SYNTHESES AND STUDIES OF MOLYBDENUM AND TUNGSTEN COMPLEXES FOR DINITROGEN REDUCTION By
advertisement

SYNTHESES AND STUDIES OF MOLYBDENUM AND TUNGSTEN COMPLEXES FOR
DINITROGEN REDUCTION
By
JIA MIN CHIN
B.A. in Chemistry, magna cum laude
Columbia College, Columbia University
May 2004
Submitted to the Department of Chemistry
In Partial Fulfillment of the Requirements
for the Degree of
DOCTOR OF PHILOSOPHY
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2010
© Massachusetts Institute of Technology, June 2010
All rights reserved.
Signature of Author
Department of Chemistry
April 22, 2010
Certified by
Richard R. Schrock
Thesis Supervisor
Accepted by
Robert W. Field
Chairman, Departmental Committee on Graduate Students
This doctoral thesis has been examined by a Committee of the Department of Chemistry as
follows:
Professor Richard R. Schrock
Thesis Supervisor
Professor Daniel G. Nocera
Chairman
Professor Stephen J. Lippard
2
SYNTHESES AND STUDIES OF MOLYBDENUM AND TUNGSTEN COMPLEXES AND
THEIR RELEVANCE TO DINITROGEN REDUCTION
BY
JIA MIN CHIN
Submitted to the Department of Chemistry on April 22, 2010
in Partial Fulfilment of the Requirements for the
Degree of Doctor of Philosophy in
Chemistry
ABSTRACT
A
series
of
monopyrroletriamine
ligands
[Arpyr(Ar’)2]H3
of
the
form
ArC4H2NHCH2N(CH2CH2NHAr’)2 (Ar = 2,4,6-mesityl (Mes), 2,4,6-triisopropylphenyl (TRIP);
Ar’ = C6F5, 2-tolyl (o-tol), naphthyl, 3,5-(2,4,6-triisopropylphenyl)phenyl (HIPT), 3,5dimethylphenyl,
3,5-di-tert-butylphenyl
were
synthesized.
[Mespyr(C6F5)2]MoCl,
([Mespyr(C6F5)2]Mo = MesitylC4H2NCH2N(CH2CH2NC6F5)2) was prepared by reaction of
[Mespyr(C6F5)2]H3 with MoCl4(THF)2 and base and [Mespyr(3,5-t-Bu)2]MoCl and [Mespyr(3,5Me)2]MoCl (3,5-t-Bu=3,5-di-tert-butylphenyl, Me = 3,5-dimethylphenyl) were synthesized
likewise. All three monochlorides are paramagnetic. [Mespyr(C6F5)2]MoNMe2, [[Mespyr(otol)2]MoNMe2, [Mespyr(3,5-t-Bu)2]MoNMe2, [Mespyr(3,5-Me)2]MoNMe2 were synthesized by
reaction of the ligands with Mo(NMe2)4. The resulting compounds are diamagnetic and range in
color from teal blue to emerald green. These low spin monodimethylamide complexes exist in
rapid equilibria with their high spin forms. [Mespyr(C6F5)2]MoN and [Mespyr(3,5-t-Bu)2]MoN
were synthesized by reaction of their respective monochlorides with NaN3 and are yellow
diamagnetic species. Reaction of [Mespyr(3,5-t-Bu)2]MoN with Et3OBF4 leads to {[Mespyr(3,5t-Bu)2]MoNEt}BF4, also a diamagnetic yellow species. [Mespyr(C6F5)2]MoOTf is synthesized
by the reaction of [Mespyr(C6F5)2]MoCl with AgOTf. Reduction of [Mespyr(3,5-t-Bu)2]MoCl
with Na under N2 led to [Mespyr(3,5-t-Bu)2]MoNNNa(THF)x, several species with varying
numbers of THF coordination, x. A single species can be obtained when [Mespyr(3,5-tBu)2]MoNNNa(THF)x is reacted with either NBu4Cl or 15-crown-5 ether to yield purple green
3
{[Mespyr(3,5-t-Bu)2]MoNN}NBu4 or [Mespyr(3,5-t-Bu)2]MoNNNa(15-c-5). All the diazenide
species are diamagnetic. Oxidation of the diazenide with AgOTf yields [Mespyr(3,5-tBu)2]Mo(N2). [Mespyr(3,5-t-Bu)2]Mo(CO) is synthesized by exposure of [Mespyr(3,5-tBu)2]Mo(N2) to CO. Reaction of [Mespyr(3,5-t-Bu)2]MoCl with NaBPh4 and NH3 yields
{[Mespyr(3,5-t-Bu)2]Mo(NH3)}BPh4. Catalytic runs employing [Mespyr(3,5-t-Bu)2]MoN as the
catalyst yielded one equivalent of NH3.
A triamidoamine ligand [(HIPTNCH2CH2CH2)3N]3- was synthesized and metalated with
MoCl4(THF)2 to produce [(HIPTNCH2CH2CH2)3N]MoCl ([HIPTtrpn]MoCl). Reduction of
[HIPTtrpn]MoCl by KC8 under an atmosphere of dinitrogen leads to the green species
[HIPTtrpn]MoNNK which can be oxidized by ZnCl2(dioxane) to produce [HIPTtrpn]Mo(N2).
Other complexes synthesized include {[HIPTtrpn]Mo(NH3)}+ salts and [HIPTtrpn]Mo(CO). Xray studies were carried out on [HIPTtrpn]MoN and {[HIPTtrpn]Mo(NH3)}BAr'4. This system is
not catalytic for the reduction of dinitrogen to ammonia and studies were carried out to elucidate
the reasons.
Oxidation studies were carried out on [HIPTN3N]Mo(N2) and [HIPTN3N]W(N2)
([HIPTN3N] = [(HIPTNCH2CH2)3N]3-). The rate of conversion of [HIPTN3N]Mo(NH3) to
[HIPTN3N]Mo(N2) was studied and found to be increased in the presence of BPh3.
[HIPTN3N]Mo(N2) conversion to [HIPTN3N]Mo(CO) was found to be dependent on CO
pressure. Protonation studies of [HIPTN3N]Mo(N2) were also carried out. Studies of
[HIPTN3N]MoNNH decomposition showed that decomposition is not base-catalyzed.
[HIPTN3N]W(CO) was synthesized by exposure of [HIPTN3N]W(N2) to CO. It is a green,
paramagnetic compound and its use as a standard (for determining relative concentrations of
other compounds in the IR sample) in IR spectroscopic studies appears to be promising.
[HIPTN3N]MoCNH2 was synthesized by addition of acid and reducing agent to
[HIPTN3N]MoCN and is a yellow, diamagnetic compound.
Two triamidophosphine ligands, triHIPTamine and tri-n-Buamine were synthesized.
Metalation of Zr(NMe2)4 with these ligands leads to formation of pn3HIPTZrNMe2 and pn3-nBuZrNMe2, both diamagnetic, pale yellow complexes.
Thesis Supervisor:
Richard R. Schrock
Title:
Frederick G. Keyes Professor of Chemistry
4
Table of Contents
Title Page………………………………………………………………………………………….1
Signature Page…………………………………………………………………………………….2
Abstract……………………………………………………………………………………………3
Table of Contents………………………………………………………………………………….6
List of Figures ................................................................................................................................. 8
List of Schemes ............................................................................................................................. 12
List of Tables ................................................................................................................................ 13
List of Abbreviations Used in the Text ......................................................................................... 14
Chapter 1 Introduction .................................................................................................................. 17
Introduction ............................................................................................................................... 18
References ................................................................................................................................. 25
Chapter 2 Syntheses and Studies of Monopyrroletriamine Ligands and their Molybdenum
Complexes..................................................................................................................................... 27
Introduction ............................................................................................................................... 28
Results and Discussion ............................................................................................................. 29
Syntheses and studies of Mespyr(C6F5)2H3 and its Mo complexes ..................................................................... 29
Other monopyrroletriamine ligands and their Mo complexes ........................................................................... 39
Conclusions ............................................................................................................................... 46
Experimental ............................................................................................................................. 46
References ................................................................................................................................. 56
Chapter 3 Synthesis and studies of some diamidomonopyrrolylamine Mo complexes ............... 58
Introduction ............................................................................................................................... 59
Results and Discussion ............................................................................................................. 60
Synthesis of 3,5-di-tert-Buphenyltriamine (1a), 3,5-di-Mephenyltriamine (1b)................................................. 60
5
Synthesis of [Mespyr(3,5-t-Bu)2]H3, (3a); [Mespyr(3,5-Me)2]H3 (3b) and their metal complexes ................... 62
Catalytic studies ................................................................................................................................................. 89
Conclusions ............................................................................................................................... 90
Experimental ............................................................................................................................. 90
References ............................................................................................................................... 102
Chapter 4 Molybdenum Complexes that Contain the [(HIPTNCH2CH2CH2)3N]3- Ligand (HIPT =
3,5-(2,4,6-i-Pr3C6H2)2C6H3) and Studies Relevant to Catalytic Reduction of Dinitrogen ......... 103
Introduction ............................................................................................................................. 104
Results and discussion ............................................................................................................ 105
Synthesis and characterization of [pMo] complexes ([pMo] = [HIPTtrpn]Mo) ............................................. 105
Relevant studies for dinitrogen reduction ........................................................................................................ 118
Conclusions ............................................................................................................................. 124
Experimental ........................................................................................................................... 125
References ............................................................................................................................... 129
Chapter 5 Continuing syntheses and studies on [HIPTN3N]Mo and [HIPTN3N]W complexes 131
Introduction ............................................................................................................................. 132
Results and discussion ............................................................................................................ 132
Oxidation of [Mo](N2) and [W](N2)................................................................................................................. 132
Synthesis of [W]CO.......................................................................................................................................... 135
Kinetics of [Mo](NH3) conversion to [Mo](N2) ............................................................................................... 136
Conversion of [Mo](N2) to [Mo](CO).............................................................................................................. 138
Studies of [Mo](N2) reactivity with acid .......................................................................................................... 140
Studies of effect of base on [Mo]NNH decomposition ..................................................................................... 144
Conclusions ............................................................................................................................. 147
Experimental ........................................................................................................................... 148
Appendix 1
Some triamidophosphine ligands and their metal complexes ............................. 151
Introduction ............................................................................................................................. 152
Results and discussion ............................................................................................................ 152
6
Synthesis of triHIPTamine ............................................................................................................................... 158
Synthesis of Metal Complexes of triHIPTamine............................................................................................... 161
Synthesis of tri-n-Buamine ............................................................................................................................... 163
Synthesis of Metal Complexes of tri-n-Buamine .............................................................................................. 164
Conclusions ............................................................................................................................. 165
Experimental ........................................................................................................................... 165
Appendix 2
Crystallographic Tables ...................................................................................... 173
Crystallographic Studies ......................................................................................................... 174
Crystallographic data for {[HIPTtrpn]Mo(NH3)}BAr'4 ......................................................... 175
Crystallographic Data for [Mespyr(C6F5)2]MoCl ................................................................... 177
Crystallographic Data for [Mespyr(C6F5)2]MoNMe2 ............................................................. 179
Crystallographic Data for [HIPTtrpn]MoN ............................................................................ 181
Crystallographic Data for Mespyr(3,5-t-Bu)2]MoN ............................................................... 184
References ............................................................................................................................... 185
Acknowledgements ................................................................................................................. 186
7
List of Figures
Figure 1.1 Examples of transition metal dinitrogen complexes. A) A ruthenium pentaamine
dinitrogen complex.13 B) trans-[Mo(N2)2(dppe)2] studied by the groups of Hidai, Leigh and
Chatt.15,16,17 C) An iron dinitrogen complex supported by a tris(phosphino)silyl ligand. .... 20
Figure 1.2 Formation of oxamide from an ansa-hafnocene dinitrogen complex.14 ...................... 20
Figure 1.3 Drawing of a triamidoamine metal complex ............................................................... 21
Figure 1.4 A) [HIPTN3N]H3 B) [HIPTN3N]MoN2....................................................................... 22
Figure 1.5 Chatt cycle for the reduction of dinitrogen to ammonia on a Mo center (Mo represents
[HIPTN3N]Mo) ..................................................................................................................... 23
Figure 1.6 Set-up of a catalytic run ............................................................................................... 24
Figure 2.1 [Ar(Ar')2]H3 ................................................................................................................. 28
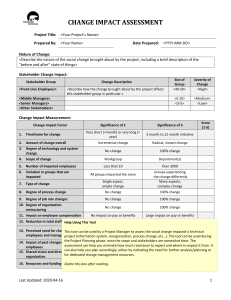
Figure 2.2 POV-ray rendering (Thermal ellipsoids shown at 50% probability) of the solid state
structure of [Mespyr(C6F5)2]MoNMe2. H atoms are omitted for clarity. Selected bond
distances (Å) and angles (°): N(5)-Mo(1) = 1.9383(12), Mo(1)-N(3) = 1.9738(12), Mo(1)N(2) = 1.9885(12), Mo(1)-N(1) = 2.0807(12), Mo(1)-N(4) = 2.2630(12), C(21)-N(2)-Mo(1)
= 129.36(10), C(31)-N(3)-Mo(1) = 128.3(3), N(1)-Mo(1)-N(4) = 77.26(5), N(2)-Mo(1)N(4) = 78.61(5), N(3)-Mo(1)-N(4) = 77.51(5), N(5)-Mo(1)-N(4) = 176.92(5). .................. 32
Figure 2.3 Triamidoamine dimethylamide Mo complexes ........................................................... 33
Figure 2.4 Temperature-dependent 1H NMR spectrum of [Mespyr(C6F5)2]MoNMe2; the arrow
indicates the position of the dimethylamide chemical shift .................................................. 34
Figure
2.5
Temperature-dependent
1
H
NMR
shift
of
the
NMe2
group
of
[Mespyr(C6F5)2]MoNMe2 ..................................................................................................... 35
Figure 2.6 POV-ray rendering (Thermal ellipsoids shown at 50% probability) of the solid state
structure of [Mespyr(C6F5)2]MoCl. H atoms are omitted for clarity. Selected bond distances
(Å) and angles (°): Cl(2)-Mo(1) = 2.3583(4), Mo(1)-N(3) = 1.9539(12), Mo(1)-N(2) =
1.9688(12), Mo(1)-N(1) = 2.0184(12), Mo(1)-N(4) = 2.1737(12), C(21)-N(2)-Mo(1) =
125.99(10), C(31)-N(3)-Mo(1) = 123.41(10), N(1)-Mo(1)-N(4) = 79.05(5), N(2)-Mo(1)N(4) = 79.49(5), N(3)-Mo(1)-N(4) = 79.98(5), N(4)-Mo(1)-Cl(2) = 174.98(3). ................. 37
Figure 2.7 HIPTBr ........................................................................................................................ 40
Figure 2.8 Temperature-dependent
1
H NMR shift of the NMe2 group of [Mespyr(o-
tol)2]MoNMe2 ....................................................................................................................... 42
8
Figure 2.9 Temperature-dependent 1H NMR spectrum of [Mespyr(o-tol)2]MoNMe2; the arrow
indicates the position of the dimethylamide chemical shift .................................................. 43
Figure 2.10 DTBTBr ..................................................................................................................... 45
Figure 3.1 Triamidoamine dimethylamide Mo complexes ........................................................... 64
Figure 3.2 Diamidomonopyrrolylamine dimethylamide Mo complexes ...................................... 64
Figure 3.3 Temperature-dependent 1H NMR shift for the dimethylamide group of [Mespyr(3,5-tBu)]MoNMe2 (4a) ................................................................................................................ 65
Figure 3.4 Temperature-dependent 1H NMR shift for the dimethylamide group of [Mespyr(3,5Me)]MoNMe2 (4b)................................................................................................................ 66
Figure 3.5 Comparison of temperature-dependent 1H NMR shifts for the dimethylamide group 66
Figure 3.6 POV-ray rendering (Thermal ellipsoids shown at 50% probability) of the solid state
structure of [Mespyr(3,5-t-Bu)2]MoN (6a). H atoms are omitted for clarity. Selected bond
distances (Å) and angles (°): Mo(1)-N(5) = 1.6746(13), Mo(1)-N(4) = 2.4134(13), Mo(1)N(1) = 2.0565(12), Mo(1)-N(2) = 1.9857(13), Mo(1)-N(3) = 1.9751(13), C(21)-N(2)-Mo(1)
= 126.27(10), C(31)-N(3)-Mo(1) = 127.25(10), N(1)-Mo(1)-N(4) = 75.66(5), N(2)-Mo(1)N(4) = 76.08(5), N(3)-Mo(1)-N(4) = 80.99(5), N(4)-Mo(1)-N(5) = 176.43(5) ................... 70
Figure 3.7 Electrochemical behavior of [Mespyr(3,5-t-Bu)2]MoN2TBA (9a) in 0.1M
[NBu4]BAr'4 in PhF recorded at a glassy carbon electrode at 100mV/s to 900mV/s scan
rates, referenced to Cp2Fe+/0 ................................................................................................. 72
Figure 3.8 Electrochemical behavior of [Mespyr(3,5-t-Bu)2]MoN2TBA (9a) in 0.1 M
[NBu4]BAr'4 in PhF recorded at a glassy carbon electrode at 900 mV/s, referenced to
Cp2Fe+/0, showing the irreversible oxidation of [Mespyr(3,5-t-Bu)2]Mo(N2) (10a) ............ 73
Figure 3.9 [HIPTtrpnN3N]Mo(N2)................................................................................................ 74
Figure 3.10 IR absorption spectrum of [Mespyr(3,5-t-Bu)2]Mo14N2/[Mespyr(3,5-t-Bu)2]Mo14N2
Na(THF)x after 15N2 exposure for 2.5 hours in C6D6 ........................................................... 75
Figure 3.11 Plot of area of A14 (area of ν14N14N absorption band) over Atotal (area of ν14N14N +
area of ν14N14N absorption bands) against time at 22 °C and one atm N2 for exchange
reaction of [Mespyr(3,5-t-Bu)2]Mo(15N2) to [Mespyr(3,5-t-Bu)2]Mo(N2) ........................... 77
Figure 3.12 Plot of Ln(A15/Atotal) against time (s) for exchange reaction[Mespyr(3,5-tBu)2]Mo(15N2) to [Mespyr(3,5-t-Bu)2]Mo(N2) ..................................................................... 77
Figure 3.13 Reaction set-up for increased pressure 15N2/N2 exchange study ............................... 79
9
Figure 3.14 Plot of area of A14 (area of ν14N14N absorption band) over Atotal (area of ν14N14N + area
of ν14N14N absorption bands) against time at 22 °C, two atm
14
N2 for exchange
15
reaction[Mespyr(3,5-t-Bu)2]Mo( N2) to [Mespyr(3,5-t-Bu)2]Mo(N2) ................................ 79
Figure 3.15 Plot of Ln(A15/Atotal) vs against time (s) for exchange reaction[Mespyr(3,5-tBu)2]Mo(15N2) to [Mespyr(3,5-t-Bu)2]Mo(N2) ..................................................................... 80
Figure 3.16 Electrochemical behavior of {[Mespyr(3,5-t-Bu)2]Mo(NH3)}BPh4 (11a) in 0.1 M
[NBu4]BAr'4 in PhF recorded at a glassy carbon electrode, referenced to Cp2Fe+/0 ............. 82
Figure 3.17 Electrochemistry of 11a under N2 atmosphere .......................................................... 83
Figure 3.18 Appearance of 10a at scan rates of 10 and 50 mV/sec .............................................. 83
Figure 3.19 Conversion of 12a to 10a against time ...................................................................... 86
Figure 3.20 IR spectrum of [HIPTN3N]Mo(CO) and [Mespyr(3,5-t-Bu)2]Mo(CO) (13a) when
exposed to increasing equivalents of [Collidinium]BAr'4 .................................................... 87
Figure 3.21 IR spectrum of [Mespyr(3,5-t-Bu)2]Mo(13CO) when exposed to 0.5 equivalents of
[Collidinium]BAr'4 in DME.................................................................................................. 88
Figure 4.1 Chatt cycle for the reduction of dinitrogen to ammonia on a Mo center (Mo represents
[HIPTN3N]Mo) ................................................................................................................... 105
Figure 4.2 POV-ray rendering of [pMo]Cl with thermal ellipsoids at 50% probability.7 Hydrogen
atoms and isopropyl groups are omitted for clarity. Selected bond lengths (Å): Cl(1)-Mo(1)
= 2.3843(5), Mo(1)-N(1) = 1.9688(17), Mo(1)-N(2) = 1.9709(18), Mo(1)-N(3) =
1.9743(18), Mo(1)-N(4) = 2.3230(18). Selected bond angles (°):N(1)-Mo(1)-N(4) =
89.03(7), N(2)-Mo(1)-N(4) = 89.26(7), N(3)-Mo(1)-N(4) = 87.76(7) ............................... 106
Figure 4.3 Cyclic voltammogram of {[pMo](NH3)]BAr'4 at a 3.0 mm glassy carbon electrode at
room temperature with 0.1 M [NBu4]BAr'4 in PhF and scan rate of 100 mV/sec, referenced
to Cp2Fe+/0 ........................................................................................................................... 108
Figure 4.4 POV-ray rendering of {[pMo](NH3)}BAr'4 with thermal ellipsoids at 50% probability
and hydrogens and isopropyl groups omitted for clarity. The BAr'4 anion is also omitted.
Selected bond distances (Å): Mo(1)-N(3) = 1.960(4), Mo(1)-N(2) = 1.964(4), Mo(1)-N(1) =
1.972(4), Mo(1)-N(5) = 2.238(4), Mo(1)-N(4) = 2.256(4). Selected angles (°): N(3)-Mo(1)N(4) = 92.10(15), N(2)-Mo(1)-N(4) = 90.92(14), N(1)-Mo(1)-N(4) = 92.25(16), N(5)Mo(1)-N(4) = 178.92(16).................................................................................................... 109
Figure 4.5 Unrefined POV-ray rendering of [pMo]NNK with hydrogens and isopropyl groups
omitted for clarity. .............................................................................................................. 111
10
Figure 4.6 Cyclic voltammogram of [pMo](N2) at a 3.0 mm glassy carbon electrode at room
temperature with 0.1 M [NBu4]BAr'4 in PhF and scan rate of 100 mV/sec , referenced to
Cp2Fe+/0. .............................................................................................................................. 114
Figure 4.7 Cyclic voltammogram of [pMo](CO) at a 3.0 mm glassy carbon electrode at room
temperature with 0.1 M [NBu4]BAr'4 in PhF and scan rate of 100 mV/sec , referenced to
Cp2Fe+/0 ............................................................................................................................... 115
Figure 4.8 POV-Ray rendering of [pMo]N with thermal ellipsoids at 50% probability. Isopropyl
groups, hydrogen atoms, and solvent molecules are omitted for clarity. Selected bond
distances (Å): Mo(1)-N(5) = 1.651(5), Mo(1)-N(1) = 2.004(5), Mo(1)-N(2) = 1.999(5),
Mo(1)-N(3) = 1.961(5). Selected angles (°): N(5)-Mo(1)-N(1) = 99.9 (2), N(5)-Mo(1)-N(2)
= 103.3(2), N(5)-Mo(1)-N(3) = 97.3(2), C(115)-N(1)-Mo(1) = 131.0(4), C(215)-N(2)Mo(1) = 127.2(4), C(315)-N(3)-Mo(1) = 121.5(4) ............................................................. 116
Figure 4.9 Area of νNN against Time (min) ................................................................................. 119
Figure 4.10 Area of νCO against equivalents of [Collidinium]BAr'4 ........................................... 122
Figure 4.11 Area of νCO against equivalents of [Collidinium]BAr'4 ........................................... 123
Figure 5.1 Hypothetical {[Mo](N2)}+ species with side-on bound N2........................................ 133
Figure 5.2 IR spectrum of [W](N2) before oxidation (solid line) and after oxidation (dashed line)
............................................................................................................................................. 134
Figure 5.3 IR spectra of [Mo](N2) (solid line), [Mo](N2) after Cp2FeBAr'4 addition (dotted line)
and[Mo](N2) after Cp2FeBAr'4 and then THF addition (dashed line) ................................ 135
Figure 5.4 Formation of [W]CO ................................................................................................. 136
Figure 5.5 Chatt-like cycle for reduction of dinitrogen to ammonia on a Mo center ................. 136
Figure 5.6 Area of νNN of Mo(N2) against time. t1/2 = 179 minutes ............................................ 137
Figure 5.7Area of νNN of Mo(N2) against time.The half-life, t1/2 = 39 minutes in presence of ten
equivalents of BPh3. ............................................................................................................ 138
Figure 5.8 Area of νCO of [Mo](CO) against time (min) at 0.44 atm pressure of CO ................ 139
Figure 5.9 Area of νCO of [Mo](CO) and νNN of [Mo](N2) against time (min) at 0.09 atm CO,
where νCO is shown in red, νNN is in blue. ........................................................................... 140
Figure 5.10 Hypothetical hydrogen bonded HB adduct. B is the conjugate base of the acid..... 141
Figure 5.11 Area of N2 peak with increasing [2,6-LutH]BAr'4 addition .................................... 142
Figure 5.12 Plot of [Mo]NNH proton peak against Time. 0.01 equivalents of base were utilized.
Delay time = two seconds. .................................................................................................. 145
11
Figure 5.13 Plot of [Mo]NNH proton peak against time. Ten equivalents of base were utilized.
Delay time = two seconds. .................................................................................................. 146
Figure A 1.1 Phosphines utilized in synthetic attempts of 2-bromo-N-HIPTaniline.................. 153
Figure A 1.2 Synthesis of a PN3 calix[6]arene-based ligand8 (Figure from reference) .............. 158
Figure A 1.3 a) TriHIPTimine b) TriHIPTamine ....................................................................... 161
List of Schemes
Scheme 2.1 Synthesis of [Arpyr(Ar’)2]H3 .................................................................................... 41
Scheme 3.1 Synthesis of [Mespyr(3,5-t-Bu)2]H3, (3a) and [Mespyr(3,5-Me)2]H3 (3b).............. 63
Scheme 3.2 Postulated 14N2 for 15N2 exchange in [ArN3N]Mo system ........................................ 78
Scheme 3.3 Pathways for 14N2/15N2 exchange .............................................................................. 81
Scheme 3.4 Exchange of N2 for NH3 ............................................................................................ 84
Scheme A 1.1 Proposed synthesis of triamidophosphine ligand with a phenylene backbone. .. 152
Scheme A 1.2 Attempt at nucleophilic substitution of tris(2-fluorophenyl)phosphine oxide .... 157
Scheme A 1.3 Synthesis of TriHIPTamine ligand ...................................................................... 159
Scheme A 1.4 Synthesis of 2,2’,2”-phosphinetriyltribenzaldehyde ........................................... 160
Scheme A 1.5 Synthesis of tri-n-Buamine.................................................................................. 163
12
List of Tables
Table 3.1 Equivalents of NH3 obtained ........................................................................................ 90
Table 4.1 Comparison of structural parameters between [pMo]Cl and [HTBTN3N]MoCl ....... 107
Table 4.2 Comparison of bond lengths and angles between [pMo(NH3)]BAr'4 and
{[Mo](NH3)}BAr'4 .............................................................................................................. 109
Table 4.3 Comparison of solid state structure of [pMo]NNK with that of [Mo]NNMgCl(THF)3
............................................................................................................................................. 112
Table 4.4 Comparison of bond lengths and angles between [pMo]N and [Mo]N...................... 117
Table 4.5 Values of νCO for Mo carbonyl complexes ................................................................. 121
Table 5.1 Area of νNN with increasing acid ................................................................................ 142
Table 5.2 Area of νNN upon addition of different acids............................................................... 143
Table A 2.1 Crystal data and structure refinement for {[HIPTtrpn]Mo(NH3)}BAr'4 ................ 175
Table A 2.2 Selected Bond lengths [Å] and angles [°] for {[HIPTtrpn]Mo(NH3)}BAr'4 .......... 176
Table A 2.3 Crystal data and structure refinement for [Mespyr(C6F5)2]MoCl ........................... 177
Table A 2.4 Selected Bond lengths [Å] and angles [°] for [Mespyr(C6F5)2]MoCl..................... 178
Table A 2.5 Crystal data and structure refinement for [Mespyr(C6F5)2]MoNMe2 ..................... 179
Table A 2.6 Selected Bond lengths [Å] and angles [°] for [Mespyr(C6F5)2]MoNMe2 ............... 180
Table A 2.7 Crystal data and structure refinement for [HIPTtrpn]MoN .................................... 181
Table A 2.8 Selected Bond lengths [Å] and angles [°] for [HIPTtrpn]MoN .............................. 182
Table A 2.9 Crystal data and structure refinement for [Mespyr(3,5-t-Bu)2]MoN ...................... 184
Table A 2.10 Selected Bond lengths [Å] and angles [°] for [Mespyr(3.5-t-Bu)2]MoN.............. 185
13
List of Abbreviations Used in the Text
Å
Angstrom
Ar
Aryl
Atm
Atmosphere(s)
BAr'4
Tetra-bis(trifluoromethyl)phenyl borate [3,5bis(CF3)C6H3]4B]-
BINAP
2,2’-bis(diphenylphosphino)-1,1’-binaphthyl
br
broad
Calcd
calculated
Cipso
ipso carbon in an aryl group
Collidine
2,4,6-trimethylpyridine
Cp*
Pentamethylcyclopentadienyl (Me5C5)
CV
Cyclic voltammetry
d
Doublet
dba
Dibenzylideneacetone
dd
Doublet of doublets
deg
Degree
DME
1,2-dimethoxyethane
DMF
N,N-dimethylformamide
DMSO
Dimethylsulfoxide
EI
Electron impact (mass spectrometry)
eq, or equiv.
Equivalent(s)
Et
Ethyl
Et2O
Diethyl ether
14
eV
Electron volts
ηx
Hapticity of a ligand bound to a metal
through x atoms
g
Grams
h or hr
Hour(s)
Hz
Hertz
HRMS
High resolution mass spectrometry
i
Pr
Isopropyl (CH(CH3)2)
IR
Infrared
JAB
Coupling constant between atoms A and B
Kcal
Kilocalories
L
Liter
Lutidine or 2,6-lutidine
2,6-dimethylpyridine
Lutidinium or 2,6-lutidinium
2,6-dimethylpyridinium
M
Molar
m
Multiplet
Me
Methyl
Mesityl
2,4,6-trimethylbenzene
Mg
Milligram(s)
Min
Minute(s)
mL
Millilitres(s)
mmol
Millimoles
Bu
Butyl (-CH2CH2CH2CH3)
Να
Nitrogen directly bound to the metal
15
Nβ
Nitrogen two bonds from the metal
NMR
Nuclear magnetic resonance
OAc
Acetate, ([-CO2CH3]-)
OTf
Triflate,
trifluoromethanesulfonate
([-O3SCF3]-)
Ph
Phenyl (-C6H5)
ppm
Parts per million
PSI
Pounds per square inch
q
Quartet
RT
Room temperature
rac
Racemic
s
Singlet
T
Temperature
t
Triplet
t-Bu
Tertiary butyl (-C(CH3)3)
THF
Tetrahydrofuran
TMS
Tetramethylsilane
TREN
N(CH2CH2NH2)3
TRPN
N(CH2CH2CH2NH2)3
TsOH
p-toluenesulfonic acid
UV
Ultraviolet
V
Volts
VT
Variable temperature
[RN3N]
[(RNCH2CH2)3N]316
Chapter 1
Chapter 1
Introduction
17
Chapter 1
Introduction
Nitrogen is extremely important, both biologically and industrially. This is apparent from
its incorporation in amino acids to its widespread uses in fertilizers, dyes and explosives.1 While
the abundance of molecular nitrogen makes it an attractive feedstock for such uses, its inertness
due to its strong triple bond (BDE of 225 kcal/mol),2 high ionization potential (15.058 eV) and
low electron affinity (-1.8eV) presents a fundamental problem.3,4,5 As a result, before nitrogen
can be used, it usually must be converted into an activated form such as ammonia. The challenge
then arises: the formation of ammonia from inert dinitrogen.
Nature’s answer to this challenge is biological nitrogen fixation, where approximately 1.7
x 108 tons/year of ammonia are formed by various nitrogenases (Equation 1.1).5,67 The most
common nitrogenase found in known nitrogen-fixing organisms is the FeMo nitrogenase
although alternative nitrogenases like FeV, FeW or all Fe nitrogenase exist. 8 , 9 Nitrogenase
isolated from Azotobacter vinelandii is composed of two proteins. The first is an Fe-protein,
which utilizes energy from ATP hydrolysis for electron transfer to the second protein. The
second protein is the FeMo protein, where dinitrogen reduction to ammonia is catalyzed.10 The
FeMo protein is an α2β2 tetramer which contains two copies of the P-cluster, thought to be
involved in electron transfer between proteins, as well as two copies of the FeMo-cofactor,
which is the active site for dinitrogen reduction.11
Equation 1.1
Industrially, the Haber-Bosch process produces a similar amount of ammonia per year.
However, this process requires high temperatures (350 – 550 °C) and pressures (150 – 350 atm)
(Equation 1.2) and a heterogeneous Fe or FeRu catalyst, a stark contrast to biological nitrogen
18
Chapter 1
fixation which takes place under ambient conditions.12 Moreover, this process consumes about 12% of the world’s energy.13 Therefore, there is a huge impetus for industrial nitrogen fixation
that is much more energy efficient.
350 - 550 °C, 150 - 350 atm
N 2 + 3 H2
2 NH3
Fe or FeRu catalyst
Equation 1.2
Unfortunately, despite intense study, the mechanistic details of both these processes have
not been completely elucidated. Chemists have long sought to reduce dinitrogen at ambient
conditions, and to understand the intricacies of this process. The first characterized dinitrogen
complex is [Ru(NH3)5N2]2+, synthesized by Allen and Senoff in 1965 (Figure 1.1).14 Following
that discovery, many dinitrogen-containing metal complexes have been found, including some
examples shown in Figure 1.1 below. Intensive efforts have been focused on the activation and
functionalization of dinitrogen using transition metal catalysts. For example, Chirik et al. have
shown that oxamide is produced on reacting an ansa-hafnocene dinitrogen complex with CO and
weak acid (Figure 1.2). 15 Also, the groups of Hidai, Chatt and Leigh have studied reducing
dinitrogen to ammonia using molybdenum and tungsten complexes (Figure 1.1). 16,17,18
19
Chapter 1
Figure 1.1 Examples of transition metal dinitrogen complexes. A) A ruthenium pentaamine dinitrogen
complex.14 B) trans-[Mo(N2)2(dppe)2] studied by the groups of Hidai, Leigh and Chatt.16,17,18 C) An iron
dinitrogen complex supported by a tris(phosphino)silyl ligand.19
Figure 1.2 Formation of oxamide from an ansa-hafnocene dinitrogen complex.15
Many dinitrogen complexes of molybdenum with phosphine supporting ligands have
been synthesized, with particular focus on compounds containing chelating ligands such as
trans-[Mo(N2)2(dppe)2], which was found to yield hydrazido complexes when treated with
halogen acids. The hydrazido complexes can then be dehydrohalogenated with base to yield
diazenido complexes. 20 , 21 Chatt’s group reported the formation of ammonia from both
molybdenum and tungsten dinitrogen complexes [M(N2)2(PMenPh3-n)4], where M = Mo or W by
treatment of these complexes with a methanol solution of sulfuric acid.22 However, the highest
yield of ammonia was limited to less than two equivalents per molybdenum or tungsten molecule
20
Chapter 1
as the process involved concomitant conversion of the complexes to unidentified products that
were not active for further dinitrogen conversion to ammonia.
Our group is similarly interested in the functionalization of dinitrogen, particularly in the
reduction of dinitrogen to ammonia. We have been focusing on the chemistry of triamidoamine
transition metal complexes, particularly those of molybdenum and tungsten. 23 The chelating
nature of the triamidoamine ligand, [(RNCH2CH2)3N]3- (Figure 1.3) prevents the ligand from
coming off the metal center completely even after a protonation of the ligand. This creates a
pocket for binding to other ligands, such as dinitrogen, for example. We think that the ligand is
capable of donating up to twelve electrons to the transition metal center of C3-symmetry, the last
lone pair being in a non-bonding orbital centered on the amido nitrogens. A variety of
triamidoamine ligands can be synthesized, where R is a silyl group or an aryl group. The
development of various C-N coupling methods, particularly the Pd catalyzed cross-coupling of
aryl halides to amines, has opened up the possibilities of installing various aryl substituents on
the amido nitrogens. Utilizing tris(2-aminoethyl)amine (tren) and aryl bromides, the desired
ligand can be obtained via a Buchwald-Hartwig Pd cross-coupling reaction.24
Figure 1.3 Drawing of a triamidoamine metal complex
Research efforts in our group have centered on creating a well-defined catalytic system
for the reduction of dinitrogen under mild conditions and we have recently been able to achieve
this in the [HIPTN3N]Mo system ([HIPTN3N]H3 = HexaIsoPropylTerphenyl tetraamine ligand)
21
Chapter 1
(Figure 1.4).25 However, the number of catalytic turnovers is limited to four, and continuing
efforts are being made to understand and improve on this system and similar ones.26
Figure 1.4 a) [HIPTN3N]H3 b) [HIPTN3N]MoN2
One of the important characteristics of the above system is the steric bulk of the HIPT
substituents. This is key in preventing the formation of the thermodynamically stable dimers
[ArN3N]Mo-N=N-Mo[N3NAr] during the course of dinitrogen reduction. Moreover, the steric
protection afforded by the HIPT substituents is crucial in preventing decomposition of the ligand
in the steric pocket (eg. [ArN3N]MoNNH to [ArN3N]Mo(N2) decomposition) in a manner that
does not produce ammonia.
22
Chapter 1
Mo(III) 14
+ N2
Mo
Mo(N2)
13
Mo-N=N-
Mo(NH3)
e-
H+
Mo-NH2
Mo-N=N-H
{Mo=N-NH2}+
10 {Mo-NH2}+
9
Mo=N-NH2
Mo(IV)
4
Mo(VI)
5
Mo(V)
6
Mo(V)
7
Mo(VI)
H+
{Mo=N-NH3}+
Mo=NH
e-, -NH3
eMo(VI)
3
e-
H+
Mo(V)
Mo(IV)
H+
eMo(V)
2
H+
Mo(IV) 12 {Mo(NH3)}+
Mo(IV) 11
Mo(III)
e-
-NH3
Mo(III)
1
8 {Mo=NH}+
H+
MoN
Figure 1.5 Chatt cycle for the reduction of dinitrogen to ammonia on a Mo center (Mo represents
[HIPTN3N]Mo)
Shown above (Figure 1.5) is the proposed Chatt-cycle which describes the sequence of
events taking place at the metal center for the reduction of dinitrogen to ammonia through the
addition of protons and electrons. Of the proposed intermediates, eight of them have been
synthesized and characterized (boxed, Figure 1.5). In accordance with expectations, these
compounds are extremely air-sensitive, with the exception of 7, [HIPTN3N]MoN, a Mo(VI)
species.
It should be noted that the above cycle is an extremely simplified version of the actual
events taking place. For example, in the triamidoamine molybdenum system, dihydrogen is
produced along with ammonia during the reduction of dinitrogen, which is similar to dinitrogen
reduction by nitrogenase. At one atmosphere pressure of dinitrogen, nitrogenase forms more than
one equivalent of dihydrogen per molecule of dinitrogen reduced. The efficiency of ammonia
formation ranges from 40 to 60% in terms of reducing equivalents present.
