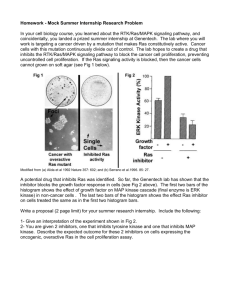
/ K-ras
advertisement

? ) / Functional Analysis of K-ras in the Mouse by Gene Targeting by Leisa Kay Johnson B.A., Chemistry Hastings College, 1989 Submitted to the Department of Biology in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy at the Massachusetts Institute of Technology May 1996 © 1996 Leisa K. Johnson All rights reserved The author hereby grants to MIT permission to reproduce and to distribute publicly copies of this thesis document in whole or in part. Signature of Author: ':"~~ " ~ rt ' ;.::r-o~ •.~ .. Y..~.Y..x;I.'w'" •• ~••••••••••••••• Department of Biology May 20,1996 Certified by: *••••••••••••••••• ., .,•••••,. ••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••• Tyler E. Jacks Assistant Professor of Biology Thesis Supervisor / Accepted by: -.;•.. 1."'.; •••••••••••••••••••••••••••••••••••••••••••••••• •••••••••••••••••••••••• Frank Solomon Chairman of the Biology Graduate Committee r ARCHIVES iw'IASSACHUSETTS INSTITUTE , OF TECHNOLOGY :r,,~ JUL 0 81996 LIBRARIES 1 FUNCTIONAL ANALYSIS OF K-RAS IN THE MOUSE BY GENE TARGETING by Leisa Kay Johnson Submitted to the Department of Biology on May 28, 1996 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy ABSTRACT Mutations in the ras oncogenes are one of the most frequent alterations in human cancer. It is estimated that approximately 30% of all human tumors carry mutations in one of the three ras genes, H-, N-, and K-ras. As such, pharmaceutical companies have focused on activated Ras as a potential chemotherapeutic target. To characterize the normal role of K-ras in growth and development, I have mutated it through gene targeting in the mouse. I now show that K-ras is the only essential member of this gene family for mouse embryogenesis. Embryos homozygous for the null mutation die between days 12 and 14 of gestation, with defects in the fetal liver and evidence of anemia. In addition, I have demonstrated partial functional compensation between the ras genes as multiple mutations within the ras gene family can affect phenotype. Most embryos lacking N-ras function and heterozygous for the K-ras mutation exhibit abnormal hematopoietic development and die between days 10 and 12 of embryogenesis. Embryos lacking both K-ras and N-ras function fail yet earlier in development. Thus, a critical threshold level of overall Ras activity must be achieved during early murine development, with a unique requirement for K-ras function arising still later in embryogenesis. In human tumors, K-ras is the most frequently mutated member of the ras gene family. Therefore, an accurate animal model to study the role of oncogenic K-ras in tumorigenesis is important. I now demonstrate a novel method of activation of an oncogene in vivo. Using a variation of the hit-and-run gene targeting strategy, I have created a strain of mice carrying a spontaneously activatable allele of the K-ras oncogene. As expected, these animals exhibit multiple lesions. In particular, they are highly susceptible to lung tumors and lymphomas. I believe that this K-ras mutant strain will prove invaluable in the study of K-ras activation in tumorigenesis as well as in evaluating the efficacy of chemotherapeutic drugs. In addition, this innovative approach of oncogene activation may be used to generate other improved animal models of cancer. Thesis Supervisor: Tyler Jacks Title: Assistant Professor of Biology Acknowledgements First and foremost, I would like to thank my adviser, Tyler. Thanks for adopting this orphan and allowing me to re-discover the joys and excitement that science can offer. Thanks for countless hours of advice, support, and for being laid back and easy to approach. Thanks for respecting my opinion and for providing such a great place to work. I hope you never lose that great sense of humor! Thanks for being such a good sport at quarters and don't ever forget what a great place Nebraska is (Lincoln-Omaha, what's the dif?) or those Cornhuskers! Maybe we'll cross paths again some day in SF! A very special thanks to my mom! Thanks for instilling in me my values and the courage to go after my dreams. Thanks for being my friend and for always being there for me, through all the good and bad times. You've been the best role model that a person could ever have. Thanks to my sister and very best friend, Ellen! I feel as though you're like a twin and can share anything with you. You're the true picture of a "big sister" and have always guided me down the right path. Thanks for giving me a little Godchild and a great brother-in-law, Darryn. I only hope that you guys have used that can of whipped cream by now (from one ---less wonder to another!). To my brother, David, thanks for everything over the years. Thanks to my old roommate and fellow orphan, Christina Scherer. Thanks for being my second mom and sending me care packages while writing this thesis! You've been a great friend and I can't wait until we're back together again on the West Coast. T and L-here we come! I just hope they're ready for us, girl. To my friend and baymate, Kay Macleod-thanks for being a great friend, for your help and advice, both personal and scientific. We will always have Patty Burke's, and we can never forget CSH and GE and JR! We will definitely have to hook up at another meeting and share the good life that Scotland has to offer. To Doron Greenbaum, thanks for putting up with my anal retentiveness and moods. You've been an immense help to me over the past two years and I will always being thankful for that. Thanks to Andi McClatchey, Laura Attardi and Lee Remington for friendship, support, and advice. Laura you've been a great friend and someone that I can easily share common hardships with. I'll miss our shopping excursions and seminar art classes! To the only other hick in the lab, Bart (Barto) Williams--thanks for the advice and help with the ole mice! Just remember--NEBRASKA rules and the Cornhuskers are #1!! Thanks to all of the other members of the lab, past and present, for being great friends and people to work with and making our second home such a great place to work! And to Rod Bronson, the pathologist of the hour! The only person I know who can bring such a humerous light to tumors! I've learned a lot from you. Thanks for taking the time to teach me and for listening to me when I was at my wits end! A special thanks to all of my friends over the years on the fifth floor, especially the Sharpies and Housmanites of old! Our 5th floor beer hours were famous! And to Margarita, thanks for always being there to help and for always having that kind smile on your face. Thanks to Neptune Mizrahi (what a great name!) for becoming such a special friend over the years. We can always thank Tufts for allowing us to cross paths in this world. To the men of our bay--Brad and Mel!! What would we have done without them? To Melrose Place, for providing endless hours of amusement for Laura, Reuben, and myself and always giving us something to look forward to on Monday nights and give George a hard time about! A very warm and special thanks to Neal, a friend and confidant of old. You gave me years of joy, laughter, love and support. And most importantly, I want to thank Devin. Thanks for giving me the time of my life and for being the pot of gold at the end of my rainbow. Thanks for brightening my days just by hearing your voice thousands of miles away. And thanks for keeping the dream alive-our time has finally come! Table of Contents Title page 1 Abstract 2 Acknowledgements 3 Table of Contents 5 List of Figures and Tables 8 Chaper 1: Overview of Ras Function and Signaling 10 I. Primary Structure Mammalian ras Genes and their Evolutionary Conservation ras-Related Genes 11 11 12 II. Biochemical Properties of Ras Guanine Nucleotide Binding Post-translational Modifications and Membrane Association The Effector Domain 14 14 16 20 III. Functions of ras Genes Non-mammalian Systems Mammalian Systems A Causative Role for Oncogenic ras in Tumorigenesis Oncogenic ras and Multistep Tumorigenesis 21 21 22 24 26 IV. Regulators of the Ras GTP/GDP Cycle Guanine Nucleotide Exchange Factors GTPase Activating Proteins Guanine Nucleotide Dissociation Inhibitors Ras Regulators as Targets for Tumorigenesis 29 30 32 37 38 V. The Ras Signal Transduction Pathway Regulation of the Ras GDP/GTP Cycle by Mitogenic Stimuli Ras Transmits a Signal to the Nucleus via a Cascade of Serine/Threonine Kinases The Rho-subfamily of Small GTP-binding Proteins: Their Signaling Pathways and Requirement for Ras-mediated Transformation 39 39 43 VI. Ras Signaling as a Target for Drug Therapy Famesyltransferase Inhibitors The FTase Inhibitor Paradox: Efficacy without Cytotoxicity 51 51 53 References: 57 46 Chapter 2: K-ras is an Essential Gene in Mouse Embryogenesis with Partial Functional Overlap with N-ras Abstract Introduction Results: Disruption of the Murine K-ras Gene K-ras is an Essential Gene Fetal Liver Defect in K-ras-1 - Embryos Functional Analysis of K-ras-/ - Hematopoiesis K-ras-/- ES Cells have Reduced Contribution to Hematopoietic Compartments No Upregulation of Ras Family Members in K-ras-1-Embryos Genetic Interaction Between K-ras and N-ras N-ras-/-; K-ras+/-Embryos Die with Severe Anemia Hematopoietic Colony Formation in Cultures of N-ras-/-; K-ras+l- Yolk Sacs N-ras-l-; K-ras+l-Embryos Die Between E3.5 and E9.5 85 89 91 94 96 99 102 104 114 116 Discussion: K-ras is an Essential Gene in the Mouse K-ras and N-ras Interact Genetically Functional Requirement for Ras in Hematopoiesis Implications for Oncogenesis and Therapy 118 120 121 122 Experimental Procedures 124 References 131 Chapter 3: Analysis of an Oncogenic Allele of K-ras in the Initiation and Progression of Tumorigenesis in the Mouse via a Novel Approach 137 Abstract 138 Introduction 139 Results: Generation of ES Cells carrying a Spontaneously Activatable Allele of K-ras Creation of ES Cells Heterozygous for an Oncogenic Allele of K-ras Through Intrachromosomal Recombination of the K-rasD12 -A Allele in vitro Generation of Mice Carrying a Latent Oncogenic K-ras Allele, K-rasD12 -L, and its Tumorigenic Consequences 144 152 159 Discussion: Strategies for Analyzing Functional Mutations in the Mouse Examining the Multistep Nature of Cancer in the Mouse 165 169 Experimental Procedures 173 Chapter 4: References 177 Overview and Future Directions 182 Appendix 1: The PDGF Inducible Factor SIF is Related to the Interferon Activated p91 Transcription Factor 187 Abstract 188 Introduction/Results/Discussion 189 Experimental Procedures 199 References 202 Biographical Sketch 205 List of Figures and Tables The Ras Superfamily Post-translational processing of Ras proteins Conservation of the Ras signal transduction pathway Ras signaling pathways Effectors of Ras 13 18 23 40 44 Chapter 1: Table 1.1. Figure 1.1. Table 1.2. Figure 1.2. Table 1.3 Chapter 2: Figure 2.1. Disruption of K-ras in ES cells Figure 2.2. Viability plot for K-ras-1- embryos Figure 2.3. Phenotypic comparison of K-ras-1- embryos and control littermates Figure 2.4. GPI isoenzyme analysis on K-ras+l- and K-ras-l chimeras Figure 2.5. N-ras and H-ras are not upregulated in response to the loss of K-ras Table 2.1. Summary of dissection analysis for N-ras-l-; K-ras+lembryos Figure 2.6. Comparison of N-ras-l-; K-ras+/-and control littermates at E10.5 Figure 2.7. Histology of E9.5 and E10.5 N-ras-/-; K-ras+/ - and control embryos Figure 2.8. N-ras-1 -; K-ras+/ - embryos exhibit morphological abnormalities late in gestation Figure 2.9. Histology of E15.5-16.5 N-ras--'; K-ras+l- fetal livers Table 2.2. Summary of dissection analysis for N-ras-/-; K-ras-1embryos 86-88 90 92-93 Figure 3.1. Figure 3.2. 145-146 148-149 Chapter 3: Strategy for creating activatable alleles of K-ras Genomic configurations of the K-rasD12 -L and K-rasD12-A alleles following insertional recombination Table 3.1. Summary of targeting frequencies and integration patterns Figure 3.3. Observed integration patterns in targeted K-ras clones Figure 3.4. The aspartic acid and HindIII mutations appear to co-integrate Table 3.2. Summary of in vitro run step on K-rasD12 -A allele Figure 3.5. Functional intrachromosomal recombination in the K-rasD12-A allele Figure 3.6. Generation of ES cells carrying a functional oncogenic allele of K-ras Figure 3.7. Germ line transmission of the K-rasD12 -L allele Figure 3.8. Histopathology of Tumors in K-rasD12 -L allele mice 97-98 100-101 103 105-106 107-108 110-111 112-113 115 150 151 153 155 156 158 160 163-164 Appendix 1: Figure 1.1. Specific crosslinking of a 91 kD protein to the SIE in extracts of v-sis conditioned media treated cells Figure 1.2. Antisera to the ISGF3 p91 supershifts SIF complexes Table 1.1. Comparison of the sequences of the c-fos SIE and interferon responsive elements Figure 1.3. The c-fos SIE and IFN-y responsive elements cross compete for binding to IFN-y and PDGF induced complexes Figure 1.4. Inducible binding to the SIE is diminished in cell lines deficient in ISGF3-p91 190 192 194 195 197 Chapter 1 Overview of Ras Function and Signaling 10 The ras oncogenes were first identified as the transforming principle of the Harvey and Kirsten rat sarcoma viruses (1). Scientists rediscovered these genes while characterizing the existence of dominant oncogenes in human and animal tumors through the use of gene transfer assays (2-8). Most of these transforming genes were later identified as mutated alleles of cellular ras protooncogenes (3, 7-12). Ever since their discovery and association with human tumors, the ras genes have been the focus of intense research trying to understand the role that they play in neoplastic development. In addition, a large and ever expanding family of ras-related genes have been identified (13). Interestingly, none of these other members has been shown to be mutated in human cancers and only recently has it been demonstrated that some of these genes possess transforming ability in vitro (14, 15). This introduction will give an overview on the structural and biochemical properties of ras genes, the proteins regulating Ras activity, the function of Ras and its signaling pathway in normal and neoplastic cell growth, and finally, the implications of Ras as a target for chemotherapeutic drug strategies. I. Primary Structure Mammalian ras Genes and their Evolutionary Conservation Three closely related ras gene have been identified in the mammalian genome, H-, N-, and K-ras, with all three genes residing on separate chromosomes (1). The ras proto-oncogenes encode highly related proteins with a molecular weight of 21 kD. The coding sequences for each of these genes are equally distributed among four exons except for K-ras, which possesses two alternative fourth coding exons (exon 4A and 4B) (16-18). K-ras4B is the predominant form of K-ras in all tissues examined (19). Mammalian ras genes also contain a 5' noncoding exon, termed exon 0, which is located immediately downstream of their respective promoters (16, 17, 20, 21). In addition, the splice junctions for each member are highly conserved, suggesting that they arose from a common ancestral origin. However, their intronic structures vary substantially, with K-ras exhibiting the greatest complexity. The Ras proteins can be divided into four domains. The first domain encompasses the amino-terminal 86 amino acids of the proteins and is identical among all three genes in both humans and rodents (1). The next 80 amino acids comprise a second domain which is 85% conserved between each member. The region of greatest divergence defines a third domain and is commonly referred to as the hypervariable domain. This region ends with a conserved CAAX motif (C, cysteine; A, aliphatic; X, any amino acid) at the carboxy terminus and is present in all members of the Ras - and Rho-related subfamilies (see below). ras genes are highly conserved throughout evolution and have been identified in a number of eukaryotic organisms including fruit flies, plants, and yeasts (22-27). Comparatively, these genes exhibit a high degree of similarity at the protein level in both their structural domains and sequence (1). The highest degree of conservation lies within the amino terminal domain (not less than 84%) and CAAX motif. The most important feature of this evolutionary conservation of ras genes is their ability to function in heterologous systems. For example, mammalian ras genes can restore viability to rasl ras2 yeast mutants and can induce phenotypic alterations in yeast cells (19, 28-30). Similarly, a yeast RAS gene (carrying a deletion in the long hypervariable domain) as well as chimeric yeast-mammalian ras genes are able to efficiently transform NIH/3T3 cells (28). These studies provided the first example of the interchangeability between functional genes of yeast and mammalian systems. ras-Related Genes In the last decade, a number of Ras-related proteins have been identified and are collectively referred to as the Ras superfamily (13). Over 50 mammalian Ras-related proteins have been described so far and are an ever-expanding family. Based on sequence homologies, they can be grouped into five subfamilies: Ras, Rho, Rab, Ran, and ARF (Table 1.1). These genes are 50% or less similar to ras genes, with the highest conservation again occurring at the protein level in the 12 Table 1.1. The Ras Superfamily a Subfamily Members Biological Functions Ras H-ras, K-ras4A/4B, N-ras, R-ras, Rap lA/B, Rap2A/B, RalA/B, TC21 growth, survival, differentiation Rho RhoA/RhoB/RhoC, Rac 1/2, Cdc42/G25K, RhoG, TC10 actin cytoskeleton, integrin activity, stress response, NADPH oxidase Rab Rabl to Rab26 vesicle transport ARF ARF1 to ARF6 vesicle transport stimulation of PLD Ran Ranl nuclear import Table lists the members of the mammalian Ras superfamily of small GTP binding proteins. aAdapted from Hall, A. (1994). Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu. Rev. Cell Biol. 10:31-54 13 amino terminal and CAAX domains (1). In addition, some members (e.g., rho and rac) have been demonstrated to be as well conserved as the ras genes in the phylogenetic scale. The Ran subfamily has evolved to function in nuclear import, while the Rab and ARF subfamilies control various aspects of vesicular transport. The Ras- and Rho-related proteins, on the other hand, regulate signal transduction pathways coupling plasma membrane receptors to various biological responses (13). While most research has centered around Ras because of its role in growth control and tumorigenesis, this is rapidly changing. Recent studies have demonstrated a role for Rhorelated proteins in controlling the organization of the actin cytoskeleton, and more importantly, a link between these processes and ras-associatedtransformation (31-34). A more thorough analysis of the Ras and Rho-mediated signal transduction pathways is discussed below (see The Ras Signal Transduction Pathway). H. Biochemical Properties of Ras Guanine Nucleotide Binding Members of the Ras superfamily belong to a class of proteins that bind guanine nucleotides (GTP and GDP) and exhibit intrinsic GTPase activity. The essential feature of these proteins is their ability to function as a molecular switch that is turned on and off by the regulated cycle of bound GTP and GDP, respectively (1, 35). In this sense, the Ras proteins are similar to other two major families of GTP binding proteins, the elongation and initiation factors involved in protein synthesis and the heterotrimeric G proteins (35-37). The relevance of this activity to Ras biological function has been demonstrated by the following lines of evidence: 1) Ras mutants that fail to bind guanine nucleotides are no longer transforming in NIH/3T3 assays (38, 39); 2) the malignant phenotype of NIH/3T3 cells transformed by oncogenic ras can be reversed by microinjecting antibodies that inhibit guanine nucleotide binding or GDP to GTP exchange (40, 41); and 3) oncogenic mutants of Ras are severely impaired in their ability to hydrolyze GTP (42-46). 14 Through a combination of mutagenesis and X-ray crystallography studies as well as sequence comparison to other G proteins, the amino acids in Ras critical for guanine nucleotide binding and hydrolysis have been deciphered (1, 47, 48). Oncogenic mutations at positions 12, 13, 59, 61, and 117 result in impaired GTPase activity (both intrinsic and stimulated), thereby resulting in the accumulation of Ras in the GTP-bound or active state. In contrast, altered guanine nucleotide exchange rates result from mutations in residues 16, 17, 116, 117, 119, 144, and 146. These transforming mutations increase the rate at which Ras exchanges guanine nucleotide with the surrounding medium due to an increased dissociation constant for GDP. Since the intracellular concentration of GTP is much higher than GDP, this too, results in an increase in the amount of Ras bound to GTP (47). The three-dimensional structures of Ras-GDP and Ras-GTP have provided significant understanding of the effects of Ras mutations and the mechanisms of action of Ras regulatory proteins (47). There are five regions of the Ras polypeptide that are associated with loops on one side of the protein. These regions are designated as G-1 to G-5 and are critical in GDP/GTP exchange, the GTP-induced conformational changes, and GTP hydrolysis. Comparison of the GDP- and GTP- bound forms revealed prominent GTP-induced changes in two regions of the protein: the G-2 loop (amino acids 30-38) which is implicated in effector interaction, and the G-3 loop together with the adjacent a-helix (specifically, residues 60-76) (49-51). These two regions are commonly referred to as Switch I and Switch II, respectively. Furthermore, it is these two loops that are most strongly affected by oncogenic mutations (52, 53). Analysis of the active site has yielded insight into the mechanistic action of oncogenic mutations on GTP hydrolysis. The side chain carbonyl of Gln 6 1 is postulated to activate a water molecule which mounts a nucleophilic attack on the y-phosphate of GTP (47). Importantly, the side chain of Gln 6 1 must be in the proper orientation to facilitate this process. Substitution of this residue by most all other amino acids is believed to impair activation of the attacking water molecule and subsequent nucleophilic attack. In contrast, oncogenic Ras mutants derived by substitution of Gly 1 2 or Gly 13 with any other amino acid besides Pro are believed to block the 15 entrance of the guanine nucleotide pocket through steric hindrance (via their side chains) or loop distortion, respectively. In either case, the entry of a nucleophilic attacking group is prevented. The end result of all three mutations is the impairment of GTPase activity, and hence, constitutive activation of the Ras protein. Two inhibitory molecules have been widely used to block normal Ras activation in cells in an effort to determine the role of normal Ras in signaling pathways. Therefore, their mechanism of action deserves special mention. The first inhibitor contains a single amino acid substitution at position 17 (N17) in Ras. This protein is thought to bind normally to guanine nucleotide exchange factors (see Regulators of the Ras GTP/GDP Cycle) and promote GDP dissociation; however, due to a reduced affinity for GTP, this mutant remains bound to the exchange factor (54-56). Thus, this sequestration of Ras activators will prevent the activation of normal Ras proteins and effectively inhibit their signaling. The second inhibitor is an anti-Ras monoclonal antibody, Y13259, that binds to residues 63-73 in the Switch II region (57). This antibody does not inhibit guanine nucleotide binding or GTP hydrolysis, but rather has been proposed to neutralize Ras function by preventing a conformational change in Switch II that is necesssary for GDP to GTP exchange (49). Binding of the antibody to Ras proteins is believed to freeze the conformation of this region such that the release of bound GDP or the exchange to bound GTP can not proceed, thereby impairing Ras activation and signaling. Post-translational Modifications and Membrane Association A substantial amount of experimentation has demonstrated that Ras proteins are translocated to the inner face of the plasma membrane (58). This localization is critical for Ras biological activity and is mediated through a series of closely linked post-translational modifications at the carboxy terminus (C-terminus) (58-60). The first modification is the addition of an isoprenoid moiety to the cysteine residue of the CAAX motif. Subsequently, the three teminal AAX residues are removed by proteolytic cleavage, and carboxyl methylation of the now C-terminal cysteine residue occurs. Mutant Ras proteins that lack either the cysteine or AAX 16 residues of the CAAX motif fail to undergo these processing steps and, as a result, are not associated with the plasma membrane and are biologically inactive. These modifications impart a hydrophobic nature to Ras; however, the exact contribution and functional role of each event has been difficult to assess. Utizilizing mutants that were blocked in one or more steps, researchers concluded that isoprenylation was sufficient to promote plasma membrane association and biological activity of both normal and oncogenic Ras proteins, although at significantly reduced levels (-50%) (61-63). Thus, all three processing events are necessary for optimal membrane association and activity. Although these CAAX-signaled modifications are critical for plasma membrane association, studies of Ras mutants and other similarly modified proteins suggested that additional modifications were necessary to confer both optimal avidity and specificity for the plasma membrane. First, a number of other proteins containing a CAAX motif are also isoprenylated, yet either localize to the nuclear membrane or remain cytosolic (58). Second, mutational analysis demonstrated that sequences directly upstream of the CAAX motif provide an additional membrane targeting signal that complements the CAAX modifications to promote avid and specific plasma membrane binding (64-66). In the cases of H-, N-, and K-ras4A, these proteins are subsequently palmitoylated at an upstream cysteine residue(s) (Figure 1.1). K-ras4B, however, has substituted this palmitylation site with a polylysine domain that serves an analogous function (65). This positively charged polylysine domain is believed to promote interaction with negatively charged phospholipid head groups, as it can be substituted with similarly charged arganine residues but not with neutral glutamines. Mutant Ras proteins that lack either these upstream cysteine residues or the polylysine domain are rendered cytosolic, despite retaining the modifications at the CAAX motif. Moreover, only the addition of the carboxy terminal residues from either H-ras or K-ras4B which included either the palmitoylated cysteines or lysine-rich domain, respectively, was capable of promoting efficient plasma membrane association to Protein A (64, 66). In summary, all of the above modifications are both necessary and sufficient to promote full and specific association to the plasma membrane. In addition, K-ras4B can be 17 1 Q) 1 C~JD _IL I .......c...~....~ 0 c t-( 0 I 0 0 ..... ... . ..· . . .. 