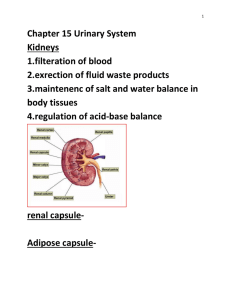
Mechanistic Investigation of Anticancer Agents that Damage
advertisement

Mechanistic Investigation of Anticancer Agents that Damage DNA and Interact with the Estrogen Receptor By Sreeja Gopal B.Tech. Chemical Engineering (2003) Indian Institute of Technology, Madras SUBMITTED TO THE DEPARTMENT OF BIOLOGICAL ENGINEERING IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF ARCHIVES DOCTOR OF PHILOSOPHY IN BIOLOGICAL ENGINEERING MASSACHUSET-S INSTITUTE- at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY L-N3( IL"I JUNE 2009 @2009 Sreeja Gopal. All rights reserved. The author hereby grants to MIT permission to reproduce and to distribute publicly paper and electronic copies of this thesis document in whole or in part in any medium now known or hereafter created. Signature of Author: Departmedt of Biological Engineering May 22, 2009 Certified by: John M. Essigmann William and sy Leitch Professor of Chemistry and Toxicology Thesis Supervisor Accepted by: - /Alan J. Grodzinsky Biological Engineering Professor of EVctrical, Mech nical,: hair, Course XX Graduate Program Committee EI2F? THIS DOCTORAL THESIS HAS BEEN EXAMINED BY A COMMITTEE OF THE DEPARTMENT OF BIOLOGICAL ENGINEERING AS FOLLOWS: A Peter C. Dedon Professor of Toxicology dnd Biological Engineering Chairman John M. Essigma William and Betsy Leitch Phge sor of Chehtry and Toxicology Thesis Supervisor Ram Sasisekharan . Professor of Biological Engineering and Health Sciences & Technology Robert G. Croy _ _ _r Department of Biological Engineering (i --., Mechanistic Investigation of Anticancer Agents that Damage DNA and Interact with the Estrogen Receptor by Sreeja Gopal Submitted to the Department of Biological Engineering on May 22, 2009 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biological Engineering ABSTRACT One of the primary goals of cancer chemotherapy is the design of antitumor agents that achieve selective targeting of tumor cells while minimizing toxicity to normal tissues. We have synthesized a series of DNA damaging agents that are designed to disrupt selectively the DNA repair and signaling pathways in tumor cells. My dissertation focuses on the mechanistic studies of the rationally designed genotoxicants E2-7aX and 2Pl, which target cancer cells that express the estrogen receptor (ER). The studies on the biological activity of E2-7ca in ovarian cancer cells reveal that the compound induces levels of cytotoxicity comparable to those of cisplatin, which is the current front-line therapeutic for ovarian cancer. E2-7ca induces crosslink formation in the ovarian cell line SKOV-3, which potentially leads to the S-phase arrest observed in these cells. Further, the arrest is persistent even after drug removal, indicating that the persistence might be one of the reasons for the enhanced toxicity of E2-7ca compared to the control compound chlorambucil. E2-7ca induces cell death in SKOV-3 cells through autophagy, as opposed to the classical cell death pathway of apoptosis. Mechanistic studies on the role of ER in E2-7a toxicity in SKOV-3 cells show that the absence of the ER (facilitated by the knockdown of ER protein through RNA interference in SKOV-3 cells) makes the cells less sensitive to the drug. In addition, the ER(-) population displays a lower level of drugDNA adducts than its ER(+) counterpart, as detected by Accelerator Mass Spectrometry (AMS). The combination of these pieces of evidence strongly suggests that as per its intended mechanism of action, E2-7a. mediates its effects through the involvement of the ER, and appears to be a promising candidate as an antitumor agent in the clinical setting. In addition, the toxicity and DNA damage caused by the 2-phenylindole compounds 2PI and 2PI(OH) were studied in ovarian cancer cell lines; these studies underscore the differences in the mechanism of action of the two compounds. Both the compounds demonstrate promise as potent anti-tumor agents, and the selectivity of these compounds in different cellular contexts (ER (+)/(-) or p53 (+)/(-)), once established, could help strengthen the basis of their therapeutic efficacy. Thesis Supervisor: John M. Essigmann Title: William and Betsy Leitch Professor of Chemistry and Toxicology ACKNOWLEDGMENTS To Professor John Essigmann, for his invaluable support and guidance. I have been inspired by his outlook, both towards science and towards life. I am grateful to him for having provided an environment conducive for intellectual growth and collaborative learning in his lab. John's passion for teaching and his zest for perfection in oral and written presentations have helped hone my skills in this regard as well. I thank John for providing the opportunity to learn from him and work with him. To my thesis committee members, Professor Pete Dedon, Professor Ram Sasisekharan, and Dr. Bob Croy, for their advice and insight. I appreciate the time each of you has put in for regular meetings, and for valuable suggestions to address several aspects of my research. To Dr. Bob Croy, who has been amazing as a mentor and advisor to my project. His extensive knowledge in the field and his guidance with experimental design and troubleshooting have been crucial for my progress in this project. I really appreciate his time and his willingness to spend time on my research. To the members of the Essigmann laboratory, for being great colleagues and friends over the past five years. A special thanks to Kyle Proffitt, my first teacher in the lab. Thanks Kyle, for showing me the ropes in setting up experiments and for teaching me how to do tissue culture and westerns. Your tenacity and your exhaustive approach to problem solving have been an inspiration. I also wish to thank John Marquis, Shawn Hillier and Pei-Sze Ng for our collaborative efforts, and Jeannette Fiala and Leslie Woo for their wonderful jobs editing my thesis drafts. Our seamless interactions at the Essigmann lab are much appreciated and have made my graduate career so much more enjoyable. I also thank Kim Schaefer, for her willingness to give administrative assistance, and for helping things run smoothly in the lab. The Essigmann lab has been a close-knit community, and I will definitely miss our interactions and our conversations on both research and on life. I also wish to thank Rosa Liberman and Paul Skipper at the Tannenbaum laboratory for our collaborations on the AMS front. To my fellow BE graduate students, for being great friends, and for making my transition to the US an enjoyable experience. The fun, the conversations, and the mutual support have been truly memorable. To my friends, both at MIT and outside, for believing in me, and for listening to me even when I go on endlessly about the woes of research. I really appreciate your support and your encouragement. Most importantly, to my family - my parents, my sister, my grandparents and my husband - for always believing in me, for inspiring me to be a better person, and for encouraging me in all my endeavors. Now that my journey here is almost drawing to a close, it is as much your achievement as it is mine. I forever will appreciate all the time you have put in on my behalf and your unconditional love and support. What I am today, I owe to you. Dedicatedto my lovingfamily This work would not have been possible without your constant support, encouragement and love TABLE OF CONTENTS COMMITTEE PAGE 2 ABSTRACT 3 ACKNOWLEDGEMENTS 4 TABLE OF CONTENTS 7 LIST OF ABBREVIATIONS 9 LIST OF TABLES 11 LIST OF FIGURES 12 CHAPTER 1: Introduction 15 1.1 1.2 CANCER ..................................................................................... . . 16 1.1.1 Cancer Therapy and Therapeutics 19 1.1.2 Ovarian Cancer 21 1.1.3 Current Therapy for Ovarian Cancer 23 26 NUCLEAR HORMONE RECEPTORS .................................................. 1.2.1 27 Estrogen Receptor 31 1.3 ER AND OVARIAN CANCER ............................................................. 1.3 RATIONAL DESIGN OF NOVEL GENOTOXICANTS THAT TARGET ER(+) T UMO RS .......................................................................................... 33 1.3.1 E2-7a-C6NC2-Mustard 34 1.3.2 Phenylindole-C6NC2-Mustard 35 1.4 FIG UR ES ..................................................................................... 1.5 REFER EN CES ................................................................................ . . 37 42 CHAPTER 2: E2-7a - Biological Consequences in Ovarian Cancer Cells: Cytotoxicity, DNA Damage and Cell Death 2.1 IN T R O DUCT IO N ................... ............. 7 59 ............................................ 60 2.2 MATERIALS AND METHODS ........................................................... 2 .3 RESULT S ....................................................................................... 2.4 DISCUSSION ......... 2.5 CONCLUSIONS ........................................ 2 .6 T AB L ES ............................................................................................ 91 2.7 FIGURES ........................................................................................ 93 2.8 REFERENCES ............................................ ..................................... 68 . 73 80 ....89 107 CHAPTER 3: Role of ER in E2-7a toxicity: Studies in ER(+) and ER(-) cell populations 139 3.1 INTRODUCTION 3.2 MATERIALS AND METHODS ............................................................. 146 3 .3 RESULT S ........................................................................................ 14 9 3.4 DISCUSSION ................................................................................... 153 3.5 CONCLUSIONS ........................................... 159 3.6 FUTURE DIRECTIONS ...................................................................... 160 3 .7 TA B LES .......................................................................................... 16 2 3 .8 FIG URES ......................................................................................... 16 4 3.9 REFERENCES ............................................ 170 ........................ ......... ................... 140 CHAPTER 4: Investigation of cytotoxicity and other biological properties of phenylindole compounds in ovarian cancer cells 203 4.1 INTRODUCTION ................................................................................ 204 4.2 MATERIALS AND METHODS ............................................................. 206 4 .3 RESUL TS ........................................................................................ 2 11 4.4 DISCUSSION ................ ..... 4.5 CONCLUSIONS ............. ................... 4.6 TABLES ....................................................................................... 225 4.7 FIG URES ......................................................................................... 226 4.8 REFERENCES ................ ... ............................... ...... ....................... 217 ........................................... 224 ......................................... 237 LIST OF ABBREVIATIONS AF Activation Function AMS Accelerator Mass Spectrometry BRCA1/ BRCA2 Breast Cancer 1/2 CA-1 25 Cancer Antigen 125 Cdk Cyclin-dependent kinase CDT Charcoal Dextran-treated cpm Counts per minute DBD DNA Binding Domain dpm Disintegrations per minute DMSO Dimethyl Sulphoxide E2-7a Estradriol-C6NC2-mustard EB Ethidium Bromide EOC Epithelial Ovarian Cancer ER Estrogen Receptor ER-LBD Estrogen Receptor Ligand Binding Domain ERE Estrogen Response Element ESI-MS Electrospray Ionization Mass Spectrometry HPLC High Performance Liquid Chromatography HNPCC Hereditary Non-Polyposis Colon Cancer HRE Hormone Response Element HRT Hormone Response Therapy HSP Heat Shock Protein ICL Interstrand Crosslink LBD Ligand Binding Domain MDC monodansylcadaverine MEM Minimum Essential Medium NLS Nuclear Localization Signal NER Nucleotide Excision Repair NHR Nuclear Hormone Receptor OC Ovarian Cancer OSE Ovarian Surface Epithelium OTM Olive Tail Moment PBS Phosphate Buffered Saline 2PI 2-Phenylindole PR Progesterone Receptor SDS Sodium Dodecyl Sulphate siRNA Short interfering RNA XPA Xeroderma Pigmentosum group A LIST OF TABLES Table 2.1 Table 2.2 G150 values of test compounds in SKOV-3 and OVCAR-3 ce lls .......................................................................... . . 91 Comet assay detects E2-7x induced crosslinks in SKOV-3 ce lls .......................................................................... . . 92 Table 3.1 Doubling times of SKOV-3 cells with estrogen and antiestrogen tre atm e nt........................................................................16 2 Table 3.2 Adducts, cell numbers, and [14C]-E2-7a distributions in ER(+) and ER(-) SKO V-3 cells...........................................................163 Table 4.1 Percentage decrease in OTM in Caov-3 and OVCAR-3 cells.....225 LIST OF FIGURES Figure 1.1 Structures of Relevant Chemotherapeutic Agents................. 37 Figure 1.2 Structural Organization of Nuclear Hormone Receptors and Estrogen Receptors a and P..............................................38 Figure 1.3 Possible Mechanisms of Action of Bifunctional Genotoxicants.....39 Figure 1.4 A Novel Agent Designed to Inhibit Repair Processes in a Cancer Cell ............................................................................ . . 40 Figure 1.5 Structures of Bifunctional Genotoxicants that Target the ER........41 Figure 2.1 Mechanism of Reaction of E2-7a with DNA...........................93 Figure 2.2 E2-7a Treatment is Cytotoxic to SKOV-3 Cells following 24 and 36 Hours of Treatm ent........................................................ 94 Figure 2.3 E2-7a Treatment is Cytotoxic to OVCAR-3 Cells following 24 and 36 Hours of Treatment.................................................... 95 Figure 2.4 E2-7ax Arrests SKOV-3 Cells in S-Phase.............................96 Figure 2.5 E2-7a Arrests OVCAR-3 Cells in S-Phase...........................97 Figure 2.6 Chlorambucil Treated Cells Recover from Arrest once Drug is Rem oved .................................................................... . . 98 Figure 2.7 E2-7a Treated Cells Do Not Recover from Arrest once Drug is Rem oved .................................................................... . . 99 Figure 2.8 Figure 2.9 E2-7a Induces Activation of Checkpoint Proteins in SKOV-3 cells ............................................................................... 100 Comet Assay Indicates Presence of Interstrand Crosslinks after E2-7c Treatm ent..............................................................101 Figure 2.10 XPA cells are more sensitive to E2-7c than HeLa cells.............102 Figure 2.11 Apoptotic markers are not activated with E2-7a treatment.......103 Figure 2.12 AO/EB Staining of Live SKOV-3 Cells (5 pM, 12 hours)............104 Figure 2.13 AO/EB Staining of Live SKOV-3 Cells (2.5 pM, 24 hours)..........105 Figure 2.14 MDC Staining of Live SKOV-3 Cells (2.5 pM, 24 hours)............106 Figure 3.1 SKOV-3 Cells are not Growth Sensitive to Estradiol or ICI 18 2,780.......................................................................... 16 4 Figure 3.2 ER is Downregulated with siRNA Treatment for up to 72 Hours..165 Figure 3.3 ER(+) and ER(-) SKOV-3 Cells are Equally Sensitive to Melphalan. They are Equally Sensitive to Cisplatin as well ........................ 166 Figure 3.4 ER(+) Cells are More Sensitive to E2-7a than ER(-) Cells.......167 Figure 3.5 E2-7a Forms DNA Adducts in SKOV-3 Cells..........................168 Figure 3.6 E2-7a Adduct Levels are Higher in ER(+) compared to ER(-) SKOV-3 Cells..................................................................169 Figure 4.1 Synthetic Scheme of 2-Phenylindole Compounds....................226 Figure 4.2 Structures and Binding Affinities of Phenylindole Compounds... .228 Figure 4.3 ER Status of Relevant Cell Lines.........................................229 Figure 4.4 2PI and 2PI(OH) Cause Growth Inhibition of Caov-3 and OVCAR-3 cells ............................................................................... 230 Figure 4.5 Cell Cycle Distribution in Caov-3 Cells Treated with Test Compo unds.....................................................................2 31 Figure 4.6 Cell Cycle Distribution in OVCAR-3 Cells Treated with Test Compo unds ..................................................................... 232 Figure 4.7 Detection of Interstrand Crosslinks in Caov-3 Cells Using Comet A ss ay ............................................................................. 233 Figure 4.8 Detection of Interstrand Crosslinks in OVCAR-3 Cells Using Comet A ssa y ............................................................................. 234 Figure 4.9 Drug-DNA Adducts in Caov-3 and OVCAR-3 cells Detected Using A MS.............................................................................. 235 Figure 4.10 Caspase-3 Activation by Cisplatin and Phenylindole Compounds.....................................................................236 Chapter 1 Introduction Cancer Cancer is a complex disease that results from the coordinated dysregulation of cellular regulatory circuits. It is characterized by compromised genomic integrity and manifests as excessive proliferation of a group of cells that invades surrounding tissue and in some cases, metastasizes to other parts of the body. The process of transformation - the genetic alteration of a normal cell to a cancer cell - occurs when a cell loses the ability to regulate its growth and proliferative pathways. It is apparent from current studies that a normal cell, during its life cycle, could progressively accumulate mutations that provide it certain growth and survival advantages that enable the cell to transform to a cancer cell. Studies on cancer incidence and its genomic characterization reinforce the theory that the transformation from a normal cell to a cancer cell is a multi-step sequence of stochastic events (Armitage and Doll, 1954; Foulds, 1954; Renan, 1993). The idea is that each of these steps corresponds to a genomic insult that takes the normal cell one step closer to malignancy (Bishop, 1991). For example, a normal cell could acquire a mutation in a tumor suppressor gene that confers it an enhanced rate of proliferation, and then, in an independent step, acquire the capability to resist apoptosis. Hanahan and Weinberg (2000) have proposed a framework to explain the steps involved in cancer progression. There are six characteristic traits of cancer cells that represent the cellular control mechanisms that normal cells have overcome in their path to becoming malignant. These traits consist of: (i) self-sufficiency in growth signals, (ii) insensitivity to anti-growth signals, (iii) ability to evade apoptosis, (iv) limitless replicative potential, (v) sustained angiogenesis, and (vi) tissue invasion and metastasis. These traits result from a variety of physical changes in the genome, including somatic mutations, chromosomal loss and duplication, genomic rearrangements, methylation of DNA, and changes in chromatin structure. Virtually all cancers display these six traits, but the order or the mechanism of acquiring these traits is usually not the same in all cells. One mutation could result in more than one trait at a time, or on the other hand, a single trait might need two or more coordinated steps to be acquired. These parallel pathways to malignancy account for the high levels of heterogeneity found amongst the vast array of cancers. The susceptibility of a cell towards accumulating mutations is affected by several endogenous factors, such as levels of DNA repair, immune conditions, hormone levels, and fidelity of replication. Inherited mutations in key tumor suppressor genes, oncogenes, and genome maintenance genes are also potential endogenous risk factors. The most significant causative link to cancer risk comes through the route of exogenous agents, which include environmental carcinogens, occupational chemicals, radiation and tobacco smoke, and exposure to pathogens (for example, certain viruses and parasites). Exposure to these agents can occur through exogenous routes such as air, food, and water or endogenous processes such as byproducts of inflammation and metabolic conversion of otherwise innocuous environmental agents. The existence of such a wide variety of risk factors for cancer could explain the high number of cases diagnosed each year. According to Cancer Facts and Figures, cancer is the second leading cause of death in the US and over 500,000 people are expected to die of cancer this year (American Cancer Society, 2008). Better treatment options over the last few decades have increased the five-year survival rate among Americans to 66% for all cancers diagnosed between 1996 and 2003, up from 50% in 1975-77. The numbers of new cases this year of the five most common cancer types - lung cancer (215,000), prostate cancer (186,000), breast cancer (184,000), colorectal cancer (148,000) and lymphoma (74,000) - reflect the urgency in investigating more effective treatment options for the disease. Cancers diagnosed at earlier stages result in better prognoses, and currently, there are several options for detecting and treating early stage malignancies. Some cancers can be screened using specific markers or visual evidence. For instance, elevated level of Prostate Specific Antigen (PSA) in patients is used as a biomarker for prostate cancer. Another tumor marker protein Cancer Antigen 125 (CA-1 25) is sometimes used to screen women with increased risk for ovarian cancer (Bandera et al., 2003; Jacobs and Bast, Jr., 1989). Mammography is used as a visual tool to assess breast cancer, sigmoidoscopy to inspect the colon, and Pap test for cervical cancer (Rimer and Schildkraut, 1997). However, several cancers do not possess well-defined screens to detect early signs of the disease. Malignancies such as epithelial ovarian cancer are not detected until the disease is at an advanced stage, and this leads to extremely poor prognosis for the patient. Once the cancer is detected and its extent determined, there is a constantly expanding array of options for therapy. However, it is especially challenging to develop effective treatment options given the enormous heterogeneity observed in tumor populations the response to treatment can vary significantly depending on the extent of the disease, its severity and subtype (Rimer and Schildkraut, 1997). The next section discusses current therapy landscape and the development of newer, improved therapeutics. Cancer Therapy and Therapeutics The main goal of cancer therapy is to reduce the tumor cell population to zero cells. This outcome is best achieved by surgical removal of the tumor to prevent the tumor from recurring or metastasizing. Surgical resection of the tumor is usually followed by adjuvant therapy that consists of one or more of treatments such as radiation, chemotherapy, immunotherapy, hormone therapy, and/or bone marrow transplantation(Chabner and Longo, 2009). Of these, chemotherapy is historically one of the oldest known treatments and is often the first choice of therapeutic intervention in patients with several types of solid tumors and hematopoietic neoplasms (Papac, 2001); it is almost always used as a systemic treatment and has been shown to cure some disseminated cancers. Although rarely curative, chemotherapeutics have been effective in decreasing tumor volume, alleviating symptoms, and even prolonging life in certain metastatic cancers (Chabner and Longo, 2009; DeVita et al., 1997 ). Nitrogen mustards - mechlorethamine ("nitrogen mustard", HN2; Figure 1.1) and tris(p-chloroethyl)amine (HN3) - were the first known chemotherapeutic agents. These compounds were originally used in chemical warfare in the 1930s; in a serendipitous turn of events, they were effectively used to treat non-Hodgkin's lymphoma, thus demonstrating for the first time that chemotherapy could induce tumor regression (Papac, 2001). The clinical use of nitrogen mustards as therapeutic agents was followed by the use of several other classes of antineoplastic agents such as antifolates, purine analogs, vinca alkaloids, microtubule inhibitors and topoisomerase inhibitors (Chabner and Roberts, Jr., 2005). Subsequently, combination chemotherapy and adjuvant therapy were introduced, and these regimens are now the standard of care treatment of human malignancy. In fact, combination of drugs have proved to be more effective than single agents both in metastatic cancer and as adjuvant therapy in cases with a high risk of relapse (Bonadonna et al., 1976; Jaffe et al., 1974; Li et al., 1958). The task of development of chemotherapeutics has been arduous - at almost every step, researchers have encountered significant problems because of long-term and acute toxicities that affect other organs in the body. A slight respite to this problem came with the improvements in molecular and genetic understanding of cell biology, which in turn led to more targeted therapeutics. A variety of compounds, including designed small molecules, monoclonal antibodies, peptidomimetics, short interfering RNAs (siRNAs), antisense oligonucleotides and other targeted therapies are being evaluated for the treatment of cancer (Clark, 2006). Tools are now available to manipulate molecular systems for therapeutic purposes. The goal of current therapy development is to target specific proteins that elicit the desired response from cancer cells while circumventing off-target effects. We at the Essigmann lab are interested in creating next generation chemotherapeutics by designing mechanism-based drugs - a series of agents that combine a molecularly targeted moiety with the proven efficacy of DNA alkylating agents, thus bringing a layer of selectivity to the DNA alkylating capability of the agent. With this combination, we aim to augment the efficacy of the antitumor agent by honing in on a selected population of cancer cells and killing these cells. DNA damaging agents used as cancer therapeutics are one of the most effective treatments in clinical use, and have demonstrated improved efficacy when used in combination with drugs that have a different mechanism of action (Hurley, 2002). A targeted DNA damaging agent would provide a means of specifically killing malignant cells while sparing normal tissues. The antitumor agents designed in the Essigmann laboratory have shown promising results that implicate a component of selectivity in their toxicity (Mitra et al., 2002; Rink et al., 1996). Understanding the mechanism of action of these drugs would help assess the potential of these agents as clinically effective chemotherapeutics. My dissertation is focused on probing the mechanism of action of two such rationally designed drug candidates that target ovarian cancer. Ovarian Cancer Ovarian Cancer (OC) is the leading cause of gynecological death worldwide - there were 204,000 estimated cases in 2002 and 125,000 deaths due to the disease in the world (Parkin et al., 2005). According to statistics provided by the American Cancer Society, 69% of the patients with advanced OC survive but this rate drops to 29% in patients with distant metastases (American Cancer Society, 2008). OCs can be subdivided into three major types: surface epithelial-stromal tumors, germ cell tumors and sex cord-stromal tumors. Of these, epithelial ovarian carcinoma dominate the cancer registries by accounting for -90% of ovarian tumors in the Western world (Greenlee et al., 2000). There are approximately 22,000 new cases and 15,000 deaths associated with this disease every year (Jemal et al., 2008). The relative lack of specific early signs and symptoms combined with inefficient screening results in overall low cure rates for epithelial ovarian cancers (EOC). The etiology of EOC is currently unknown, although there are several risk factors that have been identified by epidemiological studies. The major risk factors (Yap et al., 2009) consist of : (i) Environmental and hormonal factors: The origin and progression of ovarian carcinogenesis are still highly debated. Epidemiological studies show that the risk factors that increase risk are age, infertility, whereas increasing parity, oral contraceptive use, hysterectomy or tubal ligation decrease risk (Arai et al., 2004; Riman et al., 2001; Riman et al., 2002b; Riman et al., 2002a; Riman et al., 2004; Risch, 2002; Schildkraut et al., 1996 ). One theory regarding the origin of EOC is that frequent ovulations disrupt the ovarian surface epithelium (OSE) with repeated cycles of wounding, proliferating, and healing and this may lead to malignant transformation (Rao and Slotman, 1991). A second cause might be the persistent stimulation of the ovary by the pituitary gonadotropins luteinizing hormone and follicle-stimulating hormone - which act directly or mediate their effect through the action of estrogen. (ii) Hereditary factors: Family history of breast or ovarian cancer is a substantial risk factor for EOC - 10-20% EOCs have a hereditary predisposition (Pal et al., 2005). Women with BRCA1 or BRCA2 mutations are at a higher risk, with an average cumulative risk of -40% and -10% respectively at the age of 70 (Lukanova and Kaaks, 2005). Remarkably enough, up to 70% of ovarian cancers show mutations in the BRCA1 gene, in contrast to 20-40% in inherited breast cancers (Merajver et al., 1995; Miki et al., 1994; Zheng et al., 2001). Early onset of breast cancer, presence of ovarian cancer, and male breast cancer also point to a hereditary risk for EOC. Hereditary nonpolyposis colon cancer (HNPCC) is an observed risk factor in EOC as well (Lindor et al., 2006). Uk Current Therapy for Ovarian Cancer In spite of the knowledge of the risk factors discussed above, in most cases, it is difficult to detect the disease at an early stage. Since early-stage EOC has the best prognosis with appropriate treatment, it is indeed desirable to detect signs of the disease early. The primary marker used for the assessment of the pelvic mass is CA-125 (Bast, Jr. et al., 1983). Malignancies other than EOC can also upregulate the levels of CA-125, but the highest levels are usually observed with ovarian cancer. However, only 50% of stage-I cases were found to have elevated levels of the marker in serum (Jacobs and Bast, Jr., 1989). Hence, efforts are still ongoing to find more reliable markers and therapies for the disease, using high throughput techniques such as protein microarrays (Bandera et al., 2003; Chatterjee et al., 2006). Accurate diagnosis and stage determination are the first steps in the treatment of women with EOC. Several clinical trials over the past few decades have made improvements in treatment of the disease- surgery followed by adjuvant chemotherapy is currently the standard treatment regimen (Mutch, 2002). Surgical cytoreduction of metastatic disease is crucial in disease management. Post-surgery, patients who have a significant risk of recurrence are usually treated with a combination therapy of taxanes and platinumbased drugs (DeVita et al., 1997). While a majority of the patients respond initially to treatment, most relapse and, unfortunately, in the relapsed population there is only a 2025% five year survival rate for advanced stage EOC (Ozols, 2005). There are two measures of treatment efficiency - overall survival and progression-free survival (PFS). Recent studies have shown that chemotherapy has been able to improve PFS statistics and overall survival in recurrent cases, and therefore, chemotherapeutic regimens have been used as a benchmark to compare new therapies in Phase Ill trials (Ozols, 2005). 23 Recurrent cases, however, usually develop resistance to chemotherapy, and the patient's response to the agent decreases with each incidence of relapse after the development of resistance. Despite the efforts over several years to develop improved therapeutics, clinical responses have been short-lived, and have only led to marginal improvements in the treatment of platinum-resistant EOC (Agarwal and Kaye, 2003; Bookman et al., 2003). Given this therapy landscape, better understanding of the origins of the disease would be invaluable in developing better therapies. The view that EOC is an endocrine-related disease has been supported by several observations. The increase in incidence of EOC with the use of Hormone Replacement Therapy (HRT) or with the onset of climacterium and the decrease in incidence in women whose ovulations were suppressed by pregnancy or oral contraceptive use support the involvement of hormone levels in disease etiology (Argento et al., 2008; Borini and Rebellato, 2008; Lukanova and Kaaks, 2005; Riman et al., 2004). The primary hormone involved in the growth and differentiation in normal ovaries is estrogen. This hormone plays a key role in dictating the physiology of ovarian cells and is also implicated in the development and progression of ovarian carcinoma, but the mechanism of estrogen action is unclear. The effectors of estrogen action in cells are estrogen receptors c and p (ERax and ERp). ERa and ERp are members of the family of nuclear hormone receptors (also known as steroid receptors) and their transcriptional activity is mediated by ligand binding and subsequent binding to Estrogen Response Elements (ERE) in target sequences (Pearce and Jordan, 2004). Studies suggest that ERP, which is the principal form of ER found in normal OSE, exerts a protective effect against carcinogenesis whereas ERa, which is the predominant receptor present in ovarian cancers, promotes tumor progression. As tumors progress to malignancy, ERp expression decreases, resulting in ERa becoming the increasingly predominant form 24 found in malignant ovarian tumors (Bardin et al., 2004; Kuiper et al., 1997; Lazennec, 2006; Lindgren et al., 2004; O'Donnell et al., 2005; Rutherford et al., 2000). The significance of estrogen in the etiology of ovarian cancer is underscored by the efficacy of estrogen-directed treatments against ovarian cancer. Estrogen has been shown to induce proliferation in some ER(+) ovarian cancer cells, and antiestrogens have been shown to inhibit the growth of ovarian carcinoma (Langdon et al., 1993; Nash et al., 1989). Intervention with aromatase inhibitors has shown some clinical benefits for EOC patients as well (Krasner, 2007; Rao and Miller, 2005; Walker et al., 2007). As twothirds of EOCs express ERa at the time of diagnosis (Slotman and Rao, 1988), a promising route to target the endrocrine-dependent EOC is via ERa. Currently, the treatment options for primary and recurrent cancers are limited, and research focuses on developing new modalities of selectively targeting ovarian cancer and its metastases. Several molecular therapies have been developed based on our enhanced understanding of the disease. Therapies that target characteristics such as angiogenesis, survival, growth and metastases are now entering clinical trials (Burger, 2007; Hanahan and Weinberg, 2000). The work I have described in this dissertation focuses on the development of novel chemotherapeutics that selectively target cancer cells that express ERa and these drug candidates have been studied in the context of ovarian cancer. I shall first discuss in detail the role of ERa in ovarian cancer, and then illustrate how the genotoxicants have been designed to attack cancer cells by exploiting the role of ERa in the cell. Nuclear Hormone Receptors Nuclear Hormone Receptors (NHRs), also known as steroid receptors, are ligandinducible transcription factors that modulate gene expression by binding to specific recognition sequences in DNA. They regulate multiple biological functions during embryonic development and in adult tissues, and they are centrally involved in the physiological basis of many diseased states (Noy, 2007). Members of the NHR family include receptors for lipophilic substances such as steroid hormones (estrogen, progestins, androgens, glucocorticoids, mineralocorticoids, and vitamin D), retinoic acids, and thyroid hormones (Olefsky, 2001; Parker and Jensen, 1991). The primary structures of most of the NHRs have been elucidated by sequencing their cDNAs (Parker, 1991). Such molecular cloning and structure/function analyses have shown that all NHRs have common structural features, as shown in Figure 1.2A (adapted from Kumar and Thompson, 1999). As can be seen from the Figure, NHRs possess a C-terminal Ligand Binding Domain (LBD) and a highly conserved central DNA Binding Domain (DBD). The LBD is responsible for binding to the signaling ligand and allows for recruiting coactivator complexes and translocation to the nucleus. In their unliganded state, nuclear receptors can be found either in the cytoplasm or the nucleus, and they may be bound to co-repressors in the absence of the ligand (Olefsky, 2001). The DBD binds to specific Hormone Response Element (HRE) sequences that exist as half-sites separated by variable length nucleotide repeats. The receptors can exist as homo or heterodimers with each partner binding to specific HRE sequences. The DBD consists of two zinc fingers that set the NHR apart from all other DNA binding proteins (Mangelsdorf et al., 1995). The N-terminal and C-terminal domains are variable, as is the hinge region between the LBD and DBD. The transcriptional activity of the hormone receptor is mediated through Activation Function 1 and Activation Function 2 domains (AF-1 and AF-2) present on the C- and N- termini respectively. Ligand binding to the NHR-LBD results in a conformational change and leads to the activation of downstream transcriptional pathways. Several other molecules such as co-activators, co-repressors, co-integrators, chromatin remodeling proteins, and histone deacetylation proteins are recruited at the site of DNA binding in order to facilitate or inhibit the transcription of the target DNA into mRNA. These proteins regulate the interaction of the NHRs with their target genes involved in development, cell differentiation and organ physiology (Mangelsdorf et al., 1995). The following section addresses a specific type of NHR - the Estrogen Receptor (ER) - and its role in cellular function. Estrogen Receptor The estrogen receptor (ER), a member of the nuclear receptor superfamily, is an estrogen activated transcription factor that binds to short DNA sequences called estrogen response elements (EREs) located in the vicinity of estrogen regulated genes (Parker, 1991). There are two known isoforms of the ER, ERa and ERp, and these proteins are encoded by separate genes. The ERa isoform (66 KDa) was the first to be cloned in the 1980s from MCF-7 cells (Green et al., 1986; Greene et al., 1986; Walter et al., 1985). The P isoform (54 KDa) was cloned in the 1990s from rat prostate and ovary (Kuiper et al., 1996). The distribution of these isoforms varies from tissue to tissue, and each isoform may also localize to specific cellular subtypes within a tissue. Several studies show that ERa mRNA is predominant in the uterus, liver, mammary gland, testis, pituitary, kidney, heart and skeletal muscle, whereas ERp transcripts are significantly expressed in the ovary and the prostate (Bardin et al., 2004; Brandenberger et al., 1998; Brandenberger et al., 1999; Couse et al., 1997; Lazennec, 2006). The relative level of the two isoforms in a particular tissue type likely determines the impact of estrogen in that tissue, because the two ERs differ in their functional response to estrogen, as described below. ERa and ERp have similar structural modularity (Figure 1.2B, adapted from Ogawa et al., 1998), with a highly conserved DBD and a moderately conserved LBD. However, there are significant differences in other domains that are expected to affect the transcriptional activity of the receptors. Figure 1.2B shows the structural aspects of the two isoforms, and the percentage identity between ERa and ERp is also denoted. The A/B domain shows 30% identity, and also contains the N-terminal AF-1, a constitutive contributor to the transcriptional activity of the ERs. The activation domains within the two isoforms are not functionally equivalent - ERa displays AF-1 activity, whereas the ERP AF-1 is shown to be functionally inactive (Hall and McDonnell, 1999; Kuiper et al., 1998a; Kuiper et al., 1998b). The hinge region (D domain), which contains the first Nuclear Localization Signal (NLS) is not well conserved. Lastly, the E/F region displays 53% homology, and contains the LBD, a coregulator binding surface, a dimerization domain, a second NLS and AF-2, a ligand-dependent activation function (Pearce and Jordan, 2004). In spite of the 53% homology in the LBD, differences in hormone action arise due to differences in hormone-LBD interactions. ERa and ERP demonstrate similar binding affinities for estradiol (KD of 0.05 nM for ERa and 0.07 nM for ERP), but their binding affinities for other agonists, partial agonists and antagonists show subtle differences (Kuiper et al., 1997; Kuiper et al., 1998a). Variations in these interactions likely result in functional consequences for ERa and ERp. Although the DBDs of the isoforms are highly conserved, it has been observed that ERa has a higher affinity for DNA than ERp. This has been postulated to be due to the differences in the ability of the isoforms to dimerize (Cowley et al., 1997; Pettersson et al., 1997). The isoforms have been shown to interact with DNA as dimers; depending on the relative levels of the isoforms in cells, the receptor can form ERa homodimers, ERP homodimers, or ERa-ERp heterodimers. Depending on which one of the dimers is formed, there can exist three potential alternative pathways of estrogen signaling (Pettersson et al., 1997). It is likely that there are target genes that interact preferentially with either of the homodimers or the heterodimer, and this would implicate a larger regulatory potential for the ligand. This would also imply a mechanism by which the relative isoform levels modulate the target gene activity in tissues. The distinct transcriptional profiles of the two isoforms likely dictate the differential responses to agonists and antagonists in target cells and tissues. In the absence of the hormone, ER is associated with the nuclear compartment of the cell (Htun et al., 1999; King and Greene, 1984), indicating that the receptor contains an NLS that is constitutively active independent of ligand binding. In addition, reports from the laboratories of Stenoien and Htun have shown that there is a dramatic change in the pattern of nuclear distribution of the ER from reticular when unliganded to punctuate in the presence of either an agonist or an antagonist (Htun et al., 1999; Stenoien et al., 2000; Stenoien et al., 2001). However, recent evidence suggests that a portion of the ER population might also localize to the plasma membrane where it regulates protein kinase cascades in response to estrogen (Razandi et al., 2004; Xu et al., 2004). The supposed interactions between the plasma membrane bound liganded ERs and membrane-associated