27 , 28 , 29
Correspondingly, under one atmosphere pressure of N2, the [HIPTN3N]Mo system produces
23
Chapter 1
ammonia at about 55-65% efficiency in terms of reducing equivalents.30,31 Recently, it has been
found that dihydrogen reacts with [HIPTN3N]Mo(N2) and also [HIPTN3N]Mo(NH3) to form
poorly defined [HIPTN3N]Mo(H2) and that the presence of dihydrogen does significantly inhibit
the reduction of dinitrogen.32
The set-up of a catalytic run is shown in Figure 1.6. The proton source, [2,4-6Collidinium]BAr'4,or [2,6-Lutidinium] BAr'4 is placed in the flask with the metal catalyst with
heptane as the solvent. The compound Cp*2Cr, which is the electron source, is slowly added to
the catalyst as a solution in heptane. It is crucial to minimize direct reaction between the acid and
the reducing agent, as this would simply result in dihydrogen production (about eighteen
equivalents, based on the equivalents of electrons). The choice of [Collidinium]BAr'4 and
heptane is not incidental. The low solubility of the acid in heptane and the slow addition of
Cp*2Cr solution means that at any given time, the concentration of protons and electrons present
in solution should be low compared to the catalyst.
Figure 1.6 Set-up of a catalytic run
24
Chapter 1
Further studies recently on this system have unravelled some of the intricacies of
dinitrogen reduction as pertaining to triamidoamine Mo complexes, but much remains to be
learned. It appears that dinitrogen reduction is an extremely finely balanced process – an
unsurprising fact given the number of steps involved. It is therefore very useful to understand the
factors that are crucial for dinitrogen reduction to ammonia. This work attempts to examine that
both through studies on the [HIPTN3N]Mo system, and also through synthetically varying the
supporting ligand in the metal complexes. The synthetic variants described in this thesis focus on
the effects of varying the ligand arms themselves rather than on the effects of varying the arylsubtituents on the TREN amido nitrogens, which has been studied in depth in previous work.
References
1
Schlögl, R. Angew. Chem. Int. Ed. 2003, 42, 2004.
2
Bazhenova, T.A.; Shilov, A.E. Coord. Chem. Rev. 1995, 144, 69.
3
Shaver, M. P.; Fryzuk, M..D. Adv. Synth. Catal. 2003, 345, 1061.
4
Pool, J. A.; Lobkovsky, E.; Chirik, P. J. Nature. 2004, 427, 527.
5
Sellmann, D.; Sutter, J. Acc. Chem. Res. 1997, 30, 460.
6
Hidai, M.; Mizobe, Y. Chem Rev. 1995, 95, 1115.
7
Smil, V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food
Production; MIT Press: Cambridge,MA, 2004.
8
Eady, R.R. Chem. Rev. 1996, 96, 3013.
9
10
11
Benemann, J.R.; Smith, G.M.; Kostel, P.J.; McKenna, C.E. FEBS Letters 1973, 29, 219.
Georgiadis, M.M.; Komiya, H.; Chakrabarti, P.; Kornuc, J.J.; Rees, D.C. Science 1992, 257, 1653.
Einsle, O.; Tezcan, F.A.; Andrade, S.L.A.; Schmid, B.; Yoshida, M.; Howard, J.B.; Rees, D.C. Science,
2002, 297, 5587.
25
Chapter 1
12
Schrock, R.R. Acc. Chem. Res. 2005, 38, 955.
13
Leigh, G.J. Science 1998, 279, 506.
14
Allen, A.D.; Senoff, C.V. Chem. Commun. 1965, 621.
15
Knobloch, D.J.; Lobkovsky, E.; Chirik, P.J. Nature Chemistry 2009, 2, 30.
16
Hidai, M.; Mizobe, Y. Chem. Rev. 1995, 95, 1115.
17
Chatt, J.; Dilworth, J.R.; Richards, R.L. Chem. Rev. 1978, 78, 589.
18
Leigh, G.J. Acc. Chem. Res. 1992, 25, 177.
19
Whited, M.T.; Mankad, N.P.; Lee, Y.; Oblad, P.; Peters, J.C. Inorg. Chem.2009, 48, 2507 .
20
Chatt, J.; Heath, G.A.; Richards, R.L. J. Chem. Soc., Dalton,Trans., 1974, 2074 .
21
Chatt, J.; Pearman, A.J.; Richards, R.L. J. Chem. Soc., Dalton trans., 1976, 1520.
22
Chatt,J.; Pearman, A.J.; Richards, R.L. Nature, 1975, 253, 39.
23
Schrock, R.R. Acc. Chem. Res. 1997, 30, 9.
24
Greco, C.E.; Popa, A.I.; Schrock, R.R. Organometallics 1998, 17, 5591.
25
Yandulov, D. V.; Schrock, R. R. Science 2003, 301, 76.
26
Ritleng, V.; Yandulov, D. V.; Weare, W. W.; Schrock, R. R.; Hock, A. R.; Davis, W. M.
J. Am. Chem. Soc. 2004, 126, 6150.
27
Wherland, S.; Burgess, B.K.; Stiefel, E.I.; Newton, W.E. Biochemistry, 1981, 20, 5132.
28
Burgess, B.K.; Wherland, S.; Newton, W.E.; Stiefel, E.I. Biochemistry, 1981, 20, 5140.
29
Fisher, K.; Dilworth, M.J.; Newton, W.E. Biochemistry, 2000, 39, 15570.
30
Schrock, R.R. Angew. Chem., Int. Ed. 2008, 47, 5512.
31
Weare, W.W.; Dai, X.; Byrnes, M.J.; Chin, J.M.; Schrock, R.R.; Müller, P. Proc. Nat. Acad. Sci. 2006,
103, 17099.
32
Hetterscheid, D.; Hanna, B.S.; Schrock, R.R. Inorg. Chem., 2009, 48, 8569.
26
Chapter 2
Syntheses and Studies of Monopyrroletriamine Ligands and their
Molybdenum Complexes
Chapter 2
Introduction
Equation 2.11
We have earlier shown that the symmetric trispyrrolylamine ligand in Equation 2.1 is not
sufficiently electron donating to form a stable molybdenum complex.1 For example, reaction of
the ligand with Mo(NMe2)4 gave the resulting complex shown above, with only two of the
dimethylamides displaced (Equation 2.1). Pyrrolyl nitrogens, with their lone pairs involved in the
aromatic π-cloud of each pyrrolyl ring, are likely to be less electron donating than the
dimethylamide nitrogens.
Figure 2.1 [Ar(Ar')2]H3
Perhaps returning to the triamidoamine ligand framework that our group has studied, and
replacing just one of the arms with a pyrrolyl group, would result in a ligand which has sufficient
electron donating ability to produce stable molybdenum complexes (Figure 2.1). We think that in
triamidoamine molybdenum complexes, there is a non-bonding linear combination of the amide
28
Chapter 2
nitrogen lone pairs in the plane created by the amide nitrogens and the molybdenum center. This
non-bonding orbital may increase the likelihood of ligand arm protonation during catalytic
reduction of dinitrogen, leading to possible decomposition of the molybdenum complex. Since
the nitrogen lone pair on a pyrrolyl is part of the aromatic π-cloud of the pyrrolyl ring, and is
therefore
a
poorer
base
for
protons,
the
likelihood
of
protonation
of
a
diamidomonopyrrolylamine molybdenum complex should decrease.
Results and Discussion
Syntheses and studies of Mespyr(C6F5)2H3 and its Mo complexes
Equation 2.2
We decided to start with a ligand that should be synthetically easily accessible. The
monopyrroletriamine ligand can be broken up into two synthons – a triamine fragment and a
pyrrole fragment. We sought a triamine with a synthetic route where the primary amines were
preferentially
arylated
over
the
secondary
amines.
N1-(perfluorophenyl)-N2-(2-
(perfluorophenylamino)ethyl)ethane-1,2-diamine is synthesized by reacting hexafluorobenzene
with diethylene triamine and potassium carbonate. 2 The perfluorophenyl substituents on the
amide nitrogens should confer crystallinity to the resulting complexes. Therefore, we decided to
look into the chemistry of molybdenum complexes with diamidomonopyrrolylamine ligands
containing perfluorophenyl substituents on the amide nitrogens as an entry point into this new
ligand system.
29
Chapter 2
Equation 2.3
Substituted pyrroles can be synthesized via a Pd cross-coupling of sodium pyrrolide and
the desired aryl bromide.3 The final ligand Mespyr(C6F5)2H3 is then synthesized via a Mannich
reaction
between
the
arylated
pyrrole
N1-(perfluorophenyl)-N2-(2-
and
(perfluorophenylamino)ethyl)ethane-1,2-diamine (Equation 2.3). The triamine is first stirred with
a mixture of formaldehyde and a catalytic amount of HCl. Using a stoichiometric amount of HCl
appears to lead to decomposition. In the case of phenyl-substituted pyrrole, the final ligand is
very sensitive to oxidation, and appears to decompose rapidly during column chromatography.
Replacing the phenyl substituent with the bulkier mesityl substituent greatly increases the
stability of the resulting ligand, decreasing ligand decomposition during chromatography.
Mesityl-substituted pyrrole and its derivatives also have the advantage of being very crystalline,
thus increasing their ease of purification over that for some other aryl-substituted pyrroles. The
ligand can be further purified by recrystallization from a mixture of toluene and pentane, giving a
white powder. Mespyr(C6F5)2H3 was characterized by 1H NMR,
19
F NMR,
13
C NMR and mass
spectrometry.
30
Chapter 2
Equation 2.4
Reacting Mespyr(C6F5)2H3 with Mo(NMe2)4 leads to immediate formation of an emerald
green solution at room temperature and recrystallization from toluene and pentane affords
[Mespyr(C6F5)2]MoNMe2 (Equation 2.4). This compound appears to be diamagnetic by 1H
NMR, presumably due to donation of the lone pair from the dimethylamide nitrogen to the metal
center, increasing the orbital energy difference between the SOMOs, causing the electrons to pair
up and create a low spin complex. A single crystal x-ray diffraction study shows that the central
molybdenum is approximately trigonal bipyramidal, and that the dimethylamide ligand displays
a trigonal planar geometry and occupies a plane parallel to the mesityl substituent (Figure 2.2).
The mesityl substituent on the pyrrolyl arm is tilted less from the central apical site than are the
perfluorophenyl substituents on the amido nitrogens. This suggests that a smaller substituent on
the pyrrolyl arm would afford a similar amount of steric protection to a larger substituent on a
corresponding amido arm. The Mo(1)-N(1) bond length (2.081 Å) in [Mespyr(C6F5)2]MoNMe2
is slightly longer than the average Mo(1)-Npyrrolyl bond length (2.007 Å) in tris(pyrrolyl-αmethyl)amido molybdenum chloride [Trip3TPA]MoCl,4 possibly due to the more electron rich
nature of the central metal.
31
Chapter 2
N5
N1
N3
N2
N4
Figure 2.2 POV-ray rendering (Thermal ellipsoids shown at 50% probability) of the solid state structure
of [Mespyr(C6F5)2]MoNMe2. H atoms are omitted for clarity. Selected bond distances (Å) and angles (°):
N(5)-Mo(1) = 1.9383(12), Mo(1)-N(3) = 1.9738(12), Mo(1)-N(2) = 1.9885(12), Mo(1)-N(1) =
2.0807(12), Mo(1)-N(4) = 2.2630(12), C(21)-N(2)-Mo(1) = 129.36(10), C(31)-N(3)-Mo(1) = 128.3(3),
N(1)-Mo(1)-N(4) = 77.26(5), N(2)-Mo(1)-N(4) = 78.61(5), N(3)-Mo(1)-N(4) = 77.51(5), N(5)-Mo(1)N(4) = 176.92(5).
Earlier studies in our group on the triamidoamine complexes [TMSN3N]MoNMe2 and
[C6F5N3N]MoNMe2 showed that both these compounds (shown in Figure 2.3) are mostly
diamagnetic at room temperature, but exist in equilibrium with their paramagnetic counterparts.4
32
Chapter 2
Si
N
Si
C6F5
N
Mo
N
Si
N
N
C6F5
N
Mo
C6F5
N
N
N
N
[TMSN3N]MoNMe2
[(C6F5)N3N]MoNMe2
Figure 2.3 Triamidoamine dimethylamide Mo complexes
The chemical shift of the dimethylamido 1H NMR resonances were found to be
temperature dependent, and we proposed that the dimethylamido resonance is contact-shifted by
the paramagnetic form. It has been postulated that the extent of the contact shift of the 1H NMR
resonance for the dimethylamide group is an indicator of the energy difference between the high
and low spin forms of the Mo complex, with a larger shift indicating a smaller energy difference,
according to Equation 2.5.4
ߜ = ߜ݀݅ܽ +
ܥ
∆ܪ° ∆ܵ°
−
ܶ(1 + exp ቀ
ܴܶ
ܴ ቁ)
Equation 2.5
In this equation, δ is the observed chemical shift at temperature T, δdia is the chemical
shift of the diamagnetic form, and C is a constant. It is important to note, however, that the above
equation only describes situations where the interconversion between the high and low spin
forms is fast at all temperatures in the temperature range observed. Applying a similar argument
to [Mespyr(C6F5)2]MoNMe2, we can obtain a qualitative comparison of the energy difference
between the high and low spin forms in [Mespyr(C6F5)2]MoNMe2 versus [TMSN3N]MoNMe2
and [CF5N3N]MoNMe2.
33
Chapter 2
Figure 2.4 Temperature-dependent 1H NMR spectrum of [Mespyr(C6F5)2]MoNMe2; the arrow indicates
the position of the dimethylamide chemical shift
34
Chapter 2
2.95
δ / ppm
2.85
2.75
2.65
2.55
2.45
230
250
270
290
310
330
350
370
Temperature / K
Figure 2.5 Temperature-dependent 1H NMR shift of the NMe2 group of [Mespyr(C6F5)2]MoNMe2
The changes in the chemical shift of the dimethylamide ligand for [TMSN3N]MoNMe2
(~ 9 ppm from 180 to 304 K) and [C6F5N3N]MoNMe2 (~2.8 ppm from 259 – 367 K)4 are larger
than that for [Mespyr(C6F5)2]MoNMe2 (0.12 ppm from 233 – 302 K) which suggests that the
energy difference between the high and low spin forms is larger in [Mespyr(C6F5)2]MoNMe2
than in both [TMSN3N]MoNMe2 and [C6F5N3N]MoNMe2. Despite the presence of only two
electron-withdrawing perfluorophenyl substituents compared to three in [C6F5N3N]MoNMe2, it
appears that [Mespyr(C6F5)2]MoNMe2 has a more electron-poor molybdenum center due to the
replacement of an amide ligand arm with a pyrrolyl group. Based on Equation 2.5, the value of
∆H° for [Mespyr(C6F5)2]MoNMe2 (26.5(3.5) kJ/mol) was calculated, but the calculated error for
∆S° is too large relative to the calculated value for ∆S°, therefore we do not report it here.
35
Chapter 2
Equation 2.6
Stirring [Mespyr(C6F5)2]H3 with MoCl4(THF)2 in THF leads to the formation of an
orange red solution, which is postulated to contain the ligand-metal adduct. Addition of
LiN(TMS)2 to the reaction mixture causes rapid color change of the mixture to magenta. A
paramagnetic reddish-pink powder, [Mespyr(C6F5)2]MoCl is obtained after reaction work-up.
Red needle-like x-ray quality crystals were grown from a mixture of diethyl ether and pentane.
[Mespyr (C6F5)2]MoCl crystallizes in the P21/C space group. The solid-state structure shows a
molecule of approximately Cs symmetry. The bond length of Mo(1)-N(1) (pyrrolyl N) is
approximately 0.06 Å longer than that for Mo(1)-N(2) and Mo(1)-N(3), not unexpectedly, since
the lone pair of the pyrrolyl nitrogen should donate less to the molybdenum center than the lone
pairs on the amido nitrogens. The Mo(1)-N(1) bond (2.018 Å) in [Mespyr(C6F5)2]MoCl is
slightly shorter than that in [Mespyr(C6F5)2]MoNMe2 (2.081 Å), probably because the less
electron rich metal center in [Mespyr(C6F5)2]MoCl requires more electron donation from the
pyrrolyl nitrogen.
36
Chapter 2
Figure 2.6 POV-ray rendering (Thermal ellipsoids shown at 50% probability) of the solid state structure
of [Mespyr(C6F5)2]MoCl. H atoms are omitted for clarity. Selected bond distances (Å) and angles (°):
Cl(2)-Mo(1) = 2.3583(4), Mo(1)-N(3) = 1.9539(12), Mo(1)-N(2) = 1.9688(12), Mo(1)-N(1) = 2.0184(12),
Mo(1)-N(4) = 2.1737(12), C(21)-N(2)-Mo(1) = 125.99(10), C(31)-N(3)-Mo(1) = 123.41(10), N(1)Mo(1)-N(4) = 79.05(5), N(2)-Mo(1)-N(4) = 79.49(5), N(3)-Mo(1)-N(4) = 79.98(5), N(4)-Mo(1)-Cl(2) =
174.98(3).
Reacting [Mespyr(C6F5)2]MoCl in THF with excess sodium (smeared as a sodium mirror
on sides of vial) led to the formation of a black solid that was insoluble in common glovebox
solvents such as THF, dichloromethane, diethyl ether, pentane, benzene or toluene. Reacting
[Mespyr(C6F5)2]MoCl in benzene with 2.3 equivalents of KC8 led to the formation of a brown
orange solution. 1H NMR of the reaction mixture showed that it contained at least one
paramagnetic species. IR spectroscopy of the mixture showed no presence of a diazenide or N2
peak, however.
37
Chapter 2
It was thought that perhaps both KC8 and Na were too strongly reducing, and may lead to
decomposition. Therefore, Mg was utilized as a weaker reducing agent. Reacting
[Mespyr(C6F5)2]MoCl with Mg powder (activated with 1,2-dichloroethane) and stirring
overnight gave a color change from magenta to orange-red and the formation of some black solid
similar to that obtained when Na was used as the reducing agent. However, extracting the
mixture with benzene and filtering off the salts afforded a magenta solution. 1H NMR showed
that the mixture appeared to be diamagnetic, and IR spectroscopy (C6D6) revealed small peaks at
both 2026 cm-1 and 1830 cm-1, which may be due to N-N stretching. However, the products did
not appear to be stable and their isolation could not be achieved.
It has earlier been shown that reduction of [C6F5N3N]Mo(OTf) ([C6F5N3N] =
[(C6F5NCH2CH2)3N]3-) results in the dinuclear dinitrogen bridged species [C6F5N3N]Mo-N=NMo[C6F5N3N] in the presence of one equivalent of reducing agent and the diazenide species
[C6F5N3N]Mo-N=N-Na(ether)x when two equivalents of reducing agent is used. 5 Since the
[Mespyr(C6F5)]3- ligand, like [C6F5N3N]3-, is also very electron-withdrawing, we attempted
reduction from [Mespyr(C6F5)2]MoOTf rather than from [Mespyr(C6F5)2]MoCl as triflate is a
better leaving group than chloride. Stirring [Mespyr(C6F5)2]MoCl with two equivalents of
TMSOTf over two days did not lead to any visible reaction. However, reacting
[Mespyr(C6F5)2]MoCl with AgOTf led
to the formation of paramagnetic orange
[Mespyr(C6F5)2]MoOTf in approximately 40% yield.
Reducing [Mespyr(C6F5)2]MoOTf with Mg powder (activated with 1,2-dichloroethane)
led to the formation of a pink solid which exhibited peaks at 2030 cm-1 and 1830 cm-1 by IR
spectroscopy. The peak at 1830 cm-1 may be the N-N stretch for the diazenide anion
[Mespyr(C6F5)2]MoN2MgOTf(THF)x whereas the peak at 2030 cm-1 is likely due to N-N
stretching of some [Mespyr(C6F5)2]Mo(N2). Moreover, exposure of the IR spectroscopy sample
38
Chapter 2
to air led to a decrease in the peak at 1830 cm-1 and an increase in the peak at 2030 cm-1, which
is postulated to be due to air oxidation of the diazenide species to [Mespyr(C6F5)2]Mo(N2).
However, the products are not stable and isolation has not proved possible. Moreover, the
observations are not reproducible. We believe the instability of the diazenide species may arise
from reaction of the diazenide with the fluorine substituents on the perfluorophenyl ligands.
We then reacted [Mespyr(C6F5)2]H3 with MoCl3(THF)3 instead of MoCl4(THF)2, in the
hopes that the [Mespyr(C6F5)2]Mo “naked” species or [Mespyr(C6F5)2]Mo(N2) species could be
obtained, which would allow us to avoid formation of the diazenide species. However, reaction
of the ligand [Mespyr(C6F5)2]H3 with MoCl3(THF)3 and LiN(TMS)2 led to the formation of
[Mespyr(C6F5)2]MoCl instead, which is similar to other systems.
Reacting [Mespyr(C6F5)2]MoCl with NaN3 in acetonitrile at 70 ºC over 72 hours led to
the formation of a diamagnetic yellow product, [Mespyr(C6F5)2]MoN. Triamidoamine
molybdenum nitrides are also generally yellow. 6 , 7 This assignment is also supported by
elemental analysis and 1H NMR.
Other monopyrroletriamine ligands and their Mo complexes
Due to the sensitivity of the perfluorophenyl groups to strong reducing agents, 8 we
decided to explore ligands with other aryl groups on the amido nitrogens instead.
The synthesis of N1-aryl-N2-(2-(arylamino)ethyl)ethane-1,2-diamine is challenging, due
to the lack of selectivity for the primary amines over the secondary amine. However, it has been
shown that sufficiently hindered aryl bromides (such as aryl rings with a methyl group in the
ortho position or HexaIsoPropyl Terphenyl bromide (HIPTBr, Figure 2.7) can be coupled to the
primary amine selectively via a Pd-coupling reaction when a bulky phosphine ligand such as racBINAP is used.9
39
Chapter 2
Figure 2.7 HIPTBr
Equation 2.7
Unfortunately, attempts to perform a Mannich reaction on 2-mesityl-1H-pyrrole, 2(2,4,6-triisopropylphenyl)-1H-pyrrole or 2-(3,5-bis(trifluoromethyl)phenyl)-1H-pyrrole with 1b,
1c or 1d did not lead to the desired products. The reactions did not proceed even at elevated
temperatures of 60 °C, with prolonged stirring of the reaction mixtures (over a few days) leading
to decomposition of the pyrrole substrates. Addition of Lewis acids such as metal triflates as
catalysts did not lead to desired products either, but rather, the decomposition of the pyrroles.
It was initially thought that perhaps the triamines 1b-d were not reactive enough to form
the corresponding iminium cation during the Mannich reaction, but thin layer chromatography
shows that within 30 minutes of reacting 1b-d with formaldehyde, a new compound forms, most
likely the iminium compound. In 1a, the non-aryl substituted amine nitrogen is significantly
more nucleophilic than the aryl substituted nitrogens, unlike in 1b-d. Therefore, the formation of
3a together with 2a is unlikely. However, in the case of 1b-d, we expected to see (assuming that
the nucleophilicities of the amines within the molecule were comparable) a mixture of iminium
40
Chapter 2
cations 2b-2d and 3b-3d, and hence a mixture of Mannich reaction products, rather than no
product formation at all.
Fortunately, we came across a different synthetic route for a compound similar to our
desired
ligands,
N1,N2-bis((1H-pyrrol-2-yl)methyl)-N1,N2-dimethylethane-1,2-diamine.
10
Applying a similar synthetic process for the ligands we desired, 2-mesityl-1H-pyrrole was
aminomethylated via a Mannich reaction, with formaldehyde and Me2NH2Cl. The resulting
product was then methylated with an equivalent of MeI. Reaction of 6a with 1b and 1d led to
evolution of NMe3 and formation of the desired ligands.
Scheme 2.1 Synthesis of [Arpyr(Ar’)2]H3
Reaction of Mespyr(o-tol)2H3 (7a) with Mo(NMe2)4 led to the formation of dark green
[Mespyr(o-tol)2]MoNMe2. Like [Mespyr(C6F5)2]MoNMe2, [Mespyr(o-tol)2]MoNMe2 appears
diamagnetic by NMR spectroscopy. Unfortunately, the compound appears less stable than
[Mespyr(C6F5)2]MoNMe2, and the synthesis produces brown side products. However, extraction
of the mixture with pentane and recrystallization therefore affords the desired product free of the
brown impurities. It is suspected that perhaps the ortho-methyl group on the aryl substituent of
the amide arms reacts with the metal center, leading to decomposition. Performing VT 1H NMR
on this compound allows us a qualitative comparison with [Mespyr(C6F5)2]MoNMe2. The 1H
41
Chapter 2
NMR shift of the dimethylamido protons ranges from 2.54 – 3.51 ppm for [Mes(o-tol)2]MoNMe2
versus 2.46 – 2.87 ppm for [Mespyr(C6F5)2]MoNMe2, over the temperature range of 263 to 353
K which is in keeping with expectations, as the metal center in [Mespyr(o-tol)2]MoNMe2 should
be more electron rich than in [Mespyr(C6F5)2]MoNMe2. The donation of the lone pair of the
dimethylamide ligand in [Mespyr(o-tol)2]MoNMe2 should therefore be weaker, and the
difference in the energies between the low and high spin forms would be slightly smaller, leading
to a larger range for the 1H NMR shift of the dimethylamide protons. Based on Equation 2.5, the
value of ∆H° for [Mespyr(o-tol)2]MoNMe2 (26.09 (3.3) kJ/mol) was calculated, but the
calculated error for ∆S° is again too large relative to the calculated value for ∆S°, therefore we do
not report it here.
3.9
3.7
δ / ppm
3.5
3.3
3.1
2.9
2.7
2.5
235
255
275
295
315
335
355
375
Temperature / K
Figure 2.8 Temperature-dependent 1H NMR shift of the NMe2 group of [Mespyr(o-tol)2]MoNMe2
42
Chapter 2
Figure 2.9 Temperature-dependent 1H NMR spectrum of [Mespyr(o-tol)2]MoNMe2; the arrow indicates
the position of the dimethylamide chemical shift
43
Chapter 2
Unfortunately, reaction of 7a with MoCl4(THF)2 and subsequently LiN(TMS)2 led to
mixtures of [Mespyr(o-tol)2]MoCl and unidentified side products. A possible reason for the
formation of side products may be C-H activation of the ortho methyl substituent. Assuming that
the resulting metal complex [Mespyr(o-tol)2]MoCl would have a similar geometry to
[Mespyr(C6F5)2]MoCl, the mesityl group on the pyrrole ring does not point into the apical cavity
of the complex and hence the methyl substituents on the mesityl group does not lead to
instability of the compound. Should the ortho-tolyl substituents behave like the perfluorophenyl
substituents of [Mespyr(C6F5)2]MoCl, then the ortho-methyl substituents would be poised for CH activation by the metal center since presumably they point towards the metal center. Similar
attempts to metalate TRIPpyr(HIPT)2H3 (7b) with Mo(NMe2)4 or MoCl4(THF)2 and LiN(TMS)2
led to non-isolable products, presumably due to the extremely high solubility of the expected
products. Compounds containing hexaisopropylterphenyl (HIPT) substituents are generally very
soluble in most solvents, and moving down from C3v symmetry in [HIPTN3N]Mo complexes to
Cs symmetry in [Trippyr(HIPT)2]Mo complexes also increases the solubility of the resulting
compounds, therefore, the high solubility of the products was not completely unexpected.
Due to the difficulties mentioned, we decided to look into replacing the Ar’ groups with
aryl groups that were both less solubilizing and did not contain ortho substituents. Replacing the
isopropyl groups in HIPT with protons (abbreviated as 3,5-terphenyl (3,5-TP)) would decrease
the solubility of the resulting complex and since the analogous compounds had been synthesized
in the triamidoamine parent system, it appeared to be an obvious choice. Moreover, 3,5-TP may
be sterically bulky enough for arylation of diethylene triamine to be selective for the primary
over the secondary amine. Unfortunately, repeated attempts to synthesize the 3,5-TP substituted
triamine via a Buchwald-Hartwig cross-coupling led to little or no desired product, despite
various alterations to the reaction conditions. It was thought that perhaps protecting the extra
44
Chapter 2
secondary amine would prevent possible shut down of catalytic activity by that amine. We first
reacted diethylene triamine with ethyl trifluoroacetate to selectively protect the primary amines.
This selectivity is presumably due to ethyl trifluoroacetate’s sensitivity to steric bulk. The
resulting doubly protected triamine is then reacted with BOC2O (BOC = tert-Butyloxycarbonyl)
to protect the secondary amine. The trifluoroacetamide groups are cleaved by refluxing the
protected amine with a weak base, leaving the desired BOC-protected triamine. However Pdcatalyzed C-N cross coupling of HPTPBr with this BOC-protected triamine still resulted in little
conversion of starting material and also various side products. We then looked to a different aryl
substituent, DiTertButylTerphenylBr (DTBTBr, Figure 2.10) where two protons on the 4,4”
positions of the meta-phenyls in HPTPBr are replaced with tert-butyl groups. However, many
inseparable side products result from the cross-coupling reactions.
Figure 2.10 DTBTBr
We then turned to naphthyl substituents as replacements for HIPT, since naphthyl groups
should lead to low solubility of the metal complex, and were expected to sterically fit around the
resulting metal center. Moreover, a similar synthesis, the Pd catalyzed cross coupling reaction of
naphthyl bromide to N1-(3-aminopropyl)propane-1,3-diamine, is published and the naphthyl
susbtituents selectively coupled to the primary over the secondary amines. 11 Since naphthyl
substituents are highly activated towards such couplings, synthesis of the naphthyl substituted
triamine (1e) proceeded smoothly. Synthesis of 7c (Scheme 2.1), where Ar’ = naphthyl and Ar =
mesityl proceeded similarly to the synthesis of 7a and 7b. The compound 1e was heated with 6a
45
Chapter 2
for approximately 72 hours at 40 ºC to afford the ligand Mes(Napht)2H3 (7c) in moderate (54%)
yield.
However, the metalation of 7c with MoCl4(THF)2 and LiN(TMS)2 gave many side
products that complicated isolation of the desired [Mespyr(Napht)2]MoCl. Also, 1H NMR
appears to show only decomposition products. Attempts to metalate 7c with Mo(NMe2)4 did not
lead to the desired monodimethylamide product either. It may be that naphthyl substituents,
being redox-active, conferred redox-active properties to the resulting metal complexes, leading
to decomposition of the molybdenum compounds.
Conclusions
A new system of Mo complexes, diamidomonopyrrolyl Mo complexes, was synthesized
and
explored.
The
TRIPpyr(HIPT)2H3
monopyrroletriamine
and
Mespyr(Napht)2H3
ligands
were
Mespyr(C6F5)2H3,
synthesized
and
Mespyr(o-tol)2H3,
characterized.
The
monodimethylamide Mo complexes [Mespyr(C6F5)2]MoNMe2 and [Mespyr(o-tol)2]MoNMe2
appear diamagnetic by 1H NMR at room temperature, but appear to exist in equilibrium with
their high spin form. [Mespyr(C6F5)2]MoCl and [Mespyr(C6F5)2]MoOTf were synthesized and
found to be paramagnetic species. [Mespyr(C6F5)2]MoN was also synthesized and characterized
and is a diamagnetic, yellow compound.
Experimental
General. All air and moisture sensitive compounds were handled under N2 atmosphere using
standard Schlenk and glove-box techniques, with flame or oven-dried glassware. Ether, pentane,
dichloromethane and toluene were purged with nitrogen and passed through activated alumina
columns. Pentane was freeze-pump-thaw degassed three times and tetrahydrofuran (THF),
46
Chapter 2
benzene, deuterated benzene and toluene were distilled from dark purple Na/benzophenone ketyl
solutions. Ether and dichloromethane were stored over molecular sieves in solvent bottles in a
nitrogen-filled glovebox while pentane, THF, PhF, benzene, deuterated benzene and toluene
were stored in Teflon-sealed solvent bulbs. Molecular sieves (4 Å) and Celite were activated at
230 °C in vacuo over several days. (Me3Si)2NLi (sublimed) (Strem), (Me3Si)2NNa
(Recrystallized from anhydrous benzene) (Aldrich), anhydrous ZnCl2 (Aldrich) (Purified by
dissolving in diethyl ether and adding 1 equivalent of 1,4-dioxane to give ZnCl2(dioxane).),
MoCl5 (Strem) was used as obtained, unless indicated otherwise. MoCl4(THF)2,12 Mo(NMe2)4,13
2-mesityl-1H-pyrrole,3 2-(2,4,6-triisopropylphenyl)-1H-pyrrole3 were synthesized as referenced.
IR spectra were recorded on a Nicolet Avatar 360 FT-IR spectrometer in 0.2 mm KBr solution
cells. NMR spectra were recorded on a Varian Mercury or Varian Inova spectrometer operating
at 300 or 500 MHz respectively. 1H and
13
C NMR Spectra are referenced to the residual 1H or
13
C peaks of the solvent. 19F NMR spectra were referenced externally to fluorobenzene (-113.15
ppm upfield of CFCl3). HRMS was performed on a Bruker Daltonics APEXIV 4.7 Tesla Fourier
Transform Ion Cyclotron Resonance Mass Spectrometer at the MIT Department of Chemistry
Instrumentation Facility. Combustion analyses were performed by Midwest Microlabs,
Indianapolis, Indiana, U.S.
Mespyr(C6F5)2H3: A 40 mL scintillation vial equipped with a stirbar was charged with N1(perfluorophenyl)-N2-(2-(perfluorophenylamino)ethyl)ethane-1,2-diamine (1.918 g, 4.4 mmol).
To formaldehyde (35% wt, 0.372 mL) was added c.HCl (5 µL). THF (1 mL) was added to the
formaldehyde mixture, which was then transferred to the 40 mL scintillation vial. To the vial was
added THF (2 mL) and iPrOH (2 mL). The mixture was stirred for 20 minutes and the reaction
mixture added to a vial charged with 2-mesityl pyrrole (0.807 g, 4.4 mmol). The mixture was
47
Chapter 2
stirred at room temperature for 16 hours, then washed with 10% KOH solution (40 mL), and
extracted with diethyl ether and dried over Na2SO4. The volatiles were removed in vacuo and the
residue purified by column chromatography using 9 : 1 hexanes : ethyl acetate as the eluent. The
desired product is the second product from the column, with an Rf value of 0.158. Yield: 0.1545
g, 55%. 1H NMR (CDCl3): δ 7.97 (s, 1H, pyrrole NH), 6.92 (s, 2H, Mes 3,5-H), 6.13 (t, JHH = 3.1
Hz, 1H, pyrrole -CH), 5.96 (t, JHH = 3.1 Hz, 1H, pyrrole -CH), 4.08 (s, 2H, amine NH), 3.72 (s,
2H, C-CH2-N), 3.35 (q, JHH = 5.9 Hz, 4H, C6F5NHCH2), 2.78 (t, JHH = 5.9 Hz, 4H,
C6F5NHCH2CH2), 2.32 (s, 3H, Mes 4-CH3), 2.09 (s, 6H, Mes 2,6 –CH3).ppm 13C NMR (CDCl3):
δ 139.2, 138.2, 137.8, 137.2, 134.7, 132.7, 130.3, 129.8, 128.3, 127.0, 124.0, 108.9, 108.7, 53.9,
51.8, 43.8, 21.2, 20.6 ppm
19
F NMR (282 MHz, CDCl3): δ -159.8 (d, JFF = 20.0 Hz, 2,6 -F), -
164.1 (t, JFF = 21.2 Hz, 3,5 -F), -171.3 (tt, JFF = 5.3, 21.9 Hz, 4 -F) ppm HRMS (ESI m/z): Cald
for C30H27F10N4+ 633.207, found 633.2056
[Mespyr(C6F5)2]MoNMe2: Under a N2 atmosphere, a 20 mL scintillation vial was charged with
Mo(NMe2)4 (671.8 mg, 2.468 mmol) and [Mes(C6F5)2]H3 (1337.8 mg, 2.115 mmol) and toluene
(15 mL). The reaction mixture rapidly turned dark blue (from deep purple) and eventually
became emerald green. It was stirred for approximately 48 hours at room temperature, with the
vial periodically uncapped to facilitate loss of HNMe2. The solvent was decreased to
approximately 5 mL, and pentane (approximately 2 mL) was added. The reaction mixture was
then left at -35 ºC overnight and the product collected on a glass frit as a dark green solid. Yield:
1.347 g, 83%. 1H NMR (Toluene-d8): δ 6.77 (s, 2H, mesityl 3,5-H), 6.11 (d, JHH = 3.2 Hz, 1H,
pyrrole -H), 6.02 (d, JHH = 3.2 Hz, 1H, pyrrole, -H), 3.76 (s, 2H, -NCH2), 3.72 (m, 2H, C6F5NCH(H)), 3.23 (m, 2H, - C6F5NCH(H)), 2.97 (m, 2H, - C6F5NCH2CH(H)), 2.63 (m, 2H, C6F5NCH2CH(H)), 2.55 (s, 6H, -N(CH3)2), 2.17 (s, 3H, mesityl 4-CH3), 2.04 (s, 6H, mesityl 2,648
Chapter 2
CH3) ppm.
13
C NMR (Toluene-d8): δ 143.6, 139.7, 137.2, 136.9, 135.2, 129.6, 128.9, 128.8,
128.6 125.8, 113.2, 105.7, 66.2, 61.2, 55.9, 55.1, 21.6, 21.5 19F NMR (Toluene-d8): δ -148.0 (d),
-162.8 (t), -164.7 (t) ppm. Anal. Calcd for C32H29F10MoN5: C, 49.95; H, 3.80; N, 9.10. Found: C,
49.67; H, 3.90; N, 9.22.
[Mespyr(C6F5)2]MoCl: Under a N2 atmosphere, a 20 mL scintillation vial equipped with a stir
bar was charged with MoCl4(THF)2 (1191mg, 3.092 mmol), [Mes(C6F5)2]H3 (2000 mg, 3.162
mmol) and THF (5 mL). This caused the reaction mixture to turn from an orange suspension to
an orange red solution. The mixture was stirred for 30 minutes, and LiNTMS2 (1587 mg, 9.484
mmol) was added, leading to a darkening of the mixture to magenta. After stirring for another 30
minutes, volatiles were removed in vacuo and the mixture extracted with toluene and filtered
through celite. The product was recrystallized from toluene and pentane at -35 ºC and collected
on a glass frit. Yield: 0.989 g, 42% 1H NMR (C6D6): δ 41.03 (s), 12.38 (s), 8.31 (s), 6.84 (s),
6.02 (s), 5.23 (s), 4.34-2.91 (m), 2.12 (s), 1.80 (s), 1.27 (s), 0.88 (s), 0.30 (s), 0.01 (s), -1.21(s), 20.04 (s), -78.50 (s), -92.10 (s).
19
F NMR (C6D6): δ -71.02 (s), -96.37 (s), -121.893 (s), -122.80
(s), -148.27 (s) ppm. HRMS (ESI m/z): calcd for C30H23F10N4MoClNa+: 785.0405, found
785.0412
[Mespyr(C6F5)2]MoOTf: Under N2 atmosphere, a scintillation vial was charged with
[Mes(C6F5)2]MoCl (100 mg, 0.1314 mmol), AgOTf (33.6 mg, 0.1308 mmol) and CH2Cl2 (5
mL). The mixture was stirred overnight at RT and then filtered through celite. Volatiles were
removed in vacuo. An orange-red compound is recrystallized from a mixture of toluene, pentane
and CH2Cl2. Yield: 47.5 mg, 42% 1H NMR (C6D6): δ 39.86 (s), 14.85 (s), 12.07 (s), 7.12 (s),
7.06 (s), 7.05 (s), 7.01 (s), 6.35 (s), 4.72 (br s), 2.06 (s), 1.30 -1.24 (m), -21.47 (s), -85.28 (br s), 49
Chapter 2
108.85 (br s) ppm. Anal Calcd. for C31H23F13MoN4O3S: C, 42.58; H, 2.65; N, 6.41. Found: C,
42.40; H, 2.79; N, 6.25.