0~ II 0 z Z 0 C C 4" C E 0 0 w Lu II El x II U~ C oE 0 Q) (TJ L. Q. I C m Em a. 0 0 E o 0 III II o II II "O E p r II IL c\i 0 0u 06 2 0, co r cu coJ U- (D 0 E 0ca Cu v, phosphorylated by protein kinase A or protein kinase C at a serine residue located between the polylysine domain and the CAAX motif (67). The significance of this is not clear, but may be involved in mediating the interaction with a specific regulatory exchange factor (see Guanine Nucleotide Dissociation Stimulators). Interestingly, oncogenic Ras proteins that lack palmitoylated cysteines or the polylysine domain but retain the CAAX-signaled modifications still retain strong transforming activity in NIH/3T3 cells despite the absence of observable plasma membrane association (64, 65). Therefore, this suggests that plasma membrane association may not be critical in mediating the transforming function of Ras, and that the required CAAX modifications facilitate transformation through a role unrelated to promoting plasma membrane association. Protein prenylation is currently known to be signaled by at least three distinct carboxyterminal motifs, CAAX, CC, or CXC (60, 68). Proteins terminating in a CAAX sequence are modified by the addition of either a farnesyl or geranylgeranyl group, through the enzymatic activities of farnesyltransferase (FTase) and geranylgeranyltransferases I (GGTase I), respectively. In contrast, CC and CXC motifs are specifically geranylgeranylated by yet a third enzyme, geranylgeranyltransferase II (GGTase II). The variable amino acid (X) of the CAAX motif was originally thought to be the sole determinant of the relative specificity for FTase and GGTase I. A number of studies examining the substrate specificity of prenyltransferases used either short peptides corresponding to various CAAX sequences or Ras proteins mutagenized at this motif (69-72). These studies concluded that FTase strongly preferred a methionine or serine residue at the X position, whereas geranylgeranylated proteins typically ended in leucine. Since all four Ras proteins end in either serine or methionine, it was inferred that they only underwent farnesylation in vivo. However, it has recently been demonstrated that K-ras4B is geranylgeranylated as well as farnesylated both in vitro and in vivo (73, 74). This dual substrate property of K-ras4B has been attributed to the combined effects of the CAAX (CVIM) motif and the polylysine domain (74). Since all of the initial studies focused exclusively on the CAAX sequence and did not examine authentic K-ras4B, this geranylgeranylation of K-ras4B had previously been unnoticed. At least 19 two other Ras-related proteins, R-ras2/TC21 and RhoB, have been shown to be modified in vitro by both farnesyl and geranylgeranyl isoprenoids, and RhoB has been shown to incorporate both moieties in vivo as well (75, 76). The significance of this alternative processing for K-ras4B is discussed below (see sections Guanine Nucleotide Exchange Factors and The FTase Inhibitor Paradox: Efficacy without Cytotoxicity). The Effector Domain Deletion mutants of mammalian ras genes have defined five independent domains that are absolutely required for Ras function: residues 5-63, 77-92, 109-119, 139-165, and the C-terminal CAAX motif (39). While it was clear that some of these residues possessed roles in guanine nucleotide binding or membrane localization, extensive mutational analysis also revealed the presence of residues critical in mediating the effector activity of Ras proteins. Thus, certain mutations destroyed the ability of oncogenic Ras to signal, and yet did not compromise their ability to bind to either GTP or the plasma membrane (57). Most of these mutations map to the Switch I domain (residues 30-38), thus supporting a regulated effector interaction based on the conformational change associated with this region upon GTP binding (47, 49). In addition, the accessibility of this domain on the external surface of the molecule supported its involvement in the interaction with putative cellular targets. The recent identification and characterization of Ras effector molecules has confirmed and extended this analysis. Both biochemical and genetic studies employing allele-specific suppressors have defined interacting residues in Ras as well as in the Ras-binding domain (RBD) of effector molecules (77). Interestingly, residues 32-38 are conserved among all members of the Rassubfamily, indicating that determinants controlling specificity must lie outside of these core conserved residues. Due to its flexibility, the SwitchlI region (residues 60-76) can exist in multiple conformations (50). Because of this, it has been suggested that this region may be involved in mediating specificity through these different conformations. This idea was recently supported 20 through the demonstration that mutations within SwitchlI can compromise Ras function and selectively disrupt the interactions with a subset of known effectors (Table 1.3), including phosphatidylinositol 3-kinase (PI3K), neurofibromin (NF1), and the cysteine-rich domain of Raf1 (78-80). III. Functions of ras Genes Non-Mammalian Systems Due largely to the power of genetics, the Ras pathways in Saccaromyces cerevisiae, Caenorhabditiselegans, and Drosophila melanogasterhave yielded significant insight into the mechanism of Ras regulation and signaling (81-84). The Ras genes identified in these organisms show high sequence conservation with one another as well as with mammalian ras genes (1). This structural conservation underscores their ability to function in heterologous systems. In addition, many of the regulatory proteins and downstream signaling molecules that comprise the signaling pathway are highly conserved and will be discussed in more detail below (see The Ras Signal Transduction Pathway). In D. melanogaster and C. elegans, Ras proteins serve essential roles and mediate signaling cascades downstream of a number of receptor tyrosine kinases (RTKs) (82-84). In D. melanogaster, the Ras genes are ubiquitously expressed as they are in mammalian systems, but are predominantly expressed in neural cells. These RTK-Ras mediated signal transduction pathways are important in patterning, segmentation, and photoreceptor development. The best characterized pathway has demonstrated a critical role for Ras downstream of the Sevenless RTK in determining the cell fate of the R7 photoreceptor cell in the Drosophilaeye (83, 84). In C. elegans, the let-60 gene encodes a Ras homologue that participates in several developmental pathways. Loss-of-function of let-60 results in larval lethality, aberrant vulval development in hermaphrodites, abnormal male spicule development, sterility, and mis-specification of a cell type in the posterior ectoderm (82, 85). Extensive genetic analysis of let-60 function in vulval 21 development has lead to the identification of critical mediators in this pathway and has revealed its conservation with the RTK-Ras signaling pathway critical for specification of the R7 photoreceptor cell in Drosophila (Table 1.2). In S. cerevisiae,two RAS genes have been identified, RAS1 and RAS2 (25, 27). Mutation in either gene individually is compatible with growth, however, raslras2double mutations are lethal (86). These genes are essential for stimulating production of cAMP via adenylate cyclase in a nutrient-response pathway. This signaling pathway is distinct from the RTK pathways used in other organisms (Table 1.2). However, many of the kinases downstream of Ras in worms and flies are still present in S. cerevisiae, but appear to serve a critical function that is RAS-independent in the pheromone-response pathway (81). In addition to these systems, ras genes also serve critical roles in Xenopus laevis, Schizosaccharomyces pombe, and Dictyostelium discoideum (1, 48). Mammalian Systems The prevalence of oncogenic ras mutations in human cancers signifies the important role that Ras proteins play in regulating normal cell growth (87, 88). The phenotypic effects of Ras range from the transformation of fibroblasts to the differentiation of neural cells (1). The mitogenic response of NIH/3T3 cells to serum growth factors can be blocked by microinjection of the Ras monoclonal antibody Y13-259 or by the expression of dominant inhibitory ras mutants, thus demonstrating a critical requirement for Ras activity in normal cell growth and proliferation (89, 90). The constitutive output of this growth-promoting signal by oncogenic ras mutants can induce morphological transformation of fibroblasts, which is accompanied by membrane ruffling, pinocytosis and the activation of immediate early (e.g., c-fos and c-jun) and other cellular genes (48, 91). In other cell types, however, microinjection of oncogenic Ras can mimic the effects of nerve growth factor (NGF) and induce neuronal differentiation, as has been demonstrated in the PC12 pheochromocytoma cell line (1). Furthermore, the differentiation of neuronal cells in 22 Cu cu C) 0 Cu I-. Cu 0 0 Cu cri 0= qý *o1ý rz q6) o0 t_ 3e C) Ic) C)~o OC) -aa bL~ S 0 ed "- "om" C) "X~a C)o as Cu 'S~U ~ C)) ~ c) o CO2c- -e 4 IC2C) i n"C SSE OC c Cu2.2 -ee t~~ 0I U U c'- a, 04 -~ F?'O oe ~1 W a+4' 0'" OClb obI~` o0u 0a C) ~a Cd03 c x ~00 C) I· - ci cc c Cu " o~ vl 2, S1~~f~ C4c -C 4C) C) Vc C a .9 CISu e*_ V) , v, r c 2< co'-4 - " IC d0 C· 2 C .Ae -~ '-4 ) C 25 <-e 0 ' ab C,3 , er • 5 C) 0 .c, 4o' C C)C)aj response to NGF can be blocked by dominant inhibitory ras mutants or anti-Ras antibodies (92, 93). A wide variety of extracellular stimuli have been shown to lead to a rapid and transient increase in the amount of active GTP-bound Ras proteins (94, 95). These stimuli include factors such as those which promote the growth of fibroblasts (e.g., PDGF, EGF), the proliferation and differentiation of hematopoietic cells (e.g., IL-3, GM-CSF), and the differentiation of neuronal cells (e.g., NGF). A key feature that many of these growth/survival factors have in common are receptors that either act as tyrosine kinases themselves (e.g., PDGF and EGF receptors) and/or associate non-receptor tyrosine kinases (e.g., IL-2 Receptor). In addition, transformation of fibroblasts with a variety of tyrosine kinase oncogenes (e.g., src, fms, and fes) results in chronically elevated levels of Ras-GTP. The transforming function of these oncogenes is dependent upon Ras activity, as co-expression of dominant inhibitory ras mutants or microinjection of anti-Ras monoclonal antibodies blocks their transforming potential (96). Thus, as in flies and worms, Ras proteins are essential components of tyrosine kinase-mediated signaling pathways that regulate cellular growth, survival, and differentiation. A Causative Role for Oncogenic ras in Tumorigenesis While the precise contribution of oncogenic Ras function to the initiation and progression of tumorigenesis is not known, there is considerable epidemiological and experimental evidence that has suggested an important role for oncogenic Ras in the development of human tumors (87, 88). Oncogenic ras mutations have been identified in approximately 30% of all human tumors, with Kras mutations occurring most frequently. A number of tumor types have been associated with ras mutations, although the incidence of mutation varies strongly among different tumor types. The highest incidence occurs in pancreatic adenocarcinomas, where greater than 80% of tumors carry a mutation in K-ras (97-99). Carcinomas associated with the colon and thyroid also exhibit frequent alterations, with approximately 50% of these tumors harboring a mutant ras gene (100-102). In contrast, a number of tumor types (e.g., neuroblastoma) have not been or are very rarely associated 24 with a mutated ras gene (88). Some correlation can be drawn between a given tumor type and the ras gene that is mutated. For example, adenocarcinomas of the pancreas, colon, and lung predominantly carry mutations in the K-ras gene, whereas myeloid disorders tend to selectively carry N-ras mutations (88). However, some tumor types (e.g., thyroid tumors) seem to lack specificity for a particular ras gene mutation. This observed specificity in ras mutation may be associated with differences in Ras function, or alternatively, may simply reflect differences in the level of expression. The ras genes have been shown to be ubiquitously expressed; however, certain tissues preferentially overexpress one or two members of the family (88, 103-105). In the colon and lung, for example, K-ras is abundantly expressed and is the most frequently mutated member in carcinomas of these tissues. H-ras, on the other hand, is overexpressed in the skin and is the only ras gene mutated in rodent skin carcinogenesis models (106). It is important to note that all of the expression levels were measured in the whole tissue and, therefore, do not necessarily reflect the level of ras expression in the target cell. However, the correlation drawn between observed expression levels and the specificity of ras mutation may be very important as the level of oncogenic Ras can influence its transforming potency and is often times augmented during tumor progression (see below and Chapter 3). Rodent carcinogenesis models have also underscored the importance of ras gene mutations in tumorigenesis (106, 107). The frequent and reproducible activation of ras oncogenes in these animal tumors supports the concept that ras mutation plays a causative role in neoplastic development. In mice, for example, H-ras mutations are associated with 90% of skin papillomas and carcinomas that have been initiated by treatment with demethylbenz(a)anthracene (DMBA) and followed by promotion with the phorbol ester, TPA (8, 108). K-ras mutations, on the other hand, are frequently associated (up to 100% in some studies) with lung tumors in mice that have been induced with a variety of chemical mutagens (1, 106). This reproducible activation of the ras genes has made it possible to correlate their activating mutation with the known mutagenic effects of particular carcinogens. For example, the induction of rat mammary carcinomas by nitrosomethylurea (NMU), but not DMBA, treatment resulted in activation of the H-ras oncogene via a 25 G to A transition (109, 110). This transition results from the miscoding properties of 0 6methylguanosine, which is one of the adducts generated by the methylating activity of NMU. In contrast, DMBA is known to form large adducts with both adenine and guanosine residues, but the repair of these adducts very rarely leads to the generation of G to A transitions (111). This concept that ras oncogenes can be the direct target of chemical mutagens is supported further by observations drawn from skin carcinogenesis studies in mice. The induction of skin carcinomas by treatment with DMBA and phorbol esters involved the specific activation of H-ras oncogenes by A-to-T transitions at the second base of codon 61 (108). However, when the initiating mutagen was replaced by an alkylating agent, this mutation was not observed. Taken together, these findings indicate that the ras oncogenes can be a direct target of the mutagenic properties of inititating carcinogens and support a causative role of ras activation in tumorigenesis. The importance of oncogenic Ras in cancer has been supported further by the generation of mice carrying mutated ras sequences either through retroviral infection or the creation of transgenic mouse strains (112). These mouse strains display increased incidences of tumor formation, which are generally associated with their tissue-specific expression. For example, mice carrying a mutated H-ras gene whose expression is driven by the LTR of the mouse mammary tumor virus (MMTV) are predisposed to the development of tumors in mammary, salivary, and lymphoid tissues (113). Importantly, however, these tumors arise only after a relatively long latency period and are focal in nature. Since the MMTV promoter directs expression of the H-ras oncogene to every cell in the mammary gland, this suggests that secondary cooperating genetic events are necessary for tumor development. Oncogenic ras and Multistep Carcinogenesis The idea that ras mutations cooperate with other genetic events in the initiation and progression of tumorigenesis was first conceptualized by analyzing the in vitro transforming properties of ras oncogenes in tissue culture. Cellular ras oncogenes can not transform primary rodent fibroblasts alone (114-116). Instead, transformation of these cells requires the presence of cooperating genes, 26 such as myc, adenovirus E1A, SV40 TAg, or p53 (114, 116-121). Interestingly, ras oncogenes were sufficient to transform primary rodent fibroblasts if the inhibitory effect of neighboring normal cells was eliminated. This was achieved by cotransfection with the neomycin selectable marker followed by selection with geneticin (122-124). The molecular basis of this inhibitory effect remains to be elucidated, but does help to explain the observation that retroviruses carrying oncogenic ras genes can transform primary rat fibroblasts but only at a high multiplicity of infection (1). These initial observations suggested that activation of ras oncogenes was insufficient to trigger transformation and has since been supported in a number of animal and human tumor models. The cooperation observed in tissue culture between the ras and myc oncogenes was very elegantly demonstrated in vivo through the use of transgenic mice. Transgenic mice carrying either the v-H-ras or c-myc gene driven by the MMTV promoter/enhancer were analyzed individually and in combination (113). As discussed above, MMTV/v-H-ras transgenic mice develop focal lesions in the mammary, salivary, and lymphoid tissues after a long latency period. When these mice were crossed into the MMTV/c-myc strain, a dramatic and synergistic acceleration in tumor formation was observed. However, these tumors still arose stochastically and suggesed that additional somatic events were necessary for full malignant progression. The mouse skin carcinogenesis model has provided an excellent system for dissecting the role of Ras in the initiation and progression of skin carcinomas. Because these tumors evolve through a series of well-defined pathological stages, it has been possible to establish an order in which specific genetic alterations occur during the development of these tumors (125, 126). As discussed earlier, H-ras mutations are associated with greater than 90% of mouse skin papillomas and carcinomas (8, 108). DMBA treatment of the skin of mice is believed to result in mutational activation of the H-ras gene. These "initiated" cells are known to remain dormant until they are induced to proliferate by treatment with tumor promoters such as phorbol esters. Direct support of this model came from the observation that mouse epidermal skin cells infected with Harvey-MSV did not exhibit neoplastic properties unless they were treated with the phorbol ester, TPA (127). In 27 mice, the benign papillomas that result following promotion with TPA are known to regress to small hyperkeriotic lesions, with only a small percentage progressing to a malignant carcinoma. This suggests that ras activation is insufficient for the development of the full malignant phenotype and that additional genetic events are required to cooperate with oncogenic H-ras for tumor progression. Indeed, skin grafts of cultured keratinocytes infected with Harvey-MSV resulted in the formation of papillomas but not carcinomas (128). Transgenic mice expressing oncogenic Hras from a suprabasal keratin promoter have demonstrated further a direct involvement of oncogenic Ras in the development of skin papillomas (129). The genetic characterization of papillomas and carcinomas has revealed a number of alterations associated with progression to a malignant state. Interestingly, an increase in the copy number of the mutated H-ras allele or loss of the wild-type H-ras allele is frequently associated with skin tumor progression (130). In addition, it has recently been demonstrated that the c-fos proto-oncogene is required for progression of papillomas to a malignant carcinoma (131). This multistep pathway of tumorigenesis has also been demonstrated in human colorectal cancer (132). In contrast to mouse skin carcinogenesis, colon cancer appears to be initiated by mutations in the APC tumor suppressor gene, which are associated with more than 60% of adenomas and carcinomas (133). Mutation of a ras oncogene (K-ras) also occurs early in the development of colorectal neoplasia and is mutated in 50% of adenomas and carcinomas (100102). Extensive analysis of numerous benign lesions exhibiting varying degrees of dysplasia has led to a model in which mutation of the APC gene leads to the formation of benign adenomas (134). Mutation of a ras gene frequently occurs in one these benign tumor cells, leading to a further clonal expansion. The order in which APC and ras mutations occurs appears to be a critical determinant for the ability of these "initiated" tumor cells to progress (135). Those correctly "initiated" cells will subsequently acquire additional cooperating mutations (e.g., mutations in DCC and p53) which will lead to the clonal expansion of cells with an even greater transforming potency and ultimately result in the progression from a benign to malignant state. 28 Importantly, the level of oncogenic Ras expression appears to modulate its transforming ability. It has repeatedly been observed that the ratio between mutant and normal ras alleles is frequently increased during tumor progression (1, 87). This can occur through either amplification of the mutated allele or loss of the wild-type allele (101, 130, 136). Augmentation of oncogenic ras expression has also been demonstrated to result from mutations in intronic regulatory sequences that result in increased expression (137, 138). In addition, the expression of wild-type ras genes at artificially high levels can lead to transformation (139-143). Because many tumors contain a normal ras allele in addition to a mutated allele, oncogenic ras was believed to exert its effect in a dominant fashion. However, the above observations have questioned this. To address this issue directly, Finney and Bishop generated Ratl fibroblasts carrying an oncogenic allele of H-ras through gene targeting (144). Their results demonstrated that cells heterozygous for an activated H-ras allele were neither transformed nor tumorigenic. predisposed to transformation at a low frequency. Rather, these cells were Interestingly, greater than 90% of the transformed cell lines had amplified the mutant allele. Thus, these results confirm all of the above observations that oncogenic ras is insufficient for transformation, and that at least one additional event must occur, of which increasing the expression of a mutant ras allele may be one. IV. Regulators of the Ras GTP/GDP Cycle Although Ras proteins exhibit intrinsic GTPase and GDP/GTP exchange activities, these rates are too low to account for the rapid and transient GDP/GTP cycling that occurs following mitogenic stimulation (35). Rather, the interconversion of inactive GDP-bound and active GTP-bound Ras proteins is modulated by the concerted action of guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) (48, 145-147). GEFs bind to the GDP-bound form of the GTPase and catalyze the dissociation of GDP and subsequent replacement with GTP, thereby activating the G protein and its signaling cascade. In contrast, GAPs serve to downregulate the GTPase signal by stimulating the intrinsic rate of hydrolysis of bound GTP to GDP. 29 GuanineNucleotide Exchange Factors Regulatory factors that enhance and control the exchange of GDP to GTP on Ras would be expected to function/act directly upstream of Ras in a signaling pathway. The first identification of such factors came from genetic dissection of the Ras mediated adenylate cyclase pathway in S. cerevisiae. CDC25 was isolated as an upstream activator of this pathway (148). A genetic screen for suppressors of cdc25 mutations yielded a homologue known as SDC25 (149). Both genes were later shown to promote GDP/GTP exchange on the yeast Ras proteins (150, 151). Unlike SDC25, CDC25 is an essential gene in yeast (152). Since then, homologues of CDC25 have been identified in S. Pombe (Ste6) and D. melanogaster(Sos, for the Son-of-sevenless gene product) (153, 154). The first mammalian homologues of CDC25 were isolated from either rat, mouse or human brain libraries using degenerate oligonucleotides and by functional complementation assays of yeast cdc25 mutants with a mouse expression library (155-157). Ras-GRF (also known as mCdc25) was isolated and shown to be specifically expressed in brain tissue (158). Moreover, this GEF specifically catalyzed the GDP/GTP exchange on Ras, but not on other Ras-related proteins. Similarly, two mammalian counterparts of the D. melanogasterSos gene were isolated from human (hSOS1) and mouse (mSos-1 and mSos-2) libraries and shown to possess Ras exchange activity (159, 160). In contrast to Ras-GRF, these exchange factors are widely expressed during development and in adult tissues. Thus, at least three distinct CDC25-related Ras GRFs are present in mammalian cells. Over the past few years, a number of other GRFs have been isolated in various systems and characterized with regard to their substrates (145, 161). In mammals, a subset of these exchange factors, including Ras-GRF, Sos, Vav, Dbl, and Tiam- 1, all share a region of homology known as the Dbl domain. Many proteins for which no GEF activity has been demonstrated also contain a Dbl-homologous domain, such as the GTPase stimulating proteins Bcr and Abr. Therefore, the presence of a Dbl-domain does not appear to equate with GEF activity. Interestingly, several of these Dbl-like proteins (e.g., Vav, Dbl, Tiam-1, and Ect2) were originally 30 isolated by virtue of their oncogenic activity (162-164). Both Dbl and Tiam-1 have been shown to function as GEFs for Rho-related GTPases (165, 166). In contrast, Ras-GRF, Vav, and Sos have all been implicated as GEFs specifically for Ras and not Rho-related proteins (145, 167-169). Thus, sequence relationship between given exchange factors does not allow one to predict their substrate specificity. Perhaps, the most interesting GEF with respect to this thesis, is Smg GDS. This exchange factor does not share homology with other known GEFs and is the only exchange factor that has been shown to distinguish among the Ras proteins (170, 171). Smg GDS is active on K-ras4B and several Ras-related proteins (e.g., Rapla and Ib, RhoA and B, Racl and 2, and Cdc42) yet is inactive on H- and N-ras (171). Interestingly, all of the known substrates for Smg GDS contain a polylysine domain immediately upstream of the CAAX motif, and this result suggests that the determinants for interaction are contained within these sequences. Indeed, it has been demonstrated that all post-translational processing steps as well as the presence of the polylysine domain in the C-terminus of Rapl are important in mediating the interaction with Smg GDS (171, 172). In addition, many of these substrates contain a serine residue positioned between the polylysine domain and the CAAX motif. Phosphorylation of this residue makes Rap sensitive to the action of Smg GDS and is, therefore, believed to initiate Rap activation (173). It is tempting to speculate that this mode of regulation also operates in K-ras4B, since it too, can be phosphorylated at this residue (67). Detailed kinetic analysis using K-ras4B as a substrate has demonstrated additional unique properties of Smg GDS. First, Smg GDS specifically interacts with and stimulates the GDP/GTP exchange reaction of only the lipid-modified form of K-ras (169). In contrast, mCdc25 (and its homologues) is active on both the lipid-modified and unmodified forms of all Ras proteins. Second, both Smg GDS and mCdc25 stimulate the dissociation of GDP from K-ras and form a stable binary complex with K-ras. However, only Smg GDS forms a stable ternary complex with K-ras-GTP (174). Third, mCdc25 stimulates the GDP/GTP exchange reaction on both membrane-bound and soluble forms of K-ras, whereas Smg GDS is far less active on the 31 membrane bound form (174). And finally, Smg GDS translocates membrane K-ras-GTP to the cytoplasm as a stable ternary complex of Smg GDS-K-ras-GTP (174). The significance of this translocation is that it may allow K-ras4B to interact with a subset of effector molecules distinct from those of H- and N-ras. The functional significance of multiple Ras GEFs is not fully understood. Because Ras proteins mediate a signaling pathway downstream of a wide range of cell surface receptors (as discussed above), this degree of complexity in Ras activators may provide the necessary coordination of signals emanating from different activated receptors. Moreover, many of the GEFs appear to be restricted in their expression profiles (e.g., mCDC25 in the brain, Vav in hematopoietic cells), suggesting that additional Ras GEFs may exist in other tissues. GTPase Activating Proteins To date, four distinct mammalian GAPs specific for Ras proteins have been identified, these being pl20-GAP, neurofibromin (NF1), and two members of the GAPI family (175-181). The first GAP to be cloned and characterized was p120-GAP (175, 176, 182). This protein is ubiquitously expressed, predominantly cytosolic, and stimulates Ras GTPase activity by more than 10,000 fold. In addition, pl20-GAP is alternatively spliced at the N-terminus, resulting in the removal of hydrophobic sequences (175, 183). The functional significance of this isoform (p100-GAP) is unknown and it appears to exist only in the placenta. Based on sequence homologies, p120-GAP can be divided into two functional domains. The catalytic domain is located at the C-terminus and contains the Ras-binding domain. This domain interacts with the effector domain of Ras (AA 3240) to stimulate GTP hydrolysis (184, 185). The N-terminal domain contains many key structural features, including one SH3 and two SH2 domains, a pleckstrin homology domain (PH), and a calcium-dependent phospholipid-binding (CaLB) domain (145). The SH2, SH3 and PH domains are common features of many signaling proteins and