1 −x2 )F 77 , where E is the DFT energy of the structure and F is the Faraday constant. The oxidation states were determined by comparing calculated magnetizations (average net moments) of Ni, Ti, Mo and oxygen ions and the number of unpaired spins of Ni2+ (2), Ni3+ (1), Ni4+ (0), Ti3+ (1), Ti4+ (0), Mo5+ (1), Mo6+ (0), O2- (0), and O- (1).111,112 The contributions of Ni, Ti, Mo and oxygen ions to redox reaction were determined by the change of their magnetization as follows; contribution of A= ΔM ΔMA , Ni+ΔMTi+ΔMMo+ΔMO where ΔM is the change of the magnetization between x1 and x2. The oxygen loss potential of Li1.2-xNi0.33Ti0.33Mo0.13O2-y was also calculated with following equation; ⟨𝑉⟩ = − y 2 𝑒𝑥𝑝 2 E[Lix1 MO2 ]−E[Lix2 MO2−𝑦 ]− (E𝑐 [O2 ]−TSO (300K))−(x1 −x2 )E[Li] (x1 −x2 )F , where E is the DFT energy, Ec[O2] is the corrected DFT energy of O2,113 T is temperature (300 K) and SO𝑒𝑥𝑝 (300 K) is the entropy of O2 gas at 300 K and 1 atm as obtained from 2 experiments.114,115 We considered two possible transformed structures for the oxygen loss potential as follows;116 (a) Li0.867Ni0.33Ti0.33Mo0.13O2 0.4 Li + 0.1 O2 + Li0.519Ni0.37Ti0.37Mo0.15O2 : Oxygen loss with lattice densification (b) Li0.867Ni0.33Ti0.33Mo0.13O2 0.4 Li + 0.1 O2 + Li0.467Ni0.33Ti0.33Mo0.13O1.8 : Oxygen loss with oxygen vacancy formation 78 3.3 Experimental results 3.3.1 Design strategy from percolation theory Percolation theory predicts that sufficient Li excess is necessary to obtain high capacity from cation-disordered oxides.70,72 Thus, to design high capacity cation-disordered materials, we need to find a disordered host structure that can accommodate Li excess. In this work, LiNi0.5Ti0.5O2 was chosen as the host material for Li excess, containing Ni2+ and Ti4+. This is because LiNi0.5Ti0.5O2 is a disordered rocksalt phase with an active Ni2+/Ni4+ redox couple,117,118 which tends to deliver high voltage of ~3.8 V in the oxide cathodes.13,67,119,120 However, LiNi0.5Ti0.5O2 shows limited reversible capacity,117,118 which is due to poor Li diffusion in its structure by the absence of 0-TM percolation.70,72 Thus, introducing Li excess for 0-TM percolation can transform the material into promising cation-disordered cathode materials that not only deliver a high capacity but also high voltage (Fig. 3-1a). Figures 3-1b to 1d plot the theoretical specific capacities of the hypothetical solid solution compounds between LiNi0.5Ti0.5O2 and Li1+xM1-xO2 (M = Ti4+, Nb5+, Mo6+), all of which lead to LiNi0.5Ti0.5O2-based Li-excess cation-disordered materials. Each figure plots three different capacities: Li capacity that assumes full extraction of available Li ions, Ni2+/Ni4+ redox capacity, and 0-TM capacity, which is defined as the Li capacity accessible by the percolating 0-TM network. In all cases, there is a crossover between the Ni2+/Ni4+ redox capacity and the 0-TM capacity. By a high-valence ‘M’ cation as a charge compensator (Li1+xM1-xO2), additional Li-excess can be accommodated, making the percolating 0-TM network more extensive. Hence, the 0-TM capacity increases. However, as excess Li and ‘M’ cation replace some of the Ni sites, the Ni2+/Ni4+ capacity decreases. From Figures 3-1B to 1D, 79 it is seen that charge-compensating excess Li+ with Mo6+ sacrifices the least Ni2+/Ni4+ capacity. This is because Mo6+ has the highest valency among the charge compensators, thus can accommodate excess Li with the least amount of Mo6+, preserving the most Ni sites for the Ni2+/Ni4+ capacity. Therefore, to maintain both the Ni2+/Ni4+ and 0-TM capacity high, we selected Mo6+ as the charge compensator and investigated the performance of the cationdisordered Li-Ni-Ti-Mo oxides. Figure 3-3 (a) The crystal structure of disordered rocksalt-type solid solution compounds between LiNi0.5Ti0.5O2 and Li1+xM1-xO2 (M = Ti4+, Nb5+, Mo6+) (b-d) Theoretical capacities are given when M is (b) Ti4+ (c) Nb5+ and (d) Mo6+. Each figure plots three different capacities: Li capacity that assumes full extraction of available Li-ions, Ni2+/Ni4+ redox capacity, and 0-TM capacity that is the Li capacity accessible by the percolating 0-TM network. 80 3.3.2 Characterization of Li-Ni-Ti-Mo oxides Figure 3-4 The X-ray diffraction patterns of LiNi0.5Ti0.5O2 (LNTO), Li1.05Ni11/24Ti11/24Mo1/30O2 (LNTMO5), Li1.1Ni5/12Ti5/12Mo1/15O2 (LNTMO10), Li1.15Ni3/8Ti3/8Mo1/10O2 (LNTMO15), and Li1.2Ni1/3Ti1/3Mo2/15O2 (LNTMO20). Insets are the a-lattice parameters of the compounds. 81 To synthesize the Li-Ni-Ti-Mo oxides (Li1+x/100Ni1/2-x/120Ti1/2-x/120Mox/150O2), we applied standard solid-state methods as described in the experimental methodology section. Figure 3-2 shows the X-ray diffraction (XRD) patterns of Li1+x/100Ni1/2-x/120Ti1/2-x/120Mox/150O2 (x = 0, 5, 10, 15, 20). Hereafter, LiNi0.5Ti0.5O2 (x = 0) will be referred to as LNTO, and Li1+x/100Ni1/2-x/120Ti1/2x/120Mox/150O2 with x = 5, 10, 15, and 20 will be referred to as LNTMO5, LNTMO10, LNTMO15, and LNTMO20, respectively. The XRD patterns of a disordered rocksalt in Figure 3-2 and the elemental analysis on the compounds in Table 3-1 show that the target phases are successfully synthesized (Fig. 3-3, Table 3-2). Insets in Figure 3-2a are the a-lattice parameters of each compound. The lattice parameter increases slightly with Li excess. This trend is consistent with the hypothetical Li1.6Mo0.4O2 having bigger average cationic radius (0.726 Å ) than LiNi0.5Ti0.5O2 (0.704 Å ). Thus, introducing excess Li to LiNi0.5Ti0.5O2 by incorporating Li1.6Mo0.4O2 should increase the a-lattice parameter. Table 3-1 Target vs. actual Li:Ni:Ti:Mo atomic ratio as determined by direct current plasma emission spectroscopy. Li excess % 0 Target ratio Li:Ni:Ti:Mo 1:0.5:0.5:0 Actual ratio Li:Ni:Ti:Mo 0.99:0.51:0.5:0 5 1.05:0.458:0.458:0.033 1.04:0.45:0.47:0.035 10 1.1:0.417:0.417:0.067 1.08:0.42:0.43:0.069 15 1.15:0.375:0.375:0.1 1.15:0.365:0.385:0.1 20 1.2:0.333:0.333:0.133 1.2:0.32:0.35:0.135 82 Figure 3-3 Rietveld refinements on the XRD patterns of (a) LiNi0.5Ti0.5O2 [LNTO], (b) Li1.05Ni11/24Ti11/24Mo1/30O2 [LNTMO5] (c) Li1.1Ni5/12Ti5/12Mo1/15O2 [LNTMO10], (d) Li1.15Ni3/8Ti3/8Mo1/10O2 [LNTMO15], and (d) Li1.2Ni1/3Ti1/3Mo2/15O2 [LNTMO20]: structural parameters from the refinements are listed in Table 3-2. 83 Li excess (%) Space group Rwp Site 4a (x, y, z) = (0, 0, 0) Li occupancy Ni occupancy Ti occupancy Mo occupancy Site 4b O occupancy (x, y, z) = (0.5, 0.5, 0.5) a (Å ) Volume (Å 3) Derived density (kg/l) 0 5 10 15 20 3.38 0.497 0.262 0.254 0 6.40 0.5160 0.2269 0.2345 0.0163 Fm-3m 3.79 0.5488 0.2109 0.2153 0.0342 3.73 0.5750 0.1899 0.1925 0.0507 2.13 0.5985 0.1582 0.1708 0.0671 1 1 1 1 1 4.1426 71.094 4.39 4.1444 71.186 4.43 4.145 71.216 4.27 4.1451 71.22 4.22 4.1452 71.226 4.11 Table 3-2 Structural parameters from the Rietveld refinements in Figure 3-3: crystallographic information file of Fm-3m LiFeO2 (ICSD collection code 51208) was first used as an input file. The atomic occupancies were initially set to the atomic ratio obtained from the elemental analysis by direct current plasma emission spectroscopy, based on which the lattice parameters were initially refined. Then, we further refined the lattice parameters and the atomic occupancies simultaneously: TM occupancies were first refined, and then Li occupancy was refined. O occupancy did not change after the refinement for all the compounds. Scanning electron microscopy (SEM) shows that small primary particles, less than 200 nm in diameter (d), are highly agglomerated in secondary particles in all the compounds (Fig. 3-4). The average primary particle size is the smallest for LNTO (d~80 nm) and the largest for LNTMO20 (d~150 nm). After high-energy ball milling the compounds with carbon black for the electrode fabrication, the primary particle size becomes slightly less than d~100 nm on average and the size distribution becomes wider, as can be seen from the image of high-energy ball milled LNTMO20. 84 Figure 3-4 Scanning electron microscope (SEM) images of LNTO, LNTMO5, LNTMO10, LNTMO15, LNTMO20, and high-energy ball-milled LNTMO20 (HB-LNTMO20). 