[Mespyr(C6F5)2]MoN: Under N2 atmosphere, a 25 mL solvent bulb was charged with
[Mespyr(C6F5)2]MoCl (200 mg, 0.2628 mmol), NaN3 (13.6 mg, 0.1178 mmol) and acetonitrile (5
mL). The reaction mixture was heated at 70 ºC for 72 hours. The volatiles were removed in
vacuo and the residue extracted with toluene and filtered through celite. Diamagnetic yellow
brown needle-like crystals were recrystallized from the filtrate at -35 ºC overnight and collected
on a glass frit. Yield: 103 mg, 53%. 1H NMR (C6D6): δ 6.86 (s, 2H, mesityl 3,5-H), 6.25 (dd, JHH
= 3.0, 0.6 Hz, 1H, pyrrole –H), 6.20 (dd, JHH = 3.0, 0.6 Hz, 1H, pyrrole –H), 3.35 (s, 2H, NCH2), 3.25 (m, 2H, -(NCH(H)CH2)3N), 3.04 (m, 2H, -(NCH(H)CH2)3N), 2.48 (s, mesityl 2,6CH3), 2.25 (m, 2H, -(NCH2CH(H))3N), 2.18 (m, 2H, -(NCH2CH(H))3N), 1.92 (s, 3H, mesityl 4CH3) ppm
13
C NMR (C6D6): δ 142.8, 142.5, 140.9, 139.9, 139.0, 139.0 (overlapping), 137.9,
137.8, 137.2, 136.9, 135.1, 133.6, 128.4, 128.1, 111.2, 108.3, 58.3, 51.3, 51.0, 21.1, 21.0, 21.0
ppm
19
F NMR (C6D6): δ -150.18 (dd, 1F), -150.40 (t, 1F), -159.44 (t, 1F), -163.63 (d, 1F), -
163.60 (d, 1F, overlapping) ppm. Anal Calcd. For C30H23F10MoN5: C, 48.73; H, 3.14; N, 9.47.
Found: C, 48.51; H, 3.28; N, 9.48.
N1-(naphthalen-1-yl)-N2-(2-(naphthalen-1-ylamino)ethyl)ethane-1,2-diamine (1f): A Schlenk
flask was charged with 1-bromonaphthalene (20.09 g, 97.02 mmol), diethylene triamine (5.00 g,
48.46 mmol), Pd2(dba)3 (0.89 g, 0.97 mmol), 1,1’-Bis(diphenylphosphino)ferrocene (1.08 g, 1.95
mmol), NaOtBu (9.61 g, 100 mmol) and dioxane (50 mL). The reaction mixture was heated at 90
ºC for ~ 2 days. The mixture was allowed to cool, then was filtered through celite and washed
with acetone. Volatiles from the filtrate were removed in vacuo. The products were eluted on a
50
Chapter 2
silica gel column using Et2O to wash off impurities, then THF to elute the remaining desired
product. Yield: 15.92 g, 92%. 1H NMR (CDCl3): δ 7.82 (t, JHH = 8.5 Hz, 4H, Aryl -H), 7.45 (m,
2H, Aryl –H), 7.35 (m, 4H, Aryl –H), 7.27 (m, 2H, Aryl –H), 6.64 (d, JHH = 8.5 Hz, 2H, Aryl –
H), 5.00 (s, 2H, ArNCH2(H)), 3.41 (q, 4H, ArN(H)CH2), 3.14 (t, 4H, ArN(H)CH2CH2), 2.19 (s,
1H, CH2N(H)CH2) ppm.
1-(5-mesityl-1H-pyrrol-2-yl)-N,N-dimethylmethanamine (5a) A 100 mL RB flask was
charged with Me2NH2Cl (0.968 g, 11.87 mmol), formaldehyde (1.016 mL, 37% solution in
water, 12.52 mmol) and isopropanol (10 mL). The mixture was stirred for approximately 30
minutes. 2-mesityl-1H-pyrrole (2.200 g, 11.88 mmol) was then added to the RB flask. The
mixture was stirred for 16 hours at RT. 10 mL of 10% KOH solution was added and the mixture
stirred for 30 minutes. Volatiles were removed in vacuo and 100mL water was added. The
mixture was extracted three times with CH2Cl2 (100 mL) and the organic layer dried over
Na2SO4. Volatiles were removed in vacuo and the residue used without further purification.
Yield: 2.81 g, 98% 1H NMR (CDCl3): δ 8.245 (1H, s, pyrrole -NH), 6.915 (2H, s, mesityl 3,5-H),
6.079 (1H, t, JHH = 3.0 Hz, pyrrole C-H), 5.927 (1H, t, JHH = 3.0 Hz, pyrrole C-H), 3.456 (2H, s,
-CH2), 2.309 (3H, s, mesityl 4-CH3), 2.221 (6H, s, mesityl 2,6 –CH3), 2.146 (6H, -N(CH3)2) ppm
13
C NMR (CDCl3): δ 138.5, 137.4, 131.5, 130.0, 128.2, 128.1, 108.3, 107.5, 57.0, 44.8, 21.3,
20.8 ppm HRMS (ESI, m/z): Calcd for C16H21N2- 241.1710, found 241.1706
N,N-dimethyl-1-(5-(2,4,6-triisopropylphenyl)-1H-pyrrol-2-yl)methanamine (5b) A 100 mL
RB flask was charged with Me2NH2Cl (1.592 g, 19.52 mmol), formaldehyde (1.670 mL, 37%
solution in water, 20.58 mmol) and isopropanol (10 mL). The mixture was stirred for
approximately 30 minutes. 2-(2,4,6-triisopropylphenyl)-1H-pyrrole (5.260 g, 19.52 mmol) was
51
Chapter 2
then added to the RB flask. The mixture was stirred for approximately 70 hours at 40 ºC. 300 mL
of 10% KOH solution was added and the mixture stirred for 30 minutes. Volatiles were removed
in vacuo and 200 mL water was added. The mixture was extracted three times with CH2Cl2 (200
mL) and the organic layer dried over Na2SO4. Volatiles were removed in vacuo and the residue
used without further purification. Yield: 4.35g, 68% 1H NMR (CDCl3): δ 8.206 (1H, s, pyrrole NH), 7.037 (2H, s, aryl 3,5-H), 6.069 (1H, t, JHH = 3.0 Hz, pyrrole C-H), 5.953 (1H, t, JHH = 3.0
Hz, pyrrole C-H), 3.465 (2H, s, -CH2), 2.927 (1H, septet, JHH = 6.7 Hz, 4-CHMe2), 2.789 (2H,
septet, JHH = 6.7 Hz, 2,6-CHMe2), 2.223 (6H, s, -N(CH3)2) ppm
13
C NMR (CDCl3): δ 149.6,
149.1, 129.4, 128.7, 128.3, 120.6, 108.7, 108.3, 56.8, 45.0, 34.6, 30.7, 24.7, 24.3. ppm HRMS
(ESI, m/z): Cald for C22H33N2- 325.2659, found 325.2650.
1-(5-mesityl-1H-pyrrol-2-yl)-N,N-trimethylammonium iodide (6a) A 250 mL RB flask was
charged with 1-(5-mesityl-1H-pyrrol-2-yl)-N,N-dimethylmethanamine (1.750 g, 7.221 mmol)
and THF (150 mL). A vial with a septum sealed cap was charged with MeI (1.025 g, 7.221
mmol) and THF (15 mL). The contents of the vial were syringed out and added slowly to the
stirring solution of 1-(5-mesityl-1H-pyrrol-2-yl)-N,N-dimethylmethanamine. The reaction
mixture was stirred for 1 hour at RT, during which a very thick white suspension formed. The
white precipitate was collected on a glass frit, washed with THF and recrystallized from acetone.
Yield: 2.63 g, 95%. 1H NMR (CDCl3): δ 10.304 (1H, s, pyrrole -NH), 6.902 (2H, s, mesityl 3,5H), 6.417 (1H, t, JHH = 2.8 Hz, pyrrole C-H), 6.071 (1H, t, JHH = 2.8 Hz, pyrrole C-H), 5.148
(2H, s, -CH2), 3.271 (9H, s, N(CH3)3), 2.297 (3H, s, mesityl 4-CH3), 2.153 (6H, s, mesityl 2,6CH3) ppm
13
C NMR ((CD3)2SO): δ 137.37, 137.10, 132.15, 130.10, 127.98, 117.28, 114,56,
109.00, 54.48, 30.80, 20.76, 20.46. ppm HRMS (ESI, m/z): Calcd for C19H25N2- 257.2012, found
257.2005
52
Chapter 2
1-(5-(2,4,6-triisopropylphenyl)-1H-pyrrol-2-yl)-N,N-trimethylammonium iodide (6b) A 250
mL
RB
flask
was
charged
with
1-(5-(2,4,6-triisopropylphenyl)-1H-pyrrol-2-yl)-N,N-
dimethylmethanamine (3.950 g, 12.098 mmol) and THF (150 mL). A vial with a septum sealed
cap was charged with MeI (1.717 g, 12.098 mmol) and THF (15 mL). The contents of the vial
were syringed out and added slowly to the stirring solution of 1-(5-(2,4,6-triisopropylphenyl)1H-pyrrol-2-yl)-N,N-dimethylmethanamine. The reaction mixture was stirred for 1 hour at RT,
during which a very thick white suspension formed. The white precipitate was collected on a
glass frit, washed with THF and recrystallized from acetone. Yield: 3.121 g, 55%. 1H NMR
(CDCl3): δ 10.05 (1H, s, pyrrole -NH), 7.02 (2H, s, mesityl 3,5-H), 6.42 (1H, t, JHH = 2.9 Hz,
pyrrole C-H), 6.01 (1H, t, JHH = 2.9 Hz, pyrrole C-H), 5.20 (2H, s, -CH2), 3.30 (9H, s, N(CH3)3),
2.92 (1H, septet, JHH = 6.6 Hz, 4-CHMe2), 2.60 (2H, septet, JHH = 6.6 Hz, 2,6-CHMe2), 1.30
(6H, d, JHH = 7.1 Hz, 4-CH(CH3)2), 1.15 (6H, d, JHH = 7.1 Hz, 2,6-CH(CH3)2), 1.12 (6H, d, JHH =
7.1 Hz, 2,6-CH(CH3)2) ppm
13
C NMR (CDCl3): δ 149.6, 149.2, 133.6, 127.5, 120.8, 116.9,
114.8, 110.4, 62.4, 52.4, 34.5, 30.9, 25.1, 24.3, 24.2 ppm HRMS (ESI, m/z): Cald for C23H37N2341.2951, found 341.2937
Mes(o-tol)2H3 (7a) A 100 mL RB flask was charged with 1-(5-mesityl-1H-pyrrol-2-yl)-N,Ntrimethylammonium iodide (2.547 g, 6.628 mmol), N1-o-tolyl-N2-(2-(o-tolylamino)ethyl)ethane1,2-diamine (2.066 g, 7.290 mmol) and K2CO3 (9.160 g, 66.276 mmol) and stirred for 48 hrs at
RT, then 48 hrs at 40 ºC. The salts were filtered off and the filtrate dried in vacuo. The product
was purified by column chromatography with diethyl ether as the eluent (Rf value 0.64). Yield:
3.07 g, 96% 1H NMR (CDCl3): δ 7.893 (1H, s, pyrrole –NH), 7.090 (2H, dt, tolyl 5-H), 6.999
(2H, dd, tolyl 3-H), 6.907 (2H, s, mesityl 3,5-H), 6.640 (2H, dt, tolyl 4-H), 6.581 (2H, dd, tolyl
53
Chapter 2
6-H), 6.147 (1H, t, JHH = 2.9 Hz, pyrrole –CH), 5.968 (1H, t, JHH = 2.9 Hz, pyrrole –CH), 3.924
(2H, s, -NH), 3.756 (2H, s, -NCH2C), 3.262 (4H, t, JHH = 6.0 Hz, -NHCH2CH2N), 2.868 (4H, t,
JHH = 6.0 Hz, -NCH2CH2NH), 2.320 (3H, s, mesityl 4-CH3), 2.095 (6H, s, mesityl 2,6-CH3),
2.033 (6H, s, tolyl 2-CH3).ppm
13
C NMR (CDCl3): δ 146.2, 138.4, 137.7, 130.7, 130.2, 129.5,
128.2, 127.6, 127.3, 122.1, 117.2, 110.0, 108.6, 108.4, 53.4, ppm HRMS (ESI, m/z): Cald
C32H41N4+ 481.3326, found 481.3316.
[Mes(Otol)]MoNMe2: Under N2 atmosphere, a 50 mL solvent bulb equipped with a PTFE screw
valve was charged with Mo(NMe2)4 (334 mg, 1.2284 mmol) and [Mes(Otol)2]H3 (620 mg,
1.2898 mmol) and toluene (~5 mL). The reaction mixture was heated with stirring at 60 ºC
overnight during which the reaction mixture turned from purple to brownish green. Toluene was
removed in vacuo and the residue extracted with pentane (~200 mL) and filtered through celite
giving a blue-green solution. The solution was decreased in vacuo and stored at -30 ºC overnight.
The blue-green precipitate was then collected on a glass frit and dried. Yield: 150 mg, 20%. 1H
NMR (500MHz, C6D6): δ 7.00 (dd, 4H, o-tolyl –H), 6.91 (t, JHH = 7.6 Hz, 2H, o-tolyl –H), 6.80
(t, JHH = 7.9 Hz, 2H, o-tolyl –H). 6.80 ( overlapping s, 2H, mesityl 3,5-H), 6.33 (d, JHH = 3.0 Hz,
1H, pyrrole -H), 6.20 (d, JHH = 3.0 Hz, 1H, pyrrole, -H), 4.10 (s, 2H, -NCH2), 3.80 (br m, 2H,
o-tolylNCH(H)), 3.12 (br m, 2H, o-tolylNCH(H)), 2.97 (br m, 2H, o-tolylNCH2CH(H)), 2.81 (br
m, 2H, o-tolylNCH2CH(H)), 2.97-2.59 (overlapping br s, 6H, -N(CH3)2), 2.20 (s, 6H, o-tolyl 2CH3), 2.14 (s, 3H, mesityl 4-CH3), 1.99 (s, 6H, mesityl 2,6-CH3) ppm
13
C NMR (126 MHz
C6D6): δ 139.2, 137.2, 136.2, 132.7, 130.5, 128.3, 127.8, 127.6, 126.7, 124.3, 117.5, 112.2,
110.3, 104.6, 67.9, 64.7, 56.8, 55.8, 21.6, 21.1, 18.5 ppm. Anal Calcd. for C34H43N5Mo: C,
66.11; H, 7.02; N, 11.34. Found: C, 66.08; H, 7.02; N, 11.21.
54
Chapter 2
TRIPpyr(HIPT)2H3 (7b) A 100 mL RB flask was charged with N1-HIPT-N2-(2(HIPT)ethyl)ethane-1,2-diamine (1.194 g, 1.121 mmol), 6b (0.525 g, 1.121 mmol) and K2CO3
(1.2 g, 8.682 mmol). The flask was purged with N2, and the reaction mixture stirred overnight at
RT under N2. The product was purified by column chromatography and eluted with 2:1 hexanes:
ethyl acetate. Yield: 0.7893 g, 52% 1H NMR (CDCl3): δ 7.87 (1H, s, pyrrole –NH), 7.01 (2H, s,
TRIP aryl 3,5-H), 7.01 (8H, HIPT aryl 3,5,3”,5”-H), 6.39 (4H, d, HIPT aryl 2’,6’-H), 6.37 (2H, t,
HIPT aryl 4’-H), 6.08 (1H, t, JHH = 2.9 Hz, pyrrole C-H), 5.96 (1H, t, JHH = 2.9 Hz, pyrrole CH), 3.92 (2H, t, JHH = 5.1 Hz, NHCH2CH2), 3.75 (2H, s, NCH2C), 3.20 (4H, q, JHH = 5.8 Hz,
NHCH2CH2N), 2.92 (4H, sept, JHH = 7.0 Hz, HIPT 4,4”-CHMe2), 2.91 (1H, sept, JHH = 7.0 Hz,
TRIP 4-CHMe2), 2.81 (12H, sept, JHH = 6.9 Hz, HIPT 2,6,2”,6” -CHMe2 overlapping with
NHCH2CH2N), 2.73 (2H, sept, JHH = 6.9 Hz, TRIP 2,6-CHMe2), 1.29 (28H, d, JHH = 6.7 Hz,
HIPT 4,4”-CH(CH3)2, TRIP 4-CH(CH3)2), 1.12 (24H, d, JHH = 6.7 Hz, HIPT 2,6,2”,6”CH(CH3)2), 1.08 (12H, d, JHH = 6.7 Hz, TRIP 2,6-CH(CH3)2), 1.04 (24H, d, JHH = 6.7 Hz, HIPT
2,6,2”,6”-CH(CH3)2) ppm.
13
C NMR (126 MHz CDCl3): δ 149.5, 147.6, 147.3, 146.5, 141.6,
137.5, 129.0, 128.8, 126.9, 121.5, 120.7, 120.5, 112.5, 109.2, 108.4, 52.7, 51.1, 41.6, 34.6, 30.8,
30.4, 24.7, 24.4, 24.3, 24.2 ppm. HRMS (ESI, m/z) cald for C96H137N4+: 1347.0917, found
1347.0863
N1-((5-mesityl-1H-pyrrol-2-yl)methyl)-N2-(naphthalen-1-yl)-N1-(2-(naphthalen-1ylamino)ethyl)ethane-1,2-diamine (7c): A round bottom flask was charged with 1-(5-mesityl1H-pyrrol-2-yl)-N,N-trimethylammonium iodide (6a) (11.72 g, 30.49 mmol), N1-(naphthalen-1yl)-N2-(2-(naphthalen-1-ylamino)ethyl)ethane-1,2-diamine (1f) (10.84g, 30.49 mmol), K2CO3 (
36.37 g, 263 mmol) and acetone (300 mL). The reaction mixture was stirred for 48 hrs at RT
and then 72 hours at 40 ºC, with repeated additions of acetone solvent. Volatiles were removed
55
Chapter 2
in vacuo. Some product was recrystallized from Et2O/Acetone and collected on a glass frit.
Column chromatography was performed on the filtrate purify remaining product present in the
filtrate. Yield: 9.07 g, 54%. 1H NMR (CDCl3, 20 °C): δ 8.00 (s, 1H, pyrrole N-H), 7.74 (d, JHH =
8.1 Hz, 2H, Naphthyl –H), 7.56 (d, JHH = 8.1 Hz, 2H, Naphthyl –H), 7.33 (d, JHH = 7.4 Hz, 2H,
Naphthyl –H), 7.31 (t, JHH = 7.8 Hz, 2H, Naphthyl –H), 7.22 (s, 1H, Naphthyl –H), 7.22 (s, 1H,
Naphthyl –H), 6.87 (s, 2H, Mesityl 3,5 –H), 6.81 (t, JHH = 7.4 Hz, 2H, Naphthyl –H), 6.58 (d,
JHH = 7.4 Hz, 2H, Naphthyl –H), 6.23 (t, JHH = 3.1 Hz, 1H, pyrrole C-H), 6.02 (t, JHH = 3.1 Hz,
1H, pyrrole C-H), 4.91 (s, 2H, NaphtylN(H)CH2), 3.80 (s, 2H, -NCH2C), 3.41 (s, 4H,
NaphthylNCH2), 3.04 (s, 4H, NaphtylNCH2CH2), 2.32 (s, 3H, Mes 4-CH3), 2.04 (s, 6H, Mes 2,6CH3) ppm 13C NMR (CDCl3, 20 °C): δ 143.56, 138.37, 137.56, 134.29, 128.23, 126.61, 125.80,
124.86, 123.42, 119.89, 117.52, 108.71, 104.38, 52.99, 51.40, 41.76, 31.05, 30.44, 21.19, 20.80
ppm HRMS (ESI, m/z) Anal calcd for C38H41N4+: 553.3326, found 553.3320.
References
1
Wampler, K.M.; Schrock, R.R. Inorg. Chem. 2007, 46, 8463.
2
Schrock, R. R.; Lee, J.; Liang, L.; Davis, W.M. Inorg. Chim. Acta. 1998, 270, 353.
3
Rieth, R.D.; Mankad, N.P.; Calimano, E.; Sadighi, J.P. Org Lett. 2004, 6, 3981.
4
Mosch-Zanetti, N.C.; Schrock, R.R.; Davis, W.M.; Wanniger, K.; Seidel, S. W.; O’Donoghue,
M.B. J. Am. Chem. Soc. 1997, 119, 11037.
5
Kol, M.; Schrock, R.R; Kempe, R.; Davis, W.M. J. Am. Chem. Soc. 1994, 116, 4382.
6
Weare, W.W.; Schrock, R.R.; Hock, A.S.; Müller, P. Inorg. Chem. 2006, 45, 9185.
7
Yandulov, D.V.; Schrock, R. R. Inorg. Chem. 2003, 42, 796.
8
Burdeniuc, J.; Jedlicka, B.; Crabtree, R.H. Ber. 1997, 130, 145.
9
Hong, Y.; Senanayake, C.H.; Vandenbossche, C.P.; Tanoury, G.J.; Bakale, R.P.; Wald, S.A.
56
Chapter 2
Tetrahedron Lett. 1998, 39, 3121.
10
Banerjee, S.; Barnea, E.; Odom, A.L. Organometallics 2008, 27, 1005.
11
Beletskaya, I.P; Bessmertnykh, A.G.; Averin, A.D.; Denat, F.; Guilard, R. Eur. J. Org. Chem.
2005, 2, 261.
12
Stoffelbach, F.; Saurenz, D.; Poli, R. Eur. J. Inorg. Chem. 2001, 10, 2699.
13
Bradley, D. C.; Chisholm, M. H. J. Chem. Soc. A 1971, 2741.
57
Chapter 3
Chapter 3
Synthesis and studies of some diamidomonopyrrolylamine Mo
complexes
58
Chapter 3
Introduction
Work discussed in the previous chapter showed that the diamidomonopyrrolylamine
ligand can be synthesized through a Mannich reaction between the triamine and the pyrrole when
the aryl group on the amine nitrogens is perfluorophenyl ([Mespyr(C6F5)2]H3). Substituted
pyrroles can be synthesized via a Pd catalyzed cross-coupling of sodium pyrrolide and the aryl
bromide of choice1.
Equation 3.1 Synthesis of [Mes(C6F5)2]H3
The synthesis of a series of Mo complexes ([Mes(C6F5)2]MoX) with this ligand
framework was discussed. However, the dinitrogen complex [Mes(C6F5)2]Mo(N2) eluded our
various synthetic attempts. The diazenide complex {[Mes(C6F5)2]Mo(N2)}- may be unstable due
to the presence of fluorine atoms on the aryl subtituents. This led to the quest for
diamidomonopyrrolylamine ligands that were not fluorinated. A series of such ligands were
synthesized where Ar = Triisopropylphenyl (TRIP), Ar’ = HexaIsoPropylTerphenyl (HIPT),
([TRIPpyr(HIPT)2]H3); Ar = Mesityl, and Ar’ = o-tolyl, ([Mespyr(o-tol)2]H3) and Ar = Mesityl
and Ar’ = Naphthyl ([Mespyr(Napht)2]H3). However, each of these ligand frameworks presented
problems upon attempted synthesis of their corresponding Mo complexes. The extremely high
solubility of the [TRIPpyr(HIPT)2]Mo complexes made it virtually impossible in our hands to
isolate these compounds, whereas the [Mespyr(o-tol)2]Mo complexes were unstable, perhaps
because the ortho-methyl groups on the aryl substituents undergo some form of C-H activation.
59
Chapter 3
Attempts to synthesize [Mespyr(Napht)2]Mo complexes also resulted in decomposition, possibly
due to the ligand being redox-active.
Hence, it is important to choose ligands that 1) would not lead to extremely soluble Mo
complexes; 2) do not contain ortho-substituents on the aryl groups of the amido nitrogens and 3)
are not redox-active. Therefore, it was decided to look at ligands employing 3,5-xylyl and 3,5-ditert-butylphenyl substituents on the amido nitrogens.
Results and Discussion
Synthesis of 3,5-di-tert-Buphenyltriamine (1a), 3,5-di-Mephenyltriamine (1b)
The synthesis of 3,5-di-tert-Buphenyltriamine (1a), 3,5-di-Mephenyltriamine (1b) were
initially complicated by the lack of selective arylation of primary amines over the secondary
amine during Pd-catalyzed C-N cross-coupling reactions. Cu-catalyzed Ullmann-type couplings
were studied as an alternative route to synthesize the desired triamine. Buchwald et al. had
reported the Cu-catalyzed amination of aryl iodides utilizing CuI as the Cu source and ethylene
glycol as the ligand. 2 Moreover, they report that spermidine (N1-(3-aminopropyl)butane-1,4diamine) was selectively arylated only at the primary amine positions. Another advantage
provided by this Cu-catalyzed cross coupling is the affordability of CuI. We found that utilizing
ethylene glycol as the supporting ligand for the copper catalyst in this Ullmann-type C-N
coupling reaction is not as efficient as desired. When ethylene glycol is replaced by N,Ndiethylsalicylamide, which had been utilized as a supporting ligand for the Cu-catalyzed
amination of aryl bromides by primary amines,3 the reaction proceeds much more smoothly. As
in the reaction using ethylene glycol as the ligand, it appears important that the Cu catalyst is
pre-formed by stirring the CuI with N,N-diethylsalicylamide in DMF for at least 30 minutes
before adding diethylene triamine. Possibly, diethylene triamine competes with N,N60
Chapter 3
diethylsalicylamide to chelate to Cu and thus shuts down catalytic ability when the catalyst is not
pre-formed. Addition of the diethylene triamine to the reaction mixture last leads to the rapid
disproportionation of Cu(I) to Cu(0) and Cu(II) as can be seen from the development of a light
blue-green hue (presumably due to amine coordinating to Cu(II)) and Cu(0) powder. Heating the
reaction at 80-90 °C leads to a color change from blue-green to brown within a few hours. Thin
layer chromatography analysis of the reaction mixture every few hours shows the formation of
arylated amine with time. The aryl bromide 1-bromo-3,5-di-tert-butylbenzene was synthesized
via a Friedel-Crafts alkylation of benzene to 1,3,5-tri-tert-butylbenzene 4 and then subsequent
reaction with Fe and Br2.5,6
10% CuI,
40% N,N-diethylsalicylamide
4 K3PO4 / DMF
90 °C, 36 h
NH2
HN
+
H
N
HN
2
2
72%
Br
2
1a
Equation 3.2
Equation 3.3
1b
N1-(3,5-di-tert-butylphenyl)-N2-(2-(3,5-di-tert-butylphenylamino)ethyl)ethane-1,2diamine (3,5-t-butylphenyltriamine,1a) is a dark yellow oil. It is rather air-sensitive, since
overnight exposure to air leads to its conversion to an unidentifiable mixture of solid products.
Minimization of air exposure of the reaction mixture during the work-up significantly improves
yields. N1-(3,5-dimethylphenyl)-N2-(2-(3,5-dimethylphenylamino)ethyl)ethane-1,2-diamine (3,561
Chapter 3
methylphenyltriamine, 1b) does not appear to be as air-sensitive as 1a, probably as a
consequence of being less electron rich.
Synthesis of [Mespyr(3,5-t-Bu)2]H3, (3a); [Mespyr(3,5-Me)2]H3 (3b) and their metal
complexes
[Mespyr(3,5-t-Bu)2]H3 (3a) is synthesized in a manner similar to the synthesis of
[Mespyr(o-tol)]H3, via a nucleophilic substitution reaction between 1a and 2a (Scheme 3.1). 2a
was synthesized first via an aminomethylation using a Mannich reaction, with formaldehyde and
Me2NH2Cl. The resulting product was then methylated with one equivalent of MeI to produce
2a. Reaction of 2a with 1a led to evolution of NMe3 and formation of the desired ligand. It is
important to keep the reaction under an inert atmosphere due to the air-sensitivity of the triamine
and the resulting ligand, otherwise the formation of side products complicates purification and
reduces the yield. The ligand is a pale yellow foam after purification by column chromatography
and drying in vacuo. Further purification is carried out by triturating 3a with tetramethylsilane,
giving a white powder. [Mespyr(3,5-Me)]H3 (3b) is also synthesized in a similar manner to 3a.
However, neither 1b nor 3b appears to be as sensitive to air as 1a and 3a. As such, no special
precaution to exclude air appeared necessary in the nucleophilic substitution reaction for the
synthesis of 3b. Purification of 3b yields a pale yellow and extremely viscous oil.
62
Chapter 3
Scheme 3.1 Synthesis of [Mespyr(3,5-t-Bu)2]H3, (3a) and [Mespyr(3,5-Me)2]H3 (3b)
Metalation of [Mespyr(3,5-t-Bu)]H3 with Mo(NMe2)4 leads to a color change from purple
to an ultramarine blue over the course of a few hours at room temperature. Recrystallization
affords a bright teal blue powder, assigned as [Mespyr(3,5-t-Bu)]MoNMe2 (4a), which is
supported by elemental analysis. 1H NMR shows this compound to be diamagnetic, presumably
due to donation of the lone pair from the dimethylamide nitrogen to the metal center and pairing
up of the two d electrons in Mo(IV) to yield a low spin complex. This is similar to the family of
Mo complexes with a dimethylamide ligand in the central apical site, such as
[(C6F5)N3N]MoNMe2 in the triamidoamine system 7 and [Mespyr(C6F5)2]MoNMe2 in the
diamidomonopyrrolylamine system. Earlier studies in our group on the triamidoamine
complexes [TMSN3N]MoNMe2 and [(C6F5)N3N]MoNMe2 (Figure 3.1) showed that both these
compounds are essentially diamagnetic at room temperature, but exist in equilibrium with their
high spin counterparts.7 It was found that the dimethylamido
1
H NMR resonances of
[TMSN3N]MoNMe2 and [(C6F5)N3N]MoNMe2 are temperature dependent, and we proposed that
the dimethylamido resonance is contact-shifted by the paramagnetic form. It has been shown
63
Chapter 3
that [Mespyr(C6F5)2]MoNMe2 and [Mespyr(Otol)2]MoNMe2 (Figure 3.2) also exhibit
temperature dependent dimethylamido 1H NMR resonances. A qualitative comparison of the
energy difference between the high and low spin forms in [Mes(C6F5)2]MoNMe2 and [Mespyr(otol)2]MoNMe2 versus [TMSN3N]MoNMe2 and [C6F5N3N]MoNMe2 had also been obtained. As
such, it was decided to perform similar VT NMR studies on 4a (Figure 3.3).
Figure 3.1 Triamidoamine dimethylamide Mo complexes
Figure 3.2 Diamidomonopyrrolylamine dimethylamide Mo complexes
As one can observe from the Figure 3.3, the 1H NMR peak for the dimethylamide ligand
of 4a does shift with temperature, but this shift is relatively small (~ 0.4 ppm from 263 – 363 K)
when compared to the shift in [TMSN3N]MoNMe2 (~ 9 ppm from 180 to 304 K) )and
[C6F5N3N]MoNMe2 (~2.8 ppm from 259 – 367 K)7 but is comparable to that of
[Mespyr(C6F5)2]MoNMe2 (~0.45 ppm from 263 – 358 K).
64
Chapter 3
3.35
3.3
3.25
δ (ppm)
3.2
3.15
3.1
3.05
3
2.95
2.9
2.85
260
280
300
320
340
360
T (K)
Figure 3.3 Temperature-dependent 1H NMR shift for the dimethylamide group of [Mespyr(3,5-tBu)]MoNMe2 (4a)
Metallation of [Mespyr(3,5-Me)2]H3 with Mo(NMe2)4 led to a colour change from purple
to dark green after heating at 60 °C overnight. This reaction is a little more sluggish than that in
which 4a is prepared. Recrystallization afforded a diamagnetic dark green compound, assigned
as [Mespyr(3,5-Me)2]MoNMe2 (4b) on the basis of its 1H NMR resonances and similarity to the
related compounds [Mespyr(C6F5)2]MoNMe2 and [Mespyr(o-tol)2]MoNMe2. A VT 1H NMR
study of this compound shows that the shift of the dimethylamide resonance in 1H NMR is rather
insignificant (∆δ ≈ 0.1 ppm) over the temperature range of 263 to 373 K (Figure 3.4) when
compared to the shifts for the other diamidomonopyrrolylamine Mo complexes studied (Figure
3.5).
65
Chapter 3
2.88
2.86
2.84
δ(ppm)
2.82
2.8
2.78
2.76
2.74
2.72
260
280
300
320
340
360
380
T (K)
Figure 3.4 Temperature-dependent 1H NMR shift for the dimethylamide group of [Mespyr(3,5Me)]MoNMe2 (4b)
[Mespyr(3,5-Me)2]MoNMe2
3.8
[Mespyr(Otol)2]MoNMe2
3.6
[Mespyr(C6F5)2]MoNMe2
δ (ppm)
3.4
[Mespyr(3,5-t-Bu)2]MoNMe2
3.2
3
2.8
2.6
2.4
260
280
300
320
340
360
380
T (K)
Figure 3.5 Comparison of temperature-dependent 1H NMR shifts for the dimethylamide group
66
Chapter 3
ߜ = ߜ݀݅ܽ +
ܥ
∆ܪ° ∆ܵ°
−
ܶ(1 + exp ቀ
ܴܶ
ܴ ቁ)
Equation 3.4
It has been postulated that the extent of the contact shift of the 1H NMR resonance for the
dimethylamide group is an indicator of the energy difference between the high and low spin
forms of the Mo complex, with a larger shift indicating a smaller energy difference, according to
Equation 3.4.7 In this equation, δ is the observed chemical shift at temperature T, δdia is the
chemical shift of the diamagnetic form, and C is a constant. It is important to note, however, that
the above equation only describes situations where the interconversion between the high and low
spin forms is fast at all temperatures in the temperature range observed. This was in keeping with
expectations in earlier studies comparing [(C6F5)N3N]MoNMe2 and [TMSN3N]MoNMe2 where
[TMSN3N]MoNMe2 exhibited a larger shift in the 1H NMR resonance of its dimethylamide
group with a change in temperature than that for [(C6F5)N3N]MoNMe2. The lone pair on the
dimethylamide nitrogen is expected to donate more strongly to the more electron poor Mo center
in [(C6F5)N3N]MoNMe2 compared to [TMSN3N]MoNMe2 and hence the energy difference
between the high and low spin species is greater for [(C6F5)N3N]MoNMe2. Since the metal
center in 4a and 4b is expected to be more electron rich than that in [Mespyr(C6F5)2]MoNMe2,
we had expected to see a larger 1H NMR resonance shift with change in temperature than that for
[Mespyr(C6F5)2]MoNMe2 but we do not observe this larger resonance shift. Our assumption that
4a and 4b are more electron rich at Mo than [Mespyr(C6F5)2]MoNMe2
or that the
interconversion between the high and low spin forms of 4a and 4b is fast may therefore be
incorrect. Another possibility is that the presence of the methyl or tert-butyl groups in the 3,5
positions on the aryl substituents may sterically prevent the central dimethylamide ligand from
bending away from the vertical axis, therefore increasing the energy difference between the high
67
Chapter 3
and low spin species. Due to the uncertainty about the applicability of Equation 3.4 to 4a and 4b,
we do not calculate the energy differences between the high and low spin forms for these
compounds based on that equation here.
The reaction between MoCl4(THF)2 and [Mespyr(3,5-t-Bu)2]H3 in THF produced a dark
brown solution after 30 minutes at room temperature which presumably contains an adduct
between the ligand and MoCl4(THF)2. If the ligand was dissolved in THF first and then
MoCl4(THF)2 was added slowly over a course of 30 minutes, the yield of the monochloride
[Mespyr(3,5-t-Bu)]MoCl (5a) could be improved. Addition of NaN(TMS)2 to the reaction
mixture then led to a color change from brown to orange-brown. LiN(TMS)2 is typically used in
the synthesis of the triamidoamine monochlorides [ArN3N]MoCl
8,9
but due to the lower
solubility of 5a compared to such monochlorides, LiN(TMS)2 was avoided for fear of
contamination with lithium chloride during extraction of the residue. 5a is obtained in moderate
yields as a pink-tan powder after recrystallization from pentane and toluene. Similarly to
[HIPTN3N]MoCl8 or [Mespyr(C6F5)2]MoCl. 5a is paramagnetic, exhibiting paramagnetically
shifted backbone (δ (ppm) = -24.6, -83.7, -115.2) and aryl resonances (δ (ppm) = 18.8, 11.7).
Elemental analysis supports its assignment. Like the Mo complexes of the triamidoamine
system,8,9 5a is extremely sensitive to air and moisture.
The reaction of MoCl4(THF)2 with [Mespyr(3,5-Me)2]H3 in THF produced a dark brown
solution after fifteen minutes at room temperature which presumably contains the adduct
between the ligand and MoCl4(THF)2. Addition of NaN(TMS)2 to the solution caused a color
change from brown to orange-red. The product [Mespyr(3,5-Me)]MoCl (5b) can be
recrystallized from a mixture of pentane and toluene as a paramagnetic pink powder. Compound
5b displays paramagnetically shifted resonances at δ (ppm) 24.73 and 12.90, assigned to aryl
resonances, and at δ (ppm) -17.29, -92.61 and -98.27, which are assigned to the methylene
68
Chapter 3
protons of the backbone arms. The compound 5b is extremely air-sensitive and moisturesensitive. Unfortunately, attempts to reduce 5b under a N2 atmosphere led to decomposition.
Attempts to synthesize [Mespyr(3,5-Me)2]MoOTf as a precursor for entry into dinitrogen
chemistry were also not promising. Hence, we decided to focus on the dinitrogen chemistry of
the [Mespyr(3,5-t-Bu)2]Mo system instead.
Reaction of [Mespyr(3,5-t-Bu)2]MoCl (5a) with NaN3 in MeCN at room temperature
causes a color change from orange-brown to dark purple with the concomitant formation of a
bright yellow solid. After heating the reaction to 80 °C to ensure its completion, [Mespyr(3,5-tBu)2]MoN (6a), a bright yellow diamagnetic powder, can be isolated from the reaction mixture
in moderate yields. 1H NMR spectroscopy and elemental analysis support the assignment. X-ray
quality crystals were grown from PhF at -35 °C. Figure 3.6 shows the solid state structure of 6a
which crystallizes in the P2(1)/n space group under these conditions. The mesityl substituent on
the pyrrolyl arm is tilted less from the central apical site than are the aryl substituents on the
amido nitrogens, which suggests that a smaller substituent on the pyrrolyl arm would afford a
similar amount of steric protection to a larger substituent on a corresponding amido arm. The
slightly longer Mo(1)-N(1) bond length than the Mo(1)-N(2) andMo(1)-N(3) bond lengths
supports the idea that the lone pair on the pyrrolyl nitrogen is indeed less available for donation
to the metal center since it should be located mostly in the aromatic π-cloud of the pyrrolyl ring.