are believed to mediate molecular recognition (186, 187). The target sequences of src-homology domains have been defined through rigorous biochemical characterization. SH2 domains recognize specific sequences containing 32 phosphorylated tyrosines residues, whereas SH3 domains interact with specific proline-rich sequences. The binding sites of PH domains, on the other hand, are currently unknown (188, 189). Interestingly, though, other proteins in the Ras regulatory pathway contain PH domains, including the guanine nucleotide exchange factors Ras-GRF, mSOS, and Vav. The importance of Ras GAP activity in the regulation of cell growth has been underscored by the discovery that the human neurofibromatosis type I (NFl) gene encodes a protein with associated Ras GAP activity. Neurofibromatosis type I is a hereditary disease associated with germline mutations in one allele of the NF1 gene and is one of the most common genetic diseases that predisposes humans to cancer (1 in 3500 affected individuals worldwide) (190). Patients with NF1 develop benign neurofibromas at a high frequency, have a greatly increased risk of malignant tumors primarily of neural crest origin (e.g., neurofibrosarcomas and malignant Schwannomas), and usually exhibit other abnormalities as well. The NF1 gene, like pl20-GAP, undergoes alternative splicing (191). The product of the NF1 gene, neurofibromin, shares sequence homology with the catalytic domain of p120-GAP, but exhibits an even more extensive homology overall with the IRA1 and IRA2 GAPs from S. cerevisiae(177, 192). Like pl20-GAP, NF1-GAP also stimulates the intrinsic GTPase activity of Ras (178, 179, 193). Interestingly, NFl-GAP binds to wild-type Ras proteins with a 30-fold higher affinity than does p120-GAP, yet the specific activity of pl20-GAP is about 30-fold higher than that of NF1-GAP (178). Thus, the NF1-GAP protein might be expected to inactivate Ras more effectively than pl20-GAP at lower concentrations. The significance of this in the whole cell is not yet clear. The negatively regulatory role for GAPs has been supported through numerous criteria. The strongest evidence is the fact that they convert Ras into an inactive state by stimulating the GTPase activity. In addition, oncogenic Ras mutations (primarily at residues 12, 13, and 61) reduce or abolish GAP-stimulated GTPase activity and result in the accumulation of Ras in an active GTP-bound form (53, 176, 194). This constitutive activation of Ras can ultimately lead to unregulated cellular proliferation. Further evidence supporting a role in the downregulation of Ras activity comes from the observation that overexpression of p 20-GAP can block transformation 33 by wild-type Ras (195-197). Finally, genetic analysis in numerous systems seems to indicate that Ras GAPs negatively regulate Ras function by increasing the level of Ras-GDP. Various cell lines obtained from human NF1 patients or mice deficient in either Nfl or pl20-GAP have been examined for their sensitivity to growth factor-induced changes in Ras-GTP levels. NF1 mutant Schwann cells, for example, respond normally to growth factor stimulation, yet they exhibit an increased basal level of Ras.GTP (198). In contrast, pl20-GAP-deficient fibroblasts were more sensitive to growth factor induced changes in Ras-GTP levels than were NFl-deficient or wild-type fibroblasts (199). This type of analysis has suggested a model in which NFl-GAP may play a more important role in regulating the basal level of Ras-GTP, whereas pl20-GAP functions to downregulate Ras following the activation of receptor tryosine kinases. However, this may depend on the cell type and/or specific growth factor, as the loss of NFI was recently shown to sensitize cells to GM-CSF (200, 201). In addition to their negative regulatory roles in Ras signaling, it is also believed that both pl20-GAP and NF1 may possess a second role as downstream effectors of Ras activity (202, 203). This idea was first suggested by the observation that GAPs interacted with the effector domain of Ras in a GTP-dependent manner. Furthermore, mutations in the Ras effector domain that prevented transforming activity simultaneously prevented the association with GAP (176, 184, 185). In addition, oncogenic Ras mutants retain GAP binding activity even though they are unresponsive to stimulation by GAPs. In fact, some oncogenic Ras mutants bind to GAP with a higher affinity than do wild-type Ras proteins (53, 176, 194). Thus, GAP fulfills all of the expected criteria of an effector, in that it binds well to activated Ras and poorly to proteins exhibiting impaired signaling function. Further support has come from the finding that pl20-GAP interacts with a suppressor of Ras-induced transformation, Krev-1/Rapla. Krev-1 was isolated in a screen for proteins that could revert the transformed phenotype of K-ras transformed cells (204). This protein belongs to the Ras- subfamily and predominantly localizes to the Golgi apparatus (205). Interestingly, Krev1 binds to pl20-GAP with a higher affinity than does wild-type Ras, yet is insensitive to GTPase 34 stimulation by pl20-GAP (206, 207). Thus, it had been proposed that Krev-1 suppresses Ras transformation by binding to and sequestering p 120-GAP. If the role of p120-GAP were to solely downregulate Ras activity, then its inhibition would result in the accumulation of Ras-GTP, and therefore, Ras proteins should be activated by Krev-1 overexpression. Since the opposite phenotype was observed, it was speculated that Krev-1 suppressed transformation through the sequestration of an effector(s) of Ras activity. At the time this was proposed, pl20-GAP was the only protein known to interact with the Ras effector domain and exhibit all of the criteria expected of an effector molecule. Since then, numerous effectors have been identified and characterized, and it is now postulated that members of the Raf protein kinase family are more likely to be the critical targets of Krev-1 overexpression (208); however, this does not exclude the possibility that GAP may still play an important role in the suppressive phenotype. While all of the above observations provide indirect evidence for an effector function of pl20-GAP, direct evidence has been obtained in guinea pig atrial membranes. In this system, Ras and pl20-GAP collaborated to inhibit the G protein (Gk) mediated coupling of a muscarinic receptor to a potassium channel (209). Oncogenic mutants of Ras were more effective inhibitors and were dependent upon GAP for this function, as antibodies to GAP blocked the Ras effect. Importantly, the N-terminal domain of pl20-GAP (i.e. a noncatalytic fragment) relieved the requirement for Ras in this system (210). Based on the above observations, the effector function of p120-GAP has been attributed to its N-terminal domain. The SH2 and SH3 domains residing within this region mediate complex formation with a number of receptor and non-receptor tyrosine kinases (e.g., PDGFR, EGFR, CSF-1R, and v-src) (147). Many of these interactions result in the phosphorylation of pl20-GAP, although at generally low stoichiometry and with no apparent alteration of its activity. Moreover, the majority of pl20-GAP does not enter into these complexes. However, it has been speculated that the amount that does interact with membrane-bound kinases will result in its translocation from the cytosol to the plasma membrane, thereby increasing the effective concentration and ease of interaction of p120-GAP with membrane-bound Ras. 35 Two other cellular phosphoproteins, p62 and p190, have been found associated with the same N-terminal domain of p120-GAP. Both of these proteins were first identified as tyrosinephosphorylated, pl20-GAP associated proteins in v-src transformed or EGF treated fibroblasts (211). Only a small portion of pl20-GAP associates with p62 and vice versa, whereas up to half of cytoplasmic pl20-GAP is found in a complex with p1 9 0 (212). In contrast, very little of this complex is found in normally proliferating fibroblasts. The association of p120-GAP with p190 attenuates its catalytic activity on Ras-GTP, suggesting that this complex might promote Ras activation (212). The p190 protein has several key features including an N-terminal GTP-binding domain, a C-terminal Rho-GAP domain, and a central domain related to a proposed transcriptional repressor of the glucocorticoid receptor gene (213). The p190 protein has been demonstrated to exhibit GAP activity on Rho-related proteins, including Rho, Rac, and Cdc42Hs, while in a complex with the N-terminal domain of pl20-GAP (214, 215). This strongly suggests that the interaction of pl20-GAP and p190 may serve to functionally link the Ras and Rho signaling pathways. Indeed, expression of the N-terminus of pl20-GAP in fibroblasts results in a constitutive association with p190 and correlated with changes in the actin cytoskeleton and cell adhesion that were similar to those observed following downregulation of Rho in Swiss 3T3 cells (215). The importance of this link between the Ras and Rho pathways is discussed below (see The Rho-subfamily of Small GTP-binding Proteins: Their Signaling Pathways and Requirement for Ras-mediated Transformation). Further insight into the function of p62 awaits its cloning as the cDNA originally isolated for p62 has since been shown to encode a different protein, called Sam68 (216, 217). Recently, a number of investigators have demonstrated critical roles for the N-terminal domain of p120-GAP in Ras-dependent signaling. For example, Duchesne et al. have shown that the SH3 domain of pl20-GAP is essential for Ras-mediated germinal vesicle breakdown in Xenopus oocytes (218). It has also been demonstrated that the N-terminal SH2/SH3 domain of pl20-GAP (nGAP), but not the full length protein, can induce transcription from afos promoter (219). However, this was dependent upon Ras activity and suggests that additional signals from 36 Ras are required in order for nGAP to mediate this function. Similarly, as discussed above, the ability of nGAP to inhibit muscarinic K+ channel activity in a Ras-independent manner suggests that this domain of GAP transmits some type of signal (210). In addition, it has been shown that the C-terminal catalytic domain of pl20-GAP (cGAP) can inhibit the transcriptional stimulation of a polyoma enhancer produced by both normal and oncogenic Ras (220). Interestingly, while wildtype GAP did not affect oncogenic Ras signaling, it did reverse the inhibitory effect of cGAP on transcription induced by oncogenic Ras. This suggests, therefore, that the N-terminal domain of GAP is involved in mediating this particular Ras induced transcriptional stimulus. Taken together, the above observations suggest a model in which the binding of Ras-GTP to pl20-GAP results in a conformational change that exposes the N-terminal SH2 and SH3 domains, thereby promoting their interaction with downstream target(s) to mediate Ras signaling. It is worth noting that expression of nGAP suppresses the transforming potential of oncogenic Ras but not normal Ras (221). Therefore, it is possible that the downstream targets of oncogenic and normal Ras may be different and would have important ramifications on future drug strategies for inhibiting Ras transformation. Much less known about the putative effector function of NF1-GAP. Since the two GAPs only share homology in the catalytic Ras-binding domain, it is reasonable to assume that they mediate distinct pathways (177, 192). Tubulin has been demonstrated to interact with and inhibit the catalytic activity of NF1-GAP on Ras (222). Thus, both GAPs share connections between Ras function and cytoskeletal organization. Guanine Nucleotide DissociationInhibitors A third class of regulatory proteins that control the Ras GDP/GTP exchange cycle includes the recently identified guanine nucleotide dissociation inhibitor, Ras GDI (95, 145). Ras GDI negatively regulates Ras activity by inhibiting the dissociation of bound GDP, thereby antagonizing the action of GEFs to stimulate GDP/GTP exchange on Ras. 37 Functionally related GDIs have been cloned and more extensively characterized for the Ras-related Rab (Rab GDI) and Rho (Rho GDI) proteins (223). These GDIs have been shown to inhibit the release of GDP and to antagonize the action of GEFs specific for Rab and Rho-related proteins. In addition, Rab and Rho GDIs have been shown to inhibit the binding of their GDP bound substrates to membranes as well as to induce their dissociation from the membrane (224). Because the Rab and Rho GDIs only interact with the lipid modified forms of their respective substrates, it has been proposed that these GDIs serve to mask the hydrophobic lipid groups in order to promote membrane dissociation (225, 226). Therefore, these GDIs appear to play an even more important role in regulating the cycling of Ras-related proteins between the membrane and cytosol by promoting their release from membranes. This role is consistent with the apparent need for Rab and Rho proteins to cycle between compartments in order to regulate vesicular transport and organization of the actin cytoskeleton, respectively (35, 75). Ras Regulators as Targets for Tumorigenesis Analysis of tumors has clearly implicated an important role for Ras in oncogenesis. As discussed earlier, numerous examples have demonstrated the presence of ras mutations in a variety of human tumor types. Since these mutations result in constitutive activation of Ras function, it is conceivable that the inactivation of Ras GAPs or the chronic activation of Ras GEFs may also trigger constitutive Ras activation. Hence, GEFs would be predicted to be oncogenes, whereas GAPs may represent putative tumor suppressor genes. The loss of NF1-GAP activity in some although not all human tumors has beeen shown to result in elevated Ras-GTP levels, thereby providing support to this possibility (196, 197). In addition, somatic mutations in the NF1 gene have been found in human tumors which reduce its GTPase stimulatory activity by up to 200-fold (227). Targeted disruption of the Nfl gene in mice has also demonstrated that mice heterozygous for the mutation are predisposed to tumors, with -50% of tumors showing loss of heterozygosity at the Nfl locus (228). Interestingly, no mutations leading to the loss of p 20-GAP activity have been identified in human tumors. 38 The mutation or overexpression of Ras GEFs has not yet been described in human tumors. However, numerous experimental observations suggest that aberrant GEF activity can contribute to transformation. Overexpression of truncated forms of many GEFs (e.g., mSOS and Cdc25) caused morphological transformation of NIH/3T3 cells (229-231). In addition, smg GDS has also been described to exhibit weak transforming potential on its own, but strongly cooperated with wild-type K-ras to transform NIH/3T3 cells (232). V. The Ras Signal Transduction Pathway Although it has been known for some time that Ras proteins are critical in mediating a growth regulatory signal, the precise biochemical pathways regulated by Ras activity have only recently been identified (Figure 1.2). The combined knowledge gathered from biochemical studies in mammalian cells and genetic studies from C.elegans, D. melanogaster, S. cerevisiae, and S. pombe has revealed remarkable conservation of the signal transduction molecules and the pathways that they mediate (Table 1.2) (81, 84). It is now clear that Ras proteins are critical in eliciting a signal downstream of tyrosine kinases following mitogenic stimulation (233, 234). This signal is then propagated, in part, through a series of dual specificity serine/threonine kinases, which ultimately regulate the activities of nuclear transcription factors (235, 236). In addition, Ras has recently been linked to the regulation of a cascade of small GTP-binding proteins critical in regulating the organization of the actin cytoskeleton, thereby implicating an important role for Ras function in cell growth, motility, adhesion, and cell-cell interactions (13). Regulation of the Ras GDP/GTP Cycle by Mitogenic Stimuli As discussed above, Ras proteins have been implicated in eliciting a signal downstream of a number of receptor and non-receptor tyrosine kinases (see Mammalian Systems), as well as some G protein-regulated serpentine receptors (94, 95). The rapid and transient elevation of Ras-GTP levels is believed to occur through either stimulation of GEF activity or the inhibition of GAP activity. The inhibition of GAP activity has been observed following phorbol ester induced T cell 39 Q) 1 · rl E4 I IO I Ia I~eI Zf E0 I~w -- 1Kp c' P--_ oi . CA C,, 5-i n Cu ~o activation and in erythropoietin-treated erythroleukemic cells (237, 238). In contrast, fibroblasts stimulated with epidermal growth factor (EGF) or insulin as well as NGF-stimulated PC12 cells exhibit enhanced GEF activity (239, 240). Thus, the mode of regulation may depend upon either the cell type and/or specific stimulus. The precise mechanism governing GAP downregulation is not known. For example, protein kinase C (PKC) stimulation in T cells following phorbol ester treatment did not result in the phosphorylation of p120-GAP (237). An alternative target for PKC (a serine/threonine kinase) may be the pl20-GAP associated protein, p190. Consistent with this possibility, p190 has been shown to be predominantly phosphorylated on serine and threonine residues (211, 212). As discussed above (see GTPase Activating Proteins), p190 association with pl20-GAP is induced during tyrosine kinase-mediated transformation and cellular proliferation and attenuates the GTPase stimulatory activity of pl20-GAP. Therefore, it is possible that downregulation of GAP activity may be regulated through p190. Alternatively, GAP activity (both p120-GAP and NF1) may be downregulated through the interaction with certain mitogenically-stimulated lipids (e.g., arachidonic acid, phosphatidic acid and phosphatidyl inositol phosphates) (194, 241-244). However, this inhibitory effect has only been demonstrated in vitro and required such high concentrations of lipids, that its physiological relevance is questionable. Upregulation of GEF activity has recently been demonstrated to occur through at least two distinct mechanisms following extracellular stimulation. First, it has been demonstrated that tyrosine phosphorylation of the Vav GEF following T cell activation correlated with increased exchange activity (167). However, this mechanism of activation appears to be unique to this GEF, as the phosphorylation of other GEFs (e.g., CDC25 and mSos) has been postulated to serve in a negative feedback regulatory loop (245, 246). The second mechanism of activation involves the interaction of the Sos GEF with an "adapter" molecule called Grb2 (233, 234, 247). Grb2 contains SH2 and SH3 domains and directly couples activated tyrosine kinases with Ras activation by triggering the translocation of Sos to the plasma membrane where Ras stimulation can then occur. 41 The identification of this "adapter" component of the Ras signaling pathway came about through a number of different approaches. Grb2 was first implicated in Ras signaling by genetic analysis of vulval development in C. elegans. Genetic dissection of this RTK-mediated, Rasdependent pathway identified Sem-5 as a critical upstream activator of Let-60 (Ras homologue) (248). The mammalian homologue of Sem-5, designated Grb2, was independently isolated by virtue of its ability to bind to an activated (i.e., phosphorylated) EGF-R (249). A Grb-2 homologue, Drk, was simultaneously identified in D. melanogaster as an important upstream regulator of Ras activation (250). The Grb2/Sem-5/Drk proteins consist only of an SH2 domain and two SH3 domains and appear to lack any catalytic activity. Thus, Grb2 was believed to function as an "adapter" molecule, with its SH2 domain binding to activated tyrosine phosphoproteins and its SH3 domains binding to a Ras activator (i.e., Sos) (251). This hypothesis was subsequently confirmed by a series of interrelated biochemical studies from several groups (159, 231, 250, 252-256). Grb2 and Sos were shown to exist in a cytoplasmic complex. This interaction was mediated by the Grb2 SH3 domains, which were shown to recognize proline-rich sequences (SH3 binding motif) at the C-terminus of Sos proteins. Following ligand stimulation, this complex was shown to associate with autophosphorylated RTKs via the SH2 domain of Grb2. Since the GDP/GTP exchange activity of Sos is not regulated during this translocation step, the role of Grb2 is to mediate RTK activation of Ras by regulating the association of Sos with plasma membrane-bound Ras. A second SH2 domain containing protein, termed Shc, has also been implicated in linking activated tyrosine kinases with Ras activation (251). This protein was originally identified as an SH2-domain containing protein (257). Overexpression of Shc was sufficient to transform NIH/3T3 cells as well as induce neurite outgrowth in PC12 cells in a manner that depended upon Ras activity (251). Thus, She was implicated as an upstream regulator of Ras function. This was confirmed by studies which showed that Shc was transiently phosphorylated following RTK activation and constitutively in cells transformed by non-receptor tyrosine kinases such as src (258) The binding of Shc to these activated tyrosine kinases resulted in tyrosine phosphorylation 42 of Shc, which in turn, promoted its association with the Grb2/Sos complex (231, 251). Thus, the association of activated RTKs with Grb2, either directly or indirectly through Shc, results in the translocation of Sos to the plasma membrane where it promotes activation of the Ras signaling pathway. Ras Transmits a Signal to the Nucleus via a Cascade of Serine/Threonine Kinases In the past few years, a number of putative effector molecules for Ras have been identified (Table 1.3) (77). Several lines of evidence have now shown that Ras activates a series of serine/threonine kinases following extracellular stimulation (Figure 1.2), that are a critical component of oncogenic Ras-mediated transformation (236, 259). The first member of this kinase cascade is Raf and, as discussed below, has now been demonstrated to directly interact with Ras-GTP. Many observations had positioned Raf downstream of Ras signaling. First, activated Ras results in hyperphosphorylation of the Raf kinase in the absence of mitogenic stimuli (260). It was later shown that Ras-mediated transformation could be inhibited by a dominant negative version of Raf1 (261). Conversely, dominant negative Ras (N17) could effectively block the ability of RTKs to activate Raf kinase activity (262). In addition, genetic studies in D. melanogasterand C. elegans implicated Raf homologues in Ras-dependent signaling cascades and positioned them downstream of Ras (263, 264). Evidence for a direct interaction between Ras and Raf was revealed through a series of independent lines of investigation. The first suggestion of a direct physical association came from the observation that recombinant Ras and Raf proteins were able to interact in a GTP-dependent manner in vitro (265, 266). This was subsequently confirmed through the yeast two-hybrid system (267), which revealed an association between the Ras effector domain and the N-terminal regulatory domain of Raf- 1 (268). Simultaneously, Raf was isolated independently using the yeast two-hybrid system to screen expression libraries for proteins that bound to Ras (269). The converse experiment, in which Raf was used as the bait, resulted in the identification of the Rasrelated Rapl GTP-binding protein (270). 43 Table 1.3. Effectors of Rasa Organism Effector Molecule Function Vertebrates pl120-GAP Neurofibromin Raf-1, A-Raf, and B-Raf p110PI3K Ral-GDS RGL (or Rsb3) Rin Rsbl (or AF6) Ras-GAP Ras-GAP MAPKKKs in ERK-dependent signaling from RTKs 3' phosphoinositol-lipid kinase GEF for p27Ral GTPase Ral-GDS-like GEF Unknown Unknown Protein kinase C family member MAPKKK in Jun kinase pathway PKC- MEKK-1 D. melanogaster Draf Canoe? MAPKKK in ERK-dependent signaling from RTKs Involved in Notch signaling C. elegans Lin45 MAPKKK in ERK-dependent signaling from RTKs S. pombe Byr2 MAPKKK in pheromone response pathway Putative GEF for Cdc42sp; regulates actin cytoskeleton Scdl S. cerevisiae CDC35/CYR 1 Control of cAMP levels (adenyl cyclase) for vegetative growth Table summarizes the molecules that have been demonstrated to date to interact with Ras-GTP. aAdapted from Marshall, C. (1996) Ras effectors. Curr Opin Cell Biol. 8, 197-204. 