85 3.3.3 The electrochemical properties of Li-Ni-Ti-Mo oxides Figure 3-5 (a) The first-cycle voltage profile of LNTO, LNTMO5, LNTMO10, LNTMO15 and LNTMO20 [1.5−4.5 V, 20 mA/g], and (b) 20-cycle capacity retention of the compounds. 86 To test the cycling performance of the materials, we performed the galvanostatic chargedischarge tests. Figure 3-5a shows the first-cycle voltage profiles of LNTO, LNTMO5, LNTMO10, LNTMO15, and LNTMO20 when they are cycled between 1.5-4.5 V at 20 mA/g. The charge-discharge capacity increases with Li excess from ~110 mAh/g to ~225 mAh/g. The shape of the voltage curves also evolves with Li excess, with the beginning of the first charge coming at lower voltage and the 4.3 V plateau becoming longer with higher Li excess, all of which lead to higher charge capacity. Then, substantial increase in the discharge capacity is achieved with higher Li excess. The first discharge capacity of LNTO is only 109 mAh/g, but that of LNTMO20 is as high as 223 mAh/g. Such increase in the reversible capacity with Li excess is consistent with the increase of 0-TM capacity from percolation theory (Fig. 3-1d).70,72 It is notable that the capacity of LNTMO20 exceeds its theoretical Ni2+/Ni4+ capacity (= 201.6 mAh/g). This indicates that not only Ni2+/Ni4+ but also other redox couples are active in LNTMO20. The trend of higher capacity with Li excess continues upon further cycles as shown in Figure 3-5b. As LNTMO20 delivers the best performance among the Li-Ni-Ti-Mo oxides, we chose LNTMO20 as a representative and compared it with LNTO. Figures 3-6a and 6b show the 10cycle voltage profiles of LNTO and LNTMO20 when cycled between 1.5-4.5 V at 20 mA/g. LNTMO20 delivers much higher capacity (~230 mAh/g) and energy density (~680 Wh/kg, ~2800 Wh/l) than LNTO (~110 mAh/g, ~350 Wh/kg, ~1540 Wh/l). While the capacity above 3 V is higher for LNTMO20, most gains in the discharge capacity come at voltages lower than 3 V, particularly from the ~2.2 V plateau that becomes more obvious with cycles. This results in the average discharge voltage of ~3 V for LNTMO20. It is notable that the charge-discharge profile of LNTMO20 is asymmetric with polarization especially near the end of discharge. This indicates some degree of kinetic limitation in LNTMO20,121 although its performance is still much better than that of LNTO. 87 Figure 3-6 The voltage profiles for 10 cycles of (a) LNTO, and (b) LNTMO20 [1.5−4.5 V, 20 mA/g, room temperature]. 88 Figure 3-7 The voltage profile of (a) LNTO and (b) LNTMO20 when charged and discharged once at 10 mA/g, and then at 20, 40, 100, 200, and 400 mA/g for the subsequent cycles. 89 Figures 3-7 and 7b show the rate capability of LNTO and LNTMO20, respectively. Cells made of each compound were charged and discharged once at 10 mA/g, and then at 20, 40, 100, 200, and 400 mA/g for the subsequent cycles. From the profiles, we find that LNTMO20 delivers higher capacity than LNTO at all rates. As the rate increases from 10 to 400 mA/g, the discharge capacity decreases from 250 mAh/g (750 Wh/kg) to 120 mAh/g (365 Wh/kg) for LNTMO20 and from 120 mAh/g (366 Wh/kg) to 50 mAh/g (145 Wh/kg) for LNTO. Note that the capacity of LNTMO20 at 400 mA/g is comparable to that of LNTO at 10 mA/g. This shows that LNTMO20 has improved rate capability than that of LNTO, which is consistent with the percolation theory.70,72 However, a notable decrease in the capacity of LNTMO20 with higher rates implies that its rate capability is somewhat limited as well. To analyze the kinetics in LNTMO20, we performed the galvanostatic intermittent titration test (GITT). Figure 3-8a shows the first discharge voltage profile of LNTMO20 from the GITT. Upon first charge to 270 mAh/g and discharge to 270 mAh/g, every step of 9 mAh/g was galvanostatically charged or discharged at 20 mA/g, and then the test cell was relaxed for five hours between each step. Time-dependent polarization, inversely correlated with Li diffusion, is most significant at the end of discharge.122,123 The polarization appears to depend on the charge cutoff voltage (Fig. 3-8b). When the cutoff voltage is 4.1 V (black solid), the galvanostatic charge-discharge profiles are symmetric with only minor polarization. When the material is charged to 4.5 V (blue dash), discharge comes with substantial polarization as in Figures 3-6b and 8a. This indicates that Li diffusion in LNTMO20 depends on the structural changes that occur at high voltage. 90 Figure 3-8 (a) The first discharge voltage profile of LNTMO20 from a galvanostatic intermittent titration test after charging to 270 mAh/g: the inset zooms in the time range between 240 h and 270 h. (b) The voltage profile of LNTMO20 when charged-discharged three times between 2.0−4.1 V (black) then between 1.5−4.5 V (blue) at 20 mA/g. 91 3.3.4 The structural evolution of Li1.2Ni1/3Ti1/3Mo2/15O2 during cycling Figure 3-9 (a) The in situ XRD patterns of LNTMO20 upon two galvanostatic chargedischarge cycles between 1.5−4.8 V at 10 mA/g (b) The corresponding voltage profile (c) and the a-lattice parameter from single phase XRD refinements are shown. (d) The (002) peak is zoomed in for the intensity comparison. We performed in situ X-ray diffraction (XRD) to investigate the structural evolution of LNTMO20 upon charge and discharge. Figure 3-9a shows the in situ XRD patterns of LNTMO20 upon two galvanostatic charge-discharge cycles between 1.5−4.8 V at 10 mA/g. The corresponding voltage profile and the a-lattice parameters from single-phase XRD refinements are shown in Figures 3-9b and 9c, respectively. Upon first charge, the (002) peak shifts to a higher angle, indicating a decrease in the a-lattice parameter, but roughly in three steps. Upon first charge to ~110 mAh/g, which accompanies a sloped voltage profile, the peak 92 continuously shifts to a higher angle. However, further peak shift is negligible in charging to ~215 mAh/g, which occurs at the 4.3 V plateau. After this region, the peak further shifts to a higher angle with charging. This indicates that the disordered lattice shrinks at the beginning and end of the first charge, but there is an interval in the middle where it barely shrinks. Upon first discharge, the (002) peak shifts to a lower angle and gains intensity continuously as in Figures 3-9a and 9d, indicating Li insertion to LNTMO20 upon discharge. Two features are seen in the XRD patterns upon first discharge. First, the (002) peak quickly shifts to a lower angle by discharging to ~100 mAh/g, but further shift is small. Second, after the first discharge, the peak is at a lower angle (~19.6°) than where it was before cycle (~19.8°), showing the lattice expansion of LNTMO20 after the first cycle. During the second cycle, the a-lattice parameter decreases upon charge and increases upon discharge until the 2.2 V plateau is reached, after which the lattice expansion is small. 