molecules such as ion channels, G-proteins, and growth factor 29 receptors lead to the activation of downstream cascades such as MAP Kinase and P13 Kinase (Fu and Simoncini, 2008). Unliganded ER in the nucleus is maintained in a high affinity hormone binding conformation by the Heat Shock Protein 90 (HSP90) chaperone complex (Fliss et al., 2000; Oxelmark et al., 2003; Segnitz and Gehring, 1995; Segnitz and Gehring, 1997). Upon ligand binding, the classical ER response pathway is activated - dissociation of HSPs from the ER, receptor dimerization and association with specific EREs to modulate transcription initiation from nearby promoters. EREs in several genes have been identified and most of them conform to a core consensus sequence, a thirteen nucleotide segment with ten nucleotides forming an inverted repeat (Driscoll et al., 1998). The perfect ERE was determined to have the consensus sequence 5'-GGTCANNNTGACC-3', where N represents any nucleotide (Driscoll et al., 1998; Klein-Hitpass et al., 1988). However, many other EREs with one or two deviations from the core sequence still displayed ER binding provided there were appropriate flanking sequences, as demonstrated by Driscoll et al. (1998). In addition to responding via the classical pathway, the ER can also act via by proteinprotein tethering interactions at AP-1 and Sp1 sites (Kushner et al., 2000b; Kushner et al., 2000a; Saville et al., 2000; Webb et al., 1999), which would obviate the need for strict adherence to the consensus ERE sequence. In cells transfected with ERa or ERP and an AP-1 or Sp1 reporter, ligand stimulation induced reporter activity to different extents, indicating that this non-classical pathway does get activated by estrogens and antiestrogens (Saville et al., 2000; Webb et al., 1995). ER also gets phosphorylated at various sites in AF1 and DBD by kinases and ligands, inducing a downstream cascade of growth regulatory pathways (Ali et al., 1993; Lahooti et al., 1994; Le et al., 1994). It is clear that cell specific regulation of the estrogenic response is achieved through various means - the ratio of ERa to ERp, pathways activated (classical or non-classical), ligandreceptor affinity and ER phosphorylation. The genes activated through estrogen response pathways include progesterone receptor (PR) (Osborne et al., 1980), pS2 (Jakowlew et al., 1984; Masiakowski et al., 1982), the early response proto-oncogene c-myc (Schuchard et al., 1993), cathepsin D (Morisset et al., 1986; Rochefort, 1990), HSP 27 (Moretti-Rojas et al., 1988), aldolase A, a-tubulin and glyceraldehyde-3-phosphate (May and Westley, 1988). Some of the above genes contain the classical palindromic ERE sequences upstream of their promoter region, whereas some others contain non-consensus ERE half-sites and GC-rich motifs, which might be sites where the non-classical activation of ER dominates (Pearce and Jordan, 2004). In addition to these genes, a number of estrogen-induced mRNAs have been identified whose function is yet unknown (May and Westley, 1995). ER and Ovarian Cancer Ovaries are the site of estrogen biosynthesis, and they are the main target tissue of estrogen action. Estrogens are essential to the healthy functioning of the female reproductive system. In addition to their functions related to reproductive activity, estrogens are also necessary for the differentiation and proliferation of healthy breast epithelium (Russo and Russo, 1998). The benefits of estrogen in the cardiovascular system and in promoting bone health have been well-documented (Huang and Kaley, 2004). In fact, estrogen deficiency has been strongly correlated with age-dependent bone loss in both sexes (Riggs et al., 2002). Estrogens have also been shown to possess neurotrophic and neuroprotective effects, in additional to influencing memory mechanisms, cognition, and fine motor skills (Maggi et al., 2004; Morissette et al., 2008). The predominant intra-cellular estrogen is 17p-estradiol. Estradiol is mainly produced from follicular thecal cells and regulates several functional aspects of ovarian cells. ERa and ERp - the mediators of estrogen action - are expressed in normal OSE cells as well as in malignant cells. Although the etiological origins of EOC have not been fully identified, several studies point to the role of estrogen in promoting tumorigenesis. Estrogen has been shown to induce proliferation in many ER(+) cancer cells, and antiestrogens have been shown to inhibit the mitogenic effect of estrogen in those cells (Langdon et al., 1994; Nash et al., 1989). However, there appears to be a noteworthy variation in estrogen-mediated gene expression between ER(+) breast cancer and ovarian cancer cells. For instance, pS2 is not estrogen regulated in several ER(+) ovarian cancer cells, unlike in breast cancer cell lines. HER2, which is downregulated by estrogen in breast cancer cells, does not seem to respond similarly in ovarian cancer cells (Rochefort, 1998; Rochefort, 1999; Schaner et al., 2003). The study of the levels of effectors of estrogen action - ERa and ERp - is crucial in order to understand how estrogen regulation is carried out in ovarian cancer cells. The levels of the receptor isoforms in normal and malignant ovaries have been studied extensively, but many of the studies yield conflicting results (Lau et al., 1999; Reviewed in Pearce and Jordan, 2004). However, a majority of the studies show that the ERa/ERp ratio has been upregulated in ovarian cancer compared to the normal OSE cells (Cunat et al., 2004; Pujol et al., 1998). This interesting result supports a scenario where ERa becomes the dominant player in ovarian cancer, and ERp plays a protective role presumably by decreasing growth stimulation of cells or by inhibiting ERa mediated proliferation. It is 32 possible that cells that are ERa dominant have a selective growth advantage and proceed towards malignancy. Consequently, in the context of therapy, the isoforms will have to be targeted using different approaches. My dissertation is focused on probing the mechanism of novel antitumor agents that have been developed in the Essigmann laboratory to target the ER. Since ERx expression seems to be dominant in EOC (compared to ERP), the mechanistic studies described subsequently in this dissertation are focused on the ERa isoform. Henceforth, any references to the ER will implicitly mean ERa, unless specified otherwise. Rational Design of Novel Genotoxicants that Target ER(+) Tumors There is evidence from numerous studies to suggest that EOC growth could actually be estrogen sensitive, and this finding holds much promise for ER as a potential target for therapeutic development (Cunat et al., 2004; Ho, 2003; Masood et al., 1989; Nash et al., 1989; Rose et al., 1990). We have synthesized novel genotoxicants that leverage the presence -of ER in tumor cells to- subject selectively ER(+) cells to the consequences of interacting with the bifunctional antitumor agents (Mitra et al., 2002; Rink et al., 1996). The intellectual origins of this synthesis have been based on the lessons learned from studying the mechanisms of action of drugs such as cisplatin (Figure 1.1) in order to develop better, more selective anticancer agents. One of the cisplatin mechanisms suggests that the combination of a DNA damaging functionality with a ligand for a tumorspecific protein could provide a novel way to improve the therapeutic index of antitumor agents. The hypothesized mechanisms of action of this class of drugs are shown in Figure 1.3. Research has shown that DNA lesions formed in cells by certain carcinogens and drugs have the capability to attract transcription factors, often displaying very tight binding (Donahue et al., 1990; MacLeod et al., 1995). Such tight binding can potentially give rise to one or more of the two scenarios: (i) the interaction inhibits access of DNA repair proteins to the lesion, leading to adduct persistence and toxicity (Brown et al., 1993; McA'Nulty et al., 1996; McA'Nulty and Lippard, 1996; Treiber et al., 1994), or (ii) the interaction leads to decreased cellular access to the transcription factors sequestered by the adduct (hijacking hypothesis, explained in Figure 1.3), which can compromise cellular processes, eventually leading to cell death (Treiber et al., 1994). The genotoxicants synthesized in the Essigmann lab contain a DNA-alkylating aniline mustard moiety that is tethered to a ligand for the ER. The mechanism of action of this class of agents is shown in Figure 1.4. The aniline mustard moiety is based on FDA approved drugs such as chlorambucil and melphalan (Figure 1.1). The linker that connects the steroid moiety to the nitrogen mustard has been developed so as to permit the DNA adducts formed by the mustard moiety to present the steroid domain in such a way to enable efficient interaction with the ER. The drug candidates have also been optimized for ER binding, enhanced solubility and stability against esterases. The structural and the design aspects of the two compounds are described in the following sections. E2-7a-C6NC2-Mustard E2-7a combines a DNA damaging aniline mustard moiety with an estradiol moiety that serves as a ligand for the ER, as shown in Figure 1.4. The synthesis of the drug is described in detail in Mitra et al. (2002). The attachment between the two moieties was 34 made at the 7a position of estradiol. This choice was based on reports that large alkyl groups could be attached to the 7a position without loss of ER affinity (Bowler et al., 1989; Bucourt et al., 1978; DaSilva and van Lier, 1990). I shall describe the background studies that have been done with E2-7a in Chapter 2, in order to put in perspective my studies with the toxicant in an ovarian cancer background. In the next two chapters, I shall report some of the very interesting findings we observed with our study on E2-7a in an ovarian cancer background. Phenylindole-C6NC2-Mustard Chronologically, this compound is first of the two programmable genotoxicants that was developed in the Essigmann lab (Rink et al., 1996). The drug consists of a 2-phenylindole (2PI) moiety that serves as a ligand for the ER as shown in Figure 1.4. Derivatives of 2PI have been shown to interact well with the ER in competition assays with the natural ligand estradiol (von Angerer et al., 1984). The 2PI moiety in this compound is linked to an aniline mustard whose chemistry and ability to alkylate DNA are well understood (Lawley, 1966). The synthesis and preliminary toxicological studies of this drug have been described in the publication by Rink et al. (1996). My studies on this compound are in the ovarian cancer scenario, and the experiments described in this dissertation that focus on the 2PI compounds have been done in collaboration with Dr. Pei-Sze Ng (Essigmann laboratory). E2-7a and 2PI compounds have been discussed in separate chapters because they seem to differ in their mechanism of action - Chapters 2 and 3 will focus on my work on E2-7a, its biological consequences followed by mechanistic understanding of the effects of the genotoxicant. Chapters 4 will focus on the study of the phenylindole compound. For each of the drugs, I shall discuss effects on cultured cells (ability to inhibit cell growth, biological pathways activated and DNA adduct formation), discuss whether the mechanism of action of the compounds aligns with our hypothesis, and assess their potential as chemotherapeutic agents. I shall predominantly focus on the work that I have done, but in order to make a complete story, I will occasionally discuss the work done by my colleagues and reference accordingly. -Chapter 1- Structures of relevant chemotherapeutic agents CL ,,NH 3 CPtN. Cisplatin C1 NH3 H 3 C-N~ Mechlorethamine I Melphalan Chlorambucil 0 HO" ' Figure 1.1: Structures of chemotherapeutic agents of interest Structural organization of Nuclear Hormone Receptors C A/B D E/F LBD DBD Hinge Figure 1.2A: Structure and functional domains of Nuclear Hormone Receptors. A through F are the functional domains. The C domain isthe DBD, D is the hinge region, and E/F contains the LBD. Structure/ Function modularity of the ER isoforms C A/B ERax 595aa 302 263 180 1 D ERp E/F -I 1 149 214 248 530aa Figure 1.2B: Structure and functional domains of ERa (595 aa) and ERp (530aa). A through F are the functional domains of the ER. The numbers inthe figure refer to the amino acid numbers. The percentages refer to the percentage identity between Functions AF1 and AF2 sequences are inthe A/B domain and E/F domain respectively. The C domain is DNA Binding Domain, D is the hinge region, and E/F contains the LBD. -Chapter 1- Possible mechanisms of action of bifunctional toxicants e ...... =s.. e.......e..eem...s.a...s.e.......esse........e......es...es...e.e.g...a e40...... e.........e......em.a...se.. ee.... Nuclear A. Undamaged Cell DNA RepairG? Enzyme Enzyme Protein Promoteri enome 11111 B.Adduct Shielc ed From Repair DNA Repair . m....... . E[4* Expression of Essential Gene 4c,.C Adduct Persists C.Adduct Hijacl TranscriptiornFactor ICellD eath U 1111 ...- Diminished_ Expression Figure 1.3: DNA adducts act as decoy binding sites for nuclear proteins. Possible cellular outcomes are outlined. A. Cell with nuclear protein interacting with DNA B. Repair Shielding - Drug-DNA adducts (lollipop) attract the nuclear protein and are shielded from DNA repair enzymes, leading to adduct persistence and cell death. C. Hijacking Model - The adducts titrate the nuclear protein away from its normal site of binding, resulting in reduced expression of a critical gene. -Chapter 1- A novel agent designed to inhibit repair processes in a cancer cell Protein Recognition Domain DNA Damaging Moie Drug-DNA Adduct Repair Enzyme NORMAL CELL CANCER CELL ER Adduct is repaired by repair enzyme and is rendered nontoxic Adduct is "shielded" by ER, and the complex is toxic to cell Figure 1.4: The compounds we designed consist of a protein recognition domain linked to a DNA warhead (top left). This compound can covalently modify DNA within a cell (top right). In non-cancerous cells, the DNA adduct is removed by the repair machinery (lower left). Incancerous cells, a tumor specific protein is aberrantly expressed, as is the case with the ER inmany breast cancers. An interaction between the ER and the protein recognition domain of the designed molecules could inhibit the repair of the adduct, render the complex toxic to the cancer cell and cause cell death (lower right). -Chapter 1- Structures of bifunctional genotoxicants that target the ER 2PI-C6NC2-mustard HO HN HNHO A.C1 0 E2-7a -C6NC2-mustard Figure 1.5: Structures of 2PI (top) and E2-7a (bottom).The bifunctional toxicants have a steroid moiety that serves as a ligand for the ER linked to an aromatic mustard. The aromatic mustard iscapable of DNA alkylation. The linker has a carbamate group that provides a relatively rigid connection between the moieties and makes the compound resistant to hydrolytic enzymes. The secondary amino group inthe linker increases water solubility. Reference List 1. Ali S, Metzger D, Bornert JM, Chambon P (1993) Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J 12:1153-1160 2. American Cancer Society (2008) Cancer Facts and Figures. 3. Arai M, Utsunomiya J, Miki Y (2004) Familial breast and ovarian cancers. Int J Clin Oncol 9:270-282 4. Argento M, Hoffman P, Gauchez AS (2008) Ovarian cancer detection and treatment: current situation and future prospects. Anticancer Res 28:3135-3138 5. Armitage P, Doll (1954) The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer 8:1-12 6. Bandera CA, Ye B, Mok SC (2003) New technologies for the identification of markers for early detection of ovarian cancer. Curr Opin Obstet Gynecol 15:51-55 7. Bardin A, Hoffmann P, Boulle N, Katsaros D, Vignon F, Pujol P, Lazennec G (2004) Involvement of estrogen receptor beta in ovarian carcinogenesis. Cancer Res 64:5861-5869 8. Bast RC, Jr., Klug TL, St JE, Jenison E, Niloff JM, Lazarus H, Berkowitz RS, Leavitt T, Griffiths CT, Parker L, Zurawski VR, Jr., Knapp RC (1983) A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N EngI J Med 309:883-887 9. Bishop JM (1991) Molecular themes in oncogenesis. Cell 64:235-248 10. Bonadonna G, Brusamolino E, Valagussa P, Rossi A, Brugnatelli L, Brambilla C, De LM, Tancini G, Bajetta E, Musumeci R, Veronesi U (1976) Combination chemotherapy as an adjuvant treatment in operable breast cancer. N Engl J Med 294:405-410 11. Borini A, Rebellato E (2008) Focus on breast and ovarian cancer. Placenta 29 Suppl B:184-190 12. Bowler J, Lilley TJ, Pittam JD, Wakeling AE (1989) Novel steroidal pure antiestrogens. Steroids 54:71-99 13. Brandenberger AW, Lebovic DI, Tee MK, Ryan IP,Tseng JF, Jaffe RB, Taylor RN (1999) Oestrogen receptor (ER)-alpha and ER-beta isoforms in normal endometrial and endometriosis-derived stromal cells. Mol Hum Reprod 5:651-655 14. Brandenberger AW, Tee MK, Jaffe RB (1998) Estrogen receptor alpha (ER-alpha) and beta (ER-beta) mRNAs in normal ovary, ovarian serous cystadenocarcinoma and ovarian cancer cell lines: down-regulation of ER-beta in neoplastic tissues. J Clin Endocrinol Metab 83:1025-1028 15. Brown SJ, Kellett PJ, Lippard SJ (1993) lxrl, a yeast protein that binds to platinated DNA and confers sensitivity to cisplatin. Science 261:603-605 16. Bucourt R, Vignau M, Torelli V (1978) New biospecific adsorbents for the purification of estradiol receptor. J Biol Chem 253:8221-8228 17. Burger RA (2007) Experience with bevacizumab in the management of epithelial ovarian cancer. J Clin Oncol 25:2902-2908 18. Chabner BA, Longo DL (2009) Cancer Chemotherapy and Biotherapy 19. Chabner BA, Roberts TG, Jr. (2005) Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer 5:65-72 20. Chatterjee M, Mohapatra S, lonan A, Bawa G, li-Fehmi R, Wang X, Nowak J,Ye B, Nahhas FA, Lu K, Witkin SS, Fishman D, Munkarah A, Morris R, Levin NK, Shirley NN, Tromp G,Abrams J, Draghici S, Tainsky MA (2006) Diagnostic markers of ovarian cancer by high-throughput antigen cloning and detection on arrays. Cancer Res 66:1181-1190 21. Clark (2006) Molecular targeted drugs and growth factor receptor inhibitors. In: Chabner BA, Longo DL (eds) 22. Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS (1997) Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology 138:4613-4621 23. Cowley SM, Hoare S, Mosselman S, Parker MG (1997) Estrogen receptors alpha and beta form heterodimers on DNA. J Biol Chem 272:19858-19862 24. Cunat S, Hoffmann P, Pujol P (2004) Estrogens and epithelial ovarian cancer. Gynecol Oncol 94:25-32 25. DaSilva JN, van Lier JE (1990) Synthesis and structure-affinity of a series of 7 alpha-undecylestradiol derivatives: a potential vector for therapy and imaging of estrogen-receptor-positive cancers. J Med Chem 33:430-434 26. DeVita VT, Hellman S, Rosenberg SA (1997) Princples and Practice of Oncology, 5 edn 27. Donahue BA, Augot M, Bellon SF, Treiber DK, Toney JH, Lippard SJ, Essigmann JM (1990) Characterization of a DNA damage-recognition protein from mammalian cells that binds specifically to intrastrand d(GpG) and d(ApG) DNA adducts of the anticancer drug cisplatin. Biochemistry 29:5872-5880 28. Driscoll MD, Sathya G,Muyan M, Klinge CM, Hilf R, Bambara RA (1998) Sequence requirements for estrogen receptor binding to estrogen response elements. J Biol Chem 273:29321-29330 29. Fliss AE, Benzeno S, Rao J, Caplan AJ (2000) Control of estrogen receptor ligand binding by Hsp90. J Steroid Biochem Mol Biol 72:223-230 30. Foulds (1954) The experimental study of tumor progression: a review. Cancer Res 14:327-339 31. Fu XD, Simoncini T (2008) Extra-nuclear signaling of estrogen receptors. IUBMB Life 60:502-510 32. Green S, Walter P, Greene G, Krust A, Goffin C, Jensen E, Scrace G,Waterfield M, Chambon P (1986) Cloning of the human oestrogen receptor cDNA. J Steroid Biochem 24:77-83 33. Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J (1986) Sequence and expression of human estrogen receptor complementary DNA. Science 231:11501154 34. Greenlee RT, Murray T, Bolden S, Wingo PA (2000) Cancer statistics, 2000. CA Cancer J Clin 50:7-33 35. Hall JM, McDonnell DP (1999) The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140:5566-5578 36. Hanahan D,Weinberg RA (2000) The hallmarks of cancer. Cell 100:57-70 37. Ho SM (2003) Estrogen, progesterone and epithelial ovarian cancer. Reprod Biol Endocrinol 1:73 38. Htun H, Holth LT, Walker D, Davie JR, Hager GL (1999) Direct visualization of the human estrogen receptor alpha reveals a role for ligand in the nuclear distribution of the receptor. Mol Biol Cell 10:471-486 39. Huang A, Kaley G (2004) Gender-specific regulation of cardiovascular function: estrogen as key player. Microcirculation 11:9-38 40. Hurley LH (2002) DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer 2:188-200 41. Jacobs I, Bast RC, Jr. (1989) The CA 125 tumour-associated antigen: a review of the literature. Hum Reprod 4:1-12 42. Jaffe N, Frei E, Ill, Traggis D, Bishop Y (1974) Adjuvant methotrexate and citrovorum-factor treatment of osteogenic sarcoma. N Engl J Med 291:994-997 43. Jakowlew SB, Breathnach R, Jeltsch JM, Masiakowski P, Chambon P (1984) Sequence of the pS2 mRNA induced by estrogen in the human breast cancer cell line MCF-7. Nucleic Acids Res 12:2861-2878 44. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ (2008) Cancer statistics, 2008. CA Cancer J Clin 58:71-96 45. King WJ, Greene GL (1984) Monoclonal antibodies localize oestrogen receptor in the nuclei of target cells. Nature 307:745-747 46. Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato AC (1988) A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res 16:647-663 47. Krasner C (2007) Aromatase inhibitors in gynecologic cancers. J Steroid Biochem Mol Biol 106:76-80 48. Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138:863-870 49. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA (1996) Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A 93:5925-5930 50. Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der BB, Gustafsson JA (1998a) Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 139:4252-4263 51. Kuiper GG, Shughrue PJ, Merchenthaler 1,Gustafsson JA (1998b) The estrogen receptor beta subtype: a novel mediator of estrogen action in neuroendocrine systems. Front Neuroendocrinol 19:253-286 52. Kushner PJ, Agard D, Feng WJ, Lopez G, Schiau A, Uht R,Webb P, Greene G (2000a) Oestrogen receptor function at classical and alternative response elements. Novartis Found Symp 230:20-26 53. Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P (2000b) Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol 74:311317 54. Lahooti H,White R, Danielian PS, Parker MG (1994) Characterization of liganddependent phosphorylation of the estrogen receptor. Mol Endocrinol 8:182-188 55. Langdon SP, Hirst GL, Miller EP, Hawkins RA, Tesdale AL, Smyth JF, Miller WR (1994) The regulation of growth and protein expression by estrogen in vitro: a study of 8 human ovarian carcinoma cell lines. J Steroid Biochem Mol Biol 50:131135 56. Langdon SP, Ritchie A, Young K, Crew AJ, Sweeting V, Bramley T, Hillier S, Hawkins RA, Tesdale AL, Smyth JF, . (1993) Contrasting effects of 17 betaestradiol on the growth of human ovarian carcinoma cells in vitro and in vivo. Int J Cancer 55:459-464 57. Lau KM, Mok SC, Ho SM (1999) Expression of human estrogen receptor-alpha and -beta, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci U S A 96:5722-5727 58. Lawley PD (1966) Effects of some chemical mutagens and carcinogens on nucleic acids. Prog Nucleic Acid Res Mol Biol 5:89-131 59. Lazennec G (2006) Estrogen receptor beta, a possible tumor suppressor involved in ovarian carcinogenesis. Cancer Lett 231:151-157 60. Le GP, Montano MM, Schodin DJ, Katzenellenbogen BS (1994) Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem 269:44584466 61. Li M, HERTZ R,BERGENSTAL DM (1958) Therapy of choriocarcinoma and related trophoblastic tumors with folic acid and purine antagonists. N Engl J Med 259:66-74 62. Lindgren PR, Cajander S, Backstrom T, Gustafsson JA, Makela S, Olofsson JI (2004) Estrogen and progesterone receptors in ovarian epithelial tumors. Mol Cell Endocrinol 221:97-104 63. Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, Lynch P, Burke W, Press N (2006) Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 296:15071517 ~jI 64. Lukanova A, Kaaks R (2005) Endogenous hormones and ovarian cancer: epidemiology and current hypotheses. Cancer Epidemiol Biomarkers Prev 14:98107 65. MacLeod MC, Powell KL, Tran N (1995) Binding of the transcription factor, Spl, to non-target sites in DNA modified by benzo[a]pyrene diol epoxide. Carcinogenesis 16:975-983 66. Maggi A, Ciana P, Belcredito S, Vegeto E (2004) Estrogens in the nervous system: mechanisms and nonreproductive functions. Annu Rev Physiol 66:291-313 67. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM (1995) The nuclear receptor superfamily: the second decade. Cell 83:835-839 68. Masiakowski P, Breathnach R, Bloch J,Gannon F, Krust A, Chambon P (1982) Cloning of cDNA sequences of hormone-regulated genes from the MCF-7 human breast cancer cell line. Nucleic Acids Res 10:7895-7903 69. Masood S, Heitmann J, Nuss RC, Benrubi GI (1989) Clinical correlation of hormone receptor status in epithelial ovarian cancer. Gynecol Oncol 34:57-60 70. May FE, Westley BR (1988) Identification and characterization of estrogenregulated RNAs in human breast cancer cells. J Biol Chem 263:12901-12908 71. May FE, Westley BR (1995) Estrogen regulated messenger RNAs in human breast cancer cells. Biomed Pharmacother 49:400-414 72. McA'Nulty MM, Lippard SJ (1996) The HMG-domain protein lxrl blocks excision repair of cisplatin-DNA adducts in yeast. Mutat Res 362:75-86 73. McA'Nulty MM, Whitehead JP, Lippard SJ (1996) Binding of Ixrl, a yeast HMGdomain protein, to cisplatin-DNA adducts in vitro and in vivo. Biochemistry 35:6089-6099 74. Merajver SD, Pham TM, Caduff RF, Chen M, Poy EL, Cooney KA, Weber BL, Collins FS, Johnston C, Frank TS (1995) Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nat Genet 9:439-443 75. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, . (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66-71 76. Mitra K, Marquis JC, Hillier SM, Rye PT, Zayas B, Lee AS, Essigmann JM, Croy RG (2002) A rationally designed genotoxin that selectively destroys estrogen receptor-positive breast cancer cells. J Am Chem Soc 124:1862-1863 77. Moretti-Rojas I, Fuqua SA, Montgomery RA, 111, McGuire WL (1988) A cDNA for the estradiol-regulated 24K protein: control of mRNA levels in MCF-7 cells. Breast Cancer Res Treat 11:155-163 78. Morisset M, Capony F, Rochefort H (1986) Processing and estrogen regulation of the 52-kilodalton protein inside MCF7 breast cancer cells. Endocrinology 119:2773-2782 79. Nash JD, Ozols RF, Smyth JF, Hamilton TC (1989) Estrogen and anti-estrogen effects on the growth of human epithelial ovarian cancer in vitro. Obstet Gynecol 73:1009-1016 80. Noy N (2007) Ligand specificity of nuclear hormone receptors: sifting through promiscuity. Biochemistry 46:13461-13467 81. O'Donnell AJ, Macleod KG, Burns DJ, Smyth JF, Langdon SP (2005) Estrogen receptor-alpha mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocr Relat Cancer 12:851-866 82. Olefsky JM (2001) Nuclear receptor minireview series. J Biol Chem 276:3686336864 83. Osborne CK, Yochmowitz MG, Knight WA, Ill, McGuire WL (1980) The value of estrogen and progesterone receptors in the treatment of breast cancer. Cancer 46:2884-2888 84. Oxelmark E, Knoblauch R, Arnal S, Su LF, Schapira M, Garabedian MJ (2003) Genetic dissection of p23, an Hsp90 cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J Biol Chem 278:36547-36555 85. Ozols RF (2005) Treatment goals in ovarian cancer. Int J Gynecol Cancer 15 Suppl 1:3-11 86. Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K,Wilbanks G, Nicosia S, Cantor A, Sutphen R (2005) BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104:2807-2816 52 87. Papac RJ (2001) Origins of cancer therapy. Yale J Biol Med 74:391-398 88. Parker MG (1991) Nuclear Hormone Receptors 89. Parker, Jensen EV (1991) Overview of the Nuclear Receptor Family. 90. Pearce ST, Jordan VC (2004) The biological role of estrogen receptors alpha and beta in cancer. Crit Rev Oncol Hematol 50:3-22 91. Pettersson K, Grandien K, Kuiper GG, Gustafsson JA (1997) Mouse estrogen receptor beta forms estrogen response element-binding heterodimers with estrogen receptor alpha. Mol Endocrinol 11:1486-1496 92. Pujol P, Rey JM, Nirde P, Roger P, Gastaldi M, Laffargue F, Rochefort H, Maudelonde T (1998) Differential expression of estrogen receptor-alpha and -beta messenger RNAs as a potential marker of ovarian carcinogenesis. Cancer Res 58:5367-5373 93. Rao GG, Miller DS (2005) Clinical applications of hormonal therapy in ovarian cancer. Curr Treat Options Oncol 6:97-102 94. Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER (2004) Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol 18:2854-2865 95. Renan MJ (1993) How many mutations are required for tumorigenesis? Implications from human cancer data. Mol Carcinog 7:139-146 96. Riggs BL, Khosla S, Melton LJ, Ill (2002) Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev 23:279-302 53 97. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Persson IR (2001) Risk factors for epithelial borderline ovarian tumors: results of a Swedish case-control study. Gynecol Oncol 83:575-585 98. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Persson IR (2002a) Risk factors for invasive epithelial ovarian cancer: results from a Swedish case-control study. Am J Epidemiol 156:363-373 99. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Weiderpass E, Persson IR (2002b) Hormone replacement therapy and the risk of invasive epithelial ovarian cancer in Swedish women. J Natl Cancer Inst 94:497504 100. Riman T, Dickman PW, Nilsson S, Nordlinder H,Magnusson CM, Persson IR (2004) Some life-style factors and the risk of invasive epithelial ovarian cancer in Swedish women. Eur J Epidemiol 19:1011-1019 101. Rink SM, Yarema KJ, Solomon MS, Paige LA, Tadayoni-Rebek BM, Essigmann JM, Croy RG (1996) Synthesis and biological activity of DNA damaging agents that form decoy binding sites for the estrogen receptor. Proc Natl Acad Sci U S A 93:15063-15068 102. Risch HA (2002) Hormone replacement therapy and the risk of ovarian cancer. Gynecol Oncol 86:115-117 103. Rochefort H (1990) Cathepsin D in breast cancer. Breast Cancer Res Treat 16:3- 13 104. Rochefort H (1998) [Estrogens, cathepsin D and metastasis in cancers of the breast and ovary: invasion or proliferation?]. C R Seances Soc Biol Fil 192:241-251 105. Rochefort H (1999) [Estrogen-induced genes in breast cancer, and their medical importance]. Bull Acad Natl Med 183:955-968 106. Rose PG, Reale FR, Longcope C, Hunter RE (1990) Prognostic significance of estrogen and progesterone receptors in epithelial ovarian cancer. Obstet Gynecol 76:258-263 107. Russo IH, Russo J (1998) Role of hormones in mammary cancer initiation and progression. J Mammary Gland Biol Neoplasia 3:49-61 108. Rutherford T, Brown WD, Sapi E, Aschkenazi S, Munoz A, Mor G (2000) Absence of estrogen receptor-beta expression in metastatic ovarian cancer. Obstet Gynecol 96:417-421 109. Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S (2000) Ligand-, cell-, and estrogen receptor subtype (alpha/beta)dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem 275:53795387 110. Schaner ME, Ross DT, Ciaravino G, Sorlie T, Troyanskaya 0, Diehn M, Wang YC, Duran GE, Sikic TL, Caldeira S, Skomedal H, Tu IP, Hernandez-Boussard T, Johnson SW, O'Dwyer PJ, Fero MJ, Kristensen GB, Borresen-Dale AL, Hastie T, Tibshirani R, van de RM, Teng NN, Longacre TA, Botstein D, Brown PO, Sikic BI (2003) Gene expression patterns in ovarian carcinomas. Mol Biol Cell 14:43764386 111. Schildkraut JM, Schwingl PJ, Bastos E, Evanoff A, Hughes C (1996) Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet Gynecol 88:554-559 112. Schuchard M, Landers JP, Sandhu NP, Spelsberg TC (1993) Steroid hormone regulation of nuclear proto-oncogenes. Endocr Rev 14:659-669 113. Segnitz B, Gehring U (1995) Subunit structure of the nonactivated human estrogen receptor. Proc Natl Acad Sci U S A 92:2179-2183 114. Segnitz B, Gehring U (1997) The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J Biol Chem 272:18694-18701 115. Slotman BJ, Rao BR (1988) Ovarian cancer (review). Etiology, diagnosis, prognosis, surgery, radiotherapy, chemotherapy and endocrine therapy. Anticancer Res 8:417-434 116. Stenoien DL, Mancini MG, Patel K, Allegretto EA, Smith CL, Mancini MA (2000) Subnuclear trafficking of estrogen receptor-alpha and steroid receptor coactivator-1. Mol Endocrinol 14:518-534 117. Stenoien DL, Patel K, Mancini MG, Dutertre M, Smith CL, O'Malley BW, Mancini MA (2001) FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat Cell Biol 3:15-23 118. Treiber DK, Zhai X, Jantzen HM, Essigmann JM (1994) Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor). Proc Natl Acad Sci U S A 91:5672-5676 119. von Angerer E, Prekajac J, Strohmeier J (1984) 2-Phenylindoles. Relationship between structure, estrogen receptor affinity, and mammary tumor inhibiting activity in the rat. J Med Chem 27:1439-1447 120. Walker G, MacLeod K, Williams AR, Cameron DA, Smyth JF, Langdon SP (2007) Estrogen-regulated gene expression predicts response to endocrine therapy in patients with ovarian cancer. Gynecol Oncol 106:461-468 121. Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, Staub A, Jensen E, Scrace G, Waterfield M, . (1985) Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci U S A 82:7889-7893 122. Webb P, Lopez GN, Uht RM, Kushner PJ (1995) Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol 9:443-456 123. Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, Kushner PJ (1999) The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol 13:1672-1685 124. Xu Y, Traystman RJ, Hurn PD, Wang MM (2004) Membrane restraint of estrogen receptor alpha enhances estrogen-dependent nuclear localization and genomic function. Mol Endocrinol 18:86-96 125. Yap TA, Carden CP, Kaye SB (2009) Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer 9:167-181 126. Zheng L, Annab LA, Afshari CA, Lee WH, Boyer TG (2001) BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci U S A 98:9587-9592 Chapter 2 E2-7a - Biological Consequences in Ovarian Cancer Cells: Cytotoxicity, DNA Damage and Cell Death Introduction Estradiol-C6NC2-N,N-bis-(2-chloroethyl)-aniline (E2-7a) is a programmable therapeutic that has been designed to target estrogen receptor positive (ER(+)) tumors such as breast and ovarian cancers. The intellectual origins of E2-7a design stem from the mechanistic understanding of cisplatin action. Cisplatin (cis-diamminedichloroplatinum(II) or cis-DDP) has shown spectacular efficacy in the treatment of testicular and ovarian malignancies and offers great opportunities for probing new mechanisms of inducing toxicity. The success of cisplatin used alone or in combination with drugs such as paclitaxel, docetaxel, gemcitabine and doxorubicin in treating testicular tumors and delaying the recurrence of ovarian cancers has interested researchers world-wide, and has stimulated extensive research to understand cellular response to the drug (Boulikas and Vougiouka, 2004). It is known that cisplatin reacts with DNA (Bruhn et al., 1990), and induces apoptosis in treated cells. There is evidence, both from our laboratory and researchers elsewhere, that DNA-lesions formed by cisplatin have the ability to attract cellular proteins such as High Mobility Group (HMG) box proteins and bind them with very high affinity (Donahue et al., 1990; Pil and Lippard, 1992). In addition, proteins such as hMSH2 that bind to the cisplatin-DNA lesions are selectively expressed in organs in which cisplatin is clinically effective as an anticancer agent (Wilson et al., 1995). There are two routes by which such DNA-drug-protein complexes can exert their effect. First, the tight binding might "shield" the adduct and prevent DNA repair enzymes from accessing the lesion (Huang et al., 1994). The alternative mechanism arises from the observation that the tight binding proteins are usually critical transcription factors and by "hijacking" these factors away from their sites of action, the adduct causes reduction in expression levels of genes essential for survival. We have tailored the idea of a dual mode of action - 1) alkylation and 2) ability to interact with tumor-specific proteins - to synthesize a series of alkylating compounds that also contain a tumor-specific protein recognition moiety. The evolution of a newer generation of drugs based on the cisplatin mechanism of action will be invaluable in order to achieve efficacy similar to or better than cisplatin, yet circumvent the toxic side effects and development of resistance associated with cisplatin therapy. Cisplatin shows high efficacy in the treatment of testicular tumors, but concerns regarding its clinically significant toxicity and its side effects such as nephrotoxicity and central and peripheral neurotoxicity still remain (Reed, 2006). In addition, several cancers including ovarian cancers develop resistance after the first treatment regimen of cisplatin, and this consequently leads to very high relapse rates (Reed, 2006). Combination therapies of platinum compounds with agents such as taxol, doxorubicin, and cyclophosphamide have their own dose-limiting side effects as well (DeVita et al., 1997). In an effort to overcome the above mentioned undesirable off-target effects of cisplatin, we have extrapolated the extensive knowledge available on the drug to synthesize a series of "re-programmed" genotoxicants that are designed to target tumors more selectively and are therefore more efficacious than cisplatin. E2-7a: Synthesis Strategy Our strategy was to select a DNA damaging agent that could also attract a tumorspecific protein. This approach led us to tether an alkylating aniline mustard moiety to the ER, which functioned as the protein recognition domain of our compound of interest E2-7a. E2-7a was synthesized as per the steps outlined by Mitra et al. (2002). The linker group connecting estradiol and the aniline mustard was attached at the 7a position of estradiol based on reports that large groups could be attached to that position without affecting binding affinity to the ER (Bowler et al., 1989; Bucourt et al., 1978; DaSilva and van Lier, 1990). The aniline mustard N,N-bis-(2-chloroethyl)-aniline was chosen over an alkyl nitrogen mustard such as mechlorethamine, because the aniline mustard reacts relatively slowly (it has lower toxicity compared to the more reactive alkyl mustards) and the DNA adducts formed by the aniline mustard are readily repaired (Essigmann et al., 2001b; Pratt et al., 1994). Therefore, using the aniline mustard as the alkylating moiety would potentially reduce the toxicity of the compound in non-target tissues. Sharma and others (2004) from the Essigmann lab synthesized a series of variants of the parent E27a compound in order to fine-tune the biophysical properties of the compound. This series of compounds contained the same alkylation moiety and estradiol moiety as did E2-7a, but differed in their linker functional groups and lengths. These compounds were tested for their binding affinity to the ER, their ability to covalently modify DNA and their (differential) toxicity towards ER(+) MCF-7 and ER(-) MDA-MB-231 breast cancer cell lines. The systematic effort to optimize linker parameters by Sharma et al. helped us select the compound best suited for our further studies - the parent E2-7aX compound displayed the optimal balance between solubility, DNA reactivity, ER binding and differential toxicity between ER(+) and ER(-) cell lines. The linker portion of E2-7a contains the carbamate and amine functional groups and is stable to hydrolytic cleavage and enhances the solubility of the drug. E2-7a: Background Studies The Essigmann lab has conducted extensive research on E2-7at to characterize its biophysical and toxicological properties. The findings from these studies have been summarized in this section, some of which can also be found in the publication by Mitra et al. (2002). The history of E2-7a described here leads directly to the research goals discussed in this dissertation. The aniline mustard moiety of E2-7a is predicted to react with DNA in vitro to alkylate predominantly at the N7 position of guanine because of the highly nucleophilic nature of the nitrogen atom on the guanine. To determine the identity of DNA adducts formed by E2-7a, DNA treated with drug was subjected to reversed-phase HPLC and analyzed by full scan electrospray ionization mass spectrometry (ESI-MS) (Mitra et al., 2002). A prominent ion signal was observed at a mass consistent with the structure where one arm of the mustard is attached to guanine, and the other arm contains a -OH in place of the -Cl atom, similar to what has been reported with chlorambucil (Haapala et al., 2001). This study provides evidence that E2-7a alkylates DNA in vitro. The next key step in validating our hypothesis was to determine whether E2-7a can interact with the ER, and whether interaction existed between DNA adducted to E2-7a and the ER. The following results have also been discussed in Mitra et al. (2002). Competitive binding studies showed that E2-7a had a favorable affinity for the rabbit uterine ER protein, binding the receptor with 46% of the affinity of estradiol (Sharma et al., 2004). Gel shift experiments demonstrated that E2-7a-DNA adducts are also capable of forming a complex with ER-Ligand Binding Domain (ER-LBD). This complex was detected by the presence of a slowly migrating band in an Electrophoretic Mobility Shift Assay (EMSA). The binding specificity of the DNA adduct to ER-LBD was verified by adding increasing amounts of the competitor estradiol to the DNA adduct-ER-LBD complex. The slow migrating band corresponding to this complex disappeared, strongly suggesting that estradiol binding to the ER might be preventing the DNA adduct from interacting with the ER-LBD. The cytotoxic effect of E2-7a has been studied in ER(+) and ER(-) breast cancer cell lines (MCF-7 and MDA-MB-231 respectively), as described in Mitra et al.