44 The binding of Ras to Raf in mammalian cells may be regulated by at least two mechanisms. In the first mechanism, Rap1 appears to compete with Ras for Raf interaction, and thus blocks Ras signaling (271). In the other, cyclic AMP levels appear to prevent the association of Raf with Ras by an inhibitory phosphorylation event that is presumably mediated through protein kinase A (272, 273). It is still not clear, however, how the binding of Raf to Ras activates its kinase activity, but it appears that this step requires additional components located at the plasma membrane (274, 275). Raf activation appears to be dependent upon its tyrosine phosphorylation (276). In addition, deletion of the N-terminal regulatory domain results in the constitutive activation of Raf kinase activity (277). Therefore, it had been proposed, that the role of Ras was to recruite Raf to the plasma membrane, where Raf would be brought into association with a resident tyrosine kinase (e.g., Src) that would covalently modify Raf and relieve the inhibitory effects of the N-terminal regulatory domain. In support of this, targeting Raf-1 to the plasma membrane by expression constructs containing Ras membrane targeting signals lead to activation of Raf-1 kinase activity independently of Ras function (278, 279). This constitutive activation of Raf-l following membrane targeting occurred in the absence of mitogenic stimulation. Importantly, however, this membrane-targeted Raf-1 could be further activated by tyrosine kinases (279). This synergistic activation of Raf through Ras and tyrosine kinase-mediated events at the plasma membrane has recently been demonstrated (280). Interestingly, though, these investigators demonstrated that in addition to tyrosine phosphorylation, there was yet another activation step resulting from the recruitment of Raf-1 to the plasma membrane by Ras-GTP. Thus, the role of Ras in Raf-1 activation appears to bring Raf-1 to the plasma membrane for at least two different activation steps, one of which is tyrosine phosphorylation. Using the same yeast two-hybrid approach described above, Raf-1 was demonstrated to directly interact with its substrate MAPK kinase (also known as MEK), and suggested that Raf was acting as a bridge between Ras and a MAPK signaling cascade (268). Activated MAPK kinase then catalyzes the dual phosphorylation and activation of at least two MAPKs, designated 45 Erkl and Erk2 (Figurel.2). Numerous biochemical and genetic studies have provided evidence for the order of this signaling cascade (236, 281). This cascade of serine/threonine kinases is remarkably conserved and is a recurring theme downstream of a number of extracellular stimuli and small GTP-binding proteins (Table 1.2). For example, the pheromone response pathways in both S. pombe and S. cerevisiae are mediated through a MAPK cascade that is regulated by a G protein (81). Similar cascades have also been implicated in RTK signaling in D. melanogaster and C. elegans (82-84). The identification of numerous serine/threonine kinases and their substrate specificities has defined distinct signaling cascades in response to specific extracellular stimuli. For example, the Ras/Raf/MAPK cascade is believed to be important in regulating cellular proliferation, survival, and differentiation. In contrast, the recently identified stress response pathways appear to be involved in regulating apoptosis (282, 283). Although a linear scheme of signaling has been identified in each of these pathways, it is clear that this is an oversimplification and cross talk between these pathways can occur. The activation of MAPKs subsequently results in the activation of a number of different transcription factors as well as other kinases that are critical components of protein synthesis (235). For example, MAPKs have been demonstrated to activate the nuclear transcription factors p62TCF, c-Jun, c-Myc, and TALl (95, 235). In addition, MAPKs also phosphorylate and regulate the ribosomal S6 kinase (RSK) and MAP kinase activated protein kinase-2 (MAPKAPK2), which are believed to then phosphorylate and activate ribosomal subunits (262, 284). Elucidation of the exact roles of MAPKs in gene expression will provide insight into the critical target genes downstream of Ras activation and their potential roles in mediating cellular transformation. The Rho-subfamily of Small GTP-binding Proteins: Their Signaling Pathways and Requirementfor Ras-mediated Transformation In addition to the MAPK cascade described above, Ras has also been implicated in regulating a signaling pathway involving the Rho-subfamily of proteins (13). This family consists of nine 46 proteins: RhoA/B/C/G, Racl/2, Cdc42Hs/G25K, and TC10. They share -50-55% homology with each other and around 30% homology to Ras, and are believed to function as molecular switches in a manner analogous to that of Ras. Indeed, a number of GEFs (e.g., Dbl, Ost, and Tiam-1) and GAPs (e.g., p190, Bcr, and Cdc42-GAP) specific to Rho proteins have been identified and shown to regulate their GDP/GTP cycle (161, 285). Recently, an intensive area of research has been focused on identifying targets specific for these Rho-related GTPases and have lead to the identification of the PAK65 and PKN kinases as well as other putative effector molecules (286-290). The Rho-subfamily of proteins have been demonstrated to serve critical regulatory roles in organization of the actin cytoskeleton. The first evidence for this function came from studies using the C3 exotransferase of C. botulinum, which inactivates the RhoA, B, and C proteins specifically through an ADP ribosylation event (13). Cells that were microinjected with C3 underwent a dramatic morphological change in which they rounded up and lost their stress fibers (291, 292). In contrast, microinjection of constitutively activated Rho (V14 ) into cells resulted in drastic changes in cell shape and stimulated the formation of actin stress fibers and focal adhesions (292, 293). Microinjection of constitutively activated forms of Rac and Cdc42, on the other hand, lead to dramatic effects on the actin cytoskeleton that were quite distinct from that of Rho. Rac-GTP stimulated the rapid polymerization of actin at the plasma membrane to produce membrane ruffling and lamellipodia (294), whereas Cdc42-GTP promoted the formation of filopodia (295). In addition to these independent actions, these GTPases function as though they are linked (295, 296). Activated Cdc42 induces filopodia, but in addition, will subsequently stimulate the production of lamellipodia, and then focal adhesions and stress fibers (in that temporal order). The formation of these latter structures in response to Cdc42 activation is dependent upon Rac and Rho function. Similarly, activated Rac produces lamellipodia, followed by focal adhesions and stress fibers. Thus, Cdc42, Rac and Rho appear to function in a hierarchical cascade, with Cdc42 activating Rac which in turn leads to the activation of Rho. 47 These GTPases have also been implicated in regulating nuclear as well as cytoplasmic effects. The first evidence in support of this came from the observation that chronic activation of Cdc42, Rac, and Rho by deregulated exchange factors resulted in malignant transformation as well as morphological alterations (166, 297). A direct link from these proteins to the nucleus as well as to a MAPK cascade was recently demonstrated by three independent groups. Coso et al. and Minden et al. were able to show that activation of the stress response pathway was dependent upon Cdc42 and Rac 1, but interestingly, this activation was independent of RhoA (298, 299). The stress response pathway is induced in response to UV irradiation, pro-inflammatory cytokines, and environmental stress and is mediated by a series of MAPKs (JNK subgroup) that are distinct from those downstream of Raf (282). The JNK protein kinases were first identified as a kinase activity that phosphorylated the c-Jun transcription factor within its N-terminal transactivation domain in cells exposed to UV irradiation. Both Cdc42 and Racl were capable of activating MEKK1 (a JNK kinase kinase) and the subsequent downstream members of the kinase cascade (i.e., MKK4/SEK/JNKK and SAPK/JNK) in much the same way that Ras activates the Raf/MAPK cascade described above (298, 299). The PAK65 serine/threonine kinase is the homologue of STE20 in S. cerevisiae and has been proposed to link Cdc42 and Racl to MEKK1, due largely to STE20's role in activating a similar MAPK cascade in the response to pheromone (Table 1.2) (81, 290, 300). A requirement for RhoA in signaling to the serum response factor (SRF) in response to serum and lysophosphatidic acid (LPA) was recently demonstrated (301). The c-fos serum response element (SRE) has two distinct binding activities, SRF and the p62TCF ternary complex factor, which cooperate to maximally activate c-fos transcription (302). The Ras/Raf/MAPK pathway had been previously demonstrated to signal directly to p62TCF (303, 304). However, activation of the SRE by serum or LPA is only dependent upon SRF and can occur independently of the Ras/Raf/MAPK pathway (although suboptimally) (302). Hill et al. were able to show that Rho family members mediated this p62TCF-independent, SRF-dependent activation. Activated versions of Cdc42, Racl and RhoA were all capable of inducing transcription from a p6 2 TCF - 48 independent reporter (301). Interestingly, activation of SRF by Cdc42 and Rac 1 appeared to occur independently of RhoA, yet stimulation by LPA still depended upon functional RhoA. Thus, it appears that two distinct signaling pathways are mediated by the Rho-related proteins and converge on SRF. In addition, these two pathways are likely to converge with the Ras/Raf/MAPK-activated pathway to activate the SRE synergistically and in this way lead to maximal c-fos gene expression. The identification of Rho family members as activators of MAPK cascades as well as the conservation of this same signaling structure throughout numerous organisms suggests that many, if not all, of the MAPK pathways will be controlled by small GTPases. This link between GTPases and MAPK cascades is likely to invoke cooperating "second signals" that are provided by other kinases or small molecules. As discussed above, these "coactivators" have been implicated in regulating the activation of Raf by Ras-GTP (279). It is tempting to speculate that the very recently identified and novel kinase, designated Ksr-1, may prove to be this long sought coactivator (305-308). In much the same way, Cdc42/Racl.GTP may localize PAK65 to an appropriate compartment in the cell, where a second signal may be delivered. A requirement for a second signal may explain the apparent roles that Ras seems to play in JNK activation as well as RhoA-mediated SRF activation (301). It has been known for some time that growth factors and constitutively active Ras will stimulate the formation of stress fibers (through Rac and Rho) and membrane ruffling (Racdependent) and suggested that the activities of these GTPases were linked (293, 294). Direct support of this interrelationship was obtained when it was demonstrated that dominant negative mutants of either Racl (N17 ) or RhoA (N19 ) blocked Ras-mediated transformation (31-33). In addition, RhoA (N 19 ), but not Racl (N17 ), was also capable of inhibiting transformation by constitutively activated RafCAAX. However, activated versions of either Rho (V14 ) or Rac (V 12 ) were able to synergize strongly with RafCAAX in focus formation assays, indicating that oncogenic Ras drives at least two distinct and cooperating pathways to cause transformation. Interestingly, oncogenic Ras-mediated transformation is accompanied by the loss of stress fibers and can be restored by coexpression of dominant negative RhoA (31, 32, 297). Taken together, 49 the above data implicate Rho in the regulation of at least two separate pathways, one that induces stress fiber formation and another that is important in mediating transformation by oncogenic Ras (and Raf). The p190 protein provides a possible link between Ras and Rho activity. As discussed earlier, p190 may function to antagonize Ras pl20-GAP function (see GTPase Activating Proteins), while promoting inactivation of the Rho-mediated pathway regulating stress fiber formation through its Rho-GAP catalytic activity. It is tempting to speculate that the association of p190 with Rho may serve a second functional role in positively regulating a growth-promoting signal, which may be mediated through either the GTP-binding domain and/or transcriptional repressor domain that are also present in p190 (213). Thus, both the pl20-GAP and p190 RhoGAP proteins may serve bifunctional activities: one which downregulates their respective GTPase substrate, while the second serves an effector function that promotes cellular proliferation through the interaction with accessory proteins. The Rho-driven signaling pathway that is involved in the control of cellular proliferation remains to be elucidated. As discussed above, Rho plays an important role in regulating the c-fos proto-oncogene. Rho function is also necesssary for the activation of focal adhesion kinase and MAP kinases by growth factors (309). Furthermore, Rho has been shown to regulate a variety of signal transducers, including phosphatidylinositol 4-phosphate 5-kinase, phosphatidylinositol (4,5) 3-kinsae, and phospholipase D (310-312). It is unclear which of these activities (or as yet undetermined pathways) are important in mediating cellular proliferation. Importantly, further characterization of these Rac/Rho-signaling pathways may identify alternative, and perhaps more specific, targets for chemotherapeutic strategies designed to inhibit Ras-induced transformation. 50 VI. Ras Signaling as a Target for Drug Therapy Farnesyltransferase Inhibitors The ras genes are mutated in approximately 30% of all human tumors, thereby making them one of the most frequent alterations in cancer (88). Because of this, much effort has focused on developing chemotherapeutic drugs to inhibit the transforming activity of oncogenic Ras. As discussed earlier, Ras must be post-translationally modified at the C-terminus in order to acquire biological activity (58-60). Each step in the processing pathway is dependent upon the preceding one, such that no post-translational modifications occur if the first step, famesylation, is blocked. Blocking farnesylation totally abolishes the transforming activity of Ras as measured in NIH/3T3 cells (64, 313). In contrast, blocking steps subsequent to farnesylation, such as palmitylation, has a much less dramatic effect on oncogenic Ras function despite the lack of significant association with the plasma membrane (64, 65). Therefore, these observations have made prenyltransferase molecules excellent targets for the development of anti-Ras chemotherapeutic drugs. Because it was believed that all Ras proteins were modified by the farnesyl isoprenoid, the main target of drug research has been the farnesyltransferase (FTase) enzyme (59, 314). The classical approach of screening compound libraries has yielded several classes of compounds that inhibit FTase (315). However, despite giving promising results in vitro, all of these compounds have proven ineffective when tested on intact cells. Therefore, investigators have focused their efforts on the rational design of inhibitors based on the knowledge of the enzyme's substrates. The initial goal of these research programs was to identify drugs which would be sufficiently potent so as to inhibit Ras-mediated transformation, yet specific enough to alleviate toxicity effects on normal cells. This issue of specificity is important because a single FTase is required by multiple substrates for appropriate biological function (314). At least eight other proteins, including the nuclear lamins A and B, are known to be famesylated in addition to the four Ras proteins (58, 75, 314). Furthermore, any potential drugs should specifically inhibit FTase and not the geranylgeranyltransferase type I and type II enzymes (GGTase I and GGTase II). This is 51 particularly important given the fact that geranylgeranlyation is five to ten times more prevalent than farnesylation (59, 316, 317). Thus, an inhibitor exhibiting the above specificity would be expected to elicit fewer cytotoxic effects. The initial templates for the design of FTase inhibitors were the substrates for the reaction, namely farnesyl diphosphate and the CAAX tetrapeptide. The conversion of these charged and metabolically labile compounds into stable and lipid soluble inhibitors was a major obstacle that had to be overcome. A number of compounds representing a broad structure base have since been designed and shown to be potent as well as selective FTase inhibitors (314). Two examples of farnesyl diphosphate analogs, (at-Hydroxyfarnesyl) phosphonic acid and manumycin, have been shown to inhibit FTase and are active in cells (318). However, the inhibition of Ras farnesylation in intact cells is only partial. Furthermore, the absolute specificity of these compounds in vivo is uncertain due to the large number of enzymes in the cell which utilize farnesyl diphosphate (316, 317). CAAX-based inhibitors, on the other hand, have proven to be much more effective in their ability to inhibit FTase in the intact cell. Early versions of these inhibitors had a number of problems associated with them, including protease degradation, lipid solubility, and potency. Within the past few years, significant strides have been made in the design of tetrapeptide inihibitors and have overcome many of these issues (314, 319-322). These modifications have provided a set of inhibitors that exhibit potency toward FTase at nanomolar concentrations in vitro, while retaining selectivity against GGTase I. These compounds have since proven to be very effective inhibitors of FTase activity in the living cell, as indicated by the inhibition of Ras processing as well as the absence of [3 H]mevalonate (a metabolic precursor of both farnesyl diphosphate and geranylgeranyl diphosphate) incorpation into farnesylated proteins (319-321). Moreover, the degree of inhibition was essentially complete, in contrast with the farnesyl diphosphate analog inhibitors. However, higher concentrations (micromolar) of the drug molecules were required to block Ras processing in vivo than were needed to inhibit FTase activity 52 in vitro (nanomolar). This is an extremely important observation, the implications of which will be discussed below. Based on [3H]mevalonate labeling experiments, the FTase inhibitors have clearly been shown to block the farnesylation of several proteins in addition to Ras (320). Therefore, these inhibitors are not Ras-specific. However, as was shown in vitro, these inhibitors still retain selectivity in the cell, as the geranylgeranylation of cellular proteins was not inhibited at concentrations sufficient to fully inhibit farnesylation (319-321). The FTase Inhibitor Paradox: Efficacy without Cytotoxicity Recently, a number of FTase inhibitors have been demonstrated to revert the malignant phenotype of Ras-transformed cells both in tissue culture and in the animal (319-321, 323-325). In addition, these effects were selective for ras-inducedphenotypes, as they did not inhibit the growth of cells transformed by raformos (both of which are downstream of Ras in the signaling cascade) (321, 323, 326, 327). Surprisingly, though, no anti-proliferative or toxic effects on normal cells were observed in tissue culture or in vivo. These results raise questions as to the biological mechanism of inhibition, since Ras as well as other farnesylated proteins are critical in normal cell growth and function (89, 328, 329). One possible explanation for the above paradox is that while the function of FTase may be impaired in the presence of these inhibitors, they may not abolish the ability of GGTase I to crossprenylate protein substrates with either geranylgeranyl or farnesyl isoprenoids. It is not known whether GGTase I can transfer a farnesyl isoprenoid in vivo. In addition, while these FTase inhibitors do not affect GGTase I-dependent geranylgeranylation, their effects on GGTase Idependent farnesylation have not been examined. Thus, despite an efficient block in FTase activity, compensatory regulation by GGTase I would enable these substrates (e.g., Ras and nuclear lamins A and B) to still associate with the appropriate membrane and retain biological activity. This possibility, however, is difficult to reconcile with the specific inhibition of 53 transformed cells as cross-prenylation of oncogenic Ras molecules would be expected to activate the transforming signal. Recent observations have demonstrated that K-ras4B can be modified by geranylgeranylation as well as farnesylation both in vitro and in vivo (73, 74). K-ras4B has been shown to be a substrate for geranylgeranylation by GGTase I with an affinity for the enzyme equal to that of RaplB, an authentic GGTase I substrate which is solely modified by geranylgeranylation (74). This substrate specificity was dictated by both the CAAX sequence as well as the polylysine domain in K-ras4B. In addition, a GGTase I inhibitor has recently been designed which is 10 to 25 fold more potent than FTase inhibitors towards inhibiting geranylgeranylation both in vitro and in vivo.(73) While H-ras processing in intact cells was found to be very sensitive to FTase inhibitors, that of K-ras4B was 100 fold more resistant. In contrast, K-ras4B processing and signaling was much more sensitive to the GGTase I inhibitor. In fact, significant inhibition of oncogenic K-ras4B signaling (as measured by MAP kinase stimulation) by the GGTase I inhibitor occurred at concentrations that did not affect oncogenic H-ras signaling. Thus, this unique processing of K-ras4B helps to explain earlier observations showing that higher concentrations of FTase inhibitors were required to suppress the transforming activity of oncogenic K-ras than were necessary for H-ras (73, 74, 325, 330-332). Because K-ras4B processing is more resistant to FTase inhibitors, this also serves to explain, at least in part, why FTase inhibitors do not inhibit the growth or transient mitogenic responses of non-transformed cells at concentrations that are sufficient to prevent the farnesylation of endogenous proteins and revert malignant phenotypes (319-321, 323, 324). Furthermore,the specific inhibition of transformed cells can be explained through the sequestration of effectors into inactive cytoplasmic complexes with Ras.GTP as has been demonstrated for the Raf effector (332, 333). The above observation suggests that K-ras4B is sufficient to support the growth and mitogenic responses of normal cells in which H- and N-ras have been functionally inhibited by FTase inhibitors. Alternatively, other members of the Ras superfamily may functionally compensate for the absence of Ras function. If such a protein(s) existed, it would not only have to 54 be resistant to FTase inhibitors, but would also have to be able to utilize components of the Ras signaling cascade(s). In addition, since cell growth has been shown to require Ras function, this protein would also have to be affected to the same degree as Ras proteins to the dominant inhibitory growth effects of microinjected Ras antibodies (i.e., Y13-259) and dominant negative Ras mutants (89, 328, 329). It is possible, however, that this protein may support cell growth only in collaberation with K-ras4B, and therefore, may not be subject to the same inhibitory effects of Y13-259 and dominant negative Ras mutants. A strong candidate for such a role is TC21/R-ras2. This member shares 55% amino acid identity with Ras proteins, is expressed in all human tissues examined so far, and has partial, but significant, overlap in the Ras signaling pathway (14, 334, 335). In addition, TC21 is recognized by Y13-259, and residues critical for the effects of dominant negative Ras mutants are identical between Ras and TC21. Finally, TC21 is geranylgeranylated as well as farnesylated in vitro, and cells transformed by an oncogenic version of TC21 are resistant to the inhibitory effects of FTase inhibitors (76). Thus, it is conceivable that TC21 and/or K-ras4B may allow non-transformed cells to continue to proliferate in the presence of FTase inhibitors. Interestingly, the kinetics of morphological reversion induced by FTase inhibitors are too rapid to easily be explained solely by the suppression of Ras activity (326). Phenotypic reversion is detectable within 18 to 24 hours. Since fully modified Ras is very stable (half-life of 24 hours) (336), this amount of time is insufficient to explain depletion of Ras activity from cells treated with FTase inhibitors. Instead, the inhibitory effects of these drug molecules correlated well with effects on the regulation of the cytoskeletal architecture, namely the restoration of actin stress fiber formation. This observation implicated the Rho-subfamily of proteins as targets of the effects mediated by FTase inhibitors. One member of this family, RhoB, exists in both geranylgeranylated and farneslyated forms in vivo and is rapidly turned over in the cell (half life of -2 hours) (75, 337). Therefore, RhoB represented a potential direct target for FTase inhibitors. This hypothesis was supported when it was demonstrated that dominant negative mutants of Rho, including RhoB, mimicked the ability of FTase inhibitors to impair Ras transformation (as 55 discussed above) (31, 32, 34). In addition, Lebowitz et al. have recently demonstrated that FTase inhibitors suppressed the transformed phenotype, at least in part, by either directly or indirectly interfering with Rho function (337). Importantly, the effects of these pharmalogical inhibitors could be ablated by ectopic expression of farnesyl-independent (i.e., myristylated) forms of RhoB. In contrast, cells transformed by a myrstylated Ras construct remained susceptible to growth inhibition by these same FTase inhibitors. Together, the above data stress the importance of designing molecules specific for Ras family members, in particular, K-ras4B. This is underscored by the fact that K-ras is the most frequently mutated member of the ras gene family (88). Further characterization of the signals emanating downstream of oncogenic and normal Ras proteins may reveal distinct signaling pathways that are critical in mediating transformation, some of which may or may not overlap with the Rac/Rho signaling pathways. The identification of such mediators may ultimately provide more specific targets for future drug applications. 56 References 1. Barbacid, M., (1987). ras genes. Annu Rev Biochem 56: p. 779-827. 2. Shih, C., et al., (1981). Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature 290(5803): p. 261-4. 3. Sukumar, S., et al., (1983). Induction of mammary carcinomas in rats by nitrosomethylurea involves malignant activation of H-ras-1 locus by single point mutations. Nature 306(5944): p. 658-61. 4. Pulciani, S., et al., (1982). Oncogenes in human tumor cell lines: molecular cloning of a transforming gene from human bladder carcinoma cells. Proc Natl Acad Sci U S A 79(9): p. 2845-9. 5. Perucho, M., et al., (1981). Human-tumor-derived cell lines contain common and different transforming genes. Cell : p. 467-76. 6. Krontiris, T.G. and G.M. Cooper, (1981). Transforming activity of human tumor DNAs. Proc Natl Acad Sci U S A 78(2): p. 1181-4. 7. Eva, A. and S.A. Aaronson, (1983). Frequent activation of c-kis as a transforming gene in fibrosarcomas induced by methylcholanthrene. Science 220(4600): p. 955-6. 8. Balmain, A. and I.B. Pragnell, (1983). Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature 303(5912): p. 724. 9. Shimizu, K., et al., (1983). Isolation and preliminary characterization of the transforming gene of a human neuroblastoma cell line. Proc Natl Acad Sci U S A 80(2): p. 383-7. 10. Santos, E., et al., (1982). T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature 298(5872): p. 343-7. 11. Parada, L.F., et al., (1982). Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature 297(5866): p. 474-8. 12. Der, C.J., T.G. Krontiris, and G.M. Cooper, (1982). Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci U S A 79(11): p. 3637-40. 13. Hall, A., (1994). Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu Rev Cell Biol 10(31): p. 31-54. 14. Graham, S.M., et al., (1994). Aberrant function of the Ras-related protein TC21/R-Ras2 triggers malignant transformation. Mol Cell Biol 14(6): p. 4108-15. 57 15. Cox, A.D., et al., (1994). R-Ras induces malignant, but not morphologic, transformation of NIH3T3 cells. Oncogene 9(11): p. 3281-3288. 16. Shimizu, K., et al., (1983). Structure of the Ki-ras gene of the human lung carcinoma cell line Calu- 1. Nature 304(5926): p. 497-500. 17. McGrath, J.P., et al., (1983). Structure and organization of the human Ki-ras protooncogene and a related processed pseudogene. Nature 304(5926): p. 501-6. 18. Capon, D.J., et al., (1983). Activation of Ki-ras2 gene in human colon and lung carcinomas by two different point mutations. Nature 304(5926): p. 507-13. 19. George, D.L., et al., (1985). Structure and expression of amplified cKi-ras gene sequences in Y1 mouse adrenal tumor cells. Embo J 4(5): p. 1199-203. 20. Nakano, H., et al., (1984). Isolation of transforming sequences of two human lung carcinomas: structural and functional analysis of the activated c-K-ras oncogenes. Proc Natl Acad Sci U S A 81(1): p. 71-5. 21. Cichutek, K. and P.H. Duesberg, (1986). Harvey ras genes transform without mutant codons, apparently activated by truncation of a 5' exon (exon -1). Proc Natl Acad Sci USA 83(8): p. 2340-4. 22. Schejter, E.D. and B.Z. Shilo, (1985). Characterization of functional domains of p21 ras by use of chimeric genes. Embo J 4(2): p. 407-12. 23. Neuman, S.F., et al., (1984). The Drosophila ras oncogenes: structure and nucleotide sequence. Cell 37(3): p. 1027-33. 24. Mozer, B., et al., (1985). Characterization and developmental expression of a Drosophila ras oncogene. Mol Cell Biol 5(4): p. 885-9. 25. Powers, S., et al., (1984). Genes in S. cerevisiae encoding proteins with domains homologous to the mammalian ras proteins. Cell 36(3): p. 607-12. 26. Goldfarb, M., et al., (1982). Isolation and preliminary characterization of a human transforming gene from T24 bladder carcinoma cells. Nature 296(5856): p. 404-9. 27. DeFeo, J.D., et al., (1983). ras-Related gene sequences identified and isolated from Saccharomyces cerevisiae. Nature 306(5944): p. 707-9. 28. DeFeo, J.D., et al., (1985). Mammalian and yeast ras gene products: biological function in their heterologous systems. Science 228(4696): p. 179-84. 29. Kataoka, T., et al., (1985). Functional homology of mammalian and yeast RAS genes. Cell 40(1): p. 19-26. 58 30. Clark, S.G., J.P. McGrath, and A.D. Levinson, (1985). Expression of normal and activated human Ha-ras cDNAs in Saccharomyces cerevisiae. Mol Cell Biol 5(10): p. 2746-52. 31. Khosravi, F.R., et al., (1995). Activation of Rac 1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol 15(11): p. 6443-6453. 32. Qiu, R.G., et al., (1995). A role for Rho in Ras transformation. Proc Natl Acad Sci U S A 92(25): p. 11781-11785. 33. Qiu, R.G., et al., (1995). An essential role for Rac in Ras transformation. Nature 374(6521): p. 457-459. 34. Prendergast, G.C., et al., (1995). Critical role of Rho in cell transformation by oncogenic Ras. Oncogene 10(12): p. 2289-2296. 35. Bourne, H.R., D.A. Sanders, and F. McCormick, (1990). The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348(6297): p. 125-32. 36. Thompson, R.C., (1988). EFTu provides an internal kinetic standard for translational accuracy. Trends Biochem Sci 13(3): p. 91-3. 37. Gilman, A.G., (1987). G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56: p. 615-49. 38. Lacal, J.C., P.S. Anderson, and S.A. Aaronson, (1986). Deletion mutants of Harvey ras p21 protein reveal the absolute requirement of at least two distant regions for GTP-binding and transforming activities. Embo J 5(4): p. 679-87. 39. Willumsen, B.M., et al., (1986). Mutational analysis of a ras catalytic domain. Mol Cell Biol 6(7): p. 2646-54. 40. Feramisco, J.R., et al., (1985). Transient reversion of ras oncogene-induced cell transformation by antibodies specific for amino acid 12 of ras protein. Nature 314(6012): p. 639-42. 41. Clark, R., et al., (1985). Antibodies specific for amino acid 12 of the ras oncogene product inhibit GTP binding. Proc Natl Acad Sci U S A 82(16): p. 5280-4. 42. Gibbs, J.B., et al., (1984). Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc Natl Acad Sci U S A 81(18): p. 5704-8. 43. Manne, V., E. Bekesi, and H.F. Kung, (1985). Ha-ras proteins exhibit GTPase activity: point mutations that activate Ha-ras gene products result in decreased GTPase activity. Proc Natl Acad Sci U S A 82(2): p. 376-80. 