3.3.5 Investigation on the redox mechanism To study the redox mechanism of LNTMO20, we performed X-ray absorption near edge spectroscopy (XANES) measurements. Figures 3-10a, 10b, and 10c show the Ni K-edge, Ti Kedge, and Mo K-edge XANES spectra of LNTMO20, respectively. Each figure shows spectra before cycling (black), after the first charge to 4.8 V (blue: ~300 mAh/g charged), and after the first discharge to 1.5 V (red: ~250 mAh/g discharged). From Figure 3-10a, it is seen that the Ni edge shifts from an energy close to in LiNi2/3Sb1/3O2 used as a standard for Ni2+ to a higher energy similar to Ni3+ in NaNiO2 upon first charge to 4.8 V. After the first discharge to 1.5 V, the Ni edge returns to its starting position.119 This indicates that Ni2+ is oxidized up to Ni~3+ upon first charge to 4.8 V, then reduces back to Ni2+ after the first discharge. As the Ni2+/Ni3+ 93 capacity corresponds to ~100 mAh/g, our finding suggests that the remaining charge capacity comes from either oxygen loss and/or oxygen oxidation, both of which are known to occur in Li-excess materials.45,97,124,125 Figure 3-10 The X-ray absorption near-edge structures of (a) Ni, (b) Ti, and (c) Mo in LNTMO20 before cycle [black], after the first charge to 4.8 V [blue, ~300 mAh/g charged], and after the first discharge to 1.5 V [red, ~250 mAh/g discharged] at 20 mA/g. 94 From the absorption spectra in Figures 3-10b and 10c, it is seen that the Ti and Mo edges barely shift during charging and discharging, indicating that changes in the Mo and Ti oxidation states during cycles, if any, are small. However, the pre-edge peak of Mo XANES increases after the first charge, and remains significantly increased after the first discharge. This shows that Mo environment has deviated from the regular octahedral coordination during the cycle, which might originate from a distortion of the Mo-O octahedral or from some degree of Mo6+ migration from octahedral to tetrahedral sites.126. Comparison with the spectra of MoO2 and MoO3 shows that the Mo edge position does not shift down in energy after the first discharge. This observation strongly suggests that the majority of the Mo ions remain 6+. Likewise, any reduction in the Ti oxidation state on discharge is small on average. To investigate if oxygen loss occurs from LNTMO20, we performed electron energy loss spectroscopy (EELS) on the surface of the LNTMO20 particles before and after cycling. Figure 3-11a shows the Ti L-edge and O K-edge from the EELS spectrums of LNTMO20 before cycling (black) and after 20 cycles (red) between 1.5-4.5 V at 20 mAh/g. Comparing the EELS quantifications of the atomic ratio of O to Ti, we find a considerable decrease in the ratio by ~39 % after cycling. This indicates that oxygen loss has occurred from the surface of LNTMO20 upon cycling, which can contribute to additional charge capacity beyond the Ni2+/Ni~3+ capacity. In addition, we observe that the Ti L-edge is chemically shifted to low energy direction by ~1.5 eV relative to O K-edge after cycling (the inset in Fig. 3-11a). This indicates Ti reduction below 4+ at the surface region,127 which is not captured by the Ti XANES. 95 Figure 3-11 Electron energy loss spectra of Ti L-edge and O K-edge in LNTMO20 before cycling [black] and after 20 cycles between 1.5−4.5 V [red] at 20 mA/g: the inset focuses on the Ti L-edge. Figure 3-12 The first-cycle cyclic voltammetry profiles of LNTMO20 when voltage-swept between 1.5−4.5 V [black solid] and 1.5−4.1 V [red dash] at 0.1 mV/s. 96 Oxygen loss from LNTMO20 can also be inferred from the cyclic voltammetry (CV) tests. Figure 3-12b shows the first cycle CV profile of LNTMO20. When the oxidation cutoff voltage is 4.1 V (red), we observe a main reduction peak at ~3.7 V and a minor reduction peak at ~2.7 V. However, when the cutoff is increased to 4.5 V (black), an additional reduction peak at ~2.2 V is observed in the CV profile, which is likely associated with reduction of a second TM species. This shows that charging above 4.1 V triggers a reaction which allows reduction of species that were previously not reducible. In the case of LNTMO20, reduction of Mo6+ or Ti4+ upon discharge is likely triggered by oxygen loss, similar to reduction of Mn4+ that becomes possible in Li-excess Ni-Mn-Co oxides after oxygen loss.40,97,128 Although the Mo or Ti XANES do not show a clear evidence of the decrease in the average Mo or Ti oxidation states after the first discharge (Figs. 3-10b, 10c), the apparent discrepancy between CV (or EELS) and XANES implies that oxygen loss may be significant at the surface but not in the bulk. Otherwise, XANES should detect the overall decrease in the average Mo or Ti oxidation state after the first discharge. Based on the information from the XANES spectra, we can approximate the limit for the oxygen loss capacity of LNTMO20 during the first cycle. The Ni XANES shows that Ni2+ is oxidized to Ni~3+ upon first charge to 4.8 V, which gives ~100 mAh/g in capacity (Fig. 310a). The remaining first charge capacity (~200 mAh/g) can originate from both oxygen loss and oxygen oxidation. Depending on the oxygen loss capacity, the average Mo or Ti oxidation state should change after the first charge to 300 mAh/g then discharge to 250 mAh/g (Fig. 313). For example, if we assume uniform oxygen loss in a LNTMO20 particle, no loss in the TM content, and that Ni, Ti, and O remain as Ni2+, Ti4+, and O2- after the first discharge, the average Mo oxidation state after the first discharge should be 5.92+, 5.53+, 5.13+, and 4.35+ with increasing oxygen loss capacity of 70, 90, 110, and 150 mAh/g, respectively (Fig. 3-13). According to the Mo and Ti XANES, neither Mo nor Ti is reduced on average after the first 97 discharge (Figs. 3-10b, 10c). Thus, it is likely that oxygen loss does not account for all the extra capacity beyond the Ni2+/Ni3+ capacity, possibly less than 90 mAh/g, and the remaining first charge capacity originates from oxygen oxidation. However, note that the local oxygenloss level can be different between the surface and the bulk of a LNTMO20 particle: our estimation is on the overall oxygen loss capacity. Figure 3-13 Back-of-the-envelope calculation: Average Mo oxidation state expected after the first charge to 300 mAh/g then discharge to 250 mAh/g as a function of the oxygen-loss capacity during the first charge. It was assumed that Ni, Ti, and O stay as Ni2+, Ti4+, and O2after the first discharge, and that there is no loss in the TM content during the first cycle. 98 3.4 Computational results 3.4.1 Oxygen loss mechanisms As we have discussed, oxygen loss occurs from LNTMO20, which likely is most substantial near the surface of the LNTMO20 particles (Fig. 3-11). Two oxygen loss mechanisms have been proposed in the literature. When oxygen is released from the particle surface, either (i) oxygen vacancies or (ii) under-coordinated TM ions at the surface may diffuse into the bulk of the crystal structure.97,124,129,130 The former mechanism introduces oxygen vacancies in the bulk lattice after oxygen loss.97 The latter mechanism results in an increased TM content in the bulk, and is therefore commonly referred to as lattice densification.124,129,130 To study which mechanism accompanies oxygen loss from LNTMO20, we performed DFT calculations on the densified Li0.52Ni0.37Mo0.15O2 structures and Li0.47Ni0.33Ti0.33Mo0.13O1.8 structures with oxygen vacancies, both of which represent possible structures after partially delithiated LNTMO20 loses oxygen by following equations. (a) Li0.867Ni0.33Ti0.33Mo0.13O2 0.4 Li + 0.1 O2 + Li0.519Ni0.37Ti0.37Mo0.15O2 : Oxygen loss with lattice densification (b) Li0.867Ni0.33Ti0.33Mo0.13O2 0.4 Li + 0.1 O2 + Li0.467Ni0.33Ti0.33Mo0.13O1.8 : Oxygen loss with oxygen vacancy formation Based on our calculations, we find that the densified Li0.519Ni0.37Ti0.37Mo0.15O2 structures are energetically more stable than the Li0.467Ni0.33Ti0.33Mo0.13O1.8 structures with oxygen vacancies even though they have same composition of supercell as Li 14Ti10Ni10Mo4O54: The energy difference is 105 meV per LixMO2 formula unit (Fig. 3-14). Thus, the oxygen loss 99 potential in Figure 3-15a is derived from the reaction (a): oxygen loss with lattice densification. Figure 3-14 The calculated energies of densified Li0.52Ni0.37Ti0.37Mo0.15O2 structures and Li0.47Ni0.33Ti0.33Mo0.13O1.8 structures with oxygen vacancies: From the plot, it is seen that the energies of the densified Li0.52Ni0.37Ti0.37Mo0.15O2 structures are lower than those of Li0.47Ni0.33Ti0.33Mo0.13O1.8 structures with oxygen vacancies. This indicates that oxygen loss with densification is thermodynamically more favorable than that with oxygen vacancies in the lattice. 