(2002). It was observed that the colony forming ability was much lower in MCF-7 cells than in the ER(-) MDA cell lines after treatment with the same dose of E2-7a. Chlorambucil was used as a control compound, because it contains the same DNA damaging moiety as E2-7a. Chlorambucil did not show a difference in toxicity profile between the ER(+) and ER(-) cells. The fact that ER status affected the toxicity of E2-7a, but not that of chlorambucil, is consistent with the role of the ER as an effector of selective toxicity of E2-7a. E2-7a shows reasonable toxicity in vivo as well. HeLa cells engineered to express the ER-LBD were employed to form xenografts tumors in mice. Shawn Hillier (Essigmann laboratory) showed that E2-7a effectively inhibited the growth of ER(+) HeLa xenografts by as much as 82%, and the treated mice showed no signs of organ specific toxicity, indicating that the drug dose was well- tolerated (Unpublished results). E2-7a in the Context of Ovarian Cancer Evidence from several randomized trials has now established combination regimens of paclitaxel with cisplatin as the front line therapy against advanced ovarian cancer (Agarwal and Kaye, 2003; Ozols et al., 2003), but despite several efforts at optimizing doses and durations of treatment, clinical responses remain short-lived and the development of resistance is a huge obstacle to be overcome (Agarwal and Kaye, 2003; Yap et al., 2009). Given the current therapy landscape in ovarian cancer, there is a compelling need to develop more targeted approaches in order to circumvent these issues. E2-7a seems to have tremendous potential to provide a targeted approach to treat ER(+) ovarian tumors, potentially reducing off-target side effects as well. This chapter focuses on the studies I have performed with E2-7a, assessing its cytotoxicity in ovarian cancer cells and examining the cellular response to E2-7a treatment. The biological consequences of E2-7a treatment have also been compared to those of cisplatin treatment. Two established epithelial ovarian cancer cell lines, OVCAR-3 (Hamilton et al., 1983) and SKOV-3 (Hua et al., 1995c) have been chosen in order to study the cytotoxicity of the drug. OVCAR-3 cells are derived from a progressive adenocarcinoma of the ovary and are resistant to clinically relevant concentrations of cisplatin, adriamycin, and melphalan (Hamilton et al., 1984). Both cultured cells and xenografts exhibit the ER (Hamilton et al., 1983). This cell line has been chosen because it is a suitable model system of a cisplatin-resistant ovarian cell population in which one can study the effects of alternative therapeutics. SKOV-3 cells are epithelial cells derived from adenocarcinoma of the ovary, and are resistant to several cytotoxic drugs including cisplatin, adriamycin and diphtheria toxin (Morimoto et al., 1991; Morimoto et al., 1993). This cell line was specifically chosen because the ER present in these cells retains its ligand binding ability, but seems to be deficient in the transcriptional regulation of certain downstream growth regulatory gene products (Hua et al., 1995b; Lau et al., 1999). As such, the cells do not respond to estrogens such as estradiol or antiestrogens such as ICI 164,384 (Hua et al., 1995a). This cell line serves as an appropriate model system to understand the interaction between ER and E2-7a without involving the confounding effects of the transcriptional activity of the receptor (these mechanistic studies will be described in detail in Chapter 3). This system helps address the mechanistic aspects of E2-7aX action and answer the question whether ER is involved in modulating E2-7a toxicity. OVCAR-3 and SKOV-3 cells were tested to determine their sensitivity to E2-7a and changes in cell cycle patterns were identified in these two cell lines. In addition, some important and interesting biomarkers of cellular responses in SKOV-3 cells were examined and E2-7c-treated cells were probed for evidence of DNA damage. The nitrogen mustard moiety of E2-7a is known to exert its cytotoxicity through reaction with DNA (Sunters et al., 1992). A majority of these reaction products are monoadducts, which result from a single alkylation event involving one of the chloroethyl arms of the mustard reacting with DNA through the formation of an aziridinium ion intermediate (Figure 2.1) (Balcome et al., 2004; Kohn et al., 1987; Kohn, 1996; Kundu et al., 1994). As shown in Figure 2.1, persistence of the monoadducts could result in the formation of interstrand or intrastrand DNA crosslinks (Hartley et al., 1991; Sunters et al., 1992), and previous studies have shown that crosslinks are more cytotoxic than monoadducts (O'Connor and Kohn, 1990; Sunters et al., 1992). DNA damage analysis in treated cells will be valuable in understanding the basis of E2-7a toxicity. DNA damage triggers many rescue/repair responses in cells, including activation of key damage signal transducers, priming of the repair machinery and phosphorylation of checkpoint proteins. The cellular response to the drugs E2-7a, cisplatin and chlorambucil have been examined along these lines. In addition to the studying the cytotoxicity in ovarian cancer cells as outlined above, I employed a system of two cell lines (HeLa and XPA) which differ in their DNA repair capability in order to estimate if DNA damage repair is involved in sensitizing the cells to E2-7a. These cell lines demonstrate a significant difference in their ability to perform Pr'-. nucleotide excision repair (NER). HeLa cells used in this study are derived from adenocarcinoma of the cervix and are proficient in NER (He and Ingles, 1997); these cells were used as a control to evaluate the contribution of DNA repair to toxicity. The XPA-deficient cells used are SV-40 transformed fibroblasts that were obtained from a donor carrying a mutation in Xeroderma Pigmentosum Complementation Group A (XPA) protein (Satokata et al., 1992). XPA protein is involved in excision repair of DNA and mutation in this gene renders the cells extremely sensitive to certain kinds of DNA damaging agents (such as UV radiation) that are dependent on NER for repair; the cells are also sensitive to certain alkylating agents such as cisplatin and melphalan (Araujo et al., 2001; Koberle et al., 2006; States et al., 1998). This sensitivity can be used to evaluate whether DNA damage and its repair are involved in the toxicity towards a given agent. Another interesting aspect of E2-7a toxicity that is discussed in this chapter is the mode of cell death activated by the genotoxicant in SKOV-3 cells. Cell death occurs when the damage in the cell exceeds the threshold for its tolerance, and there are several modes by which a cell might possibly die upon treatment with DNA damaging agents (Brown and Attardi, 2005). The form of cell death induced by a particular agent depends on the cell type, the type of DNA damage to which the cell is exposed, and the dose of the agent used (Abend, 2003). Cytotoxic agents such as chlorambucil, cisplatin and melphalan have been known to induce cell death through apoptosis, a programmed cell death pathway (Gibb et al., 1997; Roy et al., 2000). The mode of cell death is crucial to the action of a chemotherapeutic agent, and in several cases, resistance to apoptosis in the tumor population leads to drug-resistant tumors (Brown and Attardi, 2005). In this context, targeted therapies that overcome apoptosis-resistance in tumor cells by modulating cell death pathways might be extremely effective in treating drug-resistant tumors. Previous studies with E2-7a in breast cancer cells have shown that these cells exhibited typical signs of apoptosis in their response to the drug, such as caspase cleavage and DNA fragmentation (John Marquis, Essigmann laboratory, unpublished results). We were interested in testing whether SKOV-3 cells demonstrated a similar response to E27a; the endpoint of E2-7a treatment in ovarian cancer cells would give us handle on the chemotherapeutic potential of the compound. The studies probing cell death pathways activated in SKOV-3 cells are described in detail in this chapter. Materials and Methods Chemicals: The bifunctional compound E2-7a used in these studies was synthesized in our laboratory. Details of the synthetic steps and characterization of the compound have been described in a previous publication (Mitra et al., 2002). Cisplatin (Cis), chlorambucil (CHL), acridine orange (AO), propidium iodide (PI), ethidium bromide (EB) and monodansylcadaverine (MDC) were obtained from Sigma-Aldrich. Cell Culture: SKOV-3 and NIH:OVCAR-3 (referred to as OVCAR-3) cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD). Fetal Bovine Serum (FBS) was obtained from Thermo Fisher Scientific (Logan, UT). OVCAR-3 cells were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) containing 2.5 mg/mL glucose, 2 mM GlutaMax, 1 mM sodium pyruvate and 100 mM HEPES buffer, supplemented with 0.01 mg/mL bovine insulin (Invitrogen) and 20% FBS. SKOV-3 cells were maintained in McCoy's 5a Medium (ATCC), supplemented with 10% FBS. HeLa cells were obtained from ATCC and XPA cells (GM04312) were from Coriell Institute for Medical Research. HeLa and XPA cells were grown in Minimum Essential Medium (MEM) from Invitrogen supplemented with 5 mM L-Glutamine and 10% FBS. All cell lines were grown in a humidified 5%CO2/air atmosphere at 37*C. For growth inhibition assays, cells were seeded in 6-well plates at 5x10 4 cells per well. Forty eight hours later, the test compound dissolved in DMSO was added to the medium. Cells were detached using trypLE Express (Invitrogen) at times indicated and the number of cells in control and drug-treated wells were determined using a Beckman Coulter Counter. The percent growth inhibition was calculated as the ratio of cell number in treated to that in control wells multiplied by 100. Relative sensitivities were assessed by the concentration of compound required to inhibit growth by 50% (GI50 ; G150 is the concentration of test drug that causes 50% growth inhibition in treated cells compared to the untreated control cells). G150 was calculated using exponential regression analysis of growth inhibition data. For clonogenic assays, cells were seeded at 103 per well in 6-well plates and allowed to attach for 24 hours. Cells were then treated with desired drug doses for 24 hours, and the medium was replaced with fresh growth medium. Colonies were allowed to form for 4-7 days. Colonies were washed with PBS and were fixed with methanol/acetic acid and stained with crystal violet. Colonies of 50 or more cells were counted and results were expressed as percentage of colony formation in untreated cells. Immunoblot Analysis: Whole cell extracts were prepared cell lysis using RIPA buffer (Santacruz) (1XTBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1 mM sodium orthovanadate, 1 mM NaF, 50 mM Tris-HCI (pH 7.4) and protease inhibitor cocktail (Sigma-Aldrich)). Protein concentrations were determined using Bio-Rad Bradford reagent with Bovine Serum Albumin (BSA) standard. Equal amounts of protein were loaded and separated on pre-cast 4-12% SDS-polyacrylamide gradient gels (Invitrogen) in MOPS buffer system and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Membranes were blocked with 5% milk in Tris-Buffered Saline - Tween20 (0.1% Tween20, 10 mM Tris (pH 7.4), 150 mM NaCl) and incubated with primary antibodies overnight at 40C. The antibodies against phospho-cdc2 (rabbit polyclonal), phosphoBRCA1 (rabbit polyclonal), caspase-3 (rabbit polyclonal), caspase-9 (rabbit polyclonal) and PARP (rabbit polyclonal) were obtained from Cell Signaling Technology (Beverly, MA). Actin antibody (C-11, rabbit polyclonal) was obtained from Santa Cruz Biotechnology. Bcl-2 antibody (mouse monoclonal) was obtained from BD Pharmingen (San Jose, CA). Anti-mouse and anti-rabbit secondary antibodies conjugated to horseradish peroxidase were obtained from Cell Signaling Technology. Membranes were incubated with secondary antibodies for 1-2 hours, and antibody complexes formed secondary antibodies were visualized using Enhanced Chemiluminescence reagents (PicoWest, Pierce, Rockford, IL). Cell-Cycle Analysis: Cells were treated with test compounds for the times indicated. Both adherent and nonadherent cells were pooled, washed with cold PBS and fixed in 100% ethanol overnight at 40C. Ethanol-fixed cells were washed with 1% FBS in PBS, resuspended at 106 cells per mL, and stained with 10 pg/mL Pl. Stained cells were then incubated with 0.5 pg/mL RNase A (DNase free; Roche Diagnostics) for 30 minutes at 370C. DNA content was determined using a Becton-Dickinson flow cytometer (BD FACScan). Cell cycle data was analyzed using the MODFIT-LT 2.0 program to estimate the percentage of cells in G1, S and G2/M phases of the cell cycle. Single-Cell Alkaline Gel Electrophoresis (Comet assay): Cells were treated with test compounds for indicated times. Alkaline comet assay was performed as outlined in the Trevigen comet assay protocol using the reagents provided in the Trevigen kit (Trevigen, Gaithersburg, MD). Following treatment, cells were trypsinized, resuspended in PBS at 105 cells/mL and irradiated on ice using a GammaCell 40 Irradiator with a dose of 10 Gy. Irradiated cells were suspended in liquid Low Melting Agarose (LMA) at 370C and plated onto a CometSlide (Trevigen). After the agar solidified, cells were incubated in lysis solution (2.5 M NaCl, 100 mM EDTA, 10mM Tris base (pH 10), 1%Sodium Lauryl Sarcosine, 0.01% Triton X-100) for an hour at 40C, followed by incubation with 100 pL of 1 mg/mL Proteinase K (Roche Diagnostics) for an hour at room temperature. Slides were then treated for 30 minutes with alkaline solution (0.3 M NaOH, 1 mM EDTA, pH >13), followed by electrophoresis under alkaline conditions (0.3 M NaOH, 1 mM EDTA). Slides were then dried, stained with SYBR Green dye, and photographed using a Nikon Eclipse epifluorescence microscope. Images were analyzed using the Komet 4.2 Single Cell Gel Electrophoresis software (Kinetic Imaging Limited, U.K.). A total of 100 cells per sample were analyzed, 50 per duplicate slide. The Olive Tail Moment (OTM), defined as the product of the tail length and the fraction of total DNA in the tail, was calculated for each cell by the software. OTM incorporates a measure of both the smallest detectable size of migrating DNA (reflected in the comet tail length) and the number of relaxed or broken pieces which is represented by the intensity of DNA in the tail (Olive et al., 1990; Olive and Banath, 2006). Comet data were represented using boxplots plotted using SSC-Stat, a statistical add-in for Excel. Boxplot was used to give a graphical representation of the heterogeneity of the comet data. The box contains the middle 50% of the data; the upper boundary of the box indicates the 7 5 th percentile of the data, and the lower boundary the 2 5 th percentile. The range of the middle two quartiles is called the interquartile range. The line in the box is the median. The ends of the "whiskers" on the boxplot extend to a maximum of 1.5 times the interquartile range. The average OTM values are also indicated on the boxplot. Percentage decrease in the average OTM was calculated for each treatment relative to the untreated irradiated control, and significance values were calculated using the MannWhitney statistic (P < 0.001 was considered significant). Autophagy detection and staining: Cells were grown in 4-well chamber slides at 105 per well and treated with various drugs. After treatment, the chambers were washed with PBS. To detect acidic vesicular organelles, drug-treated cells were stained with 4 pg/mL acridine orange for 15 min. Chambers were also co-stained with 4 pg/mL ethidium bromide to detect dying/ dead cells. Samples were observed under a Nikon Eclipse epifluorescence microscope. Visualization of autolysosomes: Following treatment with drug, autolysosomes were labeled with monodansylcadaverine (MDC) by incubating cells grown in chamber slides with 50pM MDC in PBS at 370C for 15 min. After incubation, cells were washed three times with PBS and immediately analyzed by fluorescence microscopy for live cell staining. Results E2-7a inhibits the growth of ovarian cancer cells in culture. SKOV-3 and OVCAR-3 cells were treated with a range of doses of E2-7a, cisplatin and chlorambucil for 24 hours and 36 hours. The results are shown in Figures 2.2 and 2.3, and the G150 values are summarized in Table 2.1. In SKOV-3 cells, E2-7a was found to be more toxic than cisplatin at both the durations of treatment. After 24 hours of continuous exposure, E2-7a was over four times as toxic as cisplatin to SKOV-3 cells, as can be seen from the G150 values. After 36 hours of drug treatment, the difference in toxicity between the two drugs was less pronounced - the G150 of cisplatin was 16 pM compared to 6.5 ptM for E2-7ax. In OVCAR-3 cells, a similar trend was observed - E2-7c was more toxic than cisplatin at both the durations of treatment. Roughly twice the dose of cisplatin was required to produce a growth inhibition of 50% compared to E2-7x at both 24 and 36 hours of treatment. The extent of growth inhibition observed in cells treated with E2-7a was similar in both the cell lines. These data show that E2-7a is comparable to, if not more effective than, cisplatin in inducing toxicity in ovarian cancer cells. It is very interesting to note that in both cell lines, E2-7a was more toxic than chlorambucil, despite the structural similarity of the molecules. This trend was observed at both durations of treatment. E2-7a induces S-phase arrest in SKOV-3 and OVCAR-3 cells. SKOV-3 cells were treated with E2-7c, chlorambucil and cisplatin at 10 pM dose for 24 and 36 hours. Figure 2.4 shows the cell cycle patterns after treatment with the test drugs. In SKOV-3 cells, E2-7a induced a potent S-phase arrest, which became more pronounced with increased duration of treatment. Compared to untreated cells which had 21 % cells in S-phase, E27a treated cells had over two thirds of the population arrested in S-phase, which increased to 76% with 36 hours treatment. Under these conditions, cisplatin treatment at 10 pM did not demonstrate a significant arrest pattern. However, cisplatin at certain other doses and times of exposure was able to induce S-phase arrest in these cells (data not shown). Chlorambucil induced a prominent G2/M arrest; over 48% cells accumulated in G2/M phase after the 24 hour treatment, compared to untreated population which had 12% cells in G2/M. In OVCAR-3 cells, as seen in Figure 2.5, a similar trend as in SKOV-3 cells was observed. E2-7a induced a strong arrest in S-phase, and the treated cells had almost twice as many cells in S-phase compared to the untreated cells, both at 24 hours and 36 hours after treatment. Cisplatin induced a potent G1 arrest in these cells: -84% and 93% of the treated cells were arrested in G1 phase after the 24 hour and the 36 hour treatment respectively, compared to -64% G1 population in the untreated control. S-phase arrest persists even after drug is removed. We wanted to test if the differential toxicity between E2-7a and chlorambucil was due to the persistence of the replication block (S-phase arrest) in E2-7a treated SKOV-3 cells. SKOV-3 cells were treated with 10piM E2-7a or chlorambucil for 36 hours, and were then allowed to recover in fresh medium for 28 hours. Treatment with chlorambucil for 36 hours caused a G2/M arrest, as can be seen in Figure 2.6. On removal of the drug, the G2/M arrest disappeared over the course of 28 hours, and the cells returned to a distribution similar to untreated cells after 28 hours of recovery in drug-free medium. Figure 2.7 shows the cell cycle patterns of cells exposed to E2-7a. E2-7a treatment for 36 hours resulted in an S-phase arrest as can be seen in the second panel on the top row. Once the drug was removed and cells grown in fresh medium, the arrest persisted, and after 20 hours of recovery time, 100% of the cells seemed to be arrested in S-phase, indicating a persistent replication block. This response was in contrast to that of chlorambucil, where the G2/M block was effectively removed after drug removal. E2-7a induces phosphorylation of cdc2 and BRCAI. Various cell cycle checkpoint proteins were studied to examine whether they were activated to signal cell cycle arrest. Immunoblotting experiments (Figure 2.8) showed that the phosphorylation of cdc-2 (also known as CDK1) at Tyrosine 15 (Tyrl5) was seen with chlorambucil, cisplatin and E2-7a treatment. It must be noted here that the antibody that was used for cdc-2 also recognizes another cell cycle checkpoint regulator CDK2 that is closely related to cdc-2. Cdc-2 activation is an essential checkpoint protein that regulates entry into cell cycle. Phosphorylation of cdc-2 at Tyr1 5 results in inhibiting the progression of cell cycle predominantly from G2 to M, resulting in cell cycle arrest. The inhibitory phosphorylation of CDK2 results in G1 arrest and S-phase arrest. The results that are observed are consistent with the roles of cdc-2 and CDK2 in inducing cell cycle arrest, and all the three compounds show activation of checkpoint proteins. E2-7a, in particular, induced S-phase arrest, and this finding is consistent with the phosphorylation of cdc-2 marker. BRCA1 phosphorylation at Serine 1524 (Ser 524) was also tested with the three drugs. BRCA1 is a DNA damage recognition protein and is also actuvely involved in cell cycle regulation. It is phosphorylated at Ser 1524 as a response to DNA damage, and controls the S-phase checkpoint through CDK2 and G2/M checkpoint via cdc-2. Cisplatin was the only drug that showed the phosphorylation of BRCA1 at 24 hours, but with 36 hours of treatment, all the three drugs induced BRCA1 phosphorylation at Ser 524. E2-7a forms interstrand crosslinks in SKOV-3 cells. SKOV-3 cells treated with E2-7a were subjected to the modified comet assay (Olive and Banath, 1995; Olive and Banath, 2006) to determine the presence of interstrand crosslinks (ICLs). Drug treated or mock treated cells were irradiated with 10 Gy dose to induce random DNA strand breakage, followed by cell lysis and alkaline unwinding. The presence of ICLs retards the migration of the irradiated DNA during alkaline electrophoresis, resulting in a reduced Olive Tail Moment (OTM) compared to the untreated irradiated control. The results are shown in Figure 2.10 and Table 2.2. The percent decrease in OTM values as seen in Table 2.2 represents the extent of ICL formation. We observed that the cells responded to the positive control mechlorethamine (ME) by a significant decrease in OTM of -45% compared to untreated, indicating that ME was able to form ICLs in SKOV-3 cells. E2-7a treatment with various doses for six hours also showed a significant decrease in OTM compared to untreated cells. Doses of 0.5 pM, 1 pM and 2 pM resulted in ~ 20%, 25% and 14% decrease in OTM, respectively. There was no particular dose response observed in the "extent" of crosslink formation as quantified by the percent decrease in OTM. Also, E2-7ax did not induce ICL formation as much as ME did, possibly because of the difference in the kinetics of reaction of the two compounds. E2-7a is more toxic to NER deficient XPA cells compared to NER proficient HeLa cells. In order to examine whether the repair of DNA damage was a player in the toxicity of E2-7ax, a system of NER deficient and proficient cells (XPA and HeLa, respectively) were treated with test drugs, and the colony formation ability of the cells was studied after treatment. The results are shown in Figure 2.10. E2-7a was toxic to both HeLa and XPA cells, but it was clear that XPA cells were much more sensitive to the drug than HeLa cells as seen in Figure 2.10. As estimated from the G150 values, XPA cells were approximately six times more sensitive to E2-7a than HeLa cells. At doses above 0.5 pM, there were no surviving XPA colonies, whereas ~ 10% of HeLa cells formed colonies even at 1.25 and 2.5 pM. The two sets of cells were also treated with chlorambucil, and the results showed a modest differential in toxicity between XPA and HeLa cells. XPA cells were significantly more sensitive to E2-7aC than to chlorambucil; at 1.25 puM, about 10% of the colonies in chlorambucil treated cells survived, as opposed to zero survivors in the E2-7a treated cells. The differential in sensitivity between E2-7aX and chlorambucil in HeLa cells was not as pronounced as in the XPA population. These results suggest that NER is likely to be involved in the repair of E2-7a lesions that are produced in these cells. Cell death caused by E2-7a does not proceed through caspase-3 cleavage pathway in SKOV-3 cells; E2-7a upregulates level of BcI-2, and does not induce PARP cleavage. Since E2-7a showed promising cellular toxicity, the next step was to investigate whether SKOV-3 cells undergo apoptosis, as they do with several DNA damaging agents such as cisplatin, mechlorethamine and melphalan (Ahn et al., 2004; Chen et al., 1999; Gibb et al., 1997). SKOV-3 cells were immunoblotted for caspase-3 (the antibody also detects the cleaved form). Results are shown in 2.11. None of the drug treatments demonstrated the cleavage of caspase-3. With cisplatin, literature shows apoptosis induction at -80 pM dose in SKOV-3 cells, which probably explains why we did not see caspase cleavage at 20 pM dose of cisplatin (Gibb et al., 1997). At 20 gM dose of E2-7a, there were no adherent cells remaining on the culture dish after 48 hours of treatment, and it was surprising that the floating dead cells did not display caspase-3 cleavage. In order to examine whether earlier players in the caspase pathway were involved, caspase-9 cleavage was also examined - caspase-9 is the earliest caspase that initiates the process of apoptosis and is upstream of caspase-3. The results showed the same trend as with caspase-3. No cleavage product was detected with any of the drug treatments. Other participants in the classical apoptosis pathway were examined in SKOV-3 cells (Figure 2.11). Bcl-2 is an anti-apoptotic protein that prevents apoptosis in response to various stimuli, and downregulation of Bcl-2 levels upon treatment with drug usually facilitates apoptosis (Newton and Strasser, 1998). Surprisingly, E2-7a at 20 pM dose upregulated the level of Bcl-2 in these cells. Bcl-2 upregulation has been correlated with resistance to apoptosis in several systems (Igney and Krammer, 2002; Newton and Strasser, 1998). The increased level of Bcl-2 with E2-7a. treatment probably explains the observation that the classical signs of apoptosis were not observed in SKOV-3 cells, as indicated by the lack of caspase-3 cleavage. In addition, PARP cleavage was not detected with any of the treatments. PARP is a nuclear enzyme that is involved predominantly in apoptosis and in DNA repair. It is downstream of caspase-3 in the apoptosis pathway, and usually gets cleaved by caspase-3 during the process of apoptosis. We were interested in testing whether PARP cleavage occurred as a response to drug treatment. From Figure 2.11, it can be seen that PARP cleavage did not occur, indicating that the classical apoptosis pathway resulting in PARP cleavage leading to DNA fragmentation was not activated in these cells. Alternative pathways to cell death seemed to involved, which were investigated further. E2-7a induces cell death by autophagy. When E2-7a treated SKOV-3 cells were observed under the microscope, there were no visible signs of the classical apoptosis pathway, such as nuclear condensation or membrane blebbing, even at doses that induced over 70% cell death. Instead, several cytoplasmic granular bodies were observed. This result, in combination with the lack of activation of specific apoptotic markers, strongly indicated that non-apoptotic death mechanisms might be involved. Non-apoptotic programmed cell death is principally attributed to autophagy (Reggiori and Klionsky, 2002). The process of autophagy starts with the autophagosome formation, leading to the formation of autophagolysosomes (or autolysosomes) by the fusion of acidic lysosomes with autophagosomes (Hippert et al., 2006). It was hypothesized that the cytoplasmic granular bodies that were observed were acidic autophagosomes, and were part of the autophagosome-lysosome degradation pathway. Treated cells were stained with acridine orange (AO), which fluoresces orange in acidic vesicles, to visualize acidic autophagolysosomes (AO puncta have been used as a measure of autophagy (Paglin et al., 2001)). Cells are also co-treated with ethidium bromide (EB) to detect dead/ dying cells. EB is excluded by viable cells, but can enter cells containing compromised membranes, and stain the nuclei orange, and hence EB is a marker for cells that are in the process of dying. As seen in Figure 2.12 and Figure 2.13, the untreated cells had a background level of AO fluorescence, indicating the presence of lysosomal compartments. Untreated cells also excluded EB, and this was confirmed by the absence of orange EB-stained nuclei. Cisplatin treatments for 12 hours at 5 pM (Figure 2.12) and 24 hours at 2.5 pM (Figure 2.13) looked similar to the untreated cells. E2-7a treatment, on the other hand, markedly increased the number and size of the acidic compartments in the cell, indicating that autophagic process may be activated by E2-7ax. The second E2-7a panel in Figure 2.12 also showed several cells with orange nuclei, indicating cell death. It was also seen that the nuclei were still intact, and did not exhibit signs of fragmentation. Figure 2.13 represents the AO/EB staining at 2.5 pM dose of drug for 24h and this time point showed a similar trend as the 12h treatment. Very prominent orange punctuate staining in the cytoplasm was seen with E2-7a treatment. Cisplatin did not induce such staining even at doses as high as 10 pM (data not shown), but cellular nuclei did start fluorescing orange with EB staining of cisplatin treated cells, indicating that cell membrane did get compromised, but even at the 10 ptM dose, cisplatin treated cells did not demonstrate accumulation of acidic vesicles. Cells were also stained with MDC, which is a specific marker for late stage autolysosomes (Biederbick et al., 1995; Klionsky et al., 2008). MDC staining was performed in addition to AO staining to determine independently whether E2-7a triggered autophagy. Figure 2.14 shows the results of MDC staining. E2-7C at 2.5 pM dose for 24 hours induced intense punctuate fluorescent pattern with MDC in the cell, which is an indicator of autophagic pathway being activated. The untreated cells displayed an even background green staining with MDC, as did cells treated with 2.5 pM cisplatin for 24 hours. Taken together, the AO staining and the MDC staining suggest an autophagic mechanism of cell death with E2-7a treatment. Discussion Alkylating agents, in general, have two major impacts on living mammalian cells: cytotoxicity and delay in cell progression through the cell cycle (Meyn and Murray, 1986). In the first part of this chapter, the biological outcome of E2-7a action within the framework of toxicity and cell cycle effects has been studied in ovarian cancer cells. The remaining chapter deals with the characterization of cell death pathway in SKOV-3 cells in response to E2-7ax treatment. The cytotoxicity of E2-7a was tested in cisplatin-resistant ovarian cancer cell lines OVCAR-3 and SKOV-3 (Hamilton et al., 1983; Morimoto et al., 1993). OVCAR-3 and SKOV-3 cells were treated with the test compounds cisplatin, chlorambucil, and E2-7aX. In both the cell lines, E2-7a was found to be more toxic than cisplatin and chlorambucil. The G150 of cisplatin in SKOV-3 cells treated for 36 hours was 16 pM whereas that of E27a was found to be 6.5 pM. A similar trend was seen with OVCAR-3 cells, with E2-7a showing promising results in inducing cytotoxicity. E2-7a had almost the same G150 in OVCAR-3 cells as in SKOV-3 cells, and cisplatin was slightly more toxic to these cells than to SKOV-3 cells (G150 of 12 pM in OVCAR-3 vs. 16 pM in SKOV-3). The IC50 of cisplatin from literature studies in SKOV-3 cells is estimated to be ~ 6.7 gM and in OVCAR-3 cells to be 4.6 pM (Roberts et al., 2005). The difference in the numbers can be explained by the fact that this study was conducted in a 96-well plate format and the numbers were estimated using an MTT assay (which is a measure of metabolic activity of cells), as opposed to growth inhibition by cell counting. However, the trend that cisplatin is slightly more toxic to OVCAR-3 cells than to SKOV-3 cells as demonstrated by Roberts et al. (2005) is also observed in the experiments described in this chapter. When comparing the efficacies of the cisplatin and E2-7c, the fold difference in G150 values, if not the absolute values, might be more useful. At the higher duration of treatment, E2-7a is twice as effective as cisplatin in inhibiting cell growth by 50% in SKOV-3 and OVCAR-3 cells under the conditions of our experiments. This study gives us an initial indication of the therapeutic potential of our compound compared to cisplatin, currently the most established chemotherapeutic for ovarian cancer. Compared to chlorambucil, E2-7a is significantly more toxic to both cell lines. As seen from Table 2.1, the G150 of the control compound chlorambucil is almost four times as much as E2-7a in SKOV-3 cells. In OVCAR-3 cells, chlorambucil treatment results in a G150 of 17 piM, over twice as much as that of E2-7ca. Chlorambucil was chosen for comparison with E2-7a because it contains the same NN-bis-(2-chloroethyl)-aniline moiety and is expected to form the same types of DNA damage as does E2-7a. Therefore, the observation that E2-7a is more toxic that chlorambucil implies that the presence of the estradiol domain tethered to the DNA damaging warhead plays a major role in the additional toxicity seen with E2-7ax. E2-7a contains a nitrogen mustard moiety capable of forming monoadducts and crosslinks (Brookes and Lawley, 1961), and the repair of such kinds of DNA damage can lead to delay in cell cycle progression. The experiments described in this chapter show that E2-7a induced a strong S-phase arrest in both SKOV-3 and OVCAR-3 cells, suggesting that the drug might be creating a replication block in these cells. Such cell cycle pattern changes were not observed when the compound was used to treat MCF-7 and MBA-MB-231 breast cancer cells (John Marquis, unpublished data). This indicates that the S-phase arrest pattern is cell line specific, and possibly depends on the extent and kind of damage occurring in the cell line. Cisplatin induced a strong G1 arrest in OVCAR-3 cells, and this is in agreement with studies in OVCAR-3 cells treated with varying doses of cisplatin (Gibb et al., 1997). Checkpoint protein activation could help explain the observed block in cell cycle in SKOV-3 cells treated with various test compounds. E2-7a-induced S-phase arrest in SKOV-3 cells was also accompanied by the phosphorylation of cell cycle checkpoints proteins cdc-2 and BRCA1. Cell cycle entry into mitotic phase is initiated by the dephosphorylation of two inhibitory residues Tyr1 5 and Thr1 4 of cdc-2 in G2 phase. The dephosphorylation is carried out by the cell cycle regulatory protein cdc25c, followed by activation of cdc-2 - cyclinB1 complex (Zhou and Elledge, 2000). Phosphorylation of cdc-2 at Tyr15 inhibits the formation of the cdc-2 - cyclinB1 complex, and prevents the cell cycle from progressing through G2/M phase. Cdc-2 also seems to act as an S-phase checkpoint protein - elevated levels of phospho-cdc2 (Tyr1 5) are associated with S-phase arrest after DNA damage in several systems (Barth et al., 1996; Tyagi et al., 2005). Therefore, the phosphorylation of cdc-2 at Tyr1 5 seems to be a marker for both S-phase and G2/M phase arrest in cell cycle progression. Another factor to be considered is that the antibody to phospho-cdc-2 also recognizes endogenous levels of the phosphorylated checkpoint protein CDK2, which is actively involved in G1 to S progression and S to G2 progression. Cdc-2 phosphorylation was seen to occur with treatment with all the three drugs, even though the treatments arrested cells in different phases of the cell cycle. This can be explained in one or more ways - (i) the role of cdc-2 in both the G2/M and S-phase checkpoints can directly explain the results that we observed. If inhibitory phosphorylation of cdc-2 was detected, it could indicate either S-phase arrest or G2/M arrest. (ii) The cross-reactivity of the phospho-cdc-2 antibody with phospho-CDK2 implies that the result we obtain could either be cdc-2 or CDK2 activation. Since CDK2 phosphorylation is associated with S-phase arrest, this helps explain why we see the phosphorylated protein band with E2-7a treatment. These reasons help explain why chlorambucil and cisplatin are also able to show the presence of phosphorylated protein in the western blot. BRCA1 is found to be phosphorylated at Ser1524 in response to several DNA damaging agents (Deng, 2006), and the phosphorylation of BRCA1 is responsible for controlling the progression through S-phase. E2-7ca treatment resulted in Ser1524 phosphorylation of BRCA1, which could explain the S-phase arrest in SKOV-3 cells. BRCA1 is also found to be involved in controlling the G2/M checkpoint in response to ionizing radiationinduced DNA damage (Narod and Foulkes, 2004; Venkitaraman, 2004) and is found to be phosphorylated at Ser1524 by the damage sensor protein ATR, as shown by Deng et al. (2006). BRCA1, similar to cdc-2 is activated in both S phase and G2/M checkpoints, and this could explain why all the three drugs, which arrested cells in different phases of the cell cycle, showed phosphorylation of both these proteins. The phospho-proteins activated by E2-7a are consistent with the pattern of cell cycle arrest observed with the drug. Mechanistically, it is very interesting to note that E2-7a and chlorambucil arrest cells in different phases of the cell cycle. Furthermore, the S-phase arrest by E2-7a persisted even 28 hours after the drug was removed, whereas following chlorambucil treatment, cells recovered from the arrest within this time period. This gives rise to several possibilities, one being that the arrest induced by E2-7a is irreversible, which leads to cell death and accounts for the differential toxicity between E2-7a and chlorambucil. Given that the aniline mustard moiety is capable of forming crosslinks, the block to replication by E2-7a could be attributed to the presence of unrepaired interstrand crosslinks in the treated cells. This hypothesis was supported by the comet assay, which was able to detect a modest yet measurable level of cross-linking by E2-7ax. The nitrogen mustard mechlorethamine (ME) was used as a positive control. There is evidence from several reports that ME-induced crosslink formation can be easily detected using the comet assay (Hartley et al., 1991; Sunters et al., 1992). This was the case in SKOV-3 cells as well, and ME induced a significant decrease in OTM compared to the control cells, indicative of ICL formation. The three doses of E2-7ax were also able to show the presence of ICLs, although to a lesser extent than ME. The kinetics of reaction with DNA and crosslink formation of E2-7a are likely to be different from those of ME (Essigmann et al., 2001a; Pratt et al., 1994), which probably explains the lower level of crosslinks observed with E2-7a. Another finding with E2-7a treated cells was that there was no particular dose response in crosslink formation. Also, several other doses of E2-7a and longer treatment times were assessed with this technique but crosslinks could not be detected at these doses and times of treatment (data not shown). In fact, in many cases, the effect of single strand breaks likely confounded the results of the assay in such a way that the treated, irradiated cells had a higher tail moment than the untreated, irradiated cells. Inthis light, it is possible that the tail moments observed in Table 2.2 and Figure 2.9 represent the net result of crosslinks and single strand breaks, but the finding that these OTM values are significantly lesser than those of the irradiated cells provides strong evidence that crosslink formation does occur with E2-7a treatment (Further direct measurement experiments using Triple Quadrupole Mass Spectrometry are being conducted to detect and measure directly the levels of monoadducts and crosslinks in SKOV-3 cells treated with E2-7a). The next piece of the puzzle in the role of DNA damage in toxicity fell in place through indirect experimental evidence with HeLa and XPA cells - NER proficient and NER deficient cell lines, respectively. NER is one of the several repair pathways by means of which a cell defends itself from exogenous and endogenous DNA damage, and it has been shown to be the primary repair pathway involved in the removal of cyclobutane pyrimidine dimers and (6-4) photoproducts (Friedberg et al., 1995). NER has also been implicated in the repair of bulky adducts formed by alkylating agents such as cisplatin and melphalan (Furuta et al., 2002; Grant et al., 1998; Reardon et al., 1999; Wu et al., 2003). XPA protein is the major DNA damage recognition protein in the NER pathway, and plays a central role in the formation of the excision complex (States et al., 1998). XPA is required for the initial step of damage recognition and DNA binding (Asahina et al., 1994; Jones and Wood, 1993). XPA cells are deficient in this key damage recognition protein, and therefore are an ideal model system of NER deficient cells. XPA cells are known to be hypersensitive to the DNA damage induced by cisplatin (Stevens et al., 2008). We were interesting in testing whether DNA damage repair mediated by NER was involved in the toxicity of E2-7c. Shawn Hiller has performed a similar experiment in an in vitro system to determine the contribution of NER in repairing E2-7a - DNA adducts in HeLa whole cell extracts. It was found that the addition of XPA antibody to a reaction mixture containing Hela cell extracts and either E2-7a-damaged or UV-damaged pGEM plasmid inhibited the repair of both the plasmids, although by different extents. The role of XPA in the repair of the damage in the plasmid was confirmed when the addition of XPA protein to the reaction mix restored the repair of both E2-7a-damaged and UV-damaged plasmids. This experiment indicates that NER is one of the key pathways involved in the repair of E2-7a-induced DNA damage. In my cell culture experimental set-up, I have made a comparison between the sensitivities of HeLa cells and XPA cells; HeLa cells are NER proficient and contain ~ 200,000 XPA protein molecules per cell as compared to zero XPA protein molecules in the NER deficient cell line (Koberle et al., 2006), and it is, therefore, not surprising that XPA cells are hypersensitive to the E2-7ca. The clonogenic studies I have conducted do not rule out the contribution of other repair pathways - in fact, the experiment only indicates that NER is one of the means by which E2-7ax DNA damage might be repaired. Several other mechanisms are plausible, such as Base Excision Repair (BER) and depurination of E2-7a adducts to produce apurinic (or apyrimidinic) (AP) sites which can be then be processed by AP endonucleases (Friedberg et al., 1981; Lindahl, 1979). My experiments demonstrate that XPA cells are several folds more sensitive to E2-7a than HeLa cells, in spite of being only marginally more sensitive than HeLa cells to chlorambucil, and this result is strongly suggestive of a role for NER in the repair of DNA lesions associated with E2-7a. A unique aspect of E2-7a that has not been observed with any of our other rationally designed drugs was unfolded in the experiments described later in this chapter - E2-7a induced cell death by regulated cellular self-digestion or autophagy rather than by the classical apoptotic pathway. Based on morphology, cell death can be classified into apoptotic, necrotic and autophagic cell death (Zakeri et al., 1995). Apoptotic cell death is generally characterized by caspase activation, membrane blebbing and DNA fragmentation (Clarke, 2002). Necrosis is accompanied by cell swelling, plasma membrane damage, and organelle breakdown (Festjens et al., 2006). The hallmark of autophagy is the formation of autophagosomes, which eventually fuse with lysosomes to form autophagolysosomes (also called autolysosomes) (Levine and Klionsky, 2004). These cell death pathways are not as distinct as they were once though to be and it appears that they have several common players and closely associated regulatory pathways (Vandenabeele et al., 2006). In certain cases, mitochondria are also known to move into autophagic compartments. Thus, mitochondrial dysfunction might be a common pathway that overlaps between apoptosis and autophagy; in addition, in a given cell type, different kinds of cell death might co-exist (Zakeri et al., 1995). Many signals originally studied in the context of apoptosis are now known to induce autophagy, and the pathways have some common inhibitory signals as well (Botti et al., 2006; Gozuacik and Kimchi, 2007; Meijer and Codogno, 2006). Anti-apoptotic proteins such as Bcl-2 family members inhibit the autophagy protein Beclin-1, and consequently inhibit autophagy (Pattingre et al., 2005). Although traditionally considered a pro-survival mechanism, autophagy can lead to cell death in certain settings where it is uncontrollably upregulated (Mizushima et al., 2008). Also, in situations where tumor cells cannot die by apoptosis upon exposure to metabolic stress, autophagy might occur to prevent death by necrosis. The main protein involved in controlling autophagy is the kinase mammalian Target of Rapamycin (mTOR), which is downstream of the Class Ill phosphotidylinositol-3-kinase (P13K) pathways (Mizushima, 2007). Cytotoxic drugs trigger autophagy, especially in apoptosis-defective or apoptosisresistant cells, and excessive damage and metabolic stress can promote cell death. Some common autophagy inducers include rapamycin and tamoxifen; 3-methyladenine, wortmannin and bafilomycin are known to inhibit autophagy (Mizushima, 2007). E2-7ca induced cell death through autophagy; under the same conditions, cisplatin did not show any detectable signs of autophagy. The standard markers of apoptosis such as caspase cleavage, PARP cleavage and DNA fragmentation were not observed with E27ax treatment. Instead, we noticed the appearance of granular bodies in the cytoplasm after drug treatment, and this prompted us to research further into the characterization of cell death. Accumulation of acidic vesicles in treated cells suggested that the vesicles might represent autolysosomes, the lysosomal degradative vesicles characteristic of autophagy. Punctuate staining observed with autolysosomal marker MDC supports the theory that the granular bodies observed are autolysosomes. The finding that E2-7ax induces autophagy gives us a new channel to target apoptosisresistant tumors. The caveat that should be kept in mind, as with any other targeted therapy, is that there are still many unknowns in the autophagy regulation mechanism which might determine the ultimate outcome of treatment with an autophagy inducer in a specific system. Conclusions E2-7a has been shown to exhibit properties that make it a favorable drug candidate with the mechanisms of action that we had intended it to possess (Mitra et al., 2002). In this chapter, the cytotoxicity and downstream biological response to the compound has been investigated in ovarian cancer cells. OVCAR-3 and SKOV-3 cells showed similar toxicity to the compound, and overall, E2-7c was more toxic to the cell lines than the currently used chemotherapeutic, cisplatin, and was also more toxic to both cell lines than chlorambucil. E2-7a induced S-phase arrest through the phosphorylation of checkpoint proteins cdc-2 and BRCA-1, and the cells remained arrested in S-phase several hours after drug removal. The presence of interstrand crosslinks as detected by comet assay could explain the persistent S-phase arrest that -was seen with E2-7a. Interstrand crosslinks can cause a block in the replication, and if left unrepaired, lead to cell death. The presence of interstrand crosslinks can help understand the basis for increased cytotoxicity seen with E2-7a compared to the control compound chlorambucil. In order to address whether DNA damage repair through NER was a factor in E2-7a toxicity, XPA (NER deficient) and HeLa (NER proficient) cell lines were treated with E27a and the control compound chlorambucil. Although both HeLa and XPA cells were more sensitive to E2-7a than to chlorambucil, the differential toxicity was much steeper in the repair deficient XPA cells, indicating that the XPA cell line might not be able to repair the DNA damage caused by E2-7ca, which in turn results in the increased sensitivity to the compound. These data indicate that NER might be one of the routes by which the DNA damage caused by E2-7a is repaired. Another interesting observation that was made following drug treatment was the formation of cytoplasmic granular bodies that accumulated as early as eight hours after treatment. Staining with the dyes AO and MDC revealed that there was a profusion of lysosomal fusion bodies after drug treatment; the bodies might represent late stage autophagosomes, and it seemed that cell death proceeded through the autophagic route of cellular self-digestion. This pathway represents a novel target for E2-7a, and is especially advantageous in the ovarian cancer setting where several tumors are known to be apoptosis-resistant. In conclusion, evidence has been provided in support of the formation of interstrand crosslinks, which could lead to the replication block which manifests as an S-phase arrest. Unrepaired DNA damage can result in persistent S-phase arrest, leading to cell death. E2-7a has been shown to induce cell death through the non-classical programmed cell death pathway autophagy. We are optimistic about the efficacy of this compound in a clinical setting. -Chapter 2- G150 values of test compounds in SKOV-3 and OVCAR-3 cells G150 (in pM) . "... """... ""... "".. """'""......"