44. McGrath, J.P., et al., (1984). Comparative biochemical properties of normal and activated human ras p21 protein. Nature 310(5979): p. 644-9. 59 45. Sweet, R.W., et al., (1984). The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature 311(5983): p. 273-5. 46. Temeles, G.L., et al., (1985). Yeast and mammalian ras proteins have conserved biochemical properties. Nature 313(6004): p. 700-3. 47. Bourne, H.R., D.A. Sanders, and F. McCormick, (1991). The GTPase superfamily: conserved structure and molecular mechanism. Nature 349(6305): p. 117-27. 48. Bollag, G. and F. McCormick, (1991). Regulators and effectors of ras proteins. Annu Rev Cell Biol 7: p. 601-32. 49. Milburn, M.V., et al., (1990). Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 247(4945): p. 939-45. 50. Pai, E.F., et al., (1990). Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. Embo J 9(8): p. 2351-9. 51. Brunger, A.T., et al., (1990). Crystal structure of an active form of RAS protein, a complex of a GTP analog and the HRAS p21 catalytic domain. Proc Natl Acad Sci U S A 87(12): p. 4849-53. 52. Tong, L.A., et al., (1989). Structural differences between a ras oncogene protein and the normal protein. Nature 337(6202): p. 90-3. 53. Krengel, U., et al., (1990). Three-dimensional structures of H-ras p21 mutants: molecular basis for their inability to function as signal switch molecules. Cell 62(3): p. 539-48. 54. Sigal, I.S., et al., (1986). Mutant ras-encoded proteins with altered nucleotide binding exert dominant biological effects. Proc Natl Acad Sci U S A 83(4): p. 952-6. 55. Powers, S., K. O'Neill, and M. Wigler, (1989). Dominant yeast and mammalian RAS mutants that interfere with the CDC25- dependent activation of wild-type RAS in Saccharomyces cerevisiae. Mol Cell Biol 9(2): p. 390-5. 56. Feig, L.A. and G.M. Cooper, (1988). Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol 8(8): p. 3235-43. 57. Sigal, I.S., et al., (1986). Identification of effector residues and a neutralizing epitope of Ha- ras-encoded p21. Proc Natl Acad Sci U S A 83(13): p. 4725-9. 58. Khosravi, F.R., et al., (1992). Protein prenylation: key to ras function and cancer intervention? Cell Growth Differ 3(7): p. 461-9. 59. Gibbs, J.B., (1991). Ras C-terminal processing enzymes--new drug targets? Cell 65(1): p. 1-4. 60 60. Glomset, J.A. and C.C. Farnsworth, (1994). Role of protein modification reactions in programming interactions between ras-related GTPases and cell membranes. Annu Rev Cell Biol 10(181): p. 181-205. 61. Hancock, J.F., K. Cadwallader, and C.J. Marshall, (1991). Methylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B). Embo J 10(3): p. 641-6. 62. Hrycyna, C.A., et al., (1991). The Saccharomyces cerevisiae STE14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and RAS proteins. Embo J 10(7): p. 1699-709. 63. Kato, K., et al., (1992). Isoprenoid addition to Ras protein is the critical modification for its membrane association and transforming activity. Proc Natl Acad Sci U S A 89(14): p. 6403-7. 64. Hancock, J.F., et al., (1989). All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57(7): p. 1167-77. 65. Hancock, J.F., H. Paterson, and C.J. Marshall, (1990). A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63(1): p. 133-9. 66. Hancock, J.F., et al., (1991). A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. Embo J 10(13): p. 4033-9. 67. Ballester, R., M.E. Furth, and O.M. Rosen, (1987). Phorbol ester- and protein kinase Cmediated phosphorylation of the cellular Kirsten ras gene product. J Biol Chem 262(6): p. 2688-95. 68. Cox, A.D. and C.J. Der, (1992). Protein prenylation: more than just glue? Curr Opin Cell Biol 4(6): p. 1008-16. 69. Reiss, Y., et al., (1991). Sequence requirement for peptide recognition by rat brain p2lras protein farnesyltransferase. Proc Natl Acad Sci U S A 88(3): p. 732-6. 70. Moores, S.L., et al., (1991). Sequence dependence of protein isoprenylation. J Biol Chem 266(22): p. 14603-10. 71. Gibbs, J.B., et al., (1989). Xenopus oocyte germinal-vesicle breakdown induced by [Vall2]Ras is inhibited by a cytosol-localized Ras mutant. Proc Natl Acad Sci U S A 86(17): p. 6630-4. 72. Pompliano, D.L., et al., (1993). Isoprenoid diphosphate utilization by recombinant human farnesyl:protein transferase: interactive binding between substrates and a preferred kinetic pathway. Biochemistry 32(32): p. 8341-7. 73. Lerner, E.C., et al., (1995). Disruption of oncogenic K-Ras4B processing and signaling by a potent geranylgeranyltransferase I inhibitor. J Biol Chem 270(45): p. 26770-26773. 61 74. James, G.L., J.L. Goldstein, and M.S. Brown, (1995). Polylysine and CVIM sequences of K-RasB dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J Biol Chem 270(11): p. 6221-6226. 75. Adamson, P., et al., (1992). Post-translational modifications of p2lrho proteins. J Biol Chem 267(28): p. 20033-8. 76. Carboni, J.M., et al., (1995). Farnesyltransferase inhibitors are inhibitors of Ras but not RRas2/TC21, transformation. Oncogene 1085(1080): p. 1905-1913. 77. Marshall, C.J., (1996). Ras effectors. Curr Opin Cell Biol 8: p. 197-204. 78. Moodie, S.A., et al., (1995). Different structural requirements within the switch II region of the Ras protein for interactions with specific downstream targets. Oncogene 11(3): p. 447-454. 79. Brtva, T.R., et al., (1995). Two distinct Raf domains mediate interaction with Ras. J. Biol. Chem. 270(17): p. 9809-12. 80. Drugan, J.K., et al., (1996). Ras interaction with two distinct binding domains in Raf-1 may be required for Ras transformation. J Biol Chem 271(1): p. 233-237. 81. Wittenberg, C. and S.I. Reed, (1996). Plugging it in: signaling circuits and the yeast cell cycle. Curr Opin Cell Biol 8: p. 223-230. 82. Kayne, P.S. and P.W. Sternberg, (1995). Ras pathways in Caenorhabditis elegans. Curr Opin Genet Dev 5(1): p. 38-43. 83. Wassarman, D.A., M. Therrien, and G.M. Rubin, (1995). The Ras signaling pathway in Drosophila. Curr Opin Genet Dev 5(1): p. 44-50. 84. Duffy, J.B. and N. Perrimon, (1996). Recent advances in understanding signal transduction pathways in worms and flies. Curr Opin Cell Biol 8: p. 231-238. 85. Han, M. and P.W. Sternberg, (1990). let-60, a gene that specifies cell fates during C. elegans vulval induction, encodes a ras protein. Cell 63(5): p. 921-31. 86. Kataoka, T., et al., (1984). Genetic analysis of yeast RAS 1 and RAS2 genes. Cell 37(2): p. 437-45. 87. Bos, J.L., (1988). The ras gene family and human carcinogenesis. Mutat Res 195(3): p. 255-71. 88. Bos, J.L., (1989). ras oncogenes in human cancer: a review [published erratum appears in Cancer Res 1990 Feb 15;50(4):1352]. Cancer Res 49(17): p. 4682-9. 62 89. Mulcahy, L.S., M.R. Smith, and D.W. Stacey, (1985). Requirement for ras protooncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 313(5999): p. 241-3. 90. Cai, H., J. Szeberenyi, and G.M. Cooper, (1990). Effect of a dominant inhibitory Ha-ras mutation on mitogenic signal transduction in NIH 3T3 cells. Mol Cell Biol 10(10): p. 5314-23. 91. Bar-Sagi, D. and J.R. Feramisco, (1986). Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233(4768): p. 1061-8. 92. Hagag, N., S. Halegoua, and M. Viola, (1986). Inhibition of growth factor-induced differentiation of PC12 cells by microinjection of antibody to ras p21. Nature 319(6055): p. 680-2. 93. Szeberenyi, J., H. Cai, and G.M. Cooper, (1990). Effect of a dominant inhibitory Ha-ras mutation on neuronal differentiation of PC12 cells. Mol Cell Biol 10(10): p. 5324-32. 94. Satoh, T., M. Nakafuku, and Y. Kaziro, (1992). Function of Ras as a molecular switch in signal transduction. J Biol Chem 267(34): p. 24149-52. 95. Khosravi, F.R. and C.J. Der, (1994). The Ras signal transduction pathway. Cancer Metastasis Rev 13(1): p. 67-89. 96. Smith, M.R., S.J. DeGudicibus, and D.W. Stacey, (1986). Requirement for c-ras proteins during viral oncogene transformation. Nature 320(6062): p. 540-3. 97. Almoguera, C., et al., (1988). Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53(4): p. 549-54. 98. Pellegata, N.S., et al., (1994). K-ras and p53 gene mutations in pancreatic cancer: ductal and nonductal tumors progress through different genetic lesions. Cancer Res 54(6): p. 1556-60. 99. Smit, V.T., et al., (1988). KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 16(16): p. 7773-82. 100. Vogelstein, B., et al., (1988). Genetic alterations during colorectal-tumor development. N Engl J Med 319(9): p. 525-32. 101. Bos, J.L., et al., (1987). Prevalence of ras gene mutations in human colorectal cancers. Nature 327(6120): p. 293-7. 102. Forrester, K., et al., (1987). Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature 327(6120): p. 298-303. 103. Muller, R., et al., (1982). Differential expression of cellular oncogenes during pre- and postnatal development of the mouse. Nature 299(5884): p. 640-4. 63 104. Muller, R., et al., (1983). Transcription of c-onc genes c-rasKi and c-fms during mouse development. Mol Cell Biol 3(6): p. 1062-9. 105. Leon, J., I. Guerrero, and A. Pellicer, (1987). Differential expression of the ras gene family in mice. Mol Cell Biol 7(4): p. 1535-40. 106. Mangues, R. and A. Pellicer, (1992). ras activation in experimental carcinogenesis. Semin Cancer Biol 3(4): p. 229-39. 107. Guerrero, I. and A. Pellicer, (1987). Mutational activation of oncogenes in animal model systems of carcinogenesis. Mutat Res 185(3): p. 293-308. 108. Quintanilla, M., et al., (1986). Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 322(6074): p. 78-80. 109. Loechler, E.L., C.L. Green, and J.M. Essigmann, (1984). In vivo mutagenesis by 06methylguanine built into a unique site in a viral genome. Proc Natl Acad Sci U S A 81(20): p. 6271-5. 110. Eadie, J.S., et al., (1984). Mechanism of mutagenesis by 06-methylguanine. Nature 308(5955): p. 201-3. 111. Singer, B. and J.T. Kusmierek, (1982). Chemical mutagenesis. Annu Rev Biochem: p. 655-93. 112. Adams, J.M. and S. Cory, (1991). Transgenic models of tumor development. Science 254(5035): p. 1161-7. 113. Sinn, E., et al., (1987). Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 49(4): p. 465-75. 114. Land, H., L.F. Parada, and R.A. Weinberg, (1983). Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304(5927): p. 596602. 115. Newbold, R.F. and R.W. Overell, (1983). Fibroblast immortality is a prerequisite for transformation by EJ c-Ha- ras oncogene. Nature 304(5927): p. 648-51. 116. Ruley, H.E., (1983). Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature 304(5927): p. 602-6. 117. Lee, W.M., et al., (1985). Augmented expression of normal c-myc is sufficient for cotransformation of rat embryo cells with a mutant ras gene. Mol Cell Biol 5(12): p. 334556. 118. Weinberg, R.A., (1985). The action of oncogenes in the cytoplasm and nucleus. Science 230(4727): p. 770-6. 64 119. Schwab, M., H.E. Varmus, and J.M. Bishop, (1985). Human N-myc gene contributes to neoplastic transformation of mammalian cells in culture. Nature 316(6024): p. 160-2. 120. Parada, L.F., et al., (1984). Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 312(5995): p. 649-51. 121. Eliyahu, D., et al., (1984). Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 312(5995): p. 646-9. 122. Dotto, G.P., L.F. Parada, and R.A. Weinberg, (1985). Specific growth response of rastransformed embryo fibroblasts to tumour promoters. Nature 318(6045): p. 472-5. 123. Pozzatti, R., et al., (1986). Primary rat embryo cells transformed by one or two oncogenes show different metastatic potentials. Science 232(4747): p. 223-7. 124. Spandidos, D.A. and N.M. Wilkie, (1984). Malignant transformation of early passage rodent cells by a single mutated human oncogene. Nature 310(5977): p. 469-75. 125. Yuspa, S.H., (1994). The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis--thirty-third G. H. A. Clowes Memorial Award Lecture. Cancer Res 54(5): p. 1178-89. 126. Hennings, H., et al., (1993). Critical aspects of initiation, promotion, and progression in multistage epidermal carcinogenesis. Proc Soc Exp Biol Med 202(1): p. 1-8. 127. Brown, K., et al., (1986). v-ras genes from Harvey and BALB murine sarcoma viruses can act as initiators of two-stage mouse skin carcinogenesis. Cell 46(3): p. 447-56. 128. Roop, D.R., et al., (1986). An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature 323(6091): p. 822-4. 129. Bailleul, B., et al., (1990). Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoter. Cell 62(4): p. 697-708. 130. Bremner, R. and A. Balmain, (1990). Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell 61(3): p. 407-17. 131. Saez, E., et al., (1995). c-fos is required for malignant progression of skin tumors. Cell 82(5): p. 721-732. 132. Fearon, E.R. and B. Vogelstein, (1990). A genetic model for colorectal tumorigenesis. Cell 61(5): p. 759-67. 133. Powell, S.M., et al., (1992). APC mutations occur early during colorectal tumorigenesis. Nature 359(6392): p. 235-7. 134. Vogelstein, B. and K.W. Kinzler, (1993). The multistep nature of cancer. Trends Genet 9(4): p. 138-41. 65 135. Jen, J., et al., (1994). Molecular determinants of dysplasia in colorectal lesions. Cancer Res 54(21): p. 5523-5526. 136. Yokota, J., et al., (1986). Alterations of myc, myb, and rasHa proto-oncogenes in cancers are frequent and show clinical correlation. Science 231(4735): p. 261-5. 137. Cohen, J.B. and A.D. Levinson, (1988). A point mutation in the last intron responsible for increased expression and transforming activity of the c-Ha-ras oncogene. Nature 334(6178): p. 119-24. 138. Chen, B., et al., (1994). The second intron of the K-ras gene contains regulatory elements associated with mouse lung tumor susceptibility. Proc Natl Acad Sci U S A 91(4): p. 1589-93. 139. Westaway, D., et al., (1986). Identification of a provirally activated c-Ha-ras oncogene in an avian nephroblastoma via a novel procedure: cDNA cloning of a chimaeric viral- host transcript. Embo J 5(2): p. 301-9. 140. George, D.L., et al., (1986). Enhanced c-Ki-ras expression associated with Friend virus integration in a bone marrow-derived mouse cell line. Proc Natl Acad Sci U S A 83(6): p. 1651-5. 141. Chang, E.H., et al., (1982). Tumorigenic transformation of mammalian cells induced by a normal human gene homologous to the oncogene of Harvey murine sarcoma virus. Nature 297(5866): p. 479-83. 142. McKay, I.A., et al., (1986). Transformation and stimulation of DNA synthesis in NIH3T3 cells are a titratable function of normal p21N-ras expression. Embo J 5(10): p. 261721. 143. Pulciani, S., et al., (1985). ras gene Amplification and malignant transformation. Mol Cell Biol 5(10): p. 2836-41. 144. Finney, R.E. and J.M. Bishop, (1993). Predisposition to neoplastic transformation caused by gene replacement of H-rasl. Science 260(5113): p. 1524-7. 145. Boguski, M.S. and F. McCormick, (1993). Proteins regulating Ras and its relatives. Nature 366(6456): p. 643-54. 146. Downward, J., (1992). Regulation of p21ras by GTPase activating proteins and guanine nucleotide exchange proteins. Curr Opin Genet Dev 2(1): p. 13-8. 147. Downward, J., (1992). Regulatory mechanisms for ras proteins. Bioessays 14(3): p. 17784. 148. Broek, D., et al., (1987). The S. cerevisiae CDC25 gene product regulates the RAS/adenylate cyclase pathway. Cell 48(5): p. 789-99. 66 149. Boy-Marcotte, E., et al., (1989). The C-terminal part of a gene partially homologous to CDC25 gene suppresses the cdc25-5 mutation in Saccharomyces cerevisiae. Gene 77(1): p. 21-30. 150. Crechet, J.B., et al., (1990). Enhancement of the GDP-GTP exchange of RAS proteins by the carboxyl- terminal domain of SCD25 [see comments]. Science 248(4957): p. 866-8. 151. Jones, S., M.L. Vignais, and J.R. Broach, (1991). The CDC25 protein of Saccharomyces cerevisiae promotes exchange of guanine nucleotides bound to ras. Mol Cell Biol 11(5): p. 2641-6. 152. Damak, F., et al., (1991). SDC25, a CDC25-like gene which contains a RAS-activating domain and is a dispensable gene of Saccharomyces cerevisiae. Mol Cell Biol 11(1): p. 202-12. 153. Hughes, D.A., Y. Fukui, and M. Yamamoto, (1990). Homologous activators of ras in fission and budding yeast. Nature 344(6264): p. 355-7. 154. Bonfini, L., et al., (1992). The Son of sevenless gene product: a putative activator of Ras. Science 255(5044): p. 603-6. 155. Shou, C., et al., (1992). Molecular cloning of cDNAs encoding a guanine-nucleotidereleasing factor for Ras p21 [see comments]. Nature 358(6384): p. 351-4. 156. Wei, W., et al., (1992). Identification of a mammalian gene structurally and functionally related to the CDC25 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 89(15): p. 7100-4. 157. Martegani, E., et al., (1992). Cloning by functional complementation of a mouse cDNA encoding a homologue of CDC25, a Saccharomyces cerevisiae RAS activator. Embo J 11(6): p. 2151-7. 158. Cen, H., et al., (1992). Isolation of multiple mouse cDNAs with coding homology to Saccharomyces cerevisiae CDC25: identification of a region related to Bcr, Vav, Dbl and CDC24. Embo J 11(11): p. 4007-15. 159. Chardin, P., et al., (1993). Human Sosl: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260(5112): p. 1338-43. 160. Bowtell, D., et al., (1992). Identification of murine homologues of the Drosophila son of sevenless gene: potential activators of ras. Proc Natl Acad Sci U S A 89(14): p. 6511-5. 161. Ridley, A.J., (1995). Rho-related proteins: actin cytoskeleton and cell cycle. Curr Opin Genet Dev 5(1): p. 24-30. 162. Katzav, S., Z.D. Martin, and M. Barbacid, (1989). vav, a novel human oncogene derived from a locus ubiquitously expressed in hematopoietic cells. Embo J 8(8): p. 2283-90. 67 163. Eva, A. and S.A. Aaronson, (1985). Isolation of a new human oncogene from a diffuse Bcell lymphoma. Nature 316(6025): p. 273-5. 164. Habets, G.G., et al., (1994). Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell 77(4): p. 537-49. 165. Hart, M.J., et al., (1991). Catalysis of guanine nucleotide exchange on the CDC42Hs protein by the dbl oncogene product. Nature 354(6351): p. 311-4. 166. Michiels, F., et al., (1995). A role for Rac in Tiaml-induced membrane ruffling and invasion. Nature 375(6529): p. 338-340. 167. Gulbins, E., et al., (1993). Tyrosine kinase-stimulated guanine nucleotide exchange activity of Vav in T cell activation [see comments]. Science 260(5109): p. 822-5. 168. Gulbins, E., et al., (1994). Activation of Ras in vitro and in intact fibroblasts by the Vav guanine nucleotide exchange protein. Mol Cell Biol 14(2): p. 906-13. 169. Orita, S., et al., (1993). Comparison of kinetic properties between two mammalian ras p2 1 GDP/GTP exchange proteins, ras guanine nucleotide-releasing factor and smg GDP dissociation stimulation. J Biol Chem 268(34): p. 25542-6. 170. Kaibuchi, K., et al., (1991). Molecular cloning of the cDNA for stimulatory GDP/GTP exchange protein for smg p2 1s (ras p21-like small GTP-binding proteins) and characterization of stimulatory GDP/GTP exchange protein. Mol Cell Biol 11(5): p. 287380. 171. Mizuno, T., et al., (1991). A stimulatory GDP/GTP exchange protein for smg p21 is active on the post-translationally processed form of c-Ki-ras p21 and rhoA p21. Proc Natl Acad Sci U S A 88(15): p. 6442-6. 172. Hiroyoshi, M., et al., (1991). Role of the C-terminal region of smg p21, a ras p21-like small GTP- binding protein, in membrane and smg p21 GDP/GTP exchange protein interactions. J Biol Chem 266(5): p. 2962-9. 173. Hata, Y., et al., (1991). Enhancement of the actions of smg p21 GDP/GTP exchange protein by the protein kinase A-catalyzed phosphorylation of smg p21. J Biol Chem 266(10): p. 6571-7. 174. Nakanishi, H., et al., (1994). Different functions of Smg GDP dissociation stimulator and mammalian counterpart of yeast Cdc25. J. Biol. Chem. 269(21): p. 15085-91. 175. Trahey, M., et al., (1988). Molecular cloning of two types of GAP complementary DNA from human placenta. Science 242(4886): p. 1697-700. 176. Vogel, U.S., et al., (1988). Cloning of bovine GAP and its interaction with oncogenic ras p21. Nature 335(6185): p. 90-3. 68 177. Xu, G.F., et al., (1990). The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62(3): p. 599-608. 178. Martin, G.A., et al., (1990). The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63(4): p. 843-9. 179. Ballester, R., et al., (1990). The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 63(4): p. 851-9. 180. Cullen, P.J., et al., (1995). Identification of a specific Ins(1,3,4,5)P4-binding protein as a member of the GAP1 family. Nature 376(6540): p. 527-530. 181. Maekawa, M., et al., (1994). A novel mammalian Ras GTPase-activating protein which has phospholipid- binding and Btk homology regions. Mol Cell Biol 14(10): p. 68796885. 182. Trahey, M., et al., (1987). Biochemical and biological properties of the human N-ras p2 1 protein. Mol Cell Biol 7(1): p. 541-4. 183. Halenbeck, R., et al., (1990). Purification, characterization, and western blot analysis of human GTPase-activating protein from native and recombinant sources. J Biol Chem 265(35): p. 21922-8. 184. Cales, C., et al., (1988). The cytoplasmic protein GAP is implicated as the target for regulation by the ras gene product. Nature 332(6164): p. 548-51. 185. Adari, H., et al., (1988). Guanosine triphosphatase activating protein (GAP) interacts with the p21 ras effector binding domain. Science 240(4851): p. 518-21. 186. Pawson, T. and G.D. Gish, (1992). SH2 and SH3 domains: from structure to function. Cell 71(3): p. 359-62. 187. Cohen, G.B., R. Ren, and D. Baltimore, (1995). Modular binding domains in signal transduction proteins. Cell 80(2): p. 237-248. 188. Gibson, T.J., et al., (1994). PH domain: the first anniversary. Trends Biochem Sci 19(9): p. 349-353. 189. Musacchio, A., et al., (1993). The PH domain: a common piece in the structural patchwork of signalling proteins. Trends Biochem Sci 18(9): p. 343-8. 190. Viskochil, D., R. White, and R. Cawthon, (1993). The neurofibromatosis type 1 gene. Annu Rev Neurosci : p. 183-205. 191. Andersen, L.B., et al., (1993). A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol Cell Biol 13(1): p. 487-95. 69 192. Buchberg, A.M., et al., (1990). Sequence homology shared by neurofibromatosis type-1 gene and IRA-1 and IRA-2 negative regulators of the RAS cyclic AMP pathway [see comments]. Nature 347(6290): p. 291-4. 193. Xu, G.F., et al., (1990). The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 63(4): p. 83541. 194. Bollag, G. and F. McCormick, (1991). Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature 351(6327): p. 576-9. 195. Zhang, K., et al., (1990). Suppression of c-ras transformation by GTPase-activating protein [see comments]. Nature 346(6286): p. 754-6. 196. DeClue, J.E., et al., (1992). Abnormal regulation of mammalian p2 1ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 69(2): p. 265-73. 197. Basu, T.N., et al., (1992). Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients [see comments]. Nature 356(6371): p. 713-5. 198. Kim, H.A., et al., (1995). Schwann cells from neurofibromin deficient mice exhibit activation of p2lras, inhibition of cell proliferation and morphological changes. Oncogene 11(2): p. 325-335. 199. Henkemeyer, M., et al., (1995). Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 377(6551): p. 695-701. 200. Largaespada, D.A., et al., (1996). Nfl deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nature Genet 12(2): p. 137-143. 201. Bollag, G., et al., (1996). Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nature Genet 12(2): p. 144-148. 202. McCormick, F., (1989). ras GTPase activating protein: signal transmitter and signal terminator. Cell 56(1): p. 5-8. 203. McCormick, F., (1990). The world according to GAP. Oncogene 5(9): p. 1281-3. 204. Kitayama, H., et al., (1989). A ras-related gene with transformation suppressor activity. Cell 56(1): p. 77-84. 205. Beranger, F., et al., (1991). Association of the Ras-antagonistic Rapl/Krev- 1 proteins with the Golgi complex. Proc Natl Acad Sci U S A 88(5): p. 1606-10. 206. Frech, M., et al., (1990). Inhibition of GTPase activating protein stimulation of Ras-p21 GTPase by the Krev-1 gene product. Science 249(4965): p. 169-71. 70 207. Hata, Y., et al., (1990). Inhibition of the ras p21 GTPase-activating protein-stimulated GTPase activity of c-Ha-ras p21 by smg p21 having the same putative effector domain as ras p21s. J Biol Chem 265(13): p. 7104-7. 208. McCormick, F., (1994). Raf: the holy grail of Ras biology. Trends Cell Biol 4: p. 347-50. 209. Yatani, A., et al., (1990). ras p21 and GAP inhibit coupling of muscarinic receptors to atrial K+ channels. Cell 61(5): p. 769-76. 210. Martin, G.A., et al., (1992). GAP domains responsible for ras p21-dependent inhibition of muscarinic atrial K+ channel currents [see comments]. Science 255(5041): p. 192-4. 211. Ellis, C., et al., (1990). Phosphorylation of GAP and GAP-associated proteins by transforming and mitogenic tyrosine kinases. Nature 343(6256): p. 377-81. 212. Moran, M.F., et al., (1991). Protein-tyrosine kinases regulate the phosphorylation, protein interactions, subcellular distribution, and activity of p2lras GTPase- activating protein. Mol Cell Biol 11(4): p. 1804-12. 213. Settleman, J., et al., (1992). Molecular cloning of cDNAs encoding the GAP-associated protein p190: implications for a signaling pathway from ras to the nucleus. Cell 69(3): p. 539-49. 214. Settleman, J., et al., (1992). Association between GTPase activators for Rho and Ras families. Nature 359(6391): p. 153-4. 215. McGlade, J., et al., (1993). The N-terminal region of GAP regulates cytoskeletal structure and cell adhesion. Embo J 12(8): p. 3073-81. 216. Wong, G., et al., (1992). Molecular cloning and nucleic acid binding properties of the GAP- associated tyrosine phosphoprotein p62. Cell 69(3): p. 551-8. 217. Lock, P., et al., (1996). The human p62 cDNA encodes Sam68 and not the RasGAPassociated p62 protein. Cell 84(1): p. 23-24. 218. Duchesne, M., et al., (1993). Identification of the SH3 domain of GAP as an essential sequence for Ras-GAP-mediated signaling. Science 259(5094): p. 525-8. 219. Medema, R.H., et al., (1992). GTPase-activating protein SH2-SH3 domains induce gene expression in a Ras-dependent fashion. Mol Cell Biol 12(8): p. 3425-30. 220. Schweighoffer, F., et al., (1992). Implication of GAP in Ras-dependent transactivation of a polyoma enhancer sequence. Science 256(5058): p. 825-7. 221. Clark, G.J., et al., (1993). Differential antagonism of Ras biological activity by catalytic and Src homology domains of Ras GTPase activation protein. Proc Natl Acad Sci U S A 90(11): p. 4887-91. 71 222. Bollag, G., F. McCormick, and R. Clark, (1993). Characterization of full-length neurofibromin: tubulin inhibits Ras GAP activity. Embo J 12(5): p. 1923-7. 223. Takai, Y., et al., (1995). Effects of prenyl modifications on interactions of small G proteins with regulators. Methods Enzymol 250(122): p. 122-133. 224. Isomura, M., et al., (1991). Regulation of binding of rhoB p20 to membranes by its specific regulatory protein, GDP dissociation inhibitor. Oncogene 6(1): p. 119-24. 225. Hori, Y., et al., (1991). Post-translational modifications of the C-terminal region of the rho protein are important for its interaction with membranes and the stimulatory and inhibitory GDP/GTP exchange proteins. Oncogene 6(4): p. 515-22. 226. Araki, S., et al., (1991). Role of the C-terminal region of smg p25A in its interaction with membranes and the GDP/GTP exchange protein. Mol Cell Biol 11(3): p. 1438-47. 227. Li, Y., et al., (1992). Somatic mutations in the neurofibromatosis 1 gene in human tumors. Cell 69(2): p. 275-81. 228. Jacks, T., et al., (1994). Tumour predisposition in mice heterozygous for a targeted mutation in Nfl. Nat Genet 7(3): p. 353-361. 229. Chevallier, M.M., et al., (1993). Saccharomyces cerevisiae CDC25 (1028-1589) is a guanine nucleotide releasing factor for mammalian ras proteins and is oncogenic in NIH3T3 cells. J Biol Chem 268(15): p. 11113-8. 230. Barlat, I., et al., (1993). The Saccharomyces cerevisiae gene product SDC25 C-domain functions as an oncoprotein in NIH3T3 cells. Oncogene 8(1): p. 215-8. 231. Egan, S.E., et al., (1993). Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation [see comments]. Nature 363(6424): p. 45-51. 232. Fujioka, H., et al., (1992). Transforming and c-fos promoter/enhancer-stimulating activities of a stimulatory GDP/GTP exchange protein for small GTP-binding proteins. J Biol Chem 267(2): p. 926-30. 233. McCormick, F., (1993). Signal transduction. How receptors turn Ras on [news; comment]. Nature 363(6424): p. 15-6. 234. Feig, L.A., (1993). The many roads that lead to Ras [comment]. Science 260(5109): p. 767-8. 235. Treisman, R., (1996). Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol 8: p. 205-215. 236. Daum, G., et al., (1994). The ins and outs of Raf kinases. Trends Biochem. Sci. 19(11): p. 474-480. 