69
Chapter 3
N(5)
Mo(1)
N(1)
N(4)
Figure 3.6 POV-ray rendering (Thermal ellipsoids shown at 50% probability) of the solid state structure
of [Mespyr(3,5-t-Bu)2]MoN (6a). H atoms are omitted for clarity. Selected bond distances (Å) and angles
(°): Mo(1)-N(5) = 1.6746(13), Mo(1)-N(4) = 2.4134(13), Mo(1)-N(1) = 2.0565(12), Mo(1)-N(2) =
1.9857(13), Mo(1)-N(3) = 1.9751(13), C(21)-N(2)-Mo(1) = 126.27(10), C(31)-N(3)-Mo(1) = 127.25(10),
N(1)-Mo(1)-N(4) = 75.66(5), N(2)-Mo(1)-N(4) = 76.08(5), N(3)-Mo(1)-N(4) = 80.99(5), N(4)-Mo(1)N(5) = 176.43(5)
Reduction of [Mespyr(3,5-t-Bu)2]MoCl under a N2 atmosphere and subsequent oxidation
The reaction between 5a and 2.3 equivalents of Na (smeared as a mirror on the vial wall)
under N2 atmosphere in THF led to a color change from orange-brown to purple to red overnight.
A red solid was isolated from the reaction mixture after removal of NaCl and unreacted Na. This
red solid is extremely reactive to even trace amounts of oxygen, and is proposed to be the
diazenide complex [Mespyr(3,5-t-Bu)2]MoN2Na(THF)x (7a). However there appear to be several
diamagnetic species in the 1H NMR spectrum. IR spectroscopy shows that in C6D6 as a solvent,
there are two absorption bands (1761 cm-1 and 1751 cm-1) which can be assigned to the NN
stretch of the diazenide ligand. Looking back at previous work, in the IR spectrum of
70
Chapter 3
[HIPTN3N]MoNNMgCl(THF)3 in Nujol, several bands assigned to NN stretches were observed
and the relative strengths of the different bands changed depending on the extent of THF
exposure or length of time in vacuo the sample was subjected to. This observation was explained
as stemming from the existence of [HIPTN3N]MoNNMgCl(THF)n<3 species.8 Therefore, the
observation of multiple species by IR and NMR spectroscopy for 7a is postulated to be due to
varying
amounts
of
THF
coordinated
to
the
sodium
cation
in
[Mespyr(3,5-t-
Bu)2]MoN2Na(THF)x. The number of THF molecules coordinated to sodium may depend on the
length of time the solid was left in vacuo or in a different solvent. Unsurprisingly, in THF as a
solvent, a single absorption band is observed (1766 cm-1) as a consequence of the sodium cation
being completely saturated with THF molecules.
In an attempt to isolate a single species, 7a was reacted with one equivalent of 15-crown5 at -35 °C. The orange-red solution immediately became green. A lilac powder, assigned as
[Mespyr(3,5-t-Bu)2]MoN2Na(15-c-5) (8a), can be isolated from the mixture in low yields.
Compound 8a is diamagnetic and exhibits a νNN absorption band at 1855 cm-1 in THF and is
excruciatingly sensitive to even trace amounts of moisture or O2. When 8a is completely
dissolved in THF, the solution is emerald green. It is likely that in solution, THF molecules
coordinate to the sodium cation, but in the solid state, THF is not coordinated.
In another attempt to isolate a single diazenide species, 5a was reduced with sodium
under an atmosphere of N2, then reacted with NBu4Cl (TBACl) directly to produce [Mespyr(3,5t-Bu)2]MoN2TBA (9a), a diamagnetic purple solid. Like 7a and 8a, this compound is again
excruciatingly air and moisture-sensitive. IR spectroscopy reveals a dinitrogen stretch of the
diazenide ligand at 1840 cm-1 in C6D6. In comparison, [HIPTN3N]MoNNTBA exhibits νNN at
1855 cm-1 in C6D6.8
71
Chapter 3
40.0
30.0
20.0
10.0
100
300
500
700
900
I/µA
0.0
-10.0
mV/s
mV/s
mV/s
mV/s
mV/s
-20.0
-30.0
-40.0
-50.0
-1.0
-1.2
-1.4
-1.6
-1.8
-2.0
-2.2
-2.4
-2.6
E/V vs. FeCp2+/0
Figure 3.7 Electrochemical behavior of [Mespyr(3,5-t-Bu)2]MoN2TBA (9a) in 0.1M [NBu4]BAr'4 in PhF
recorded at a glassy carbon electrode at 100mV/s to 900mV/s scan rates, referenced to Cp2Fe+/0
The compound 9a undergoes a reversible oxidation at -1.96 V in 0.1 M [NBu4]BAr'4 in
PhF as shown in Figure 3.7. In the parent system, the neutral dinitrogen species
[HIPTN3N]Mo(N2) undergoes a reversible reduction at -2.01 V under similar conditions (0.1M
[NBu4]BAr'4 in PhF recorded at a glassy carbon electrode).8 The pyrrolyl nitrogen lone pair does
not donate effectively to the Mo metal center, being part of the aromatic π–cloud of the pyrrolyl
ring. Therefore, the metal center in [Mespyr(3,5-t-Bu)2]Mo(N2) is expected to be more electronpoor than that in
[HIPTN3N]Mo(N2), and thus 9a should be harder to oxidize than
{[HIPTN3N]MoN2}-. More positive potentials reveal an irreversible oxidation of [Mespyr(3,5-tBu)2]Mo(N2) (Figure 3.8). In contrast, the oxidation of [HIPTN3N]Mo(N2) appears to be
reversible in PhF, although it is irreversible in THF as a consequence of displacement of N2 by
THF in {[HIPTN3N]MoN2}+.8,11 We conclude that N2 is lost much more easily in the
[Mespyr(3,5-t-Bu)2]Mo system than in the [HIPTN3N]Mo system.
72
I/μA
Chapter 3
[Mespyr(3,5-t-Bu)2]Mo(N2)0/-
E/V vs. FeCp2+/0
Figure 3.8 Electrochemical behavior of [Mespyr(3,5-t-Bu)2]MoN2TBA (9a) in 0.1 M [NBu4]BAr'4 in PhF
recorded at a glassy carbon electrode at 900 mV/s, referenced to Cp2Fe+/0, showing the irreversible
oxidation of [Mespyr(3,5-t-Bu)2]Mo(N2) (10a)
The reaction between 7a and 0.66 equivalents of Zn(OAc)2 in Et2O was incomplete, even
with stirring overnight, as evidenced by the diazenide NN absorption band in IR spectroscopy.
However, the appearance of a new absorption band at 2012 cm-1 (in C6D6) is assigned to νNN of
the neutral dinitrogen species, [Mespyr(3,5-t-Bu)2]Mo(N2) (10a). When the stronger oxidant
AgOTf was reacted with 9a instead, with care taken to ensure the reaction was set-up and carried
out in the dark, the oxidation of the diazenide went to completion. 10a was isolated as a
paramagnetic red solid. The value of νNN for 10a is intermediate between that of the
triamidoamine “parent” [HIPTN3N]Mo(N2) (at 1990 cm-1 in C6D6)8 and that of the
triamidoamine “propyl” [HIPTtrpnN3N]Mo(N2) complex (2030 cm-1 in C6D6)10 (Figure 3.9). We
73
Chapter 3
infer that the N2 ligand is not coordinated as strongly in 10a as in the parent dinitrogen species,
although N2 appears to be more activated in 10a than in the propyl dinitrogen species.
Figure 3.9 [HIPTtrpnN3N]Mo(N2)
A C6D6 solution mixture of 10a and 7a (from the incomplete oxidation of 7a by
Zn(OAc)2) was freeze-pump-thaw degassed and then exposed to
15
N2. After 2.5 hours, IR
spectroscopy of the solution revealed that some conversion of 10a to [Mespyr(3,5-tBu)2]Mo(15N2) had taken place (Figure 3.10). A new νNN absorption band at 1944 cm-1 is
assigned to [Mespyr(3,5-t-Bu)2]Mo(15N2). The band occurs almost exactly as predicted according
to the harmonic oscillator equation (1944 cm-1) (Equation 3.5). Interestingly, the νNN absorption
band belonging to 7a (1751 cm-1) also shows a shifted band at 1692 cm-1 which is assigned to
[Mespyr(3,5-t-Bu)2]Mo15N2Na(THF)x. Again, this band occurs very closely to what is predicted
by the equation, which is 1692 cm-1.
ν=
1 k
√
2π µ
Equation 3.5
74
Chapter 3
Figure 3.10 IR absorption spectrum of [Mespyr(3,5-t-Bu)2]Mo14N2/[Mespyr(3,5-t-Bu)2]Mo14N2
Na(THF)x after 15N2 exposure for 2.5 hours in C6D6
Earlier work showed that exposure of a C6D6 solution of [HIPTN3N]Mo(15N2) to a large
excess of 14N2 led to conversion over the course of a couple days, with t½ ≈ 35 hours at 22 °C.8 In
contrast, [Mespyr(3,5-t-Bu)2]Mo(14N2) conversion to [Mespyr(3,5-t-Bu)2]Mo(15N2), occurs much
more quickly, based on the observation of a significant amount of [Mespyr(3,5-t-Bu)2]Mo(15N2)
after exposure of [Mespyr(3,5-t-Bu)2]Mo(N2) to
15
N2 for less than three hours. It has been
postulated that the exchange goes through the “naked” species in the parent system, the
formation of which is slow and is the rate-determining step (Scheme 3.2).11 The
14
N2/15N2
exchange in [Mespyr(3,5-t-Bu)2]Mo(14N2) does not appear to occur through the “naked” species
at all. Also, N2 does not appear to be bound as tightly to the [Mespyr(3,5-t-Bu)2]Mo center,
based on νNN which shows that the N2 ligand is activated to a lesser extent.
75
Chapter 3
To explain the observation of [Mespyr(3,5-t-Bu)2]Mo15N2Na(THF)x, we turn to earlier
work where it has also been shown that the exchange of [HIPTN3N]Mo(N2) with
[HIPTN3N]MoN2TBA is fast (a rate similar to or faster than the NMR time scale) and it is
postulated that the 14N2/15N2 exchange observed for [HIPTN3N]MoN2TBA occurs at least partly
(if not entirely) through the neutral dinitrogen species, rather than as [HIPTN3N]MoN2TBA.8 It
is likely that our observation of [Mespyr(3,5-t-Bu)2]Mo15N2Na(THF)x after just 2.5 hours of 15N2
exposure is due to a similar process.
Exchange of 14N2 for 15N2 in [Mespyr(3,5-t-Bu)2]Mo(15N2)
Equation 3.6
[Mespyr(3,5-t-Bu)2]Mo(15N2) can be synthesized by exposure of [Mespyr(3,5-tBu)2]Mo(N2) to 15N2. A study of the exchange reaction of [Mespyr(3,5-t-Bu)2]Mo(15N2) under N2
atmosphere in C6D6 solvent and in a nitrogen-filled glovebox at 22 °C showed kobs (where kobs =
k[N2]) to be 1.97 x 10-4 s-1 (t½ ≈ one hour). Aliquots from a stirred solution of [Mespyr(3,5-tBu)2]Mo(15N2) in a vial were periodically obtained for IR spectroscopy, by which the νNN
absorption bands of the labelled and unlabelled dinitrogen species could be observed. The rate of
exchange was calculated by least-squares regression analysis of a plot of ln(A15/Atotal) (A15 =
area of absorption band of ν15N15N, Atotal = Area of absorption band of ν15N15N and ν14N14N)
against time.
76
Chapter 3
A14/Atotal against Time (s)
1
0.9
0.8
A14/Atotal
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0
0
2000
4000
6000
8000
10000 12000 14000 16000 18000
Time ( s)
Figure 3.11 Plot of area of A14 (area of ν14N14N absorption band) over Atotal (area of ν14N14N + area of
ν14N14N absorption bands) against time at 22 °C and one atm N2 for exchange reaction of [Mespyr(3,5-tBu)2]Mo(15N2) to [Mespyr(3,5-t-Bu)2]Mo(N2)
Ln(A15/Atotal) against T(s)
0
-0.5 0
5000
10000
15000
20000
-1
-1.5
-2
-2.5
-3
-3.5
-4
Figure 3.12 Plot of Ln(A15/Atotal) against time (s) for exchange reaction[Mespyr(3,5-t-Bu)2]Mo(15N2) to
[Mespyr(3,5-t-Bu)2]Mo(N2) at one atm N2
77
Chapter 3
In contrast, the analogous exchange in the parent [HIPTN3N]Mo system has a rate
constant of 6 x 10-6 s-1 and t½ ≈ 35 hours.8 Moreover, in the even more sterically hindered
[HTBTN3N]Mo
system
(where
HTBT
is
hexa-t-butylterphenyl),
the
exchange
of
[HTBTN3N]Mo(15N2) under N2 exhibits a t½ ≈ 750 hours (k ~ 3 x 10-7s-1). 11 The rate constant of
the exchange in both [HIPTN3N]Mo and [HTBTN3N]Mo systems changes little with the
pressure of N2, pointing to a reaction where the rate determining step is unimolecular, and is
hypothesized to be the formation of the “naked” species or some weakly solvated species
(Scheme 3.2).
Scheme 3.2 Postulated 14N2 for 15N2 exchange in [ArN3N]Mo system
We think that even though formation of the “naked” species is slow, the alternative
reaction pathway, via a bimolecular reaction going through a bis(dinitrogen) intermediate, is
presumably higher in energy. Even though the electronics of the system differ from that of the
triamidoamine Mo complexes, it is expected that the readily available frontier orbitals in the
apical coordination cavity remain similar, and cannot easily accommodate two dinitrogen
ligands. However we decided to investigate the diamidomonopyrrolyl Mo complexes to verify
that the rate-determining step for the exchange goes through a dissociative pathway.
The
15
N2 for N2 exchange was repeated under an overpressure of fifteen PSI
(approximately two atm N2 pressure). Aliquots of the reaction mixture were periodically
withdrawn for IR spectroscopy to measure the
14
N2 and
15
N2 absorption bands and the reaction
flask re-pressurized to ~ two atmospheres of N2 after each aliquot was removed. The reaction
78
Chapter 3
set-up is as shown in Figure 3.13, with a modified side-arm which has a 14/20 joint. The joint is
stoppered with a rubber septum and aliquots were periodically withdrawn by a syringe.
14
N2, ~ 2 atm
Joint is stoppered with
septum
Figure 3.13 Reaction set-up for increased pressure 15N2/N2 exchange study
A14/Atotal against Time (s)
1
0.9
0.8
A14/Atotal
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0
0
1000
2000
3000
4000
5000
6000
Time (s)
Figure 3.14 Plot of area of A14 (area of ν14N14N absorption band) over Atotal (area of ν14N14N + area of ν14N14N
absorption bands) against time at 22 °C, two atm 14N2 for exchange reaction[Mespyr(3,5-t-Bu)2]Mo(15N2)
to [Mespyr(3,5-t-Bu)2]Mo(N2)
79
Chapter 3
Ln(A15/Atotal) against T(s)
0
0
1000
2000
3000
4000
5000
6000
-0.5
-1
-1.5
-2
-2.5
Figure 3.15 Plot of Ln(A15/Atotal) vs against time (s) for exchange reaction[Mespyr(3,5-t-Bu)2]Mo(15N2) to
[Mespyr(3,5-t-Bu)2]Mo(N2) at two atm N2
When the pressure of 14N2 was increased to two atmospheres, the corresponding t½ for the
exchange reaction decreased from ~ one hour to ~ 30 minutes. This decrease strongly supports a
rate-determining step in which 14N2 is implicated and suggests that instead of a dissociative step,
as shown in Scheme 3.2, we have an associative step, with the formation of a six-coordinate
species. We theorize that the decrease in steric hindrance around the metal center makes the
molybdenum center in [Mespyr(3,5-t-Bu)2]Mo(N2) much more accessible compared to that in
[HIPTN3N]Mo(N2), therefore allowing for an associative exchange mechanism, which is
otherwise not allowed in the case of [HIPTN3N]Mo(N2). Exchange may still occur through a
dissociative step, albeit much more slowly. The reaction is first order in [Mespyr(3,5-t-Bu)2]Mo
but is overall pseudo-first order because 14N2 is in large excess during the course of the reaction.
80
Chapter 3
Scheme 3.3 Pathways for 14N2/15N2 exchange, [Mo] = [Mespyr(3,5-t-Bu)]Mo
{[Mespyr(3,5-t-Bu)2]Mo(NH3)}BPh4 (11a):
Exposure of a PhF solution of 5a with NaBAr'4 to an atmosphere of NH3 (dried over Na)
led to a rapid color change from orange-red to burgundy. 1H NMR shows the formation of a new
paramagnetic species. However, attempts to isolate what should be {[Mespyr(3,5-tBu)2]Mo(NH3)}BAr'4 were unsuccessful as it appears to be an oil immisicible with Et2O,
pentane, toluene and CH2Cl2. The compound is soluble in THF, but does not precipitate out of it
even at -35 °C. A paramagnetic yellow solid was isolated from the reaction mixture when
NaBAr'4 is replaced with NaBPh4. This solid is assigned as {[Mespyr(3,5-t-Bu)2]Mo(NH3)}BPh4
(11a). Elemental analysis supports this assignment. After purification, 11a is relatively insoluble
in toluene. Addition of Cp*2Co to a THF solution of 11a under an Ar atmosphere leads to a color
change from yellow-brown to green with the concomitant formation of yellow [Cp*2Co]BPh4.
Based on the color of the resulting solution, we think that [Mespyr(3,5-t-Bu)2]Mo(NH3) (12a) is
green, although it was not isolated. When THF is removed in vacuo, and the mixture redissolved
in C6D6 and exposed to one atm N2, the stirred solution changed color to red over the course of a
day and IR spectroscopy of the mixture showed the formation of 10a, as seen from the
distinctive absorption band at 2012 cm-1. It is interesting to note that during the first 30 minutes
81
Chapter 3
of a catalytic run utilizing [Mespyr(3,5-t-Bu)2]MoN (6a) as the Mo reagent, the reaction mixture
also turns an emerald green. Thereafter, the mixture remains the same. Presumably, the nitride is
converted to 12a after the addition of protons and electrons, but this then does not convert to any
visible extent to 10a, which is red.
70.0
60.0
50.0
40.0
100 mV/s
30.0
I/µA
300 mV/s
500 mV/s
20.0
700 mV/s
900 mV/s
10.0
0.0
-10.0
-20.0
-30.0
-0.8
-1.0
-1.2
-1.4
-1.6
-1.8
-2.0
-2.2
-2.4
+/0
E/V vs. FeCp2
Figure 3.16 Electrochemical behavior of {[Mespyr(3,5-t-Bu)2]Mo(NH3)}BPh4 (11a) in 0.1 M
[NBu4]BAr'4 in PhF recorded at a glassy carbon electrode, referenced to Cp2Fe+/0
Compound 11a undergoes an irreversible reduction under Ar at approximately -1.68 V at
a scan rate of 900 mV/sec. In contrast, {[HIPTN3N]Mo(NH3)}+ is reduced at -1.63 V and is fully
reversible in both PhF and THF. Rapid loss of ammonia possibly causes this electrochemical
reduction of 11a to be irreversible. Compared to [HIPTN3N]Mo(NH3), the apical site in 12a is
much more “open” and NH3 is may be lost more easily. However, this also means that NH3
could back-react more easily as well. We were curious as to whether 10a could be observed at
progressively slower scan rates under N2 atmosphere and it can be seen from Figure 3.18 that the
{[Mespyr(3,5-t-Bu)2]Mo(N2)}0/- couple appears at scan rates of 10 mV/sec and 50 mV/sec. A
82
Chapter 3
similar appearance of the {[HIPTN3N]Mo(N2)}0/- couple was observed in the electrochemical
reduction of {[HIPTN3N]Mo(NH3)}+.
70.0
60.0
50.0
40.0
30.0
10 mV/s
I/µA
50 mV/s
100 mV/s
20.0
300 mV/s
500 mV/s
10.0
700 mV/s
900 mV/s
0.0
-10.0
-20.0
-30.0
-40.0
-0.2
-0.7
-1.2
-1.7
E/V vs.
-2.2
-2.7
+/0
FeCp2
Figure 3.17 Electrochemistry of 11a under N2 atmosphere
20.0
{[Mespyr(3,5-t-Bu)2]Mo(N2)}0/couple
15.0
10.0
I/µA
10 mV/s
50 mV/s
5.0
0.0
-5.0
-0.2
-0.7
-1.2
-1.7
-2.2
-2.7
+/0
E/V vs. FeCp2
Figure 3.18 Appearance of 10a at scan rates of 10 and 50 mV/sec
83
Chapter 3
We were interested in monitoring the exchange reaction of N2 with NH3 in the apical
cavity of [Mespyr(3,5-t-Bu)2]Mo as a comparison with the [HIPN3N]Mo system. Typical kinetic
studies of this exchange in the parent [HIPTN3N]Mo system are undertaken by reducing the
ammonium cation with Cp*2Cr in C6D6 and then obtaining IR spectra of the aliquots of the
reaction over a period of time to chart the growth of νNN against time. Due to the low solubility
of 11a in benzene and its incomplete reduction by Cp*2Cr, Cp*2Co was utilized as the reducing
agent and THF as the solvent. In consideration of the different solubilities of N2 and NH3 in THF
versus benzene, we wanted to monitor the exchange for the [Mespyr(3,5-t-Bu)2]Mo system in
C6D6 to keep the experimental conditions similar to conditions present during the exchange
studies on the parent system. Therefore, 11a was first reduced in THF by Cp*2Co under an Ar
atmosphere, then THF was removed and the reaction mixture dissolved in C6D6 and exposed to
one atm N2 (Scheme 3.4).
Scheme 3.4 Exchange of N2 for NH3
84
Chapter 3
The exchange reaction appears to occur extremely slowly – a very small growth in νNN is
observed even after approximately five hours. On the other hand, if a Lewis acid is added to trap
NH3 that is released, the exchange is much faster, with an estimated t½ of less than twenty
minutes and approaches completion in about two hours. Figure 3.19 plots the area of νNN of 10a
at 2012 cm-1 against time in both the absence of BPh3 and in the presence of ten equivalents of
BPh3. The back reaction is likely to depend on the concentration of NH3 in solution and in the
absence of a Lewis acid trap for NH3, NH3 that is released from the metal center back-reacts with
10a such that the reaction mixture remains in equilibrium between 10a and 12a. Figure 3.19
shows the conversion between 12a and 10a. Both exchanges were carried out with the same
starting concentration of 11a but as can be seen from Figure 3.19, the extent of 10a formation is
much less in the absence of BPh3. Hence, the equilibrium appears to greatly favour 12a over 10a
in the absence of a Lewis acid trap. This helps to explain the observation mentioned earlier that
in the duration of a catalytic run, the reaction mixture does not appear by visual inspection to
form an appreciable amount of 10a.
85
Chapter 3
Area of N2 absorpion band
3
2.5
2
no BPh3
1.5
10 BPh3
1
0.5
0
0
50
100
150
200
250
300
350
Time (min)
Figure 3.19 Conversion of 12a to 10a against time
Equation 3.7
Exposure of a solution of 10a to an atmosphere of CO led to a colour change from red to
brown. Recrystallization affords a brown powder that is assigned as [Mespyr(3,5-t-Bu)2]Mo(CO)
(13a). Similarly to [Mespyr(3,5-t-Bu)2]Mo(N2), the compound is paramagnetic, in accordance
with expectations. IR spectroscopy reveals an absorption band at 1902 cm-1 which is assigned to
νCO. [Mespyr(3,5-t-Bu)2]Mo(13CO) exhibits an absorption band at 1856 cm-1, which is close to
the expected absorption band based on the harmonic oscillator equation. It is of utmost
86
Chapter 3
importance that the CO used is free of impurities such as water and oxygen because this leads to
only the formation of undefined products with several CO absorption bands in the IR spectrum.
Elemental analysis also supports the assignment of 13a.
Protonation studies of [Mespyr(3,5-t-Bu)2]Mo(CO)
To study the relative susceptibility of this system to protonation compared to the parent
system, we decided to look into the protonation of [Mespyr(3,5-t-Bu)2]Mo(CO) versus that of
[HIPTN3N]Mo(CO).
No [CollH]BAr'4
1 equiv. [CollH]BAr'4
2 equiv. [CollH]BAr'4
Figure 3.20 IR spectrum of [HIPTN3N]Mo(CO) and [Mespyr(3,5-t-Bu)2]Mo(CO) (13a) when exposed to
increasing equivalents of [Collidinium]BAr'4
Reaction of a 1:1 mixture of 13a and [HIPTN3N]Mo(CO) in DME with increasing
amounts of [Collidinium]BAr'4 showed that the CO absorption band for 13a (1896 cm-1 in DME)
decreased much more rapidly than that for [HIPTN3N]MoCO (1884 cm-1 in DME) (Figure 3.20).
When two equivalents of [Collidinium]BAr'4 were added to a 1:1 mixture of [Mespyr(3,5-t87
Chapter 3
Bu)2]Mo(CO) and [HIPTN3N]MoCO in DME, the CO absorption band for [Mespyr(3,5-tBu)2]Mo(CO) was no longer discernible, although that for [HIPTN3N]MoCO decreased only by
about half. However, a new absorption band at 1925 cm-1 was observed, which is assigned to νCO
of protonated [Mespyr(3,5-t-Bu)2]MoCO. When [Mespyr(3,5-t-Bu)2]Mo(13CO) was mixed with
0.5 equivalents of [Collidinium]BAr'4, a new absorption band is observed at 1875 cm-1, which is
21 cm-1 blue-shifted from ν13CO at 1853 cm-1. This blue shift is similar in magnitude to the blue
shift observed upon protonation of 13a.
Figure 3.21 IR spectrum of [Mespyr(3,5-t-Bu)2]Mo(13CO) when exposed to 0.5 equivalents of
[Collidinium]BAr'4 in DME
[{[Mespyr(3,5-t-Bu)2]MoNEt}]BAr'4 (14a)
Attempts
to
synthesize
[{[Mespyr(3,5-t-Bu)2]MoNH}]BAr'4
were
unsuccessful,
therefore, we decided to look into alkylation of 6a by reacting it with [Et3O]BAr'4. Unfortunately
it appears that the presence of the [BAr'4]- counter anion causes the compound to occur as an
88
Chapter 3
immiscible oil in a variety of solvents. However, the product can be dried in vacuo to give a
foam. The product appears diamagnetic in 1H NMR, in accordance with expectations and the
reaction is rather clean since there is very little impurity visible in the spectrum.
{[Mespyr(3,5-t-Bu)2]MoNEt}]BF4 (15a)
We decided to replace the [BAr'4]- counter-anion with another counter-anion in the hopes
of circumventing the difficulties associated with isolating the ethylated salt. Reacting the nitride
6a with [Et3O]BF4 in Et2O overnight affords the bright yellow ethylated nitride 15a. This
compound is diamagnetic, as expected. The low solubility of [Et3O]BF4 in Et2O allows the
reaction to occur very slowly, hence cooling of the reagents or slow addition of [Et3O]BF4 was
unnecessary. Moreover, the product precipitates out of Et2O as it is formed, helping to drive the
reaction forward. Despite its low solubility in Et2O, however, it is rather soluble in less polar
solvents such as toluene and benzene. Combustion analysis together with 1H NMR supports the
assignment of this species.
Catalytic studies
To determine if the [Mespyr(3,5-t-Bu)2]Mo system is capable of catalytic dinitrogen
reduction, catalytic runs utilizing 6a were carried out, where protons and electrons were slowly
added to a heptane solution of 6a in the presence of N2, similarly to the procedure utilized in
determining the catalytic ability of [HIPTN3N]Mo. 12 The amount of NH3 produced was then
quantified by the indophenol method. Approximately one equivalent of NH3 was produced in
each run, which indicates that this system does not catalyze the reduction of dinitrogen to
ammonia. One of the possible reasons has been mentioned earlier – the slow overall exchange of
N2 for NH3 in the apical cavity. We think that another reason is the instability of the postulated
diazenido species “[Mespyr(3,5-t-Bu)2]MoNNH”, if it should form at all during the course of the
catalytic run. We know from previous work that the formation and stability of this species is a
89
Chapter 3
crucial factor in the catalytic ability of the Mo system. However, attempts to synthesize the
diazenido species [Mespyr(3,5-t-Bu)2]MoNNH
have been unsuccessful. The lack of steric
protection around the metal center likely causes instability of the diazenido species in this
system. Table 3.1 summarizes the equivalents of NH3 obtained from several catalytic studies.
Catalytic run
Equivalents of NH3
1
1) 0.99
2) 0.92
2
1) 1.2
2) 1.2
3
1) 0.95
2) 0.96
Average equivalents of NH3
1.02 ± 0.12
Table 3.1 Equivalents of NH3 obtained
Conclusions
Two new ligands have been synthesized as well as several Mo complexes of two
diamidomonopyrrolylamine Mo systems. Dinitrogen activation by Mo complexes in the last
several years has largely been performed by triamidoamine or trisanilide Mo complexes, but we
show here that moving away from the TREN ligand framework to a diamidomonopyrrolylamine
ligand framework can also afford dinitrogen activation.
Experimental
General. All air and moisture sensitive compounds were handled under N2 atmosphere using
standard Schlenk and glove-box techniques, with flame or oven-dried glassware. Ether, pentane
and toluene were purged with nitrogen and passed through activated alumina columns.
90
Chapter 3
Dichloromethane was distilled from a CaH2 suspension. Pentane was freeze-pump-thaw
degassed three times and tetrahydrofuran (THF), benzene, tetramethylsilane, benzene-d6, THF-d4
and toluene-d8 were distilled from dark purple Na/benzophenone ketyl solutions. Ether and
dichloromethane were stored over molecular sieves in solvent bottles in a nitrogen-filled
glovebox while pentane, THF, PhF, benzene, benzene-d6, THF-d8 and toluene-d8 were stored in
Teflon-sealed solvent bulbs. Molecular sieves (4 Å) and Celite were activated at 230 °C in vacuo
over several days. (Me3Si)2NLi (sublimed) (Strem), (Me3Si)2NNa (Recrystallized from THF and
dried) (Aldrich), anhydrous ZnCl2 (Aldrich) (Purified by dissolving in diethyl ether and adding 1
equivalent of 1,4-dioxane to give ZnCl2(dioxane).), MoCl5 (Strem) was used as obtained, unless
indicated otherwise. MoCl4(THF)2, 13 Mo(NMe2)4,14 2-mesityl-1H-pyrrole3 were synthesized as
referenced. 1-bromo-3,5-dimethylbenzene and 1-bromo-3,5-di-tert-butylbenzene were obtained
from Sigma Aldrich. 1-bromo-3,5-di-tert-butylbenzene was also synthesized as referenced.4,5,6
IR spectra were recorded on a Nicolet Avatar 360 FT-IR spectrometer in 0.2 mm KBr solution
cells. NMR spectra were recorded on a Varian Mercury or Varian Inova spectrometer operating
at 300 or 500 MHz (1H), respectively. 1H and 13C NMR Spectra are referenced to the residual 1H
or
13
C peaks of the solvent.
19
F NMR spectra were referenced externally to fluorobenzene (-
113.15 ppm upfield of CFCl3). HRMS was performed on a Bruker Daltonics APEXIV 4.7 Tesla
Fourier Transform Ion Cyclotron Resonance Mass Spectrometer at the MIT Department of
Chemistry Instrumentation Facility. Combustion analyses were performed by Midwest
Microlabs, Indianapolis, Indiana, U.S.
3,5-t-Buphenyltriamine (1a): Under N2 atmosphere, a 1 L Schenk flask was charged with 3,5di-tert-butylbromobenzene (20.781 g, 77.2 mmol), CuI (0.646 g, 3.6 mmol), N,Ndiethylsalicylamide (2.821 g, 14.6 mmol), K3PO4 (30.544 g, 143.9 mmol) and a magnetic stirbar.
91
Chapter 3
DMF (70 mL) was added and resulting suspension stirred for 30-45 minutes. Diethylene triamine
(3.714 g, 36.0 mmol) was added and washed down with DMF (30 mL). The reaction flask was
then heated to 90 °C for 36 h with stirring. The initially blue-green mixture turns brown with
concomitant formation of reddish Cu powder as the mixture is heated after approximately 2
hours. The mixture was allowed to cool to room temperature, then aqueous NH3 (100 mL) and
H2O (300 mL) was added with stirring. Extraction was performed with CH2Cl2 (4x 200 mL) and
the organic layers dried over Na2SO4. The product was purified via column chromatography first
eluting with 4:1 Hexanes: Ethyl acetate and then subsequently with Et2O. Care was taken to limit
exposure of the product to O2. The product was obtained as a yellow oil. Yield: 12.44 g, 72%. 1H
NMR (C6D6): δ 7.022 (2H, s, Aryl 4-H), 6.589 (4H, s, Aryl 2,6-H), 3.776 (2H, ArNH), 2.986
(4H, t, ArNHCH2), 2.509 (4H, t, ArNHCH2CH2), 1.374 (36H, s, -C(CH3)3) ppm. HRMS (ESI,
m/z): Cald for C32H54N3+: 480.4312, found 480.4294.
3,5-Mephenyltriamine (1b): A 300 mL Schlenk flask was charged with CuI (0.4507 g, 2.4
mmol), N,N-diethylsalicylamide (1.826 g, 9.5 mmol), K3PO4 (20.067 g, 94.5 mmol) and DMF
(50 mL). The mixture was stirred for 30 minutes. 1-bromo-3,5-dimethylbenzene (10.0 g, 54.4
mmol) was added and the the mixture stirred for 5 minutes before subsequent addition of
diethylene triamine (2.438 g, 23.6 mmol) was added and washed down with DMF (30 mL). The
reaction flask was then heated to 90 °C for 96 h with stirring. The initially blue-green mixture
turns brown with concomitant formation of reddish Cu powder as the mixture is heated after
approximately 2 hours. The mixture was allowed to cool to room temperature, then aqueous
NH3 (100 mL) and H2O (200 mL) was added with stirring. Extraction was performed with ethyl
acetate (4x 200 mL) and the organic layers dried over Na2SO4. The product was purified via
column chromatography first eluting with Et2O to remove impurities and then THF. The product
92
Chapter 3
was obtained as a brown-yellow oil. Yield: 7.200 g, 98%. 1H NMR (C6D6): δ 6.442 (2H, s, xylyl
4-H), 6.268 (4H, s, xylyl 2,6-H), 3.791 (2H, br s, ArNHCH2), 2.903 (4H, t, ArNHCH2), 2.448
(4H, t, ArNHCH2CH2). HRMS (ESI, m/z): Cald for C20H30N3+: 312.2434, found 312.2442
[Mespyr(3,5-t-Bu)2]H3, (3a): Under N2 atmosphere, a 500 mL Schlenk flask was charged with
1a (12.44 g, 25.9 mmol), 1-(5-mesityl-1H-pyrrol-2-yl)-N,N-trimethylammonium iodide (2)
(9.848 g, 25.6 mmol), K2CO3 (35.88 g, 259.6 mmol) and THF (250 mL). The reaction was
heated to 50 °C for 72 hours, with the flask periodically vented to an atmosphere of N2. The
mixture was cooled to room temperature, then filtered and extracted with Et2O. The volatiles
were removed in vacuo and the resulting mixture purified via column chromatography using 4:1
hexanes: ethyl acetate as the eluent. Yield: 10.44 g, 60%. 1H NMR (C6D6): δ 7.325 (1H, s,
pyrrole N-H), 7.009 (2H, t, JHH=1.7 Hz, Aryl 4-H), 6.834 (2H, s, mesityl 3,5-H), 6.596 (4H, d,
JHH=1.7 Hz, Aryl 2,6-H), 6.214 (1H, dd (apparent triplet), pyrrole CH), 6.087 (1H, dd (apparent
triplet), pyrrole CH), 3.867 (2H, s, ArylNH), 3.413 (2H, s, pyrroleCH2N), 3.041 (4H, t, JHH=5.8
Hz, ArylNHCH2), 2.498 (4H, t, JHH=5.8 Hz, ArylNHCH2CH2), 2.187, 2.181 (overlapping, 12H,
s, mesityl 2,4,6-CH3), 1.285 (36H, s, Aryl 3,5-C(CH3)3).
13
C NMR (C6D6): δ 152.18, 148.05,
138.70, 137.67, 131.784, 130.223, 128.827, 128.68, 128.29, 112.57, 109.39, 108.94, 108.45,
53.46, 52.33, 42.62, 35.35, 32.15, 21.52, 21.32. HRMS (ESI, m/z): Cald for C46H67N4+:
677.5517, found 677.5504
[Mespyr(3,5-Me)2]H3 (3b): A 500 mL flask was charged with 1b (7.200 g, 23.1 mmol), 2
(8.797 g, 22.9 mmol), Cs2CO3 (16.78g, 2.3mmol) and THF. The flask was stoppered with a cap
equipped with a needle to prevent pressure build-up. The reaction mixture was stirred for 48h at
70°C. The volatiles were removed in vacuo, and the mixture extracted with Et2O and filtered.
93
Chapter 3
The volatiles were removed in vacuo, and the resulting oil purified via column chromatography
(3:1 hexanes: ethyl acetate as eluent) to produce a viscous yellow oil. Yield: 4.181g, 36%. 1H
NMR (C6D6): δ 7.540 (1H, s, pyrrole NH), 6.823 (2H, s, Aryl 4-H), 6.393 (2H, s, mesityl 3,5-H),
6.227 (4H, s, Aryl 2,6-H), 6.201 (1H, t, JHH= 2.6 Hz, pyrrole-H), 6.081 (1H, t, JHH= 2.6 Hz,
pyrrole-H), 3.691 (2H, s, ArylNH), 3.342 (2H, s, pyrroleCH2N), 2.864 (4H, t, JHH= 5.4 Hz,
ArylNHCH2), 2.356 (4H, t, JHH= 5.4 Hz, ArylNHCH2CH2), 2.203 (12H, s, Aryl 3,5-CH3), 2.198
(3H, s, mesityl 4-CH3), 2.158 (6H, s, mesityl 2,6-CH3).
13
C NMR (C6D6):δ 149.10, 138.83,
138.41, 137.22, 131.46, 129.94 (quaternary carbons, 1 carbon overlapping with C6D6); 128.48,
119.94, 111.43, 108.75, 108.53 (tertiary carbons), 52.96, 51.75, 41.88 (secondary carbons),
21.74, 21.20, 20.94 (primary carbons). HRMS (ESI, m/z): Cald for C34H45N4+: 509.3639, found
509.3640
[Mespyr(3,5-t-Bu)2]MoNMe2 (4a): In a N2 atmosphere glovebox, a 25 mL solvent bulb
equipped with a PTFE screw valve was charged with 3a (635 mg, 0.938 mmol) and Mo(NMe2)4
(313 mg, 1.15 mmol) and toluene. The reaction mixture turned from purple to ultramarine blue
within a couple of hours, but was left to stir at room temperature overnight. The mixture was
brought back into the glovebox and volatiles were removed in vacuo. The desired product was
purified via recrystallization from pentane/toluene at -35 °C giving a bright teal blue diamagnetic
powder. Yield: 588 mg, 77.0%. 1H NMR (C6D6): δ 7.178 (2H, s, Aryl 4-H), 6.938 (2H, s,
mesityl 3,5-H), 6.553 (4H, s, Aryl 2,6-H), 6.283 (1H, d, JHH= 2.8 Hz, pyrrole CH), 6.266 (1H, d,
JHH= 2.8 Hz, pyrrole CH), 3.963 (2H, dt, ArNCH2CH2), 3.828 (2H, s, pyrroleCH2N), 3.813 (2H,
dt, ArNCH2CH2), 3.200 (2H, dt, ArNCH2CH2), 3.006 (6H, s, MoN(CH3)2), 2.749 (2H, dt,
ArNCH2CH2), 2.261 (6H, s, mesityl 2,6-CH3), 2.239 (3H, s, mesityl 4-CH3), 1.288 (36H, s, Aryl
94
Chapter 3
3,5-C(CH3)3) ppm. Anal Calcd. for C48H71N5Mo: C, 70.82; H, 8.79; N, 8.60. Found: C, 70.47; H,
8.41; N, 8.46.