* "'"""-"'"-'""""*"""*" -v --"--".".""""-"-"""-"""""" - --------------------- - ---------- Treatment SKOV-3 6h 24h OVCAR-3 24h 36h Chlorambucil 22 24 19 17 Cisplatin 19 16 18 12 E2-7a 6.4 9.0 6.5 .............. ;................. ........................ ...................................... 4.0 Table 2.1: G150 values of chlorambucil, cisplatin and E2-7a in ovarian cancer cells. These values correspond well with the trends observed in the cytotoxicity curves. -Chapter 2- Comet assay detects E2-7a induced crosslinks in SKOV-3 cells .............................................................................................................................................................................. Avg OTM Percent Decrease in OTM ME 5 pM 6.4 44 E2-7a 0.5 pM 9.2 20 E2-7a 1 pM 8.6 25 E2-7a 2 pM 9.9 14 Treatment Table 2.2: Comet assay results. OTM values of treated samples are shown. The percent decrease in OTM indicates extent of crosslink formation. E2-7a is shown to induce a modest amount of crosslink formation. -Chapter 2- Mechanism of Reaction of E2-7a with DNA CI CI RO + N R N CI~ (CI Aziridinium ion Bifunctional alkylating agent CI NH _ R N Guanine CI- Second aziridi nium ion formation N NNH 2 H N7 guanine adduct 0 0 Guanine IN>NH H 2 OH R= HO HO *0Hs HN HN 0 N N NH2 H G-G crosslink Figure 2.1: Activation of the aniline mustard of E2-7a occurs via aziridinium ion formation. Reaction with guanine results in an N7 guanine monoadduct; formation of a second aziridinium ion and reaction with guanine results in a G-G crosslink. E2-7a treatment is cytotoxic to SKOV-3 cells following 24 hours and 36 hours of treatment 1001 CHL -+-CCis c 80- -A- E2-7 0 24 hour treatment 60- D c40CU ,* 20 0 5 10 15 20 15 20 Dose in uM 100 80 36 hour treatment 60 40 20 0 5 10 Dose in uM Figure 2.2: Growth inhibition assay in SKOV-3 cells treated with cisplatin, chlorambucil and E2-7a. Cells were more sensitive to E2-7a than to chlorambucil or cisplatin. GI5 values are listed in Table 2.1 E2-7a treatment is cytotoxic to OVCAR-3 cells following 24 hours and 36 hours of treatment 100 CHL i ~80 80 +Cis -A E2-7c - 60aI) 24 hour treatment 21 24020- 0 5 10 15 20 15 20 Dose in uM 100 80 60 36 hour treatment 40 20 0 5 10 Dose in uM Figure 2.3: Growth inhibition assay in OVCAR-3 cells treated with cisplatin, chlorambucil and E2-7ax. Cells were more sensitive to E2-7ax than to chlorambucil or cisplatin. G150 values are provided inTable 2.1. E2-7a arrests SKOV-3 cells in S-phase UNTREATED 13 Cisplatin 24h 6 Cisplatin 36h 0 - 25 21 73 75 CHL36h CHL24h 23 G1 42 48 42 G2/M E2-7a 24h E2-7a 36h 0 0 24 33 66 76 Figure 2.4: Cell cycle patterns in SKOV-3 cells treated with 10 pM dose of cisplatin, chlorambucil and E2-7ax. E2-7ax induces a potent S-phase arrest, whereas chlorambucil induces a G2/M arrest. There was no striking arrest pattern observed with cisplatin under these conditions E2-7a arrests cells in S-phase in OVCAR-3 cells UNT 24h 14 65 Cisplatin 48h Cisplatin 24h 7 16 31 93 84 G2/M E2-7a 24h 8 40 E2-7a 48h 8 52 48 Figure 2.5: Cell cycle patterns in OVCAR-3 cells treated with 10 pM dose of cisplatin and E2-7ax. E2-7a induces a potent S-phase arrest, whereas cisplatin is shown to halt cells in G1. Chlorambucil treated cells recover from G2/M arrest once drug is removed UNTREATED Oh post treatment 15 43 21 45 12 8h post treatment 12h post treatment 32 G1 20 59 59 G2/M 20h post treatment 28h post treatment 17 17 64 Figure 2.6: Cells treated for 36 hours with 10 pM chlorambucil and allowed to recover for 28 hours in fresh medium. After 28h, cell cycle patterns are similar to those of untreated cells. C atr2- E2-7a treated cells do not recover from S-phase arrest after drug is removed UNTREATED Oh post treatment 2 15 21 57 8h post treatment 12h post treatment 15 35 41 45 G2/M 58 20h post treatment 28h post treatment 100 100 Figure 2.7: Cells treated for 36 hours with 10 ptM E2-7ca and allowed to recover for 28 hours in fresh medium. After 28 hours, treated cells are still accumulated in S-phase. -Chapter 2- E2-7a induces activation of checkpoint proteins in SKOV-3 cells 24 hour treatment 1 2 3 4 6 5 p-cdc2 Actin 24 hour treatment 1 2 3 Lane 4 5 6 Treatment Untreated 2 Cis 20M p-BRCA1 3 CHL 10M Actin 4 CHL 20pM 5 E2-7ax 1OpM 6 E2-7ca 20M 36 hour treatment 1 2 3 4 5 6 p-BRCA1 Actin Figure 2.8: Checkpoint proteins activated upon treatment with test drugs. Cdc-2 and BRCA1 are activated by E2-7a. 100 -Chapter 2- Comet assay indicates presence of interstrand crosslinks after E2-7a treatment OTM vs. Treatment (6h) 20 16 # Treatment E12 Untreated Irradiated 10 Gy ME 5 gM+Irr E2-7a 0.5 pM+Irr E2-7cc 1.0 pM+Irr E2-7o 2.0 pM+Irr 08 0 1 2 3 4 5 6 Figure 2.9: Boxplot showing distribution of tail moment in SKOV-3 cells. Numbers above the boxes represent average OTM value. Both ME and E2-7a treatments (starred) result ina significant decrease in OTM compared to the irradiated control sample (P<0.001) . The OTM values are tabulated inTable 2.2. 101 XPA cells are more sensitive to E2-7a than HeLa cells Clonogenic survival: XPA vs. HeLa 100 ----- ------------ 'A 10 Cl) U) Cu 4-' c U) 0~ HeLa -E2-7ca -0-U- 1 0.1 XPA -E2-7a - -A - HeLa-CHL - -+ - XPA-CHL 0.01 0 1.5 1 0.5 2 2.5 Dose in uM Treatment G1 50 in_M HeLa-E2-7a 0.55 XPA-E2-7ca 0.08 HeLa-CHL 1.32 XPA-CHL 0.44 Figure 2.10: Clonogenic survival response of XPA and HeLa cells to drugs after 24 hour treatment, followed by recovery and colony formation. XPA cells are much more sensitive to E2-7a than HeLa cells. Chlorambucil induces a marginal increased sensitivity in XPA compared to HeLa cells. 102 Apoptotic markers are not activated with E2-7a treatment 48 hour treatment 1 2 3 4 5 6 Caspase-3 Caspase-9 Actin 36 hour treatment 1 2 3 4 5 BcI-2 6 Lane Treatment 1 2 3 Untreated Cis 20pM CHL 10pM 4 CHL20pM 5 6 E2-7o 1OpM E2-7a 20pM PARP Actin Figure 2.11: Western blot showing apoptotic markers. Caspase-3, caspase-9 or PARP cleavage are not observed with E2-7ax treatment. Bcl-2 levels seem to be upregulated in response to E2-7a treatment. 103 -Chapter 2- AO/ EB staining of live SKOV-3 cells (treatment for 12 hours a t 5M dos Figure 2.12: Treatments as indicated. AO can be seen as punctuate orange staining inacidic compartments and green stain inthe nuclei of live cells. Orange nuclei represent staining by EB inmembrane compromised cells. 104 11 _ _ , __ __ - -1- -- .. .... .. ...... .. ....................... . ...... 1. _. ,,,...... .... -Chapter 2- AO/ EB staining of live SKOV-3 cells (treatment for 24 hours at 2.5 M dos TR I"I I",\TIIo Figure 2.13: Treatments as indicated. AO can be seen as punctuate orange staining inacidic compartments and green stain inthe nuclei of live cells. Orange nuclei represent staining by EB inmembrane compromised cells. 105 ................ ..... ........ ... ... ...... .. -Chapter 2- MDC staining of live SKOV-3 cells (treatment for 24 hours at 2.5 pM dose) GCI IA TVIN Figure 2.14: Treatments as indicated. MDC can be seen as punctuate green staining in E2-7ca treated cells and as a diffuse green stain inuntreated and cisplatin treated cells. 106 Reference List 1. Abend M (2003) Reasons to reconsider the significance of apoptosis for cancer therapy. Int J Radiat Biol 79:927-941 2. Agarwal R, Kaye SB (2003) Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer 3:502-516 3. Ahn HJ, Kim YS, Kim JU, Han SM, Shin JW, Yang HO (2004) Mechanism of taxolinduced apoptosis in human SKOV3 ovarian carcinoma cells. J Cell Biochem 91:1043-1052 4. Ali S, Metzger D, Bornert JM, Chambon P (1993) Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J 12:1153-1160 5. American Cancer Society (2008) Cancer Facts and Figures. 6. Arai M, Utsunomiya J, Miki Y (2004) Familial breast and ovarian cancers. Int J Clin Oncol 9:270-282 7. Araujo SJ, Nigg EA, Wood RD (2001) Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol Cell Biol 21:2281-2291 8. Argento M, Hoffman P, Gauchez AS (2008) Ovarian cancer detection and treatment: current situation and future prospects. Anticancer Res 28:3135-3138 107 9. Armitage P, Doll (1954) The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer 8:1-12 10. Asahina H, Kuraoka I, Shirakawa M, Morita EH, Miura N, Miyamoto I, Ohtsuka E, Okada Y, Tanaka K (1994) The XPA protein is a zinc metalloprotein with an ability to recognize various kinds of DNA damage. Mutat Res 315:229-237 11. Balcome S, Park S, Quirk Dorr DR, Hafner L, Phillips L, Tretyakova N (2004) Adenine-containing DNA-DNA cross-links of antitumor nitrogen mustards. Chem Res Toxicol 17:950-962 12. Bandera CA, Ye B, Mok SC (2003) New technologies for the identification of markers for early detection of ovarian cancer. Curr Opin Obstet Gynecol 15:51-55 13. Bardin A, Hoffmann P, Boulle N, Katsaros D, Vignon F, Pujol P, Lazennec G (2004) Involvement of estrogen receptor beta in ovarian carcinogenesis. Cancer Res 64:5861-5869 14. Barth H, Hoffmann 1,Klein S, Kaszkin M, Richards J, Kinzel V (1996) Role of cdc25-C phosphatase in the immediate G2 delay induced by the exogenous factors epidermal growth factor and phorbolester. J Cell Physiol 168:589-599 15. Bast RC, Jr., Klug TL, St JE, Jenison E, Niloff JM, Lazarus H, Berkowitz RS, Leavitt T, Griffiths CT, Parker L, Zurawski VR, Jr., Knapp RC (1983) A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N Engl J Med 309:883-887 16. Bernstein E, Caudy AA, Hammond SM, Hannon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409:363-366 108 17. Biederbick A, Kern HF, Elsasser HP (1995) Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 66:3-14 18. Bishop JM (1991) Molecular themes in oncogenesis. Cell 64:235-248 19. Biswas DK, Singh S, Shi Q, Pardee AB, Iglehart JD (2005) Crossroads of estrogen receptor and NF-kappaB signaling. Sci STKE 2005:e27 20. Bonadonna G, Brusamolino E, Valagussa P, Rossi A, Brugnatelli L, Brambilla C, De LM, Tancini G, Bajetta E, Musumeci R,Veronesi U (1976) Combination chemotherapy as an adjuvant treatment in operable breast cancer. N Engl J Med 294:405-410 21. Borini A, Rebellato E (2008) Focus on breast and ovarian cancer. Placenta 29 Suppl B:184-190 22. Botti J, Djavaheri-Mergny M, Pilatte Y, Codogno P (2006) Autophagy signaling and the cogwheels of cancer. Autophagy 2:67-73 23. Boulikas T, Vougiouka M (2004) Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review). Oncol Rep 11:559-595 24. Bowler J, Lilley TJ, Pittam JD, Wakeling AE (1989) Novel steroidal pure antiestrogens. Steroids 54:71-99 25. Brandenberger AW, Lebovic DI, Tee MK, Ryan IP,Tseng JF, Jaffe RB, Taylor RN (1999) Oestrogen receptor (ER)-alpha and ER-beta isoforms in normal endometrial and endometriosis-derived stromal cells. Mol Hum Reprod 5:651-655 109 26. Brandenberger AW, Tee MK, Jaffe RB (1998) Estrogen receptor alpha (ER-alpha) and beta (ER-beta) mRNAs in normal ovary, ovarian serous cystadenocarcinoma and ovarian cancer cell lines: down-regulation of ER-beta in neoplastic tissues. J Clin Endocrinol Metab 83:1025-1028 27. Brookes, Lawley PD (1961) The reaction of mono- and di-functional alkylating agents with nucleic acids. Biochem J 80:496-503 28. Brown JM, Attardi LD (2005) The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 5:231-237 29. Brown SJ, Kellett PJ, Lippard SJ (1993) lxrl, a yeast protein that binds to platinated DNA and confers sensitivity to cisplatin. Science 261:603-605 30. Bruhn, Toney JH, Lippard SJ (1990) Biological processing of DNA modified by platinum compounds., 38 edn 31. Bucourt R, Vignau M, Torelli V (1978) New biospecific adsorbents for the purification of estradiol receptor. J Biol Chem 253:8221-8228 32. Burger RA (2007) Experience with bevacizumab in the management of epithelial ovarian cancer. J Clin Oncol 25:2902-2908 33. Chabner BA, Longo DL (2009) Cancer Chemotherapy and Biotherapy 34. Chabner BA, Roberts TG, Jr. (2005) Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer 5:65-72 35. Chatterjee M, Mohapatra S, lonan A, Bawa G, li-Fehmi R, Wang X, Nowak J, Ye B, Nahhas FA, Lu K, Witkin SS, Fishman D, Munkarah A, Morris R, Levin NK, Shirley 110 112 NN, Tromp G,Abrams J, Draghici S, Tainsky MA (2006) Diagnostic markers of ovarian cancer by high-throughput antigen cloning and detection on arrays. Cancer Res 66:1181-1190 36. Chen Z, Fadiel A, Jia JF, Sakamoto H, Carbone R, Naftolin F (1999) Induction of apoptosis and suppression of clonogenicity of ovarian carcinoma cells with estrogen mustard. Cancer 85:2616-2622 37. Clark (2006) Molecular targeted drugs and growth factor receptor inhibitors. In: Chabner BA, Longo DL (eds) 38. Clarke PG (2002) Apoptosis: from morphological types of cell death to interacting pathways. Trends Pharmacol Sci 23:308-309 39. Clinton GM, Hua W (1997) Estrogen action in human ovarian cancer. Crit Rev Oncol Hematol 25:1-9 40. Coezy E, Borgna JL, Rochefort H (1982) Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res 42:317-323 41. Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS (1997) Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology 138:4613-4621 42. Cowley SM, Hoare S, Mosselman S, Parker MG (1997) Estrogen receptors alpha and beta form heterodimers on DNA. J Biol Chem 272:19858-19862 111 43. Cunat S, Hoffmann P, Pujol P (2004) Estrogens and epithelial ovarian cancer. Gynecol Oncol 94:25-32 44. DaSilva JN, van Lier JE (1990) Synthesis and structure-affinity of a series of 7 alpha-undecylestradiol derivatives: a potential vector for therapy and imaging of estrogen-receptor-positive cancers. J Med Chem 33:430-434 45. De BK, Vanden BW, Haegeman G (2006) Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene 25:6868-6886 46. Deng CX (2006) BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 34:1416-1426 47. DeVita VT, Hellman S, Rosenberg SA (1997) Princples and Practice of Oncology, 5 edn 48. Donahue BA, Augot M, Bellon SF, Treiber DK, Toney JH, Lippard SJ, Essigmann JM (1990) Characterization of a DNA damage-recognition protein from mammalian cells that binds specifically to intrastrand d(GpG) and d(ApG) DNA adducts of the anticancer drug cisplatin. Biochemistry 29:5872-5880 49. Dong L, Wang W, Wang F, Stoner M, Reed JC, Harigai M, Samudio I, Kladde MP, Vyhlidal C, Safe S (1999) Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J Biol Chem 274:3209932107 50. Driscoll MD, Sathya G, Muyan M, Klinge CM, Hilf R, Bambara RA (1998) Sequence requirements for estrogen receptor binding to estrogen response elements. J Biol Chem 273:29321-29330 112 '1K) K) 51. Elbashir SM, Harborth J, Lendeckel W, Yalcin A,Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498 52. Essigmann JM, Rink SM, Park HJ, Croy RG (2001b) Design of DNA damaging agents that hijack transcription factors and block DNA repair. Adv Exp Med Biol 500:301-313 53. Essigmann, Rink SM, Park HJ, Croy RG (2001a) Design of DNA damaging agents that hijack transcription factors and block DNA repair. In: Dansette (ed) Biological Reactive Intermediates VI, 6 edn 54. Festjens N, Vanden BT, Vandenabeele P (2006) Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 1757:1371-1387 55. Fliss AE, Benzeno S, Rao J, Caplan AJ (2000) Control of estrogen receptor ligand binding by Hsp90. J Steroid Biochem Mol Biol 72:223-230 56. Foulds (1954) The experimental study of tumor progression: a review. Cancer Res 14:327-339 57. Friedberg EC, Bonura T, Love JD, McMillan S, Radany EH, Schultz RA (1981) The repair of DNA damage: recent developments and new insights. J Supramol Struct Cell Biochem 16:91-103 58. Friedberg EC, Walker G, Siede W (1995) DNA repair and mutagenesis 113 59. Fu XD, Simoncini T (2008) Extra-nuclear signaling of estrogen receptors. IUBMB Life 60:502-510 60. Furuta T, Ueda T, Aune G, Sarasin A, Kraemer KH, Pommier Y (2002) Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res 62:4899-4902 61. Gibb RK, Taylor DD, Wan T, O'Connor DM, Doering DL, Gercel-Taylor C (1997) Apoptosis as a measure of chemosensitivity to cisplatin and taxol therapy in ovarian cancer cell lines. Gynecol Oncol 65:13-22 62. Gozuacik D, Kimchi A (2007) Autophagy and cell death. Curr Top Dev Biol 78:217245 63. Grant DF, Bessho T, Reardon JT (1998) Nucleotide excision repair of melphalan monoadducts. Cancer Res 58:5196-5200 64. Green S, Walter P, Greene G, Krust A, Goffin C, Jensen E, Scrace G, Waterfield M, Chambon P (1986) Cloning of the human oestrogen receptor cDNA. J Steroid Biochem 24:77-83 65. Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J (1986) Sequence and expression of human estrogen receptor complementary DNA. Science 231:11501154 66. Greenlee RT, Murray T, Bolden S, Wingo PA (2000) Cancer statistics, 2000. CA Cancer J Clin 50:7-33 114 67. Haapala E, Hakala K, Jokipelto E, Vilpo J, Hovinen J (2001) Reactions of N,Nbis(2-chloroethyl)-p-aminophenylbutyric acid (chlorambucil) with 2'deoxyguanosine. Chem Res Toxicol 14:988-995 68. Hall JM, McDonnell DP (1999) The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140:5566-5578 69. Hamilton TC, Young RC, McKoy WM, Grotzinger KR, Green JA, Chu EW, WhangPeng J, Rogan AM, Green WR, Ozols RF (1983) Characterization of a human ovarian carcinoma cell line (NIH:OVCAR-3) with androgen and estrogen receptors. Cancer Res 43:5379-5389 70. Hamilton TC, Young RC, Ozols RF (1984) Experimental model systems of ovarian cancer: applications to the design and evaluation of new treatment approaches. Semin Oncol 11:285-298 71. Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57-70 72. Hartley JA, Berardini MD, Souhami RL (1991) An agarose gel method for the determination of DNA interstrand crosslinking applicable to the measurement of the rate of total and "second-arm" crosslink reactions. Anal Biochem 193:131-134 73. He Z, Ingles CJ (1997) Isolation of human complexes proficient in nucleotide excision repair. Nucleic Acids Res 25:1136-1141 74. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8:193-204 115 75. Hippert MM, O'Toole PS, Thorburn A (2006) Autophagy in cancer: good, bad, or both? Cancer Res 66:9349-9351 76. Ho SM (2003) Estrogen, progesterone and epithelial ovarian cancer. Reprod Biol Endocrinol 1:73 77. Htun H, Holth LT, Walker D, Davie JR, Hager GL (1999) Direct visualization of the human estrogen receptor alpha reveals a role for ligand in the nuclear distribution of the receptor. Mol Biol Cell 10:471-486 78. Hua W, Christianson T, Rougeot C, Rochefort H,Clinton GM (1995a) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 79. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995b) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 80. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995c) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 81. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995d) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 82. Huang A, Kaley G (2004) Gender-specific regulation of cardiovascular function: estrogen as key player. Microcirculation 11:9-38 116 83. Huang JC, Zamble DB, Reardon JT, Lippard SJ, SancarA (1994) HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc Natl Acad Sci U S A 91:1039410398 84. Hurley LH (2002) DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer 2:188-200 85. Igney FH, Krammer PH (2002) Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2:277-288 86. Jacobs I, Bast RC, Jr. (1989) The CA 125 tumour-associated antigen: a review of the literature. Hum Reprod 4:1-12 87. Jaffe N, Frei E, Ill, Traggis D, Bishop Y (1974) Adjuvant methotrexate and citrovorum-factor treatment of osteogenic sarcoma. N Engl J Med 291:994-997 88. Jakowlew SB, Breathnach R,Jeltsch JM, Masiakowski P, Chambon P (1984) Sequence of the pS2 mRNA induced by estrogen in the human breast cancer cell line MCF-7. Nucleic Acids Res 12:2861-2878 89. Jemal A, Siegel R,Ward E, Hao Y, Xu J, Murray T, Thun MJ (2008) Cancer statistics, 2008. CA Cancer J Clin 58:71-96 90. Jones CJ, Wood RD (1993) Preferential binding of the xeroderma pigmentosum group A complementing protein to damaged DNA. Biochemistry 32:12096-12104 91. Kim DH, Rossi JJ (2007) Strategies for silencing human disease using RNA interference. Nat Rev Genet 8:173-184 117 92. King WJ, Greene GL (1984) Monoclonal antibodies localize oestrogen receptor in the nuclei of target cells. Nature 307:745-747 93. Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato AC (1988) A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res 16:647-663 94. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di SF, Dice JF, Difiglia M, neshKumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr., Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl 118 T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri 1,Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov 0, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanaka K, Tanida 1,Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van dK, I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk 0, Zheng X, Zhu X, Deter RL (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151-175 95. Koberle B, Roginskaya V, Wood RD (2006) XPA protein as a limiting factor for nucleotide excision repair and UV sensitivity in human cells. DNA Repair (Amst) 5:641-648 96. Kohn KW (1996) Beyond DNA cross-linking: history and prospects of DNAtargeted cancer treatment--fifteenth Bruce F. Cain Memorial Award Lecture. Cancer Res 56:5533-5546 119 97. Kohn KW, Hartley JA, Mattes WB (1987) Mechanisms of DNA sequence selective alkylation of guanine-N7 positions by nitrogen mustards. Nucleic Acids Res 15:10531-10549 98. Krasner C (2007) Aromatase inhibitors in gynecologic cancers. J Steroid Biochem Mol Biol 106:76-80 99. Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138:863-870 100. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA (1996) Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A 93:5925-5930 101. Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der BB, Gustafsson JA (1998a) Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 139:4252-4263 102. Kuiper GG, Shughrue PJ, Merchenthaler I, Gustafsson JA (1998b) The estrogen receptor beta subtype: a novel mediator of estrogen action in neuroendocrine systems. Front Neuroendocrinol 19:253-286 103. Kundu GC, Schullek JR, Wilson IB(1994) The alkylating properties of chlorambucil. Pharmacol Biochem Behav 49:621-624 104. Kurreck J (2009) RNA interference: from basic research to therapeutic applications. Angew Chem Int Ed EngI 48:1378-1398 120 105. Kushner PJ, Agard D, Feng WJ, Lopez G,Schiau A, Uht R, Webb P, Greene G (2000a) Oestrogen receptor function at classical and alternative response elements. Novartis Found Symp 230:20-26 106. Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P (2000b) Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol 74:311317 107. Lahooti H,White R, Danielian PS, Parker MG (1994) Characterization of liganddependent phosphorylation of the estrogen receptor. Mol Endocrinol 8:182-188 108. Langdon SP, Hirst GL, Miller EP, Hawkins RA, Tesdale AL, Smyth JF, Miller WR (1994) The regulation of growth and protein expression by estrogen in vitro: a study of 8 human ovarian carcinoma cell lines. J Steroid Biochem Mol Biol 50:131135 109. Langdon SP, Ritchie A, Young K, Crew AJ, Sweeting V, Bramley T, Hillier S, Hawkins RA, Tesdale AL, Smyth JF, . (1993) Contrasting effects of 17 betaestradiol on the growth of human ovarian carcinoma cells in vitro and in vivo. Int J Cancer 55:459-464 110. Lau KM, Mok SC, Ho SM (1999) Expression of human estrogen receptor-alpha and -beta, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci U S A 96:5722-5727 111. Lawley PD (1966) Effects of some chemical mutagens and carcinogens on nucleic acids. Prog Nucleic Acid Res Mol Biol 5:89-131 121 112. Lazennec G (2006) Estrogen receptor beta, a possible tumor suppressor involved in ovarian carcinogenesis. Cancer Lett 231:151-157 113. Le GP, Montano MM, Schodin DJ, Katzenellenbogen BS (1994) Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem 269:44584466 114. Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463-477 115. Li M, HERTZ R, BERGENSTAL DM (1958) Therapy of choriocarcinoma and related trophoblastic tumors with folic acid and purine antagonists. N Engl J Med 259:66-74 116. Liberman RG, Tannenbaum SR, Hughey BJ, Shefer RE, Klinkowstein RE, Prakash C, Harriman SP, Skipper PL (2004) An interface for direct analysis of (14)c in nonvolatile samples by accelerator mass spectrometry. Anal Chem 76:328-334 117. Lindahl T (1979) DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog Nucleic Acid Res Mol Biol 22:135-192 118. Lindgren PR, Cajander S, Backstrom T, Gustafsson JA, Makela S, Olofsson JI (2004) Estrogen and progesterone receptors in ovarian epithelial tumors. Mol Cell Endocrinol 221:97-104 119. Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, Lynch P, Burke W, Press N (2006) Recommendations for the care of individuals with an 122 inherited predisposition to Lynch syndrome: a systematic review. JAMA 296:15071517 120. Lukanova A, Kaaks R (2005) Endogenous hormones and ovarian cancer: epidemiology and current hypotheses. Cancer Epidemiol Biomarkers Prev 14:98107 121. MacLeod MC, Powell KL, Tran N (1995) Binding of the transcription factor, Sp1, to non-target sites in DNA modified by benzo[a]pyrene diol epoxide. Carcinogenesis 16:975-983 122. Maggi A, Ciana P, Belcredito S, Vegeto E (2004) Estrogens in the nervous system: mechanisms and nonreproductive functions. Annu Rev Physiol 66:291-313 123. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM (1995) The nuclear receptor superfamily: the second decade. Cell 83:835-839 124. Masiakowski P, Breathnach R, Bloch J, Gannon F, Krust A, Chambon P (1982) Cloning of cDNA sequences of hormone-regulated genes from the MCF-7 human breast cancer cell line. Nucleic Acids Res 10:7895-7903 125. Masood S, Heitmann J, Nuss RC, Benrubi GI (1989) Clinical correlation of hormone receptor status in epithelial ovarian cancer. Gynecol Oncol 34:57-60 126. May FE, Westley BR (1988) Identification and characterization of estrogenregulated RNAs in human breast cancer cells. J Biol Chem 263:12901-12908 123 127. May FE, Westley BR (1995) Estrogen regulated messenger RNAs in human breast cancer cells. Biomed Pharmacother 49:400-414 128. McA'Nulty MM, Lippard SJ (1996) The HMG-domain protein lxrl blocks excision repair of cisplatin-DNA adducts in yeast. Mutat Res 362:75-86 129. McA'Nulty MM, Whitehead JP, Lippard SJ (1996) Binding of lxrl, a yeast HMGdomain protein, to cisplatin-DNA adducts in vitro and in vivo. Biochemistry 35:6089-6099 130. Meijer AJ, Codogno P (2006) Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 27:411-425 131. Merajver SD, Pham TM, Caduff RF, Chen M, Poy EL, Cooney KA, Weber BL, Collins FS, Johnston C, Frank TS (1995) Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nat Genet 9:439-443 132. Meyn, Murray D (1986) Cell cycle Effects of alkylating agents. 133. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, . (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66-71 134. Mitra K, Marquis JC, Hillier SM, Rye PT, Zayas B, Lee AS, Essigmann JM, Croy RG (2002) A rationally designed genotoxin that selectively destroys estrogen receptor-positive breast cancer cells. J Am Chem Soc 124:1862-1863 135. Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861-2873 124 136. Mizushima N, Levine B,Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069-1075 137. Moretti-Rojas I, Fuqua SA, Montgomery RA, Ill, McGuire WL (1988) A cDNA for the estradiol-regulated 24K protein: control of mRNA levels in MCF-7 cells. Breast Cancer Res Treat 11:155-163 138. Morimoto H, Safrit JT, Bonavida B (1991) Synergistic effect of tumor necrosis factor-alpha- and diphtheria toxin-mediated cytotoxicity in sensitive and resistant human ovarian tumor cell lines. J Immunol 147:2609-2616 139. Morimoto H, Yonehara S, Bonavida B (1993) Overcoming tumor necrosis factor and drug resistance of human tumor cell lines by combination treatment with antiFas antibody and drugs or toxins. Cancer Res 53:2591-2596 140. Morisset M, Capony F, Rochefort H (1986) Processing and estrogen regulation of the 52-kilodalton protein inside MCF7 breast cancer cells. Endocrinology 119:2773-2782 141. Morissette M, Le SM, D'Astous M, Jourdain S, Al SS, Morin N, Estrada-Camarena E, Mendez P, Garcia-Segura LM, Di PT (2008) Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J Steroid Biochem Mol Biol 108:327-338 142. Narod SA, Foulkes WD (2004) BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 4:665-676 125 143. Nash JD, Ozols RF, Smyth JF, Hamilton TC (1989) Estrogen and anti-estrogen effects on the growth of human epithelial ovarian cancer in vitro. Obstet Gynecol 73:1009-1016 144. Newton K, Strasser A (1998) The Bcl-2 family and cell death regulation. Curr Opin Genet Dev 8:68-75 145. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA (2001) Mechanisms of estrogen action. Physiol Rev 81:1535-1565 146. Noy N (2007) Ligand specificity of nuclear hormone receptors: sifting through promiscuity. Biochemistry 46:13461-13467 147. O'Connor PM, Kohn KW (1990) Comparative pharmacokinetics of DNA lesion formation and removal following treatment of L1210 cells with nitrogen mustards. Cancer Commun 2:387-394 148. O'Donnell AJ, Macleod KG, Burns DJ, Smyth JF, Langdon SP (2005) Estrogen receptor-alpha mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocr Relat Cancer 12:851-866 149. Olefsky JM (2001) Nuclear receptor minireview series. J Biol Chem 276:3686336864 150. Olive PL, Banath JP (1995) Sizing highly fragmented DNA in individual apoptotic cells using the comet assay and a DNA crosslinking agent. Exp Cell Res 221:19-26 126 151. Olive PL, Banath JP (2006) The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1:23-29 152. Olive PL, Banath JP, Durand RE (1990) Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the "comet" assay. Radiat Res 122:86-94 153. Osborne CK, Yochmowitz MG, Knight WA, Ill, McGuire WL (1980) The value of estrogen and progesterone receptors in the treatment of breast cancer. Cancer 46:2884-2888 154. Oxelmark E, Knoblauch R,Arnal S, Su LF, Schapira M, Garabedian MJ (2003) Genetic dissection of p23, an Hsp90 cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J Biol Chem 278:36547-36555 155. Ozols RF (2005) Treatment goals in ovarian cancer. Int J Gynecol Cancer 15 Suppl 1:3-11 156. Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM, Baergen R (2003) Phase Ill trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage IlIl ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 21:3194-3200 157. Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61:439-444 127 158. Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K, Wilbanks G, Nicosia S, Cantor A, Sutphen R (2005) BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104:2807-2816 159. Papac RJ (2001) Origins of cancer therapy. Yale J Biol Med 74:391-398 160. Parker MG (1991) Nuclear Hormone Receptors 161. Parker, Jensen EV (1991) Overview of the Nuclear Receptor Family. 162. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1dependent autophagy. Cell 122:927-939 163. Pearce ST, Jordan VC (2004) The biological role of estrogen receptors alpha and beta in cancer. Crit Rev Oncol Hematol 50:3-22 164. Pettersson K, Grandien K, Kuiper GG, Gustafsson JA (1997) Mouse estrogen receptor beta forms estrogen response element-binding heterodimers with estrogen receptor alpha. Mol Endocrinol 11:1486-1496 165. Pil PM, Lippard SJ (1992) Specific binding of chromosomal protein HMG1 to DNA damaged by the anticancer drug cisplatin. Science 256:234-237 166. Pratt WB, Ruddon RW, Ensminger WD, Maybaum J (1994) The Anticancer drugs 167. Pujol P, Rey JM, Nirde P, Roger P, Gastaldi M, Laffargue F, Rochefort H, Maudelonde T (1998) Differential expression of estrogen receptor-alpha and -beta 128 messenger RNAs as a potential marker of ovarian carcinogenesis. Cancer Res 58:5367-5373 168. Rao BR, Slotman BJ (1991) Endocrine factors in common epithelial ovarian cancer. Endocr Rev 12:14-26 169. Rao GG, Miller DS (2005) Clinical applications of hormonal therapy in ovarian cancer. Curr Treat Options Oncol 6:97-102 170. Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER (2004) Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol 18:2854-2865 171. Reardon JT, Vaisman A, Chaney SG, Sancar A (1999) Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamineplatinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res 59:39683971 172. Reed (2006) Cisplatin, carboplatin and oxaliplatin. In: Chabner BA, Longo DL (eds) Cancer chemotherapy and biotherapy, 4 edn 173. Reggiori F, Klionsky DJ (2002) Autophagy in the eukaryotic cell. Eukaryot Cell 1:11-21 174. Renan MJ (1993) How many mutations are required for tumorigenesis? Implications from human cancer data. Mol Carcinog 7:139-146 129 175. Reynolds A, Anderson EM, Vermeulen A, Fedorov Y, Robinson K, Leake D, Karpilow J, Marshall WS, Khvorova A (2006) Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA 12:988-993 176. Riggs BL, Khosla S, Melton U, 111(2002) Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev 23:279-302 177. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Persson IR (2001) Risk factors for epithelial borderline ovarian tumors: results of a Swedish case-control study. Gynecol Oncol 83:575-585 178. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Persson IR (2002a) Risk factors for invasive epithelial ovarian cancer: results from a Swedish case-control study. Am J Epidemiol 156:363-373 179. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Weiderpass E, Persson IR (2002b) Hormone replacement therapy and the risk of invasive epithelial ovarian cancer in Swedish women. J Natl Cancer Inst 94:497504 180. Riman T, Dickman PW, Nilsson S, Nordlinder H, Magnusson CM, Persson IR (2004) Some life-style factors and the risk of invasive epithelial ovarian cancer in Swedish women. Eur J Epidemiol 19:1011-1019 181. Rink SM, Yarema KJ, Solomon MS, Paige LA, Tadayoni-Rebek BM, Essigmann JM, Croy RG (1996) Synthesis and biological activity of DNA damaging agents that form decoy binding sites for the estrogen receptor. Proc Natl Acad Sci U S A 93:15063-15068 130 182. Risch HA (2002) Hormone replacement therapy and the risk of ovarian cancer. Gynecol Oncol 86:115-117 183. Roberts D, Schick J, Conway S, Biade S, Laub PB, Stevenson JP, Hamilton TC, O'Dwyer PJ, Johnson SW (2005) Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br J Cancer 92:1149-1158 184. Rochefort H (1990) Cathepsin D in breast cancer. Breast Cancer Res Treat 16:313 185. Rochefort H (1998) [Estrogens, cathepsin D and metastasis in cancers of the breast and ovary: invasion or proliferation?]