72 237. Downward, J., et al., (1990). Stimulation of p2lras upon T-cell activation [see comments]. Nature 346(6286): p. 719-23. 238. Torti, M., et al., (1992). Erythropoietin induces p21ras activation and pl20GAP tyrosine phosphorylation in human erythroleukemia cells. J Biol Chem 267(12): p. 8293-8. 239. Medema, R.H., et al., (1993). Ras activation by insulin and epidermal growth factor through enhanced exchange of guanine nucleotides on p21ras. Mol Cell Biol 13(1): p. 15562. 240. Li, B.Q., et al., (1992). Nerve growth factor stimulation of the Ras-guanine nucleotide exchange factor and GAP activities. Science 256(5062): p. 1456-9. 241. Tsai, M.H., C.L. Yu, and D.W. Stacey, (1990). A cytoplasmic protein inhibits the GTPase activity of H-Ras in a phospholipid-dependent manner. Science 250(4983): p. 982-5. 242. Tsai, M.H., et al., (1989). The effect of GTPase activating protein upon ras is inhibited by mitogenically responsive lipids. Science 243(4890): p. 522-6. 243. Yu, C.L., M.H. Tsai, and D.W. Stacey, (1990). Serum stimulation of NIH 3T3 cells induces the production of lipids able to inhibit GTPase-activating protein activity. Mol Cell Biol 10(12): p. 6683-9. 244. Serth, J., et al., (1991). The inhibition of the GTPase activating protein-Ha-ras interaction by acidic lipids is due to physical association of the C-terminal domain of the GTPase activating protein with micellar structures. Embo J 10(6): p. 1325-30. 245. Buday, L., P.H. Warne, and J. Downward, (1995). Downregulation of the Ras activation pathway by MAP kinase phosphorylation of Sos. Oncogene 11(7): p. 1327-1331. 246. de, V., et al., (1995). Shc associates with an unphosphorylated form of the p21ras guanine nucleotide exchange factor mSOS. Oncogene 10(5): p. 919-25. 247. Egan, S.E. and R.A. Weinberg, (1993). The pathway to signal achievement [news]. Nature 365(6449): p. 781-3. 248. Clark, S.G., M.J. Stem, and H.R. Horvitz, (1992). C. elegans cell-signalling gene sem-5 encodes a protein with SH2 and SH3 domains [see comments]. Nature 356(6367): p. 3404. 249. Lowenstein, E.J., et al., (1992). The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70(3): p. 431-42. 250. Simon, M.A., G.S. Dodson, and G.M. Rubin, (1993). An SH3-SH2-SH3 protein is required for p21Rasl activation and binds to sevenless and Sos proteins in vitro. Cell 73(1): p. 169-77. 73 251. Rozakis, A.M., et al., (1992). Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature 360(6405): p. 689-92. 252. Rozakis, A.M., et al., (1993). The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSosl [see comments]. Nature 363(6424): p. 83-5. 253. Olivier, J.P., et al., (1993). A Drosophila SH2-SH3 adaptor protein implicated in coupling the sevenless tyrosine kinase to an activator of Ras guanine nucleotide exchange, Sos. Cell 73(1): p. 179-91. 254. Li, N., et al., (1993). Guanine-nucleotide-releasing factor hSosl binds to Grb2 and links receptor tyrosine kinases to Ras signalling [see comments]. Nature 363(6424): p. 85-8. 255. Gale, N.W., et al., (1993). Grb2 mediates the EGF-dependent activation of guanine nucleotide exchange on Ras [see comments]. Nature 363(6424): p. 88-92. 256. Buday, L. and J. Downward, (1993). Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 73(3): p. 611-20. 257. Pelicci, G., et al., (1992). A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 70(1): p. 93-104. 258. McGlade, J., et al., (1992). Shc proteins are phosphorylated and regulated by the v-Src and v-Fps protein-tyrosine kinases. Proc Natl Acad Sci U S A 89(19): p. 8869-73. 259. Roberts, T.M., (1992). Cell biology. A signal chain of events [news; comment]. Nature 360(6404): p. 534-5. 260. Morrison, D.K., et al., (1988). Signal transduction from membrane to cytoplasm: growth factors and membrane-bound oncogene products increase Raf-1 phosphorylation and associated protein kinase activity. Proc Natl Acad Sci U S A 85(23): p. 8855-9. 261. Heidecker, G., et al., (1992). The role of Raf-1 phosphorylation in signal transduction. Adv Cancer Res : p. 53-73. 262. Wood, K.W., et al., (1992). ras mediates nerve growth factor receptor modulation of three signal- transducing protein kinases: MAP kinase, Raf- 1, and RSK. Cell 68(6): p. 1041-50. 263. Han, M., et al., (1993). C. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature 363(6425): p. 133-40. 264. Dickson, B., et al., (1992). Raf functions downstream of Rasl in the Sevenless signal transduction pathway [see comments]. Nature 360(6404): p. 600-3. 265. Moodie, S.A., et al., (1993). Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase [see comments]. Science 260(5114): p. 1658-61. 74 266. Warne, P.H., P.R. Viciana, and J. Downward, (1993). Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 364(6435): p. 352-5. 267. Fields, S. and 0. Song, (1989). A novel genetic system to detect protein-protein interactions. Nature 340(6230): p. 245-6. 268. Van, A.L., et al., (1993). Complex formation between RAS and RAF and other protein kinases. Proc Natl Acad Sci U S A 90(13): p. 6213-7. 269. Vojtek, A.B., S.M. Hollenberg, and J.A. Cooper, (1993). Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 74(1): p. 205-14. 270. Zhang, X.F., et al., (1993). Normal and oncogenic p21ras proteins bind to the aminoterminal regulatory domain of c-Raf-1. Nature 364(6435): p. 308-13. 271. Cook, S.J., et al., (1993). RapV12 antagonizes Ras-dependent activation of ERK1 and ERK2 by LPA and EGF in Rat-1 fibroblasts. Embo J 12(9): p. 3475-85. 272. Wu, J., et al., (1993). Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3',5'-monophosphate [see comments]. Science 262(5136): p. 1065-9. 273. Cook, S.J. and F. McCormick, (1993). Inhibition by cAMP of Ras-dependent activation of Raf [see comments]. Science 262(5136): p. 1069-72. 274. Williams, N.G., T.M. Roberts, and P. Li, (1992). Both p2lras and pp60v-src are required, but neither alone is sufficient, to activate the Raf-1 kinase. Proc Natl Acad Sci U S A 89(7): p. 2922-6. 275. Kikuchi, A. and L.T. Williams, (1994). The post-translational modification of ras p2 1 is important for Raf-1 activation. J Biol Chem 269(31): p. 20054-20059. 276. Fabian, J.R., .LO.Daar, and D.K. Morrison, (1993). Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol Cell Biol 13(11): p. 7170-9. 277. Rapp, U.R., (1991). Role of Raf-1 serine/threonine protein kinase in growth factor signal transduction. Oncogene 6(4): p. 495-500. 278. Stokoe, D., et al., (1994). Activation of Raf as a result of recruitment to the plasma membrane [see comments]. Science 264(5164): p. 1463-7. 279. Leevers, S.J., H.F. Paterson, and C.J. Marshall, (1994). Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 369(6479): p. 411-4. 280. Marais, R., et al., (1995). Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. Embo J 14(13): p. 3136-3145. 281. Marshall, C.J., (1994). MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr. Opin. Genet. Dev. 4(1): p. 82-89. 75 282. Davis, R.J., (1994). MAPKs: new JNK expands the group. Trends Biochem Sci 19(11): p. 470-473. 283. Xia, Z., et al., (1995). Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270(5240): p. 1326-1331. 284. Stokoe, D., et al., (1992). MAPKAP kinase-2; a novel protein kinase activated by mitogen-activated protein kinase. Embo J 11(11): p. 3985-94. 285. Lamarche, N. and A. Hall, (1994). GAPs for rho-related GTPases. Trends Genet 10(12): p. 436-40. 286. Symon, M., et al., (1996). Wiskott-aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell 84: p. 723-734. 287. Watanabe, G., et al., (1996). Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science 271(5249): p. 645-648. 288. Amano, M., et al., (1996). Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science 271(5249): p. 648-650. 289. Diekmann, D., et al., (1994). Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science 265(5171): p. 531-3. 290. Martin, G.A., et al., (1995). A novel serine kinase activated by racl/CDC42Hs-dependent autophosphorylation is related to PAK65 and STE20. Embo J 14(17): p. 4385. 291. Chardin, P., et al., (1989). The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. Embo J 8(4): p. 1087-92. 292. Paterson, H.F., et al., (1990). Microinjection of recombinant p2lrho induces rapid changes in cell morphology. J Cell Biol 111(3): p. 1001-7. 293. Ridley, A.J. and A. Hall, (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70(3): p. 389-99. 294. Ridley, A.J., et al., (1992). The small GTP-binding protein rac regulates growth factorinduced membrane ruffling. Cell 70(3): p. 401-10. 295. Nobes, C.D. and A. Hall, (1995). Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81(1): p. 53-62. 296. Chant, J. and L. Stowers, (1995). GTPase cascades choreographing cellular behavior: movement, morphogenesis, and more. Cell 81(1): p. 1-4. 76 297. Khosravi, F.R., et al., (1994). Dbl and Vav mediate transformation via mitogen-activated protein kinase pathways that are distinct from those activated by oncogenic Ras. Mol. Cell. Biol. 14(10): p. 6848-57. 298. Minden, A., et al., (1995). Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 81(7): p. 1147-1157. 299. Coso, O.A., et al., (1995). The small GTP-binding proteins Rac 1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 81(7): p. 1137-1146. 300. Herskowitz, I., (1995). MAP kinase pathways in yeast: for mating and more. Cell 80(2): p. 187-197. 301. Hill, C.S., J. Wynne, and R. Treisman, (1995). The Rho family GTPases RhoA, Racl, and CDC42Hs regulate transcriptional activation by SRF. Cell 81(7): p. 1159-1170. 302. Treisman, R., (1994). Ternary complex factors: growth factor regulated transcriptional activators. Curr Opin Genet Dev 4(1): p. 96-101. 303. Gille, H., A.D. Sharrocks, and P.E. Shaw, (1992). Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature 358(6385): p. 414-7. 304. Gille, H., et al., (1995). ERK phosphorylation potentiates Elk- -mediated ternary complex formation and transactivation. Embo J 14(5): p. 951-962. 305. Downward, J., (1995). KSR: a novel player in the RAS pathway [comment]. Cell 83(6): p. 831-834. 306. Sundaram, M. and M. Han, (1995). The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 83(6): p. 889-901. 307. Kornfeld, K., D.B. Hom, and H.R. Horvitz, (1995). The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 83(6): p. 903-913. 308. Therrien, M., et al., (1995). KSR, a novel protein kinase required for RAS signal transduction. Cell 83(6): p. 879-888. 309. Kumagai, N., et al., (1993). ADP-ribosylation of rho p21 inhibits lysophosphatidic acidinduced protein tyrosine phosphorylation and phosphatidylinositol 3-kinase activation in cultured Swiss 3T3 cells. J Biol Chem 268(33): p. 24535-8. 310. Zhang, J., et al., (1993). Activation of platelet phosphatidylinositide 3-kinase requires the small GTP-binding protein Rho. J Biol Chem 268(30): p. 22251-4. 311. Malcolm, K.C., et al., (1994). Activation of rat liver phospholipase D by the small GTPbinding protein RhoA. J. Biol. Chem. 269(42): p. 25951-4. 77 312. Chong, L.D., et al., (1994). The small GTP-binding protein Rho regulates a phosphatidylinositol 4- phosphate 5-kinase in mammalian cells. Cell 79(3): p. 507-13. 313. Jackson, J.H., et al., (1990). Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc Natl Acad Sci U S A 87(8): p. 3042-6. 314. Gibbs, J.B., A. Oliff, and N.E. Kohl, (1994). Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell 77(2): p. 175-8. 315. Hancock, J.F., (1993). Anti-Ras drugs come of age. Curr Biol 3(11): p. 770-772. 316. Glomset, J.A., M.H. Gelb, and C.C. Farnsworth, (1990). Prenyl proteins in eukaryotic cells: a new type of membrane anchor. Trends Biochem Sci 15(4): p. 139-42. 317. Maltese, W.A., (1990). Posttranslational modification of proteins by isoprenoids in mammalian cells. Faseb J 4(15): p. 3319-28. 318. Tamanoi, F., (1993). Inhibitors of Ras farnesyltransferases. Trends Biochem. Sci. 18(9): p. 349-353. 319. Garcia, A.M., et al., (1993). Peptidomimetic inhibitors of Ras farnesylation and function in whole cells. J Biol Chem 268(25): p. 18415-8. 320. James, G.L., et al., (1993). Benzodiazepine peptidomimetics: potent inhibitors of Ras farnesylation in animal cells [see comments]. Science 260(5116): p. 1937-42. 321. Kohl, N.E., et al., (1993). Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor [see comments]. Science 260(5116): p. 1934-7. 322. Nigam, M., et al., (1993). Potent inhibition of human tumor p2lras farnesyltransferase by A1A2- lacking p21ras CA1A2X peptidomimetics. J Biol Chem 268(28): p. 20695-8. 323. Kohl, N.E., et al., (1994). Protein farnesyltransferase inhibitors block the growth of rasdependent tumors in nude mice. Proc Natl Acad Sci U S A 91(19): p. 9141-9145. 324. Kohl, N.E., et al., (1995). Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med 1(8): p. 792-797. 325. Manne, V., et al., (1995). Bisubstrate inhibitors of farnesyltransferase: a novel class of specific inhibitors of ras transformed cells. Oncogene 10(9): p. 1763-1779. 326. Prendergast, G.C., et al., (1994). Farnesyltransferase inhibition causes morphological reversion of ras- transformed cells by a complex mechanism that involves regulation of the actin cytoskeleton. Mol Cell Biol 14(6): p. 4193-202. 327. Cox, A.D., et al., (1994). The CAAX peptidomimetic compound B581 specifically blocks farnesylated, but not geranylgeranylated or myristylated, oncogenic ras signaling and transformation. J Biol Chem 269(30): p. 19203-19206. 78 328. Stacey, D.W., et al., (1991). Dominant inhibitory Ras mutants demonstrate the requirement for Ras activity in the action of tyrosine kinase oncogenes. Oncogene 6(12): p. 2297-304. 329. Stacey, D.W., L.A. Feig, and J.B. Gibbs, (1991). Dominant inhibitory Ras mutants selectively inhibit the activity of either cellular or oncogenic Ras. Mol Cell Biol 11(8): p. 4053-64. 330. Reiss, Y., et al., (1990). Inhibition of purified p21ras farnesyl:protein transferase by CysAAX tetrapeptides. Cell 62(1): p. 81-8. 331. Sun, J., et al., (1995). Ras CAAX peptidomimetic FTI 276 selectively blocks tumor growth in nude mice of a human lung carcinoma with K-Ras mutation and p53 deletion. Cancer Res 55(19): p. 4243-4247. 332. Lerner, E.C., et al., (1995). Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem 270(45): p. 26802-26806. 333. Miyake, M., et al., (1996). Unfarnesylated transforming Ras mutant inhibits the Rassignaling pathway by forming a stable Ras.Raf complex in the cytosol. Febs Lett 378(1): p. 15-18. 334. Chan, A.M., et al., (1994). A human oncogene of the RAS superfamily unmasked by expression cDNA cloning. Proc Natl Acad Sci U S A 91(16): p. 7558-7562. 335. Drivas, G.T., et al., (1990). Characterization of four novel ras-like genes expressed in a human teratocarcinoma cell line. Mol Cell Biol 10(4): p. 1793-8. 336. Ulsh, L.S. and T.Y. Shih, (1984). Metabolic turnover of human c-rasH p21 protein of EJ bladder carcinoma and its normal cellular and viral homologs. Mol Cell Biol 4(8): p. 1647- 52. 337. Lebowitz, P.F., J.P. Davide, and G.C. Prendergast, (1995). Evidence that farnesyltransferase inhibitors suppress Ras transformation by interfering with Rho activity. Mol Cell Biol 15(12): p. 6613-6622. 79 Chapter 2 K-ras is an Essential Gene in Mouse Embryogenesis with Partial Functional Overlap with N-ras 80 Abstract Mammalian ras genes are critical in the regulation of cellular proliferation and differentiation and are mutated in -30% of all human tumors. It has been demonstrated that N-ras and H-ras are dispensable for mouse development. To characterize the normal role of K-ras in growth and development, I have mutated it via gene targeting in the mouse. On an inbred genetic background, embryos homozygous for this mutation die between 12 and 14 days of gestation, with fetal liver defects and evidence of anemia. Thus, K-ras is the only member of the ras gene family essential for mouse embryogenesis. I also demonstrate that multiple mutations within the ras gene family can affect phenotype. Most animals lacking N-ras function and heterozygous for the K-ras mutation exhibit abnormal hematopoietic development and die between days 10 and 12 of embryogenesis. Complete elimination of K-ras and N-ras function is compatible with development through the blastocyst stage, but double mutant embryos do not develop to 9.5 days of gestation. Thus, partial functional compensation appears to occur within the ras gene family, but K-ras provides a unique and essential function. 81 Introduction Over the past decade, a convergence of genetic and biochemical data from diverse systems has supported a critical role for Ras-mediated signal transduction pathways in multiple aspects of growth control and development (Satoh et al., 1992; Khosravi and Der, 1994; McCormick, 1994). Through a combination of genetic studies in Drosophila melanogaster and Caenorhabditis elegans and biochemical studies using mammalian cells, many of the components in the Ras signaling cascade have been elucidated (Daum et al., 1994; Davis, 1994; Kayne and Sternberg, 1995; Wassarman et al., 1995; Duffy and Perrimon, 1996; Wittenberg and Reed, 1996). These studies have revealed that many of the components responsible for transducing the extracellular signals are shared by many receptor tyrosine kinases and are highly conserved throughout evolution as well. A variety of genetic, cell biological, and biochemical approaches have provided significant insight into the ras family of proto-oncogenes and their role in normal growth and tumorigenesis. Although it has been known for some time that Ras proteins function as molecular switches regulated at the level of GDP/GTP binding (Barbacid, 1987; Bourne et al., 1990), only in the past few years have many of the components in the Ras signaling cascade been identified, cloned, and biochemically characterized. A critical equilibrium exists in the cell between inactive (GDPbound) and active (GTP-bound) Ras proteins, and the interconversion between these two forms is regulated by the concerted action of guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) following the appropriate extracellular stimuli (Bollag and McCormick, 1991; Downward, 1992b; Downward, 1992a; Boguski and McCormick, 1993). Many of the oncogenic mutations in ras either reduce the rate of hydrolysis of GTP on Ras and its stimulation by GAPs or increase the guanine nucleotide exchange rate, both leading to an accumulation of Ras in the active, GTP-bound state. In mammals there are three functional ras genes located on different chromosomes that encode four highly homologous 21 kD proteins: H-, N-, K4A-, and K4B-ras (Barbacid, 1987). 82 The coding sequences of all three genes are distributed amongst four exons with precisely corresponding splice junctions, suggesting a common origin from one ancestral gene. K-ras is unique in that it possesses two alternative fourth coding exons, thereby allowing the synthesis of two p21 isoforms which differ only in their carboxy terminal (C-terminal) residues (Capon et al., 1983; McGrath et al., 1983; Shimizu et al., 1983). The first 86 amino acids of the mammalian Ras proteins, which harbors the putative effector domain, are 100% identical. Only the C-termini of these proteins differ significantly from one another in a region known as the hypervariable domain. Despite considerable progress in elucidating the signal transduction pathways involving Ras, it is still not known what individual roles, if any, the different ras family members play. Many observations suggest that these proteins possess overlapping functions. For example, all three ras genes are expressed ubiquitously (Chesa et al., 1987; Furth et al., 1987; Leon et al., 1987). All cell types analyzed, including those which are terminally differentiated and postmitotic, have been found to express the Ras proteins. In addition, certain tumor types (e.g., thyroid) show no absolute specificity for which ras family member is mutated (Bos, 1989; Lemoine et al., 1989). Finally, it has been demonstrated in yeast as well as in mice that ras gene function is dispensable. In S. cerevisiae, two RAS genes, RASI andRAS2, have been identified (Powers et al., 1984). Strains carrying mutations in both genes are inviable, whereas inactivation individually is compatible with growth (Kataoka et al., 1984). Importantly, mice homozygous null for either N-ras (Umanoff et al., 1995) or H-ras (M. Katsuki, personal communication) are viable and exhibit no overt abnormalities in development and post-natally. However, several lines of evidence suggest the existence of unique roles for the three mammalian ras genes. For example, despite ubiquitous ras expression, levels of ras mRNA appear to be regulated both temporally and spatially, with certain tissues expressing one or more members of the family preferentially (Muller et al., 1982; Muller et al., 1983; Leon et al., 1987). K-ras and N-ras exhibit similar expression profiles. In addition, many tumor types exhibit mutation of one member of the family more frequently than the other two (e.g., K-ras in colon, 83 lung, and pancreatic tumors; N-ras in myeloid disorders; and H-ras in skin and mammary tumors), suggesting a unique oncogenic role for each of these genes in specific tissues (Bos, 1988b; Bos, 1988a; Bos, 1989). As mentioned above, K-ras is alternatively spliced at its last coding exon, resulting in two isoforms with distinct C-termini, K-ras4A and 4B. K-ras4B, the predominant isoform (Capon et al., 1983; George et al., 1985), is distinct from the other three Ras proteins in the type of post-translational modifications that occur at the C-terminus (Hancock et al., 1989; Hancock et al., 1990; James et al., 1995). This difference is believed to be responsible for the specific association of K-ras4B with the GEF, Smg GDS (Mizuno et al., 1991; Orita et al., 1993). Interestingly, this GEF not only catalyzes the exchange of bound GDP by GTP, but also translocates small G proteins (as shown for K-ras4B and RaplB) from the membrane to the cytoplasm (Kawamura et al., 1993; Nakanishi et al., 1994). Therefore, it is tempting to speculate that this specific interaction and translocation may allow K-ras4B to associate with a distinct subset of potential effector molecules not shared by the other Ras molecules. Because activating ras mutations have been detected in up to 30% of all human tumors analyzed (Barbacid, 1987; Bos, 1988a; Bos, 1989), considerable research has been directed towards rational drug design aimed at selectively inhibiting Ras function in transformed cells (Hancock, 1993; Gibbs et al., 1994). The efficacy of these drugs will depend, in part, on the physiological consequences of the inhibition of Ras in normal cells, including whether the loss of Ras function is compatible with cell viability. Gene targeting experiments have demonstrated that neither N-ras nor H-ras function are essential in the mouse. In contrast, microinjection experiments suggest that Ras activity is required for mouse embryos to develop beyond the twocell stage (Yamauchi et al., 1994). Together, these data suggest that K-ras has a unique and essential function or that some critical threshold level of overall Ras activity is required during early embryogenesis. As a means of addressing these and related issues, I have generated mice carrying a null allele of K-ras by gene targeting. In addition, I have crossed this mutation with the N-ras null allele to examine possible genetic interactions between these two family members. 84 Results Disruption of the Murine K-ras Gene I targeted one allele of the murine K-ras gene in mouse embryonic stem (ES) cells using the positive-negative selection method of Mansour et al. (Mansour et al., 1988). The K-ras targeting vector was constructed by replacing exon 1 sequence with the bacterial neomycin gene (neo) expression cassette as shown in Figure 2.1A. Loss of exon 1 sequences should result in a nonfunctional allele of K-ras as it contains a critical portion of the effector domain (George et al., 1985; Willumsen et al., 1986). The herpes simplex virus thymidine kinase (HSV-tk) gene was also placed downstream of the K-ras sequences in order to select against ES cells that had randomly integrated the entire DNA targeting vector. Following introduction of the K-ras targeting vector into 129/Sv D3 ES cells, the resulting G418- and gancyclovir-resistant clones were screened by Southern blot analysis using a probe located 3' to the sequences present in the targeting vector. This probe detects an 8.1 kilobase (kb) StuI fragment from wild-type DNA and an additional 7.0 kb BamHI to StuI mutant-specific fragment (Figure 2.1A and 2.1B). Eight out of 380 ES cell clones screened had acquired the exogenous K-ras sequences by homologous recombination as detected by the 3' probe and were therefore heterozygous for the mutation (K-ras+/-). Counter-selection by gancyclovir resulted in a 13-fold reduction in the number of ES cell colonies compared to G418 selection alone. Further analysis using a 5' external probe revealed that 2 of these 8 clones had undergone aberrant recombination at the 5' side (data not shown), and these were not used in the generation of chimeras. Interestingly, Southern blot analysis on the K-ras+/ - clones using a probe derived from neo sequences revealed that all 8 targeted clones contained an additional 6.9 kb band as well as the expected 8.4 kb fragment in a Stul (Figure 2.1C). Extensive Southern blot and polymerase chain reaction (PCR) analysis of these clones showed that the neo and 3' fragment of K-ras carried in the targeting vector were duplicated in a head-to-tail fashion one or more times (data not shown). The genomic configuration of K-ras mutant alleles is depicted in Figure 2.1A. These anomalous 85 Figure 2.1. Disruption of K-ras in ES cells. (A) The K-ras targeting vector, pK-ras KO, was constructed by inserting fragments from intron 0 and intron 1 of the mouse K-ras gene into the plasmid pPNT. The regions of homology consist of a 2.8 kb NotI-Sall fragment and a 5.1 kb HindIll-KpnI fragment. Both the pkg-neo and HSV-tk cassettes were positioned such that they were transcribed in the same transcriptional orientation as K-ras. (B) Southern blot analysis of BamHI plus Stul-digested genomic DNA from ES cell clones using a probe 3' to the region of homology (3' ext probe). Lanes 3, 4, 5, and 6 represent four independent K-ras+l-ES cells clones, as they possess both an 8.1 kb wild-type (wt) allele and a 7.0 kb mutant-specific K-ras allele. The DNA in Lane 1 is from wt ES cells, Lane 2 from a nonhomologous integrant, and Lanes 7 and 8 represent two independent K-ras-1 - mutant ES cell clones which were obtained following exposure to increasing concentrations of G418. (C) Southern blot analysis of StuI-digested genomic DNA using a probe specific for neo. On a parallel set of samples to (B), the neo probe detected the expected 8.4 kb StuI fragment, as well as an additional 6.9 kb fragment in all K-ras+l- ES cell clones (Lanes 3-6). As expected, no signal was detected in the wt ES cells (Lane 1), and the lower band depicted in Lane 2 represents the random integration pattern for this non-homologous integrant. A number of different digests were performed and screened with the above probes as well as with additional 5' external and internal probes to determine the actual configuration of the mutant allele. Both the expected and actual K-ras mutant allele configurations are shown in (A). This was confirmed further by PCR analysis using the primer pairs shown in (A). Primer pairs 3'neo + 5'neo and 3'hom + 5' hom specifically amplify a 5.1 kb and 1.8 kb fragment, respectively, as would be expected for this configuration. This head-to-tail integration pattern occurred either one time (see Lanes 3 and 5 in (B)) or multiple times (see Lanes 4 and 6 in (B)). (D) PCR analysis of E12.5 embryos derived from a K-ras+/- intercross showing the presence of K-ras-1- embryos (lanes marked with an asterisk (*)). The two alleles can be distinguished using primer pairs that specifically amplify a 360 bp wt fragment (5'-IO + 3'-Ex1) or a 270 bp mutant specific fragment (5'-IO + 3'-neo). 86 oc C) I-I A, n I ·J Z EP O W Z 1?C o h bD E •o · 1• .0 0 o r" .0 .oj Figure 2.1 B C 1 2 3 4 5 6 7 1 8 8.4kb 8.1kb b 7.0kb 2 3 5 4 p.