100 3.4.2 Calculated voltage profiles from possible redox mechanisms Figure 3-15 (a) The voltage profiles of Li1.2-xNi0.33Ti0.33Mo0.13O2: The black curve is an experimental profile from the galvanostatic intermittent titration test (GITT) during the first charge, and the red line is calculated voltage profile of Li1.2-xNi0.33Ti0.33Mo0.13O2 that assumes no oxygen loss during the first charge: the dotted red arrows specify the region of Ni oxidation and O oxidation. Finally, the dashed blue line indicates the oxygen-loss potential of Li1.2xNi0.33Ti0.33Mo0.13O2-y. (b) The calculated contribution of Ni [black] and oxygen [red] oxidation upon delithiation in Li1.2-xNi0.33Ti0.33Mo0.13O2 when no oxygen loss is assumed (the red curve in Figure 3-15a) 101 To further understand the behavior of Li1.2-xNi0.33Ti0.33Mo0.13O2 (LNTMO20) during cycling, we investigated the redox mechanism in LNTMO20 with density functional theory (DFT) calculations. Figure 3-15a compares the computed first charge voltage profiles of LNTMO20 against the experimental profile. The red curve is the computed profile assuming no oxygen loss in the crystal structure of LNTMO20 upon first charge. The blue line is the computed profile assuming that partially-delithiated LNTMO20 (Li0.867Ni0.33Ti0.33Mo0.13O2) loses oxygen (−0.1 O2) upon further charge (−0.4 Li), leading to a densified disordered phase of Li0.52Ni0.37Mo0.15O2 that is thermodynamically more stable than a disordered phase with oxygen vacancies (Li0.47Ni0.33Ti0.33Mo0.13O1.9) (Fig. 3-14). Finally, the black curve is the experimental profile from GITT. Figure 3-15b shows the contribution of Ni and oxygen oxidation upon delithiation in LNTMO20 when no oxygen loss is assumed, which results in the red profile in the Figure 3-15a. In general, the experimental voltage (black) is lower than the calculated voltage without oxygen loss (red) and higher than the calculated voltage with oxygen loss (blue). The much lower (calculated) voltage from oxygen loss than without oxygen loss shows that there is a clear driving force for oxygen loss from LNTMO20 after a certain level of delithiation. The calculated voltage with oxygen loss is quite lower that the experimental profile, implying that the actual first charge mechanism is likely to include some degree of oxygen loss based on the thermodynamic driving force. Note that the oxygen loss mechanism in experiments can be more complicated than what is assumed in our calculations. Therefore, there can be a discrepancy in the oxygen loss voltage between the calculations and experiments. Although there exists a clear driving force for oxygen loss after a certain level of delithiation, the computed voltage without oxygen loss (thus with Ni and O oxidation only) is within the experimental voltage window of 1.5−4.5 V (or 1.5−4.8 V). Thus, not to mention the Ni oxidation which is experimentally confirmed by XANES (Fig. 3-10a), oxygen oxidation can 102 also take place at high voltage along with oxygen loss. It is notable that the oxygen oxidation is predicted to take place before Ni2+ becomes completely oxidized to Ni4+ (Fig. 3-15b). In theory, full Ni2+/Ni4+ capacity allows for 0.66 Li delithiation. However, oxygen oxidation already takes place after ~0.35 Li delithiation, which demonstrates an overlap between Ni 3d and O 2p bands in LNTMO20. 3.4.3 Reduction of Mo and Ti after oxygen loss After oxygen loss, not only Ni3+, Ni4+, and O-, but also Mo6+ and Ti4+ can be reduced upon discharge. To study which species (Mo6+ vs. Ti4+) is first reduced after oxygen loss, we computationally studied the redox mechanism of Li1.11-xNi0.37Ti0.37Mo0.15O2 which represents a densified disordered phase of LNTMO20 after some degree of oxygen loss. Figure 3-16a shows the voltage profile of Li1.11-xNi0.37Ti0.37Mo0.15O2, and Figure 3-16b shows the average net moments of Ni, Ti, Mo and oxygen ions in Li1.11-xNi0.37Ti0.37Mo0.15O2 at each composition. The average net moment of Ni ions continuously increases from 1.06 to 1.77 as x decreases from 1.04 to 0.22, indicating reduction of Ni~3+ to Ni2+ in average. Simultaneously, the average net moment of O ions decreases from 0.17 to 0.03 as x decreases from 1.04 to 0.44, indicating reduction of O~1.8- to O2- in average. Although the net moments of Ti ions remain unchanged for whole compositions in Li1.11-xNi0.37Ti0.37Mo0.15O2, the average net moment of Mo ions rapidly increases as x decreases from 0.22 to 0. This indicates that Mo reduction occurs before Ti reduction but after O and Ni reduction. Thus, for the Ti reduction to occur after oxygen loss, it is likely that Mo reduction has already taken place to some degree. 103 Figure 3-16 (a) The voltage profiles of Li1.11-xNi0.37Ti0.37Mo0.15O2. (b) The average net moments of Ni, Ti, Mo and oxygen ions in Li1.11-xNi0.37Ti0.37Mo0.15O2 (x = 0, 0.074, 0.148, 0.222, 0.296, 0.370, 0.444, 0.519, 0.593, 0.667, 0.741, 0.815, 0.889, 0.963 and 1.037). 104 3.5 Discussions 3.5.1 Redox mechanism When we designed the Li-Ni-Ti-Mo oxides, full Ni2+/Ni4+ oxidation was assumed to be possible. In experiments, LNTMO20 can be charged beyond the full Ni2+/Ni4+ capacity. However, the Ni XANES suggests that Ni2+ can be oxidized to only up to Ni~3+ (Fig. 3-10a), and the rest of the first charge capacity originates from either oxygen loss or/and oxygen oxidation. Because the Mo and Ti XANES do not show significant reduction of the average Mo and Ti oxidation state after the first discharge, we suspect that oxygen loss does not account for all the extra capacity beyond the Ni2+/Ni~3+ capacity, and oxygen oxidation may be responsible for the remaining charge capacity. Based on this understanding, we propose the following first charge mechanism for LNTMO20 as a representative of the Li-Ni-Ti-Mo oxides: after the Ni2+/Ni~3+ oxidation, oxygen loss mainly occurs until the surface becomes passivated against the oxygen loss, and then oxygen oxidation dominantly takes place at higher voltage. Note that clear distinction between the oxygen loss region and the oxygen oxidation region may not exist as both can happen simultaneously, such as oxygen loss at the surface and oxygen oxidation in the bulk.129,130 The proposed mechanism is consistent with the change in the lattice parameter of LNTMO20 during the first charge (Fig. 3-17). Upon first charge to ~110 mAh/g, the lattice parameter decreases continuously. This can be explained with the Ni2+/ Ni~3+ oxidation (~100 mAh/g) because Ni3+ (r = 0.56 Å ) and Ni4+ (r = 0.48 Å ) are smaller than Ni2+ (r = 0.69 Å ), thus the lattice parameter decreases with the smaller ions. Upon further charge to ~215 mAh/g, the lattice parameter barely decreases. This can be related to oxygen loss because 105 charging with oxygen loss slows down the increase in the oxidation state of the remaining ions in the crystal structure. As the oxidation state thus the size of the remaining ions stays similar, the lattice shrink can be retarded.30,39 Note that the capacity from this region is ~105 mAh/g, which roughly agrees with our maximum estimated oxygen loss capacity (~90 mAh/g) from the XANES results. Finally, charging beyond ~215 mAh/g decreases the lattice parameter. This can be explained by oxygen oxidation that can shrink the oxygen framework either by making the oxygen ions smaller in size or by introducing peroxo-like species whose oxygen-to-oxygen bond distance is shorter.45,125 Although the Mo and Ti XANES do not show clear evidence of Mo or Ti reduction in LNTMO20 after the first discharge (Figs. 3-10b, 10c), we believe that after oxygen loss, Mo6+ and Ti4+ can be reduced upon discharge in addition to Ni3+, Ni4+ and O−, especially near the surface. First, EELS Ti Li-edge collected from the surface region shows a chemical shift to low energy direction by ~1.5 eV relative to O K-edge after cycling, indicating Ti reduction below 4+ (Fig. 3-11).127 Furthermore, the activation of the Mo or Ti redox couples can be inferred from the CV test, which shows reduction of an additional species when the oxidation cutoff voltage is increased to 4.5 V and LNTMO20 shows oxygen loss (Fig. 3-12). Finally, LNTMO20 delivers ~50 % of its discharge capacity below 3 V during the 1.5−4.5 V cycling test (Fig. 3-6), and its discharge plateau at ~2.2 V matches to Ti4+ reduction in the literature.117 As our calculations predict Mo6+ reduction to occur before Ti4+ reduction (Fig. 3-16), it is likely that oxygen loss allows for both Mo6+ and Ti4+ reduction upon discharge, particularly near the surface. The apparent discrepancy between EELS/CV (showing reduction of Mo6+ or Ti4+) and XANES (showing no change in Mo6+ or Ti4+) may be explained by significant oxygen loss near the surface but not in the bulk. More careful studies to characterize the redox activity are underway. 