. C R Seances Soc Biol Fil 192:241-251 186. Rochefort H (1999) [Estrogen-induced genes in breast cancer, and their medical importance]. Bull Acad Natl Med 183:955-968 187. Rose PG, Reale FR, Longcope C, Hunter RE (1990) Prognostic significance of estrogen and progesterone receptors in epithelial ovarian cancer. Obstet Gynecol 76:258-263 188. Roy G, Horton JK, Roy R, Denning T, Mitra S, Boldogh 1(2000) Acquired alkylating drug resistance of a human ovarian carcinoma cell line is unaffected by altered levels of pro- and anti-apoptotic proteins. Oncogene 19:141-150 189. Russo IH,Russo J (1998) Role of hormones in mammary cancer initiation and progression. J Mammary Gland Biol Neoplasia 3:49-61 131 190. Rutherford T, Brown WD, Sapi E, Aschkenazi S, Munoz A, Mor G (2000) Absence of estrogen receptor-beta expression in metastatic ovarian cancer. Obstet Gynecol 96:417-421 191. Satokata 1,Tanaka K, Miura N, Narita M, Mimaki T, Satoh Y, Kondo S, Okada Y (1992) Three nonsense mutations responsible for group A xeroderma pigmentosum. Mutat Res 273:193-202 192. Saville B,Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S (2000) Ligand-, cell-, and estrogen receptor subtype (alpha/beta)dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem 275:53795387 193. Sawyers C (2004) Targeted cancer therapy. Nature 432:294-297 194. Schaner ME, Ross DT, Ciaravino G, Sorlie T, Troyanskaya 0, Diehn M, Wang YC, Duran GE, Sikic TL, Caldeira S, Skomedal H, Tu IP, Hernandez-Boussard T, Johnson SW, O'Dwyer PJ, Fero MJ, Kristensen GB, Borresen-Dale AL, Hastie T, Tibshirani R, van de RM, Teng NN, Longacre TA, Botstein D, Brown PO, Sikic BI (2003) Gene expression patterns in ovarian carcinomas. Mol Biol Cell 14:43764386 195. Schildkraut JM, Schwingl PJ, Bastos E, Evanoff A, Hughes C (1996) Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet Gynecol 88:554-559 196. Schuchard M, Landers JP, Sandhu NP, Spelsberg TC (1993) Steroid hormone regulation of nuclear proto-oncogenes. Endocr Rev 14:659-669 132 197. Segnitz B, Gehring U (1995) Subunit structure of the nonactivated human estrogen receptor. Proc Natl Acad Sci U S A 92:2179-2183 198. Segnitz B, Gehring U (1997) The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J Biol Chem 272:18694-18701 199. Sharma U, Marquis JC, Nicole DA, Hillier SM, Fedeles B, Rye PT, Essigmann JM, Croy RG (2004) Design, synthesis, and evaluation of estradiol-linked genotoxicants as anti-cancer agents. Bioorg Med Chem Lett 14:3829-3833 200. Shuey DJ, McCallus DE, Giordano T (2002) RNAi: gene-silencing in therapeutic intervention. Drug Discov Today 7:1040-1046 201. Slotman BJ, Rao BR (1988) Ovarian cancer (review). Etiology, diagnosis, prognosis, surgery, radiotherapy, chemotherapy and endocrine therapy. Anticancer Res 8:417-434 202. States JC, McDuffie ER, Myrand SP, McDowell M, Cleaver JE (1998) Distribution of mutations in the human xeroderma pigmentosum group A gene and their relationships to the functional regions of the DNA damage recognition protein. Hum Mutat 12:103-113 203. Stenoien DL, Mancini MG, Patel K,Allegretto EA, Smith CL, Mancini MA (2000) Subnuclear trafficking of estrogen receptor-alpha and steroid receptor coactivator-1. Mol Endocrinol 14:518-534 204. Stenoien DL, Patel K, Mancini MG, Dutertre M, Smith CL, O'Malley BW, Mancini MA (2001) FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat Cell Biol 3:15-23 133 205. Stevens EV, Nishizuka S, Antony S, Reimers M, Varma S, Young L, Munson PJ, Weinstein JN, Kohn EC, Pommier Y (2008) Predicting cisplatin and trabectedin drug sensitivity in ovarian and colon cancers. Mol Cancer Ther 7:10-18 206. Sunters A, Springer CJ, Bagshawe KD, Souhami RL, Hartley JA (1992) The cytotoxicity, DNA crosslinking ability and DNA sequence selectivity of the aniline mustards melphalan, chlorambucil and 4-[bis(2-chloroethyl)amino] benzoic acid. Biochem Pharmacol 44:59-64 207. Tew, Colvin OM, Jones RB (2006) Clinical and High-Dose Alkylating Agents. In: Chabner BA, Longo DL (eds) Cancer Chemotherapy and Biotherapy, 4 edn 208. Treiber DK, Zhai X, Jantzen HM, Essigmann JM (1994) Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor). Proc Natl Acad Sci U S A 91:5672-5676 209. Turteltaub KW, Dingley KH (1998) Application of accelerated mass spectrometry (AMS) in DNA adduct quantification and identification. Toxicol Lett 102-103:435439 210. Turteltaub KW, Felton JS, Gledhill BL, Vogel JS, Southon JR, Caffee MW, Finkel RC, Nelson DE, Proctor ID, Davis JC (1990) Accelerator mass spectrometry in biomedical dosimetry: relationship between low-level exposure and covalent binding of heterocyclic amine carcinogens to DNA. Proc Natl Acad Sci U S A 87:5288-5292 211. Tyagi A, Singh RP, Agarwal C, Siriwardana S, Sclafani RA, Agarwal R (2005) Resveratrol causes Cdc2-tyrl5 phosphorylation via ATM/ATR-Chkl/2-Cdc25C 134 pathway as a central mechanism for S phase arrest in human ovarian carcinoma Ovcar-3 cells. Carcinogenesis 26:1978-1987 212. Vandenabeele P, Vanden BT, Festjens N (2006) Caspase inhibitors promote alternative cell death pathways. Sci STKE 2006:e44 213. Venkitaraman AR (2004) Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat Rev Cancer 4:266-276 214. von Angerer E, Prekajac J, Strohmeier J (1984) 2-Phenylindoles. Relationship between structure, estrogen receptor affinity, and mammary tumor inhibiting activity in the rat. J Med Chem 27:1439-1447 215. Walker G, MacLeod K, Williams AR, Cameron DA, Smyth JF, Langdon SP (2007) Estrogen-regulated gene expression predicts response to endocrine therapy in patients with ovarian cancer. Gynecol Oncol 106:461-468 216. Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, Staub A, Jensen E, Scrace G,Waterfield M, . (1985) Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci U S A 82:7889-7893 217. Webb P, Lopez GN, Uht RM, Kushner PJ (1995) Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol 9:443-456 218. Webb P, Nguyen P,Valentine C, Lopez GN, Kwok GR, McInemey E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, Kushner PJ (1999) The estrogen receptor enhances AP-1 activity by two distinct mechanisms with 135 different requirements for receptor transactivation functions. Mol Endocrinol 13:1672-1685 219. Wilson TM, Ewel A, Duguid JR, Eble JN, Lescoe MK, Fishel R, Kelley MR (1995) Differential cellular expression of the human MSH2 repair enzyme in small and large intestine. Cancer Res 55:5146-5150 220. Wu X, Fan W, Xu S, Zhou Y (2003) Sensitization to the cytotoxicity of cisplatin by transfection with nucleotide excision repair gene xeroderma pigmentosun group A antisense RNA in human lung adenocarcinoma cells. Clin Cancer Res 9:58745879 221. Xu Y, Traystman RJ, Hurn PD, Wang MM (2004) Membrane restraint of estrogen receptor alpha enhances estrogen-dependent nuclear localization and genomic function. Mol Endocrinol 18:86-96 222. Yap TA, Carden CP, Kaye SB (2009) Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer 9:167-181 223. Zakeri Z, Bursch W, Tenniswood M, Lockshin RA (1995) Cell death: programmed, apoptosis, necrosis, or other? Cell Death Differ 2:87-96 224. Zang XP, Pento JT (2008) SiRNA inhibition of ER-alpha expression reduces KGFinduced proliferation of breast cancer cells. Anticancer Res 28:2733-2735 225. Zheng L,Annab LA, Afshari CA, Lee WH, Boyer TG (2001) BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci U S A 98:9587-9592 136 226. Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408:433-439 137 138 i) 1(1 Chapter 3 Role of ER in E2-7a toxicity: Studies in ER(+) and ER(-) cell populations 139 Introduction Nitrogen mustards (mechlorethamine and tris(p-chloroethyl)amine) were the first nonhormonal agents to show significant antitumor activity in humans (Chabner and Roberts, Jr., 2005). These agents have been a ubiquitous element of chemotherapy ever since, and several modifications have been made to the parent mechlorethamine molecule to develop a range of more effective therapeutics. Four of the nitrogen mustard derivatives have been found to be superior to their parent compound - melphalan, chlorambucil, cyclophosphamide and ifosfamide. They display a higher therapeutic index, broader range of clinical activity and can be administered both orally and intravenously (Tew et al., 2006). These alkylating agents have found their niche in the chemotherapeutic domain, but the quest for an anticancer agent that is selective for tumor tissues and spares normal tissues continues. Targeted therapy involves the development of a new generation of chemotherapeutics meant to interfere specifically with a molecular target that is believed to play a crucial role in tumor growth and progression (Hurley, 2002; Sawyers, 2004). The vast increase of knowledge of the biological, genetic and molecular aspects of neoplastic disease has resulted in more options to pursue. The Essigmann lab is particularly interested in developing selective genotoxicants that interact with DNA; we have leveraged the lessons learned from the study of the mechanisms of action of DNA damaging drugs such as cisplatin in order to develop better, more selective antitumor agents. One of the cisplatin mechanisms suggests that the combination of a DNA damaging functionality with a ligand for a tumor-specific protein provides a novel way to improve the therapeutic index of antitumor agents. In this regard, steroid hormones and their receptors make attractive targets for drug development. Hormone-receptor interactions 140 play a crucial role in the development and progression of many hormonally-responsive cancers including those of the prostate, breast and ovary. We have synthesized a series of such agents that are designed to introduce DNA damage in cancer cells while sparing non-target tissues. The premise behind the design is that selectivity can be conferred by combining a steroid receptor recognition moiety and a bi-functional DNA damaging group (Figure 1.3). Our lab has previously reported the synthesis of an agent designed to target estrogen receptor (ER) positive cancers (Mitra et al., 2002). This agent, estradiol-C6NC2-N,N-bis-(2-chloroethyl)-aniline (E2-7a), contains a DNA damaging aniline mustard moiety tethered to the estradiol through a stable linker. The hypothesis for the mechanism of action of E2-7a is that the drug-DNA adduct interacts with the ER through the estradiol moiety of E2-7a, thereby mediating cell death selectively in cancer cells through one or more of the following routes: (i) Because they are shielded by the ER, the DNA adducts formed by E2-7a are difficult for ER(+) tumor cells to repair; the adducts therefore persist, and selectively disrupt growth and survival mechanisms in ER(+) tumors, thereby sensitizing cells to the cytotoxic effects of the drug. (ii) Drug-DNA adducts sequester the ER away from the protein's site of transcriptional activity, and thereby disrupt the transcription of downstream ER-regulated genes (transcription factor hijacking) (Rink et al., 1996). In other words, formation of the receptor-adduct complex antagonizes the expression of downstream ER-responsive genes and causes the disruption of cellular processes in ER(+) cells, leading to cell death. We have detected a promising indication of the differential toxicity of E2-7a in ER(+) vs. ER(-) breast cancer cell lines, as described in Mitra et al. (2002). There was significantly increased sensitivity towards E2-7a in MCF-7 cells (ER(+)) compared to the ER(-) breast 141 2 ~ cancer cell line MDA-MB-231. This difference was not observed when the two cell lines were treated with chlorambucil. Chlorambucil contains the same DNA damaging moiety as does E2-7a and is expected to form similar kinds of DNA damage. Therefore, it can be deduced that the tethering of estradiol moiety to the aniline mustard might be responsible for the selectivity towards E2-7a. This prompted further studies into the mechanism of action of E2-7a in the breast cancer cell lines. In order to understand the possible contribution of repair shielding to E2-7a toxicity, Accelerator Mass Spectrometry (AMS) was employed by researchers in the Essigmann and Tannenbaum laboratories. AMS can be used to measure very low levels of radioactivity associated with DNA; its sensitivity is almost five orders of magnitude greater than conventional decay counting techniques (Liberman et al., 2004). AMS enables the quantification of drug-DNA adducts, and correlations can be drawn between the levels of adducts formed in culture (for which we can rapidly measure biological effects) and adduct levels in target tissues of animal models (Turteltaub et al., 1990; Turteltaub and Dingley, 1998). Shawn Hillier and John Marquis (Essigmann laboratory; unpublished results) employed AMS to determine the level of DNA adducts in E2-7atreated cells. Their experiments show that the removal of [14C]-E2-7a-DNA adducts is much slower in the ER(+) breast cancer cell line MCF-7 compared to the ER(-) MDAMB-231 cells. Under the same conditions, there was no difference in the level of [14C]-melphalan-DNA adducts in MCF-7 vs. MDA-MB-231 cells. These preliminary results indicate a favorable role for ER in retaining the drug-DNA adducts in cells, and in inducing selective toxicity in ER(+) cells. This experiment is a step in the right direction towards studying the mechanism of toxicity of E2-7a but the results must be interpreted with the caveat in mind that the breast cancer cell lines possess other biochemical 142 differences besides ER status that might determine the ultimate outcome of drug action. It would be more definitive if we could perform the same study with cell lines that have varying levels of ER but are otherwise isogenic, thereby diminishing the above biochemical differences. One of the main objectives of my research is to understand whether receptor-drug interaction affects the toxicity of E2-7ax. This chapter focuses on testing a pivotal mechanistic element of E2-7a action - the role of ER in E2-7a toxicity. Towards this end, we needed a system of ER(+) and ER(-) cell lines that are otherwise isogenic. Our strategy to create such a system was to use the potential of RNA interference (RNAi), by the means of which a chosen ER(+) cell population could be converted transiently into an ER(-) population by depleting the ER levels in the cells. RNAi is a fundamental pathway in eukaryotic cells where a sequence-specific small interfering RNA (siRNA) molecule targets and degrades its complementary mRNA (Elbashir et al., 2001). Naturally occurring siRNA molecules (21-23 nucleotides long) are produced in cells by the cleavage of long pieces of double-stranded RNA by the enzyme Dicer (Bernstein et al., 2001). The siRNA-Dicer complex recruits additional components to form an RNA-induced silencing complex (RISC), which then targets a specific complementary mRNA strand for enzymatic degradation, leading to post-transcriptional gene silencing (Shuey et al., 2002). In addition to being produced in vivo, siRNA molecules can also be synthetically produced, and introduced into cells to knockdown the gene of choice. Synthetic siRNA molecules have an advantage over long doublestranded RNA molecules in that introducing long double-stranded RNA into cells results in the interaction between the RNA molecules and intracellular RNA receptors, leading to immune interferon response and shutdown of cellular protein production in target cells 143 (Reynolds et al., 2006). The siRNA molecules, on the other hand, are too short to cause an interferon response. RNA interference pathway has been employed extensively to study gene functions and the physiological effect of a gene product (Kim and Rossi, 2007; Kurreck, 2009). Commercially available validated siRNA duplexes can be used to knockdown the gene product of interest - ER, in our case. The estrogen receptor is a central protein in the cellular network and is a transcription factor that regulates gene expression in response to ligand binding (Nilsson et al., 2001; Pearce and Jordan, 2004). Therefore, knocking down the ER could have several consequences on cell behavior - for instance, cell growth might be affected since ER is involved in the regulation of proliferation. When the receptor was knocked down in MCF-7 breast cancer cells, it significantly reduced cell proliferation (Zang and Pento, 2008). By knocking down the ER in target cells using RNAi, the role of the receptor in mediating drug toxicity can be studied very effectively. At the same time, it is also desirable that the growth profile of the ER-knocked down cells remain unaltered compared to the parental cell line so that ER(-) cells are not inherently growth compromised even before drug treatment. With these considerations in mind, the SKOV-3 cell line has been used for the RNAi-based knockdown of ERa. These cells provide a model for ERa positive, cisplatin resistant ovarian tumor, and more importantly, they are not growth responsive to hormones in culture (Hua et al., 1995d), suggesting that titrating down the levels of ER may not affect their growth profiles significantly. SKOV-3 cells possess the ER, but do not respond to estrogens such as estradiol or antiestrogens such as ICI 164,384. Although estrogen binding to ER occurs with a Kd 144 similar to that of canonical estrogen receptors such as those in MCF-7 cells (0.2 nM in SKOV-3 cells compared to 0.4 nM in MCF-7; Coezy et al., 1982; Hua et al., 1995d) and in various other systems (Kd ranging from 0.1-1nM; Kuiper et al., 1997), the ER present in SKOV-3 seems to be deficient in the transcriptional regulation of selected growth regulatory gene products. Hence, knocking down the ER would be expected to not affect any of the growth characteristics of the cells. Thus, this cell line serves as an appropriate model system to understand the interaction between ER and E2-7a without involving the confounding effects of the transcriptional activity of the receptor. This system helps address one of the two proposed mechanisms of action of E2-7a (repair shielding) and understand the involvement of the ER in modulating E2-7a toxicity. Studies describing the biological response of SKOV-3 cells to E2-7a were described in detail in Chapter 2. We found that E2-7a was cytotoxic to SKOV-3 cells, and induced cell death through autophagy. We also found evidence of DNA damage that might be responsible for the persistent S-phase arrest observed with E2-7a treatment. In this chapter, we extend our investigations of E2-7a to gain a more in-depth understanding of the mechanism of action of E2-7a. The relative toxicity of E2-7a towards the parent SKOV-3 cells has been tested and compared to the ER knockdown SKOV-3 cells. In order to correlate the observed toxicity towards E2-7a with DNA adduct formation, drugDNA adduct levels have been measured in these distinct cell populations using AMS. The results of these studies are described subsequently. 145 Materials and Methods Chemicals: Radiolabeled E2-7a (specific activity 20 mCi/mmol) used in these studies was synthesized in our laboratory. Radiolabeled melphalan (specific activity 50 mCi/mmol) was purchased from Moravek and re-purified using HPLC. Radiolabeled drugs were diluted with unlabeled material where necessary. Cisplatin, melphalan, and 17p-estradiol were obtained from Sigma-Aldrich. ICI 182,780 was a kind gift from Professor Robert Hanson at Northeastern University. Antibodies to ER (sc-8002, mouse monoclonal) and Actin (C-1 1, rabbit polyclonal) were obtained from Santa Cruz Biotechnology. Secondary anti-mouse IgG and anti-rabbit IgG antibodies, both conjugated to Horseradish Peroxidase, were obtained from Cell Signaling Technology (Beverly, MA). Cell Culture: SKOV-3 cells were purchased from the American Type Culture Collection (ATCC) (Rockville, MD). Fetal Bovine Serum (FBS) and Charcoal-Dextran Treated FBS (CDTFBS) was obtained from Thermo Fisher Scientific (Logan, UT). SKOV-3 cells were maintained in McCoy's 5a Medium (ATCC), supplemented with 10% FBS. Cells were grown in a humidified 5%CO2/air atmosphere at 370 C. For cell growth assays in the presence of estrogens/antiestrogens, cells were grown in full medium for 24 hours, and transferred to phenol-red free medium with 10% CDT-FBS 146 and grown an additional 36 hours. E2-7c, melphalan, E2 and ICI 182,780 were dissolved in DMSO and all treatments were carried out in phenol-red free medium with CDT-FBS. Transient ERa knockdown using siRNA: Validated siRNA sequence from Stealth RNAi DuoPaks (ERa validated Stealth Select RNAi, duplex 2; Invitrogen, Carlsbad, CA) was used to target ER. The Stealth RNAi negative control (medium GC content; Invitrogen) was utilized as a control to ensure the specificity of each targeted knockdown. Cells were plated at 40,000 per well in 6-well plates, grown for 24 hours, followed by siRNA/NC treatment (25 nM) for 7 hours. Lipofectamine 2000 (Invitrogen) was used as the transfection reagent, and the procedure for transfection was as outlined in the Invitrogen product manual. Cells were treated with test compounds one day after siRNA treatment. Gene knockdown was verified using immunoblotting with antibody against ER. For growth inhibition assays, cells were treated with the test compounds dissolved in DMSO. Cells were detached using trypLE Express (Invitrogen) at times indicated and the number of cells in control and drug-treated wells was determined using a Coulter Counter. The percent growth inhibition was calculated as the ratio of cell number in treated and control wells multiplied by 100. Significance was estimated by two-tailed Pvalues obtained from unpaired t-test calculated using GraphPad software with the mean and standard deviation of growth inhibition value at each treatment point. Comparisons with P<0.05 were considered to be significant. 147 Immunoblot Analysis: Whole cell extracts were prepared by cell lysis using RIPA buffer (Santacruz; 1XTBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1 mM sodium orthovanadate, 1 mM NaF, 50 mM Tris-HCI (pH 7.4) and protease inhibitor cocktail (Sigma-Aldrich)). Protein concentrations were determined using Bradford reagent with BSA standard. Equal amounts of protein were separated on pre-cast 4-12% SDSpolyacrylamide gradient gels (Invitrogen) in MOPS buffer system and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Membranes were blocked with 5% milk in Tris-Buffered Saline - Tween 20 (0.1% Tween 20, 10 mM Tris [pH 7.4], 150 mM NaCl) and incubated with primary antibodies (against ERa or actin) overnight at 40C. Membranes were incubated with secondary antibodies for 1-2 hours, and antibody complexes formed with horseradish peroxidase-conjugated secondary antibodies were visualized using Enhanced Chemiluminescence reagents (PicoWest, Pierce, Rockford, IL). Adduct measurement using Accelerator Mass Spectrometry: SKOV-3 cells were exposed to various doses of [14C]-labeled test compounds (E2-7a or melphalan). At the end of the incubation period, wells were washed once with fresh medium, followed by a wash with PBS, and cells were detached from the plates using trypLE express (Invitrogen), pelleted, and washed with phosphate buffered saline (PBS). The cells were lysed in 0.5 mL of extraction buffer containing HEPES (50 mM), NaCl (100 mM), EDTA (10 mM) and 0.8% SDS, followed by incubation with Proteinase K (Roche Diagnostics; 0.6 mg/mL for 2 hours at 370C). After phenol-chloroform extraction 148 I I~ the aqueous phase was extracted with chloroform and the nucleic acids were precipitated with 100% ethanol overnight at -200C, and subsequently pelleted by centrifugation at 7000g for 15 min at 40C. The pellet was washed with 70% ethanol and dried in vacuo. The dried pellet was reconstituted with 0.3 mL of the extraction buffer, and incubated with RNase A (Roche Diagnostics; 0.9 mg/mL, 30 minutes at 37*C). DNA was extracted by subsequent phenol:chloroform:isoamyl alcohol and chloroform:isoamyl alcohol extractions. The aqueous phase from the second extraction was collected and the DNA precipitated with 3 volumes of ice-cold ethanol, centrifuged and washed twice with 70% ethanol. DNA was dissolved in 0.2 mL water and the concentration was estimated by absorbance at 260 nm. All samples were adjusted to the same concentration (4 pg/mL). AMS analyses were conducted at the Biological Engineering Accelerator Mass Spectrometry (BEAMS) Lab at MIT by Paul Skipper and Rosa Liberman as described in Liberman, et al. (2004). DNA solutions were applied directly to a CuO matrix used for sample combustion. A standard consisting of a solution of [14Cmethyl] bovine serum albumin was used to calibrate the instrument. The amount of [14 C] in each sample was calculated from the peak area ratio of the sample to the standard. All samples were analyzed at least twice. Readings were obtained from AMS instrument as dpm/pg DNA and converted to adducts per million bases. Results SKOV-3 cells are not growth sensitive to E2 or ICI 182,780. SKOV-3 cells were grown in the presence of either the pure estrogen estradiol or the pure antiestrogens ICI 182,780 at various doses. The growth patterns are shown in Figure 3.1, and the corresponding doubling times are shown in Table 3.1. There was no significant 149 difference in the cell numbers between the untreated cells growing in medium supplemented with CDT-FBS and cells treated with various doses of estradiol or ICI 182,780. All the seven treatments resulted in similar doubling times, as can be seen in Table 3.1. In addition, the cells grown in medium containing FBS grew at a similar rate as the cells that were supplemented with CDT-FBS (data shown in Table 3.1). These results indicate that as reported in the literature, SKOV-3 cells are not growth sensitive to estradiol or pure antiestrogen in our system as well. E2-7a is more toxic to the ER(+) SKOV-3 cells compared to ER(-) SKOV-3 cells. Following treatment with siRNA against the ER or NC, cells were analyzed for their ER levels by immunoblotting. Figure 3.2 shows a representative western blot of ER levels with NC/siRNA treatments. It can be seen that 25 nM siRNA treatment effectively reduced the ER levels by 92% within 24 hours of treatment (Figure 3.2). From the Figure, it can be seen that the ER levels remained knocked down up to 72 hours after treatment with siRNA (79% knockdown after 48 hours, and 90% knockdown after 72 hours). The timeframe of subsequent ER knockdown experiments is well within 72 hours, and this ensured that the levels of the receptor were low enough to create a transient ER(-) population during the time course of all further experiments. Figure 3.3 shows the results of treatment of the ER knockdown cells compared to the NC-treated SKOV-3 cells with cisplatin and melphalan. As seen in Figure 3.3A, the growth inhibition pattern of SKOV-3 ER(+) and ER(-) populations was identical following cisplatin treatment, and the percent growth inhibition in the two populations was the same at each dose in the range of doses from 1.25-20 pM. It appeared that knocking down ER did not lead to a noticeable change in toxicity towards cisplatin. A similar pattern was observed with melphalan, as can be seen from Figure 3.3B. There was no 150 significant difference in the toxicity of melphalan at various doses from 1.25-20 piM, and as was the case with cisplatin, the ER status did not seem to affect the toxicity towards melpahalan. From Figure 3.4, we observe that E2-7a treatment, as opposed to cisplatin and melphalan treatments, was able to induce a difference in cytotoxicity in ER(+) versus ER(-) cells - there was a significant decrease in the toxicity of E2-7ca after ER knockdown at 2.5, 5 and 10 gM doses. This difference in toxicity is not likely due to the differences in growth rates of the ER(+) and ER(-) cells; preliminary experiments with these populations showed that they grew with similar doubling times (ER(+) cells -39 hours and ER(-) cells-38 hours, data not shown). This clearly indicates a role for the presence of the ER in SKOV-3 cells in inducing the additional toxicity observed in the ER(+) cells compared to the ER knockdown cells. E2-7a forms DNA-adducts in SKOV-3 cells. We wanted to examine whether there was a correlation between the observed toxicity of E2-7a towards ER(+) and ER knockdown SKOV-3 cells and the DNA-adduct levels in these populations. The first step of this study was to test whether E2-7x-DNA adducts could be detected in these cells. This was done by treating cells with [14C]-E2-7ax, isolating their DNA and measuring the radioactivity associated with the DNA using AMS. The result of this experiment is shown in Figure 3.5. We were able to detect E2-7a-DNA adducts at doses as low as 0.3 pM. The interesting observation with the dose-response curve was that it was not a linear response. When the dose was doubled from 0.31 to 0.62 pM, the adduct levels increased more than tenfold. Similarly, when the dose was doubled from 1.25 to 2.5 pM, the adduct level 151 increased from 14.8 to 68.0, demonstrating the non-linear nature of the dose-response curve. E2-7a adducts are higher in ER(+) vs. ER(-) treated SKOV-3 cells, whereas melphalan treated cells show similar number of adducts in both cell populations. A possible explanation for the observed differential toxicity of E2-7a between the ER(+) and ER(-) populations is the repair shielding pathway by which E2-7a was designed. If repair shielding were to occur in SKOV-3 cells, then the adduct levels would be expected to be higher in those cells than in the SKOV-3 cells in which the siRNA had suppressed the level of the ER. In order to address the question whether the presence of the ER affected adduct levels, we exposed siRNA-treated and NC-treated SKOV-3 cells to [14C]-E2-7c and measured the DNA-adduct levels after 8, 24 and 36 hours of drug exposure. The results are shown in Figure 3.6A. Various relevant parameters associated with the treatment, such as adduct levels, cell number and drug intake into cells, are listed in Table 3.2. We observed that the adduct levels were significantly higher in the ER(+) SKOV-3 cells compared to the siRNA-treated population (P <0.05 at all treatment durations). Following an eight hour treatment, there were twice as many adducts in the ER(+) as in the ER(-) cells. When the treatment duration was increased to 24 hours, the ER(+) cells had almost four times as many adducts as the ER(-). A similar trend was observed at 36 hours as well. This difference in adduct levels was not likely due to decreased accumulation or transport of E2-7a into ER(-) cells, since we determined that the amount of drug accumulating in the cells was the same in the ER(+) versus the ER(-) populations (data is shown in Table 3.1; this finding was based on scintillation counting of the distribution of the radioactive drug in the cells and medium). In fact, at all the durations of treatment, both ER(+) and ER(-) had similar amounts of radioactivity associated with the medium and with the cells. This indicated that (i) there was no 152 differential retention of the drug in the cells in the ER(+) compared to the ER(-) cells, and (ii) there was an equilibrium between the amounts of drug in the cells and in the medium over the course of treatment and furthermore, increasing the duration of treatment did not lead to increased accumulation of the drug in the cells. In contrast to E2-7a, melphalan produced a similar adduct burden in ER(+) and ER(-) cells, as can be seen in Figure 3.6B. In addition, there was no change in the adduct levels over time of treatment, and the adduct levels were lower than those observed with E2-7a. The experiment with melphalan helps rule out artifacts in the siRNA treatment that might have caused the differential sensitivity to E2-7a. Discussion DNA alkylating agents are very effective in killing cells that proliferate rapidly, and consequently have demonstrated high efficacy as chemotherapeutics in the treatment of potentially fatal tumors (Helleday et al., 2008; Hurley, 2002). One approach to improving the therapeutic index of conventional alkylating agents is to target DNA repair machinery selectively in cancer cells. We have chosen this route to develop a compound E2-7a that alkylates DNA and contains a tumor-specific protein recognition moiety. The synthesis and design strategy of this compound has been described in detail elsewhere (Mitra et al., 2002; Sharma et al., 2004). E2-7a was designed to target cancers that aberrantly express the estrogen receptor (ER), such as breast and ovarian cancers. This compound was shown to be effective in inducing selective toxicity towards ER(+) cells compared to the ER(-) breast cancer cells by Mitra et al (2002). The focus of this dissertation is the application of E2-7a to the ovarian cancer scenario, and understanding the mechanism of action of E2-7ca in ovarian cancer cells. Over 60% of 153 ovarian cancers express the ER (Brandenberger et al., 1998; Rao and Slotman, 1991) and estrogen seems to play a key role in tumorigenesis of ovarian cancers (Clinton and Hua, 1997; Langdon et al., 1994; Nash et al., 1989). Based on these pieces of evidence, we hypothesized that E2-7a might be able to selectively induce toxicity in ER(+) ovarian cancer cells compared to ER(-) cells. In this chapter, the mechanistic aspect of E2-7a action has been probed further, and we have, in particular, tried to elucidate the role of ER in the cytotoxicity of E2-7a towards ovarian cancer cells. A system of ER(+) and ER(-) cell populations that are otherwise isogenic has been produced by treating the parent ovarian cancer cell lines SKOV-3 with a negative control (NC) siRNA duplex and an siRNA duplex against ER. In order to confirm that the SKOV-3 cells were not growth-responsive to estrogens or antiestrogens (it was necessary to obtain this information before we proceeded further with the ER knockdown experiment), cells were grown in the presence of estradiol or the pure antiestrogen ICI 182,780. It was found that the cells did not show any changes in their growth rates in the presence of either ligand. Doses in the range of 1 nM - 100 nM were tested, and there were no apparent differences in growth rates or cellular morphology between control and treated cells. This result is in agreement with the work done by Hua et al. (1995d) where the authors had identified SKOV-3 cells to be an ovarian cancer cell line resistant to the effects of estrogens and antiestrogens. In their experiments, SKOV-3 cells were depleted of steroids, and then treated with 0.1, 1 and 10 nM estradiol. It was found that the cell line did not show any mitogenic effects in response to the estrogens. In addition, cells were also treated with antiestrogens ICI 164,384 (pure antiestrogen) and 40H-tamoxifen (partial antagonist) at various doses, and neither of these agents was able to induce any 154 changes in growth in these cells. Our experiments in this chapter confirmed the literature findings; these results ensure that the cell line we are working with is not growth sensitive to estrogens, and therefore it is likely that these cells will also not be growth sensitive to the knockdown of ER. In the experimental system employed in this chapter, specially designed and validated siRNA duplexes against the ER were chosen and their concentrations were titrated to confer the optimal level of knockdown. The knockdown of the receptor levels after siRNA treatment was confirmed by immunoblotting experiments. It was determined that the ER levels remained knocked down by 90% even at 72 hours after treatment with the siRNA duplex (Figure 3.2), which ensured that during the time-frame of our subsequent experiments with these cells, the ER levels remain suppressed. The ER knockdown cells (ER(-)) and the ER(+) SKOV-3 cells were treated with E2-7a and our results in Figure 3.4 clearly indicate that the absence of the ER makes the cells less sensitive to the drug. This decreased sensitivity is unlikely to be due to any inherent changes in growth patterns following ER knockdown; control experiments that measured cell doubling time after the ER siRNA/negative control (NC) treatments confirmed that the two cell populations grew at similar rates (data not shown). Therefore, the change in growth inhibition patterns between ER(+) and ER(-) cells following E2-7aC treatment is most likely a direct contribution of ER itself. Although knocking down the ER in SKOV-3 did not affect the growth of the cells, we had considered the possibility that the ER in SKOV-3 cells might be involved in determining cell survival, which might in turn affect the sensitivity towards E2-7a. Several experimental and clinical studies have implicated the ER in conferring neuroprotective 155 effects (Maggi et al., 2004; Morissette et al., 2008). Also, the ER has been shown to induce the expression of anti-apoptotic bcl-2 gene expression in response to estradiol through the interaction with cis-genomic Sp1 sites in MCF-7 and T47D breast cancer cells (Dong et al., 1999). ER-mediated inhibition of NF-KB contributes to the antiinflammatory and protective effects of estrogen in breast cancer, bone health and cardiovascular systems (Biswas et al., 2005; De et al., 2006). Based on these reports, we considered it possible that knocking down the ER might render the SKOV-3 more vulnerable to cytotoxic therapies, but the results from our control cytotoxicity experiments with cisplatin and melphalan diminish the likelihood of this possibility. Both cisplatin and melphalan treatments caused similar levels of growth inhibition in the ER(+) and ER(-) cell populations. Also, the AMS experiment with [14C]-melphalan treatment (Figure 3.6B) clearly demonstrated that a similar number of drug-DNA adducts were being formed in the SKOV-3 ER(+) and ER(-) cells. These results reinforce the conclusion that knocking down the ER did not make the cells inherently more sensitive to cytotoxic agents and that the results we observed best fit with the hypothesis that ER contributes to the change in the growth inhibition patterns between ER(+) and ER(-) cells following E2-7a treatment. This result is strongly in favor of our intended mechanism of action of E2-7a -- that is, the presence of the ER induces selective toxicity in ER(+) cells compared to the ER knockdown cells. We were interested in testing if the trend of drug-DNA adduct levels observed in each of the populations aligned with the differential toxicity in order to investigate whether the presence of the ER affected the adduct levels in treated cells. As mentioned earlier in this chapter, there are at least two means by which interaction of the ER with drug-DNA adduct can induce toxicity - repair shielding and transcription factor hijacking. In the SKOV-3 system, the hijacking model is less likely to be operative as a factor in 156 differential toxicity because the ER in this cell line has been shown to be deficient in the transcriptional activation of several of its canonical estrogen-responsive gene targets such as progesterone receptor, HER2/neu and cathepsin D (Hua et al., 1995d). In order to test whether the presence of the ER affected adduct levels, the DNA-adduct levels were measured in SKOV-3 cell populations (ER(+) and ER(-)) following continuous exposure to E2-7a. The results shown in Figure 3.6A indicate that the ER(+) population endured a higher adduct burden compared to the ER(-) cells. One difference between the ER(+) and ER(-) cells that could influence adduct levels is the greater growth observed in the ER(-) cells during the treatment. It can be seen from Table 3.2 that there were 1.3 times as many cells in the ER(-) compared to the ER(+) population following the 36 hour treatment. Given this difference in cell number, we need to ensure that the six-fold difference in adduct levels observed at this duration is still meaningful. If drug-DNA adducts were continually being formed in both the populations, then number of adducts per cell would still be six fold higher in the ER(+) compared to the ER(-) cells, independent of the actual cell numbers in both the populations. If, on the other hand, adduct formation were limited, then the existing adducts would be distributed among the growing cells. In this case, the higher growth rate in the ER(-) cells can account at the most for a 1.3 fold lower level of adducts per million bases. Therefore, in either case, the increased adduct levels in the ER(+) population is an not artifact of differential cell growth, and the significant six-fold increase in the ER(+) adduct levels is a meaningful consequence of the presence of ER in these cells. Melphalan treatment resulted in equivalent levels of adducts in both the cell populations. In addition, it could be noticed that the adduct levels did not change with increasing 157 duration of treatment. This result likely indicates continual formation of adducts in both the populations considering that the cell numbers are higher in the 36 hour time point compared to the 8 hour time point. The melphalan cytotoxicity experiment (Figure 3.3B) indicated that there was no difference in toxicity towards ER(+) and ER(-) cells. This finding was consistent with the observation of similar adduct levels in this pair of cells when treated with melphalan. Melphalan has the same DNA damaging moiety as E2-7a, and is expected to form similar kind of DNA damage as E2-7a. Therefore, this experiment makes it unlikely that inherent differences in the repair capacity of the ER(+) and ER(-) cells might be responsible for the differential adduct levels. Since the cells are expected to be isogenic except for their ER status, the result implies that the ER plays an irrefutable role in increasing the adduct levels in E2-7a treated ER(+) cells above the levels observed in the ER(-) population. The increased toxicity of the ER(+) cells towards E2-7a is best explained by the role of ER in facilitating the persistence of drug-DNA adducts in the ER(+) cells. The receptor could mediate this effect either through its role in adduct shielding, or it could, through some other means, enable increased formation of adducts in the ER(+) cells. The adduct levels observed are the net effect of adduct formation and repair, and with the continuous exposure to drug, it is not possible deconvolve the contribution of one from the other with these studies. Nevertheless, the contribution of repair shielding to E2-7aL toxicity is strongly supported by the experiments described in this chapter - ER prevents the adducts from getting repaired, thereby resulting in net increased levels of adducts. The increased adduct levels correlate well with the enhanced toxicity towards the drug in ER(+) cells. 158 Conclusions The design of novel targeted chemotherapeutics is indispensable in order to tackle the issues of off-target effects and the emergence of resistance with existing therapies. We have rationally designed a multifunctional genotoxicant that has the ability to overcome both these concerns. By combining the ability to form adducts within DNA and to disrupt cellular signaling into one agent, we have developed the compound E2-7aL that is capable of selectively targeting cancer cells - E2-7a combines the modern targeted approach with the traditional method of interfering with DNA replication. In this chapter, the mechanistic basis of E2-7a toxicity was explored further, and we found that the compound was selectively toxic to the ER(+) cells compared to the ER knockdown SKOV-3 cells. In addition, these results were supported by the observation that the E2-7a-DNA adduct levels were higher in the ER(+) compared to the ER(-) cell population. Since the two cell populations are expected to be isogenic except for their ER status, the observed difference in adduct levels is likely a direct contribution of the ER. There was no differential toxicity observed in the ER(+) and ER knockdown cells towards other alkylating agents such as melphalan and cisplatin. In addition, the drugDNA adduct levels in melphalan-treated cells were similar regardless of the ER status of the cell population, indicating that the differential adducts levels observed with E2-7a treatment was likely due to the interaction of the ER with the either E2-7a or the drugDNA complex or both. The results presented in this chapter strongly support the repair shielding mechanism of E2-7a action and present a compelling case for the potential of E2-7a as a targeted chemotherapeutic agent. 