[Mespyr(3,5-Me)2]MoNMe2 (4b): In a N2 atmosphere glovebox, a 25 mL solvent bulb equipped
with a PTFE screw valve was charged with 3b (509 mg, 1 mmol) and Mo(NMe2)4 (313 mg, 1.15
mmol) and toluene (10 mL). The bulb was sealed and brought out of the glovebox and heated at
60 °C for 48 h. The mixture was brought back into the glovebox and volatiles were removed in
vacuo. The desired product was purified via recrystallization from pentane/toluene at -35 °C
giving a dark green diamagnetic powder. Yield: 321 mg, 50.0%. 1H NMR (C6D6): δ 6.815 (2H,
s, aryl 4-H), 6.549 (2H, s, mesityl 3,5-H), 6.285 (4H, s, aryl 2,6-H), 6.246 (2H, m, pyrrole CH,
overlapping), 3.882 (2H, dt, ArylNHCH2CH2), 3.846 (2H, s, pyrroleCH2N), 3.631 (2H, dt,
ArylNHCH2CH2), 3.064 (2H, dt, ArylNHCH2CH2), 2.829 (6H, s, MoN(CH3)2), 2.701 (2H, dt,
ArylNHCH2CH2), 2.193 (12H, s, Aryl 3,5-CH3), 2.149 (9H, s, mesityl 2,4,6-CH3) ppm.
[Mespyr(3,5-t-Bu)2]MoCl (5a): In a N2 atmosphere glovebox, a 20 mL scintillation vial was
charged with 3a (890 mg, 1.3 mmol) and THF (10 mL). The solution was stirred for 5 minutes to
ensure complete dissolution of the ligand. MoCl4(THF)2 (515.6 mg, 1.4 mmol) was added very
slowly with stirring over the course of 30 minutes. The resulting dark brown solution was stirred
for 40 minutes at room temperature. NaN(TMS)2 (770.2 mg, 4.2 mmol) was added slowly over
15 minutes to the mixture, which turned from brown to dark brownish orange. The mixture was
stirred for 30 minutes and the volatiles were removed in vacuo, extracted with toluene and
filtered through Celite. The toluene was removed in vacuo and the mixture triturated with
pentane and cooled to -35°C overnight. The desired product was collected on a glass frit as a
paramagnetic pink-tan powder. Yield: 0.564g, 53% 1H NMR (C6D6): δ 18.78 (s, Aryl 4-H),
95
Chapter 3
11.71 (br s, Aryl 2,6-H), 8.20 (s), 5.94 (s), 5.20 (s), 5.10 (s, overlapping), 2.56 (s), 1.82 (36H, s,
Aryl 3,5 –C(CH3)3), -24.56 (br s, backbone CH2), -83.71 (br s, backbone CH2), -115.23 (br s,
backbone CH2) ppm. Anal Calcd. for C46H65N4MoCl: C, 68.60; H, 8.13; N, 6.96. Found: C,
68.62; H, 8.01; N, 6.86.
[Mespyr(3,5-Me)2]MoCl (5b): In a N2 atmosphere glovebox, a 20 mL scintillation vial was
charged with 3b (961.7 mg, 1.9 mmol), MoCl4(THF)2 (696.3 mg, 1.8 mmol) and THF (10mL).
The resulting dark brown solution was stirred for fifteen minutes at room temperature.
NaN(TMS)2 (770.2 mg, 4.2 mmol) was added slowly over fifteen minutes to the mixture, which
turned from brown to dark brownish orange. The mixture was stirred for two hours and the
volatiles were removed in vacuo, extracted with toluene and filtered through celite. The toluene
was removed in vacuo and the mixture triturated with pentane. The resulting suspension was
centrifuged and the supernatant decanted off. The remaining precipitate was triturated again with
pentane and the process repeated twice. Finally the precipitate was recrystallized from
toluene/pentane at -35°C to yield a paramagnetic pink powder. Yield: 561.9mg, 46.7%. 1H NMR
(C6D6): δ 24.73 (s, Aryl 4-H), 12.90 (br s, Aryl 2,6-H), 9.98 (s), 5.87 (2H, s, mesityl 3,5-H), 5.01
(6H, br s, Aryl 2,4,6-H), 4.47 (2H, s, pyrrole -H), 2.63 (12H, s, Aryl 3,5-CH3), 2.35 (6H, s,
mesityl 2,6-CH3), 2.16 (3H, s, mesityl 4-CH3), -17.29 (br s, backbone CH2), -92.61 (br s,
backbone CH2), -98.27 (br s, backbone CH2) ppm.
[Mespyr(3,5-t-Bu)2]MoN (6a): In a N2 atmosphere glovebox, a 25 mL solvent bulb equipped
with a PTFE screw valve was charged with 5a (100 mg, 0.12 mmol), NaN3 (8.1 mg, 0.12 mmol)
and MeCN (10 mL). The reaction mixture was stirred at room temperature for 10 h, turning from
orange brown to purple overnight, with formation of a yellow precipitate. It was then brought out
96
Chapter 3
of the glovebox and heated at 80 °C for 24 h. The flask was brought back into the glovebox, the
volatiles removed in vacuo and the residue extracted with toluene and filtered through celite. The
volume of the filtrate was decreased to 5mL and cooled to -35 °C overnight. The resulting
yellow precipitate as collected on a glass frit and washed with cold pentane. The product
obtained is a bright yellow diamagnetic powder. Yield: 45 mg, 46.2%. 1H NMR (C6D6): δ 7.454
(4H, d, JHH=1.7 Hz , Aryl 2,6-H), 7.262 (2H, t, JHH=1.7 Hz, Aryl 4-H), 6.913 (2H, s, mesityl 3,5H), 6.345 (2H, s, pyrrole-H), 3.594 (2H, dt, ArylNCH2CH2), 3.561 (2H, dt, ArylNCH2CH2),
3.531 (2H, s, pyrroleCH2N), 2.437 (6H, s, mesityl 2,6-CH3), 2.338 (2H, dt, ArylNCH2CH2),
2.278 (3H, s, mesityl 4-CH3), 2.261 (2H, dt, ArylNCH2CH2), 1.318 (36H, s, Aryl 3,5-C(CH3)3)
ppm. Anal Calcd. for C46H65N5Mo: C, 70.47; H, 8.36; N, 8.93. Found: C, 70.47; H, 8.44; N,
9.02.
[Mespyr(3,5-t-Bu)2]MoN2Na(15-crown-5) (8a): In a N2 atmosphere glovebox, a 20 ml
scintillation vial was charged with [Mespyr(3,5-t-Bu)2]MoN2Na(THF)x (7a) (150 mg, ~ 0.15
mmol) and Et2O (5 ml). A separate vial was charged with 15-crown-5 ether (33 mg, 0.15 mmol)
and Et2O (5 ml). Both vials were chilled at -35 °C for 1 h, then the solution of the crown ether
was slowly added with stirring to the diazenide solution. An immediate color change is observed,
from orange-red to green. A diamagnetic lilac solid is recrystallized from THF at -35 °C. Yield:
28.5 mg, 20%. 1H NMR (THF-d8): δ 7.303 (4H, s, Aryl 2,6-H), 6.927 (2H, s, Aryl 4-H), 6.627
(2H, s, mesityl 3,5-H), 5.983 (1H, d, JHH=3.0 Hz, pyrrole-CH), 5.852 (1H, d, JHH=3.0 Hz,
pyrrole-CH), 3.926 (2H, dt, ArylNCH2CH2N), 3.861 (2H, dt, ArylNCH2CH2N), 3.603 (2H, dt,
overlapping with solvent peak, ArylNCH2CH2N), 3.482 (2H, s, overlapping with 15-c-5 1H peak,
pyrrolylCH2N), 3.409 (20H, s, 15-crown-5 -OCH2CH2O-), 2.497 (2H, t, JHH=5.7 Hz,
ArylNCH2CH2N), 2.174 (3H, s, mesityl 4-CH3), 2.126 (6H, s, mesityl 2,6-CH3), 1.230 (36H, s,
97
Chapter 3
Aryl 3,5-C(CH3)3) ppm. IR (THF): νNN 1855cm-1. Anal Calcd. for C56H85N6MoNaO5: C, 64.60;
H, 8.23; N, 8.07. Found: C, 64.59; H, 8.10; N, 7.98.
[Mespyr(3,5-t-Bu)2]MoN2TBA (9a): In a N2 atmosphere glovebox, a 20ml scintillation vial was
charged with 5a (700 mg, 0.87 mmol), Na (45.9 mg, 2.00 mmol), and THF (10 ml). The reaction
mixture was stirred for 12 h at RT with a glass stirbar, with a concomitant color change from
orange red to dark purple to red. The mixture as filtered through Celite and NBu4Cl (TBACl)
(252.9 mg, 1.04 mmol) was added. The mixture turned orange, then dark green after stirring for
40 h at RT. Volatiles were removed in vacuo, then the residue was extracted with toluene and
filtered through Celite. The filtrate was decreased in vacuo, with a color change from green to
purple. The solution was chilled at -35 °C overnight and the resulting diamagnetic lavender
powder was collected on a glass frit. Yield: 550 mg, 60.7% 1H NMR (C6D6): δ 7.331 (4H, s,
Aryl 2,6-H), 7.057 (2H, s, mesityl 3,5-H), 6.962 (2H, s, Aryl 4-H), 6.771 (1H, d, JHH= 2.9 Hz,
pyrrole-H), 6.735 (1H, d, JHH= 2.9 Hz, pyrrole-H) , 4.018 (2H, dt, ArylNHCH2CH2), 3.849 (2H,
dt, ArylNHCH2CH2), 3.481 (2H, s, pyrroleCH2N), 2.740 (6H, s, mesityl 2,6-CH3), 2.399 (3H, s,
mesityl 4-CH3), 2.382 (2H, dt, ArylNHCH2CH2), 2.252 (2H, dt, ArylNHCH2CH2), 2.136 (8H, m,
N(CH2CH2CH2CH3)4), 1.490 (36H, s, Aryl 3,5-C(CH3)3), 0.996 (8H, m, N(CH2CH2CH2CH3)4),
0.755 (20H, m, N(CH2CH2CH2CH3)4) ppm. IR (C6D6): νNN 1840 cm-1
[Mespyr(3,5-t-Bu)2]MoN2 (11a): In a N2 atmosphere glovebox, a 20 ml scintillation vial was
charged with 5a (1431 mg, 1.78 mmol), Na (94 mg, 4.09 mmol), and THF (10 ml). The reaction
mixture was stirred for 12 hours at RT with a glass stirbar, then filtered through Celite. AgOTf
(457 mg, 1.78 mmol) was added and the reaction mixture stirred in the dark for 12 hours at RT.
Volatiles were removed in vacuo and the residue extracted with toluene and filtered through
98
Chapter 3
Celite. The filtrate was reduced to 5 ml and pentane (5 ml) was added. The solution was left at 35 °C overnight and the resulting paramagnetic reddish-pink precipitate was collected on a glass
frit, washed with cold pentane and dried. Yield: 746 mg, 52.6%. 1H NMR (C6D6): δ 21.855 (2H,
br s, ArylNCH2CH2N), 21.044 (2H, br s, ArylNCH2CH2N), 18.526 (2H, br s), 15.909 (2H, br s,
ArylNCH2CH2N), 14.900 (2H, br s, ArylNCH2CH2N), 8.214 (2H, s), 2.500 (2H, s), 0.601 (36H,
br s, Aryl 3,5-C(CH3)3), -4.423 (6H, br s, mesityl 2,6-CH3), -7.235 (3H, br s, mesityl 4-CH3), 23.100 (2H, br s), -42.300 (4H, br s) ppm. IR (C6D6): νNN 2012 cm-1 (C6D6), ν15N15N 1944 cm-1
(C6D6) Anal Calcd. for C46H65N6Mo: C, 69.24; H, 8.21; N, 10.53. Found: C, 69.03; H, 8.46; N,
10.18.
{[Mespyr(3,5-t-Bu)2]Mo(NH3)}BPh4 (11a): In a N2 atmosphere glovebox, a 100 mL solvent
bulb equipped with a PTFE screw valve was charged with 5a (622.5 mg, 0.77 mmol), NaBPh4
(290.9 mg, 0.85 mmol) and PhF (15 mL). The bulb was brought out of the glovebox, and freezepump-thaw degassed three times. Anhydrous NH3 (100 mL, 1 atm) which was dried over Na was
vacuum transferred into the degassed solvent bulb with the reaction mixture. The mixture
immediately changed from orange-red to burgundy. The reaction was stirred for 12 hours at RT.
The bulb was brought back into the glovebox and the volatiles removed in vacuo. The residue
was extracted with toluene and filtered through Celite. The filtrate was cooled to -35 °C
overnight, then filtered through a glass frit to remove a dark reddish solid. The volume of the
resulting yellow brown filtrate was decreased to 5 mL and pentane (15 mL) was added to
precipitate a yellow brown solid. The mixture was chilled to -35°C for 1h and then filtered
through a glass frit to collect the paramagnetic yellow solid. Yield: 267.5 mg, 31.7%. 1H NMR
(THF-d8): δ 33.011 (br s), 30.208 (br s), 9.971 (br s), 7.301 (2H, s, Aryl 4-H), 7.215 (4H, s, Aryl
2,6-H), 6.821 (4H, s) 6.691 (2H, s, pyrrole-H), 5.885 (br s), 4.848 (br s), 1.635 (s, Aryl 3,599
Chapter 3
C(CH3)3), -24.605 (br s), -91.744 (br s) ppm. Anal Calcd. for C70H88BMoN5: C, 76.00; H, 8.02;
N, 6.33. Found: C, 75.60; H, 7.90; N, 6.34.
[Mespyr(3,5-t-Bu)2]Mo(CO) (13a):
In a N2 atmosphere glovebox, a 100 mL solvent bulb
equipped with a PTFE screw valve was charged with 10a (250 mg, 0.313 mmol) and benzene.
Outside the glovebox, the bulb was freeze-pump-thaw degassed three times, and then CO (1 atm,
100 mL) was vac-transferred into this bulb from another bulb kept at -78 °C (to freeze out water
vapour that may be present in the CO gas). The mixture was warmed to RT and stirred over 12
hours. The reaction mixture was brought back into the N2 atmosphere glovebox whereby
benzene was removed in vacuo and toluene added to the residue. The toluene solution was
chilled at -35 °C overnight and the resulting paramagnetic green-brown precipitate was collected
on a glass frit, washed with pentane and dried. Yield: 159 mg, 64%. 1H NMR (C6D6): δ 20.13
(2H, br s, ArylNCH2CH2N), 17.24 (2H, s, ArylNCH2CH2N), 13.83 (1H, s, pyrrole-H), 12.25
(2H, br s, ArylNCH2CH2N), 8.69 (2H, s, Aryl 4-H), 7.88 (4H, s, Aryl 2,6-H), 3.02 (1H, s,
pyrrole-H), 1.82 (2H, s), 0.69 (36H, br s, Aryl 3,5-C(CH3)3), -0.40 (2H, s), -3.72 (6H, s, mesityl
2,6-CH3), -7.54 (3H, br s, mesityl 4-CH3), -19.63 (2H, br s), -34.28 (2H, br s) ppm. Anal Calcd.
for C47H65N4MoO: C, 70.74; H, 8.21; N, 7.02. Found: C, 70.99; H, 8.11; N, 6.96.
[{[Mespyr(3,5-t-Bu)2]MoNEt}]BAr'4 (14a): In a N2 atmosphere glovebox, a scintillation vial
was charged with 6a (120 mg, 0.153 mmol) and CH2Cl2 (5ml). A separate vial was charged with
[Et3O]BAr'4 (150 mg, 0.153mmol) and CH2Cl2 (5ml). The solutions were chilled at -35 °C for 1
hr, then the solution of [Et3O]BAr'4 was added dropwise to the solution of 6a, giving an
immediate color change from yellow to orange red. The resulting product was dried in vacuo but
was not isolated. 1H NMR (CD2Cl2): δ 7.727 (8H, t, BAr4' 2,6-H), 7.566 (4H, t, BAr4' 4-H),
100
Chapter 3
7.315 (2H, t, Aryl 4-H), 6.848 (2H, s, mesityl 3,5-H), 6.791 (4H, d, Aryl 2,6-H), 6.392 (1H, d,
pyrrole-H), 6.080(1H, d, pyrrole-H), 4.511(2H, s, pyrroleCH2N), 4.482 ( 2H, dt, ArylNCH2CH2,
overlapping), 4.458 (2H, dt, ArylNCH2CH2, overlapping), 3.723 (2H, dt, ArylNCH2CH2), 3.669
(2H, dt, ArylNCH2CH2), 2.237 (2H, q, MoNCH2CH3), 2.164 (3H, s, mesityl 4-CH3), 2.1152 (3H,
s, mesityl 2,6-CH3), 1.267 (36H, s, Aryl 3,5-C(CH3)3), -0.395 (3H, t, MoNCH2CH3) ppm.
[{[Mespyr(3,5-t-Bu)2]MoNEt}]BF4 (15a): In a N2 atmosphere glovebox, a scintillation vial was
charged with 6a (100 mg, 0.128 mmol) and Et2O (10 mL). [Et3O]BF4 (24 mg, 0.127 mmol) was
added to the solution. The mixture was stirred over 12 hours at RT leading to the formation of a
bright yellow precipitate suspended in very pale yellow solution. The diamagnetic precipitate
was collected on a glass frit, washed with Et2O (20 mL) and dried in vacuo. Yield: 112 mg, 98%.
1
H NMR (THF-d8): δ 7.244 (2H, s, Aryl 4-H), 7.182 (4H, s, Aryl 2,6-H), 6.872 (2H, s, Mesityl
3,5-H), 6.320 (1H, d, JHH= 3.0 Hz, pyrrole-H), 5.993 (1H, d, JHH= 3.0 Hz, pyrrole-H), 4.909 (2H,
td, JHH= 14.7, 5.5 Hz, ArylNCH2CH2), 4.575 (2H, s, pyrroleCH2N), 4.04 (2H, dt, JHH= 14.7, 5.5
Hz, ArylNCH2CH2), 4.198 (2H, m, ArylNCH2CH2), 3.587 (2H, m, ArylNCH2CH2 , overlaps
with THF peak), 2.299 (2H, q, JHH =7.6 Hz, Mo=NCH2CH3), 2.215 (6H, s, Mesityl 2,6-CH3),
2.169 (3H, s, Mesityl 4-CH3), 1.288 (36H, s, Aryl 3,5-C(CH3)3), -0.446 (3H, t, JHH = 7.3 Hz,
Mo=NCH2CH3) ppm. Anal. Calcd. for C48H70BF4MoN5: C, 64.07; H, 7.84; N, 7.78. Found: C,
63.72; H, 7.50; N, 7.63.
101
Chapter 3
References
1
Rieth, R.D.; Mankad, N.P.; Calimano, E.; Sadighi, J.P. Org Lett. 2004, 6, 3981.
2
Kwong, F.Y.; Klapars, A.; Buchwald, S.L. Org. Lett., 2002, 4, 581.
3
Kwong, F.Y.; Buchwald, S.L. Org. Lett., 2003, 5, 793.
4
Ditto, S.R.; Card, R.J.; Davis, P.D.; Neckers, D.C. J. Org. Chem. 1979, 44, 894.
5
Frampton, M.J.; Akdas, H.; Cowley, A.R.; Rogers, J.E.; Slagle, J.E.; Fleitz, P.A.; Drobizhev, M.;
Rebane, A.; Anderson, H.L. Org. Lett., 2005, 7, 5365.
6
Nomura, N.; Ishii, R.; Yamamoto, Y.; Kondo, T. Chemistry- A Eur. J. 2007, 13, 4433.
7
Mosch-Zanetti, N.C.; Schrock, R.R.; Davis, W.M.; Wanniger, K.; Seidel, S. W.; O’Donoghue,
M.B. J. Am. Chem. Soc. 1997, 119, 11037.
8
Yandulov, D.V.; Schrock, R. R. Inorg. Chem. 2003, 42, 796.
9
Weare, W.W.; Schrock, R.R.; Hock, A.S.; Müller, P. Inorg. Chem. 2006, 45, 9185.
10
Chin, J. PhD Thesis, Chapter 4.
11
Weare,W.W.; Dai, X.; Byrnes, M,; Chin, J,; Schrock, R.R.; Müller P. PNAS, 2006, 103, 17099.
12
Yandulov, D. V.; Schrock, R. R. Science 2003, 301, 76.
13
Stoffelbach, F.; Saurenz, D.; Poli, R. Eur. J. Inorg. Chem. 2001, 10, 2699.
14
Bradley, D. C.; Chisholm, M. H. J. Chem. Soc. A 1971, 2741.
102
Chapter 4
Chapter 4
Molybdenum Complexes that Contain the [(HIPTNCH2CH2CH2)3N]3Ligand (HIPT = 3,5-(2,4,6-i-Pr3C6H2)2C6H3) and Studies Relevant to
Catalytic Reduction of Dinitrogen
103
Chapter 4
Introduction
We have explored some variations of the [HIPTN3N]3- ligand (e.g. hexa-tbutylterphenyl), but in most cases the variations have led to a decrease in catalytic efficiency, or,
in many cases, no catalytic turnover at all.1,2 No catalytic reduction of dinitrogen was observed
by [HIPTN3N]Cr, 3 [HIPTN3N]W, 4 or [HIPTN3N]V5 species. In view of the sensitivity of the
catalytic reduction to the nature of the triamidoamine ligand and the fact that ultimate removal of
the [HIPTN3N]3- ligand during catalysis is a major problem, we became interested in a simple
variation in which the ethylene arm is replaced by a propylene arm, namely
[(HIPTNCH2CH2CH2)3N]3-, or [HIPTtrpn]3- (trpn = (NCH2CH2CH2)3N). So far, two titanium
complexes that contain a [(Me3Si)trpn]3- ligand have been reported, [(Me3Si)trpn]TiCl and
[(Me3Si)trpn]TiMe. 6 Therefore we set out to prepare the (HIPTNHCH2CH2CH2)3N and Mo
complexes that contain the [HIPTtrpn]3- ligand that are relevant to catalytic reduction of
dinitrogen (Figure 4.1). Earlier work has shown that (HIPTNHCH2CH2CH2)3N is synthesized in
a similar manner to H3[HIPTN3N], 7,1 by utilizing the Buchwald-Hartwig C-N cross coupling
reaction between HIPTBr and tripropylenetetramine. Also, the synthesis of [HIPTtrpn]MoCl has
been previously reported by Weare in his Ph.D. thesis.7 This chapter describes the continuation
of studies on the [HIPTtrpn]Mo system, including the syntheses of [HIPTtrpn]Mo complexes
and the evaluation of the catalytic ability of this system.
104
Chapter 4
Mo(III) 14
+ N2
Mo
Mo(N2)
13
Mo-N=N-
Mo(NH3)
e-
H+
Mo-NH2
Mo-N=N-H
{Mo=N-NH2}+
10 {Mo-NH2}+
9
Mo=N-NH2
Mo(IV)
4
Mo(VI)
5
Mo(V)
6
Mo(V)
7
Mo(VI)
H+
{Mo=N-NH3}+
Mo=NH
e-, -NH3
eMo(VI)
3
e-
H+
Mo(V)
Mo(IV)
H+
eMo(V)
2
H+
Mo(IV) 12 {Mo(NH3)}+
Mo(IV) 11
Mo(III)
e-
-NH3
Mo(III)
1
8 {Mo=NH}+
H+
MoN
Figure 4.1 Chatt cycle for the reduction of dinitrogen to ammonia on a Mo center (Mo represents
[HIPTN3N]Mo)
Results and discussion
Synthesis and characterization of [pMo] complexes ([pMo] = [HIPTtrpn]Mo)
The synthesis of [pMo]Cl has consistently lower yields than the synthesis of [Mo]Cl,
[pMo]Cl being considerably more soluble than [Mo]Cl. The synthesis of [pMo]Cl is also
accompanied by the formation of various side products, increasing the difficulty of its
purification. Both the high solubility of [pMo]Cl as well as side product formation are recurring
problems with the various [pMo] complexes described in this thesis. The extra methylene spacer
in each ligand arm (three instead of two, as in H3[(HIPTN)3N]) may affect the ability of the
ligand to effectively chelate the Mo center, therefore decreasing formation of the presumed
adduct prior to base addition. Replacing LiN(TMS)2 in the synthesis of [pMo]Cl with different
bases such as NaN(SiMe3)2 and KN(SiMe3)2 led to decreased yields whereas utilizing MeMgCl,
benzyl potassium and n-BuLi led to mixtures of unidentifiable compounds.
105
Chapter 4
As [pMo]Cl was the entry point into [pMo] chemistry, its low synthetic yield was a
considerable drawback. In the search for an alternative entry point, attempts to synthesize
[pMo]NMe2 were made. Our group has previously shown that aryl-substituted triamidoamine
ligands react with Mo(NMe2)4 to produce [(ArNCH2CH2)3N]MoNMe2 in high yields. 8 , 9
However, no reaction was observed in a mixture of (HIPTNHCH2CH2CH2)3N and Mo(NMe2)4
after several days at 70 °C. Increasing the temperature led to decomposition of Mo(NMe2)4 as
well as the formation of free ligand.
Figure 4.2 POV-ray rendering of [pMo]Cl with thermal ellipsoids at 50% probability.7 Hydrogen atoms
and isopropyl groups are omitted for clarity. Selected bond lengths (Å): Cl(1)-Mo(1) = 2.3843(5), Mo(1)N(1) = 1.9688(17), Mo(1)-N(2) = 1.9709(18), Mo(1)-N(3) = 1.9743(18), Mo(1)-N(4) = 2.3230(18).
Selected bond angles (°):N(1)-Mo(1)-N(4) = 89.03(7), N(2)-Mo(1)-N(4) = 89.26(7), N(3)-Mo(1)-N(4) =
87.76(7)
106
Chapter 4
[pMo]Cl7
[(HTBT)N3N]MoCl1
Average dihedral angles / °
Cipso-Namido-Mo-Cl
35.1
10.3
Displacement from amide N
plane / Å
0.046
0.332
Bond length / Å Mo-Napical
2.323
2.194
Average angles / °
Namido-Mo-Namine
88.7
80.4
Average angles / °
Cipso-Namido-Mo
118.5
129.3
Table 4.1 Comparison of structural parameters between [pMo]Cl and [HTBTN3N]MoCl
In previous work, [pMo]Cl was crystallized at RT from benzene as purple needles.7
[pMo]Cl crystallizes in the P212121 space group (Figure 4.2). In this solid state structure, the
metal center is located deeper in the cavity created by the three ligand arms, and is located nearly
within the plane created by the amide nitrogens compared to [HTBTN3N]MoCl where the Mo
center sits slightly above this plane. Notably, the bond length between the metal center and the
central donor amine Mo-Napical is more than 0.1 Å longer in [pMo]Cl than in
[HTBTN3N]MoCl.1a,7 The torsion angles Cipso-Namido-Mo-Cl are also larger by about 25 ° in
[pMo]Cl than in [HTBTN3N]MoCl, resulting in a larger cavity for ligand binding.
Synthesis and studies of [pMo(NH3)]BAr′4 and [pMo(NH3)]BPh4:
Reaction of [pMo]Cl with NH3 and NaBAr′4 in PhF leads to the formation of red orange
{[pMo](NH3)}BAr′4. Unlike other [pMo] compounds we report here, {[pMo](NH3)}BAr′4 is
very crystalline and easily purified by recrystallization from pentane. Utilizing NaBPh4 instead
of NaBAr′4 in the reaction mixture leads to the formation of red {[pMo](NH3)}BPh4. It was
expected that {[pMo](NH3)}BPh4 would be less soluble than {[pMo](NH3)}BAr′4 but the
107
Chapter 4
opposite was true. Both compounds are paramagnetic d2 species, exhibiting paramagnetically
shifted peaks in 1H NMR spectroscopy. Combustion analyses support the assignment of both
these species.
Electrochemistry of [pMo(NH3)]BAr′4:
Electrochemistry
[pMo](NH3)
CV of of
pMoNH3
10
Current (μA)
8
6
4
2
0
-2
-4
-1
-1.5
-2
+/0
Potential / V, referenced to FeCp2
-2.5
Figure 4.3 Cyclic voltammogram of {[pMo](NH3)]BAr'4 at a 3.0 mm glassy carbon electrode at room
temperature with 0.1 M [NBu4]BAr'4 in PhF and scan rate of 100 mV/sec, referenced to Cp2Fe+/0
Electrochemical studies show that {[pMo](NH3)}BAr′4 undergoes a quasi-reversible
reduction at -1.76 V (vs Cp2Fe+/0) at a glassy carbon electrode in a PhF solution of 0.1 M
[NBu4]BAr′4 at a scan rate of 100 mV/s (Figure 4.3). Although Cp*2Cr+/0 occurs at -1.63 V
versus Cp2Fe+/0 in PhF, which implies that it is not a strong enough reducing agent for
{[pMo](NH3)}BAr′4, addition of Cp*2Cr to {[pMo](NH3)}BAr′4
reduction within ten minutes as observed by
1
in C6D6 led to complete
H NMR spectroscopy. Precipitation of
[Cp*2Cr]BAr′4 from solution drives the reduction in C6D6, and it is expected that in heptane, the
solvent utilized for catalytic runs, {[pMo](NH3)}BAr′4 is also completely reduced by Cp*2Cr.
108
Chapter 4
Figure 4.4 POV-ray rendering of {[pMo](NH3)}BAr'4 with thermal ellipsoids at 50% probability.
Hydrogens, isopropyl groups and BAr'4 anion are omitted for clarity. Selected bond distances (Å): Mo(1)N(3) = 1.960(4), Mo(1)-N(2) = 1.964(4), Mo(1)-N(1) = 1.972(4), Mo(1)-N(5) = 2.238(4), Mo(1)-N(4) =
2.256(4). Selected angles (°): N(3)-Mo(1)-N(4) = 92.10(15), N(2)-Mo(1)-N(4) = 90.92(14), N(1)-Mo(1)N(4) = 92.25(16), N(5)-Mo(1)-N(4) = 178.92(16)
{[pMo](NH3)}BAr'4
{[Mo](NH3)}BAr'42b
Average torsion angles / °
Cipso-Namido-Mo-Nammonia
33.5
12.9
Displacement from amide N
plane / Å
0.061 (below plane)
0.297 (above plane)
Bond length / Å Mo-Napical
2.256
2.148
Average angles / °
Cipso-Namido-Mo
123.3
128.4
Table 4.2 Comparison of bond lengths and angles between [pMo(NH3)]BAr'4 and {[Mo](NH3)}BAr'4
Crystals of {[pMo](NH3)}BAr′4 were grown in pentane at -25 °C as red cuboids. There is
rotational disorder of the isopropyl groups as well as the –CF3 groups of the BAr'4 anion. For
clarity, isopropyl groups as well as the BAr'4 anion have been omitted from the POV-Ray
diagram of {[pMo](NH3)}BAr′4. The Mo atom sits further into the plane created by the three
109
Chapter 4
amide nitrogens in {[pMo](NH3)}BAr′4 than in {[Mo](NH3)}BAr'4.2b In fact, the Mo atom sits
below the plane rather than above it (Table 4.2). However, the bond length between the metal
center and the donor nitrogen trans to NH3 is ~ 0.1 Å longer in {[pMo](NH3)}BAr′4 than in
{[Mo](NH3)}BAr'4. Therefore, it is expected that this donor nitrogen has a weaker donation to
Mo in {[pMo](NH3)}BAr′4 than in {[Mo](NH3)}BAr'4. 2b Also, the HIPT substituents are more
tilted from the N(4)-Mo(1)-N(5) axis in {[pMo](NH3)}BAr′4 than in {[Mo](NH3)}BAr'4, as
shown by the torsion angles Cipso-Namido-Mo-Nammonia which may mean that they do not extend as
far vertically above the Mo center. As a result, there may be less steric protection for ligands in
the apical site for [pMo] rather than [Mo] complexes.
Synthesis of [pMo]NNK and pMo(N2):
Reaction of [pMo]Cl with KC8 in benzene under dinitrogen for two days led to a color
change in the solution from orange to green-brown. From this reaction, [pMo]NNK was obtained
as a lime-green, diamagnetic powder. [pMo]NNK exhibits an infrared absorption band at 1810
cm-1 in C6D6 and 1820 cm-1 in heptane. This absorption band is assigned as νNN. [pMo]15N15NK
exhibits a band at 1760 cm-1 in heptane. Due to C6D6 absorbing strongly in that spectral region,
ν15N15N in C6D6 is not reported here. As a comparison, [Mo]NNK exhibits νNN at 1798 cm-1 in
C6D6. It is interesting to note that [pMo]NNK will react with PTFE, a fact that demonstrates the
reducing nature of this diazenide.
110
Chapter 4
Figure 4.5 Unrefined POV-ray rendering of [pMo]NNK with hydrogens and isopropyl groups omitted for
clarity.
A solid-state structure of [pMo]NNK was also obtained, and despite the poor quality of
the structure, the connectivity of the molecule could still be determined (Figure 4.5). Crystals
were grown in heptane at room temperature as emerald flakes. Due to their thinness, the structure
could not be further refined. However, it can still be seen that K+ resides between the diazenido
nitrogens in the apical pocket, rather than associating only with Nß and appears to be associated
with the aromatic π-clouds of two terphenyl groups.
111
Chapter 4
[pMo]NNK
[Mo]NNMgCl(THF)32b
Average dihedral angles / °
Cipso-Namido-Mo-Napical
-37.0
-23.2
Displacement from amide N
plane / Å
0.043 (above plane)
0.353 (above plane)
Bond length / Å Mo-Napical
2.418
2.237
Average angles / °
Cipso-Namido-Mo
124.7
134.5
Table 4.3 Comparison of solid state structure of [pMo]NNK with that of [Mo]NNMgCl(THF)3
Oxidation of [pMo]NNK with ZnCl2(dioxane) in THF led to an immediate color change
to a purple brown solution. However, no absorption band in the IR region was observed as would
be expected for oxidation product, [pMo](N2). When the same reaction was performed in Et2O,
the green-brown solution of [pMo]NNK became purple immediately. Work up afforded
[pMo](N2), a dark purple paramagnetic solid (Equation 4.1).
Equation 4.1 Formation of [pMo](N2) from [pMo]NNK
IR spectroscopy showed the presence of an absorption band at 2030 cm-1 (in C6D6, 2024
cm-1 in PhF) which is ascribed to νNN for [pMo](N2). Since νNN is only observed when
[pMo]NNK is oxidized in Et2O rather than THF, it appears that THF displaces the N2 ligand
from pMo(N2). In comparison, THF does not displace N2 from [Mo](N2) (νNN = 1990 cm-1 in
C6D6) although it does so from {[Mo](N2)}+. 2b , 1c Also, νNN for [pMo](N2) is 40 cm-1 higher than
112
Chapter 4
that for Mo(N2), therefore, it can be seen that the back donation of electron density from the Mo
center toward the N2 ligand is considerably less for [pMo](N2) compared to [Mo](N2).
Initial attempts to synthesize [pMo](15N2) from pMo15N15NK in Et2O were unsuccessful.
IR spectroscopy of the reaction mixture revealed only an absorption band at 2030 cm-1 in C6D6.
It appears that [pMo](15N2) rapidly converts to [pMo](14N2) under N2 atmosphere. When a
degassed solution of [pMo](14N2) was exposed to 0.9 atm of 15N2 and stirred for approximately
one hour, the absorption band at 2024 cm-1 (ν14N14N in PhF) disappeared. A new band appeared at
1960 cm-1 in the IR spectrum (in PhF), in accordance with where a 15N2 absorption band should
occur, as calculated by the harmonic oscillator equation. The band at 1960 cm-1 is therefore
ascribed to ν15N15N. Unfortunately, elemental analyses of [pMo](N2) have so far been
unsuccessful, presumably due to the high solubility of this compound and the difficulties
associated with its purification. However, IR and reactivity studies clearly support the
assignment of [pMo](N2).
Electrochemistry of [pMo](N2):
Electrochemical studies on [pMo](N2) showed two quasi-reversible waves at -0.94 V and
-1.52 V (referenced to Cp2Fe+/0), which are postulated to correspond to [pMo](N2)+/0 and
[pMo](N2)0/- respectively.
113
Chapter 4
6.00
Current / µA
4.00
2.00
0.00
-2.00
-4.00
-6.00
0
-0.5
-1
-1.5
-2
+/0
Potential / V, vs. Cp2Fe
Figure 4.6 Cyclic voltammogram of [pMo](N2) at a 3.0 mm glassy carbon electrode at room temperature
with 0.1 M [NBu4]BAr'4 in PhF and scan rate of 100 mV/sec , referenced to Cp2Fe+/0.
Comparing the oxidation and reduction potentials of [pMo](N2) (-0.94 V and -1.52 V)
+/0
with [Mo](N2), where [Mo](N2)
0/-
occurs at –0.66 V and [Mo](N2)
occurs at –2.01 V, it can be
seen than [pMo](N2) is both easier to oxidize and easier to reduce. This suggests that its HOMOLUMO gap is smaller than that in [Mo](N2).
Synthesis of [pMo](CO):
Exposure of a degassed solution of [pMo](N2) in PhF to one atmosphere of CO converts
the solution from purple to ultramarine. A dark blue paramagnetic solid is isolated from the
reaction and is assigned as [pMo](CO). An IR absorption band is observed at 1883 cm-1 and is
ascribed to νCO. When a [pMo](N2) solution is exposed to 13CO, an absorption band at 1840 cm-1
(in PhF) is observed and assigned to ν13CO. The position of the band is in agreement with the
position of ν13CO as predicted by the harmonic oscillator equation (1841 cm-1). In the parent
system, [Mo](CO) has an absorption band at 1888 cm-1 in C6D6, whereas [Mo](13CO) exhibits a
band at 1845 cm-1 in C6D6.10 Comparing the [pMo](CO) compound to [Mo](CO) (νCO = 1885
cm-1 in PhF), it appears that back donation to CO in both systems is comparable, which seems
114
Chapter 4
surprising given the distinct difference in the extent of back-donation to N2 in the corresponding
dinitrogen complexes. However, CO is a much stronger π-acid ligand than N2, and hence subtle
differences in the back-donating abilities of [Mo] versus [pMo] may lead to more profound
Current / µA
effects for N2 than CO.
Potential / V
Figure 4.7 Cyclic voltammogram of [pMo](CO) at a 3.0 mm glassy carbon electrode at room temperature
with 0.1 M [NBu4]BAr'4 in PhF and scan rate of 100 mV/sec , referenced to Cp2Fe+/0
Cyclic voltammetry of [pMo](CO) in a 0.1 M solution of [NBu4]BAr'4 in PhF reveals the
reversible couples [pMo](CO)0/- at -2.09 V and pMo(CO)+/0 at -0.86 V (Figure 4.7). In the
parent system, [Mo](CO) displays a reversible reduction potential at -1.58 V and a quasireversible oxidation around -0.22 V.11 It appears that [pMo](CO) is harder to reduce but easier to
oxidize than [Mo](CO).
Synthesis of [pMo]N:
Reaction of [pMo]Cl and NaN3 (with acetonitrile and THF as the solvent) yielded an
orange-yellow diamagnetic compound, which was assigned as the nitride, [pMo]N. A solid state
structure of the compound confirms this assignment. Interestingly, despite being a Mo(VI)
species, [pMo]N appears to be much less stable than the corresponding parent species,
115
Chapter 4
[HIPTN3N]MoN, decomposing immediately on exposure to air. It also seems to be thermally
unstable, as it decomposes when left at room temperature overnight under N2.