106 Figure 3-17 Proposed first charge mechanism of LNTMO20: Ni2+/Ni~3+ oxidation, oxygen loss, and oxygen oxidation largely in sequence. The voltage profile of LNTMO20 from the in situ XRD test and the corresponding a-lattice parameters from single-phase XRD refinements are overlayed. 3.5.2 Electrochemical performance As the percolation theory predicts (Fig. 3-1d), the reversible capacity and the rate capability improves with Li excess. In particular, LNTMO20 delivers high capacity and energy density (250 mAh/g, 750 Wh/kg, 3080 Wh/l) at 10 mA/g, which is double the capacity and energy density of LNTO (120 mAh/g, 366 Wh/kg, 1610 Wh/l) at the same rate. Nevertheless, Li diffusion is still somewhat limited in LNTMO20, resulting in large polarization and limited rate capability (Fig. 3-8a). This can also be explained with the structural changes from oxygen loss. 107 As we have discussed, oxygen loss occurs from LNTMO20, which likely is most substantial near the surface of the LNTMO20 particles (Fig. 3-11). Two oxygen loss mechanisms have been proposed in the literature. When oxygen is released from the particle surface, either (i) oxygen vacancies or (ii) under-coordinated TM ions at the surface may diffuse into the bulk of the crystal structure.97,124,129,130 The former mechanism introduces oxygen vacancies in the bulk lattice after oxygen loss.97 The latter mechanism results in an increased TM content in the bulk, and is therefore commonly referred to as lattice densification.124,129,130 Based on our calculations and the literatures, oxygen loss with lattice densification is thermodynamically more favorable than oxygen loss with oxygen vacancies in the lattice (Fig. 3-14).124,129,130 Therefore, it is likely that the surface of LNTMO20 becomes densified after oxygen loss (Fig. 3-18). This can impede Li diffusion in LNTMO20 because densification lowers the Li-excess level by increasing the TM content, resulting in poorer 0-TM percolation in the disordered structure (Fig. 3-1d).70,72 For example, the EELS measurement on the surface of LNTMO20 particles shows a considerable decrease (~39 %) in the O/Ti intensity ratio after the cycling between 1.5−4.5 V (Fig. 3-11). If we assume no loss in the TM content upon oxygen loss with densification, the decrease in the ratio by ~39 % can be interpreted as the change in the composition at the surface from Li1.2-xTM0.8O2 (20 % Li excess) to Li0.7-xTM1.3O2 (−30 % Li excess), which is well below the threshold for 0-TM percolation (~9 % Li excess). This demonstrates how greatly the Li-excess level can decrease at the surface by oxygen loss, thus 0-TM percolation becomes poorer after oxygen loss. While further work is necessary to clearly confirm this hypothesis, it is consistent with Li transport becoming limited when LNTMO20 is charged above ~110 mAh/g, the threshold capacity after which oxygen loss occurs (Fig. 3-8b). 108 Figure 3-18 Illustrations of a LNTMO20 particle before and after oxygen loss with densification near the surface: oxygen loss with densification reduces the Li-excess level, and thus decreases the 0-TM capacity. 3.5.3 Strategies for improvements The observation that Ni2+ can be oxidized up to only Ni~3+ indicates a great overlap between the Ni 3d and O 2p bands in LNTMO20. Variations in local environments for Ni and O in the disordered structure may result in varying orbital overlap, reversing some Ni and O states in energy (Fig. 3-15). In such a scenario, high capacity could not be achieved without oxygen loss or oxidation. While oxygen oxidation can be beneficial because it delivers capacity at high voltage,45,125,130 oxygen loss with densification can be detrimental because it degrades Li diffusion by lowering the Li-excess level in the disordered structure. Therefore, avoiding the oxygen loss seems necessary for any disordered Li-excess materials. Here, we propose two different approaches to avoid oxygen loss from the Li-Ni-Ti-Mo oxides and thus improve the materials. 109 First, decreasing the lattice parameter by cation substitution can be beneficial. This is because a smaller lattice parameter results in a greater orbital overlap between the Ni 3d and O 2p orbitals, which increases the covalency of the Ni-O bonding.7,12 As the covalency increases, the Ni 3d band (eg* band) which has anti-bonding characteristic becomes higher in energy, resulting in less band overlap between the Ni 3d and O 2p bands. This can maximize the Ni2+/Ni4+ capacity so that oxygen loss or oxidation is not required to achieve high capacity. For example, if it were possible to completely use the Ni2+/Ni4+ capacity, LNTMO20 could have delivered ~200 mAh/g without oxygen loss or oxidation at all. Second, surface coating can be beneficial.131,132 If surface coating can retard oxygen loss, oxygen oxidation will immediately take over instead. In this way, high capacity and energy density may be achieved only by the Ni redox and O redox, bypassing the oxygen loss with densification. Thus, the 0-TM percolation stays intact for facile Li diffusion in the Li-NiTi-Mo oxides. 110 3.6 Conclusion In conclusion, we have designed a new class of high-capacity cation-disordered oxides: Li-NiTi-Mo oxides. As 0-TM percolation theory predicts, the reversible capacity and the rate capability improve with Li excess. In particular, Li1.2Ni1/3Ti1/3Mo2/15O2 (20 % Li excess) delivers up to 250 mAh/g and 750 Wh/kg (~3080 Wh/l) at 10 mAh/g, which is double the capacity and energy density of LiNi1/2Ti1/2O2 (0 % Li excess) at the same rate. Through a combination of the in situ XRD, XAS, EELS, and electrochemistry, we propose that first charging Li1.2Ni1/3Ti1/3Mo2/15O2 to 4.8 V is accompanied by Ni2+/Ni~3+ oxidation, oxygen loss, and oxygen oxidation largely in this sequence, after which Mo6+ and/or Ti4+ can be reduced upon discharge. Furthermore, we argue that oxygen loss with densification can impede Li diffusion in Li-Ni-Ti-Mo oxides especially near the surface, because densification lowers the Li-excess level, resulting in the poorer 0-TM percolation in the disordered materials. Finally, we proposed that preventing oxygen loss will improve the new disordered materials by preserving the 0-TM percolation for facile Li diffusion. We believe that the line of thought in this work provides important guidelines for the design of high energy density cation-disordered cathode materials for rechargeable lithium batteries. 111 -------------Part IV------------- Conclusions 112 To develop high-performance Li-ion batteries, the hunt for cathode materials with high-energy densities has been intense. This is because such cathode materials will enable smaller and lighter Li-ion batteries for various complex applications, such as for electric vehicles and grid energy storage. Most high-energy density cathodes have been sought from well-ordered close-packed oxides such as layered (rocksalt-type) Li-TM oxides, and there has been a common wisdom that a well-ordered crystal structure is necessary for facile Li diffusion in the cathode materials, leading to good cyclability of the materials. Accordingly, cation-disordered (i.e. disordered rocksalt) Li-TM oxides have received only a limited attention as cathode materials. In this thesis, it was demonstrated that cation-disordered Li-TM oxides can be promising cathode (or electrode) materials, which enlarges the search space for high-energy density cathode materials to cation-disordered oxides. In the first part of this thesis, the potential of the cation-disordered oxides was demonstrated by the counterintuitive performance of Li1.211Mo0.467Cr0.3O2. This material forms into a well-layered structure but transforms to a cation-disordered structure after several charge-discharge cycles. While a common wisdom expects poor cycling performance of this