159 Future Directions The adduct level measurements described in this chapter strongly support a role for the ER in persistence of adducts in the ER(+) cells. Approaching the hypothesis from another angle would make the results described in this chapter more substantial. We would like to measure and characterize adduct levels in a more direct manner, and towards this end, we are exploring the application of Triple Quadrupole Mass Spectrometry (TQMS) in order detect specific drug-DNA base adducts and crosslinks. This technique has shown promising results in detecting guanine adducts and guanineguanine crosslinks in preliminary in vitro experiments with E2-7a-treated calf-thymus DNA. Once this technique is optimized to detect E2-7c-DNA adducts in SKOV-3 cells, we would like to repeat the siRNA experiment and detect the adducts using the TQMS in order to confirm the differential adduct levels in the ER(+) versus the ER(-) populations. As mentioned in the previous section, the role of ER in creating more adducts in the ER(+) cells is likely a net effect of the role of ER in repair shielding and in facilitating increased rate of adduct formation. Once adducts are allowed to form, the rate of removal of adducts could be determined by measuring adduct levels at the end of a recovery period following drug treatment. Such experiments would help estimate the role of ER in adduct repair and provide the basis for a more direct role for the ER in repair shielding. Another experiment that would support the repair shielding hypothesis is along similar lines as the in vitro competition assay described by Mitra et al (2002). The authors had added increasing amounts of estradiol to verify the gel shift caused by the E2-7a-DNA- 160 ER complex, and found that estradiol could diminish the gel shift by competitively binding to the ER. We would like to perform a similar experiment by adding estradiol to the SKOV-3 cells (ER(+) and the knockdown ER(-) cells) following E2-7a treatment and observe whether the adduct levels are diminished in the ER(+) cells. This result would test the specificity of drug-DNA adduct interaction with the ER, and if we do observe diminished adduct levels in ER(+) cells, it would underscore the role of ER in repair shielding. The experiments described above would further confirm the results described in this chapter and provide additional evidence to reinforce the mechanism of action of E2-7a. 161 Doubling times of SKOV-3 cells with estrogen and antiestrogen treatment Treatment Doubling time in hours Untreated (FBS) 30 Untreated(CDT-FBS) 30 E210nM 31 E2100nM 31 E21pM 30 ICI 10 nM 31 ICI 100 nM 32 ICI 1 pM 31 Table 3.1: Doubling times of SKOV-3 cells. All cells (except untreated-FBS) were grown in phenol-red free medium + CDT-FBS, and treated with estradiol (E2) or ICI 182,780 at various doses. The growth curves were fitted to an exponential curve, and the doubling times were calculated from the equation: Doubling Time=ln(2)/kd where kd is the growth constant inthe fitted exponential curve N= Noexp(kd.t). N, No are cell numbers at time t and to (time in hours). The data in the table is derived from the growth curves in Figure 3.1 162 Adducts, cell numbers and ["4C]-E2-7a distributions in ER(+) and ER(-) SKOV-3 cells ER ER status ER(+) ER(-) 8h 24h 36h 8h 24h 36h bases 43.8 91.2 80.0 22.9 30.5 12.9 Average cell number (x 10-5) 1.02 1.49 1.54 1.07 1.77 2.02 Countscells (x 10-4) 2.71 2.65 2.96 2.54 2.72 2.87 [14C] Countsmedium (x 10-4) 11.6 11.8 12.0 11.4 11.8 12.1 Adducts per 106 [14C] Table 3.2: Adducts, cell numbers and [14C] counts in ER(+) and ER(-) cells. Cells were treated with 2 pM [14C]-E2-7a for 8, 24 or 26 hours. Adducts per million bases, cell numbers, radioactivity per well (cpm) associated with cells and with the medium are shown for each duration of treatment. The adduct levels vs. duration of treatment is also shown in Figure 3.6. 163 -Chapter 3- SKOV-3 cells are not growth-sensitive to estradiol or ICI 182,780 Growth Characteristics - SKOV-3 cells 7006000500x 400 .a E 300C i200100 00 2 3 Time in days LI Untreated ME2 1OnM ElCllOnM 4 5 ! E2 1OOnM E E2 1uM ICl1OOnM MICIl1uM Figure 3.1: SKOV-3 cells treated with various doses of 17pestradiol or pure antiestrogen ICI 182,780. The growth pattern of the cells appears to be not affected by the presence of estradiol or antiestrogen at various doses (Error bars represent the standard deviation). The doubling time of the cells with each of the treatments is listed in Table 3.1. 164 ER is downregulated after siRNA treatment for up to 72 hours NC siRNA 72 hours 48 hours 24 hours NC siRNA NC siRNA ER (66 kDa) -MEO Actin MO - Duration after siRNA treatment Percentage knockdown of ER 24 hours 92.0 48 hours 78.9 72 hours 89.9 Figure 3.2: ER levels after knockdown using siRNA. SKOV-3 cells treated with 25 nM siRNA against ER or negative control (NC) for 7 hours, followed by incubation in fresh medium for 24, 48 or 72 hours. Percentage knockdown is relative to corresponding negative control treatment and is determined by spot densitometry after normalizing the actin levels. ER levels can be seen to be knocked down up to 72 hours after siRNA treatment. 165 ER(+) and ER(-) SKOV-3 cells are equally sensitive to cisplatin; they are equally sensitive to melphalan as well Cisplatin: ER(+) vs. ER(-) SKOV-3 cells 100 80 60 40 20 0 0 5 15 10 Dose in uM 20 Melphalan: ER(+) vs. ER(-) SKOV-3 cells 100 -c .0-a 0 L~. 80 ~1) 60 CU C 'I) 0 40 (9 c,) 4-a ci) 0~ 20 0 5 15 10 Dose in uM 20 Figure 3.3: Cells treated with siRNA against ER or negative control, followed by treatment for 36 hours with A. Cisplatin, or B. Melphalan.There is no significant difference in the response of the ER(+) vs. ER(-) populations to either of the drugs. 166 ER(+) cells are more sensitive to E2-7a than ER(-) cells E2-7a: ER(+) vs. ER(-) SKOV-3 cells + ER(-) 100 0 0) --A- ER(+) 80 60 40 20 0 0 10 5 15 20 Dose inuM Figure 3.4: Cells treated with siRNA against ER or negative control for 7 hours. After 24 hours, ER(+) and ER(-) populations were treated with various doses of E2-7a for 36 hours. Adherent cells were collected, and percent growth relative to corresponding untreated control was measured and plotted. Error bars represent standard deviations. Suppression of the ER in SKOV-3 cells decreases sensitivity to E2-7a. The treatment doses of 2.5, 5 and 10 pM show significant difference in growth percentage between ER(+) and ER(-) populations (P<0.05). 167 E2-7a forms DNA adducts in SKOV-3 cells 80 C,) C Dose Response: SKOV-3 cells treated with E2-7a 68.0 70 60 0 .E 50 40 CO -o 30 14.8 20 8.6 8 10 2.6 0.3 0.6 1.3 2.5 5.0 Dose inuM Figure 3.5: Cells treated with various doses of radiolabeled E2-7a for 4 hours, DNA isolated and adduct levels measured using AMS. The values above the bars are the mean number of adducts per million bases for each dose. Error bars represent standard deviation. 168 E2-7a adduct levels are higher in ER(+) compared to ER(-) SKOV-3 cell populations Adduct levels: E2-7a treatment 40120 - ER(+) + ER(-) C-1000 A -80 60 40 20 8 12 16 20 24 28 36 32 40 Duration of treatment (hours) Adduct levels: Melphalan treatment 0020 -A ER(+) +ER(-) C 0 B E 010 0. 8 12 16 20 24 28 32 36 40 Duration of treatment (hours) Figure 3.6: Adduct levels in siRNA vs. NC treated SKOV-3 cells after treatment with A. E2-7ax, or B. Melphalan. E2-7ax treated cells show significantly higher adduct levels in ER(+) compared to ER(-) cells (P<0.05). Melphalan treated ER(+) and ER(-) cells show similar adduct levels. 169 Reference List 1. Abend M (2003) Reasons to reconsider the significance of apoptosis for cancer therapy. Int J Radiat Biol 79:927-941 2. Agarwal R, Kaye SB (2003) Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer 3:502-516 3. Ahn HJ, Kim YS, Kim JU, Han SM, Shin JW, Yang HO (2004) Mechanism of taxolinduced apoptosis in human SKOV3 ovarian carcinoma cells. J Cell Biochem 91:1043-1052 4. Ali S, Metzger D, Bornert JM, Chambon P (1993) Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J 12:1153-1160 5. American Cancer Society (2008) Cancer Facts and Figures. 6. Arai M, Utsunomiya J, Miki Y (2004) Familial breast and ovarian cancers. Int J Clin Oncol 9:270-282 7. Araujo SJ, Nigg EA, Wood RD (2001) Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol Cell Biol 21:2281-2291 170 8. Argento M, Hoffman P, Gauchez AS (2008) Ovarian cancer detection and treatment: current situation and future prospects. Anticancer Res 28:3135-3138 9. Armitage P, Doll (1954) The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer 8:1-12 10. Asahina H, Kuraoka 1,Shirakawa M, Morita EH, Miura N, Miyamoto I, Ohtsuka E, Okada Y, Tanaka K (1994) The XPA protein is a zinc metalloprotein with an ability to recognize various kinds of DNA damage. Mutat Res 315:229-237 11. Balcome S, Park S, Quirk Dorr DR, Hafner L, Phillips L,Tretyakova N (2004) Adenine-containing DNA-DNA cross-links of antitumor nitrogen mustards. Chem Res Toxicol 17:950-962 12. Bandera CA, Ye B, Mok SC (2003) New technologies for the identification of markers for early detection of ovarian cancer. Curr Opin Obstet Gynecol 15:51-55 13. Bardin A, Hoffmann P, Boulle N, Katsaros D, Vignon F, Pujol P, Lazennec G (2004) Involvement of estrogen receptor beta in ovarian carcinogenesis. Cancer Res 64:5861-5869 14. Barth H, Hoffmann I, Klein S, Kaszkin M, Richards J, Kinzel V (1996) Role of cdc25-C phosphatase in the immediate G2 delay induced by the exogenous factors epidermal growth factor and phorbolester. J Cell Physiol 168:589-599 15. Bast RC, Jr., Klug TL, St JE, Jenison E, Niloff JM, Lazarus H, Berkowitz RS, Leavitt T, Griffiths CT, Parker L,Zurawski VR, Jr., Knapp RC (1983) A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N Engl J Med 309:883-887 171 16. Bernstein E, Caudy AA, Hammond SM, Hannon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409:363-366 17. Biederbick A, Kern HF, Elsasser HP (1995) Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 66:3-14 18. Bishop JM (1991) Molecular themes in oncogenesis. Cell 64:235-248 19. Biswas DK, Singh S, Shi Q, Pardee AB, Iglehart JD (2005) Crossroads of estrogen receptor and NF-kappaB signaling. Sci STKE 2005:e27 20. Bonadonna G, Brusamolino E, Valagussa P, Rossi A, Brugnatelli L, Brambilla C, De LM, Tancini G, Bajetta E, Musumeci R, Veronesi U (1976) Combination chemotherapy as an adjuvant treatment in operable breast cancer. N Engl J Med 294:405-410 21. Bookman MA, Greer BE, Ozols RF (2003) Optimal therapy of advanced ovarian cancer: carboplatin and paclitaxel versus cisplatin and paclitaxel (GOG158) and an update on GOG0182-ICON5. Int J Gynecol Cancer 13 Suppl 2:149-155 22. Borini A, Rebellato E (2008) Focus on breast and ovarian cancer. Placenta 29 Suppl B:184-190 23. Botti J, Djavaheri-Mergny M, Pilatte Y, Codogno P (2006) Autophagy signaling and the cogwheels of cancer. Autophagy 2:67-73 24. Boulikas T, Vougiouka M (2004) Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review). Oncol Rep 11:559-595 172 I 25. Bowler J, Lilley TJ, Pittam JD, Wakeling AE (1989) Novel steroidal pure antiestrogens. Steroids 54:71-99 26. Brandenberger AW, Lebovic DI, Tee MK, Ryan IP,Tseng JF, Jaffe RB, Taylor RN (1999) Oestrogen receptor (ER)-alpha and ER-beta isoforms in normal endometrial and endometriosis-derived stromal cells. Mol Hum Reprod 5:651-655 27. Brandenberger AW, Tee MK, Jaffe RB (1998) Estrogen receptor alpha (ER-alpha) and beta (ER-beta) mRNAs in normal ovary, ovarian serous cystadenocarcinoma and ovarian cancer cell lines: down-regulation of ER-beta in neoplastic tissues. J Clin Endocrinol Metab 83:1025-1028 28. Brookes, Lawley PD (1961) The reaction of mono- and di-functional alkylating agents with nucleic acids. Biochem J 80:496-503 29. Brown JM, Attardi LD (2005) The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 5:231-237 30. Brown SJ, Kellett PJ, Lippard SJ (1993) lxr1, a yeast protein that binds to platinated DNA and confers sensitivity to cisplatin. Science 261:603-605 31. Bruhn, Toney JH, Lippard SJ (1990) Biological processing of DNA modified by platinum compounds., 38 edn 32. Bucourt R, Vignau M, Torelli V (1978) New biospecific adsorbents for the purification of estradiol receptor. J Biol Chem 253:8221-8228 33. Burger RA (2007) Experience with bevacizumab in the management of epithelial ovarian cancer. J Clin Oncol 25:2902-2908 173 34. Chabner BA, Longo DL (2009) Cancer Chemotherapy and Biotherapy 35. Chabner BA, Roberts TG, Jr. (2005) Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer 5:65-72 36. Chatterjee M, Mohapatra S, lonan A, Bawa G, li-Fehmi R, Wang X, Nowak J, Ye B, Nahhas FA, Lu K, Witkin SS, Fishman D, Munkarah A, Morris R, Levin NK, Shirley NN, Tromp G, Abrams J, Draghici S, Tainsky MA (2006) Diagnostic markers of ovarian cancer by high-throughput antigen cloning and detection on arrays. Cancer Res 66:1181-1190 37. Chen Z, Fadiel A, Jia JF, Sakamoto H, Carbone R, Naftolin F (1999) Induction of apoptosis and suppression of clonogenicity of ovarian carcinoma cells with estrogen mustard. Cancer 85:2616-2622 38. Clark (2006) Molecular targeted drugs and growth factor receptor inhibitors. In: Chabner BA, Longo DL (eds) 39. Clarke PG (2002) Apoptosis: from morphological types of cell death to interacting pathways. Trends Pharmacol Sci 23:308-309 40. Clinton GM, Hua W (1997) Estrogen action in human ovarian cancer. Crit Rev Oncol Hematol 25:1-9 41. Coezy E, Borgna JL, Rochefort H (1982) Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res 42:317-323 174 42. Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS (1997) Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology 138:4613-4621 43. Cowley SM, Hoare S, Mosselman S, Parker MG (1997) Estrogen receptors alpha and beta form heterodimers on DNA. J Biol Chem 272:19858-19862 44. Cunat S, Hoffmann P, Pujol P (2004) Estrogens and epithelial ovarian cancer. Gynecol Oncol 94:25-32 45. DaSilva JN, van Lier JE (1990) Synthesis and structure-affinity of a series of 7 alpha-undecylestradiol derivatives: a potential vector for therapy and imaging of estrogen-receptor-positive cancers. J Med Chem 33:430-434 46. De BK, Vanden BW, Haegeman G (2006) Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene 25:6868-6886 47. Deng CX (2006) BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 34:1416-1426 48. DeVita VT, Hellman S, Rosenberg SA (1997) Princples and Practice of Oncology, 5 edn 49. Donahue BA, Augot M, Bellon SF, Treiber DK, Toney JH, Lippard SJ, Essigmann JM (1990) Characterization of a DNA damage-recognition protein from mammalian cells that binds specifically to intrastrand d(GpG) and d(ApG) DNA adducts of the anticancer drug cisplatin. Biochemistry 29:5872-5880 175 50. Dong L, Wang W, Wang F, Stoner M, Reed JC, Harigai M, Samudio I, Kladde MP, Vyhlidal C, Safe S (1999) Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J Biol Chem 274:3209932107 51. Driscoll MD, Sathya G, Muyan M, Klinge CM, Hilf R, Bambara RA (1998) Sequence requirements for estrogen receptor binding to estrogen response elements. J Biol Chem 273:29321-29330 52. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498 53. Essigmann JM, Rink SM, Park HJ, Croy RG (2001b) Design of DNA damaging agents that hijack transcription factors and block DNA repair. Adv Exp Med Biol 500:301-313 54. Essigmann, Rink SM, Park HJ, Croy RG (2001a) Design of DNA damaging agents that hijack transcription factors and block DNA repair. In: Dansette (ed) Biological Reactive Intermediates VI, 6 edn 55. Festjens N, Vanden BT, Vandenabeele P (2006) Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 1757:1371-1387 56. Fliss AE, Benzeno S, Rao J, Caplan AJ (2000) Control of estrogen receptor ligand binding by Hsp90. J Steroid Biochem Mol Biol 72:223-230 176 57. Foulds (1954) The experimental study of tumor progression: a review. Cancer Res 14:327-339 58. Friedberg EC, Bonura T, Love JD, McMillan S, Radany EH, Schultz RA (1981) The repair of DNA damage: recent developments and new insights. J Supramol Struct Cell Biochem 16:91-103 59. Friedberg EC, Walker G, Siede W (1995) DNA repair and mutagenesis 60. Fu XD, Simoncini T (2008) Extra-nuclear signaling of estrogen receptors. IUBMB Life 60:502-510 61. Furuta T, Ueda T, Aune G, Sarasin A, Kraemer KH, Pommier Y (2002) Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res 62:4899-4902 62. Gibb RK, Taylor DD, Wan T, O'Connor DM, Doering DL, Gercel-Taylor C (1997) Apoptosis as a measure of chemosensitivity to cisplatin and taxol therapy in ovarian cancer cell lines. Gynecol Oncol 65:13-22 63. Gozuacik D, Kimchi A (2007) Autophagy and cell death. Curr Top Dev Biol 78:217245 64. Grant DF, Bessho T, Reardon JT (1998) Nucleotide excision repair of melphalan monoadducts. Cancer Res 58:5196-5200 65. Green S, Walter P, Greene G, Krust A, Goffin C, Jensen E, Scrace G,Waterfield M, Chambon P (1986) Cloning of the human oestrogen receptor cDNA. J Steroid Biochem 24:77-83 177 66. Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J (1986) Sequence and expression of human estrogen receptor complementary DNA. Science 231:11501154 67. Greenlee RT, Murray T, Bolden S, Wingo PA (2000) Cancer statistics, 2000. CA Cancer J Clin 50:7-33 68. Haapala E, Hakala K, Jokipelto E, Vilpo J, Hovinen J (2001) Reactions of N,Nbis(2-chloroethyl)-p-aminophenylbutyric acid (chlorambucil) with 2'deoxyguanosine. Chem Res Toxicol 14:988-995 69. Hall JM, McDonnell DP (1999) The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140:5566-5578 70. Hamilton TC, Young RC, McKoy WM, Grotzinger KR, Green JA, Chu EW, WhangPeng J, Rogan AM, Green WR, Ozols RF (1983) Characterization of a human ovarian carcinoma cell line (NIH:OVCAR-3) with androgen and estrogen receptors. Cancer Res 43:5379-5389 71. Hamilton TC, Young RC, Ozols RF (1984) Experimental model systems of ovarian cancer: applications to the design and evaluation of new treatment approaches. Semin Oncol 11:285-298 72. Hanahan D,Weinberg RA (2000) The hallmarks of cancer. Cell 100:57-70 178 73. Hartley JA, Berardini MD, Souhami RL (1991) An agarose gel method for the determination of DNA interstrand crosslinking applicable to the measurement of the rate of total and "second-arm" crosslink reactions. Anal Biochem 193:131-134 74. He Z, Ingles CJ (1997) Isolation of human complexes proficient in nucleotide excision repair. Nucleic Acids Res 25:1136-1141 75. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8:193-204 76. Hippert MM, O'Toole PS, Thorburn A (2006) Autophagy in cancer: good, bad, or both? Cancer Res 66:9349-9351 77. Ho SM (2003) Estrogen, progesterone and epithelial ovarian cancer. Reprod Biol Endocrinol 1:73 78. Htun H, Holth LT, Walker D, Davie JR, Hager GL (1999) Direct visualization of the human estrogen receptor alpha reveals a role for ligand in the nuclear distribution of the receptor. Mol Biol Cell 10:471-486 79. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995d) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 80. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995a) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 179 81. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995b) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 82. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995c) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 83. Huang A, Kaley G (2004) Gender-specific regulation of cardiovascular function: estrogen as key player. Microcirculation 11:9-38 84. Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A (1994) HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc Natl Acad Sci U S A 91:1039410398 85. Hurley LH (2002) DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer 2:188-200 86. Igney FH, Krammer PH (2002) Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2:277-288 87. Jacobs I, Bast RC, Jr. (1989) The CA 125 tumour-associated antigen: a review of the literature. Hum Reprod 4:1-12 88. Jaffe N, Frei E, Ill, Traggis D, Bishop Y (1974) Adjuvant methotrexate and citrovorum-factor treatment of osteogenic sarcoma. N Engl J Med 291:994-997 180 89. Jakowlew SB, Breathnach R, Jeltsch JM, Masiakowski P, Chambon P (1984) Sequence of the pS2 mRNA induced by estrogen in the human breast cancer cell line MCF-7. Nucleic Acids Res 12:2861-2878 90. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ (2008) Cancer statistics, 2008. CA Cancer J Clin 58:71-96 91. Jones CJ, Wood RD (1993) Preferential binding of the xeroderma pigmentosum group A complementing protein to damaged DNA. Biochemistry 32:12096-12104 92. Kim DH, Rossi JJ (2007) Strategies for silencing human disease using RNA interference. Nat Rev Genet 8:173-184 93. King WJ, Greene GL (1984) Monoclonal antibodies localize oestrogen receptor in the nuclei of target cells. Nature 307:745-747 94. Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato AC (1988) A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res 16:647-663 95. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di SF, Dice JF, Difiglia M, neshKumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, 181 Dunn WA, Jr., Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska 1,Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri 1,Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov 0, Settleman J, Shacka JJ, Shapiro IM,Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanaka K, Tanida 1,Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van dK, I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, 182 Zabirnyk 0, Zheng X, Zhu X, Deter RL (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151-175 96. Koberle B, Roginskaya V, Wood RD (2006) XPA protein as a limiting factor for nucleotide excision repair and UV sensitivity in human cells. DNA Repair (Amst) 5:641-648 97. Kohn KW (1996) Beyond DNA cross-linking: history and prospects of DNAtargeted cancer treatment--fifteenth Bruce F. Cain Memorial Award Lecture. Cancer Res 56:5533-5546 98. Kohn KW, Hartley JA, Mattes WB (1987) Mechanisms of DNA sequence selective alkylation of guanine-N7 positions by nitrogen mustards. Nucleic Acids Res 15:10531-10549 99. Krasner C (2007) Aromatase inhibitors in gynecologic cancers. J Steroid Biochem Mol Biol 106:76-80 100. Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138:863-870 101. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA (1996) Cloning of a novel receptor expressed in rat prostate and ovary. Proc Nati Acad Sci U S A 93:5925-5930 183 102. Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der BB, Gustafsson JA (1998a) Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 139:4252-4263 103. Kuiper GG, Shughrue PJ, Merchenthaler I, Gustafsson JA (1998b) The estrogen receptor beta subtype: a novel mediator of estrogen action in neuroendocrine systems. Front Neuroendocrinol 19:253-286 104. Kumar R, Thompson EB (1999) The structure of the nuclear hormone receptors. Steroids 64:310-319 105. Kundu GC, Schullek JR, Wilson IB (1994) The alkylating properties of chlorambucil. Pharmacol Biochem Behav 49:621-624 106. Kurreck J (2009) RNA interference: from basic research to therapeutic applications. Angew Chem Int Ed Engl 48:1378-1398 107. Kushner PJ, Agard D, Feng WJ, Lopez G, Schiau A, Uht R, Webb P, Greene G (2000a) Oestrogen receptor function at classical and alternative response elements. Novartis Found Symp 230:20-26 108. Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P (2000b) Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol 74:311317 109. Lahooti H, White R, Danielian PS, Parker MG (1994) Characterization of liganddependent phosphorylation of the estrogen receptor. Mol Endocrinol 8:182-188 184 110. Langdon SP, Hirst GL, Miller EP, Hawkins RA, Tesdale AL, Smyth JF, Miller WR (1994) The regulation of growth and protein expression by estrogen in vitro: a study of 8 human ovarian carcinoma cell lines. J Steroid Biochem Mol Biol 50:131135 111. Langdon SP, Ritchie A, Young K, Crew AJ, Sweeting V, Bramley T, Hillier S, Hawkins RA, Tesdale AL, Smyth JF, . (1993) Contrasting effects of 17 betaestradiol on the growth of human ovarian carcinoma cells in vitro and in vivo. Int J Cancer 55:459-464 112. Lau KM, Mok SC, Ho SM (1999) Expression of human estrogen receptor-alpha and -beta, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci U S A 96:5722-5727 113. Lawley PD (1966) Effects of some chemical mutagens and carcinogens on nucleic acids. Prog Nucleic Acid Res Mol Biol 5:89-131 114. Lazennec G (2006) Estrogen receptor beta, a possible tumor suppressor involved in ovarian carcinogenesis. Cancer Lett 231:151-157 115. Le GP, Montano MM, Schodin DJ, Katzenellenbogen BS (1994) Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem 269:44584466 116. Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463-477 185 117. Li M, HERTZ R, BERGENSTAL DM (1958) Therapy of choriocarcinoma and related trophoblastic tumors with folic acid and purine antagonists. N Engl J Med 259:66-74 118. Liberman RG, Tannenbaum SR, Hughey BJ, Shefer RE, Klinkowstein RE, Prakash C, Harriman SP, Skipper PL (2004) An interface for direct analysis of (14)c in nonvolatile samples by accelerator mass spectrometry. Anal Chem 76:328-334 119. Lindahl T (1979) DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog Nucleic Acid Res Mol Biol 22:135-192 120. Lindgren PR, Cajander S, Backstrom T, Gustafsson JA, Makela S, Olofsson JI (2004) Estrogen and progesterone receptors in ovarian epithelial tumors. Mol Cell Endocrinol 221:97-104 121. Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, Lynch P, Burke W, Press N (2006) Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 296:15071517 122. Lukanova A, Kaaks R (2005) Endogenous hormones and ovarian cancer: epidemiology and current hypotheses. Cancer Epidemiol Biomarkers Prev 14:98107 123. MacLeod MC, Powell KL, Tran N (1995) Binding of the transcription factor, Sp1, to non-target sites in DNA modified by benzo[a]pyrene diol epoxide. Carcinogenesis 16:975-983 186 124. Maggi A, Ciana P, Belcredito S, Vegeto E (2004) Estrogens in the nervous system: mechanisms and nonreproductive functions. Annu Rev Physiol 66:291-313 125. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM (1995) The nuclear receptor superfamily: the second decade. Cell 83:835-839 126. Masiakowski P, Breathnach R, Bloch J, Gannon F, Krust A, Chambon P (1982) Cloning of cDNA sequences of hormone-regulated genes from the MCF-7 human breast cancer cell line. Nucleic Acids Res 10:7895-7903 127. Masood S, Heitmann J, Nuss RC, Benrubi GI (1989) Clinical correlation of hormone receptor status in epithelial ovarian cancer. Gynecol Oncol 34:57-60 128. May FE, Westley BR (1988) Identification and characterization of estrogenregulated RNAs in human breast cancer cells. J Biol Chem 263:12901-12908 129. May FE, Westley BR (1995) Estrogen regulated messenger RNAs in human breast cancer cells. Biomed Pharmacother 49:400-414 130. McA'Nulty MM, Lippard SJ (1996) The HMG-domain protein lxr1 blocks excision repair of cisplatin-DNA adducts in yeast. Mutat Res 362:75-86 131. McA'Nulty MM, Whitehead JP, Lippard SJ (1996) Binding of lxr1, a yeast HMGdomain protein, to cisplatin-DNA adducts in vitro and in vivo. Biochemistry 35:6089-6099 132. Meijer AJ, Codogno P (2006) Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 27:411-425 187 133. Merajver SD, Pham TM, Caduff RF, Chen M, Poy EL, Cooney KA, Weber BL, Collins FS, Johnston C, Frank TS (1995) Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nat Genet 9:439-443 134. Meyn, Murray D (1986) Cell cycle Effects of alkylating agents. 135. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, . (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66-71 136. Mitra K, Marquis JC, Hillier SM, Rye PT, Zayas B, Lee AS, Essigmann JM, Croy RG (2002) A rationally designed genotoxin that selectively destroys estrogen receptor-positive breast cancer cells. J Am Chem Soc 124:1862-1863 137. Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861-2873 138. Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069-1075 139. Moretti-Rojas I, Fuqua SA, Montgomery RA, 111, McGuire WL (1988) A cDNA for the estradiol-regulated 24K protein: control of mRNA levels in MCF-7 cells. Breast Cancer Res Treat 11:155-163 140. Morimoto H, Safrit JT, Bonavida B (1991) Synergistic effect of tumor necrosis factor-alpha- and diphtheria toxin-mediated cytotoxicity in sensitive and resistant human ovarian tumor cell lines. J Immunol 147:2609-2616 188 141. Morimoto H, Yonehara S, Bonavida B (1993) Overcoming tumor necrosis factor and drug resistance of human tumor cell lines by combination treatment with antiFas antibody and drugs or toxins. Cancer Res 53:2591-2596 142. Morisset M, Capony F, Rochefort H (1986) Processing and estrogen regulation of the 52-kilodalton protein inside MCF7 breast cancer cells. Endocrinology 119:2773-2782 143. Morissette M, Le SM, DAstous M, Jourdain S, Al SS, Morin N, Estrada-Camarena E, Mendez P, Garcia-Segura LM, Di PT (2008) Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J Steroid Biochem Mol Biol 108:327-338 144. Mutch DG (2002) Surgical management of ovarian cancer. Semin Oncol 29:3-8 145. Narod SA, Foulkes WD (2004) BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 4:665-676 146. Nash JD, Ozols RF, Smyth JF, Hamilton TC (1989) Estrogen and anti-estrogen effects on the growth of human epithelial ovarian cancer in vitro. Obstet Gynecol 73:1009-1016 147. Newton K, Strasser A (1998) The Bcl-2 family and cell death regulation. Curr Opin Genet Dev 8:68-75 148. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J,Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA (2001) Mechanisms of estrogen action. Physiol Rev 81:1535-1565 189 149. Noy N (2007) Ligand specificity of nuclear hormone receptors: sifting through promiscuity. Biochemistry 46:13461-13467 150. O'Connor PM, Kohn KW (1990) Comparative pharmacokinetics of DNA lesion formation and removal following treatment of L1210 cells with nitrogen mustards. Cancer Commun 2:387-394 151. O'Donnell AJ, Macleod KG, Burns DJ, Smyth JF, Langdon SP (2005) Estrogen receptor-alpha mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocr Relat Cancer 12:851-866 152. Ogawa S, Inoue S, Watanabe T, Hiroi H,Orimo A, Hosoi T, Ouchi Y, Muramatsu M (1998) The complete primary structure of human estrogen receptor beta (hER beta) and its heterodimerization with ER alpha in vivo and in vitro. Biochem Biophys Res Commun 243:122-126 153. Olefsky JM (2001) Nuclear receptor minireview series. J Biol Chem 276:3686336864 154. Olive PL, Banath JP (1995) Sizing highly fragmented DNA in individual apoptotic cells using the comet assay and a DNA crosslinking agent. Exp Cell Res 221:19-26 155. Olive PL, Banath JP (2006) The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1:23-29 156. Olive PL, Banath JP, Durand RE (1990) Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the "comet" assay. Radiat Res 122:86-94 190 157. Osborne CK, Yochmowitz MG, Knight WA, Ill, McGuire WL (1980) The value of estrogen and progesterone receptors in the treatment of breast cancer. Cancer 46:2884-2888 158. Oxelmark E, Knoblauch R,Arnal S, Su LF, Schapira M, Garabedian MJ (2003) Genetic dissection of p23, an Hsp90 cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J Biol Chem 278:36547-36555 159. Ozols RF (2005) Treatment goals in ovarian cancer. Int J Gynecol Cancer 15 Suppl 1:3-11 160. Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM, Baergen R (2003) Phase Ill trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage Ill ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 21:3194-3200 161. Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61:439-444 162. Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K,Wilbanks G, Nicosia S, Cantor A, Sutphen R (2005) BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104:2807-2816 163. Papac RJ (2001) Origins of cancer therapy. Yale J Biol Med 74:391-398 164. Parker MG (1991) Nuclear Hormone Receptors 191 165. Parker, Jensen EV (1991) Overview of the Nuclear Receptor Family. 166. Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55:74-108 167. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1dependent autophagy. Cell 122:927-939 168. Pearce ST, Jordan VC (2004) The biological role of estrogen receptors alpha and beta in cancer. Crit Rev Oncol Hematol 50:3-22 169. Pettersson K, Grandien K, Kuiper GG, Gustafsson JA (1997) Mouse estrogen receptor beta forms estrogen response element-binding heterodimers with estrogen receptor alpha. Mol Endocrinol 11:1486-1496 170. Pil PM, Lippard SJ (1992) Specific binding of chromosomal protein HMG1 to DNA damaged by the anticancer drug cisplatin. Science 256:234-237 171. Pratt WB, Ruddon RW, Ensminger WD, Maybaum J (1994) The Anticancer drugs 172. Pujol P, Rey JM, Nirde P, Roger P, Gastaldi M, Laffargue F, Rochefort H, Maudelonde T (1998) Differential expression of estrogen receptor-alpha and -beta messenger RNAs as a potential marker of ovarian carcinogenesis. Cancer Res 58:5367-5373 173. Rao BR, Slotman BJ (1991) Endocrine factors in common epithelial ovarian cancer. Endocr Rev 12:14-26 192 174. Rao GG, Miller DS (2005) Clinical applications of hormonal therapy in ovarian cancer. Curr Treat Options Oncol 6:97-102 175. Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER (2004) Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol 18:2854-2865 176. Reardon JT, Vaisman A, Chaney SG, Sancar A (1999) Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamineplatinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res 59:39683971 177. Reed (2006) Cisplatin, carboplatin and oxaliplatin. In: Chabner BA, Longo DL (eds) Cancer chemotherapy and biotherapy, 4 edn 178. Reggiori F, Klionsky DJ (2002) Autophagy in the eukaryotic cell. Eukaryot Cell 1:11-21 179. Renan MJ (1993) How many mutations are required for tumorigenesis? Implications from human cancer data. Mol Carcinog 7:139-146 180. Reynolds A, Anderson EM, Vermeulen A, Fedorov Y, Robinson K, Leake D, Karpilow J, Marshall WS, Khvorova A (2006) Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA 12:988-993 (2002) Sex steroids and the construction and 181. Riggs BL, Khosla S, Melton U, 111 conservation of the adult skeleton. Endocr Rev 23:279-302 193 182. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Persson IR (2001) Risk factors for epithelial borderline ovarian tumors: results of a Swedish case-control study. Gynecol Oncol 83:575-585 183. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Persson IR (2002a) Risk factors for invasive epithelial ovarian cancer: results from a Swedish case-control study. Am J Epidemiol 156:363-373 184. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Weiderpass E, Persson IR (2002b) Hormone replacement therapy and the risk of invasive epithelial ovarian cancer in Swedish women. J Natl Cancer Inst 94:497504 185. Riman T, Dickman PW, Nilsson S, Nordlinder H, Magnusson CM, Persson IR (2004) Some life-style factors and the risk of invasive epithelial ovarian cancer in Swedish women. Eur J Epidemiol 19:1011-1019 186. Rimer, Schildkraut JM (1997) Cancer Screening. In: DeVita VT, Hellman S, Rosenberg SA (eds), Fifth edn pp 619-631 187. Rink SM, Yarema KJ, Solomon MS, Paige LA, Tadayoni-Rebek BM, Essigmann JM, Croy RG (1996) Synthesis and biological activity of DNA damaging agents that form decoy binding sites for the estrogen receptor. Proc Natl Acad Sci U S A 93:15063-15068 188. Risch HA (2002) Hormone replacement therapy and the risk of ovarian cancer. Gynecol Oncol 86:115-117 194 189. Roberts D, Schick J,Conway S, Biade S, Laub PB, Stevenson JP, Hamilton TC, O'Dwyer PJ, Johnson SW (2005) Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br J Cancer 92:1149-1158 190. Rochefort H (1990) Cathepsin D in breast cancer. Breast Cancer Res Treat 16:313 191. Rochefort H (1998) [Estrogens, cathepsin D and metastasis in cancers of the breast and ovary: invasion or proliferation?]. C R Seances Soc Biol Fil 192:241-251 192. Rochefort H (1999) [Estrogen-induced genes in breast cancer, and their medical importance]. Bull Acad Natl Med 183:955-968 193. Rose PG, Reale FR, Longcope C, Hunter RE (1990) Prognostic significance of estrogen and progesterone receptors in epithelial ovarian cancer. Obstet Gynecol 76:258-263 194. Roy G, Horton JK, Roy R, Denning T, Mitra S, Boldogh 1(2000) Acquired alkylating drug resistance of a human ovarian carcinoma cell line is unaffected by altered levels of pro- and anti-apoptotic proteins. Oncogene 19:141-150 195. Russo IH,Russo J (1998) Role of hormones in mammary cancer initiation and progression. J Mammary Gland Biol Neoplasia 3:49-61 196. Rutherford T, Brown WD, Sapi E,Aschkenazi S, Munoz A, Mor G (2000) Absence of estrogen receptor-beta expression in metastatic ovarian cancer. Obstet Gynecol 96:417-421 195 197. Satokata 1,Tanaka K, Miura N, Narita M, Mimaki T, Satoh Y, Kondo S, Okada Y (1992) Three nonsense mutations responsible for group A xeroderma pigmentosum. Mutat Res 273:193-202 198. Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S (2000) Ligand-, cell-, and estrogen receptor subtype (alpha/beta)dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem 275:53795387 199. Sawyers C (2004) Targeted cancer therapy. Nature 432:294-297 200. Schaner ME, Ross DT, Ciaravino G, Sorlie T, Troyanskaya 0, Diehn M, Wang YC, Duran GE, Sikic TL, Caldeira S, Skomedal H, Tu IP, Hernandez-Boussard T, Johnson SW, O'Dwyer PJ, Fero MJ, Kristensen GB, Borresen-Dale AL, Hastie T, Tibshirani R, van de RM, Teng NN, Longacre TA, Botstein D, Brown PO, Sikic BI (2003) Gene expression patterns in ovarian carcinomas. Mol Biol Cell 14:43764386 201. Schildkraut JM, Schwingl PJ, Bastos E, Evanoff A, Hughes C (1996) Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet Gynecol 88:554-559 202. Schuchard M, Landers JP, Sandhu NP, Spelsberg TC (1993) Steroid hormone regulation of nuclear proto-oncogenes. Endocr Rev 14:659-669 203. Segnitz B, Gehring U (1995) Subunit structure of the nonactivated human estrogen receptor. Proc Natl Acad Sci U S A 92:2179-2183 196 204. Segnitz B, Gehring U (1997) The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J Biol Chem 272:18694-18701 205. Sharma U, Marquis JC, Nicole DA, Hillier SM, Fedeles B, Rye PT, Essigmann JM, Croy RG (2004) Design, synthesis, and evaluation of estradiol-linked genotoxicants as anti-cancer agents. Bioorg Med Chem Lett 14:3829-3833 206. Shuey DJ, McCallus DE, Giordano T (2002) RNAi: gene-silencing in therapeutic intervention. Drug Discov Today 7:1040-1046 207. Slotman BJ, Rao BR (1988) Ovarian cancer (review). Etiology, diagnosis, prognosis, surgery, radiotherapy, chemotherapy and endocrine therapy. Anticancer Res 8:417-434 208. States JC, McDuffie ER, Myrand SP, McDowell M, Cleaver JE (1998) Distribution of mutations in the human xeroderma pigmentosum group A gene and their relationships to the functional regions of the DNA damage recognition protein. Hum Mutat 12:103-113 209. Stenoien DL, Mancini MG, Patel K, Allegretto EA, Smith CL, Mancini MA (2000) Subnuclear trafficking of estrogen receptor-alpha and steroid receptor coactivator-1. Mol Endocrinol 14:518-534 210. Stenoien DL, Patel K, Mancini MG, Dutertre M, Smith CL, O'Malley BW, Mancini MA (2001) FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat Cell Biol 3:15-23 197 211. Stevens EV, Nishizuka S, Antony S, Reimers M, Varma S, Young L, Munson PJ, Weinstein JN, Kohn EC, Pommier Y (2008) Predicting cisplatin and trabectedin drug sensitivity in ovarian and colon cancers. Mol Cancer Ther 7:10-18 212. Sunters A, Springer CJ, Bagshawe KD, Souhami RL, Hartley JA (1992) The cytotoxicity, DNA crosslinking ability and DNA sequence selectivity of the aniline mustards melphalan, chlorambucil and 4-[bis(2-chloroethyl)amino] benzoic acid. Biochem Pharmacol 44:59-64 213. Tew, Colvin OM, Jones RB (2006) Clinical and High-Dose Alkylating Agents. In: Chabner BA, Longo DL (eds) Cancer Chemotherapy and Biotherapy, 4 edn 214. Treiber DK, Zhai X, Jantzen HM, Essigmann JM (1994) Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor). Proc Natl Acad Sci U S A 91:5672-5676 215. Turteltaub KW, Dingley KH (1998) Application of accelerated mass spectrometry (AMS) in DNA adduct quantification and identification. Toxicol Lett 102-103:435439 216. Turteltaub KW, Felton JS, Gledhill BL, Vogel JS, Southon JR, Caffee MW, Finkel RC, Nelson DE, Proctor ID, Davis JC (1990) Accelerator mass spectrometry in biomedical dosimetry: relationship between low-level exposure and covalent binding of heterocyclic amine carcinogens to DNA. Proc Natl Acad Sci U S A 87:5288-5292 217. Tyagi A, Singh RP, Agarwal C, Siriwardana S, Sclafani RA, Agarwal R (2005) Resveratrol causes Cdc2-tyrl5 phosphorylation via ATM/ATR-Chk1/2-Cdc25C 198 pathway as a central mechanism for S phase arrest in human ovarian carcinoma Ovcar-3 cells. Carcinogenesis 26:1978-1987 218. Vandenabeele P,Vanden BT, Festjens N (2006) Caspase inhibitors promote alternative cell death pathways. Sci STKE 2006:e44 219. Venkitaraman AR (2004) Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat Rev Cancer 4:266-276 220. von Angerer E, Prekajac J, Strohmeier J (1984) 2-Phenylindoles. Relationship between structure, estrogen receptor affinity, and mammary tumor inhibiting activity in the rat. J Med Chem 27:1439-1447 221. Walker G, MacLeod K, Williams AR, Cameron DA, Smyth JF, Langdon SP (2007) Estrogen-regulated gene expression predicts response to endocrine therapy in patients with ovarian cancer. Gynecol Oncol 106:461-468 222. Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, Staub A, Jensen E, Scrace G,Waterfield M, . (1985) Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci U S A 82:7889-7893 223. Webb P, Lopez GN, Uht RM, Kushner PJ (1995) Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol 9:443-456 224. Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, Kushner PJ (1999) The estrogen receptor enhances AP-1 activity by two distinct mechanisms with 199 different requirements for receptor transactivation functions. Mol Endocrinol 13:1672-1685 225. Wilson TM, Ewel A, Duguid JR, Eble JN, Lescoe MK, Fishel R, Kelley MR (1995) Differential cellular expression of the human MSH2 repair enzyme in small and large intestine. Cancer Res 55:5146-5150 226. Wu X, Fan W, Xu S, Zhou Y (2003) Sensitization to the cytotoxicity of cisplatin by transfection with nucleotide excision repair gene xeroderma pigmentosun group A antisense RNA in human lung adenocarcinoma cells. Clin Cancer Res 9:58745879 227. Xu Y, Traystman RJ, Hurn PD, Wang MM (2004) Membrane restraint of estrogen receptor alpha enhances estrogen-dependent nuclear localization and genomic function. Mol Endocrinol 18:86-96 228. Yap TA, Carden CP, Kaye SB (2009) Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer 9:167-181 229. Zakeri Z, Bursch W, Tenniswood M, Lockshin RA (1995) Cell death: programmed, apoptosis, necrosis, or other? Cell Death Differ 2:87-96 230. Zang XP, Pento JT (2008) SiRNA inhibition of ER-alpha expression reduces KGFinduced proliferation of breast cancer cells. Anticancer Res 28:2733-2735 231. Zheng L,Annab LA, Afshari CA, Lee WH, Boyer TG (2001) BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci U S A 98:9587-9592 200 232. Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408:433-439 201 Chapter 4 Investigation of cytotoxicity and other biological properties of phenylindole compounds in ovarian cancer cells 203 Introduction Ovarian cancer (OC) is the leading cause of gynecological deaths in the United States, with 22,000 new cases and 15,000 deaths associated with the disease every year (American Cancer Society, 2008). The five-year survival rate of OC disease ranges from 30 to 92%, depending on the severity of the spread of the disease at the time of diagnosis (Jemal et al., 2008). The relative lack of specific early signs and symptoms of OC combined with inefficient screening techniques results in overall low cure rates for the disease. Currently, the treatment options for both primary and recurrent cancers are limited, and research focuses on developing new modalities of selectively targeting OC and its metastases. Debulking surgery remains key in OC treatment (Agarwal and Kaye, 2003); adjuvant chemotherapy has also been shown to be beneficial. Results from randomized trails have now established platinum-taxol combination regimen as the firstline treatment for advanced OC (du Bois et al., 1999; Neijt et al., 2000; Ozols et al., 2003; Piccart et al., 2000). Although the response rate to such therapies has been promising, studies point to the outcome that most of the patients will relapse with a median progression-free survival of 18 months (Greenlee et al., 2001). In this scenario, therapy based on better understanding of the disease seems to be the most effective means to a better prognosis for the patient (Yap et al., 2009). Although the etiological origins of OC have not been fully identified, several studies point to the role of estrogen in promoting tumorigenesis (Greenlee et al., 2001; Lukanova and Kaaks, 2005). Estrogen has been shown to induce proliferation in many ER(+) cancer cells, and antiestrogens have been reported to inhibit the mitogenic effect of estrogen in ER(+) cells (Langdon et al., 1994; Nash et al., 1989). Epidemiological studies have 204 found that hormonal replacement therapy can increase the risk of OC in postmenopausal women, while oral contraceptive use has been associated with decreased risk of developing OC (Cunat et al., 2004; Risch, 2002; Slotman and Rao, 1988). In addition, interventions with antiestrogens and aromatase inhibitors have displayed some clinical benefits for OC patients (Cunat et al., 2005; Krasner, 2007; Walker et al., 2007). These findings reinforce the crucial role played by estrogens and their cognate receptors in the development and progression of OC. In this context, estrogen receptor (ER), the mediator of estrogen action in ovarian systems, is an attractive target for the development of drugs to treat primary and recurrent OC. ER has been found to be expressed in 60% of ovarian cancers at the time of diagnosis (Slotman and Rao, 1988). The work that I have described in this chapter focuses on novel chemotherapeutics that selectively target cancer cells expressing the ER in the context of ovarian cancer. Our lab previously reported the synthesis of bifunctional compounds that displayed selective toxicity in breast cancer cells that expressed the ER (Rink 1996). The design of these compounds was based on the mechanistic understanding of cisplatin action. These compounds consisted of a 2-phenylindole (2PI) group which serves as a ligand for the ER, linked to N,N-bis(2-chloroethyl)aniline that can react with DNA to produce covalent adducts. These drug-DNA adducts have the unique ability to form complexes with the ER. We proposed that the formation of ER-DNA adduct complexes prevented repair of the adducts in ER(+) cells and was responsible for the selective toxicity that we observed in ER(+) cells. Since ER has been found to play a crucial role in ovarian cancer, we hypothesized that such compounds may have similar effects in ovarian cancer cell lines. 205 In this chapter the toxicity of two of our bifunctional compounds towards the ovarian cancer cell lines Caov-3 and OVCAR-3 is investigated. While both compounds contain a reactive N,N-bis(2-chloroethyl)aniline group, they have dissimilar 2-phenylindole groups with high and low affinities for the ER. These compounds - 2PI-C6NC2-mustard (2PI; high affinity for the ER) and 2PI(OH)-C6NC2-mustard (2PI(OH); low affinity for the ER) were tested in ovarian cancer cells, and by studying the responses to these compounds we hoped to distinguish the role of the ER in mediating the biological effects of the phenylindole compounds. Comparisons were also made between cellular response to these compounds and cisplatin, the current front-line therapeutic in ovarian cancer. The experiments in this chapter were a joint effort by Dr. Pei-Sze Ng and me, and references will be made wherever relevant. Materials and Methods Chemicals: The bifunctional compounds 2PI and 2PI(OH) used in these studies were synthesized in the Essigmann laboratory by Pei-Sze Ng. Radiolabeled analogs of the 2PI and 2Pl(OH) compounds were prepared by coupling [14C]-labeled nitrogen mustards (with a specific activity of 26 mCi/mmol) to phenylindole carbonates. Details of the synthetic steps and characterization of the compound have been described in a previous publication (Rink et al., 1996). The synthesis has also been outlined in Figure 4.1. Cisplatin, 17p-estradiol and propidium iodide (PI) were obtained from Sigma-Aldrich. 206 Cell Culture: Caov-3 and NIH:OVCAR-3 (referred to as OVCAR-3) cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD). Fetal Bovine Serum (FBS) was obtained from Thermo Fisher Scientific (Logan, UT). OVCAR-3 cells were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) containing 2.5 mg/mL glucose, 2 mM GlutaMax, 1 mM sodium pyruvate and 100 mM HEPES buffer, supplemented with 0.01 mg/mL bovine insulin (Invitrogen) and 10% FBS. Caov-3 cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS and 1 mM sodium pyruvate. All cell lines were grown in a humidified 5% CO2/air atmosphere at 370C. For growth inhibition assays, cells were seeded in 12-well plates at 5x10 4 cells per well. Twenty four hours later, the test compounds dissolved in DMSO were added to the medium. After 24 hours, the drug-containing medium was removed and the cells were grown in fresh medium for an additional 24 hours. Cells were detached using trypsin/EDTA solution (Invitrogen) and the number of cells in control and drug-treated wells were determined using a Z1 Coulter Counter. The percent growth inhibition was calculated as the ratio of cell number in treated to that in control wells multiplied by 100. Significance values were calculated using unpaired t tests, with a two-tailed P value less than 0.05 indicating significance. Growth inhibition assays were performed by Pei-Sze Ng. Relative Binding Affinity Assays: Relative ligand binding affinities (RBAs) were determined by a competitive radioligand binding assay using purified ER from a commercial source (ERa; Invitrogen). [3H]-estradiol at 10 nM and ERa at 1.5 nM were combined with unlabeled competitors 207 and incubated for 12-18 hours at 40C. Receptor-ligand complexes were adsorbed to hydroxyapatite and radioactivity was determined by scintillation counting. Binding affinities were expressed as a percentage relative to estradiol (estradiol RBA =100%). Binding assays were conducted by Pei-Sze Ng. Immunoblot Analysis: Whole cells extracts used for ER analyses were prepared by lysis of cells in SDS sample buffer (62.5 mM Tris-HCI (pH 6.8), 2% SDS, 41.6 mM DTT, 10% glycerol, 0.01% bromophenol blue). Whole cell extracts for cleaved caspase-3 were prepared by cell lysis using RIPA buffer (Santacruz; 1X tris-buffered saline (TBS), 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1 mM sodium orthovanadate, 1 mM NaF, 50 mM Tris-HCI (pH 7.4) and protease inhibitor cocktail (Sigma-Aldrich)). Protein concentrations were determined using Bio-Rad RC-DC protein assay with Bovine Serum Albumin (BSA) standard. Equal amounts of protein were loaded and separated on SDS-polyacrylamide gels and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Membranes were blocked with 5% milk in TBS-Tween20 (TBS-T; 0.1% Tween20, 10 mM Tris (pH 7.4), 150 mM NaCl) and incubated with primary antibodies overnight at 40C. The antibody against ER (mouse monoclonal) was obtained from Upstate Biotechnology (Lake Placid, NY) and anti-cleaved caspase-3 antibody was purchased from Cell Signaling Technologies (Beverly, MA). Anti-mouse and anti-rabbit secondary antibodies conjugated to horseradish peroxidase were obtained from Cell Signaling Technologies. Membranes were incubated with secondary antibodies for 1-2 hours, and antibody complexes formed with the secondary antibodies were visualized using Enhanced Chemiluminescence reagents (SuperSignal West Femto, Pierce, Rockford, IL). 208 Cell-Cycle Analysis: Cells were treated with test compounds for 30-32 hours. Both adherent and nonadherent cells were recovered, washed with cold phosphate-buffered saline (PBS) and fixed in 100% ethanol overnight at 4*C. Ethanol-fixed cells were washed with 1% FBS in PBS, resuspended at 106 cells/mL, and stained with 10 tg/mL Pl. Stained cells were incubated with 0.5 pg/mL RNase A (DNase free; Roche Diagnostics) for 30 minutes at 370C. DNA content was determined using a Becton-Dickinson flow cytometer (BD FACScan). Cell cycle data was analyzed using the MODFIT-LT 3.0 program to estimate the percentage of cells in G1, S and G2/M phases of the cell cycle. Single-Cell Alkaline Gel Electrophoresis (Comet assay): Cells were treated with test compounds for indicated times. Alkaline comet assay was performed as outlined in the Trevigen comet assay protocol using the reagents provided in the Trevigen kit (Trevigen, Gaithersburg, MD). Following treatment, cells were trypsinized, resuspended in PBS at 105 cells/mL and irradiated on ice using a GammaCell 40 Irradiator with a dose of 8 Gy. Irradiated cells were suspended in liquid Low Melting Agarose (LMA) at 370C and plated onto a CometSlide (Trevigen). After the agar solidified, cells were incubated in lysis solution (2.5 M NaCl, 100 mM EDTA, 10mM Tris base (pH 10), 1%Sodium Lauryl Sarcosine, 0.01% Triton X-100) for an hour at 40C, followed by incubation with 100 pL of 1 mg/mL Proteinase K (Roche Diagnostics) for an hour at room temperature. Slides were then treated for 30 minutes with alkaline solution (0.3 M NaOH, 1 mM EDTA, pH >13), followed by electrophoresis under alkaline conditions (0.3 M NaOH, 1mM EDTA). Slides were then dried, stained with SYBR Green dye, and photographed using a Nikon Eclipse epifluorescence microscope. Images 209 were analyzed using the Komet 4.2 Single Cell Gel Electrophoresis software (Kinetic Imaging Limited, U.K.). A total of 100 cells per sample were analyzed, 50 per duplicate slide. The Olive Tail Moment (OTM), defined as the product of the tail length and the fraction of total DNA in the tail, was calculated for each cell by the software. OTM incorporates a measure of both the smallest detectable size of migrating DNA (reflected in the comet tail length) and the number of relaxed or broken pieces which is represented by the intensity of DNA in the tail (Olive et al., 1990; Olive and Banath, 2006). Comet data were represented using boxplots plotted using SSC-Stat, a statistical add-in for Excel. Boxplot was used to give a graphical representation of the heterogeneity of the comet data. The box contains the middle 50% of the data, the upper boundary of the box indicates the 75 th percentile of the data, and the lower boundary the 25 th percentile. The range of the middle two quartiles is called the interquartile range. The line in the box is the median. The ends of the "whiskers" on the boxplot extend to a maximum of 1.5 times the interquartile range. The average OTM values were indicated on the boxplot. Percentage decrease in the average OTM was calculated for each treatment relative to the untreated irradiated control, and significance values were calculated using the MannWhitney statistic (P < 0.001 was considered significant). Adduct measurement using Accelerator Mass Spectrometry (AMS): Cells were exposed to various doses of [14C]-labeled test compounds. At the end of the incubation period, wells were washed once with fresh medium, followed by a wash with PBS, and cells were trypsinized and collected. They were then lysed in 0.5 mL extraction buffer containing HEPES (50 mM), NaCl (100 mM), EDTA (10 mM) and 0.8% SDS, and 210 incubated with RNase A (Roche Diagnostics; 0.9 mg/mL, 30 minutes at 370C) followed by Proteinase K (Roche Diagnostics; 0.6 mg/mL, 2 hours at 370C). Samples were subjected to phenol:chloroform extraction, and the aqueous phase was extracted once more with chloroform. DNA was precipitated with 100% ethanol overnight at -200C, and subsequently pelleted by centrifugation at 7000g for 15 min at 40C. The pellet was washed with 70% ethanol, and dried in vacuo. DNA was dissolved in 0.2 mL water and the concentration was estimated by absorbance at 260 nm. AMS analyses were conducted at the Biological Engineering Accelerator Mass Spectrometry (BEAMS) Lab at MIT by Paul Skipper and Rosa Liberman as described in Liberman, et al. (2004). DNA solutions were applied directly to a CuO matrix used for sample combustion. A standard consisting of a solution of [14C-methyl] bovine serum albumin was used to calibrate the instrument. The amount of [14 C] in each sample was calculated from the peak area ratio of the sample to the standard. All samples were analyzed at least twice. Readings were obtained from AMS instrument as dpm/pg DNA and converted to adducts per million bases. Two-tailed P values for AMS experiments were calculated using unpaired t tests, and a value of P<0.05 was considered significant. The distribution of the [14 C]-labeled drug in cells and medium was calculated using scintillation counting. Results 2PI demonstrates almost a ten-fold higher binding affinity to the ER compared to 2PI(OH). The binding affinities of the 2-phenylindole compounds relative to that of 17pestradiol were measured. The structures of the two compounds and their RBA values are shown in Figure 4.2. The 2PI compound shows excellent binding affinity for the ER, 211 binding at 57% the affinity of estradiol. The 2PI(OH) compound, which lacks the 5-hydroxy group on the ER recognition domain, only weakly interacted with the ER (RBA=6). The binding affinities of the phenylindole compounds follow the same trend as was observed with the published data on these compounds (Rink et al., 1996). Caov-3 and OVCAR-3 express the ER. In order to test whether the putative interaction between the 2-phenylindole moiety and ER could occur in treated cells, the ER expression in the two cell lines was studied. On immunoblotting OVCAR-3 and Caov-3 cells for ER levels, the protein was detected in both the cell lines, as can be seen in Figure 4.3. The breast cancer cell line MCF-7 was used as a positive control for ER expression. The relative levels of the receptor were calculated using spot densitometry. The levels of ER (relative to Caov-3 cells) were Caov-3: OVCAR-3: MCF-7 = 1: 0.8: 1.8. MCF-7 cells expressed approximately twice the level of ER as the ovarian cancer cells. This is in agreement with the levels reported in previous studies (Havrilesky et al., 2001). 2PI(OH) and 2PI exert equivalent toxicity in both Caov-3 and OVCAR-3 cells. The cytotoxicity of the compounds 2P1 and 2PI(OH) was determined in comparison to cisplatin in the two ovarian cancer cell lines. The results of the experiment are shown in Figure 4.4. The cytotoxicity data demonstrated that Caov-3 cells were more sensitive to all the compounds tested than OVCAR-3 cells. This observation is also consistent with the reported data on the sensitivity of Caov-3 cells to cisplatin relative to OVCAR-3 (Serova et al., 2006). In Caov-3 cells, all the three compounds seemed to induce similar levels of toxicity. The cells showed a slightly enhanced sensitivity towards 2PI(OH) compared to 2PI and cisplatin at the 5 pM dose, but all the other doses were equivalently cytotoxic. At doses 212 above 5 pM, the three compounds were able to achieve almost a 100% cell kill. In OVCAR-3 cells, a similar trend was observed, although the cells were less sensitive to the three compounds than Caov-3. At concentrations of 5 pM and 10 pM, 2PI(OH) was more cytotoxic than both 2PI and cisplatin. Cisplatin was unable to achieve complete cell killing even at the highest concentration tested (20 pM), in contrast to 2P and 2PI(OH). Overall, in both the cell lines, the three compounds seemed to be equivalent in inducing growth inhibition. The unexpected finding was that the 2PI(OH) compound was as toxic as the 2PI compound in both the cell lines tested. The selective increased toxicity of 2PI over 2PI(OH) that was previously seen in ER(+) breast cancer cells (Rink et al., 1996) was not observed in either of the ovarian cancer cell lines tested. 2PI induces S-phase arrest in ovarian cancer cells. In order to study whether the cytotoxicity of the phenylindole compounds was associated with changes in the cell cycle patterns, the effect of our compounds on cell cycle progression of Caov-3 and OVCAR-3 cells was evaluated by flow cytometry. The results are shown in Figure 4.5 (Caov-3) and Figure 4.6 (OVCAR-3). Both cell lines were treated with 2.5 pM of compounds for 30-32 hours (one complete cell cycle). In the control cell population, a higher S-phase fraction in Caov-3 cells (35%) than in OVCAR-3 cells (21 %) was observed. A higher S-phase population in the untreated Caov-3 cells could increase its susceptibility to DNA damage (Kolfschoten et al., 2000). This might explain the observation that Caov-3 cells were more sensitive to treatment with our compounds and cisplatin in comparison to OVCAR-3 cells. In both the cell lines, we noticed consistent trends in the response to the three compounds. 2PI and cisplatin treatments resulted in a marked increase of cells in the 213 S-phase compared to the control population. 2PI increased the S-phase population from 35% to 65% in Caov-3 cells and from 21% to 71% in OVCAR-3 cells. Cisplatin treatment arrested 80% of Caov-3 cells and 46% of OVCAR-3 cells in the S-phase. This result is consistent with the report that the cisplatin causes DNA damage and arrest cells in the S-phase or the G2/M phase of the cell cycle (Nguyen et al., 1993; Sekiguchi et al., 1996). In contrast, 2PI(OH) treatment caused only a slight increase in S-phase from 21%-39% in OVCAR-3 cells, and no changes in cell cycle patterns were observed in Caov-3 cells with drug treatment. The cytotoxic profiles of the three drugs at this dose (2.5 pM) were similar to each other, and were also equivalent in the two cell lines. The dissimilarity in the cell cycle distributions with 2PI(OH) treatment compared to 2PI and cisplatin seems to indicate that 2PI(OH) may be acting via a different mechanism from the other two compounds. Comet assay detects crosslinks induced by 2PI in both the cell lines, and by 2PI(OH) in OVCAR-3 cells. Cells treated with the phenylindole compounds were subjected to the modified alkaline comet assay (Olive and Banath, 1995; Olive and Banath, 2006) to determine the presence of interstrand crosslinks (ICLs). Drug-treated or mock-treated cells were .irradiated with 8 Gy dose to induce random DNA strand breakage, followed by cell lysis and alkaline unwinding. The presence of ICLs retards the migration of the irradiated DNA during alkaline electrophoresis, resulting in a reduced Olive Tail Moment (OTM) compared to the mock-treated irradiated control. The results of the experiment are shown in Figure 4.7 (Caov-3, 6 hours) and Figure 4.8 (OVCAR-3, 8 hours). The percent decrease in tail moments, which indicates the extent of crosslink formation, is listed in Table 4.1. 214 In Caov-3 cells, treatment with cisplatin at 2.5 pM for 6 hours showed a significant decrease in tail moment, and increasing the dose to 5 pM did not seem to change the extent of crosslinking detected by the assay - both the doses of cisplatin induced approximately 30% decrease in OTM. 2PI compound was also found to induce crosslinking, although to a lesser extent than cisplatin. The 2.5 pM dose of 2PI induced a 20.8% decrease in OTM and the 5 pM dose a 13.1 % decrease in OTM compared to the irradiated sample. The percent decrease in OTM induced by 2PI was significant at both the doses, indicating that ICLs were being formed by the 2P1 compound. In contrast, 2PI(OH) treatment did not induce any detectable crosslink formation in Caov-3 cells. The interesting observation with 2PI(OH) treatment was that both the doses induced a significant increase in OTM compared to the irradiated control. This result can be explained by the possible formation of extensive single strand breaks or induction of changes related to apoptosis such as DNA fragmentation with the treatment. Both these situations would relax the DNA strands, leading to increased OTM in the 2PI(OH) treated samples compared to the untreated irradiated control. Therefore, the effect we see is likely a net result of crosslinks and strand breaks, and an increase in OTM indicates that the effect of the single strand breaks outweighs that of any crosslinks that might have formed. The increase in OTM does not rule out ICL formation, it merely eliminates the detection of the lesion with this assay. All the three compounds formed crosslinks in OVCAR-3 cells (Figure 4.8, Table 4.1), and the crosslinking was more potent in these cells than in Caov-3 cells. Cisplatin displayed the highest extent of crosslinking - a significant decrease in OTM of 48% with 2.5 pM dose and 71% with 5 pM dose were observed. 2PI demonstrated a 54% decrease in OTM with both the doses. 2PI(OH) also produced crosslinks in these cells, 215 although to a lesser extent than cisplatin and 2PI (a decrease in OTM of 25% at 2.5 pM and 40% at 5 piM). Crosslinking caused by the drugs could lead to a block in replication, triggering checkpoint activation, and leading to cell cycle arrest. In this light, the results of the comet assay seem to be consistent with the cell cycle experiments. In Caov-3 cells, cisplatin and 2PI induced S-phase arrest and these drugs were also shown to form ICLs. 2PI(OH) does not show a change in the cell cycle distribution and no crosslinking ability could be detected using the comet assay. A similar parallel was seen between the cell cycle arrest patterns and crosslink formation in OVCAR-3 cells. Similar adduct levels are detected with the test compounds using AMS in OVCAR-3 and Caov-3 cells. We were interested in testing whether the phenylindole compounds formed similar levels of DNA adducts in treated cells. AMS studies (results shown in Figure 4.9) with radiolabeled 2PI and 2PI(OH) demonstrated that both the compounds were capable of forming DNA adducts in treated cells. Doses from 0.5 IM to 5 pM were tested, and all the doses were observed to form similar number of DNA adducts in both Caov-3 cells and OVCAR-3 cells. At the 5 IM dose there was an abnormally low number of adducts in OVCAR-3 cells treated with 2PI(OH). It was expected that the 5 pM dose would produce as many as, if not more adducts than the 2.5 pM dose of 2PI(OH). This anomalous response was not observed in the Caov-3 cells. This data could not be explained, and we regarded this anomalous reading as an outlier point. All the other doses from 0.5-3.5 pM in both cell lines seemed to demonstrate a clear dose response to the drugs. Although some doses seemed to produce more adducts with one compound than with the other, overall, we did not detect any significant changes in adduct levels with the two compounds. The analysis of formation of DNA 216 adducts demonstrated that 2PI(OH) was as effective as 2PI in the formation of DNA adducts. Both the cell lines also seemed to form an equivalent number of adducts with the phenylindole compounds. A stronger caspase-3 activation is observed with 2PI(OH) than with 2PI treatment. Cells treated with 2PI and 2PI(OH) were immunoblotted for caspase-3 in order to investigate whether apoptosis occurred as a result of treatment. The results are shown in Figure 4.10. Caov-3 cells were treated with 5 pM and OVCAR-3 cells with 10 pM of test compounds for 15 hours. In both the cell lines, it was observed that treatment with cisplatin and 2PI(OH) induced a strong activation of procaspase-3 (32 kDa) to form the cleaved 17 kDa caspase-3 product, which was specifically detected by the antibody. The 2PI compound was also able to induce the activation of cleaved caspase-3, but only weakly. The difference in the activation of caspase-3 between 2PI and 2PI(OH) is consistent with the cytotoxicity studies; in Caov-3 cells at the 5 pM dose of the compounds, and in OVCAR-3 cells at the 10 pM dose, 2PI(OH) was significantly more cytotoxic than 2Pl. This difference in cytotoxicity is likely reflected in the caspase-3 activation - 2PI(OH) induces a much stronger apoptotic signal (as detected by the cleavage of procaspase-3 to the 17 kDa fragment) than the 2PI compound. Discussion Using the lessons learned from cisplatin, the Essigmann lab sought to improve the therapeutic index of antitumor agents by targeting cancers that aberrantly expressed the ER. The principle behind the drug design was that by combining a DNA damaging functionality and a protein recognition moiety specific for a cancer cell in the same 217 molecule, selective toxicity towards cancer cells could be achieved. This chapter deals with the study of the first set of rationally designed drugs developed in the Essigmann laboratory. These agents (2-phenylindole compounds, Figure 4.2) target cancers that express the ER. Rink et al. (1996) have described in detail the synthesis and characterization of the phenylindole compounds. The compound 2PI-C6NC2-mustard (2PI) contains the 2-phenylindole moiety that serves as a ligand for the ER (RBA of 57 relative to estradiol), and a DNA alkylating aniline mustard moiety that is capable of reacting covalently with DNA. 2PI(OH)-C6NC2mustard was synthesized as a control compound that has poor affinity for the ER (RBA = 6, Figure 4.2) as a result of only a monohydroxylation of the phenylindole moiety (von Angerer et al., 1984). Our hypothesis of the mechanism of action of 2PI is that it forms DNA lesions that attract the transcription factor ER. As explained in Rink et al. (1996), association with the ER could either shield the DNA adduct from repair or titrate away the transcription factor from its normal site of binding, thereby inducing toxicity. The 2PI(OH) compound is designed to not interact with the ER, and we expected it to be less toxic than the 2PI compound for this reason. We had seen promising results consistent with the above mechanism with the phenylindole compounds in breast cancer cells; it was observed that the 2PI compound was more toxic to MCF-7 breast cancer cells than its analog that does not interact with the ER. This result supported our hypothesis that the interaction with ER is crucial to the toxicity of our compound. The favorable toxicity profiles of the phenylindole compounds in breast cancer cells prompted us to investigate whether a similar response towards the compounds could be detected in ovarian cancer cells. 218 Cytotoxicity assays revealed that overall, Caov-3 cells were more sensitive to cisplatin, 2PI and 2PI(OH) than OVCAR-3 cells. The unexpected finding from the growth inhibition studies was that the 2PI(OH) control compound was as toxic to the cells as the 2PI compound. We expected that the ovarian cells, similar to the breast cancer cells tested earlier (Rink et al., 1996), would be more sensitive to the 2PI compound than to 2PI(OH). The differences in the response of the ovarian cells indicate that the two compounds are likely acting via different mechanisms in these cells. Although similar toxicity towards the two compounds was observed in the ovarian cancer cells, the apoptotic marker caspase-3 was found to be much more strongly induced by 2Pl(OH) compared to 2PI, indicating that the 2PI(OH) compound clearly induces apoptosis. This finding shown that 2PI(OH) and 2PI are clearly different in the way they induce cell death. Alternative modes of cell death induced by 2PI (in addition to apoptosis) might be able account for the toxicity of the compound. In this regard, we can draw a parallel between E2-7a-C6NC2-mustard (which contains estradiol as the ER binding domain) and the 2PI compound. We had discussed the cytotoxicity and cell death patterns induced by E2-7a in ovarian cancer cells in Chapter 2, and it was found that the compound induces cell death predominantly through the pathway of autophagy, and that no caspase-3 activation could be detected in ovarian cells treated with E2-7a. Since 2PI and E2-7ax are similar in their binding ability to DNA and both the compounds alkylate DNA, it is reasonable to expect similar effects in their manner of inducing cell death. Since 2PI does not seem to induce apoptosis, it is possible that the compound induces another form of cell death - autophagy - which could account for the toxicity of the compound. This possibility could also account for the absence of a strong cleaved caspase-3 signal with 2PI treatment. Investigations along these lines would help piece together the caspase-3 activation and the toxicity data of the phenylindole compounds; 219 these studies would also help understand the different mechanisms of action of the phenylindole compounds. The cytotoxicity of 2PI(OH) in the cisplatin-resistant ovarian cancer cells was intriguing and its mechanism of action was investigated further by studying the progression of cell cycle. In our studies, 2PI, similar to cisplatin, caused a potent S-phase arrest in Caov-3 and OVCAR-3 cells. 2PI(OH), however, did not cause any noticeable changes in the cell cycle patterns in Caov-3 cells and caused a slight S-phase arrest in OVCAR-3 cells. These observations can be explained by one of the two possibilities: (i) 2PI(OH) resulted in little or no DNA damage in comparison to cisplatin and 2PI, or (ii) the damage induced by 2PI(OH) went undetected or was not given sufficient time to be repaired, hence it bypassed the cell cycle checkpoints to directly induce toxicity. In either case, it seems likely that 2PI and 2PI(OH) target the cells through different mechanisms, which may or may not involve the ER. Both the phenylindole compounds contain a nitrogen mustard moiety capable of forming monoadducts and crosslinks, and the repair of such kinds of DNA damage could lead to delay in cell cycle progression (Brookes and Lawley, 1961; Meyn and Murray, 1986). We considered the possibility that the two compounds might be inducing different levels of DNA damage in treated cells, leading to the difference in cell cycle responses. In order to test this hypothesis, AMS and comet assay were employed to detect DNA adducts and ICLs respectively. The AMS experiments enabled the detection of drug-DNA adducts in populations treated with [14C]-2-phenylindole compounds. It was found that the two compounds formed a similar number of DNA adducts in both the cell lines (Figure 4.9). Moreover, the dose 220 response curve showed that the level of adducts was equivalent in Caov-3 and OVCAR-3 cells. Overall, the results of the AMS experiment indicated that the difference in the mechanism of action of 2PI and 2PI(OH) is not likely to stem from the differences in their abilities to form DNA adducts. The ability of the phenylindole compounds to form ICLs in treated Caov-3 and OVCAR-3 cells was examined using the comet assay. The comet assay results in Caov-3 cells (Figure 4.7) clearly demonstrated the ability of 2PI to form ICLs in these cells. ICL formation caused by the control compound could not be detected in Caov-3 cells. In fact, our observations indicate that the presence of any ICLs in these cells following 2PI(OH) treatment might have been masked by cell changes related to apoptosis (for example, DNA fragmentation) or by the formation extensive single strand breaks. We observed a similar trend in OVCAR-3 cells - both the compounds were found to induce ICL formation in these cells, although to different extents. The results of the comet assay indicate that the compounds differed in their ability to form crosslinks in the cells, or in their ability to form single strand breaks, or a combination of both. It was not evident whether the ER played a role in the toxicity of these compounds in the ovarian cancer system. The 2PI compound displayed DNA damage characteristics that are distinctive of an aniline mustard (Sunters et al., 1992) - it caused cytotoxicity and arrested both the cell lines in the S-phase of the cell cycle. The arrest pattern can be explained by the formation of ICLs leading to a block in replication, directly causing S-phase arrest in the cells. The presence of DNA damage, crosslinks and S-phase arrest could explain the observed toxicity of the compound. 221 The control compound 2PI(OH) did not seem to function as per its intended mechanism of action. Guided by the observation that the compound damaged DNA and induced apoptosis in treated cells but did not cause any changes in the cell cycle distribution, we considered certain alternative routes by which 2PI(OH) may be inducing toxicity. One possible explanation stems from the hypothesis that the compound interferes with cell cycle checkpoint machinery to suppress the activation of checkpoint proteins that are required to halt the cell cycle in response to DNA damage. The checkpoint abrogation by 2PI(OH) could underlie the difference in modes of action of the two phenylindole compounds. It is possible that the following 2PI(OH) treatment, cells proceed through the cell cycle despite their DNA damage, and therefore, are likely to be very sensitive to the compound. There are reports in the literature that cells that have not been arrested in S or G2 phases after exposure to a damaging agent and still enter mitosis with damaged DNA undergo cell death rapidly (Shapiro and Harper, 1999). Drugs that facilitate this kind of checkpoint abrogation could be extremely cytotoxic. Agents that inhibit cell cycle arrest elicited by DNA damage, and thereby sensitize cells to cisplatin and other alkylating drugs have been reported in the literature (Lau and Pardee, 1982; Schlegel and Pardee, 1986; Sugiyama et al., 2000). Furthermore, it has been shown that the sensitization of cells is especially selective for p53-deficient cells (Waldman et al., 1996). Cells that have compromised p53 are able to enter mitosis following DNA damaging treatments, and mitotic entry before the damage repair leads to cell death. The p53 tumor suppressor protein is required for G1 arrest induced by DNA damage in most cells, but usually not for arrest in S or G2. Normal cells (p53 positive) arrest mainly in G1, whereas p53negative tumor cells arrest in S or G2 in response to DNA damage. Therefore, the 222 abrogation of these checkpoints would make the p53 deficient cells extremely sensitive to DNA damage. One of the first compounds that was shown to abrogate S and G2 checkpoints and induce selective killing in p53-deficient cells is 7-hydroxystaurosporine (UCN-01), a protein kinase inhibitor (Bunch and Eastman, 1996; Bunch and Eastman, 1997; Wang et al., 1996). UCN-01 abrogates S and G2 checkpoints reportedly due to its inhibition of both Chk1 activity and the Cdc25C pathway (Graves et al., 2000). It has been shown that UCN-01 enhances the toxicity of cisplatin by abrogating the cell cycle arrest caused by cisplatin (Bunch and Eastman, 1996; Bunch and Eastman, 1997). This report suggests a possible mechanism of 2PI(OH) action - the compound might be capable of abrogating the checkpoint response to the arrest caused by its own DNA alkylating moiety. This would lead to enhanced sensitivity to the compound especially in p53 mutant cells. In our case, both Caov-3 and OVCAR-3 cells are p53 mutant (Hagopian et al., 1999; Wolf et al., 1999), and have been shown to be vulnerable to the effects of 2PI(OH). In effect, our compound might be assuming the roles of both UCN-01 and cisplatin in this system. It could be interacting with the cell cycle checkpoint machinery to inhibit cell cycle arrest, and thereby enhancing the toxicity caused by the aniline mustard moiety. Understanding the basis for checkpoint abrogation by the 2PI(OH) compound requires further investigations. These studies will doubtless help elucidate the basis of the differences in the mechanisms of action of 2PI and 2PI(OH). We postulate that the underlying mechanism of 2PI(OH) action might involve the interaction with cell cycle regulatory proteins, leading to abrogation of cell cycle checkpoints, whereas the 2PI compound induces DNA damage, cell cycle arrest and cytotoxicity. The role of ER in the 223 toxicity of either of the compounds needs to be investigated further as well. It is possible that the ER does not play a major role in the inhibition of cell proliferation with one or more of the phenylindole compounds in these cell lines, in contrast to the ER(+) breast cancer cell line MCF-7. Since the only difference between the 2PI(OH) and 2PI is the absence of the 5-hydroxy group in the phenylindole moiety in 2PI(OH), identification of the biological interactions of this portion of the molecule with the cellular machinery could shed light on these mechanisms. Conclusions This chapter describes some interesting observations that were made with the cytotoxicity and DNA damage studies of the 2-phenylindole compounds 2PI and 2PI(OH). The DNA damage profiles and the patterns of cell cycle arrest observed with the two compounds underscore the differences in the modes of action of the two compounds in the p53 mutant ovarian cell lines tested. In this light, 2PI might be inducing toxicity as per its intended mechanism, by interacting with the ER and forming adducts that are shielded from repair, thus inducing toxicity to treated cells. The role of ER in the toxicity of 2P1 needs to be delved into in further detail, and this will be one of the main aims of future investigations with this compound. 2PI(OH) displays the potential to combine the effects of a checkpoint abrogator and a DNA alkylating agent in one molecule, and the interaction of the compound with checkpoint machinery needs to be investigated. Both the compounds demonstrate promise as potent anti-tumor agents, and the selectivity of these compounds in different cellular contexts (ER (+)/(-) or p53 (+)/(-)), if established, could help strengthen the basis of their therapeutic efficacy. 224 Detection of crosslinks using comet assay: Percentage decrease in OTM in Caov-3 and OVCAR-3 cells Percent decrease in OTM Treatment Caov-3 OVCAR-3 Cisplatin 2.5 pM 32 (*) 48(*) Cisplatin 5.0 pM 2PI 2.5 pM 35 (*) 71(*) 21 (*) 54(*) 2PI 5.0 pM 13(*) 54(*) 2PI(OH) 2.5 pM - 25(*) 2PI(OH) 5.0 pM - 40(*) (*) - significant decrease in OTM (P<0.001) Table 4.1: Percent decrease in OTM vs. treatment. The corresponding OTM values are plotted in Figure 4.7 (Caov-3) and Figure 4.8 (OVCAR-3). The percentage decrease in OTM is relative to the OTM of untreated irradiated sample. Significance is calculated using MannWhitney test. The percent decrease in OTM indicates extent of crosslink formation. 225 Synthetic scheme of 2-Phenylindole compounds: 2PI (14a) and 2PI(OH) (14b) - Part 1 R 0 / \O -i i - OH H 2N H 4 4a R1 =R2= OH iy 4bR1=H, R2=OH V + H R2 NN H 3 la-3a R R=R 2 =OCH3 lb-3bRi = H, R 2 = OCH 3 vi, vii R3 R\i R1 iii NN Br R1 R2 R1 NN R2 H 5 5a-6a Rj= R2= OCH2OCH 3 5b-6b Ri= H, R2 = OCH 2OCH 3 NH NN \2- R1A R1 C 6H12 b6H12 N-R C6 H12 I I N-R 3 N-R3 3 t 8 HO R2 9 0 10 8a-10a R1= R2= OCH 2OCH 3, R3= PO(C6 ,H5 ) 2 8b-10b R1= H, R2 =OCH 2OCH 3, R3 = PO(C 6 H 5)2 Conjugation of nitrogen mustard to ligand 0 H2N HO xi i + 10 2 N NI R N R4 Ila, lb R4=Cl R f4 12b NR 4 "1 13 13a Ri= R2 = OCH 2OCH 3, R3 = PO(C4H5 )2 , R4= Cl 13b R1 = H, R2= OCH 2OCH 3, R3 = PO(CeH5) 2 , R4 = Cl 12a, 12bR4 = Cl 1xiii R R2 NN R4 H SNCH12N 14a Rr' R2 = OH, R4 14b R1= H, R2 = OH, = C1 R4 = O ON Cl /H M 14 Figure 4.1A: Synthetic scheme of 2PI-C6NC2-mustard and 2PI(OH)-C6NC2-mustard compounds. Intermediates are labeled in Figure 4.1B 226 Synthetic scheme of 2-Phenylindole compounds: 2PI (14a) and 2PI(OH) (14b) - Part 2 (i) Compound 1 (1 eqv.), CuBr2 (2.4 eqv.), EtOAc, 900C (ii) Compound 2 (1 eqv.), p-anisidine (3.3 eqv.), N, N dimethylaniline, 2000C (iii) Compound 3 (1 eqv.), BBr 3 (3.3 eqv.), CH2Cl2 , -780C to RT overnight (iv) Compound 4 (1 eqv.), NaH (2.5 eqv.), monomethylbromoether (2.5 eqv.), THF, -780C to RT (v) Compound 5 (1 eqv.), 1,6-dibromohexane (6 eqv.), NaH (2 eqv.), DMF, -780C to RT overnight. (vi) Aminoethanol (2 eqv.), diphenylphosphinic chloride (1 eqv.), diisopropylethylamine (1.3 eqv.),CH 2Cl2, OC to RT. (vii) Tertbutyldimethylsilyl chloride (1.2 eqv.), DMF, imidazole, RT (viii) Compound 6 (1 eqv.), 7 (1.7 eqv.), NaH (2.8 eqv.), tetrabutylammonium bromide (0.16 eqv.), benzene, reflux 2 hr (ix) Compound 8 (1 eqv.), tertbutylammonium fluoride (2 eqv.), RT. (x) Compound 9 (1 eqv.), p-nitrophenylchloroformate (2 eqv.), diisopropylethylamine (2 eqv.), CH2Cl2 , RT, 3 hr.(xi) Compound 11 (1 eqv.), ethyl chloroformate (1.2 eqv.), triethylamine (1.2 eqv.), sodium azide (2 eqv.) 0CC, acetone, reflux in toluene, 8N HCI, 10 min. (xii) Compound 12 (2 eqv.), compound 10 (1 eqv.), diisopropylamine (4 eqv.), tetrahydrofuran, reflux, 3 h (xiii) Compound 13, HCI, methanol. Figure 4.1 B: Intermediates in the synthetic scheme of 2PI-C6NC2-mustard and 2PI(OH)-C6NC2-mustard compounds. 227 Structures and relative binding affinities (RBAs) of phenylindole compounds Linker I R H OYN N N-"O H N H OH I I~C Ligand for estrogen receptor Compounds R Alkylating group RBA (relative Putative cellular to estradiol) interaction (%) 2PI OH 57 DNA, ER 2PI(OH) H 6 DNA Figure 4.2:Structures and relative binding affinities of the 2-Phenylindole compounds. The two compounds differ only intheir 5-hydroxy group in the phenylindole domain. Binding assays were conducted using purified ER obtained from Invitrogen. RBAs were calculated on a percentage scale relative to 17p-estradiol. 228 ER status of relevant cell lines 0 ERa (66 kDa) e 0 C C MCF-7 W mow& W IMO Actin amme =""" o - OVCAR-3 C - Caov-3 Figure 4.3: ER status of OVCAR-3,Caov-3 cells and MCF-7 (positive control). Actin was used as a loading control.OVCAR-3 and Caov-3 cells express the ER, as does the positive control MCF-7. The relative ratio of ER in the three cell lines was determined by spot densitometry to be Caov-3 : OVCAR-3: MCF-7 = 1 : 0.8: 1.8 229 2PI and 2PI(OH) cause growth inhibition of Caov-3 and OVCAR-3 cells A Caov-3 cells 100 80 0 * 2PI(OH) -- PCpI --a- Cisplatin 60 CD, C 40 20 0 10 5 15 20 Dose inuM OVCAR-3 cells 100 0 + 2PI(OH) -A- 2P -U- Cisplatin 80 L- 0) 60 (D, 40 20 0 10 5 15 20 Dose inuM Figure 4.4: Growth inhibition curves for Caov-3 and OVCAR-3 cells. Cells were treated with test compounds for 24 hours, followed by 24 hour incubation infresh medium. Percent growth iscalculated relative to untreated control. Values represent average of three independent experiments, and error bars are standard deviations. 230 Cell cycle distribution in Caov-3 cells treated with test compounds Untreated Cisplatin 2.5 piM 0.0 6.5 19.6 34.8 58.7 80.5 G1 G2/M 2PI 2.5 IM 2PI(OH) 2.5 piM 0.0 3.7 35.5 42.2 64.5 54.1 Figure 4.5: Pie-charts representing the cell cycle patterns in Caov-3 cells treated with 2.5 pM dose of cisplatin, 2PI and 2PI(OH) for 30 hours. Numbers around the pie-charts represent percentage of cells in the corresponding phase of cell cycle. Cisplatin and 2PI induce a potent S-phase arrest, whereas 2PI(OH) does not seem to change the cell cycle distribution. 