~ = 6.9kb *. .. neo probe 3' probe 360 bp 270 bp PCR 88 6 recombination events may be due to the relatively low level of expression of the K-ras locus (Muller et al., 1983); perhaps only heterozygous ES cell clones that duplicated neo-containing sequences attained sufficient levels of protein to provide G418 resistance at the selection level used. Regardless, in each of the heterozygous ES cell clones used for further study, critical K-ras exon 1 sequences had been deleted as confirmed by Southern blot analysis on K-ras homozygous mutant ES cells (data not shown). K-ras is an Essential Gene Chimeric animals were created by injecting K-ras+l- ES cells into C57BL/6 blastocyst-stage embryos. Germline contribution was determined by breeding the chimeras to C57BL/6 mice, and BL/6:129/Sv agouti progeny were genotyped. Those mice that carried the mutant allele were initially identified by Southern blot analysis and subsequently by PCR amplification of tail DNA (data not shown). Three independent heterozygous ES cell clones produced chimeras that transmitted the K-ras mutant allele through the germline. To establish the requirements for K-ras function during mouse development, K-ras+/l mice were mated and the genotypes of offspring determined at weaning. Genotyping of the first 83 offspring from these heterozygous intercrosses revealed no K-ras-1- animals, indicating that K-ras is an essential gene for mouse embryogenesis. I collected embryos at progressively earlier times in development to delineate the timing of the death of K-ras-1 - embryos; all embryos were genotyped by PCR analysis (Figure 2. 1D). This analysis was carried out on both a mixed BL/6:129/Sv background as well as on an inbred 129/Sv background. The viability plot for both genetic backgrounds is depicted in Figure 2.2. On a mixed background, K-ras-1-embryonic viability was not affected until embryonic day 12.5 (E12.5). The number of viable K-ras-1 - embryos decreased with increased gestational age, with no mutants surviving past birth. In contrast to the broad window of lethality observed on the mixed genetic background, K-ras- 1- embryos died during a much more restricted developmental period when the mutation was examined on an inbred 129/Sv background. Lethality was again first evident at E12.5, and no K-ras-1 - embryo survived beyond E14.0. 89 v25 -I 10 5 0 9.5 10.5 11.5 12.5 13.5 14.5 15.5 16.5 17.5 18.5 19.5 gestational days Figure 2.2. Viability plot for K-ras-l- embryos. The data is graphed as the percentage of viable embryos that were K-ras-1- as a function of increasing gestational age. All embryos were derived from K-ras+l-intercrosses; thus, 25% would be expected to be K-ras-1- embryos. This analysis was performed on both a mixed BL/6:129/Sv genetic background as well as on a pure 129/Sv background. All heterozygous parents used in this analysis were the Fl progeny of chimeras bred with either pure BL/6 females (for the mixed genetic background analysis) or pure 129/Sv females (for the inbred genetic background analysis). On the mixed genetic background, approximately 600 total implants were genotyped, ranging from 30 to 170 embryos per gestational day. On the pure 129/Sv genetic background, approximately 400 total implants were genotyped. During the critical time period (E 11.5-14.5), the number of embryos genotyped ranged from 30 to 130 per gestational day. 90 On both the mixed and inbred backgrounds, K-ras-1- embryos appeared morphologically normal and were indistinguishable from their littermates up to E10.5. Beginning at E11.5, mutant embryos could be identified morphologically based on their smaller size and delayed growth. The E12.5 K-ras-1- embryo shown in Figure 2.3A exemplifies the mutant phenotype: the mutant is developmentally delayed, has a less obvious superficial vasculature, and is paler than normal littermates. Moreover, the livers of mutants were very pale and reduced in size (typically 2-8 fold fewer total cells than control livers), and the embryo exhibited signs of edema, particularly in the pericardial space. These superficial features are consistent with anemia and a defect in the production or circulation of red blood cells. The developmental delay ranged from 0.5 to 3 gestational days, with those embryos surviving late in gestation (mixed background) exhibiting the most significant delay. Typically, this delay was coordinate; however, some embryos did exhibit a non-coordinate delay as shown in Figure 2.3B. In this K-ras-1- embryo, features such as the limbs, skin, tail, and whiskers had developed to a stage associated with E17.5-18.5, whereas the eyes did not develop beyond the equivalent of E15.5 since eye closure was not attained. Moreover, histological analysis of E15.5 or older K-ras-1 - embryos on the mixed genetic background showed that a small percentage of these mutants exhibited a non-coordinate development of internal organs (data not shown). Fetal Liver Defect in K-ras-"- Embryos Morphologically, K-ras-1- exhibited a phenotype that was consistent with a defect leading to anemia. The fetal organs/tissues required at this stage in development to support a functional hematopoietic and circulatory system for the red blood cells include the placenta, extraembryonic membranes (e.g., yolk sac and chorion), liver, heart, and vasculature system. Histological examination of these and other tissues was carried out on K-ras 1/- embryos. Due to the more uniform expressivity of the homozygous mutant phenotype on the 129/Sv background, most of the histological and cellular analysis was performed on embryos from this genetic background. With 91 Figure 2.3. Phenotypic comparison of K-ras"- embryos and control littermates. (A) The wild-type littermate is on the left and the K-ras-/ - littermate is at the right. Note the slight developmentally delay (- 0.5 gestational days) and pale coloring of the liver in the K-ras-1embryo. (B) The control E18.5 K-ras+l- embryo is on the left and the K-ras-" - embryo is on the right. Note the marked reduction in size of the mutant relative to the control littermate as well as the noncoordinate development of the K-ras-1 - embryo. The eyes have not developed beyond E15.5, whereas the limbs, tail, and skin have all advanced to at least E17.5. (C) and (D) Parasagittal section through an E12.5 K-ras-1 - (C) and a control wild-type (D) fetal liver. Note the areas of hypocellularity in the K-ras mutant fetal liver, whereas the cells are densely packed in the control liver. Also, pyknotic nuclei (white arrows) in the distal portion of the K-ras-" - hepatic lobe are indicated. (E) and (F) Cell death analysis on an adjacent fetal liver section from (C) and (D). Note the presence of significant numbers of TUNEL-positive (brown staining) cells in the K-ras-1 - fetal livers (E). In more severely effected embryos, these apoptotic cells were present throughout the liver. In contrast, very few cells stain positive in the TUNEL assay from control fetal livers (F). 92 the exception of the fetal liver (see below), no other tissues of the K-ras-'- embryos were consistently defective. Histological examination of the livers from E12.5 to E13.5 K-ras-/-embryos indicated that they were smaller and lacked extensive cellularization compared with controls (Figure 2.3C and 2.3D). In addition, cell death was observed in the livers of more severely affected mutant embryos. This cell death was first apparent in the distal portions of the hepatic lobes (Figure 2.3C), but extended throughout the entire organ in the final stages of viability (data not shown). The pyknotic nuclear morphology exhibited in mutant livers is typically associated with apoptosis. To assess whether this cell death was due to apoptosis, I analyzed fetal liver sections by the TUNEL assay. Liver sections from both E12.5 and E13.5 control embryos showed a few isolated cells that stained positive for biotin-dUTP incorporation (Figure 2.3F and data not shown), whereas numerous cells were TUNEL-positive in K-ras-/ - liver sections (Figure 2.3E and data not shown). Thus, the cell death observed in K-ras-deficient embryos was due largely, if not exclusively, to apoptosis. Furthermore, this excessive cell death was specific to the liver as other tissues did not exhibit significantly enhanced levels of TUNEL-positive staining (data not shown). Functional Analysis of K-ras-1- Hematopoiesis At E12.5, the liver constitutes the major hematopoietic organ, predominantly for definitive erythropoiesis (Dzierzak and Medvinsky, 1995). Based on the timing of death for the K-ras-1embryos, it was conceivable that there may have been a specific defect in this process. Histological analysis, however, established the presence of definitive (enucleated) red cells in the embryo. Furthermore, peripheral blood smears from E12.5 to E18.5 (mixed background embryos were used for E14.5 or older) embryos showed that mutants were comparable to control embryos of the same developmental stage in the percentage of enucleated vs. nucleated red cells (data not shown). Therefore, erythroid cells of K-ras-1- embryos were capable of achieving endstage differentiation within the hepatic microenvironment. However, this analysis does not establish that hematopoiesis occurred efficiently (see below). 94 Ras has been implicated in relaying the signal downstream of a number of hematopoietic growth and survival factors (Satoh et al., 1992). In particular, Ras has been found to be important in eliciting a survival signal downstream of GM-CSF and IL-3 in hematopoietic progenitor cells (Kinoshita et al., 1995). The cell death observed in the fetal liver may, therefore, reflect an intrinsic defect of the mutant hematopoietic progenitor cells to survive, proliferate, and/or differentiate. Alternatively, the cell death may represent a failure of the mutant hepatic microenvironment to support these processes. To determine the functional potential of K-ras-l- hematopoietic progenitors and stem cells, hematopoietic assays were performed in vitro and in vivo. Single cell suspensions made from E12.5 livers of 129/Sv derived K-ras-l- and control embryos were plated at equal cell densities in vitro in methylcellulose medium optimized for hematopoietic colony differentiation. Comparable sizes, onset of differentiation, and morphologies were found for CFU-E, BFU-E, CFU-GM, and CFU-GEMM colonies derived from mutant and control fetal livers (data not shown). However, I consistently observed an -2 fold reduction in the number of colonies derived from the K-ras-lfetal livers as compared to controls (data not shown). This difference may indicate that K-ras-/ committed progenitors are partially impaired in their ability to survive and/or differentiate. Alternatively, the developmental delay of the mutant embryos may account for this difference, as control embryos of a similar developmental stage could also exhibit a 1.2-2 fold reduction in the number of colonies obtained (data not shown). To examine the presence and function of more primitive K-ras-/- multipotential progenitors as well as the long-term repopulating hematopoietic stem cell (LTR-HSC), in vivo reconstitution assays were performed. Lethally-irradiated BL/6 recipient mice were injected retro-orbitally with single cell suspensions made from the entire livers of 129/Sv derived, E13.5 mutant and control embryos. Donor cell contribution to the peripheral blood was monitored by the glucose phosphate isomerase (GPI) assay two weeks post-injection and at 1 month intervals thereafter. Both the onset and extent of contribution to the peripheral blood were equivalent for mutant (3/4 cases) and control cells (data not shown). To date, 3/4 recipients have survived 4 or more months post- 95 injection with K-ras-1 - hematopoietic cells (data not shown), indicating that both short-term (1 month) and long-term (4-6 months) repopulation has taken effect. Thus, K-ras function is not required for differentiation of the LTR-HSC. This result is consistent with the in vitro colony assays, and together, these data suggest that the apparent anemia in K-ras- / - embryos is a consequence of defects in the fetal liver microenvironment. K-ras-1- ES Cells have Reduced Contribution to Hematopoietic Compartments Analysis of K-ras-/- embryos has shown a requirement for K-ras in the normal development of, at least, the fetal liver. However, due to the attendant lethality of the K-ras-1embryo, it was not possible to determine what effects, if any, the absence of K-ras function would have later in gestation or in the adult animal. This issue is important, in part, given the recent efforts to develop chemotherapeutic strategies to inhibit Ras function by such drugs as farnesyltransferase inhibitors (Hancock, 1993; Gibbs et al., 1994). Therefore, K-ras+l-ES cells were subjected to enhanced concentrations of G418 to generate K-ras-1 - ES cells (Mortensen et al., 1992). Out of 200 clones screened, two were found to have lost the wild-type allele (Figure 2. 1B). These K-ras-1 -ES cell clones were then injected into BL/6 blastocysts to create chimeric animals. For comparison, chimeras were also generated using the parental K-ras+l-ES cell line. Only 4-6 K-ras-1 - ES cells could be injected per blastocyst to avoid the lethality associated with K-ras homozygosity. Contribution by the K-ras-deficient cells to chimeric animals was estimated from the extent of the agouti coat color and found to range from -10 to 50%. In addition, I determined the percent contribution of mutant cells to the internal tissues of four week old chimeras using GPI analysis. More than 25 tissues were examined from 8 K-ras-1- chimeras and 4 control K-ras+/- chimeras using this assay. As summarized in Figure 2.4, significant contribution by the K-ras-/- cells was observed in most tissues, suggesting that most cell types can develop and survive in the absence of K-ras function. However, since this assay measures the percent contribution to the 96 Figure 2.4. GPI isoenzyme analysis on K-ras+ /- and K-ras-1- chimeras. Tissue contribution of K-ras+/- and K-ras-/- ES cells was analyzed by the GPI isoenzyme assay as described in the experimental procedures. The graph represents the average percent contribution of K-ras+/-and K-ras-1- ES cells to representative tissues in the chimeras. The number of animals from which each tissue was examined is indicated in parentheses following the tissue (K-ras+/- / K-ras-l-). The lowest and highest values of contribution to tissues in individual chimeras are indicated in the columns to the right, with the K-ras+ /- chimeras listed above in each case. The data is shown in two graphs, with those tissues exhibiting lower contribution (except for the pancreas) by the K-ras-1- cells shown in the upper graph and those exhibiting normal contribution in the lower graph. 97 Figure 2.4 lympho/monocytes (315) %contribution Lowest Highest -M IK-ras+l" whole blood (4/8) 0 K-ras"/ - r platelets (3/6) RBC (4/6) thymus (2/7) P"Mm RMIM spleen (4/8) lung (4/8) bone marrow (4/8) 2.0 2.3 1.0 1.0 3.0 2.3 10.0 1.0 1.0 1.0 27.5 3.7 43.8 12.5 22.5 10.0 45.0 15.0 6.3 10.0 1.0 45.0 1.0 52.5 1.0 3.0 1.0 3.0 1.0 liver A (4/8) liver B (4/8) 1.0 coat color (4/8) pancreas (4/8) 40 30 20 10 ---1 -- cecum (4/8) colon (4/8) 1:8 IT:• 90.0 50.0 47.5 stomach (4/7) IK-ras+l' 36.3 U K-ras-/- 12.5 1.0 1.0 42.5 1.0 47.5 v/~·rr////~7m/rr///~///rr/r//~il 50.0 10.0 46.3 1.0 7.5 50.0 50.0 4:9 98:8 5.0 50.0 13.8 1.0 55.0 50.0 5.0 7.5 35.0 52.5 55.0 52.5 1.0 T---~- medulla (4/8) cerebrum (4/8) atrium (3/8) cerebellum (3/8)1 7.5 ventricle (4/8) 23.8 1.0 12.5 3.0 47.5 adrenal (4/8) kidney (4/8) 0 • I 10 I I I 20 30 40 average % contribution 60 43.8 43.8 17.5 28.8 JI 37.5 5.0 v~n/n/n/nnm/n~ ileum (4/8) duodenum/jujenum (4/8) bladder (4/8) 52.5 50 1.0 ~''/~'//~/~////I/~-////1~51 41.3 10.0 10.0 3.0 1.0 C€ 36.3 48.8 36.3 50.0 40.0 42.5 45.0 50.0 50.0 56.3 50.0 55.0 55.0 whole tissue, it is possible that K-ras may be required for the proper development of certain cell types. Indeed, the homozygous mutant phenotype indicates that this must be so. Interestingly, relatively low contribution by the K-ras-1- cells was observed in the lung as well as in multiple hematopoietic lineages and the tissues that support their production throughout embryogenesis and adult life (e.g., liver and spleen). Moreover, those chimeras with the highest percent contributions to the liver and spleen exhibited the lowest contribution to the hematopoietic cells of the bone marrow (data not shown). In addition, in those chimeras with a relatively high contribution of K-ras-deficient cells in the bone marrow, the mutant cells were underrepresented in the more differentiated cells of the blood. The low contribution to hematopoietic lineages stands in contrast with the in vitro and in vivo hematopoietic cell analyses and suggests that the lack of K-ras function may affect hematopoietic progenitor cells directly. This discrepancy may be explained by the fact that in the chimeras, K-ras-deficient hematopoietic cells are developing in competition with wild-type cells. Thus, any subtle intrinsic defect present in the mutant cells may be accentuated. No Upregulation of Ras Family Members in K-ras-"/ Embryos The observed differences in viability for K-ras-l-embryos on the two different genetic backgrounds suggested that it is likely that there are one or more modifier loci influencing the requirement for K-ras function. Given the common expression patterns and signaling pathways utilized by K-ras and N-ras, it was conceivable that differences in N-ras expression levels could affect the K-ras mutant phenotype. To determine if N-ras or H-ras were upregulated in the K-ras mutant background, I examined the levels of these proteins in mouse embryonic fibroblasts (MEFs) derived from E13.5 embryos on both genetic backgrounds. Lysates of subconfluent cultures of MEFs were immunoprecipitated with Y13-259 antibody (which recognizes all three forms of Ras) and subsequently immunoblotted with antibodies specific for each of the three Ras proteins. As shown in Figure 2.5A, there was no detectable K-ras protein in MEFs derived from K-ras-l embryos. Moreover, neither N-ras nor H-ras protein levels were altered in cells lacking K-ras. 99 Figure 2.5. N-ras and H-ras are not upregulated in response to the loss of K-ras. (A) Mouse embryonic fibroblasts (MEFs) were derived from E13.5 littermates on both genetic backgrounds and analyzed for the levels of K, N-, and H-ras proteins. Ras was immunoprecipitated from at least two independently derived clones for each genotype using the pan-Ras antibody Y13-259. Immunocomplexes were then analyzed by immunoblotting with monoclonal antibodies specific for each of the Ras proteins. The blot probed with K-ras (F234) was stripped and reprobed with a pan Ras (F111) antibody to confirm that the mutant lysates were intact (data not shown). Lysates from cells prepared from a mixed genetic background are shown; however, identical results were obtained on the pure 129/Sv background (data not shown). (B) Tissues were prepared from E12.5 embryos derived on the mixed genetic background and analyzed as above. One immunoblot was made and probed with the same series of Ras antibodies as used in (A). The order of analysis was as follows: N-ras (F155), H-ras (F235), K-ras (F234), and then pan Ras (F111). The slight amount of signal which is detected in the K-ras-1 - tissues by the K-ras specific antibody is due to background cross-reactivity to the faster migrating N- and Hras proteins. Probing with the pan Ras antibody mirrored the expression levels seen with each antibody (data not shown). Similar results were also obtained with heart and lung tissues (data not shown). 100 Figure 2.5 K-ras Genotype: +1+ +1+ +1- +1- -I K-ras -- I-------- N-ras H-ras ,,r•I,--------..-. Western Ab: K-ras K-ras Genotype: N-ras +/+ +/- 1ien Liver +/+ +/. ./ +/ +/- ./. n m Brain H-ras I- ]~iI, I, ViT Carcass I 1 I IP Ab: Y13-259 (pan) 101 ~ I ] 1· In order to assess whether N-ras and/or H-ras may have been upregulated in a tissuespecific manner, extracts were prepared from E12.5 mutant and control littermate tissues (BL/6:129/Sv background) and analyzed as above. Examination of 5 different tissues revealed that there was no significant change in either N-ras or H-ras protein levels in the K-ras deficient tissues (Figure 2.5B and data not shown). Ras proteins are active when bound to GTP and tight regulation of the GTP/GDP bound states of these proteins is critical for maintaining normal growth control. Therefore, it remains possible that there is functional compensation for the loss of K-ras through increases in the level of GTP-bound N-ras and/or H-ras. Genetic Interaction Between K-ras and N-ras In contrast to the requirement for K-ras in mouse embryogenesis described here, neither N-ras (Umanoff et al., 1995) nor H-ras (M. Katsuki, personal communication) are essential genes in the mouse. One possible explanation for the normal phenotype of N- and H-ras mutants, and for the relatively late onset phenotype in the K-ras mutants, is that different members of the family have at least partial overlapping function. To address a possible genetic interaction between K- and N-ras, mice carrying various combinations of the two null mutations were generated on a mixed BL/6:129/Sv genetic background. As shown in Table 2.1, double heterozygotes (N-ras+l-; K-ras+/- ) were viable with no overt abnormalities. However, animals deficient for N-ras and heterozygous for the K-ras mutation (N-ras-l-; K-ras+l- ) died during gestation. Also, heterozygosity at the N-ras locus significantly worsened the K-ras homozygous mutant phenotype (N-ras+l-;K-ras-l- ) (Table 2.2). Embryos were collected at progressively earlier timepoints in development to delineate the timing of death of N-ras-l-; K-ras+l- and N-ras+l-; K-ras -1- embryos; due to breeding considerations, a more extensive analysis was undertaken on the N-ras-l-; K-ras+/- embryos. As with the K-ras-1- embryos on a mixed background, N-ras-l-; K-ras+l- mutants exhibited a variable expressivity in phenotype and a broad window of lethality. -70% of these mutants died between E10.0 and E12.0, with the remainder dying perinatally (Table 2.1). A few embryos were found to 102 Table 2.1. Summary of Dissection Analysis for N-rasl-; K-ras+l- Embryos Embryonic Day N-ras: K-ras: 9.5-10.0 # viable recovered: Genotypes of Embryos from N+/'; K +1" x N-1'; K +/+ +/+/-/-/+/+ +/+/+ +/- 11 14 16 % abnormal: 18.2d 21.4d 31.3d 50.0 d 10.5-11.0 # viable recovered: %abnormal: 39 5.1d 22 4.6d 30 3 .3 d 20 6 5 .0d,h 11.5-12.0 # viable recovered: % abnormal: 12.5-13.0 12 13 18 12 4 7.7d,h 5.6d 8.3d 100.0d,h # viable recovered: 19 14 17 4 % abnormal: 5.3s 14.3s,p 17.6s,p 7 5 .0 d,s,p 13.5-14.0 # viable recovered: % abnormal: 11 9.1d 9 22 .2 d,p 13 15 .4d 5 80.Od,p,e 14.5-15.0 # viable recovered: 13 9 12 8 % abnormal: 7.7d 33.3d,p,e 16.7d,p 87.5d,p,e,t # viable recovered: 11 9 9 4 % abnormal: 0.0 33 .3d,s,p 11.1d 100.Od,s,p,e 16.5-17.0 # viable recovered: % abnormal: 11 9.1s 11 9.1s,p 17 11.8s,p 6 100.Od,p,e,t 17.5-18.0 # viable recovered: % abnormal: 11 0.0 13 15.4d,s 6 16.7s 3 33.3d 18.5-19.0 # viable recovered: 11 10 9 4 % abnormal: 0.0 0.0 0.0 50.0d,t 15.5-16.0 Embryos resulting from a N-ras+l-;K-ras+/- x N-ras-/-; K-ras+/+cross were dissected at various times in gestation and analyzed for their viability, gross morphological appearance, and subsequently fixed for histological purposes. Based on the parent's genotypes, four different genotypic classes of embryos should result, each with an expected frequency of 25%. Abnormalities are denoted as follows: (d) delayed by 0.5 day or more, (h) dilated heart and pericardial sac, (s) small for developmental stage, (p) pale and less vascularized, (e) asymmetrical development of the eye: either significantly smaller than its normal counterpart or the pigment of the eye was overgrown, and (t) severe shortening of the tail. survive up to 2 days past birth, but were subsequently neglected by their mothers and died shortly thereafter. No apparent differences in phenotype were noted in crosses performed with parents of different mutant genotypes. N-ras'-; K-ras+l" Embryos Die with Severe Anemia N-ras-l-; K-ras+l-embryos were indistinguishable from normal littermates at E8.5. Starting at E9.5, a higher percentage of N-ras-l-; K-ras+l- embryos were delayed by 0.5-1.0 developmental days (50% vs. -23% average of the other three genotypes). By E10.5, 65% of viable embryos were abnormal (Table 2.1). The E10.5 N-ras-l-; K-ras+l- embryo shown in Figure 2.6A exemplifies the phenotype observed at this stage: the mutant has arrested at -9.5 days of development, is markedly more pale than normal littermates, and has a dilated heart and pericardial sac. Figure 2.6B demonstrates the severity of dilatation within the heart and pericardial sac that can be observed in N-ras-l-; K-ras+l- embryos. Very few circulating red blood cells are seen within either the embryo or its yolk sac. Furthermore, yolk sacs from these embryos exhibited a wrinkled or roughened appearance when compared to the smooth yolk sacs of littermate controls (Figure 2.6C). Despite an overall growth delay in N-ras-l-; K-ras+l embryos, many developmental processes were completed, including cardiac contraction, fusion of the allantois and chorion, rotation of the embryo, and closure of the anterior neuropore. Histological examination of affected E1O.5N-ras-l-;K-ras+l- embryos demonstrated the near or complete absence of blood islands in their yolk sacs and significantly reduced numbers of circulating primitive erythrocytes in either the yolk sac (compare Figures 2.7A and 2.7B) or the embryo (data not shown). However, other tissues of mesodermal origin, such as the myocardium, somites, and blood vessels, were present and appeared to develop normally. Numerous pyknotic nuclei were observed throughout N-ras-l-; K-ras+/- embryos. Although in the yolk sac cell death appeared to be restricted to cells of mesodermal origin (data not shown), extensive cell death occurred throughout the embryo in cells derived from all three germ layers (compare Figures 2.7C with 2.7D and Figure 2.7E with 2.7F). In earlier N-ras-l-; K-ras+l- 104 Figure 2.6. Comparison of N-ras-A; K-ras + " and control littermates at E10.5. (A) Embryos were isolated at E10.5. Embryonic genotypes are as follows: N-ras-l-; K-ras+l(left) and N-ras+l-;K-ras+l- (right). The N-ras-l-; K-ras+l- embryo has only developed to a stage equivalent to E9.5, has a dilated heart and pericardial sac, and has reduced numbers of circulating red blood cells. (B) Higher magnification view of a different N-ras-/-; K-ras+l- embryo isolated at E10.5. This mutant represents the severity in dilation that can be observed in both the heart and pericardial sac. Amazingly, cardiac contraction still occurs in these embryos. (C) Visceral yolk sacs from E10.5 N-ras-l-; K-ras+l- embryos (2 on the left) and a N-ras+l-; K-ras+l- embryos (right). Note the roughened and wrinkled appearance that the N-ras-l-; K-ras+lyolk sacs take on compared to the smooth appearance of the control yolk sac. Moreover, there is a marked reduction in the number of circulating embryonic blood cells present in the yolk sacs of N-ras-l-; K-ras+/- embryos. 105 Figure 2.7. Histology of E9.5 and E10.5 N-ras-l; K-ras+l- and control embryos. (A) and (B) Histological analysis of E10.5 N-ras-'l; K-ras+l- (A) and N-ras+l+;K-ras+l- (B) visceral yolk sacs. Note the strict absence of blood islands (larger black arrow) and the presence of very limiting numbers of circulating primitive erythrocytes (small black arrows) in the N-ras-l-; K-ras+l- yolk sac. Otherwise, the yolk sac tissue and endothelial-lined blood vessels exhibit similar appearances in both yolk sacs. (C) and (D) Parasagittal sections through the forebrains of an E9.5 N-ras-l-; K-ras+l- embryo (C) and a N-ras+l+;K-ras+l - littermate control (D). Note the prevelant cell death (pyknotic nuclei (black arrows)) throughout the forebrain of the N-ras-l-; K-ras+l-embryo. (E) and (F) Parasagittal sections through the mid-portion of an E10.5 N-ras-l/; K-ras+l- embryo (E) and a N-ras+l+;K-ras+l- control littermate (F). The cell death observed at E9.5 extends throughout the entire embryo by E10.5. This section reveals prevalent cell death in the primitive mesenchyme which extends into the somitic tissue. 