material, this material delivers a very high capacity (~265 mAh/g) and energy density (~660 Wh/kg), which is very rarely achieved even by the well-layered materials. Using ab initio computations, we explained that the counterintuitive behavior originates from percolation of active 0-TM channels in disordered Li-excess materials. When diffusing through a 0-TM channel, an activated Li+ ion does not face-share high-valent transition metal ions. Therefore, the diffusion barrier through the channel can stay low (thus allows for facile Li migration) even in a cation-disordered structure in which the relaxation space for the activated Li+ ion against face-sharing species is much smaller than in a layered structure: other types of channels are inactive in the cation-disordered structure due to the small relaxation space. Nevertheless, for 113 the active 0-TM channels to support macroscopic lithium diffusion in the cation-disordered materials, the number of 0-TM channels must be great enough for the channels to percolate in the cation-disordered structure, which requires a Li-excess composition (x > 1.09 in LixTM2xO2). As the Li-excess level increases, the percolating network of 0-TM channels become more extensive, enabling a higher fraction of Li ions in a disordered structure to cycle through the network. The Li-excess level of Li1.211Mo0.467Cr0.3O2 is x ~ 1.233 (~23 % Li excess) which is high enough for the percolating 0-TM network to allow for as much as ~1 Li per f.u. (~265 mAh/g) to cycle through the network. Therefore, even as the material disorders during cycling, the materials can still cycle as much as ~1 Li per f.u. through the percolating 0-TM network. Based on the above understanding, a new class of high capacity cation-disordered oxides (Li-Ni-Ti-Mo oxides) was designed in the second part of this thesis. The Li-Ni-Ti-Mo oxides are Li-excess cation-disordered oxides with a general composition of Li1+x/100Ni1/2x/120Ti1/2-x/120Mox/150O2 (0 < x < 30). As the 0-TM percolation theory predicts, the reversible capacity and rate capability improve with Li excess. In particular, Li1.2Ni1/3Ti1/3Mo2/15O2 (20 % Li excess) delivers a high capacity and energy density up to 250 mAh/g and 750 Wh/kg (~3080 Wh/l) at 10 mA/g, which is double the capacity and energy density of LiNi0.5Ti0.5O2 (0 % Li excess) at the same rate. However, large polarization in the voltage profile and limited rate capability from Li1.2Ni1/3Ti1/3Mo2/15O2 indicate that lithium transport needs to be further improved. Combining various characterization techniques, it was proposed that first charging Li1.2Ni1/3Ti1/3Mo2/15O2 to 4.8 V is accompanied by Ni2+/Ni~3+ oxidation, oxygen loss, and oxygen oxidation largely in this sequence, after which both Mo6+ and Ti4+ can be reduced upon discharge. Furthermore, we argued that oxygen loss with densification can impede lithium diffusion in the Li-Ni-Ti-Mo oxides especially near the surface, because densification lowers the Li-excess level, leading to poorer 0-TM percolation in the disordered materials. Finally, we 114 proposed that preventing oxygen loss will improve the new disordered materials by preserving the 0-TM percolation. The search of high-capacity cation-disordered materials has just started. While much effort will be necessary for these materials to become ultimate cathode materials that replace existing materials, I believe the work in this thesis has unlocked the potential of cationdisordered oxides for rechargeable lithium battery cathodes, thus offers hope for substantial improvement in the performance of rechargeable lithium batteries. 115 116 References 1. Tarascon, J. M. & Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–67 (2001). 2. Dunn, B., Kamath, H. & Tarascon, J.-M. Electrical Energy Storage for the Grid: A Battery of Choices. Science 334, 928–935 (2011). 3. Denholm, P., Ela, E., Kirby, B. & Milligan, M. The Role of Energy Storage with Renewable Electricity Generation The Role of Energy Storage with Renewable Electricity Generation. 1–61 (2010). 4. Etacheri, V., Marom, R., Elazari, R., Salitra, G. & Aurbach, D. Challenges in the development of advanced Li-ion batteries: a review. Energy Environ. Sci. 3243–3262 (2011). 5. Ceder, G. Opportunities and challenges for first-principles materials design and applications to Li battery materials. MRS Bull. 35, 693–702 (2010). 6. Armand, M. & Tarascon, J.-M. Building better batteries. Nature 451, 652–657 (2008). 7. Goodenough, J. B. & Kim, Y. Challenges for Rechargeable Li Batteries. Chem. Mater. 22, 587–603 (2010). 8. Whittingham, M. S. Lithium batteries and cathode materials. Chem. Rev. 104, 4271–301 (2004). 9. Goodenough, J. B. & Park, K. S. The Li-ion rechargeable battery: A perspective. J. Am. Chem. Soc. 135, 1167–1176 (2013). 10. Aravindan, V., Gnanaraj, J., Madhavi, S. & Liu, H.-K. Lithium-Ion Conducting Electrolyte Salts for Lithium Batteries. Chemistry. Eur. J. 17, 14326−14346 (2011). 11. Aurbach, D. et al. Design of electrolyte solutions for Li and Li-ion batteries: A review. Electrochim. Acta 50, 247–254 (2004). 12. Aydinol, M. K., Kohan, A. F. & Ceder, G. Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Phys. Rev. B 56, 1354–1365 (1997). 13. Kang, K., Meng, Y. S., Bréger, J., Grey, C. P. & Ceder, G. Electrodes with high power and high capacity for rechargeable lithium batteries. Science 311, 977–980 (2006). 14. Kang, B. & Ceder, G. Battery materials for ultrafast charging and discharging. Nature 458, 190–193 (2009). 15. Nykvist, B. & Nilsson, M. Rapidly falling costs of battery packs for electric vehicles. Nat. Clim. Chang. 5, 100–103 (2015). 117 16. Hautier, G. et al. Phosphates as Lithium-Ion Battery Cathodes : An Evaluation Based on High-Throughput ab Initio Calculations. Chem. Mater. 23, 3495−3508 (2011). 17. Jain, A., Hautier, G., Ong, S. P., Dacek, S. & Ceder, G. Relating Q1 Q2 voltage and thermal safety in Li-ion battery cathodes: a high-throughput computational study. Phys. Chem. Chem. Phys. 17, 5942–5953 (2015). 18. Yoon, W.-S., Grey, C. P., Balasubramanian, M., Yang, X.-Q. & McBreen, J. In Situ Xray Absorption Spectroscopic Study on LiNi0.5Mn0.5O2 Cathode Material during Electrochemical Cycling. Chem. Mater. 15, 3161–3169 (2003). 19. Ohzuku, T. Electrochemistry and Structural Chemistry of LiNiO2 (R-3m) for 4 Volt Secondary Lithium Cells. J. Electrochem. Soc. 140, 1862 (1993). 20. Dahn, J., Fuller, E., Obrovac, M. & Vonsacken, U. Thermal stability of LixCoO2, LixNiO2 and λ-MnO2 and consequences for the safety of Li-ion cells. Solid State Ionics 69, 265–270 (1994). 21. Dahn, J. R., Vonsacken, U., Juzkow, M. W. & Aljanaby, H. Rechargeable LiNiO2 Carbon Cells. J. Electrochem. Soc. 138, 2207–2211 (1991). 22. Kobayashi, H., Arachi, Y., Emura, S. & Tatsumi, K. Investigation on lithium deintercalation mechanism for Li1-yNi1/3Mn1/3Co1/3O2. J. Power Sources 146, 640–644 (2005). 23. Choi, J. & Manthiram, a. Role of Chemical and Structural Stabilities on the Electrochemical Properties of Layered LiNi1/3Mn1/3Co1/3O2 Cathodes. J. Electrochem. Soc. 152, A1714 (2005). 24. Bréger, J. et al. Effect of High Voltage on the Structure and Electrochemistry of LiNi0.5Mn0.5O2: A Joint Experimental and Theoretical Study. Chem. Mater. 18, 4768– 4781 (2006). 25. Kim, D. K. et al. Spinel LiMn2O4 nanorods as lithium ion battery cathodes. Nano Lett. 8, 3948–3952 (2008). 26. Shaju, K. M. & Bruce, P. G. A stoichiometric nano-LiMn2O4 spinel electrode exhibiting high power and stable cycling. Chem. Mater. 20, 5557–5562 (2008). 27. Cho, J., Kim, T.-J., Kim, Y. J. & Park, B. Complete blocking of Mn3+ ion dissolution from a LiMn2O4 spinel intercalation compound by Co3O4 coating. Chem. Commun. 1074–1075 (2001). 28. Van Der Ven, A., Marianetti, C., Morgan, D. & Ceder, G. Phase transformations and volume changes in spinel LixMn2O4. Solid State Ionics 135, 21–32 (2000). 29. Yang, M.-C. et al. Electronic, Structural, and Electrochemical Properties of LiNixCuyMn 2–x–yO4 (0 < x < 0.5, 0 < y < 0.5) High-Voltage Spinel Materials. Chem. Mater. 23, 2832–2841 (2011). 