Attempts to synthesize [pMo]N by reacting [pMo]Cl with TMSN3 or [NBu4]N3 led to
either no reaction or decomposition. It had previously been shown that oxidizing
{[W](NH3)}BAr′4 and treating it with base led to [W]N ([W] = [HIPTN3N]W)4 but in the case of
{[pMo](NH3)}BAr′4 only decomposition was observed. We then tried to ligate compounds
where the molybdenum nitride functionality was already in place, such as NMo(NMe2)3 or
NMo(OtBu)3. However, we observed no reaction at room temperature, and decomposition at 60
°C.
N5
C315
Mo1
N3
N2
C215
N1
N4
Figure 4.8 POV-Ray rendering of [pMo]N with thermal ellipsoids at 50% probability. Isopropyl groups,
hydrogen atoms, and solvent molecules are omitted for clarity. Selected bond distances (Å): Mo(1)-N(5)
= 1.651(5), Mo(1)-N(1) = 2.004(5), Mo(1)-N(2) = 1.999(5), Mo(1)-N(3) = 1.961(5). Selected angles (°):
N(5)-Mo(1)-N(1) = 99.9 (2), N(5)-Mo(1)-N(2) = 103.3(2), N(5)-Mo(1)-N(3) = 97.3(2), C(115)-N(1)Mo(1) = 131.0(4), C(215)-N(2)-Mo(1) = 127.2(4), C(315)-N(3)-Mo(1) = 121.5(4)
Crystals of [pMo]N were obtained from a solvent mixture of THF, acetonitrile and
pentane at -35 °C as orange needles (Figure 4.8). There was a significant amount of disorder in
116
Chapter 4
the solvent molecules, but the structure of [pMo]N could be resolved. The compound crystallizes
in the P2(1)/c space group and has a trigonal-pyramidal coordination geometry at the central Mo
atom. The donor nitrogen trans to the nitride is not bonding to the metal center, with a Mo(1)N(4) inter-atomic distance of about 2.84 Å. This is explained in part by the strong trans influence
of the nitride ligand. In contrast, there is a bond between Mo and the donor amine in [Mo]N,
with a bond length of 2.395(5) Å.2c However, the Mo – nitride bond (Mo(1)-N(5)) in [pMo]N
(1.651(5) Å) is only slightly shorter than that in [Mo]N (1.656(8) Å).
[pMo]N
[HIPTN3N]MoN2d
Average torsion angles / °
Cipso-Namido-Mo-Nnitride
37.7
4.1
Bond length / Å
Mo-Namine
N/A
2.395(5)
Average angles / °
Namido-Mo-Namine
80.0
78.4
Average angles / °
Cipso-Namido-Mo
126.6
128.6
Table 4.4 Comparison of bond lengths and angles between [pMo]N and [Mo]N
Attempts to synthesize [pMo]NNH:
In the catalytic cycle for dinitrogen reduction, the diazenido species is thought to be a
crucial intermediate. Hence, we were interested in studying the stability of [pMo]NNH. The
compound [pMo]NNK was reacted with a variety of acids such as 1,1,1,3,3,3-hexafluoro-2methyl-propan-2-ol, [Et3NH]BAr′4, H(OEt2)2BAr′4. All attempts led to immediate production of
[pMo](N2) as could be seen by its characteristic IR absorption band at 2030 cm-1 (C6D6) and its
dark purple color. Attempts to synthesize [NBu4][pMoN2] as a potential precursor to [pMo]NNH
were unfortunately unsuccessful. It is interesting to note that in a hybrid system, [3,5bis(CF3)HIPT2N3N]Mo, where there is a HIPT substituent on two of the amide nitrogens and a
117
Chapter 4
3,5-bis(CF3)phenyl substituent on the third amide nitrogen, the diazenido species [3,5bis(CF3)HIPT2N3N]MoNNH decomposes about four times more rapidly than [Mo]NNH.12 Also,
[Mo]NNH decomposes to [Mo]H whereas [3,5-bis(CF3)HIPT2N3N]MoNNH decomposes to [3,5bis(CF3)HIPT2N3N]Mo(N2) instead. In systems where steric protection is insufficient, the
diazenido species decomposes much more rapidly and via a different mechanism than in the
parent system to produce the dinitrogen species rather than the molybdenum hydride.
Relevant studies for dinitrogen reduction
Conversion of [pMo](NH3) to [pMo](N2):
The rate of conversion of [pMo](NH3) to [pMo](N2) (Equation 4.2) was studied since this
conversion step is crucial for turnover within the catalytic cycle (Figure 4.1).
Equation 4.2
The conversion of [pMo](NH3) to [pMo](N2) was investigated by reducing a C6D6
solution of {[pMo](NH3)}BAr′4 to [pMo](NH3) under N2 atmosphere and measuring aliquots of
the mixture by IR spectroscopy periodically. The formation of [pMo](N2) was monitored by
measuring the area of the IR absorption band at 2030 cm-1 (νNN for [pMo](N2)) against time
(Figure 4.9).
118
Area of νNN
Chapter 4
Time / minute
With BPh3, starting {[pMo](NH3)}BAr'4 concentration 10.9 mM
Without BPh3, starting {[pMo](NH3)}BAr'4 concentration 12.7 mM
Figure 4.9 Area of νNN against Time (min)
When these measurements (presented in Figure 4.9) were repeated using similar
concentrations of {[pMo](NH3)}BAr'4 with ten equivalents of BPh3 to prevent back reaction of
NH3 with [pMo](N2), the amount of [pMo](N2) observed increased approximately ten-fold.
Figure 4.9 shows that [pMo](N2) concentration rises rapidly and reaches a plateau at
approximately 400 minutes. Assuming that back reaction of NH3 with [pMo](N2) is effectively
prevented by adduct formation of Ph3BNH3, the conversion of [pMo](NH3) to [pMo](N2) goes
to completion at approximately 400 minutes. This shows that were the conversion between
[pMo](NH3) to [pMo](N2) allowed to reach an equilibrium, this equilibrium would favor
[pMo](NH3) over [pMo](N2). Hence the amount of [pMo](N2) present and available for reaction
with protons and electrons during a catalytic run (where BPh3 is absent for NH3 uptake) may be
limited. However, back reaction of NH3 with [pMo](N2) in a standard catalytic set-up may also
119
Chapter 4
be limited by the likelihood of NH3 reacting with the large excess of [Collidinium]+ present in
the mixture.
Reaction of [pMo](N2) with acid:
As our ultimate aim for the [pMo] system is the catalytic reduction of dinitrogen to
ammonia through the addition of protons and electrons, we wanted to determine the
susceptibility of this triamidoamine Mo system to acid. Whilst the protonatability of the Mo
complexes may be important for dinitrogen reduction (we are unsure as to whether proton
transfer or electron transfer occurs first), it is also crucial to note that protonation may catalyze
the decomposition of the Mo complex.
Addition of one equivalent of [2,6-Lutidinium]BAr′4 to [pMo](N2) in PhF results in
complete disappearance of νNN at 2024 cm-1 and no appearance of a new absorption band in the
IR spectrum. In comparison with the parent system, however, addition of one equivalent of [2,6lutidinium]BAr′4 to [Mo](N2) results in only a 20% decrease in νNN. Given that even with the
addition of two equivalents of [2,6-lutidinium]BAr′4 to Mo(N2) in PhF, the original νNN at 1990
cm-1 can still be observed (along with a new peak at 2057 cm-1)13, it appears that [pMo](N2) is
more sensitive to the presence of acid than [Mo](N2).
Moreover, the absence of a new
absorption band ascribed to νNN indicates that N2 loss likely occurs upon protonation –
unsurprising since the N2 ligand appears labile even in unprotonated [pMo](N2) since it is easily
displaced by THF.
Reaction of pMo(CO) with acid:
Addition of one equivalent of [2,6-lutidinium]BAr′4 to [pMo](CO) in PhF results in the
appearance of a new absorption band at 2000 cm-1 – an increase of 115 cm-1 from νCO for
[pMo](CO). Performing the same addition to [pMo](13CO) gives a new absorption band at 1955
cm-1, also an increase of 115 cm-1 from the original ν13CO. Both these new absorption bands are
120
Chapter 4
assigned to ν12CO and ν13CO of the protonated [pMo](CO) complexes. The blue-shift of νCO of
[pMo](CO) upon protonation is much larger than for [Mo]CO where the increase is only 47 cm-1
to give a new absorption band at 1932 cm-1. The position of the absorption bands of the [pMo]
and [Mo] complexes are presented in Table 4.5
Compound
νCO(cm-1)
[Mo](CO)
1885
[pMo](12CO)
1883
[pMo](13CO)
1840
[[Mo](CO)]H+
1932
{[pMo](12CO)}H+
2000
{[pMo](13CO)}H+
1955
Table 4.5 Values of νCO for Mo carbonyl complexes
Comparison of acid susceptibility of [pMo](CO) and [Mo](CO):
Fluorobenzene solutions of [pMo](CO) and [Mo](CO) with identical starting
concentrations of the Mo complex were exposed to increasing equivalents of [2,4,6collidinium]BAr′4 and the resulting areas of their respective νCO absorption bands were measured
as an indicator of how much either compound remained unprotonated. The carbonyl complexes
were chosen rather than the dinitrogen complexes because the carbonyl complexes exhibit a
much stronger IR absorption band. Also, CO is less labile than N2, and is not so easily lost from
the apical site upon protonation of the molybdenum complex; therefore we can observe the
growth of the new νCO band. From Figure 4.10, it appears that [pMo](CO) is more susceptible to
addition of acid than [Mo](CO), as can be seen from its sharper decrease in νCO. Moreover, the
121
Chapter 4
area of the new absorption bands at 1932 cm-1 and 2000 cm-1 (νCO for protonated [Mo](CO) and
[pMo](CO) respectively) increases more steeply for [pMo](CO), as shown in Figure 4.11. When
3.5 equivalents of acid were present, we see that νCO for [pMo](CO) has almost completely
disappeared whereas that for [Mo](CO) has not. This difference in νCO decrease is attributed to
decomposition of the species by the ligand coming off Mo and loss of CO from the metal center.
Figure 4.10 Area of νCO against equivalents of [Collidinium]BAr'4
122
Chapter 4
Figure 4.11 Area of νCO against equivalents of [Collidinium]BAr'4
Catalytic runs:
Slow addition of Cp*2Cr solution via a syringe pump (~ 5 μmol/h) to a stirred mixture of
{[pMo](NH3)}BAr'4 and [2,6-lutidinium]BAr′4 in heptane yielded one equivalent of NH3 (per
equivalent of [pMo(NH3)]BAr′4) which is presumably derived from the NH3 ligand in the apical
pocket. Therefore, {[pMo](NH3)}BAr'4 is not catalytic for the reduction of dinitrogen to
ammonia. A postulated reason for the lack of catalytic activity is the conversion of [pMo](NH3)
to [pMo](N2), which as mentioned earlier, is considerably slower than for the conversion of
[Mo](NH3) to [Mo](N2). On top of this, the equilibrium appears to favor [pMo](NH3) over
[pMo](N2).
Performing a similar experiment on [pMo](N2) yielded 0.1 equivalents of NH3,
suggesting that the reason for lack of catalytic activity is not limited to NH3 exchange for N2.
[pMo]NNH is a very unstable species and converts almost immediately to [pMo](N2) upon
formation, which would prevent the [pMo] system from being catalytic for dinitrogen reduction.
123
Chapter 4
Despite the fact that like in [HIPTN3N]Mo complexes, [pMo] complexes have three sterically
bulky HIPT substituents, these substituents in [pMo] exhibit much larger torsion angles (CipsoNamido-Mo-X) which results in a more open apical cavity. Based on previous work, we surmise
that steric protection for the cavity is crucial not just to prevent the formation of bimetallic
complexes, but also for the stability of the diazenido species.12
Another problem is simply loss of the ligand in [pMo] in the presence of acid. This ligand
is very susceptible to protonation at one of the amide nitrogens and possibly also the central
amine donor when it comes off the metal. The tilting of the HIPT substituents (and as such the
twisting of the lone pairs of the amide nitrogens away from the plane amide plane) probably
causes the difference in the reactivity of [pMo] complexes. Since these lone pairs are no longer
orthogonal to the dxz and dyz orbitals there may be some occurrence of orbital mixing which
alters the reactivity and stability of these compounds. The solid state structure of
[(Me3Si)trpn]TiCl also displays the formation of “chair form” TiN2C3 rings like in pMo
complexes, causing the trimethylsilyl groups to be tilted more than in complexes with a TREN
backbone (the torsion angle increases by 30° compared to that in [TMSN3N]VCl).6,14
Conclusions
A new system of triamidoamine Mo complexes, [HIPTtrpn]Mo, has been synthesized and
studied. A series of complexes, [pMo]Cl, [pMo]NNK, [pMo](N2), {[pMo](NH3)}BAr'4 and
[pMo]N were synthesized and characterized. This system is not catalytic for the reduction of
dinitrogen to ammonia. The lack of catalytic activity is postulated to be due to the tilting of the
HIPT substituents which opens up the central cavity, leading to intermediates in the cavity being
less stable than in the parent system. Also, the exchange between [pMo](NH3) and [pMo](N2)
124
Chapter 4
may be too slow. The tilting of the HIPT substituents also causes the amido nitrogen lone pairs to
tilt and donation to the metal center is thought to be less effective.
Experimental
General.
All experiments involving air or moisture sensitive complexes or reactions were
performed under nitrogen in a Vacuum Atmospheres drybox or using standard Schlenk
techniques with glassware stored in an oven at ~190 °C for at least twelve hours prior to use.
Pentane was washed with HNO3/H2SO4 (5:95 by volume), sodium bicarbonate, and water, dried
over CaCl2, and then sparged with nitrogen and passed through an alumina column followed by
storage over Na/benzophenone and vacuum transfer prior to use. Dry and deoxygenated benzene
was purchased from Aldrich and passed through Q5 and alumina columns. Heptane, benzene-d6,
and toluene-d8 were dried over Na/benzophenone then degassed (freeze-pump-thaw) and vacuum
transferred prior to use. THF, diethyl ether, and toluene were dried by passage through an
alumina column and stored over Na/benzophenone. They were degassed (freeze-pump-thaw)
and vacuum transferred prior to use. Dichloromethane and benzene were dried by passage
through an alumina column. Fluorobenzene was distilled twice from P2O5. All solvents were
stored over 4 Å molecular sieves in a drybox after transfer.
Pd2(dba)3 (Strem), NaO-t-Bu (Aldrich), (Me3Si)2NLi (sublimed) (Strem), anhydrous
ZnCl2 (Aldrich) (Purified by dissolving in diethyl ether and adding 1 equivalent of 1,4-dioxane
to give ZnCl2(dioxane).), Cp*2Cr (sublimed), Et3N (distilled from CaH2, stored over 4Å
molecular sieves) (Aldrich), MoCl5 (Strem), NH3 (BOC, condensed onto Na sand),
15
N2 (CIL),
CO (BOC, Aldrich) and 13CO (CIL) were used as obtained unless indicated otherwise. HIPTBr2b,
MoCl4(THF)215 and [H(OEt2)2]BAr'416 were synthesized as referenced. [Et3NH] BAr'4 and [2,4,6Collidinium] BAr'4 were prepared by reacting the respective acid chloride with a slight excess of
125
Chapter 4
NaBAr'4. IR spectra were recorded on a Nicolet Avatar 360 FT-IR spectrometer in 0.2 mm KBr
solution cells. 1H NMR spectra were obtained on a Varian (300 or 500 MHz) or Bruker (400
MHz) spectrometer and were referenced to the residual protio-solvent peak. Elemental analyses
were performed by H. Kolbe Microanalytics Laboratory, Mulheim an der Ruhr, Germany.
[HIPTtrpn]MoN=NK. In a N2-filled glove-box, a 50 mL round-bottomed flask equipped with a
glass stir bar was charged with pMoCl (1.00 g, 0.568 mmol), KC8 (200 mg, 1.476 mmol) and
benzene (5 mL). The flask was sealed with a glass stopper and the mixture stirred for 3 days,
then filtered through celite. Volatiles were removed from the filtrate in vacuo and pentane was
added. The resulting solution was kept at -20 °C for 5 hours and the product was collected on a
frit, and washed with 10 mL of cold pentane, yielding a lime-green solid. Yield: 396 mg, 0.221
mmol, 38.9% IR (C6D6): 1810 cm-1, (νNN). 1H NMR (C6D6, 20 °C): δ 7.19 (s, 6H, 2’, 6’-H), 7.16
(obscured, 3,5,3”,5”-H), 6.69 (3H, 4’-H), 3.58 (septet, 3H, 2,6,2”,6”-CHMe2), 3.29 (septet, 6H,
2,6,2”,6”-CHMe2), 3.11 (septet, 3H, 2,6,2”,6”-CHMe2), 2.93 (septet, 3H, 4,4”-CHMe2),
2.48
(septet, 3H, 4,4”-CHMe2), 1.74 (t, 6H, NCH2), 1.51 (t, 6H, NCH2), 1.35 (m, 76H, 2,6,2”,6”CH(CH3)2), 1.34 (m, 36H, 4,4”-CH(CH3)2), 1.10 (s, br, 6H, NCH2CH2CH2N) ppm.
[HIPTtrpn]Mo15N=15NK. In a N2-filled glove-box, a 50 mL solvent bulb with a PTFE screw
valve was charged with pMoCl (61.0 mg, 0.035 mmol) and KC8 (12.2 mg, 0.090 mmol) and a
glass coated magnetic stir bar. The flask was evacuated, and then benzene (freeze-pump-thaw
degassed 3 times) was vacuum-transferred into the solvent bulb. The solvent bulb was
pressurized to 1 atm with
15
N2 purified with solid Na/Ph2CO and the mixture stirred at room
temperature for 48 hours. Volatiles were removed in vacuo and an IR spectrum of the residue
was taken with heptane as the solvent. IR (heptane): 1760 cm-1, (ν15N15N)
126
Chapter 4
[HIPTtrpn]Mo(N2). In a N2-filled glovebox, a scintillation vial equipped with a glass coated
magnetic stir bar was charged with pMoCl (500 mg, 0.284 mmol), KC8 (0.100 mg, 0.738 mmol)
and benzene (5 mL). The mixture was stirred for 48 hours, filtered through Celite and the
filtrates dried in vacuo. To the filtrate was added ZnCl2(dioxane) (64.4 mg, 0.286 mmol) and
diethyl ether (5 ml) and the mixture stirred for 45 minutes. An immediate color change from
orange brown to dark purple occurred. Volatiles were removed in vacuo and the residue
extracted with pentane, filtered through celite and decreased to 2 mL. The solution was placed in
the freezer for several days. Microcrystalline purple precipitate was collected on a glass frit and
dried. Yield: 73 mg, 15%. IR (C6D6): 2030 cm-1 (νNN). 1H NMR (C6D6, 20°C): δ 6.51 (br, s,
12H, 3,5,3,5”-H), 3.19 (br, sh, 12H, 2,6,2,”6”-CHMe2), 2.90 (s, 3H, 4’-H), 2.79 (br, s, 6H, 4,4”H), 1.44 (br, sh, 48H, -NCH2, 4,4”-CH(CH3)2), 1.30 (m, 78H, 2,6,2”6”-CH(CH3)2), 2’6’-H), 0.88
(m, 6H, NCH2CH2) ppm.
{[HIPTtrpn]Mo(NH3)}[BAr′4]. In a N2-filled glovebox, a 100 mL solvent bulb was charged
with pMoCl (760 mg, 0.432 mmol), NaBAr′4 (600 mg, 0.677 mmol) and 10 mL PhF. The
mixture was freeze-pump-thaw degassed 3 times, and NH3 (100 mL, 1 atm, 4.089 mmol) dried
over Na was vacuum-transferred into the bulb. The mixture was stirred for 4 hours, volatiles
were removed in vacuo at 60 °C. The residue was extracted with pentane, filtered through celite
and the filtrates concentrated to 4 mL. The filtrates were placed at -25 °C overnight and
formation of a reddish orange solid occurred. The product was isolated by filtration, washed with
cold pentane and dried in vacuo, turning red. Yield: 872 mg, 0.323 mmol, 74.8%. 1H NMR
(C6D6, 20 °C):δ 167.06 (s, 1H, NH3), 26.13 (br, s, NCH2CH2CH2N), 23.03 (br, s,
NCH2CH2CH2N), 9.48 (br s, 4’6’-H), 8.20 (s, 8H, C6H3-3,5-(CF3)2-2,4-H), 7.59 (s, 4H, C6H3127
Chapter 4
3,5-(CF3)2-2,4-H), 7.40 (s, 6H, 3,5,3”,5”-H), 7.07 (s, 6H, 3,5,3”,5”-H), 2.80 (s, 6H, 4,4”CHMe2), 2.61 (m, 12H, 2,6,2”,6”-CHMe2), 1.21, 1.02 (overlapping) ppm. Anal Calcd. for
C149H180BF24MoN5: C, 68.73; H, 6.97; B, 0.42; F, 17.51; N, 2.60. Found: C, 68.54; H, 7.08; B,
0.42; F, 17.56; N, 2.60
{[HIPTtrpn]Mo(NH3)}BPh4. In a N2-filled glovebox, a 100 mL solvent bulb was charged with
pMoCl (555 mg, 0.315 mmol), NaBPh4 (119 mg, 0.347 mmol) and 10 mL PhF. The mixture was
freeze-pump-thaw degassed 3 times, and NH3 (100 mL, 1 atm, 4.089 mmol) dried over Na was
vacuum-transferred into the bulb. The mixture was stirred overnight and volatiles were removed
in vacuo at 60 °C. The residue was extracted with pentane, filtered through celite and the filtrates
concentrated to 2 mL. The filtrates were placed at -25 °C for a few weeks and formation of a
reddish purple solid occurred. The product was isolated by filtration, washed with cold pentane
and dried in vacuo. Yield: 130 mg, 0.063 mmol, 20%. 1H NMR (C6D6, 20 °C): δ 180.00 (br s,
1H, NH3), 28.57 (br s, 2H, NCH2CH2CH2N), 24.79 (br s, 2H, NCH2CH2CH2N), 9.02 (s, 3H, 2’H), 8.00 (s, 8H, -B(C6H5)4 ), 7.47 (br s, 3,5,3”,5’-H), 2.89 (br s, 2,6,2”,6”-CHMe2), 2.76 (br s,
4,4”-CHMe2), 1.30 (br, 72H, 2,6,2”,6”-CH(CH3)2), 1.14 (br s, 36H, 4,4”-CH(CH3)2) ppm.
Anal.Calcd. for C141H188BMoN5: C, 82.22; H, 9.20; N, 3.40. Found: C, 82.05; H, 9.35; N, 3.29.
[HIPTtrpn]Mo(CO). In a N2-filled glovebox, A scintillation vial equipped with a glass coated
stirbar was charged with [HIPTtrpn]MoCl (500 mg, 0.284 mmol) and KC8 (95.9 mg, 0.710
mmol) and benzene (5 mL). The mixture was stirred for 2 days then the benzene removed in
vacuo and the mixture extracted with pentane and filtered through celite. The pentane was
removed in vacuo and Et2O was added (5 mL). ZnCl2(dioxane) (94.5 mg, 0.426 mmol) was
added to the Et2O solution and the mixture stirred with a glass-coated stirbar for 30 minutes.
128
Chapter 4
Et2O was removed in vacuo, the residue extracted with pentane and filtered through Celite. The
filtrate was decreased to 10 mL and placed in a 100 mL solvent bulb equipped with a PTFE
screw valve. The bulb was sealed, brought out of the glove box and freeze-pump-thaw degassed
thrice. It was then filled with one atm of CO (90 mL, 3.68 mmol) and stirred overnight at room
temperature. Volatiles were removed in vacuo at 60 °C and the reaction flask brought back into
the glovebox. Dissolving the residue in pentane and recrystallization at -25 °C overnight afforded
dark blue powder which was collected on a glass frit. Yield: 215 mg, 43%. 1H NMR (C6D6): δ
6.36 (br s), 3,34 (v br s), 2,79 (br s), 1.52 (br s), 1.32 (br s), 1.28 – 1.22 (overlapping peaks), 0.88
(t) ppm. IR (PhF): 1883 (ν12CO), 1840 (ν13CO).
References
1
(a) Ritleng, V.; Yandulov, D. V.; Weare, W. W.; Schrock, R. R.; Hock, A. R.; Davis, W. M., J. Am.
Chem. Soc. 2004, 126, 6150. (b) Weare, W. W.; Schrock, R. R.; Hock, A. S.; Müller, P. Inorg. Chem.
2006, 45, 9185. (c) Weare, W. W.; Dai, C.; Byrnes, M. J.; Chin, J.; Schrock, R. R. Proc. Nat. Acad. Sci.
2006, 103, 17099.
2
(a) Yandulov, D. V.; Schrock, R. R. J. Am. Chem. Soc. 2002, 124, 6252. (b) Yandulov, D. V.; Schrock,
R. R.; Rheingold, A. L.; Ceccarelli, C.; Davis, W. M. Inorg. Chem. 2003, 42, 796. (c) Yandulov, D. V.;
Schrock, R. R. Science 2003, 301, 76. (d) Yandulov, D.; Schrock, R. R. Inorg. Chem. 2005, 44, 1103. (e)
Schrock, R. R. Acc. Chem. Res. 2005, 38, 955.
3
Smythe, N. C.; Schrock, R. R.; Müller, P.; Weare, W. W. Inorg. Chem. 2006, 45, 7111.
4
Yandulov, D. V.; Schrock, R. R. Canad. J. Chem. 2005, 83, 341.
5
Smythe, N. C.; Schrock, R. R.; Müller, P.; Weare, W. W. Inorg. Chem. 2006, 45, 9197.
6
Schrock, R. R.; Cummins, C. C.; Wilhelm, T.; Kol, M.; Lin, S.; Reid, S.; Davis, W. M. Organometallics
1996, 15, 1470.
129
Chapter 4
7
Weare, W.W. PhD Thesis 2006.
8
Greco, G. E.; Schrock, R. R. Inorg. Chem. 2001, 40, 3850.
9
Greco, G.E.; Schrock, R.R. Organometallics. 1998, 17, 5591.
10
Byrnes, M.J.; Dai, X.; Schrock, R.R.; Hock, A.S.; Müller, P. Organometallics 2005, 24, 4437.
11
Byrnes, M.J. Unpublished results.
12
Weare, W.W.; Schrock, R.R.; Hock, A.S.; Müller, P. Inorg. Chem. 2006, 45, 9185.
13
Yandulov, D.V.; Schrock, R.R. Inorg. Chem. 2005, 44, 1103.
14
Cummins, C.C.; Schrock, R.R.; Davis, W.M. Organometallics, 1992, 11, 1452.
15
Stoffelbach, F.; Saurenz, D.; Poli, R. Eur. J. Inorg. Chem. 2001, 10, 2699.
16
Brookhart, M.; Grant, B.; Volpe, A. F., Jr. Organometallics 1992, 11, 3920.
130
Chapter 5
Chapter 5
Continuing syntheses and studies on [HIPTN3N]Mo and [HIPTN3N]W
complexes
131
Chapter 5
Introduction
Although
the N2
chemistry of
Mo
complexes containing a
triamidoamine
([(ArNCH2CH2)3N]3- = [ArN3N], Ar = Aryl) ligand has been explored extensively by our group,
there are still some areas that remained to be studied. We have shown that catalytic reduction of
N2 at a Mo center can be achieved by the [HIPTN3N]Mo system. However, this system is still
limited to only four turnovers.1 The reduction of N2 to NH3 through the sequential addition of
protons and electrons requires at least twelve steps (assuming that each proton or electron
transfer is one step), which shows how complicated and finely balanced this overall reaction
must be, especially if it were to occur without the harsh conditions of the Haber Bosch process.
This is evidenced by the fact that in our [ArN3N]Mo systems, any small steric changes reduce or
shut down catalytic reduction. It is therefore important to learn about the mechanistic details of
dinitrogen reduction at [HIPTN3N]Mo. Studies of the [HIPTN3N]W system described in this
chapter also continue on the work of previous group members.2
Results and discussion
Oxidation of [Mo](N2) and [W](N2)
Earlier work showed that oxidation of the [Mo](N2) ([Mo] = [HIPTN3N]Mo) complex by
[Cp2Fe]BAr4' (Ar' = 3,5-(CF3)2C6H3) in C6D6 gave a species which showed no observable
absorption band for the N2 stretch in the expected IR range in C6D6.3 However, oxidation of the
[Mo](15N2) complex (ratio of
15
N2/14N2 = 92:8) by [Cp2Fe]BAr4' and its subsequent reduction
back to the neutral species after approximately twenty minutes under N2 atmosphere gave a ratio
of
15
N2/14N2 of 18:82.4 A proposed explanation for the lack of observable νNN is that in this
species the N2 molecule may exhibit side-on binding (Figure 5.1) instead of the terminal binding
132
Chapter 5
found in the [Mo](N2) complex or that there may be no bound N2 in the apical cavity of
{[Mo](N2)}+.
Figure 5.1 Hypothetical {[Mo](N2)}+ species with side-on bound N2
Earlier cyclic voltammetry studies have shown that [W](N2) undergoes a reversible
electrochemical oxidation at the same potential as [Mo](N2) (–0.66 V with respect to Cp2Fe+/0).2,
5
However {[W](N2)}+ is expected to be more stable than {[Mo](N2)}+, since the W metal center
is more electron rich than the Mo center. Therefore, back-donation from W into the N2 ligand
should be greater than from Mo, binding N2 more tightly to the metal center and increasing the
chances of observing νNN. {[W](N2)}+ was synthesized for comparison by oxidation of [W](N2)
with [Cp2Fe]BAr'4. IR spectroscopy of {[W](N2)}+ in C6D6 (Figure 5.2) revealed an absorption
band at 2112 cm-1 (assigned to νNN), an increase of 224 cm-1 from the absorption band of
[W](N2) (νNN = 1888 cm-1) This result suggests that N2 in the {[W](N2)}+ species is terminally
bound.
133
Chapter 5
Figure 5.2 IR spectrum of [W](N2) before oxidation (solid line) and after oxidation (dashed line)
Based on the position of νNN for {[W](N2)}+, νNN of {[Mo](N2)}+ was extrapolated to
occur at ~ 2210 cm-1. C6D6 and toluene-d8 were therefore replaced by heptane in IR studies of
{[Mo](N2)}+ since heptane did not display strong absorption bands in that region. The IR
spectrum of {[Mo](N2)}+ in heptane showed the appearance of a peak at 2255 cm-1, which is
assigned to νNN of {[Mo](N2)}+ (Figure 5.3). Since the N2 ligand is expected to have a very small
dipole moment in {[Mo](N2)}+ due to its weak activation by Mo, and as such would only couple
weakly to IR radiation, it is unsurprising that this absorption band at 2255 cm-1 is weak
compared to νNN in [Mo](N2). In comparison, νNN of free N2 in the Raman spectrum is 2331 cm-1.
This peak was not observed in earlier work due to overlap with toluene-d8 absorption bands,
where there is a large band at ~ 2210 cm-1. The νNN suggests that N2 is terminally bound in both
the {[Mo](N2)}+ and {[W](N2)}+ species. Also, subsequent addition of THF to the solution of
{[Mo](N2)}+ led to the disappearance of the band at 2255 cm-1.
134
Chapter 5
Figure 5.3 IR spectra of [Mo](N2) (solid line), [Mo](N2) after Cp2FeBAr'4 addition (dotted line)
and[Mo](N2) after Cp2FeBAr'4 and then THF addition (dashed line)
Synthesis of [W]CO
We were interested in the synthesis of [W](CO) firstly as a comparison with [Mo](CO)6
and secondly, as a possible standard in IR solution kinetics since we expected the compound to
exhibit a strong absorption band in the IR region. [W](N2) was reacted with CO overnight at 50
°C (Figure 5.4), giving a crystalline, emerald green compound that shows a strong absorption at
1851 cm-1 in heptane (ν13CO = 1813 cm-1) in the IR spectrum. Since W should be a better backdonor to CO than Mo, the lower energy absorption band at 1851 cm-1 compared to νCO for Mo
(1888 cm-1 in C6D6) is expected. Combustion analyses also support this assignment. Given that
the absorption band does not overlap with the absorption band of [Mo](N2) (1994 cm-1 in
heptane), [W](CO) could ostensibly be utilized as a standard in IR solution kinetics studies for
the exchange of [Mo](NH3) to [Mo](N2).
135
Chapter 5
Figure 5.4 Formation of [W]CO
Kinetics of [Mo](NH3) conversion to [Mo](N2)
Figure 5.5 Chatt-like cycle for reduction of dinitrogen to ammonia on a Mo center
The Chatt-like catalytic cycle of dinitrogen reduction on a Mo center is shown above
(Figure 5.5), where the boxed intermediates have been isolated and characterized. One of the
slow steps in this cycle is the loss of NH3 from [Mo](NH3) and its replacement by N2. We were
interested in investigating the possible acceleration of this conversion in the presence of a Lewis
acid trap for NH3. The kinetics of the conversion of [Mo](NH3) to [Mo](N2) in the presence of
136
Chapter 5
varying equivalents of a Lewis acid, BPh3 was studied by measuring the area of the [Mo](N2)
νNN stretch at 1990 cm-1 against time via IR spectroscopy.
Earlier work showed that t½ of the exchange of [Mo](NH3) to [Mo](N2) was about 104 to
117 minutes when the solution is stirred,3, 7 but in the absence of stirring t½ was found to be 179
minutes (Figure 5.6).
Area of N2 absorption band against Time
4.5
4
3.5
3
Area νNN
2.5
2
1.5
1
0.5
0
0
200
400
600
800
1000
1200
Time (Min)
Figure 5.6 Area of νNN of Mo(N2) against time. t½ = 179 minutes
When ten equivalents of BPh3 were present in a solution of [Mo](NH3) under N2
atmosphere, t½ = 39 minutes (Figure 5.7). It seems that NH3 that is lost from [Mo](NH3) and
remains in solution back-reacts with [Mo](N2) and this process goes forwards and backwards
until the NH3 leaves the
solution. Presumably, BPh3 increases the rate of conversion of
[Mo](NH3) to [Mo](N2) by trapping NH3. This would help explain the longer t½ observed in the
absence of BPh3 and stirring. Also, it was found that a decrease in the stirring rate during a
catalytic run leads to a one to two equivalent drop in the yield of ammonia,8 presumably because
increasing the stirring rate accelerates NH3 loss from solution.
137
Chapter 5
Area of MoN2 peak against Time, 10 equiv BPh3
6
5
Area νNN
4
3
2
1
0
0
200
400
600
800
Time (min)
1000
1200
1400
Figure 5.7Area of νNN of Mo(N2) against time.The half-life, t½ = 39 minutes in presence of ten equivalents
of BPh3.
To determine if BPh3 is able to abstract NH3 from [Mo](NH3), a mixture of BPh3 and
[Mo](NH3) under static vacuum was monitored over three days by 1H NMR and showed little
change, suggesting that NH3 abstraction does not occur.
Conversion of [Mo](N2) to [Mo](CO)
It had earlier been observed that [Mo](15N2) converts to [Mo](14N2) with t½ ca. 35 hours
through a dissociative process and the rate-determining step is the formation of the naked [Mo]
species.5 We had thought that since only three frontier orbitals (one σ and two π-orbitals) are
easily accessible in the apical cavity, and that 15N2 for 14N2 exchange would require at least four
orbitals (two σ and two π-orbitals), therefore the exchange had to go through dissociation
process.4 Given that like N2, CO is also both a σ-donor and a π-acid ligand, presumably
conversion of [Mo](N2) to [Mo](CO) may also involve formation of the naked species as the
rate-determining step. If this was true, then t½ for [Mo](CO) formation would be ca. 35 hours as
138
Chapter 5
well. Studies of this conversion were therefore undertaken to verify this hypothesis. A degassed
solvent bulb containing [Mo](N2) in C6D6 was exposed to CO and IR spectra of aliquots of this
reaction mixture were taken periodically.
Area of CO Absorption Peak Against Time
6
Area νCO
5
4
3
2
1
0
0
200
400
600
800
1000
1200
Time (min)
Figure 5.8 Area of νCO of [Mo](CO) against time (min) at 0.44 atm pressure of CO
Exposure of an 11.7 mM solution of [Mo](N2) to CO (0.44 atm, ~70 equivalents vs.
[Mo](N2)) led to rapid formation of [Mo](CO) and was too fast for accurate determination of t½
by IR spectroscopy (Figure 5.8). Within a period of 42 minutes, virtually all [Mo](N2) had
converted to [Mo](CO) and assuming pseudo first order kinetics, t½ was approximately seven
minutes. This suggests that the conversion occurs by an associative rather than dissociative
process.
Exposure of [Mo](N2) (5.84 mM) to 0.09 atm of CO (~thirteen equivalents) gave a
pseudo first-order t½ of sixteen minutes (Figure 5.9), showing that the rate is indeed dependent
on CO concentration. The conversion of [Mo](N2) to [Mo](CO) is therefore associative.
139
Chapter 5
Figure 5.9 Area of νCO of [Mo](CO) and νNN of [Mo](N2) against time (min) at 0.09 atm CO, where νCO is
shown in red, νNN is in blue.
Studies of [Mo](N2) reactivity with acid
When [Mo](N2) is exposed to two equivalents of [2,6-LutH]BAr'4 in PhF, a new species
is observed by IR spectroscopy with νNN at 2057 cm-1,9 possibly due to protonation of an amide
nitrogen, hence lowering the electron density at Mo and decreasing back-bonding to N2.
Protonation of the metal center instead of an amide nitrogen could not be precluded, since this
would also lead to a hypsochromic shift for νNN. Another possibility is hydrogen-bonding of the
acid to Nß of the N2 ligand, since we are unsure as to how this would affect νNN.
140
Chapter 5
HB+
N
HIPT
N
HIPT
N
HIPT
Mo
N
N
N
Figure 5.10 Hypothetical hydrogen bonded HB adduct. B is the conjugate base of the acid
IR spectroscopy studies were therefore carried out on the reaction of [Mo](N2) with acid.
Reacting [Mo](N2) with [2,6-LutD]BAr'4 did not result in the appearance of a novel absorption
band ascribable to νNN besides the peak at 2057 cm-1, showing that proton transfer, if it does
occur, does not lead to protonation of the N2 ligand. To verify that the new peak at 2057 cm-1
was indeed due to N-N stretching, [Mo](15N2) was reacted with [2,6-LutH]BAr'4 which led to
the appearance of a new peak at 1990 cm-1 and the decrease of the original νNN at 1924 cm-1. The
similar shift of 66 cm-1 verifies that the new peak at 2057 cm-1 is indeed due to N-N vibration.
[Mo](CO) was also reacted with [2,6-LutH]BAr'4 and it was shown that a new peak
ppears at 1932 cm-1 which is assigned to νCO. This represents an increase of 47 cm-1 and supports
the idea that a proton transfer reaction is involved rather than the formation of a hydrogenbonded adduct which presumably would not form with the oxygen atom of the CO ligand, since
unlike Nß in an N2 ligand, the oxygen atom has a partial positive charge.
It has been shown that [Mo]CN reacts with B(C6F5)3, to form the putative
{[Mo]CN→B(C6F5)3)} adduct complex.6 It was thought that perhaps [Mo](CO) and [Mo](N2)
would react similarly. Since B(C6F5)3 is a Lewis acid, formation of the adduct may give some
insight into the properties of a hydrogen-bonded adduct. However, neither [Mo](CO) nor
[Mo](N2) reacts with B(C6F5)3.