231 Cell cycle distribution in OVCAR-3 cells treated with test compounds Cisplatin 2.5 p.M Untreated 0.0 6.0 21.5 45.9 54.2 G1 72.6 S G2/M 2PI 2.5 p.M 0.1 2PI(OH) 2.5 pM 8.2 29. 38.8 53.0 70.7 Figure 4.6: Pie-charts representing the cell cycle patterns in OVCAR-3 cells treated with 2.5 pM dose of cisplatin, 2PI and 2PI(OH) for 30 hours. Numbers around the pie-charts represent percentage of cells inthe corresponding phase of cell cycle. All the three compounds induce arrest inthe S-phase, although to different extents. 232 -Chapter 4- Detection of interstrand crosslink formation in Caov-3 cells using comet assay OTM vs. treatment: Caov-3 cells 20- 16 -9 E14 - # Treatment 0~ 12 7.7 . =710- 1 Untreated 2 Irradiated 8Gy 3 Cis 2.5 pM+Irr 4 Cis 5.0 pM+Irr 5 2PI 2.5pM+Irr T -- O- 0 7.3 10 8 2 12. * 11.3 18 62PI 5.0 pM+Irr 7 2PI(OH)2.5pM+Irr 1 8 2PI(OH)5pM+1rr - 0.8 2 3 4 5 6 7 8 Figure 4.7: Boxplots showing distribution of tail moments in Caov-3 cells. The numbers above the boxes represent average OTM value. Both cisplatin and 2PI treatments (starred entries) result in a significant decrease in OTM compared to the irradiated control sample (P<0.001). 2PI(OH) did not induce a decrease in OTM, but increased the OTM significantly (P<0.001).The percent decrease in OTM is tabulated in Table 4.1. 233 -Chapter 4- Detection of interstrand crosslink formation in OVCAR-3 cells using comet assay OTM vs. treatment: OVCAR-3 cells 20 18 10.7 16 # Treatment * 8.0 *14 E 6.4 * o 12 5.0 5.0 =10 * 3.1 . - _ 6 1 Irradiated 8Gy 2 Cis 2.5 iM+Irr 3 Cis 5.0 pM+Irr 4 2PI 2.5 pM+lrr 5 2PI5.0 gM+Irr 6 2PI(OH)2.5pM+Irr 7 2PI(OH)5pM+Irr 42- 0 1 2 3 4 5 6 7 Figure 4.8: Boxplots showing distribution of tail moments in OVCAR-3 cells.The numbers above the boxes represent average OTM value. Cisplatin, 2PI and 2PI(OH) (starred entries) treatments result ina significant induction of crosslinks, although to different extents (P<0.001) . The percent decrease in OTM istabulated inTable 4.1. 234 Drug-DNA adducts in Caov-3 and OVCAR-3 cells detected by AMS Caov-3 cells OVCAR-3 cells 25 25 C,) C,) a) C 0 20 20 4 2PIOVCAR-3 -*-2PI(OH)OVCAR-3 15 E a,) 10 10 5 5 0 0.0 1.0 2.0 3.0 4.0 5.0 0.0 1.0 2.0 3.0 4.0 5.0 Dose in uM Dose in uM Treatment dose in piM Adducts per million bases Caov-3 2PI Caov-3 2PI(OH) OVCAR-3 2PI OVCAR-3 2PI(OH) 1.52 0.55 1.29 1.71 7.43 0.5 1.0 2.5 1.15 1.15 1.66 2.58 6.96 3.5 5.0 6.96 14.7 7.43 23.6 1.48 4.01 11.3 17.4 16.1 7.79 Figure 4.9: Adducts per million bases following treatment with various radiolabeled drugs measured using AMS. Error bars represent standard deviation. The average number of adducts per million bases vs. treatment has been represented in the table above. Both the drugs form similar number of adducts in the two ovarian cell lines. 235 Caspase-3 activation by cisplatin and phenylindole compounds Cis 2PI(OH) 2PI Caov-3 Cleaved caspase-3 OVCAR-3 Figure 4.10: Western blot for anti-cleaved caspase-3 antibody in cell extracts treated with cisplatin and the compounds 2PI and 2PI(OH). Caov-3 cells were treated with 5 pM and OVCAR-3 cells with 10 pM of test compounds for 15 hours. Equal amounts of protein (25pg/lane) were loaded ineach lane. Cisplatin and 2PI(OH) induce a strong activation of caspase-3 whereas the 2Pl compound only mildly induces the formation of cleaved caspase-3. Cleavage of caspase-3 can be correlated to the extent of apoptosis inthese cells. 236 Reference List 1. Abend M (2003) Reasons to reconsider the significance of apoptosis for cancer therapy. Int J Radiat Biol 79:927-941 2. Agarwal R, Kaye SB (2003) Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer 3:502-516 3. Ahn HJ, Kim YS, Kim JU, Han SM, Shin JW, Yang HO (2004) Mechanism of taxolinduced apoptosis in human SKOV3 ovarian carcinoma cells. J Cell Biochem 91:1043-1052 4. Ali S, Metzger D, Bornert JM, Chambon P (1993) Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J 12:1153-1160 5. American Cancer Society (2008) Cancer Facts and Figures. 6. Arai M, Utsunomiya J, Miki Y (2004) Familial breast and ovarian cancers. Int J Clin Oncol 9:270-282 7. Araujo SJ, Nigg EA, Wood RD (2001) Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol Cell Biol 21:2281-2291 8. Argento M, Hoffman P, Gauchez AS (2008) Ovarian cancer detection and treatment: current situation and future prospects. Anticancer Res 28:3135-3138 237 9. Armitage P, Doll (1954) The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer 8:1-12 10. Asahina H, Kuraoka 1,Shirakawa M, Morita EH, Miura N, Miyamoto I, Ohtsuka E, Okada Y, Tanaka K (1994) The XPA protein is a zinc metalloprotein with an ability to recognize various kinds of DNA damage. Mutat Res 315:229-237 11. Balcome S, Park S, Quirk Dorr DR, Hafner L, Phillips L,Tretyakova N (2004) Adenine-containing DNA-DNA cross-links of antitumor nitrogen mustards. Chem Res Toxicol 17:950-962 12. Bandera CA, Ye B, Mok SC (2003) New technologies for the identification of markers for early detection of ovarian cancer. Curr Opin Obstet Gynecol 15:51-55 13. Bardin A, Hoffmann P, Boulle N, Katsaros D,Vignon F, Pujol P, Lazennec G (2004) Involvement of estrogen receptor beta in ovarian carcinogenesis. Cancer Res 64:5861-5869 14. Barth H, Hoffmann 1,Klein S, Kaszkin M, Richards J, Kinzel V (1996) Role of cdc25-C phosphatase in the immediate G2 delay induced by the exogenous factors epidermal growth factor and phorbolester. J Cell Physiol 168:589-599 15. Bast RC, Jr., Klug TL, St JE, Jenison E, Niloff JM, Lazarus H,Berkowitz RS, Leavitt T, Griffiths CT, Parker L, Zurawski VR, Jr., Knapp RC (1983) A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N Engl J Med 309:883-887 16. Bernstein E, Caudy AA, Hammond SM, Hannon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409:363-366 238 17. Biederbick A, Kern HF, Elsasser HP (1995) Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 66:3-14 18. Bishop JM (1991) Molecular themes in oncogenesis. Cell 64:235-248 19. Biswas DK, Singh S, Shi Q, Pardee AB, Iglehart JD (2005) Crossroads of estrogen receptor and NF-kappaB signaling. Sci STKE 2005:e27 20. Bonadonna G, Brusamolino E, Valagussa P, Rossi A, Brugnatelli L, Brambilla C, De LM, Tancini G, Bajetta E, Musumeci R, Veronesi U (1976) Combination chemotherapy as an adjuvant treatment in operable breast cancer. N Engl J Med 294:405-410 21. Bookman MA, Greer BE, Ozols RF (2003) Optimal therapy of advanced ovarian cancer: carboplatin and paclitaxel versus cisplatin and paclitaxel (GOG158) and an update on GOG0182-ICON5. Int J Gynecol Cancer 13 Suppl 2:149-155 22. Borini A, Rebellato E (2008) Focus on breast and ovarian cancer. Placenta 29 Suppl B:184-190 23. Botti J, Djavaheri-Mergny M, Pilatte Y, Codogno P (2006) Autophagy signaling and the cogwheels of cancer. Autophagy 2:67-73 24. Boulikas T, Vougiouka M (2004) Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review). Oncol Rep 11:559-595 25. Bowler J, Lilley TJ, Pittam JD, Wakeling AE (1989) Novel steroidal pure antiestrogens. Steroids 54:71-99 239 26. Brandenberger AW, Lebovic DI, Tee MK, Ryan IP,Tseng JF, Jaffe RB, Taylor RN (1999) Oestrogen receptor (ER)-alpha and ER-beta isoforms in normal endometrial and endometriosis-derived stromal cells. Mol Hum Reprod 5:651-655 27. Brandenberger AW, Tee MK, Jaffe RB (1998) Estrogen receptor alpha (ER-alpha) and beta (ER-beta) mRNAs in normal ovary, ovarian serous cystadenocarcinoma and ovarian cancer cell lines: down-regulation of ER-beta in neoplastic tissues. J Clin Endocrinol Metab 83:1025-1028 28. Brookes, Lawley PD (1961) The reaction of mono- and di-functional alkylating agents with nucleic acids. Biochem J 80:496-503 29. Brown JM, Attardi LD (2005) The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 5:231-237 30. Brown SJ, Kellett PJ, Lippard SJ (1993) lxrl, a yeast protein that binds to platinated DNA and confers sensitivity to cisplatin. Science 261:603-605 31. Bruhn, Toney JH, Lippard SJ (1990) Biological processing of DNA modified by platinum compounds., 38 edn 32. Bucourt R,Vignau M, Torelli V (1978) New biospecific adsorbents for the purification of estradiol receptor. J Biol Chem 253:8221-8228 33. Bunch RT, Eastman A (1996) Enhancement of cisplatin-induced cytotoxicity by 7hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin Cancer Res 2:791-797 240 34. Bunch RT, Eastman A (1997) 7-Hydroxystaurosporine (UCN-01) causes redistribution of proliferating cell nuclear antigen and abrogates cisplatin-induced S-phase arrest in Chinese hamster ovary cells. Cell Growth Differ 8:779-788 35. Burger RA (2007) Experience with bevacizumab in the management of epithelial ovarian cancer. J Clin Oncol 25:2902-2908 36. Chabner BA, Longo DL (2009) Cancer Chemotherapy and Biotherapy 37. Chabner BA, Roberts TG, Jr. (2005) Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer 5:65-72 38. Chatterjee M, Mohapatra S, lonan A, Bawa G, li-Fehmi R, Wang X, Nowak J, Ye B, Nahhas FA, Lu K, Witkin SS, Fishman D, Munkarah A, Morris R, Levin NK, Shirley NN, Tromp G, Abrams J, Draghici S, Tainsky MA (2006) Diagnostic markers of ovarian cancer by high-throughput antigen cloning and detection on arrays. Cancer Res 66:1181-1190 39. Chen Z, Fadiel A, Jia JF, Sakamoto H, Carbone R, Naftolin F (1999) Induction of apoptosis and suppression of clonogenicity of ovarian carcinoma cells with estrogen mustard. Cancer 85:2616-2622 40. Clark (2006) Molecular targeted drugs and growth factor receptor inhibitors. In: Chabner BA, Longo DL (eds) 41. Clarke PG (2002) Apoptosis: from morphological types of cell death to interacting pathways. Trends Pharmacol Sci 23:308-309 241 42. Clinton GM, Hua W (1997) Estrogen action in human ovarian cancer. Crit Rev Oncol Hematol 25:1-9 43. Coezy E, Borgna JL, Rochefort H (1982) Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res 42:317-323 44. Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS (1997) Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology 138:4613-4621 45. Cowley SM, Hoare S, Mosselman S, Parker MG (1997) Estrogen receptors alpha and beta form heterodimers on DNA. J Biol Chem 272:19858-19862 46. Cunat S, Hoffmann P, Pujol P (2004) Estrogens and epithelial ovarian cancer. Gynecol Oncol 94:25-32 47. Cunat S, Rabenoelina F, Daures JP, Katsaros D, Sasano H, Miller WR, Maudelonde T, Pujol P (2005) Aromatase expression in ovarian epithelial cancers. J Steroid Biochem Mol Biol 93:15-24 48. DaSilva JN, van Lier JE (1990) Synthesis and structure-affinity of a series of 7 alpha-undecylestradiol derivatives: a potential vector for therapy and imaging of estrogen-receptor-positive cancers. J Med Chem 33:430-434 49. De BK, Vanden BW, Haegeman G (2006) Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene 25:6868-6886 242 50. Deng CX (2006) BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 34:1416-1426 51. DeVita VT, Hellman S, Rosenberg SA (1997) Princples and Practice of Oncology, 5 edn 52. Donahue BA, Augot M, Bellon SF, Treiber DK, Toney JH, Lippard SJ, Essigmann JM (1990) Characterization of a DNA damage-recognition protein from mammalian cells that binds specifically to intrastrand d(GpG) and d(ApG) DNA adducts of the anticancer drug cisplatin. Biochemistry 29:5872-5880 53. Dong L, Wang W, Wang F, Stoner M, Reed JC, Harigai M, Samudio I, Kladde MP, Vyhlidal C, Safe S (1999) Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J Biol Chem 274:3209932107 54. Driscoll MD, Sathya G, Muyan M, Klinge CM, Hilf R, Bambara RA (1998) Sequence requirements for estrogen receptor binding to estrogen response elements. J Biol Chem 273:29321-29330 55. du Bois A, Neijt JP, Thigpen JT (1999) First line chemotherapy with carboplatin plus paclitaxel in advanced ovarian cancer--a new standard of care? Ann Oncol 10 Suppl 1:35-41 56. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498 243 57. Essigmann JM, Rink SM, Park HJ, Croy RG (2001b) Design of DNA damaging agents that hijack transcription factors and block DNA repair. Adv Exp Med Biol 500:301-313 58. Essigmann, Rink SM, Park HJ, Croy RG (2001a) Design of DNA damaging agents that hijack transcription factors and block DNA repair. In: Dansette (ed) Biological Reactive Intermediates VI, 6 edn 59. Festjens N,Vanden BT, Vandenabeele P (2006) Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 1757:1371-1387 60. Fliss AE, Benzeno S, Rao J, Caplan AJ (2000) Control of estrogen receptor ligand binding by Hsp90. J Steroid Biochem Mol Biol 72:223-230 61. Foulds (1954) The experimental study of tumor progression: a review. Cancer Res 14:327-339 62. Friedberg EC, Bonura T, Love JD, McMillan S, Radany EH, Schultz RA (1981) The repair of DNA damage: recent developments and new insights. J Supramol Struct Cell Biochern 16:91-103 - 63. Friedberg EC, Walker G,Siede W (1995) DNA repair and mutagenesis 64. Fu XD, Simoncini T (2008) Extra-nuclear signaling of estrogen receptors. IUBMB Life 60:502-510 244 65. Furuta T, Ueda T, Aune G, Sarasin A, Kraemer KH, Pommier Y (2002) Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res 62:4899-4902 66. Gibb RK, Taylor DD, Wan T, O'Connor DM, Doering DL, Gercel-Taylor C (1997) Apoptosis as a measure of chemosensitivity to cisplatin and taxol therapy in ovarian cancer cell lines. Gynecol Oncol 65:13-22 67. Gozuacik D, Kimchi A (2007) Autophagy and cell death. Curr Top Dev Biol 78:217245 68. Grant DF, Bessho T, Reardon JT (1998) Nucleotide excision repair of melphalan monoadducts. Cancer Res 58:5196-5200 69. Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O'Connor PM, PiwnicaWorms H (2000) The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem 275:5600-5605 70. Green S, Walter P, Greene G, Krust A, Goffin C, Jensen E, Scrace G, Waterfield M, Chambon P (1986) Cloning of the human oestrogen receptor cDNA. J Steroid Biochem 24:77-83 71. Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J (1986) Sequence and expression of human estrogen receptor complementary DNA. Science 231:11501154 72. Greenlee RT, Hill-Harmon MB, Murray T, Thun M (2001) Cancer statistics, 2001. CA Cancer J Clin 51:15-36 245 73. Greenlee RT, Murray T, Bolden S, Wingo PA (2000) Cancer statistics, 2000. CA Cancer J Clin 50:7-33 74. Haapala E, Hakala K, Jokipelto E, Vilpo J, Hovinen J (2001) Reactions of N,Nbis(2-chloroethyl)-p-aminophenylbutyric acid (chlorambucil) with 2'deoxyguanosine. Chem Res Toxicol 14:988-995 75. Hagopian GS, Mills GB, Khokhar AR, Bast RC, Jr., Siddik ZH (1999) Expression of p53 in cisplatin-resistant ovarian cancer cell lines: modulation with the novel platinum analogue (1R,2R-diaminocyclohexane)(trans-diacetato)(dichloro)platinum(IV). Clin Cancer Res 5:655-663 76. Hall JM, McDonnell DP (1999) The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140:5566-5578 77. Hamilton TC, Young RC, McKoy WM, Grotzinger KR, Green JA, Chu EW, WhangPeng J, Rogan AM, Green WR, Ozols RF (1983) Characterization of a human ovarian carcinoma cell line (NIH:OVCAR-3) with androgen and estrogen receptors. Cancer Res 43:5379-5389 78. Hamilton TC, Young RC, Ozols RF (1984) Experimental model systems of ovarian cancer: applications to the design and evaluation of new treatment approaches. Semin Oncol 11:285-298 79. Hanahan D,Weinberg RA (2000) The hallmarks of cancer. Cell 100:57-70 246 80. Hartley JA, Berardini MD, Souhami RL (1991) An agarose gel method for the determination of DNA interstrand crosslinking applicable to the measurement of the rate of total and "second-arm" crosslink reactions. Anal Biochem 193:131-134 81. Havrilesky U, McMahon CP, Lobenhofer EK, Whitaker R, Marks JR, Berchuck A (2001) Relationship between expression of coactivators and corepressors of hormone receptors and resistance of ovarian cancers to growth regulation by steroid hormones. J Soc Gynecol Investig 8:104-113 82. He Z, Ingles CJ (1997) Isolation of human complexes proficient in nucleotide excision repair. Nucleic Acids Res 25:1136-1141 83. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8:193-204 84. Hippert MM, O'Toole PS, Thorburn A (2006) Autophagy in cancer: good, bad, or both? Cancer Res 66:9349-9351 85. Ho SM (2003) Estrogen, progesterone and epithelial ovarian cancer. Reprod Biol Endocrinol 1:73 86. Htun H, Holth LT, Walker D, Davie JR, Hager GL (1999) Direct visualization of the human estrogen receptor alpha reveals a role for ligand in the nuclear distribution of the receptor. Mol Biol Cell 10:471-486 87. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995d) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 247 88. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995a) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 89. Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM (1995b) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 90. Hua W, Christianson T, Rougeot C, Rochefort H,Clinton GM (1995c) SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279-289 91. Huang A, Kaley G (2004) Gender-specific regulation of cardiovascular function: estrogen as key player. Microcirculation 11:9-38 92. Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A (1994) HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc NatI Acad Sci U S A 91:1039410398 93. Hurley LH (2002) DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer 2:188-200 94. Igney FH, Krammer PH (2002) Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2:277-288 95. Jacobs I, Bast RC, Jr. (1989) The CA 125 tumour-associated antigen: a review of the literature. Hum Reprod 4:1-12 248 96. Jaffe N, Frei E, Ill, Traggis D, Bishop Y (1974) Adjuvant methotrexate and citrovorum-factor treatment of osteogenic sarcoma. N Engl J Med 291:994-997 97. Jakowlew SB, Breathnach R, Jeltsch JM, Masiakowski P, Chambon P (1984) Sequence of the pS2 mRNA induced by estrogen in the human breast cancer cell line MCF-7. Nucleic Acids Res 12:2861-2878 98. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ (2008) Cancer statistics, 2008. CA Cancer J Clin 58:71-96 99. Jones CJ, Wood RD (1993) Preferential binding of the xeroderma pigmentosum group A complementing protein to damaged DNA. Biochemistry 32:12096-12104 100. Kim DH, Rossi JJ (2007) Strategies for silencing human disease using RNA interference. Nat Rev Genet 8:173-184 101. King WJ, Greene GL (1984) Monoclonal antibodies localize oestrogen receptor in the nuclei of target cells. Nature 307:745-747 102. Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato AC (1988) A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res 16:647-663 103. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, 249 (% Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di SF, Dice JF, Difiglia M, neshKumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr., Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov 0, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanaka K, Tanida 1,Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van dK, 250 I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L,Yue Z, Yuzaki M, Zabirnyk 0, Zheng X, Zhu X, Deter RL (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151-175 104. Koberle B, Roginskaya V, Wood RD (2006) XPA protein as a limiting factor for nucleotide excision repair and UV sensitivity in human cells. DNA Repair (Amst) 5:641-648 105. Kohn KW (1996) Beyond DNA cross-linking: history and prospects of DNAtargeted cancer treatment--fifteenth Bruce F. Cain Memorial Award Lecture. Cancer Res 56:5533-5546 106. Kohn KW, Hartley JA, Mattes WB (1987) Mechanisms of DNA sequence selective alkylation of guanine-N7 positions by nitrogen mustards. Nucleic Acids Res 15:10531-10549 107. Kolfschoten GM, Hulscher TM, Pinedo HM, Boven E (2000) Drug resistance features and S-phase fraction as possible determinants for drug response in a panel of human ovarian cancer xenografts. Br J Cancer 83:921-927 108. Krasner C (2007) Aromatase inhibitors in gynecologic cancers. J Steroid Biochem Mol Biol 106:76-80 109. Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138:863-870 251 110. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA (1996) Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A 93:5925-5930 111. Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der BB, Gustafsson JA (1998a) Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 139:4252-4263 112. Kuiper GG, Shughrue PJ, Merchenthaler I, Gustafsson JA (1998b) The estrogen receptor beta subtype: a novel mediator of estrogen action in neuroendocrine systems. Front Neuroendocrinol 19:253-286 113. Kumar R,Thompson EB (1999) The structure of the nuclear hormone receptors. Steroids 64:310-319 114. Kundu GC, Schullek JR, Wilson IB(1994) The alkylating properties of chlorambucil. Pharmacol Biochem Behav 49:621-624 115. Kurreck J (2009) RNA interference: from basic research to therapeutic applications. Angew Chem Int Ed Engl 48:1378-1398 116. Kushner PJ, Agard D, Feng WJ, Lopez G, Schiau A, Uht R,Webb P, Greene G (2000a) Oestrogen receptor function at classical and alternative response elements. Novartis Found Symp 230:20-26 117. Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P (2000b) Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol 74:311317 252 118. Lahooti H, White R, Danielian PS, Parker MG (1994) Characterization of liganddependent phosphorylation of the estrogen receptor. Mol Endocrinol 8:182-188 119. Langdon SP, Hirst GL, Miller EP, Hawkins RA, Tesdale AL, Smyth JF, Miller WR (1994) The regulation of growth and protein expression by estrogen in vitro: a study of 8 human ovarian carcinoma cell lines. J Steroid Biochem Mol Biol 50:131135 120. Langdon SP, Ritchie A, Young K, Crew AJ, Sweeting V, Bramley T, Hillier S, Hawkins RA, Tesdale AL, Smyth JF, . (1993) Contrasting effects of 17 betaestradiol on the growth of human ovarian carcinoma cells in vitro and in vivo. Int J Cancer 55:459-464 121. Lau CC, Pardee AB (1982) Mechanism by which caffeine potentiates lethality of nitrogen mustard. Proc Natl Acad Sci U S A 79:2942-2946 122. Lau KM, Mok SC, Ho SM (1999) Expression of human estrogen receptor-alpha and -beta, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci U S A 96:5722-5727 123. Lawley PD (1966) Effects of some chemical mutagens and carcinogens on nucleic acids. Prog Nucleic Acid Res Mol Biol 5:89-131 124. Lazennec G (2006) Estrogen receptor beta, a possible tumor suppressor involved in ovarian carcinogenesis. Cancer Lett 231:151-157 125. Le GP, Montano MM, Schodin DJ, Katzenellenbogen BS (1994) Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and 253 examination of their influence on transcriptional activity. J Biol Chem 269:44584466 126. Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463-477 127. Li M, HERTZ R,BERGENSTAL DM (1958) Therapy of choriocarcinoma and related trophoblastic tumors with folic acid and purine antagonists. N Engl J Med 259:66-74 128. Liberman RG, Tannenbaum SR, Hughey BJ, Shefer RE, Klinkowstein RE, Prakash C, Harriman SP, Skipper PL (2004) An interface for direct analysis of (14)c in nonvolatile samples by accelerator mass spectrometry. Anal Chem 76:328-334 129. Lindahl T (1979) DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog Nucleic Acid Res Mol Biol 22:135-192 130. Lindgren PR, Cajander S, Backstrom T, Gustafsson JA, Makela S, Olofsson JI (2004) Estrogen and progesterone receptors in ovarian epithelial tumors. Mol Cell Endocrinol 221:97-104 131. Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, Lynch P, Burke W, Press N (2006) Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 296:15071517 132. Lukanova A, Kaaks R (2005) Endogenous hormones and ovarian cancer: epidemiology and current hypotheses. Cancer Epidemiol Biomarkers Prev 14:98107 254 133. MacLeod MC, Powell KL, Tran N (1995) Binding of the transcription factor, Spl, to non-target sites in DNA modified by benzo[a]pyrene diol epoxide. Carcinogenesis 16:975-983 134. Maggi A, Ciana P, Belcredito S, Vegeto E (2004) Estrogens in the nervous system: mechanisms and nonreproductive functions. Annu Rev Physiol 66:291-313 135. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM (1995) The nuclear receptor superfamily: the second decade. Cell 83:835-839 136. Masiakowski P, Breathnach R, Bloch J, Gannon F, Krust A, Chambon P (1982) Cloning of cDNA sequences of hormone-regulated genes from the MCF-7 human breast cancer cell line. Nucleic Acids Res 10:7895-7903 137. Masood S, Heitmann J, Nuss RC, Benrubi GI (1989) Clinical correlation of hormone receptor status in epithelial ovarian cancer. Gynecol Oncol 34:57-60 138. May FE, Westley BR (1988) Identification and characterization of estrogenregulated RNAs in human breast cancer cells. J Biol Chem 263:12901-12908 139. May FE, Westley BR (1995) Estrogen regulated messenger RNAs in human breast cancer cells. Biomed Pharmacother 49:400-414 140. McA'Nulty MM, Lippard SJ (1996) The HMG-domain protein Ixr1 blocks excision repair of cisplatin-DNA adducts in yeast. Mutat Res 362:75-86 255 141. McA'Nulty MM, Whitehead JP, Lippard SJ (1996) Binding of lxrl, a yeast HMGdomain protein, to cisplatin-DNA adducts in vitro and in vivo. Biochemistry 35:6089-6099 142. Meijer AJ, Codogno P (2006) Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 27:411-425 143. Merajver SD, Pham TM, Caduff RF, Chen M, Poy EL, Cooney KA, Weber BL, Collins FS, Johnston C, Frank TS (1995) Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nat Genet 9:439-443 144. Meyn, Murray D (1986) Cell cycle Effects of alkylating agents. 145. Miki Y, Swensen J,Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, . (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66-71 146. Mitra K, Marquis JC, Hillier SM, Rye PT, Zayas B, Lee AS, Essigmann JM, Croy RG (2002) A rationally designed genotoxin that selectively destroys estrogen receptor-positive breast cancer cells. J Am Chem Soc 124:1862-1863 147. Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861-2873 148. Mizushima N, Levine B,Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069-1075 149. Moretti-Rojas I, Fuqua SA, Montgomery RA, Ill, McGuire WL (1988) A cDNA for the estradiol-regulated 24K protein: control of mRNA levels in MCF-7 cells. Breast Cancer Res Treat 11:155-163 256 150. Morimoto H, Safrit JT, Bonavida B (1991) Synergistic effect of tumor necrosis factor-alpha- and diphtheria toxin-mediated cytotoxicity in sensitive and resistant human ovarian tumor cell lines. J Immunol 147:2609-2616 151. Morimoto H,Yonehara S, Bonavida B (1993) Overcoming tumor necrosis factor and drug resistance of human tumor cell lines by combination treatment with antiFas antibody and drugs or toxins. Cancer Res 53:2591-2596 152. Morisset M, Capony F, Rochefort H (1986) Processing and estrogen regulation of the 52-kilodalton protein inside MCF7 breast cancer cells. Endocrinology 119:2773-2782 153. Morissette M, Le SM, D'Astous M, Jourdain S, Al SS, Morin N, Estrada-Camarena E, Mendez P, Garcia-Segura LM, Di PT (2008) Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J Steroid Biochem Mol Biol 108:327-338 154. Mutch DG (2002) Surgical management of ovarian cancer. Semin Oncol 29:3-8 155. Narod SA, Foulkes WD (2004) BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 4:665-676 156. Nash JD, Ozols RF, Smyth JF, Hamilton TC (1989) Estrogen and anti-estrogen effects on the growth of human epithelial ovarian cancer in vitro. Obstet Gynecol 73:1009-1016 157. Neijt JP, Engelholm SA, Tuxen MK, Sorensen PG, Hansen M, Sessa C, de Swart CA, Hirsch FR, Lund B,van Houwelingen HC (2000) Exploratory phase Ill study of 257 paclitaxel and cisplatin versus paclitaxel and carboplatin in advanced ovarian cancer. J Clin Oncol 18:3084-3092 158. Newton K, Strasser A (1998) The BcI-2 family and cell death regulation. Curr Opin Genet Dev 8:68-75 159. Nguyen HN, Sevin BU, Averette HE, Perras J, Ramos R, Donato D,Ochiai K, Penalver M (1993) Cell cycle perturbations of platinum derivatives on two ovarian cancer cell lines. Cancer Invest 11:264-275 160. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J,Andersson G, Enmark E, Pettersson K,Warner M, Gustafsson JA (2001) Mechanisms of estrogen action. Physiol Rev 81:1535-1565 161. Noy N (2007) Ligand specificity of nuclear hormone receptors: sifting through promiscuity. Biochemistry 46:13461-13467 162. O'Connor PM, Kohn KW (1990) Comparative pharmacokinetics of DNA lesion formation and removal following treatment of LI 210 cells with nitrogen mustards. Cancer Commun 2:387-394 163. O'Donnell AJ, Macleod KG, Burns DJ, Smyth JF, Langdon SP (2005) Estrogen receptor-alpha mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocr Relat Cancer 12:851-866 164. Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, Hosoi T, Ouchi Y, Muramatsu M (1998) The complete primary structure of human estrogen receptor beta (hER beta) and its heterodimerization with ER alpha in vivo and in vitro. Biochem Biophys Res Commun 243:122-126 258 165. Olefsky JM (2001) Nuclear receptor minireview series. J Biol Chem 276:3686336864 166. Olive PL, Banath JP (1995) Sizing highly fragmented DNA in individual apoptotic cells using the comet assay and a DNA crosslinking agent. Exp Cell Res 221:19-26 167. Olive PL, Banath JP (2006) The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1:23-29 168. Olive PL, Banath JP, Durand RE (1990) Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the "comet" assay. Radiat Res 122:86-94 169. Osborne CK, Yochmowitz MG, Knight WA, Ill, McGuire WL (1980) The value of estrogen and progesterone receptors in the treatment of breast cancer. Cancer 46:2884-2888 170. Oxelmark E, Knoblauch R, Arnal S, Su LF, Schapira M, Garabedian MJ (2003) Genetic dissection of p23, an Hsp9O cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J Biol Chem 278:36547-36555 171. Ozols RF (2005) Treatment goals in ovarian cancer. Int J Gynecol Cancer 15 Suppl 1:3-11 172. Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM, Baergen R (2003) Phase Ill trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage Ill ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 21:3194-3200 259 173. Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61:439-444 174. Pal T, Permuth-Wey J,Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K,Wilbanks G, Nicosia S, Cantor A, Sutphen R (2005) BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104:2807-2816 175. Papac RJ (2001) Origins of cancer therapy. Yale J Biol Med 74:391-398 176. Parker MG (1991) Nuclear Hormone Receptors 177. Parker, Jensen EV (1991) Overview of the Nuclear Receptor Family. 178. Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55:74-108 179. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1dependent autophagy. Cell 122:927-939 180. Pearce ST, Jordan VC (2004) The biological role of estrogen receptors alpha and beta in cancer. Crit Rev Oncol Hematol 50:3-22 181. Pettersson K, Grandien K, Kuiper GG, Gustafsson JA (1997) Mouse estrogen receptor beta forms estrogen response element-binding heterodimers with estrogen receptor alpha. Mol Endocrinol 11:1486-1496 260 182. Piccart MJ, du BA, Gore ME, Neijt JP, Pecorelli S, Pujade-Lauraine E (2000) A new standard of care for treatment of ovarian cancer. Eur J Cancer 36:10-12 183. Pil PM, Lippard SJ (1992) Specific binding of chromosomal protein HMG1 to DNA damaged by the anticancer drug cisplatin. Science 256:234-237 184. Pratt WB, Ruddon RW, Ensminger WD, Maybaum J (1994) The Anticancer drugs 185. Pujol P, Rey JM, Nirde P, Roger P, Gastaldi M, Laffargue F, Rochefort H, Maudelonde T (1998) Differential expression of estrogen receptor-alpha and -beta messenger RNAs as a potential marker of ovarian carcinogenesis. Cancer Res 58:5367-5373 186. Rao BR, Slotman BJ (1991) Endocrine factors in common epithelial ovarian cancer. Endocr Rev 12:14-26 187. Rao GG, Miller DS (2005) Clinical applications of hormonal therapy in ovarian cancer. Curr Treat Options Oncol 6:97-102 188. Razandi M, Pedram A, Merchenthaler 1,Greene GL, Levin ER (2004) Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol 18:2854-2865 189. Reardon JT, Vaisman A, Chaney SG, Sancar A (1999) Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamineplatinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res 59:39683971 261 190. Reed (2006) Cisplatin, carboplatin and oxaliplatin. In: Chabner BA, Longo DL (eds) Cancer chemotherapy and biotherapy, 4 edn 191. Reggiori F, Klionsky DJ (2002) Autophagy in the eukaryotic cell. Eukaryot Cell 1:11-21 192. Renan MJ (1993) How many mutations are required for tumorigenesis? Implications from human cancer data. Mol Carcinog 7:139-146 193. Reynolds A, Anderson EM, Vermeulen A, Fedorov Y, Robinson K, Leake D, Karpilow J, Marshall WS, Khvorova A (2006) Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA 12:988-993 194. Riggs BL, Khosla S, Melton U, Ill (2002) Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev 23:279-302 195. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Persson IR (2001) Risk factors for epithelial borderline ovarian tumors: results of a Swedish case-control study. Gynecol Oncol 83:575-585 196. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Persson IR (2002a) Risk factors for invasive epithelial ovarian cancer: results from a Swedish case-control study. Am J Epidemiol 156:363-373 197. Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Weiderpass E, Persson IR (2002b) Hormone replacement therapy and the risk of invasive epithelial ovarian cancer in Swedish women. J Natl Cancer Inst 94:497504 262 198. Riman T, Dickman PW, Nilsson S, Nordlinder H, Magnusson CM, Persson IR (2004) Some life-style factors and the risk of invasive epithelial ovarian cancer in Swedish women. Eur J Epidemiol 19:1011-1019 199. Rimer, Schildkraut JM (1997) Cancer Screening. In: DeVita VT, Hellman S, Rosenberg SA (eds), Fifth edn pp 619-631 200. Rink SM, Yarema KJ, Solomon MS, Paige LA, Tadayoni-Rebek BM, Essigmann JM, Croy RG (1996) Synthesis and biological activity of DNA damaging agents that form decoy binding sites for the estrogen receptor. Proc Nati Acad Sci U S A 93:15063-15068 201. Risch HA (2002) Hormone replacement therapy and the risk of ovarian cancer. Gynecol Oncol 86:115-117 202. Roberts D, Schick J, Conway S, Biade S, Laub PB, Stevenson JP, Hamilton TC, O'Dwyer PJ, Johnson SW (2005) Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br J Cancer 92:1149-1158 203. Rochefort H (1990) Cathepsin D in breast cancer. Breast Cancer Res Treat 16:313 204. Rochefort H (1998) [Estrogens, cathepsin D and metastasis in cancers of the breast and ovary: invasion or proliferation?]. C R Seances Soc Biol Fil 192:241-251 205. Rochefort H (1999) [Estrogen-induced genes in breast cancer, and their medical importance]. Bull Acad Natl Med 183:955-968 263 206. Rose PG, Reale FR, Longcope C, Hunter RE (1990) Prognostic significance of estrogen and progesterone receptors in epithelial ovarian cancer. Obstet Gynecol 76:258-263 207. Roy G, Horton JK, Roy R, Denning T, Mitra S, Boldogh I (2000) Acquired alkylating drug resistance of a human ovarian carcinoma cell line is unaffected by altered levels of pro- and anti-apoptotic proteins. Oncogene 19:141-150 208. Russo IH, Russo J (1998) Role of hormones in mammary cancer initiation and progression. J Mammary Gland Biol Neoplasia 3:49-61 209. Rutherford T, Brown WD, Sapi E, Aschkenazi S, Munoz A, Mor G (2000) Absence of estrogen receptor-beta expression in metastatic ovarian cancer. Obstet Gynecol 96:417-421 210. Satokata 1,Tanaka K, Miura N, Narita M, Mimaki T, Satoh Y, Kondo S, Okada Y (1992) Three nonsense mutations responsible for group A xeroderma pigmentosum. Mutat Res 273:193-202 211. Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S (2000) Ligand-, cell-, and estrogen receptor subtype (alpha/beta)dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem 275:53795387 212. Sawyers C (2004) Targeted cancer therapy. Nature 432:294-297 213. Schaner ME, Ross DT, Ciaravino G, Sorlie T, Troyanskaya 0, Diehn M,Wang YC, Duran GE, Sikic TL, Caldeira S, Skomedal H, Tu IP,Hernandez-Boussard T, Johnson SW, O'Dwyer PJ, Fero MJ, Kristensen GB, Borresen-Dale AL, Hastie T, 264 Tibshirani R, van de RM, Teng NN, Longacre TA, Botstein D, Brown PO, Sikic BI (2003) Gene expression patterns in ovarian carcinomas. Mol Biol Cell 14:43764386 214. Schildkraut JM, Schwingl PJ, Bastos E, Evanoff A, Hughes C (1996) Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet Gynecol 88:554-559 215. Schlegel R, Pardee AB (1986) Caffeine-induced uncoupling of mitosis from the completion of DNA replication in mammalian cells. Science 232:1264-1266 216. Schuchard M, Landers JP, Sandhu NP, Spelsberg TC (1993) Steroid hormone regulation of nuclear proto-oncogenes. Endocr Rev 14:659-669 217. Segnitz B, Gehring U (1995) Subunit structure of the nonactivated human estrogen receptor. Proc Natl Acad Sci U S A 92:2179-2183 218. Segnitz B, Gehring U (1997) The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J Biol Chem 272:18694-18701 219. Sekiguchi 1,Suzuki M, Tamada T, Shinomiya N, Tsuru S, Murata M (1996) Effects of cisplatin on cell cycle kinetics, morphological change, and cleavage pattern of DNA in two human ovarian carcinoma cell lines. Oncology 53:19-26 220. Serova M, Calvo F, Lokiec F, Koeppel F, Poindessous V, Larsen AK, Laar ES, Waters SJ, Cvitkovic E, Raymond E (2006) Characterizations of irofulven cytotoxicity in combination with cisplatin and oxaliplatin in human colon, breast, and ovarian cancer cells. Cancer Chemother Pharmacol 57:491-499 265 221. Shapiro GI, Harper JW (1999) Anticancer drug targets: cell cycle and checkpoint control. J Clin Invest 104:1645-1653 222. Sharma U, Marquis JC, Nicole DA, Hillier SM, Fedeles B, Rye PT, Essigmann JM, Croy RG (2004) Design, synthesis, and evaluation of estradiol-linked genotoxicants as anti-cancer agents. Bioorg Med Chem Lett 14:3829-3833 223. Shuey DJ, McCallus DE, Giordano T (2002) RNAi: gene-silencing in therapeutic intervention. Drug Discov Today 7:1040-1046 224. Slotman BJ, Rao BR (1988) Ovarian cancer (review). Etiology, diagnosis, prognosis, surgery, radiotherapy, chemotherapy and endocrine therapy. Anticancer Res 8:417-434 225. States JC, McDuffie ER, Myrand SP, McDowell M, Cleaver JE (1998) Distribution of mutations in the human xeroderma pigmentosum group A gene and their relationships to the functional regions of the DNA damage recognition protein. Hum Mutat 12:103-113 226. Stenoien DL, Mancini MG, Patel K,Allegretto EA, Smith CL, Mancini MA (2000) Subnuclear trafficking of estrogen receptor-alpha and steroid receptor coactivator-1. Mol Endocrinol 14:518-534 227. Stenoien DL, Patel K, Mancini MG, Dutertre M, Smith CL, O'Malley BW, Mancini MA (2001) FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat Cell Biol 3:15-23 266 228. Stevens EV, Nishizuka S, Antony S, Reimers M, Varma S, Young L, Munson PJ, Weinstein JN, Kohn EC, Pommier Y (2008) Predicting cisplatin and trabectedin drug sensitivity in ovarian and colon cancers. Mol Cancer Ther 7:10-18 229. Sugiyama K, Shimizu M, Akiyama T, Tamaoki T, Yamaguchi K, Takahashi R, Eastman A, Akinaga S (2000) UCN-01 selectively enhances mitomycin C cytotoxicity in p53 defective cells which is mediated through S and/or G(2) checkpoint abrogation. Int J Cancer 85:703-709 230. Sunters A, Springer CJ, Bagshawe KD, Souhami RL, Hartley JA (1992) The cytotoxicity, DNA crosslinking ability and DNA sequence selectivity of the aniline mustards melphalan, chlorambucil and 4-[bis(2-chloroethyl)amino] benzoic acid. Biochem Pharmacol 44:59-64 231. Tew, Colvin OM, Jones RB (2006) Clinical and High-Dose Alkylating Agents. In: Chabner BA, Longo DL (eds) Cancer Chemotherapy and Biotherapy, 4 edn 232. Treiber DK, Zhai X, Jantzen HM, Essigmann JM (1994) Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor). Proc Natl Acad Sci U S A 91:5672-5676 233. Turteltaub KW, Dingley KH (1998) Application of accelerated mass spectrometry (AMS) in DNA adduct quantification and identification. Toxicol Lett 102-103:435439 234. Turteltaub KW, Felton JS, Gledhill BL, Vogel JS, Southon JR, Caffee MW, Finkel RC, Nelson DE, Proctor ID, Davis JC (1990) Accelerator mass spectrometry in biomedical dosimetry: relationship between low-level exposure and covalent 267 binding of heterocyclic amine carcinogens to DNA. Proc Natl Acad Sci U S A 87:5288-5292 235. Tyagi A, Singh RP, Agarwal C, Siriwardana S, Sclafani RA, Agarwal R (2005) Resveratrol causes Cdc2-tyrl 5 phosphorylation via ATM/ATR-Chk 1/2-Cdc25C pathway as a central mechanism for S phase arrest in human ovarian carcinoma Ovcar-3 cells. Carcinogenesis 26:1978-1987 236. Vandenabeele P, Vanden BT, Festjens N (2006) Caspase inhibitors promote alternative cell death pathways. Sci STKE 2006:e44 237. Venkitaraman AR (2004) Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat Rev Cancer 4:266-276 238. von Angerer E, Prekajac J, Strohmeier J (1984) 2-Phenylindoles. Relationship between structure, estrogen receptor affinity, and mammary tumor inhibiting activity in the rat. J Med Chem 27:1439-1447 239. Waldman T, Lengauer C, Kinzler KW, Vogelstein B (1996) Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature 381:713-716 240. Walker G, MacLeod K, Williams AR, Cameron DA, Smyth JF, Langdon SP (2007) Estrogen-regulated gene expression predicts response to endocrine therapy in patients with ovarian cancer. Gynecol Oncol 106:461-468 241. Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, Staub A, Jensen E, Scrace G, Waterfield M, . (1985) Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci U S A 82:7889-7893 268 242. Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O'Connor PM (1996) UCN01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Nati Cancer Inst 88:956-965 243. Webb P, Lopez GN, Uht RM, Kushner PJ (1995) Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol 9:443-456 244. Webb P, Nguyen P,Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, Kushner PJ (1999) The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol 13:1672-1685 245. Wilson TM, Ewel A, Duguid JR, Eble JN, Lescoe MK, Fishel R, Kelley MR (1995) Differential cellular expression of the human MSH2 repair enzyme in small and large intestine. Cancer Res 55:5146-5150 246. Wolf JK, Mills GB, Bazzet L, Bast RC, Jr., Roth JA, Gershenson DM (1999) Adenovirus-mediated p53 growth inhibition of ovarian cancer cells is independent of endogenous p53 status. Gynecol Oncol 75:261-266 247. Wu X, Fan W, Xu S, Zhou Y (2003) Sensitization to the cytotoxicity of cisplatin by transfection with nucleotide excision repair gene xeroderma pigmentosun group A antisense RNA in human lung adenocarcinoma cells. Clin Cancer Res 9:58745879 269 248. Xu Y, Traystman RJ, Hurn PD, Wang MM (2004) Membrane restraint of estrogen receptor alpha enhances estrogen-dependent nuclear localization and genomic function. Mol Endocrinol 18:86-96 249. Yap TA, Carden CP, Kaye SB (2009) Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer 9:167-181 250. Zakeri Z, Bursch W, Tenniswood M, Lockshin RA (1995) Cell death: programmed, apoptosis, necrosis, or other? Cell Death Differ 2:87-96 251. Zang XP, Pento JT (2008) SiRNA inhibition of ER-alpha expression reduces KGFinduced proliferation of breast cancer cells. Anticancer Res 28:2733-2735 252. Zheng L, Annab LA, Afshari CA, Lee WH, Boyer TG (2001) BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci U S A 98:9587-9592 253. Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408:433-439 270