118 30. Ma, X., Kang, B. & Ceder, G. High Rate Micron-Sized Ordered LiNi0.5Mn1.5O4. J. Electrochem. Soc. 157, A925 (2010). 31. Kang, B. & Ceder, G. Electrochemical Performance of LiMnPO4 Synthesized with OffStoichiometry. J. Electrochem. Soc. 157, A808 (2010). 32. Chung, S.-Y., Bloking, J. T. & Chiang, Y.-M. Electronically conductive phosphoolivines as lithium storage electrodes. Nat. Mater. 1, 123–8 (2002). 33. Chung, S.-Y. & Chiang, Y.-M. Microscale Measurements of the Electrical Conductivity of Doped LiFePO4. Electrochem. Solid-State Lett. 6, A278 (2003). 34. Delmas, C., Maccario, M., Croguennec, L., Le Cras, F. & Weill, F. Lithium deintercalation in LiFePO4 nanoparticles via a domino-cascade model. Nat. Mater. 7, 665–71 (2008). 35. Wang, F. et al. Conversion Reaction Mechanisms in Lithium Ion Batteries: Study of the Binary Metal Fluoride Electrodes. J. Am. Chem. Soc. 133, 18828−18836 (2011). 36. Ong, S. P., Wang, L., Kang, B. & Ceder, G. Li-Fe-P-O2 Phase Diagram from First Principles Calculations. Society 77, 1798–1807 (2008). 37. Ong, S., Chevrier, V. & Ceder, G. Comparison of small polaron migration and phase separation in olivine LiMnPO4 and LiFePO4 using hybrid density functional theory. Phys. Rev. B 83, 1–7 (2011). 38. Wang, L., Maxisch, T. & Ceder, G. A first-principles approach to studying the thermal stability of oxide cathode materials. Chem. Mater. 19, 543–552 (2007). 39. Bervas, M., Yakshinskiy, B., Klein, L. C. & Amatucci, G. G. Soft-Chemistry Synthesis and Characterization of Bismuth Oxyfluorides and Ammonium Bismuth Fluorides. J. Am. Ceram. Soc. 89, 645–651 (2006). 40. Thackeray, M. M. et al. Li2MnO3-stabilized LiMO2 (M = Mn, Ni, Co) electrodes for lithium-ion batteries. J. Mater. Chem. 17, 3112 (2007). 41. Johnson, C. S., Li, N., Lefief, C., Vaughey, J. T. & Thackeray, M. M. Synthesis, Characterization and Electrochemistry of Lithium Battery Electrodes: xLi2MnO3·(1−x) LiMn0.333Ni0.333Co0.333O2 (0 ≤ x ≤ 0.7). Chem. Mater. 20, 6095–6106 (2008). 42. Li, J. et al. Synthesis and Characterization of the Lithium-Rich Core-Shell Cathodes with Low Irreversible Capacity and Mitigated Voltage Fade. Chem. Mater.27, 3366−3377 (2015). 43. Cho, J., Kim, Y.-W., Kim, B., Lee, J.-G. & Park, B. A breakthrough in the safety of lithium secondary batteries by coating the cathode material with AlPO4 nanoparticles. Angew. Chem. Int. Ed. Engl. 42, 1618–1621 (2003). 119 44. Sun, Y.-K. et al. A novel concentration-gradient Li[Ni0.83Co0.07Mn0.10]O2 cathode material for high-energy lithium-ion batteries. J. Mater. Chem. 21, 10108 (2011). 45. Sathiya, M. et al. Reversible anionic redox chemistry in high-capacity layered-oxide electrodes. Nat. Mater. 12, 827–35 (2013). 46. Lee, H. W. et al. Ultrathin spinel LiMn2O4 nanowires as high power cathode materials for Li-ion batteries. Nano Lett. 10, 3852–3856 (2010). 47. Li, L. & Cells, M. Dissolution of Spinel Oxides and Capacily Losses in 4 V. Society 143, 2204–2211 (1996). 48. Wang, L.-F., Ou, C.-C., Striebel, K. a. & Chen, J.-S. Study of Mn Dissolution from LiMn2O4 Spinel Electrodes Using Rotating Ring-Disk Collection Experiments. J. Electrochem. Soc. 150, A905 (2003). 49. Sun, Y. K., Hong, K. J., Prakash, J. & Amine, K. Electrochemical performance of nanosized ZnO-coated LiNi0.5Mn1.5O4 spinel as 5 V materials at elevated temperatures. Electrochem. commun. 4, 344–348 (2002). 50. Oh, P. et al. Superior Long-Term Energy Retention and Volumetric Energy Density for Li-Rich Cathode Materials. Nano Lett. 14, 5965−5972 (2014). 51. Kang, K. & Ceder, G. Factors that affect Li mobility in layered lithium transition metal oxides. Phys. Rev. B 74, 1–7 (2006). 52. Cho, J., Kim, Y. J. & Park, B. Novel LiCoO2 Cathode Material with Al2O3 Coating for a Li Ion Cell. Chem. Mater. 12, 3788–3791 (2000). 53. Cho, J. Correlation between AlPO4 nanoparticle coating thickness on LiCoO2 cathode and thermal stability. Electrochim. Acta 48, 2807–2811 (2003). 54. Xie, J. et al. Orientation dependence of Li–ion diffusion kinetics in LiCoO2 thin films prepared by RF magnetron sputtering. Solid State Ionics 179, 362–370 (2008). 55. Cho, J., Kim, Y. J. & Park, B. LiCoO2 Cathode Material That Does Not Show a Phase Transition from Hexagonal to Monoclinic Phase. J. Electrochem. Soc. 148, A1110 (2001). 56. Van der Ven, A. First-Principles Evidence for Stage Ordering in LixCoO2. J. Electrochem. Soc. 145, 2149 (1998). 57. Ueda, A. Solid-State Redox Reactions of LiCoO2 (R-3m) for 4 Volt Secondary Lithium Cells. J. Electrochem. Soc. 141, 2010 (1994). 58. Cho, Y. & Cho, J. Significant Improvement of LiNi0.8Co0.15Al0.05O2 Cathodes at 60°C by SiO2 Dry Coating for Li-Ion Batteries. J. Electrochem. Soc. 157, A625 (2010). 120 59. McCalla, E., Lowartz, C. M., Brown, C. R. & Dahn, J. R. Formation of layered-layered composites in the Li-Co-Mn oxide pseudoternary system during slow cooling. Chem. Mater. 25, 912–918 (2013). 60. Yabuuchi, N., Yoshii, K., Myung, S.-T., Nakai, I. & Komaba, S. Detailed studies of a high-capacity electrode material for rechargeable batteries, Li2MnO3LiCo1/3Ni1/3Mn1/3O2. J. Am. Chem. Soc. 133, 4404–4419 (2011). 61. Johnson, C. S. et al. The significance of the Li2MnO3 component in ‘composite’ xLi2MnO3·(1−x)LiMn0.5Ni0.5O2 electrodes. Electrochem. commun. 6, 1085–1091 (2004). 62. Zheng, J. et al. Mitigating voltage fade in cathode materials by improving the atomic level uniformity of elemental distribution. Nano Lett. 14, 2628–2635 (2014). 63. Croy, J. R., Gallagher, K. G., Balasubramanian, M., Long, B. R. & Thackeray, M. M. Quantifying Hysteresis and Voltage Fade in xLi2MnO3·(1-x)LiMn0.5Ni0.5O2 Electrodes as a Function of Li2MnO3 Content. J. Electrochem. Soc. 161, A318–A325 (2013). 64. Mohanty, D. et al. Unraveling the Voltage Fade Mechanism in High-Energy-Density Lithium-ion Batteries: Origin of the Tetrahedral Cations for Spinel Conversion. Chem. Mater. 26, 6272−6280 (2014). 65. Kang, S. H. & Thackeray, M. M. Enhancing the rate capability of high capacity xLi2MnO3·(1 - x)LiMO2 (M = Mn, Ni, Co) electrodes by Li-Ni-PO4 treatment. Electrochem. commun. 11, 748–751 (2009). 66. Rougier, A. Effect of cobalt substitution on cationic distribution in LiNi1 − yCoyO2 electrode materials. Solid State Ionics 90, 83–90 (1996). 67. Lu, Z., MacNeil, D. D. & Dahn, J. R. Layered Li[NixCo1−2xMnx]O2 Cathode Materials for Lithium-Ion Batteries. Electrochem. Solid-State Lett. 4, A200 (2001). 68. Rougier, A., Gravereau, P. & Delmas, C. Optimization of the Composition of the Li1zNi1+zO2 Electrode Materials: Structural, Magnetic, and Electrochemical Studies. J. Electrochem. Soc. 143, 1168–1175 (1996). 69. Van der Ven, A. Lithium Diffusion in Layered LixCoO2. Electrochem. Solid-State Lett. 3, 301 (1999). 70. Lee, J. et al. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries. Science 343, 519–522 (2014). 71. Van Der Ven, A., Bhattacharya, J. & Belak, A. a. Understanding Li diffusion in Liintercalation compounds. Acc. Chem. Res. 46, 1216–1225 (2013). 72. Urban, A., Lee, J. & Ceder, G. The Configurational Space of Rocksalt-Type Oxides for High-Capacity Lithium Battery Electrodes. Adv. Energy Mater. 4, 1400478 (2014). 121 73. Kang, K. et al. Synthesis and Electrochemical Properties of Layered Li0.9Ni0.45Ti0.55O2. Chem. Mater. 15, 4503−4507 (2003). 74. Li, J., Li, J., Luo, J., Wang, L. & He, X. Recent Advances in the LiFeO2-based Materials for Li-ion Batteries. Int. J. Electrochem. Sci. 6, 1550–1561 (2011). 75. Obrovac, M. N., Mao, O. & Dahn, J. R. Structure and electrochemistry of LiMO2 (M = Ti , Mn , Fe , Co , Ni) prepared by mechanochemical synthesis. Solid State Ionics 112, 9–19 (1998). 76. Sakurai, Y., Arai, H. & Yamaki, J. Preparation of electrochemically active α-LiFeO2 at low temperature. Solid State Ionics 113-115, 29–34 (1998). 77. Ozawa, K. et al. Structural modifications caused by electrochemical lithium extraction for two types of layered LiVO2 (R-3m). J. Power Sources 174, 469–472 (2007). 78. Pralong, V., Gopal, V., Caignaert, V., Duffort, V. & Raveau, B. Lithium-Rich RockSalt-Type Vanadate as Energy Storage Cathode: Li2-xVO3. Chem. Mater. 