Since addition of two equivalents of [2,6-LutH]BAr'4 to [Mo](N2) only led to
approximately 30% conversion to the new species,9 an experiment was conducted where
141
Chapter 5
increasing amounts of [2,6-LutH]BAr'4 were added to [Mo](N2) to determine how much [2,6LutH]BAr'4 was required to completely convert [Mo](N2) to the new species and to see the effect
of increasing [2,6-LutH]BAr'4 on [Mo](N2) (Table 5.1).
Equivalents of H+ Area of νNN at 1990 cm-1 Area of νNN at 2057cm-1
0
6.638
0.176
1
5.329
1.722
2
3.272
2.093
3
1.847
2.453
4
1.361
2.651
5
0.776
2.552
6
0.924
2.472
7
0.537
2.545
8
0.638
2.024
Table 5.1 Area of νNN with increasing acid
Area νNN
Area of N2 peak against Equivalents of Acid
7
6
5
4
3
2
1
0
0
1
2
3
4
5
6
Equivalents of Acid
vNN at 1990 cm-1
vNN at 2057 cm-1
Figure 5.11 Area of N2 peak with increasing [2,6-LutH]BAr'4 addition
142
Chapter 5
As can be seen from Table 5.1, the area of the peak at 2057 cm-1 increases with
increasing equivalents of [2,6-LutH]BAr'4 but starts to decrease after five equivalents is reached.
Increasing acid concentration increases formation of the new species but increases
decomposition of this new species by causing ligand loss via protonation.
If indeed the acid was hydrogen-bonded to [Mo](N2) in the new species, changing the
conjugate base of the acid should change the position of the observed νNN. However, if the
reaction was a proton-transfer, then the area of νNN for the new complex should vary with
varying pKa of the acid used. One equivalent of acid was added to the same amount of [Mo](N2)
solution (taken from a stock [Mo](N2) solution) and the results are shown in Table 5.2.
Acid
νNN (1990 cm-1) νNN (2057 cm-1)
[2,6-LutH]BAr'4
16.618
2.850
[2,4,6-CollH]BAr'4
18.842
1.687
[Et3NH]BAr'4
21.201
0.653
Table 5.2 Area of νNN upon addition of different acids
The position of the new absorption band does not shift from 2057 cm-1 when the
conjugate base of the acid is changed. As can be seen from Table 5.2, the amount of formation of
the new species is lowest for [Et3NH]BAr'4, the weakest acid, and highest for [2,6-LutH]BAr'4,
the strongest acid. We thus conclude that the new complex is indeed a protonated species and not
a hydrogen-bonded adduct.
[H(OEt2)2]BAr'4 was reacted with [Mo](N2) since it should only react via a proton
transfer and not through formation of a hydrogen bonded HBase adduct. Half an equivalent of
[H(OEt2)2]BAr'4 was added to [Mo]N2 at – 40 °C and the reaction mixture allowed to warm up
143
Chapter 5
slowly. However, IR spectroscopy only showed the presence of the νNN at 1990 cm-1, indicating
that [H(OEt2)2]BAr'4 addition only led to decomposition rather than proton transfer.
Moreover, hydrogen-bonding of acid to N2 should result in a lowering of the νNN because
the HOMO of N2 in [Mo](N2) is a bonding molecular orbital. Also, the reduced mass of the
oscillator should be increased by the formation of a hydrogen bonding adduct, which would also
bathochromically shift νNN as well. Thus, νNN at 2057 cm-1 is most likely due to [Mo](N2) with a
protonated amide nitrogen or metal center.
Studies of effect of base on [Mo]NNH decomposition
[Mo]NNH decomposes when heated at 61 °C to [Mo]H and this decomposition is
catalyzed by the presence of acid.5 The mechanism of this decomposition is unknown and it has
been postulated to occur perhaps by a form of β-hydride elimination. However, we are unsure as
to why the presence of acid would accelerate this decomposition. We were curious as to whether
the decomposition process could also be catalyzed by base as catalytic runs employing many
equivalents of [2,6-LutH]BAr'4 or [2,4,6-CollH]BAr'4 invariably contain some equivalents of
their conjugate bases 2,6-lutidine and 2,4,6-collidine at the end of the run. Also, since the acidcatalyzed decomposition observed earlier by Yandulov was based on [Et3NH]+ acid,5 we were
interested in whether it might actually be its conjugate base which catalyzed the decomposition.
N-methylmorpholine was also one of the bases included in the study because it is of a similar
basicity as 2,6-lutidine, but unlike 2,6-lutidine, should not be able to accept a H· radical. Samples
of [Mo]NNH with 1% base and diphenylmethane as the proton standard (δ 3.74 ppm) were
prepared in J. Young tubes and 1H NMR was used to monitor the decomposition rate by
measuring the [Mo]NNH proton peak (δ 8.57 ppm).
Unfortunately, 1H NMR showed that the area of the [Mo]NNH proton peak fluctuated
greatly with time and it was suggested that this may be due to the delay time in the collection of
144
Chapter 5
NMR data. Unfortunately, changing the delay time did not remove the fluctuation. Despite the
fluctuations in the area of the [Mo]NNH proton peak, it can be seen that the various bases do not
appear to catalyze the decomposition of [Mo]NNH at 0.01 equivalents of base (Figure 5.12) and
even on going to ten equivalents of base (Figure 5.13), we see little difference between the bases
containing samples and the control sample.
Area
Area of MoNNH Hydride Peak against Time
(0.01 equiv of base)
1.3
1.25
1.2
1.15
1.1
1.05
1
0.95
0.9
0.85
0.8
Control
2,4,6-collidine
2,6-lutidine
N-methyl morpholine
Et3N
0
2000
4000
6000
Time (min)
Figure 5.12 Plot of [Mo]NNH proton peak against Time. 0.01 equivalents of base were utilized. Delay
time = two seconds.
145
Chapter 5
MoNNH Hydride Peak against Time
(10eq. Base)
Area
1.6
1.4
Control
1.2
2,4,6-collidine
2,6-Lutidine
1
0.8
N-methyl morpholine
0.6
Et3N
0.4
0
2000
4000
6000
Time (min)
Figure 5.13 Plot of [Mo]NNH proton peak against time. Ten equivalents of base were utilized. Delay time
= two seconds.
Synthesis of [HIPTN3N]MoCNH2
The compound [HIPTN3N]MoCN had been earlier reported by our group as an orange
paramagnetic species.6 We were interested in whether the slow addition of protons and electrons
to the compound would lead to formation of [Mo](N2) by first forming ammonia and [Mo]CH
which then may undergo further protonation and reduction to give [Mo](N2). We found that the
slow addition of [2,4,6-Collidinium]BAr'4 and Cp*2Cr to [Mo]CN in a similar manner to in a
catalytic run set-up yielded a major product which we identify as an aminocarbyne complex,
[Mo]CNH2. The compound is diamagnetic, characteristic of a Mo(VI) complex, and exhibits a
singltet carbon resonance in 13C NMR at 242.2 ppm, characteristic of an alkylidyne carbon. For
comparison, the oxycarbyne compound [Mo](13CO)Na exhibits a singlet at 232.5 ppm in
13
C{1H}NMR. Despite the addition of more than nine equivalents of acid and reducing agent to
[Mo]CN, it appears that the reaction stops at [Mo]CNH2, presumably due to the stability of the
Mo-C triple bond in triamidoamine systems.
146
Chapter 5
When [2,6-LutH]BAr'4 and Cp2Co was added slowly over the course of 30 minutes to a
solution of [Mo]CN, the major product formed was again [Mo]CNH2, although this was
accompanied by the formation of various side products. The formation of more side products is
not unexpected since the faster addition of acid could lead to protonation at various sites on the
metal complex and subsequent decomposition products.
Conclusions
It has been shown that both {[W](N2)}+ and {[Mo](N2)}+ can be synthesized and that the
N2 ligand in {[Mo](N2)}+ is easily displaced. A new compound, [W](CO) had been synthesized
and characterized. The exchange of [Mo](NH3) with N2 appears to go backwards and forwards
until NH3 is lost from solution. As such, the stirring rate and presence of a Lewis acid (trapping
agent for NH3) are all factors in the rate of exchange. A higher stirring rate (and hence faster loss
of NH3 from solution) and also presence of BPh3 increases the rate of exchange and formation of
[Mo](N2). [Mo](N2) appears to exchange with CO to form [Mo](CO) through an associative
rather than a dissociative process, as the exchange rate is dependent on the partial pressure of CO
in the atmosphere. Reaction of [Mo](N2) with a Bronsted acid results in a protonated product
rather than a hydrogen-bonded adduct (Figure 5.10). Reaction of [Mo](CO) with a Bronsted acid
also results in a protonated product with a new IR absorption band νCO at 1932 cm-1 (a blue-shift
of 47 cm-1). [Mo]NNH decomposition to [Mo]H does not appear to be facilitated by the presence
of base, as no decomposition was observed compared to the control even after more than 100
hours. A new compound, [Mo]CNH2, was synthesized.
147
Chapter 5
Experimental
General. All air and moisture sensitive compounds were manipulated using standard Schlenk
and glovebox techniques under atmosphere of nitrogen in oven-dried glassware. Ether, pentane,
and toluene were purged with nitrogen and passed through activated alumina columns. Pentane
was freeze-pump-thaw degassed three times and tetrahydrofuran (THF), dimethoxyethane
(DME), benzene, deuterated benzene and toluene were distilled from dark purple
Na/benzophenone ketyl solutions. Dichloromethane and cyclopentene were distilled from CaH2.
Ether, DME, dichloromethane and cyclopentene were stored over molecular sieves in a nitrogenfilled glovebox while pentane, THF, DME, benzene, deuterated benzene and toluene were stored
in Teflon-sealed solvent bulbs in a nitrogen-filled glovebox. Molecular sieves (4 Å) and Celite
were activated at 230 °C in vacuo over several days. (Me3Si)2NLi (sublimed) (Strem) and
anhydrous ZnCl2 (Aldrich) were used as received, unless indicated otherwise. Cp*2Co was
reduced from [Cp*2Co]PF6 (Strem) and purified by sublimation. 2,6-lutidine (Aldrich), 2,4,6collidine (Aldrich), N-methyl morpholine (Aldrich) were dried over Na/benzophenone ketyl and
distilled. Quinuclidine (Aldrich) was purified by sublimation. [Mo]Cl, [Mo](N2), [Mo]NNH,
{[Mo](NH3)}[BAr'4], [Mo](NH3),5 [Mo](CO),6 [W](N2)2 were synthesized as referenced.
ZnCl2(Dioxane). In a nitrogen-filled glovebox, ZnCl2 (34 g, 0.249 mol) was dissolved in 200
mL ether. 1,4-Dioxane (20.3 g, 0.231 mol) was added causing immediate formation of white
precipitate. The precipitate was filtered off, combined with the crop from a previous synthesis
[ZnCl2 (33 g, 0.243 mol), 1,4-dioxane (21 mL, 0.243 mol)] and dried in vacuo overnight at room
temperature. Yield: 78.7 g, 0.351 mol, 73.1%
148
Chapter 5
Oxidation of [HIPTN3N]W(N2). A scintillation vial was charged with [W](N2) (40 mg, 0.0228
mmol) and Cp2FeBAr4’ (41.7 mg, 0.0437 mmol) and 0.8 mL C6D6, giving a green solution. The
mixture was allowed to stir for approximately 2 hrs giving an orange-brown solution. IR (C6D6):
2112 cm-1 (νNN for [W]N2]+), 1888 cm-1 (νNN for [W]N2)
Oxidation of [HIPTN3N]Mo(15N2). A scintillation vial was charged with [Mo]15N2 (44 mg,
0.00151 mmol) and Cp2FeBAr4’ (40 mg, 0.0419 mmol) and toluene. The mixture was stirred for
10 minutes giving a brown solution. IR (toluene): no peaks for νN-N of [[Mo]N2]+
[HIPTN3N]W(CO). In a N2-filled glovebox, a J.Y. tube was charged with [W]N2 (57.1 mg,
0.0317 mmol) and approximately 0.7 mL C6D6 to give a red-brown solution. The sample was
degassed and the J.Y. tube was filled with CO gas (0.88 atm in approx. 1.5 cm3, 0.0540 mmol).
After shaking the J.Y. tube for 5 minutes, the solution became brown-green. The mixture was
heated to 50 °C overnight and C6D6 was then removed in vacuo, leaving a green residue which
was dissolved in heptane for IR spectroscopy. 1H NMR (C6D6): δ 31.1 (br, s, 6H, NCH2), 7.12 (s,
12H, 3,5,3”,5”-H), 6.73 (s, 6H, 4’,6’-H), 2.77 (s, 6H, 4,4”-CHMe2), 2.36 (s, 12H, 2,6,2”,6”CHMe2), 1.21 (d, 36H, 2,6,2”,6”-CH(CH3)2, 0.824 (d, 18H, 4,4”-CH(CH3)2, -17.724 (s, 6H,
NCH2); IR (Heptane): 1851.3cm-1 (νCO) , 1813.3 (ν13CO) Anal Calcd. for C115H159N4OW: C,
76.85; H, 8.92; N, 3.12. Found: C, 76.66; H, 8.86; N, 3.06
[HIPTN3N]MoCNH2: In a N2-filled glovebox, a catalytic apparatus was charged with [Mo]CN
(175 mg, 0.1025 mmol), [2,4,6-collidinium]BAr'4 and heptane. A solution of Cp*2Cr (304 mg,
0.943 mmol) in heptane (10 mL) was added at a rate of 1.7 mL/h with stirring. After 8 hours, the
reaction mixture was dried in vacuo and extracted with pentane and filtered through celite. The
149
Chapter 5
filtrate was chilled overnight at -35 °C. A yellow diamagnetic compound was collected on a glass
frit and dried. Yield: 95 mg, 55% 1H NMR (C6D6): δ 7.44 (s, 6H, 3,5,3”,5”-H), 7.14 (s, 3H,
2’,6’-H), 6.54 (s, 3H, 2’,6’-H), 4.97 (s, 2H, MoCNH2), 3.67 (s, 6H, 3,5,3”,5”-H), 3.14 (septet,
JHH = 7.1 Hz, 12H, 2,6,2”,6”-CHMe2), 2.89 (septet, JHH = 6.8 Hz, 4,4”-CHMe2), 1.32 (d, JHH =
7.3 Hz, -CH3), 1.20 (d, JHH = 7.3 Hz, -CH3), 1.09 (d, JHH = 7.3 Hz, -CH3) ppm.
13
C NMR
(C6D6): δ 242.2 (MoCNH2), 161.8, 148.2, 147.3, 140.8, 138.8, 125.0, 121.1, 120.6, 57.2, 51.9,
35.3, 31.1, 25.5, 25.5, 25.1, 24.9, 24.9 ppm.
References
1
2
Yandulov, D. V.; Schrock, R. R. Science 2003, 301, 76.
Yandulov, D. V.; Schrock, R. R. Can. J. Chem. 2005, 83, 341.
3
Byrnes, M.J. Unpublished results, Final Report, 2005.
4
Weare, W.W.; Dai, X.; Byrnes, M.J.; Chin, J.; Schrock, R.R.; Müller, P. Proc. Nat. Acad. Sci. U.S.A.,
2006, 103, 17101.
5
Yandulov, D. V.; Schrock, R. R.; Rheingold, A. L.; Ceccarelli, C.; Davis, W. M. Inorg. Chem. 2003, 42,
796.
6
Byrnes, M.J.; Dai, X.; Schrock, R.R.; Hock, A.S.; Müller, P. Organometallics 2005, 24, 4437.
7
Weare, W.W. PhD Thesis 2006.
8
Hanna, B.S. Unpublished results.
9
Yandulov, D.V.; Schrock, R.R. Inorg. Chem. 2005, 44, 1103.
150
Appendix 1
\
Appendix 1
Some triamidophosphine ligands and their metal complexes
151
Appendix 1
Introduction
Earlier work in our group has focused on triamidoamine Mo complexes for the reduction
of dinitrogen. We are interested in replacing the central amine in TREN with a phosphine as the
apical donor of the phosphine ligand should have a stronger trans influence and enhance the rate
of conversion of [Mo](NH3) to [Mo](N2) (where [Mo] = [HIPTN3N]Mo). However, work by
previous group members has shown that there are considerable difficulties in the synthesis of
triamidophosphine ligands.1 Most notable amongst these is the problem of arylating the amide
nitrogens for an alkyl phosphine ligand. For the triamidoamine ligands, arylation is achieved via
a Buchwald-Hartwig Pd cross-coupling, but attempts to perform the cross-coupling reaction with
the phosphine analog of TREN, 2,2',2''-phosphinetriyltriethanamine (TRAP) showed no
reaction.1 We think this is because the alkyl phosphine ligand coordinates to the Pd catalyst and
shuts down catalytic activity. We therefore decided to explore ligands that utilize a phenylene
instead of an ethylene backbone for each of the three ligand arms. Despite aryl phosphines being
weaker electron donors than alkyl phosphines, we were interested in investigating the results of
replacing the amine with an aryl phosphine.The ligand arms can be synthesized prior to assembly
on the central phosphorus atom, similar to Fryzuk’s work.2
Results and discussion
Attempts to synthesize 2,2’,2”-phosphinetriyltris(N-HIPTaniline)
Scheme A 1.1 Proposed synthesis of triamidophosphine ligand with a phenylene backbone.
152
Appendix 1
This work looks into expanding previous attempts to synthesize the triamidophosphine
ligand 2,2’,2”-phosphinetriyltris(N-HIPTaniline). Earlier work to synthesize a phosphine ligand
with phenylene backbones studied the possibility of performing a Pd catalyzed cross-coupling of
p-tolylaniline with HIPTBr to synthesize p-tolyl(HIPT)aniline, but the subsequent bromination
of p-tolyl(HIPT)aniline in the ortho position on the aniline led to a mixture of products that were
not separable by column chromatography.1
The first route explored here was a Chan-Lam type coupling of 2-bromophenylboronic
acid with HIPTNH2 using Cu catalysts to yield 2-bromo-N-HIPTaniline. This reaction avoids an
additional bromination step later. Buchwald et al. have shown that using Cu(OAc)2 and myristic
acid, aryl boronic acids can be coupled with aryl amines. 3 Kantam et al. reported a similar
coupling utilizing CuFAP (copper exchanged fluorapatite) as the catalyst, citing that even
deactivated aryl boronic acids such as 4-(trifluoromethyl)phenylboronic acid and 2bromophenylboronic acid are active for this compound.4
However, attempts at synthesizing 2-bromo-N-HIPTaniline using either Cu(OAc)2 or
CuFAP as the catalyst led to no formation of desired product. In the case of Cu(OAc)2, the
catalyst is may not be active enough to couple deactivated aryl boronic acids such as 2bromophenylboronic acid. Also, HIPTNH2 might be too sterically hindered for the coupling to
take place easily with either Cu(OAc)2 or CuFAP.
Figure A 1.1 Phosphines utilized in synthetic attempts of 2-bromo-N-HIPTaniline
153
Appendix 1
Since the Cu catalyzed coupling reactions were unsuccessful, we turned to BuchwaldHartwig Pd catalyzed couplings as a potential alternative route. Pd catalyzed couplings between
1,2-dibromobenzene and HIPTNH2 utilizing either rac-BINAP, CyJohnPhos and Xphos (Figure
A 1.1) as the supporting ligand for the Pd catalyst gave either many inseparable side products or
no desired product. However, when DPEphos (Figure A 1.1) was utilized as the ligand, the
desired product was obtained under the reaction conditions as shown in Equation A 1.1. Mass
spectrometry showed that using a 1:1 equivalent of 1,2-dibromobenzene with HIPTNH2 led to
the formation of the doubly substituted product, N1,N2-diHIPTbenzene-1,2-diamine. To suppress
over-arylation, two equivalents of 1,2-dibromobenzene to each equivalent of HIPTNH2 were
needed. However, the product was obtained in moderate yield and purified by column
chromatography.
Equation A 1.1 Synthesis of 2-bromo-N-HIPTaniline
Reaction of 2-bromo-N-HIPTaniline with an equivalent of n-BuLi and subsequently
reacting it with 1/3 equivalents of PCl3 led to a mixture of products. In order to prevent side
reaction, the amine group was protected with a trimethylsilane group; N-SiMe3 functionalities
are easily cleaved by acid catalyzed hydrolysis and yet are resistant to lithium or Grignard
reagents. Cooling a solution of 2-bromo-N-HIPTaniline and reacting it with an equivalent of nBuLi and then an excess of TMSCl gave N-(2-bromophenyl)-N-HIPT-1,1,1-trimethylsilanamine
which was recrystallized from pentane in moderate yield as a white powder.
154
Appendix 1
Reacting the silyl-protected aniline with n-BuLi and PCl3 and then deprotection with HCl
/ Et2O again led to a mixture of products. None of the products appeared to contain the desired
species, as determined by MALDI-TOF. Attempts to react the silyl protected aniline with Mg to
form a Grignard reagent and then PCl3 also led to mixtures of many side products but no desired
product.
As synthesis of 2,2’,2”-phosphinetriyltris(N-HIPTaniline) by first synthesizing the ligand
arms before installation on the central phosphorus atom was unsuccessful, we looked into Pd CN cross-coupling of tris(2-bromophenyl)phosphine
5
with substituted aniline, despite the
possibility of the phosphine coordinating to the Pd center.
Unfortunately, even at 110 °C for three days, there was no reaction when Pd2(dba)3 was
utilized. When Pd3(OAc)6 was used, although some reaction did occur, various phosphoruscontaining products were formed that could not be separated by column chromatography.
Inamoto et al. showed that by protecting phosphine hydride with borane, the inhibition of
palladium catalyst can be avoided. 6 Adding BH3THF to tris(2-bromophenyl)phosphine and
stirring for about 30 minutes, then subsequently setting up the Pd-coupling with this mixture
again led to several phosphorus-containing products that could not be separated by column
chromatography (Equation A 1.2).
Equation A 1.2
Oxidation of the phosphine to phosphine oxide should drastically affect the coordinating
abilities of the molecule, which may prevent the molecule from shutting down catalytic activity
155
Appendix 1
during a Pd-catalyzed cross-coupling reaction. Employing a reaction procedure similar to that
reported in the synthesis of tris(2-fluorophenyl)phosphine oxide,7 tris(2-bromophenyl)phosphine
was reacted with hydrogen peroxide and glacial acetic acid under reflux for about twenty
minutes (Equation A 1.3).
31
P NMR showed that the reaction was essentially quantitative, with
the resulting white product exhibiting a single peak at 29.28 ppm, which is 31.29 ppm downfield
of the starting material. However, Pd-coupling of the phosphine oxide with HIPTNH2 did not
yield the desired product.
Equation A 1.3
156
Appendix 1
Scheme A 1.2 Attempt at nucleophilic substitution of tris(2-fluorophenyl)phosphine oxide
We then turned to the nucleophilic substitution of tris(2-fluorophenyl)phosphine oxide by
an anilide. Although in the case of tris(2-fluorophenyl)phosphine, nucleophilic substitution is
unlikely due to electron donation from the phosphorus atom, when an electron withdrawing
substituent is on the P atom, the P atom should be electron deficient, hence stabilizing a negative
charge on a carbon in the ortho position during a nucleophilic attack. The tris(2fluorophenyl)phosphine can be prepared by nucleophilic substitution of chloride on PCl3 by (2fluorophenyl)lithium.
The
oxide
is
then
easily
prepared
by
reacting
tris(2-
fluorophenyl)phosphine with hydrogen peroxide in the presence of glacial acetic acid (Scheme A
1.2). The phosphine oxide exhibits a quartet in 31P NMR due to coupling with the ortho fluorine
atoms. Although heating it with lithium (4-n-butylphenyl)amide gave a product that exhibited a
singlet in 31P NMR, mass spectrometry of the reaction products did not reveal the presence of the
desired ligand.
157
Appendix 1
Synthesis of triHIPTamine
The synthesis of a PN3 calix[6]arene-based ligand is previously reported. 8 The report
details how a calix[6]arene compound is capped with a trisaldehyde derivative of a
triphenylphosphine via a condensation reaction between the amine functionalities on the
calix[6]arene and the benzaldehyde groups on the triphenylphosphine to produce a trisimine
compound that can subsequently be reduced to the trisamine.
Figure A 1.2 Synthesis of a PN3 calix[6]arene-based ligand8 (Figure from reference)
It was thought that one could likewise condense 2,2’,2”-phosphinetriyltribenzaldehyde
with HIPTNH2 to produce a trisimine compound which could also be subsequently reduced to
158
Appendix 1
the trisamine PN3 ligand (Scheme A 1.3). This would provide one with a triamidophosphine
ligand that similarly to the [HIPTtrpn]Mo system (described in Chapter 4 of this thesis), has a
three carbon spacer between the apical donor and the amido groups. However, the ligand arms in
such PN3Mo complexes were expected to be more rigid than in the [HIPTtrpn]Mo complexes
and therefore less likely to fall off the metal center. Also, the apical phosphine is expected to
donate electron density more strongly to the Mo atom than an amine group, leading to
differences in the reactivity of these compounds. However, the apical phosphine, being a triaryl
phosphine, will likely exhibit π-acceptor properties which may affect the ability of the metal
center to bind and reduce N2. Unlike the earlier proposed 2,2’,2”-phosphinetriyltris(NHIPTaniline) ligand with a pure phenylene backbone, the amide arms on this ligand will be
benzyl rather than aryl amides which should confer similar electron donating properties on the
amides of this new proposed triamidophosphine system to the triamidoamine systems our group
has been working with.
Scheme A 1.3 Synthesis of TriHIPTamine ligand
159
Appendix 1
2,2’,2”-phosphinetriyltribenzaldehyde:
2,2’,2”-phosphinetriyltribenzaldehyde
was
synthesized
first
by
protecting
2-
bromobenzaldehyde with ethylene glycol to make 2-(2-bromophenyl)-1,3-dioxolane which was
then converted to its Grignard derivative. 9,10 The Grignard was reacted with PCl3 to afford tris[2-(1,3-dioxolan-2-yl)phenyl]phosphine. The acetal group was then deprotected to afford
2,2’,2”-phosphinetriyltribenzaldehyde. Synthesis of both the 2-(2-bromophenyl)-1,3-dioxolane
and tris-[2-(1,3-dioxolan-2-yl)phenyl]phosphine occurred smoothly in moderate yields according
to published procedures, but that of 2,2’,2”-phosphinetriyltribenzaldehyde required slight
modifications from the published procedure and the product can be obtained in good yield.
Scheme A 1.4 Synthesis of 2,2’,2”-phosphinetriyltribenzaldehyde
TriHIPTimine:
The condensation of the tribenzaldehyde with HIPTNH2 to produce triHIPTimine occurs
smoothly. The mixture of HIPTNH2 and tribenzaldehyde forms a bright yellow sticky mixture
upon heating for a few hours, after which prolonged stirring overnight gives a suspension of
yellow powder in a yellow solution. Purification by column chromatography affords the
triHIPTimine (Figure A 1.3) in moderate yield and its identity was confirmed by mass
160
Appendix 1
spectrometry and NMR spectroscopy. An excess of HIPTNH2 is required to prevent formation of
products with only one or two benzaldehyde groups replaced with the imine functionality.
Figure A 1.3 a) TriHIPTimine b) TriHIPTamine
TriHIPTamine:
TriHIPTimine was refluxed in EtOH with a large excess of NaBH4 over several hours.
After the disappearance of the yellow substrate and formation of a white suspension, the reaction
was worked up. The product was purified by column chromatography, and its identity was
confirmed by mass spectometry and 31P, 13C and 1H NMR spectroscopy. However, upon scaling
up the reaction, it was found that the reaction did not proceed to completion even after prolonged
heating over several days. Removal of EtOH, and then adding a 2:1 EtOH:THF solvent mixture
along with excess NaBH4 finally led to completion of the reaction after heating at 80 °C over
twelve hours. This was presumably due to the requirement that the substrate be completely
dissolved by the solvent and the need for a higher reaction temperature.
Synthesis of Metal Complexes of triHIPTamine
pn3HIPTMoCl:
MoCl4(THF)2 was stirred with triHIPTamine in THF, leading to a color change from an
orange suspension to a dark green solution. Addition of LiNTMS2 led to a rapid color change
from dark green to magenta. Work up and recrystallization of the product from tetramethylsilane
resulted in a dark pink powder in low yield. This powder is assigned as the monochloride species
pn3HIPTMoCl, as shown in Equation A 1.4. The low yield of this product is similar to that of
161
Appendix 1
[pMo]Cl (described in Chapter 4 of this thesis), since both compounds are exceedingly soluble
even in non-polar solvents such as pentane or tetramethylsilane. This is postulated to be due to
the three carbon nature of each ligand arm, leading to greater flexibility of the arms and greater
solubility than in ligands with two carbon backbones. Due to the high solubility of the products,
isolation and purification of this compound is very difficult and hence combustion analyses did
not pass.
Equation A 1.4 Synthesis of pn3MoCl
Attempt to synthesize [pn3HIPTMo(NH3)]BAr′4:
Reaction of pn3HIPTMoCl with NH3 (dried over Na) in the presence of NaBAr'4 with
diethyl ether as the solvent led to a color change from a magenta solution to green within fifteen
minutes. However, the green solution converted back to magenta after another fifteen minutes.
Work up and recrystallization from pentane gave a white powder which was identified by 1H
NMR spectroscopy as free ligand.
Attempt to reduce pn3HIPTMoCl:
Reacting pn3HIPTMoCl with both Na and Mg led to the formation of an orange brown
material that was paramagnetic and did not exhibit any diazenide stretches by IR spectroscopy.
Air or chemical oxidation did not reveal any N2 stretch either.
Attempt to synthesize pn3HIPTMoN:
Addition of TMSN3 to pn3HIPTMoCl results in an immediate color change from magenta
to dark violet, then to ultramarine blue. 1H NMR of the reaction mixture shows that the
162
Appendix 1
paramagnetically shifted peaks characteristic of pn3HIPTMoCl have disappeared and the reaction
mixture appears diamagnetic. IR spectroscopy does not reveal a peak assignable to the azide
stretching frequency. A possible product may be pn3HIPTMoNTMS, since 1H NMR suggests the
presence of a trimethylsilyl group. However, due to the high solubility of the products, isolation
and purification of this compound is very difficult and hence combustion analysis did not pass.
pn3HIPTZrNMe2:
Reacting triHIPTamine with Zr(NMe2)4 at 60 ºC led to the formation of a pale yellow
solution and the formation of a new diamagnetic compound with a new peak in
31
P NMR.
Recrystallization from pentane yielded a pale yellow powder which is assigned to be
pn3HIPTZrNMe2. The assignment of this species is supported by combustion analysis.
Synthesis of tri-n-Buamine
Scheme A 1.5 Synthesis of tri-n-Buamine
We think that the three carbon spacer between the phosphine donor and the amide
nitrogen lends greater flexibility to the ligand arms of pn3HIPTamide on a metal complex. This
flexibility, combined with the high solubility of HIPT-bearing compounds, leads to difficulty in
isolation and characterization of pn3HIPTMo complexes. Replacing the HIPT substituents with
less solubilizing anilines should make the corresponding compounds more isolable. The
compound
4-n-butylaniline
was
selected
for
condensation
with
2,2’,2”-
phosphinetriyltribenzaldehyde.
163
Appendix 1
Tri-n-Buimine can be synthesized similarly to triHIPTimine. Refluxing 2,2’,2”phosphinetriyltribenzaldehyde with 4-n-butylaniline in a mixture of methanol and toluene and a
catalytic amount of formic acid leads to the formation of tri-n-Buimine in moderate yield as a
yellow powder. This can be reduced to tri-n-Buamine by refluxing it with excess NaBH4 in
ethanol and THF. Purification by column chromatography affords the amine as a white powder.
Synthesis of Metal Complexes of tri-n-Buamine
Attempts to synthesize pn3-n-BuMoCl and “pn3-n-BuMo” or pn3-n-BuMo(N2):
Stirring tri-n-Buamine with MoCl4(THF)2 and subsequent addition of LiN(TMS)2 led to
the formation of dark purple intractable solids and some tiny orange crystals. The mixture is
paramagnetic by 1H NMR. Performing the same reaction with MoCl3(THF)3 instead of
MoCl4(THF)2 in the hopes of forming pn3nBuMo(N2) led to a paramagnetic mixture of purple
and green solids but no visible N2 absorption band by IR spectroscopy.
pn3-n-BuZrNMe2:
Equation A 1.5
Heating tri-n-Buamine with Zr(NMe2)4 overnight at 100 °C led to a color change from colorless
to bright yellow. 1H NMR shows that the reaction product is diamagnetic, as is expected of pn3-nBuZrNMe2. Recrystallization afforded a bright yellow powder. Unfortunately, crystals of this material
164
Appendix 1
tend to grow in a cluster of needles, making crystal growing for solid-state diffraction very difficult.
However, elemental analysis supports the assignment of the compound as pn3-n-BuZrNMe2.
Conclusions
Two new ligands, triHIPTamine and tri-n-Buamine were synthesized. Attempts to
synthesize Mo complexes of triHIPTamine and tri-n-Buamine were unsuccessful. However, the
diamagnetic monodimethylamide zirconium complexes pn3HIPTZrNMe2 and pn3nBuZrNMe2
were synthesized and characterized.
Experimental
General. All air and moisture sensitive compounds were handled under N2 atmosphere using
standard Schlenk and glove-box techniques, with flame or oven-dried glassware. Ether, pentane,
dichloromethane and toluene were purged with nitrogen and passed through activated alumina
columns. Pentane was freeze-pump-thaw degassed three times and tetrahydrofuran (THF),
benzene, deuterated benzene and toluene were distilled from dark purple Na/benzophenone ketyl
solutions. PhF was distilled twice from P2O5. Ether and dichloromethane were stored over
molecular sieves in solvent bottles in a nitrogen-filled glovebox while pentane, THF, PhF,
benzene, deuterated benzene and toluene were stored in Teflon-sealed solvent bulbs. Molecular
sieves (4 Å) and Celite were activated at 230 °C in vacuo over several days. (Me3Si)2NLi
(sublimed) (Strem), anhydrous ZnCl2 (Aldrich) (Purified by dissolving in diethyl ether and
adding one equivalent of 1,4-dioxane to give ZnCl2(dioxane).), MoCl5 (Strem), p-toluenesulfonic
acid (Fluka), NaBH4 (Aldrich, Strem), 4-n-butylaniline (Aldrich), Zr(NMe2)4 (Strem) were used
as received unless indicated otherwise. 2-(2-bromophenyl)-1,3-dioxolane and tris-[2-(1,3dioxolan-2-yl)phenyl]phosphine were synthesized according to published procedures.10
165
Appendix 1
MoCl4(THF)211and MoCl3(THF)38 were prepared according to published procedures. IR spectra
were recorded on a Nicolet Avatar 360 FT-IR spectrometer in 0.2 mm KBr solution cells. 1H
NMR spectra were obtained on a Varian (300 or 500 MHz) or Bruker (400 or 600 MHz)
spectrometer and were referenced to the residual protio-solvent peak.
Synthesis of 2-bromo-N-HIPTaniline: Under an atmosphere of N2, a 20 mL scintillation vial
equipped with a stirbar was charged with Pd2(dba)3 (0.55 g, 0.60 mmol) and DPEphos (0.97 g,
1.80 mmol) and toluene (~10 mL). The mixture was stirred overnight and filtered through celite
and washed with toluene (~10 mL). A 100 mL solvent bulb equipped with a stirbar was charged
with HIPTNH2 (5.84 g, 11.72 mmol), 1,2-dibromobenzene (5.51 g, 23.36 mmol) and the orange
Pd filtrate. NaOtBu (1.57 g, 16.33 mmol) and toluene (~10 mL) was added last. The flask was
sealed and brought out into air and heated overnight at 110 °C and monitored by thin layer
chromatography. When the reaction was complete, the reaction mixture was filtered through
celite and extracted with acetone. The volatiles were removed in vacuo and the residue was
chromatographed with 1:5 toluene:hexane as the eluent. Yield: 4.70 g, 61%. 1H NMR (C6D6): δ
7.38 (d, 1H), 7.20 (s, 4H, 3,5,3”,5”-H), 6.92 (t, 2H, 2’,6’-H), 6.80 – 6.73 (multi, 2H), 6.44 (td,
1H), 6.03 (s, 1H, 4’-H), 3.08 (sept, 4H, 2,6,2”,6”-CHMe2), 2.88 (sept, 2H, 4, 4” –CHMe2), 1.29
(d, 12H, 2,4,6,2”,4”,6”-CH(CH3)2), 1.24 (d, 12H, 2,4,6,2”,4”,6”-CH(CH3)2), 1.22 (d, 12H,
2,4,6,2”,4”,6”-CH(CH3)2) ppm.
Synthesis of N-(2-bromophenyl)-N-HIPT-1,1,1-trimethylsilanamine: Under an atmosphere of
N2, a 300 mL 3 neck RB flask equipped with two addition funnels and a stir bar was charged
with 2-bromo-N-HIPTaniline (1.000 g, 1.53 mmol) and Et2O (100 mL). The first addition funnel
was charged with nBuLi (0.9037 mL, 1.609 mmol) in hexanes and diluted with Et2O (20 mL).
166
Appendix 1
The second addition funnel was charged with TMSCl (0.250 g, 2.30 mmol) and Et2O (20 mL).
The flask was brought out of the glovebox and cooled to -78 °C. The nBuLi solution was added
dropwise over the course of 20 minutes. The reaction mixture was warmed slowly to 0 °C
leading to the formation of a pale yellow suspension, stirred for 30 minutes and then the solution
of TMSCl was added dropwise over the course of 15 minutes. The flask was warmed slowly to
room temperature. The pale yellow suspension became a solution. The reaction mixture was
stirred for ~ 18 hours. The volatiles were removed in vacuo and the flask was brought into the
glovebox, extracted with toluene and filtered through celite. Volatiles were again removed in
vacuo and the residue was dissolved in pentane. The solution was cooled to -25 °C for ~ 3 hours
leading to the appearance of microcrystalline white solid which was collected on a glass frit and
dried. Yield: 0.79g, 66% 1H NMR (C6D6): 7.28 (dd, 1H), 7.19 (s, 4H), 6.97 (dd, 1H), 6.76 (td,
1H), 6.68 (d, 2H), 6.59 (td, 1H), 6.48 (td, 1H), 3.20 (sept, 4H), 2.86 (sept, 2H), 1.32 -1.23
(multiplet, 36H), 0.212 (s, 12H, -Si(CH3)3) ppm.
Synthesis of 2,2’,2”-phosphinetriyltribenzaldehyde: A 3 L RB flask equipped with a stirbar
was charged with tris-[2-(1,3-dioxolan-2-yl)phenyl]phosphine (18.72 g, 39.1 mmol) and
ptoluenesulfonic acid (1.73 g, 10.0 mmol) and acetone (~1.9 L). The mixture was refluxed at 60
– 70 °C for ~ 3 hours giving a yellow solution. The acetone solution was decreased to 500 mL.
Deionized water (~400 mL) was added to the acetone solution and the reaction flask kept at 0 °C
overnight leading to the formation of bright yellow microcrystals that were collected on a glass
frit and dried in vacuo. Yield: 14.22 g, 92%. The spectroscopic data for this compound are the
same as previously reported.9,10
167
Appendix 1
Synthesis of triHIPTimine: A 500 mL RB flask equipped with a stirbar was charged with
HIPTNH2 (16.50 g, 33.15 mmol), 2,2’,2”-phosphinetriyltribenzaldehyde (3.28 g, 9.47 mmol),
MeOH (100 mL) and formic acid (10 drops). The mixture was refluxed overnight, resulting in a
bright yellow suspension. The volatiles were removed in vacuo, and the residue was purified by
column chromatography with 4:1 toluene:hexanes. The desired product comes off the column
with an Rf value of 0.9 as the front fraction. This was collected and the volatiles were removed
in vacuo. The residual product was dried for ~ 18 hours at 70 °C in vacuo to produce a bright
yellow amorphous solid. Yield: 9.01 g, 53.3%. 1H NMR (C6D6): δ 8.94 (d, 3H, -NCH), 7.93 (dd,
3H, -PCH), 7.41 (s, 6H, 2’,6’-H), 7.31 (t, 3H), 7.18 -7.09 (multiplet, 12H), 6.90 (t, 3H), 6.41 (d,
6H), 3.08 (sept, 6H,4,4”-CHMe2), 2.74 (sept, 12H, 2,6, 2”, 6”-CHMe2), 1.46 (d,
36H,2,4,6,2”,4”6” –CH(CH3)2), 1.22 (t, 36H, 2,4,6,2”,4”6” –CH(CH3)2), 1.11 (dd, 36H,
2,4,6,2”,4”6” –CH(CH3)2) ppm.
13
C NMR (C6D6): δ 158.80 (d), 151.97 (s), 148.37 (s), 146.76
(d), 142.15 (s), 139.13 (d), 137.23 (s), 134.81 (s), 131.13 (s), 129.69 (s), 129.12 (s), 120.78 (s),
120.58 (s), 34.91 (s), 30.88 (d), 24.873 (s), 24.731 (s), 24.644 (s), 24.569 (s), 24.484 (s) ppm. 31P
NMR (C6D6): δ -20.00 ppm.
Synthesis of triHIPTamine: A 500 mL RB flask equipped with a stirbar was charged with
triHIPTimine (7.29 g, 4.03 mmol), NaBH4 (8.50 g, 224.69 mmol), THF (100 mL) and EtOH
(200 mL). The reaction mixture was refluxed at 80 °C for 12 hours and monitored by thin layer
chromatography. Yield: 5.95 g, 82%. 1H NMR (C6D6): δ 7.54 (dd, 3H), 7.21 (s, 12H, 3,5, 3”, 5”H), 6.90 (t, 3H), 6.75 (dd, 3H) 6.43 (d, 6H), 6.39 (d, 6H), 4.75 (br s, 3H), 3.78 (s, 6H, benzylic
Hs), 3.06 (m, 12H, 2,6, 2”, 6”-CHMe2), 2.89 (sept, 6H, 4,4”-CHMe2), 1.29 (d, 36H,
2,4,6,2”,4”6” –CH(CH3)2), 1.22 (d, 36H, 2,4,6,2”,4”6” –CH(CH3)2), 1.18 (m, 36H, 2,4,6,2”,4”6”
–CH(CH3)2) ppm.
13
C NMR (C6D6): δ 147.942 (s), 147.475 (s), 147.036 (s), 144.008 (s),
168
Appendix 1
143.713 (s), 141.695 (s), 138.477 (s), 133.701 (s), 132.756 (s), 132.612 (s), 127.568 (s), 127.058
(d, P-C), 121.401 (s), 120.651 (s), 113.261 (s), 34.990 (s), 30.413 (s), 24.919 (s), 24.791 (s),
24.555 (s) ppm.
31
P NMR (C6D6): δ -41.94 ppm MS (ESI) cald m/e: 1792.7491, found:
1792.3043.
Synthesis of pn3MoCl: A 100 mL Rb flask equipped with a stirbar was charged with
MoCl4(THF)2 (408.4 mg, 1.07 mmol) and triHIPTamine (1940 mg, 1.08 mmol) and THF (50
mL). The reaction mixture was stirred for 1 hour, leading to a color change from an orange
suspension to an emerald green solution. LiN(TMS)2 (553.9 mg, 3.31 mmol) was added slowly
with stirring and caused an immediate color change to magenta. The reaction mixture was stirred
for ~ 30 minutes and then the volatiles were removed in vacuo. The residue was extracted with
pentane and filtered through celite. The filtrate was dried in vacuo and the resulting residue was
redissolved in tetramethylsilane and cooled overnight at -25 °C yielding a paramagnetic dark
pink powder which was collected on a glass frit and dried. Yield: 0.5557 g, 27%. 1H NMR
(C6D6): 119.36 (s), 13.51 (s), 13.71 (s), 3.28 (t), 3.21 (br s), 2.93 (br t), 2.81 (br s), 1.52 (s), 1.31
(d), 1.27 (t), 1.14 (br s), -7.12 (s), -48.79 (s) ppm.
Tris(2-bromophenyl)phosphine oxide: Tris(2-bromophenyl)phosphine was suspended in EtOH
(10 mL) in a 100 mL RB flask equipped with a stirbar. To the vial was added a mixture of H2O2
(35% wt, 1 mL), glacial acetic acid (1.5 mL), and EtOH (15 mL). The mixture was refluxed for
20 minutes with stirring. The mixture was then neutralized with NaHCO3 and extracted with
chloroform, then dried over Na2SO4. The volatiles were then removed, leaving a white residue.
Yield: quantitative. 1H NMR (CDCl3): δ 7.79 (m, 1H, 5-H), 7.70 (m, 1H), 7.43 (m, 2H) ppm.
31
P NMR (CDCl3): δ 29.28 (s) ppm.
169
Appendix 1
pn3HIPTZrNMe2: A 25 mL solvent bulb was charged with Zr(NMe2)4 (71 mg, 0.268 mmol),
triHIPTamine (480.1 mg, 0.268 mmol) and heated overnight at 60 ºC, giving a pale yellow
solution. Removal of volatiles and recrystallization from pentane and tetramethylsilane yielded a
diamagnetic pale yellow powder. Yield: 125 mg, 24%. 1H NMR (C6D6): δ 7.19 (m, 6H, Aryl-H),
6.92 (d, JHH= 1.2 Hz, 3H, Aryl-H, overlapping with next peak), 6.91 (t, 3H, JHH=7.8 Hz,
overlapping with previous peak), 6.85 (t, JHH= 6.9 Hz, 3H, Aryl-H), 6.79 (t, JHH=7.2 Hz, 3H,
Aryl-H), 6.48 (m, 3H, Aryl-H), 5.22 (dd, JHH= 15.2, 3.3 Hz, 6H, Aryl-H), 4.45 (dd, JHH=15.0, 2.6
Hz, 6H, Aryl-H), 3.29 (t, JHH= 6.9 Hz, 6H, NCH2), 3.12 (sept, JHH= 6.9 Hz, 6H, -CHMe2), 3.04
(s, 6H, N(CH3)2), 2.94 (sept, JHH= 6.9 Hz, 6H, -CHMe2), 2.89 (sept, JHH= 6.9 Hz, 6H, -CHMe2),
1.55 (m, 9H, -CH(CH3)2), 1.40 (m, 9H, -CH(CH3)2), 1.30 (m, 36H, -CH(CH3)2), 1.22 (d, JHH=
6.8 Hz, 12H, -CH(CH3)2), 1.17 (d, JHH= 6.8 Hz, 12H, -CH(CH3)2), 1.14 (d, JHH= 6.8 Hz, 12H, CH(CH3)2), 0.88 (t, JHH= 7.5 Hz, 18H, -CH(CH3)2) ppm.
31
P NMR (toluene-d6): δ -45.24 (s)
ppm. Anal Calc. for C131H171N4PZr: C, 81.78; H, 8.96; N, 2.91. Found: C, 81.92; H, 8.93, N,
3.07.
Tri-n-Buimine: A 500 mL RB flask was charged with 2,2’,2”-phosphinetriyltribenzaldehyde
(2.513 g, 7.36 mmol), 4-n-butylaniline (5.414 g, 36.28 mmol), toluene (50 mL), MeOH (50 mL)
and formic acid (35%, 5 drops). The reaction mixture was allowed to reflux overnight. The
volatiles were removed in vacuo and purified by column chromatography using toluene as the
eluent. The product is a bright yellow powder. Yield: 3.53 g, 66%. 1H NMR (CD2Cl2): δ 9.09 (d,
JHH=5.8 Hz, 3H, -NCH), 8.19 (m, 3H, -PCH), 7.52 (t, JHH=8.3 Hz, 3H, Aryl-H), 7.37 (t, JHH=8.3
Hz, 3H, Aryl-H), 7.21 (m, 3H, Aryl-H), 7.07 (d, JHH=8.3 Hz, 6H, aniline Aryl-H), 6.80 (d,
JHH=8.3 Hz, 6H, aniline Aryl-H), 1.55 (m, 9H, -CH2CH2CH2CH3), 1.33 (m, 3H, -CH2CH2CH2CH3),
0.92 (t, JHH=7.4 Hz, 3H, -CH2CH2CH2CH3) ppm
31
P NMR: δ-27.53 (s) ppm.
170
Appendix 1
Tri-n-Buamine: A 500 mL RB flask was charged with tritBuimine (3.53 g, 4.77 mmol), NaBH4
(8.00 g, 211 mmol), THF (100 mL) and EtOH (100 mL). The mixture was refluxed for 16 hours.
The volatiles were removed in vacuo and EtOH (100 mL) and H2O (100 mL) was added. The
organic layer was decanted off and the aqueous layer washed with diethyl ether (4x100 mL). The
organic layers were combined, dried over MgSO4 and the volatiles removed. The product was
purified by column chromatography with toluene as the eluent. Yield: 2.45 g. 69%. 1H NMR
(CD2Cl2): δ 7.51 (m, 3H, CH (ortho to methanamine group)), 7.39 (dt, 3JHH = 7.6 Hz, 4JHH= 1.4
Hz, 3H, CH (para to P), 7.25 (dt, 3JHH = 7.6 Hz, 4JHH= 1.4 Hz, 3H, CH (para to methanamine)),
6.96 (m, 3H, CH (ortho to P), 6.91 (d, JHH= 6.2 Hz, 6H, CH (ortho to N)), 6.21 (d, JHH= 6.2 Hz,
6H, CH (meta to N)), 4.34 (s, 6H, benzylic Hs), 3.52 (s, 3H, NH), 2.52 (t, JHH= 7.8 Hz, 6H, CH2CH2CH2CH3), 1.57 (m, 6H, -CH2CH2CH2CH3), 1.38 (m, 6H, -CH2CH2CH2CH3), 0.96 (t,
JHH= 7.3 Hz, 9H, -CH2CH2CH2CH3) ppm. 13C NMR (CDCl3): δ 145.60 (s, CH ipso to N), 143.80
(d, 1JCP= 25.2Hz, CH ipso to P), 135.05 (d, 2JCP= 12.8 Hz, CH ipso to methanamine), 134.05 (s,
C ortho to P), 131.75 (s, CH ipso to nBu), 129.26 (s, CH ortho to P), 129.03 (d, 3JCP= 5.16 Hz,
CH ortho to methanamine), 128.95 (s, CH ortho to n-Bu), 127.74 (s, CH para to methanamine),
112.74 (s, CH ortho to N), 47.43 (d, CH2N), 34.82 (s, CH2CH2CH2CH3), 34.14 (s,
CH2CH2CH2CH3), 22.46 (s, CH2CH2CH2CH3), 14.10 (s, CH2CH2CH2CH3) ppm. 1H and
13
C
NMR peaks were assigned on the basis of 1H, J-modulated 13C, COSY, TOCSY, HMBC, HSQC
NMR.
31
P NMR (CDCl3): δ -35.56 (s) MS: Theoretical mass: 746.4598 [M + H] Obtained:
746.4604 [M + H]
pn3-n-BuZrNMe2: A 25 mL solvent bulb equipped with a PTFE screw valve was charged with
trinBuamine (200 mg, 0.268 mmol), Zr(NMe2)4 (71 mg, 0.265 mmol) and toluene (20 mL). It
171
Appendix 1
was heated overnight at 100 ºC with stirring. Removal of volatiles in vacuo and recrystallization
from toluene and pentane at -35 ºC gave bright yellow crystals. Yield (212 mg, 91%). 1H NMR
(tol-d6): δ 7.25 (dt, 3H, aryl-H), 7.11 (dt, 3H, aryl-H), 7.03 (2 s peaks, overlapping, 12H), 6.86
(dt, 3H, aryl-H), 6.79 (dt, 3H, aryl-H), 5.3-4.2 (v br, 6H, CH2N), 3.04 (s, 6H, N(CH3)2), 2.46 (t,
JHH= 8.0Hz, 6H, -CH2CH2CH2CH3), 1.49 (m, 9H, -CH2), 1.26 (t, 9H, -CH2), 0.83 (m, 9H, -CH3)
ppm.
31
P NMR: δ -50.41 (s) ppm. Anal Calcd for C53H60N4PZr: C, 72.73; H, 6.91; N, 6.40.
Found: C, 72.77; H, 7.30; N, 6.50.
References
1
Symthe, N. PhD Thesis, Chapter 4, 2007.
2
MacLachlan, E.A.; Fryzyk, M.D. Organometallics 2005, 24, 1112.
3
Antilla, J.C.; Buchwald, S.L. Org Lett. 2001, 3, 2077.
4
Kantam, M.L.; Venkanna, G.T.; Sridhar, C.; Sreedhar, B.; Choudary, B.M. J. Org. Chem. 2006, 71,
9523.
5
Tsuji, H.; Inoue, T.; Kaneta, Y.; Sase, S.; Kawachi, A.; Tamao, K. Organomet. 2006, 25, 6142.
6
Oshiki, T; Inamoto, T. J. Am. Chem. Soc. 1992, 114, 3975.
7
Stegmann, H.B.; Kuhne, H.M.; Wax, G.; Scheffler, K. P & S. 1982, 13, 331.
8
Zeng, X.; Hucher, N.; Reinaud, O.; Jabin, I. J. Org. Chem.2004, 69, 6886.
9
Schiemenz, G.P.; Kaack, H. Liebigs Ann. Chem. 1973, 1480.
10
Whitnall, M.R.; Hii, K.K.; Thornton-Pett, M.; Kee, T.P. J. Organomet. Chem. 1997, 529, 35.
11
Stoffelbach, F.; Saurenz, D.; Poli, R. Eur. J. Inorg. Chem. 2001, 10, 2699.
172
Appendix 2
Appendix 2
Crystallographic Tables
173
Appendix 2
Crystallographic Studies
Diffraction data were collected on a Siemens Platform three-circle diffractometer or a
Bruker D8 threecircle diffractometer coupled to a Bruker-AXS Smart Apex CCD detector with
graphite-monochromated Mo Κα radiation (λ ) 0.71073 Å) or Cu Κα radiation (λ) 1.54178 Å)
performing φ and ω-scans. All structures were solved by direct methods using SHELXS and
refined against F2 on all data by full-matrix leastsquares with SHELXL-97.96 All non-hydrogen
atoms were refined anisotropically. Hydrogen atoms on any carbon atom that binds directly to
molybdenum were taken from the difference Fourier synthesis and refined semifreely with the
help of distance restraints. All other hydrogen atoms were included into the model at
geometrically calculated positions and refined using a riding model unless stated otherwise.
Data for the structures including CIF files are also available to the public at
http://www.reciprocalnet.org
174
Appendix 2
Crystallographic data for {[HIPTtrpn]Mo(NH3)}BAr'4
Table A 2.1 Crystal data and structure refinement for {[HIPTtrpn]Mo(NH3)}BAr'4
Identification code
Empirical formula
06137
C163 H212 B F24 Mo N5
Formula weight
2804.13
Temperature
Wavelength
Crystal system
100(2) K
0.71073 Å
Triclinic
Space group
P-1
Unit cell dimensions
Volume
a = 15.4574(9) Å
b = 22.5295(15) Å
c = 25.1080(17) Å
3
8261.7(9) Å
Z
Density (calculated)
2
3
1.127 Mg/m
Absorption coefficient
0.154 mm
F(000)
Crystal size
2976
3
0.50 x 0.50 x 0.05 mm
Theta range for data collection
Index ranges
Reflections collected
Independent reflections
Completeness to theta = 26.02°
Absorption correction
Max. and min. transmission
Refinement method
0.95 to 26.02°.
-19<=h<=19, -27<=k<=27, -30<=l<=30
144464
32504 [R(int) = 0.0750]
99.9 %
Semi-empirical from equivalents
0.9924 and 0.9271
2
Full-matrix least-squares on F
Data / restraints / parameters
Goodness-of-fit on F2
Final R indices [I>2sigma(I)]
R indices (all data)
Largest diff. peak and hole
32504 / 5893 / 2309
1.076
R1 = 0.0936, wR2 = 0.2540
R1 = 0.1369, wR2 = 0.2937
-3
2.190 and -0.856 e.Å
α= 82.239(2)°.
ß= 82.699(2)°.
γ= 73.285(2)°.
-1
175
Appendix 2
Table A 2.2 Selected Bond lengths [Å] and angles [°] for {[HIPTtrpn]Mo(NH3)}BAr'4
Mo(1)-N(3)
1.960(4)
N(2)-Mo(1)-N(4)
90.92(14)
Mo(1)-N(2)
Mo(1)-N(1)
Mo(1)-N(5)
1.964(4)
1.972(4)
2.238(4)
N(1)-Mo(1)-N(4)
N(5)-Mo(1)-N(4)
92.25(16)
178.92(16)
C(3)-N(4)-C(6)
106.9(4)
Mo(1)-N(4)
2.256(4)
N(4)-C(3)
N(4)-C(6)
N(4)-C(9)
1.473(7)
1.510(6)
1.529(7)
C(3)-N(4)-C(9)
C(6)-N(4)-C(9)
C(3)-N(4)-Mo(1)
108.0(4)
105.7(4)
112.2(3)
C(6)-N(4)-Mo(1)
112.9(3)
C(1)-N(1)
1.462(7)
C(9)-N(4)-Mo(1)
N(1)-C(1)-C(2)
C(1)-C(2)-C(3)
N(4)-C(3)-C(2)
N(2)-C(4)-C(5)
C(4)-C(5)-C(6)
N(4)-C(6)-C(5)
N(3)-C(7)-C(8)
C(9)-C(8)-C(7)
C(8)-C(9)-N(4)
C(115)-N(1)-C(1)
C(115)-N(1)-Mo(1)
C(1)-N(1)-Mo(1)
C(215)-N(2)-C(4)
C(215)-N(2)-Mo(1)
C(4)-N(2)-Mo(1)
C(315)-N(3)-C(7)
C(315)-N(3)-Mo(1)
C(7)-N(3)-Mo(1)
110.8(3)
113.6(5)
111.4(4)
113.4(4)
112.7(4)
110.6(5)
112.9(4)
112.4(4)
111.2(5)
113.7(4)
111.0(4)
122.8(3)
126.3(3)
109.9(3)
122.2(3)
127.8(3)
108.3(4)
124.7(3)
127.0(3)
C(1)-C(2)
C(2)-C(3)
C(4)-N(2)
C(4)-C(5)
C(5)-C(6)
C(7)-N(3)
C(7)-C(8)
C(8)-C(9)
N(1)-C(115)
N(2)-C(215)
N(3)-C(315)
N(3)-Mo(1)-N(2)
N(3)-Mo(1)-N(1)
N(2)-Mo(1)-N(1)
N(3)-Mo(1)-N(5)
N(2)-Mo(1)-N(5)
N(1)-Mo(1)-N(5)
N(3)-Mo(1)-N(4)
1.514(8)
1.528(7)
1.481(6)
1.504(7)
1.530(7)
1.476(6)
1.514(7)
1.501(8)
1.426(6)
1.440(6)
1.448(5)
119.12(17)
118.61(17)
121.99(17)
87.22(15)
88.68(14)
88.81(16)
92.10(15)
Two of the three ammonia-hydrogen atoms were taken from the difference Fourier
synthesis. The third one (H5NC) could not be located and its position was calculated instead. All
three hydrogen atoms were then refined semi-freely with the help of distance restraints.
176
Appendix 2
Crystallographic Data for [Mespyr(C6F5)2]MoCl
Table A 2.3 Crystal data and structure refinement for [Mespyr(C6F5)2]MoCl
Identification code
08134
Empirical formula
Formula weight
Temperature
C30 H23 Cl F10 Mo N4
760.91
100(2) K
Wavelength
0.71073 Å
Crystal system
Space group
Unit cell dimensions
Monoclinic
P2(1)/c
a = 10.0775(5) Å
Volume
b = 19.4734(9) Å
c = 16.1625(8) Å
3
3027.5(3) Å
Z
Density (calculated)
4
3
1.669 Mg/m
Absorption coefficient
0.612 mm
F(000)
Crystal size
1520
3
0.42 x 0.35 x 0.18 mm
Theta range for data collection
Index ranges
Reflections collected
Independent reflections
Completeness to theta = 29.57°
Absorption correction
Max. and min. transmission
Refinement method
1.68 to 29.57°.
-13<=h<=13, -25<=k<=27, -22<=l<=22
57573
8488 [R(int) = 0.0344]
100.0 %
Semi-empirical from equivalents
0.8978 and 0.7832
2
Full-matrix least-squares on F
Data / restraints / parameters
Goodness-of-fit on F2
Final R indices [I>2sigma(I)]
R indices (all data)
Largest diff. peak and hole
8488 / 0 / 418
1.069
R1 = 0.0249, wR2 = 0.0606
R1 = 0.0294, wR2 = 0.0640
-3
0.487 and -0.355 e.Å
α= 90°.
ß= 107.3480(10)°.
γ= 90°.
-1
177
Appendix 2
Table A 2.4 Selected Bond lengths [Å] and angles [°] for [Mespyr(C6F5)2]MoCl
Cl(2)-Mo(1)
2.3583(4)
N(3)-Mo(1)-N(2)
117.92(5)
Mo(1)-N(3)
Mo(1)-N(2)
Mo(1)-N(1)
1.9539(12)
1.9688(12)
2.0184(12)
N(3)-Mo(1)-N(1)
N(2)-Mo(1)-N(1)
N(3)-Mo(1)-N(4)
113.78(5)
118.56(5)
79.98(5)
Mo(1)-N(4)
2.1737(12)
N(2)-Mo(1)-N(4)
79.49(5)
N(1)-C(1)
N(1)-C(9)
N(2)-C(21)
1.4011(18)
1.4022(19)
1.4127(18)
N(1)-Mo(1)-N(4)
N(3)-Mo(1)-Cl(2)
N(2)-Mo(1)-Cl(2)
79.05(5)
98.79(4)
96.94(4)
N(2)-C(3)
1.4869(19)
N(1)-Mo(1)-Cl(2)
105.84(4)
N(3)-C(31)
N(3)-C(5)
N(4)-C(4)
N(4)-C(6)
N(4)-C(2)
1.4204(18)
1.4810(19)
1.4873(19)
1.4918(18)
1.5022(18)
C(4)-N(4)-Mo(1)
C(6)-N(4)-Mo(1)
C(2)-N(4)-Mo(1)
N(1)-C(1)-C(2)
106.51(9)
106.43(8)
107.59(8)
116.92(12)
178
Appendix 2
Crystallographic Data for [Mespyr(C6F5)2]MoNMe2
Table A 2.5 Crystal data and structure refinement for [Mespyr(C6F5)2]MoNMe2
Identification code
Empirical formula
08186
C32 H29 F10 Mo N5
Formula weight
769.54
Temperature
Wavelength
Crystal system
100(2) K
0.71073 Å
Monoclinic
Space group
P2(1)/c
Unit cell dimensions
Volume
a = 8.6519(6) Å
b = 16.6172(11) Å
c = 21.5630(15) Å
3
3100.1(4) Å
Z
Density (calculated)
4
3
1.649 Mg/m
Absorption coefficient
0.516 mm
F(000)
Crystal size
1552
3
0.48 x 0.35 x 0.04 mm
Theta range for data collection
Index ranges
Reflections collected
Independent reflections
Completeness to theta = 29.13°
Absorption correction
Max. and min. transmission
Refinement method
1.55 to 29.13°.
-11<=h<=11, -22<=k<=22, -29<=l<=29
78331
8342 [R(int) = 0.0396]
100.0 %
Semi-empirical from equivalents
0.9797 and 0.7898
2
Full-matrix least-squares on F
Data / restraints / parameters
Goodness-of-fit on F2
Final R indices [I>2sigma(I)]
R indices (all data)
Largest diff. peak and hole
8342 / 498 / 538
1.062
R1 = 0.0252, wR2 = 0.0592
R1 = 0.0317, wR2 = 0.0635
-3
0.558 and -0.374 e.Å
α= 90°.
ß=90.2090(10)°.
γ= 90°.
-1
179
Appendix 2
Table A 2.6 Selected Bond lengths [Å] and angles [°] for [Mespyr(C6F5)2]MoNMe2
N(4)-C(9)
1.4819(19)
C(52)-N(5)-Mo(1)
126.43(10)
N(4)-C(7)
N(4)-C(5)
N(4)-Mo(1)
1.4828(19)
1.4873(19)
2.2630(12)
C(51)-N(5)-Mo(1)
N(5)-Mo(1)-N(3)
124.92(10)
103.25(5)
N(5)-Mo(1)-N(2)
103.78(5)
N(5)-C(52)
1.4688(19)
N(5)-C(51)
N(5)-Mo(1)
Mo(1)-N(3)
1.4709(19)
1.9383(12)
1.9738(12)
N(3)-Mo(1)-N(2)
N(5)-Mo(1)-N(1)
N(3)-Mo(1)-N(1)
110.68(5)
99.80(5)
117.35(5)
N(2)-Mo(1)-N(1)
118.87(5)
Mo(1)-N(2)
1.9885(12)
N(5)-Mo(1)-N(4)
N(3)-Mo(1)-N(4)
N(2)-Mo(1)-N(4)
N(1)-Mo(1)-N(4)
C(2)-C(1)-N(1)
C(2)-C(1)-C(5)
N(1)-C(1)-C(5)
C(1)-C(2)-C(3)
C(4)-C(3)-C(2)
C(3)-C(4)-N(1)
C(1)-N(1)-C(4)
C(1)-N(1)-Mo(1)
C(4)-N(1)-Mo(1)
C(21)-N(2)-C(6)
C(21)-N(2)-Mo(1)
C(6)-N(2)-Mo(1)
C(31A)-N(3)-C(8)
C(31)-N(3)-C(8)
C(31A)-N(3)-Mo(1)
C(31)-N(3)-Mo(1)
C(8)-N(3)-Mo(1)
176.92(5)
77.51(5)
78.61(5)
77.26(5)
110.62(13)
131.66(14)
117.70(13)
106.60(13)
107.45(14)
109.41(13)
105.92(12)
116.23(10)
136.36(10)
112.09(12)
129.36(10)
118.55(9)
110.3(3)
109.83(18)
128.3(3)
126.06(18)
120.69(10)
Mo(1)-N(1)
C(1)-C(2)
C(1)-N(1)
C(1)-C(5)
C(2)-C(3)
C(3)-C(4)
C(4)-N(1)
C(4)-C(11)
C(6)-N(2)
C(6)-C(7)
C(8)-N(3)
C(8)-C(9)
N(2)-C(21)
N(3)-C(31)
C(9)-N(4)-C(7)
C(9)-N(4)-C(5)
C(7)-N(4)-C(5)
C(9)-N(4)-Mo(1)
C(7)-N(4)-Mo(1)
C(5)-N(4)-Mo(1)
C(52)-N(5)-C(51)
2.0807(12)
1.364(2)
1.3967(19)
1.488(2)
1.429(2)
1.376(2)
1.3985(19)
1.486(2)
1.4998(19)
1.518(2)
1.4867(19)
1.511(2)
1.4117(18)
1.435(5)
112.69(12)
110.30(12)
111.98(12)
106.04(9)
106.90(9)
108.62(9)
108.62(12)
One of the two C6F5 is disordered (in-plane!). Treated the usual way.
180
Appendix 2
Crystallographic Data for [HIPTtrpn]MoN
Table A 2.7 Crystal data and structure refinement for [HIPTtrpn]MoN
Identification code
Empirical formula
d09073
C127.95 H185.63 Mo N6.76 O1.50
Formula weight
1938.50
Temperature
Wavelength
Crystal system
100(2) K
1.54178 Å
Monoclinic
Space group
P2(1)/c
Unit cell dimensions
Volume
a = 17.3347(3) Å
b = 31.9817(7) Å
c = 23.5663(4) Å
3
13064.7(4) Å
Z
Density (calculated)
4
3
0.986 Mg/m
Absorption coefficient
1.159 mm
F(000)
Crystal size
4219
3
0.20 x 0.15 x 0.05 mm
Theta range for data collection
Index ranges
Reflections collected
Independent reflections
Completeness to theta = 56.54°
Absorption correction
Max. and min. transmission
Refinement method
2.90 to 56.54°.
-16<=h<=18, -34<=k<=34, -25<=l<=25
207153
17285 [R(int) = 0.1544]
99.7 %
Semi-empirical from equivalents
0.9443 and 0.8013
2
Full-matrix least-squares on F
Data / restraints / parameters
Goodness-of-fit on F2
Final R indices [I>2sigma(I)]
R indices (all data)
Largest diff. peak and hole
17285 / 5410 / 1641
1.092
R1 = 0.0904, wR2 = 0.2335
R1 = 0.1323, wR2 = 0.2586
-3
1.083 and -0.598 e.Å
α= 90°.
ß= 90.3620(10)°.
γ = 90°.
-1
181
Appendix 2
Table A 2.8 Selected Bond lengths [Å] and angles [°] for [HIPTtrpn]MoN
C(8)-C(9)
1.542(10)
C(8)-H(8A)
C(8)-H(8B)
0.9900
0.9900
1.651(5)
1.467(8)
1.546(10)
C(9)-H(9A)
C(9)-H(9B)
0.9900
0.9900
Mo(1)-N(3)
1.961(5)
C(1)-H(1A)
0.9900
C(1)-H(1B)
C(2)-C(3)
C(2)-H(2A)
0.9900
1.529(10)
0.9900
Mo(1)-N(2)
Mo(1)-N(1)
N(1)-C(115)
1.999(5)
2.004(5)
1.406(7)
C(2)-H(2B)
C(3)-H(3A)
C(3)-H(3B)
C(4)-N(2)
C(4)-C(5)
C(4)-H(4A)
C(4)-H(4B)
C(5)-C(6)
C(5)-H(5A)
C(5)-H(5B)
C(6)-H(6A)
C(6)-H(6B)
C(7)-C(8)
C(7)-N(3)
C(7)-H(7A)
C(7)-H(7B)
0.9900
0.9900
0.9900
1.492(8)
1.514(10)
0.9900
0.9900
1.513(10)
0.9900
0.9900
0.9900
0.9900
1.496(11)
1.501(8)
0.9900
0.9900
N(4)-C(9)
N(4)-C(6)
1.483(9)
1.489(8)
N(4)-C(3)
1.494(9)
N(5)-Mo(1)
C(1)-N(1)
C(1)-C(2)
N(2)-C(215)
N(3)-C(315)
C(9)-N(4)-C(6)
C(9)-N(4)-C(3)
C(6)-N(4)-C(3)
N(1)-C(1)-C(2)
N(5)-Mo(1)-N(3)
N(5)-Mo(1)-N(2)
N(3)-Mo(1)-N(2)
N(5)-Mo(1)-N(1)
N(3)-Mo(1)-N(1)
N(2)-Mo(1)-N(1)
C(115)-N(1)-C(1)
C(115)-N(1)-Mo(1)
C(1)-N(1)-Mo(1)
C(4)-N(2)-Mo(1)
C(7)-N(3)-Mo(1)
1.407(7)
1.437(8)
111.1(5)
106.8(5)
109.5(6)
113.5(5)
97.3(2)
103.3(2)
112.1(2)
99.9(2)
122.1(2)
116.8(2)
117.6(5)
131.0(4)
110.8(4)
113.1(4)
127.5(5)
The data is very weak, especially at higher resolution, and the usable resolution range
extends only to about 0.92 Å. Restraints were used extensively to improve the data-to-parameter
ratio. The structure contains a mixture of highly disordered solvent (THF, acetonitrile and
pentane). Partially occupied solvent molecules and different solvent molecules sharing sites in
182
Appendix 2
the unit cell lead to non-integer numbers for the elements C, H, N, and O in the empirical
formula.
183
Appendix 2
Crystallographic Data for Mespyr(3,5-t-Bu)2]MoN
Table A 2.9 Crystal data and structure refinement for [Mespyr(3,5-t-Bu)2]MoN
Identification code
Empirical formula
d09075
C55 H72.50 F1.50 Mo N5
Formula weight
928.12
Temperature
Wavelength
Crystal system
100(2) K
1.54178 Å
Monoclinic
Space group
P2(1)/n
Unit cell dimensions
Volume
a = 17.0308(4) Å
b = 14.0790(4) Å
c = 20.7053(5) Å
3
4920.1(2) Å
Z
Density (calculated)
4
3
1.253 Mg/m
Absorption coefficsient
2.537 mm
F(000)
Crystal size
1972
3
0.45 x 0.15 x 0.10 mm
Theta range for data collection
Index ranges
Reflections collected
Independent reflections
Completeness to theta = 67.73°
Absorption correction
Max. and min. transmission
Refinement method
3.61 to 67.73°.
-20<=h<=20, -16<=k<=16, -24<=l<=24
93103
8719 [R(int) = 0.0269]
97.9 %
Semi-empirical from equivalents
0.7855 and 0.3948
2
Full-matrix least-squares on F
Data / restraints / parameters
Goodness-of-fit on F2
Final R indices [I>2sigma(I)]
R indices (all data)
Largest diff. peak and hole
8719 / 143 / 610
1.026
R1 = 0.0243, wR2 = 0.0606
R1 = 0.0252, wR2 = 0.0613
-3
0.361 and -0.389 e.Å
α= 90°.
ß= 97.678(2)°.
γ= 90°.
-1
184
Appendix 2
Table A 2.10 Selected Bond lengths [Å] and angles [°] for [Mespyr(3.5-t-Bu)2]MoN
Mo(1)-N(5)
1.6746(13)
N(5)-Mo(1)-N(4)
175.43(5)
Mo(1)-N(3)
Mo(1)-N(2)
Mo(1)-N(1)
1.9751(13)
1.9857(13)
2.0565(12)
N(3)-Mo(1)-N(4)
N(2)-Mo(1)-N(4)
N(1)-Mo(1)-N(4)
80.99(5)
76.08(5)
75.66(5)
Mo(1)-N(4)
2.4134(13)
C(9)-N(1)-C(1)
105.87(12)
N(1)-C(9)
N(1)-C(1)
N(2)-C(41)
1.401(2)
1.407(2)
1.4223(19)
C(9)-N(1)-Mo(1)
C(1)-N(1)-Mo(1)
C(41)-N(2)-C(3)
132.90(10)
118.99(10)
114.90(12)
N(2)-C(3)
1.4855(19)
C(41)-N(2)-Mo(1)
126.27(10)
1.428(2)
1.4871(19)
1.4736(19)
1.474(2)
1.477(2)
103.10(6)
100.43(6)
109.82(5)
104.33(6)
112.26(5)
123.86(5)
C(3)-N(2)-Mo(1)
C(21)-N(3)-C(5)
C(21)-N(3)-Mo(1)
C(5)-N(3)-Mo(1)
C(4)-N(4)-C(2)
C(4)-N(4)-C(6)
C(2)-N(4)-C(6)
C(4)-N(4)-Mo(1)
C(2)-N(4)-Mo(1)
C(6)-N(4)-Mo(1)
118.83(9)
115.16(12)
127.25(10)
115.08(10)
112.76(12)
114.67(12)
112.88(12)
107.18(9)
107.43(9)
100.79(9)
N(3)-C(21)
N(3)-C(5)
N(4)-C(4)
N(4)-C(2)
N(4)-C(6)
N(5)-Mo(1)-N(3)
N(5)-Mo(1)-N(2)
N(3)-Mo(1)-N(2)
N(5)-Mo(1)-N(1)
N(3)-Mo(1)-N(1)
N(2)-Mo(1)-N(1)
References
96
Sheldrick, G. M. Acta Crystallogr. 2008, A64, 112.
185
Acknowledgements
I’d first like to thank Dick for having been a wonderful advisor. I appreciate the
opportunity to join his group and to work on this project. There have been many difficult times
working on this project, with what seemed to be endless insurmountable hurdles in chemistry. I
am very grateful to Dick for his patience and understanding during those times.
I have had the good fortune of being welcomed into this lab by group members who have
since left, in particular Andrea, who has been a great source of support, and also an amazing
friend. I couldn’t have survived my first year without her. With regards to members on the
dinitrogen project, I’d like to thank Brian, Michael, Nathan and Matt. The free exchange of ideas
and chemicals has made working with them extremely enjoyable. Brian’s constant supply of
candy has helped keep me chipper and Michael has been a great bike buddy – without him, I
would never have discovered my love of mountain biking. Keith (though not technically on the
dinitrogen project) has also been a wonderful source of help in the lab, in particular with
electrochemistry and glovebox rescues. Working in the group would not have been as much fun
without all these people.
I’d also like to thank the members of 6-428 for being such wonderful labmates. Victor,
though having only recently joined the lab, has been a kind and generous boxmate, and a really
good friend. I am very grateful for his support and help during his time here.
There are also various people outside this group whose lives have intersected with mine
at MIT and have been crucial in providing support or in shaping my life here. Mark and Yidong
have taught me many things and I am grateful to have met them. My SGI friends, in particular,
Woei Ling, Yi-Chun, Tanya and Donna have also been incredibly crucial in helping me get
through difficult days.
186
Most importantly, I’d like to thank my parents and little brother who have been giving me
so much love, support and daimoku.
187
Massachusetts Institute of Technology
Room 6-428
77 Massachusetts Ave
Cambridge, MA 02139
67 Mayflower Ave
Singapore 568888
(065)64587332
jiaminchin@hotmail.com
EDUCATION
Graduate Massachusetts Institute of Technology
2004-2010
Cambridge, MA
Inorganic synthesis, homogeneous catalysis with transition metal complexes, small
molecule activation (Prof. Richard R. Schrock)
B.A. Magna Cum Laude Columbia College, Columbia University
2001-2004
New York, NY
Inorganic synthesis, radical polymerization (Prof. Jack R. Norton)
AWARDS
2010
Massachusetts Institute of Technology Morse Travel Grant
2001-2010 Singapore Agency for Science Technology and Research Full Scholarship
2001-2004 Columbia College, Columbia University I.I. Rabi Fellowship
PUBLICATIONS
Chin, J.M.; Schrock, R.R., Müller, P. “ Synthesis of DiamidoPyrrolyl Molybdenum Complexes
Relevant to Reduction of Dinitrogen to Ammonia.” Inorg. Chem. Submitted.
Chin, J.M.; Weare,W.W.; Schrock, R.R.; Müller, P. “Molybdenum Complexes that Contain the
[(HIPTNCH2CH2CH2)3N]3- Ligand (HIPT = 3,5-(2,4,6-i-Pr3C6H2)2C6H3) that are Relevant to Catalytic
Reduction of Dinitrogen.” Inorg. Chem. Submitted.
McNaughton, R. L.; Roemelt, M.; Chin, J.M.; Schrock, R.R.; Neese, F.; Hoffman, B.M. “Experimental
and Theoretical EPR Study of Jahn−Teller-Active [HIPTN3N]MoL Complexes (L = N2, CO, NH3).” J.
Am. Chem. Soc.ASAP
McNaughton, R.L.; Chin, J.M.; Weare, W.W.; Schrock, R.R.; Hoffman, B.M. “EPR Study of the LowSpin [d3; S =1/2], Jahn−Teller-Active, Dinitrogen Complex of a Molybdenum Trisamidoamine.” J. Am.
Chem. Soc. 2007, 129, 3480.
Weare, W.W.; Byrnes, M.J.; Chin, J.M.; Dai, X.; Schrock, R.R.; Müller, P. “Nitrogen fixation at
a single molybdenum atom: recent advances in understanding this catalytic cycle.” PNAS, 2006,
103, 17099.
188