24, 12−14 (2012). 79. Yabuuchi, N. et al. High-capacity electrode materials for rechargeable lithium batteries: Li3NbO4-based system with cation-disordered rocksalt structure. Proc. Natl. Acad. Sci. 112, 7650−7655 (2015). 80. Park, K. S. et al. LiFeO2-Incorporated Li2MoO3 as a Cathode Additive for Lithium-Ion Battery Safety. Chem. Mater. 24, 2673–2683 (2012). 81. Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 7, 1912–1919 (1973). 82. Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965). 83. Perdew, J. P. J., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996). 84. Blochl, P. E. Projecter augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994). 85. Kresse, G. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). 86. Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996). 87. Kresse, G. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). 88. Jain, A. et al. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 50, 2295–2310 (2011). 122 89. Jonsson, H., Mills, G. & Jacobsen, W. Classical and Quantum Dynamics in Condensed Phase Simulations. (1998). 90. Henkelman, G. & Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 113, 9978– 9985 (2000). 91. Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901– 9904 (2000). 92. Newman, M. E. J. & Ziff, R. M. Efficient Monte Carlo algorithm and high-precision results for percolation. Phys. Rev. Lett. 85, 4104–4107 (2000). 93. Daulton, T. L. & Little, B. J. Determination of chromium valence over the range Cr(0)Cr(VI) by electron energy loss spectroscopy. Ultramicroscopy 106, 561–573 (2006). 94. Zhang, L. & Noguchi, H. Novel Layered Li-Cr-Ti-O Cathode Materials Related to the LiCrO2-Li2TiO3 Solid Solution. J. Electrochem. Soc. 150, A601 (2003). 95. Van der Ven, A., Aydinol, M., Ceder, G., Kresse, G. & Hafner, J. First-principles investigation of phase stability in LixCoO2. Phys. Rev. B 58, 2975–2987 (1998). 96. Shirafuji, J. & Field, J. E. CoO2, The End Member of the LiCoO2 Solid Solution. Electrochem. Soc. 143, 1114–1123 (1996). 97. Lu, Z. & Dahn, J. R. Understanding the Anomalous Capacity of Li/Li[NixLi(1/3−2x/3)Mn(2/3−x/3)]O2 Cells Using In Situ X-Ray Diffraction and Electrochemical Studies. J. Electrochem. Soc. 149, A815 (2002). 98. Zheng, J. et al. Structural and Chemical Evolution of Li- and Mn-Rich Layered Cathode Material. Chem. Mater. 27, 1381−1390 (2015). 99. Yan, P. et al. Evolution of Lattice Structure and Chemical Composition of the Surface Reconstruction Layer in Li1.2Ni0.2Mn0.6O2 Cathode Material for Lithium Ion Batteries. Nano Lett. 15, 514–522 (2015). 100. Hewston, T. a. & Chamberland, B. L. A Survey of first-row ternary oxides LiMO2 (M = Sc-Cu). J. Phys. Chem. Solids 48, 97–108 (1987). 101. Prabaharan, S. R. S., Michael, M. S., Ikuta, H., Uchimoto, Y. & Wakihara, M. Li2NiTiO4 - A new positive electrode for lithium batteries: Soft-chemistry synthesis and electrochemical characterization. Solid State Ionics 172, 39–45 (2004). 102. Yang, M. et al. Cation disordered rock salt phase Li2CoTiO4 as a potential cathode material for Li-ion batteries. J. Mater. Chem. 22, 6200 (2012). 103. Sakuda, A. et al. Rock-salt-type lithium metal sulphides as novel positive-electrode materials. Sci. Rep. 4, 4883 (2014). 123 104. Chen, R. et al. Disordered Lithium-Rich Oxyfluoride as a Stable Host for Enhanced Li+ Intercalation Storage. Adv. Energy Mater. 5, 1401814 (2015). 105. Ren, S. et al. Improved Voltage and Cycling for Li+ Intercalation in High-Capacity Disordered Oxyfluoride Cathodes. Adv. Sci. 1500128 (2015). 106. Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for Xray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005). 107. Dudarev, S. L., Savrasov, S. Y., Humphreys, C. J. & Sutton, a. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998). 108. Jain, A. et al. Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 84, 1–10 (2011). 109. Woodley, S., Battle, P., Gale, J. & Catlow, C. The prediction of inorganic crystal structures using a genetic algorithm and energy minimisation. Phys. Chem. Chem. Phys. 1, 2535–2542 (1999). 110. Woodley, S. M. & Catlow, R. Crystal structure prediction from first principles. Nat. Mater. 7, 937–946 (2008). 111. Reed, J., Ceder, G. & Van Der Ven, a. Layered-to-Spinel Phase Transition in LixMnO2. Electrochem. Solid-State Lett. 4, A78 (2001). 112. Hwang, B. J., Tsai, Y. W., Carlier, D. & Ceder, G. A combined computational/experimental study on LiNi1/3Co1/3Mn1/3O2. Chem. Mater. 15, 3676–3682 (2003). 113. Wang, L., Maxisch, T. & Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B - Condens. Matter Mater. Phys. 73, 1–6 (2006). 114. Chase, M. NIST-JANAF Thermodynamical Tables. American Chemical Society: New York 12, (1998). 115. Kang, S., Mo, Y., Ong, S. P. & Ceder, G. Nanoscale stabilization of sodium oxides: Implications for Na-O2 batteries. Nano Lett. 14, 1016–1020 (2014). 116. Tran, N. et al. Mechanisms Associated with the ‘Plateau’ Observed at High Voltage for the Overlithiated Li1.12(Ni0.425Mn0.425Co0.15)0.88O2 System. Chem. Mater. 20, 4815–4825 (2008). 117. Zhang, L. et al. Synthesis and electrochemistry of cubic rocksalt Li–Ni–Ti–O compounds in the phase diagram of LiNiO2–LiTiO2–Li[Li1/3Ti2/3]O2. J. Power Sources 185, 534–541 (2008). 118. Trócoli, R., Cruz-Yusta, M., Morales, J. & Santos-Peña, J. On the limited electroactivity of Li2NiTiO4 nanoparticles in lithium batteries. Electrochim. Acta 100, 93–100 (2013). 124 119. Twu, N. et al. Designing New Lithium-Excess Cathode Materials from Percolation Theory: Nanohighways in LixNi2-4x/3Sbx/3O2. Nano Lett. 15, 596–602 (2015). 120. Ong, S. P. et al. Voltage, stability and diffusion barrier differences between sodium-ion and lithium-ion intercalation materials. Energy Environ. Sci. 4, 3680−3688 (2011). 121. Yu, H.-C. et al. Designing the next generation high capacity battery electrodes. Energy Environ. Sci. 7, 1760−1768 (2014). 122. Weppner, W. & Huggins, R. A. Determination of the Kinetic Parameters of MixedConducting Electrodes and Application to the System Li3Sb. J. Electrochem. Soc. 124, 1569−1578 (1978). 123. Kim, J. C., Seo, D., Chen, H. & Ceder, G. The effect of antisite disorder and particle size on Li intercalation kinetics in monoclinic LiMnBO3. Adv. Energy. Mater. 5, 1–17 (2015). 124. Armstrong, A. R. et al. Demonstrating oxygen loss and associated structural reorganization in the lithium battery cathode Li[Ni0.2Li0.2Mn0.6]O2. J. Am. Chem. Soc. 128, 8694–8 (2006). 125. Sathiya, M. et al. High Performance Li2Ru1-yMnyO3 (0.2 <y<0.8) Cathode Materials for Rechargeable Lithium-Ion Batteries: Their Understanding. Chem. Mater. 25, 1121–1131 (2013). 126. Hara, D. et al. Charge–discharge reaction mechanism of manganese molybdenum vanadium oxide as a high capacity anode material for Li secondary battery. J. Mater. Chem. 13, 897–903 (2003). 127. Stemmer, S. et al. Oxidation states of titanium in bulk barium titanates and in (100) fiber-textured (BaxSr1-x)Ti1+yO3+z thin films. Appl. Phys. Lett. 79, 3149–3151 (2001). 128. Ates, M. N., Mukerjee, S. & Abraham, K. M. A high rate Li-rich layered MNC cathode material for lithium-ion batteries. RSC Adv. 5, 27375–27386 (2015). 129. Koga, H. et al. Different oxygen redox participation for bulk and surface: A possible global explanation for the cycling mechanism of Li1.20Mn0.54Co0.13Ni0.13O2. J. Power Sources 236, 250–258 (2013). 130. Koga, H. et al. Operando X-ray Absorption Study of the Redox Processes Involved upon Cycling of the Li-Rich Layered Oxide Li1.20Mn0.54Co0.13Ni0.13O2 in Li Ion Batteries. J. Phys. Chem. C 118, 5700–5709 (2014). 131. Oh, P., Ko, M., Myeong, S., Kim, Y. & Cho, J. A Novel Surface Treatment Method and New Insight into Discharge Voltage Deterioration for High-Performance 0.4Li2MnO30.6LiNi1/3Co1/3Mn1/3O2 Cathode Materials. Adv. Energy Mater. 4, 1400631 (2014). 125 132. Kim, H., Kim, M. G., Jeong, H. Y., Nam, H. & Cho, J. A New Coating Method for Alleviating Surface Degradation of LiNi0.6Co0.2Mn0.2O2 Cathode Material: Nanoscale Surface Treatment of Primary Particles. Nano Lett. 15, 2111−2119 (2015). 126 127