107 embryos (E9.5), cell death was prevalent in the forebrain (Figure 2.7C) and to a lesser extent along the neural axis. I cannot conclude if the cell death is due solely to an extreme anemia and subsequent hypoxia, or whether Ras function is required for global cell survival. Surprisingly, a significant proportion (-30%) of N-ras-1-; K-ras+l-embryos survived past this critical stage in development. As can be seen in Table 2.1, almost all of them were abnormal and readily identifiable except at the final stages of gestation. The N-ras-/-; K-ras+l- embryo shown in Figure 2.8A represents some of the phenotypes observed at these later stages in development: the embryo is developmentally delayed by -0.5 gestational days, is small for its developmental stage, and is very anemic and edematous. Most N-ras-l-; K-ras+/-embryos were delayed by only 0.5-1.0 days in development and never exhibited the non-coordinate delay that could be observed in late stage K-ras-1 - embryos. Interestingly, 22% of E13.5-18.5 N-ras-l-; K-ras+/- embryos showed an asymmetrical pattern in eye development, with only the right eye being affected, and 20% exhibited a defect in proper tail development (Table 2.1). Two different abnormalities were observed with respect to eye development: either the right eye was significantly smaller than the left eye (compare Figure 2.8D with 2.8G) or the pigmentation in the right eye was markedly over-developed in comparison with the left eye (compare Figure 2.8E with 2.8H). A control embryo is shown for comparison in Figures 2.8C and 2.8F. Histological examination, however, revealed that most structures in the eye appeared to develop correctly. Moreover, other structures within the head of the same embryo had developed symmetrically, demonstrating that this anomaly was specific to the eye. Figure 2.8B demonstrates the severe truncation in tail development, which was also observed at a fairly high frequency (20%). In addition, a higher percentage of embryos with only two of the four ras alleles (N-ras+/-;K-ras+/and N-ras-l-; K-ras+/+), as compared to those carrying three of the four ras alleles (N-ras+/-; K-ras+/+), showed similar phenotypes (see Table 1). In summary, these data suggest that a critical threshold level of Ras activity must be met in order for development to occur normally. Histological analysis of E15.5-16.5 N-ras-l-; K-ras+l- embryos revealed that the only defect that could be consistently defined was again in the fetal liver. As shown in Figures 2.9A 109 Figure 2.8. N-ras'l-; K-ras' "/ embryos exhibit morphological abnormalities late in gestation. (A) E15.5 N-ras-l-; K-ras+/- embryo (left) and a control N-ras-l-; K-ras+l + littermate (right) are shown. The N-ras-/-; K-ras+/ - embryo is developmentally delayed by 0.5 gestational days and is severely anemic and edematous. (B) E18.5 N-ras-l-; K-ras+l-embryo (right) and a control N-ras+l-;K-ras+l- littermate (left) are shown. Embryos were fixed in Bouin's and subsequently photographed. Note the severe truncation in tail development in the N-ras-/-; K-ras+l- embryo. This occurred in -20% of N-ras-l-; K-ras+l-embryos, age E14.5 or older. Right eye (C)-(E) and left eye (F)-(H) development in the same respective embryo at E15.5. Two different developmental and asymmetrical abnormalities were observed in -22% of N-ras-l-; K-ras+/- embryos between the ages of E13.5 and E16.5. Either the right eye, specifically, was significantly smaller than the left eye in N-ras-l-; K-ras+l-embryos (compare (D) and (G)), or the pigment of the eye was overgrown, with the right eye again being more affected than the left eye (compare (E) and (H)). A control N-ras-/-; K-ras+l + embryo is shown in (C) and (F) for comparison. 110 Figure 2.9. Histology of E15.5-16.5 N-ras-l-; K-ras+l- fetal livers. (A) and (B) Parasagittal sections through the fetal livers of an E15.5 N-ras-l-; K-ras+/- embryo (A) and a control E16.5 N-ras-/-; K-ras+l+ embryo (B). Note the absence of blood filled, endothelial-lined vessels (longer black arrow in (B)) in the N-ras-l-;K-ras+l- embryo. Moreover, the ratio of erythroblasts (dark, blue staining cells and smaller black arrow) to hepatocytes is significantly reduced in these embryos. (C) Parasagittal section through the fetal liver of an E16.5 N-ras-l-; K-ras+l- embryo. A similar, but more severe phenotype, was observed at this age, in which the hepatocytes would take on a very vacuolated appearance (indicated by the black arrows). (D) Higher magnification view of the fetal liver in (C) demonstrating the vacuoles (black arrows). 112 and 2.9B, livers from E15.5 N-ras-1-; K-ras+l-embryos have a significantly reduced ratio in the number of erythroblasts to hepatocytes as compared to normal control littermates. These hepatocytes also tended to become extremely vacuolated by E16.5 (Figure 2.9C and 2.9D). I did not observe elevated cell death in these livers, suggesting that this defect is either different than the defect in K-ras-1- fetal livers or not as severe. Moreover, there were very few circulating red blood cells within the embryonic tissues (compare Figures 2.9A and 2.9B; data not shown). Peripheral blood smears obtained from these animals confirmed that definitive erythropoiesis occurred, and that the ratio of enucleated to nucleated red blood cells was appropriate for their developmental stage (data not shown). Therefore, their anemia appears to be due to inefficient, but apparently normal, production of definitive erythrocytes and may reflect a defect in the survival and/or differentiation of either the erythroblasts or a more primitive progenitor cell. As in the case of the K-ras-1- phenotype, this may be due to an intrinsic defect and/or a defect within the microenvironment to support these hematopoietic processes (see Discussion). As shown in Table 2.2, N-ras+/-;K-ras-" - embryos were capable of surviving to E9.5. However, these embryos were more severely affected than N-ras-1 -; K-ras+/l- embryos, as 40% were inviable and 100% were abnormal at this developmental stage. Morphological and histological examination of N-ras+l-;K-ras-1 - embryos revealed a similar phenotype to N-ras-1-; K-ras+/l- embryos. However, N-ras+/l;K-ras-1 - embryos exhibited more extensive cell death at E9.5 than their N-ras-l-; K-ras+l-counterparts (data not shown). Thus, embryos carrying only one of the four functional alleles of N-ras and K-ras were less severely affected if the one allele was K-ras. Hematopoietic Colony Formation in Cultures of N-ras-l-; K-ras+/- Yolk Sacs Prior to the establishment of fetal liver hematopoiesis at ~E1O.5-1 1.0, the earliest erythroid cells arise in the blood islands of the yolk sac, giving rise to primitive (nucleated) red blood cells (Dzierzak and Medvinsky, 1995; Orkin, 1995). The absence of blood islands as well as the limited number of primitive erythrocytes observed in N-ras-l - , K-ras+/- yolk sacs, may reflect a 114 o c r r E i; ~ r, ;r; L r C 3 ~ o h 3 1 1 I + + + Ic Iv 0 ri 0 0 + + ++ ++ a~c r- rnIooc SN Od 'Ile T~~~''~ W > 0 Cow io 9z. i eq I Cu~ ; ~ c ki, OOOa -- j so 0I AC ~~~c od v 400 * 0 C) 0B w 14: o a l 2 ~4 03Oa) v a.) ý L Q C) li 04 oo cl u0 0 C) a) CG g "o- Ln S0 Ura $a4 ve 0 a.)V 3 bO a. >=t 0o vlo0 ya.it wC0 oca.a - 0 o a.) 4-J soo a) oc ·( C4 (L) 4-J Us a) g 150 a) r-e cn oo R - o ~8<eA oi a) a. = 0 rc COO)0 a.) cVj n~A 4- cl Q 4-) u CIS C)$ a ;l< * > 0>,ght 0ý: .'" cna. 0bJ-o S 00.) + + ed ~O <0 +, Zn~ ~l w) B 0-u a.C 0 a.'s) 0y c 4 failure in either their ability to survive or the ability of the erythroid progenitor cells to survive and/or differentiate efficiently within the yolk sac microenvironment. To examine the presence and function of N-ras-/- , K-ras+/- hematopoietic progenitors, in vitro colony assays were performed with E9.5 and E10.5 embryonic yolk sacs. Erythroid, myeloid, and mixed progenitors were present in N-ras-/-, K-ras+/- yolk sacs and capable of proliferating and differentiating normally (data not shown). However, a more pronounced reduction in the number of all three progenitor classes was observed from the yolk sacs of more severely affected N-ras-1 -, K-ras+l- embryos compared to similarly staged control embryos (data not shown). Thus, this marked reduction in colony number appeared to represent an intrinsic defect in the progenitors for their ability to survive, proliferate, and/or differentiate rather than the developmental delay of the mutant embryos. The possibility still exists that there may be an additional defect residing within the yolk sac microenvironment. N-rasrl; K-rasr" Embryos Die Between E3.5 and E9.5 Due to the difficulty in assessing the phenotype of double homozygous embryos (only 1/16 embryos generated from a N-ras+/-; K-ras+/- by N-ras+/-; K-ras+/- cross is expected to be N-ras-l-; K-ras-l-), the analysis of this genotype was limited. As summarized in Table 2.2, N-ras-l -; K-ras-1 - embryos survived to the blastocyst stage and were normal. Out of 123 blastocysts isolated and genotyped by PCR analysis, 7 were determined to be N-ras-l-; K-ras-l- . This yield was consistent with the 1/16 expected frequency for this genotypic class. Embryos were also isolated at E9.5 and scored for phenotypic abnormalities. As shown in Table 2.2, no N-ras-/-;K-ras-1- embryos were recovered out of 72 genotyped implants (4-5 were expected) and a significant proportion (12%) of implants were resorbed and could not be genotyped (data not shown). These data indicate that N-ras-1-; K-ras-1- embryos develop normally to the blastocyst stage, but subsequently fail in gestation sometime before E9.5. Thus, H-ras appears to be sufficient to support embryonic development to at least the blastocyst stage. Alternatively, given the long half-lives of Ras proteins (24 hours) (Ulsh and Shih, 1984), it is conceivable that N-ras-l-; 116 K-ras-/ - embryos survive to the blastocyst stage due to maternal contribution of these proteins. It is clear, however, that at least one allele from either K-ras or N-ras is needed for embryonic survival up to E9.5 and beyond. 117 Discussion Ras function is central in the signal transduction pathways leading downstream of many growth and survival factors (Satoh et al., 1992) and is known to be required for proper cellular proliferation, differentiation, and survival in many developmental systems (Barbacid, 1987; Khosravi and Der, 1994). Given the structural and functional similarities between the three mammalian ras genes, it has long been of interest to determine if distinct functions exist for one or more members of the gene family. To understand the role of individual ras genes in mammalian development, I and others have created targeted null alleles for each one of them in the mouse. Analyses of mice lacking N-ras and H-ras function have demonstrated that both genes are dispensable for normal development. Here, I have shown that K-ras provides an essential function in mouse embryogenesis and have demonstrated partial functional compensation between different members of the Ras gene family. K-ras is an Essential Gene in the Mouse On an inbred 129/Sv genetic background, K-ras-1 - embryos failed in gestation between E12 and E14. Thus, although the growth and differentiation of many tissues can proceed apparently normally in the absence of K-ras function through mid-gestation, completion of embryogenesis is dependent on the activity of this gene. These mutant embryos exhibited a slight developmental growth delay starting at approximately E11, which was more pronounced in embryos late in gestation on the mixed (BL/6:129/Sv) genetic background. The lethality is likely to be due to hematopoietic defects, as evidenced by the overall pale color of the mutant embryos and the increased levels of cell death observed in the fetal liver (the major site of erythropoiesis in midgestation). However, I have shown that the K-ras function is not required for the differentiation of hematopoietic progenitors in vitro or upon transplant into lethally-irradiated recipients, suggesting that the apparent anemia in the mutant embryos may be caused by an abnormal hematopoietic microenvironment in the fetal liver. Interestingly, in chimeric mice composed in part of K-ras- 118 deficient cells, the contribution of mutant cells to different hematopoietic compartments was consistently lower than to other tissues. Therefore, the absence of K-ras may cause a slight impairment in the capacity of hematopoietic cells to differentiate or survive, which is manifested when the mutant cells are in competition with wild-type cells during development and differentiation in the chimeras or in an otherwise defective fetal liver microenvironment in the homozygous mutant embryos. The phenotype of embryos mutant for both K- and N-ras also supports a direct role for Ras function in hematopoiesis (see below). The requirement for K-ras in the developing mouse may reflect either a unique function of this gene not shared by H- or N-ras or simply the expression pattern of the different members of the gene family. Although several studies have concluded that all three genes are expressed ubiquitously (Muller et al., 1982; Muller et al., 1983; Leon et al., 1987), these were conducted on whole tissues or embryos. Therefore, it is possible that a critical cell type (e.g., within the fetal liver) expresses K-ras exclusively. In the context of the K-ras mutation and without transcriptional upregulation of H- or N-ras, such a cell would be effectively devoid of Ras function and might fail to differentiate properly or die. In fact, upon examination of mutant MEFs in culture and various tissues in K-ras-1- embryos, I have seen no evidence for compensatory increases in the level of either N- or H-Ras. More precise expression analysis will be necessary to address this possibility directly. Based largely on differences in post-translational modification, it has been suggested that K-Ras, and specifically the 4B isoform, may have a unique function in cell signaling. Localization of Ras proteins to the plasma membrane is critical for their biological activity (Gibbs, 1991; Khosravi et al., 1992; Glomset and Farnsworth, 1994). Membrane association is achieved through a series of closely linked post-translational modifications, including farnesylation, proteolysis, and carboxylmethylation, which are signaled by the consensus CAAX (C, cysteine; A, aliphatic amino acid; X, any amino acid) sequence located at the C-terminus. In addition, N-, H-, and K-Ras4A are subsequently palmitoylated at upstream cysteine residues (Hancock et al., 1989). In K-ras4B, however, these upstream cysteine residues are substituted by a polylysine domain that 119 serves an analogous function to that of palmitylation (Hancock et al., 1990; Hancock et al., 1991). In addition, it was recently demonstrated that K-ras4B is geranylgeranylated as well as farnesylated both in vitro and in vivo (James et al., 1995; Lerner et al., 1995b). Together, these posttranslational modifications promote the specific interaction of Ras molecules with the plasma membrane as well as with upstream and downstream components of signaling pathways (Takai et al., 1992; Marshall, 1993). The specific association of K-ras4B with the smg-GDS exchange factor is believed to be mediated, at least in part, through its distinctive C-terminal modifications (Mizuno et al., 1991). Thus, in certain contexts in the developing and adult animal, signal transduction may be dependent specifically on K-ras4B. The lethality of the K-ras-1 - embryos may, therefore, reflect the disruption of these pathways. This possibility could be addressed by creating a mutant allele of K-ras that specifically blocks production of the 4B isoform. K-ras and N-ras Interact Genetically The normal phenotype of animals mutant for either N- or H-ras as well as the relatively late death of K-ras mutant embryos could be explained by partially overlapping function within the ras family. Furthermore, I have found that genetic background can affect the expressivity of the K-ras-1- phenotype, suggesting the existence of modifier genes that can influence the requirement for K-ras function in development. Due to the similar expression profiles of K- and N-ras, I addressed the possibility of functional compensation between these two genes. Analysis of mice carrying various combinations of the K- and N-ras null alleles strongly suggests some form of overlapping function. For example, -70% of animals deficient for N-ras and heterozygous for K-ras (N-ras-l -; K-ras+/-) died between 10 and 12 days of gestation, with the remainder having failed later in gestation or just after birth. This result shows that the normal development of N-rasdeficient mice is dependent on wild-type levels of K-ras. Similarly, the K-ras homozygous mutant phenotype was significantly worsened in embryos with only one functional N-ras allele (N-ras+/l-;K-ras-l-). Again consistent with a specific developmental function for K-ras, N-ras+/-; K-ras-1 - embryos were more severely impaired than N-ras-1 -; K-ras+/ - embryos. Finally, 120 embryos lacking both K- and N-ras function (N-ras-l-; K-ras-l-) failed at an as yet undetermined stage between the blastocyst (E3.5) and early organogenesis (E9.5). Therefore, in early murine development, there appears to be a critical threshold level of Ras activity, which may be achieved by various combinations of the three gene products. Beginning prior to organogenesis, the overall threshold appears to rise, and still later, a specific requirement for K-ras is revealed. Functional Requirement for Ras in Hematopoiesis Various lines of evidence point to a critical role for Ras function in hematopoiesis. In addition to the apparent anemia of K-ras mutant embryos and the low contribution of K-ras-1- cells to the hematopoietic compartments of chimeric animals, I observed the absence of blood islands in the yolk sacs of the majority of N-ras-l-; K-ras+l- embryos. Prior to the establishment of fetal liver hematopoiesis at -E10.5-11.0, the earliest erythroid cells arise in yolk sac blood islands, where they produce primitive (nucleated) red blood cells (Dzierzak and Medvinsky, 1995; Orkin, 1995). The absence of blood islands as well as the limited number of primitive erythrocytes observed in N-ras-l-; K-ras+l- yolk sacs may reflect an impairment in the ability of primitive erythrocytes or their progenitors to survive and/or differentiate efficiently within the yolk sac microenvironment. Targeted disruption of the GATA-2, Tal-II/SCL, and Rbtn2/LM02 genes results in a similar hematopoietic defect. In contrast to results from analysis of Tal-1 and Rbtn2 mutant embryos (Warren et al., 1994; Robb et al., 1995; Shivdasani et al., 1995), in vitro hematopoietic colony assays on E9.5 and E10.5 N-ras-l-; K-ras+l- embryonic yolk sacs revealed that erythroid, myeloid, and mixed progenitors were present and capable of proliferating and differentiating normally. However, a marked reduction in the number of all three progenitor classes was observed in the yolk sacs of severely affected N-ras-1 -; K-ras+/- embryos (data not shown). In this respect, the N-ras-1 -; K-ras+l- phenotype more closely resembles that of the GATA-2 mutant (Tsai et al., 1994). Interestingly, Towatari et al. recently demonstrated that GATA2 was phosphorylated in response to the survival factor, IL-3, via activation of a MEK/MAPK cascade in hematopoietic progenitor cells (Towatari et al., 1995). This cascade is known to lie downstream of Ras in many 121 signal transduction pathways. Therefore, the defect in N-ras-l-; K-ras+l-embryos may be due, in part, to inefficient signaling to GATA2 in response to IL-3. Ras function has also been implicated downstream of a number of other hematopoietic cytokines as well, such as GM-CSF, EPO (erythropoietin), and SCF (stem-cell factor) (Satoh et al., 1992). These factors are critical for the maintenance and proliferation of hematopoietic cells, including multipotential progenitors (Arai et al., 1990). In the absence of stromal cells or cytokines, hematopoietic cells cease to proliferate and die by apoptosis (Williams et al., 1990). Recently, the Ras signaling pathway was shown to play a pivotal role in the anti-apoptotic functions of GM-CSF and IL-3. Whereas Ras function was not required for promoting mitotic entry in IL-3 dependent hematopoietic cell lines, it was essential for long-term proliferation as well as cell survival (Kinoshita et al., 1995). Thus, a reduction in the overall level of Ras activity or the specific elimination of K-ras may lead to apoptotic elimination of hematopoietic progenitors. A cell survival function for Ras is also supported by the observation of widespread cell death in the N-ras-/-; K-ras+l- embryos. Implications for Oncogenesis and Therapy An estimated 30% of all human cancers carry mutations in one member of the ras family, with K-ras mutations occurring most frequently (Bos, 1989; Khosravi and Der, 1994). Although the predominance of K-ras mutation in certain tumor types has some correlation with the expression profile of the three ras genes in various tissues (Leon et al., 1987), my results suggest that K-ras may have specific functions in signal transduction not shared by the other family members. Thus, mutational activation of K-ras may result in the stimulation of a suite of signal transduction pathways common to all Ras proteins plus those that are singularly dependent on K-ras. The further characterization of the developmental phenotype of the K-ras mutant mouse may then provide insights into the signal transduction requirements for oncogenic transformation. Moreover, the inhibition of these K-ras-specific pathways may have therapeutic value in cancer treatment. 122 Our results have additional implications important for the design of anti-cancer drugs based on the inhibition of oncogenic or normal Ras function, including the recently described farnesyltransferase inhibitors (FTIs). Our data demonstrate that inhibition of K-ras function, at least during embryogenesis, is lethal. Moreover, analysis of the contribution of K-ras-deficient cells to various tissues in chimeric animals suggests that the gene is also important in the development or maintenance of cells at later stages of gestation or in the adult animal, including in different hematopoietic compartments and in the lung. Therefore, it is likely that drugs that inhibit Ras function nonspecifically would be highly toxic. Interestingly, higher concentrations of FTIs are required to suppress the transforming activity of oncogenic K-ras than H-ras (Reiss et al., 1990; James et al., 1995; Lerner et al., 1995a; Lerner et al., 1995b; Manne et al., 1995; Sun et al., 1995). This may be explained by the fact that K-ras4B can be modified by geranylgeranylation as well as farnesylation (James et al., 1995; Lerner et al., 1995b), and this difference in modification may account for the non-toxic effect of FTIs observed in vivo (Kohl et al., 1995). This observation also supports an important role for the K-ras4B isoform in cellular function. Based on the lack of phenotype in the H- and N-ras mutant mice, it should be possible to develop nontoxic inhibitors specific for the oncogenic forms of these proteins. Alternatively, it may be more sensible to screen for compounds that inhibit steps downstream of Ras in the signal transduction pathways leading to cellular transformation rather than cell growth/differentiation, should such a distinction in signaling pathways exist. Finally, our data do not address directly the requirement for K-ras in the adult, and, therefore, it remains possible that inhibition of its function might be tolerated during cancer treatment. To examine what cell types in the adult are dependent on K-ras (and other ras family members), it will be necessary to develop conditional mutant alleles of these genes. 123 Experimental Procedures: Construction of Targeting Vectors A genomic DNA clone corresponding to K-ras was isolated from a 129/Sv genomic library using a probe derived from pHiHi3 (Ellis et al., 1981). The genomic DNA was excised with SalIl, and the resulting two genomic fragments were subcloned into pGEM4, forming pK-ras-5' and pK-ras-3'. pK-ras-5' was found to encompass exon 0 and its flanking sequence, whereas pK-ras-3' covered exon 1 and surrounding sequence. The targeting vector was constructed by first inserting a 2.8 kb NotI-SalI fragment from pK-ras-5' into pKSII + (Stratagene), creating pKSII+/K-ras-5'. Then a 5.1 kb HindIII-KpnI fragment from pK-ras-3' was cloned into pKSII + , forming pKSII+/K-ras-3'. Next, a four piece ligation was performed with the following fragments to create pK-ras-KO: a 2.8 kb NotI-XhoI fragment from pKSII+/K-ras-5', a 1.8 kb XhoI-BamHI fragment containing PGK-neo-pA sequences from pPNT (Tybulewicz et al., 1991), a 5.1 kb BamHI-KpnI fragment from pKSII+/pK-ras-3', and a 5.4 kb KpnI-NotI fragment containing PGK-HSV-tk-pA sequences from pPNT. Electroporation, Selection of ES Cell Clones, and Southern Blot Analysis D3 ES cells (Gossler et al., 1986) were cultured and electroporated as described by Tybulewicz et al. (Tybulewicz et al., 1991), except that primary mouse embryonic fibroblasts were used as feeder cells. G418 (GIBCO-BRL) was used at 200 gg/ml active weight. Individual clones were expanded and genomic DNAs were isolated using the method of Laird et al. (Laird et al., 1991). DNAs were digested with BamHI plus Stul and resolved on 0.8% agarose gels. Samples were transferred onto Hybond-N (Amersham) and hybridized with the 32 p-labeled probes indicated in Figure 1A using Expresshyb (Clontech). ES cell clones homozygous for the null mutation (K-ras-/ -) were created by enhanced G418 selection (Mortensen et al., 1992) of the heterozygous (K-ras+l-) clones. K-ras+l-ES cell clones were plated at a density of 5 x 104 to 5 x 105 cells/p100 and 40 hours after plating, the medium was supplemented with G418 at concentrations ranging 124 from 200 gtg/ml to 1.0 mg/ml active weight. After 9-10 days of selection, individual clones were expanded from those conditions under which clear selection had occurred and analyzed as above. Confirmation of the actual configuration of the mutant allele was performed by Southern blot analysis on multiple digest patterns using the probes described in Figure 2.1. PCR analysis was also performed using Klentaq and the primer pairs indicated in Figure 2. 1A. Primer sequences were as follows: 3' hom (5'-GGGATfGCAGCAATGATTTGGGGG-3'), 5'hom (5'-CCTGAA GATCTTACTCATCAAACTG-3'), 3'neo (5'-AAGCTGACTCTAGAGGATCCCC-3'), and 5'neo (5'-ACGAGACTAGTGAGACGTGC-3'). PCR conditions were as recommended by the manufacturer. Generation of Chimeras C57BL/6 blastocyst-stage embryos were injected with 10-15 K-ras+/- ES cells or 4-6 K-ras-1- ES cells and then transferred to pseudopregnant CD1 or Swiss Webster females for further development, essentially as described (Bradley, 1987). Chimeric mice were mated to C57BL/6 and 129/Sv animals and agouti offspring were genotyped. Germline transmission of the mutant allele was detected by either Southern blot (as above) or PCR analysis of tail DNA obtained at weaning. PCR Analysis of Offspring and Embryos Tail and yolk sac DNA was prepared using the method of Laird et al. (Laird et al., 1991). All PCR reactions were performed under the following conditions: 10 mM Tris-HC1, pH8.3, 50 mM KC1, 2 mM MgC12, 0.001% gelatin, 0.01% NP40, 0.01% Tween-20, 0.2 mM dNTP's, and 0.2-0.4 gM of each primer in the reaction. The annealing temperature was 60 0C for both K-ras and N-ras PCR reactions and 30 rounds of amplification were performed. Primer sequences for K-ras genotyping were as follows: 5'-10 (5'-AGGGTAGGTGTTGGGATAGC-3'), 3'-Exl (5'-CTCA GTCATTTIICAGCAGGC-3'), and 3'-neo (5'-ACGAGACTAGTGAGACGTGC-3'). The primer sequences for N-ras genotyping were: 5'WT (5'-CCCAGGATTCTTACCGAAAGC-3'), 3'WT 125 (5'-CCTGTAGAGGTTAATATCTGC-3'), and 3'MUT (5'-AATATGCGAAGTGGACCTGGG3'). Blastocysts were isolated from superovulated females as described and collected into 10 pl of TE, pH 8.0. They were subsequently heated at 95 0C for 5 minutes, treated with Proteinase K (200 gg/ml) at 550C for 90 minutes, and were then heated at 95 0C for 5 minutes. The sample was then split in half for K-ras and N-ras PCR analysis. PCR conditions were identical to those described above, except that 40 rounds of amplification were performed and the annealing temperature was 58 0C. Histological Analysis of Embryos and Yolk Sacs Embryos were dissected free of decidua and uterine muscle and separated from the yolk sac. Depending on the experimental procedure, either the yolk sac or embryo was saved for genotyping by PCR. For histology, embryos (or yolk sacs) were fixed in 10% neutral buffered formalin, dehydrated in graded solutions of alcohol, embedded in paraffin, sectioned at 4-6 gpm and stained with hematoxylin and eosin (H & E). Cell Death Analysis Serial sections from E12.5 or E13.5 embryos were prepared as described above and every fifth slide was stained with H & E. Unstained slides were deparaffinized in xylene and rehydrated through a graded alcohol series and finally H20. Sections were then incubated with Proteinase K (10 gg/ml) in 10 mM Tris-HC1, pH 8.0, 20 mM EDTA for 15 minutes at 25 0C, rinsed in H20, incubated for 30 minutes at 25 0C in 3% H202, 10% MeOH, and then rinsed as before. Sections were then pre-treated for 15 minutes at 25 0C in 1X TdT buffer (30 mM Tris, 140 mM sodium cacodylate, 1 mM CoC12), incubated at 37 0 C for 1 hour in 1X TdT buffer, 40 pM biotin-16-dUTP (Boehringer Mannheim), 20 units TdT (Gibco-BRL), and the reaction was stopped by washes in 2X SSC followed by PBS. The incorporated biotin-dUTP was detected using the Vectastain ABC kit with DAB as the peroxidase substrate (both from Vector Laboratories) according to the 126 manufacturer's instructions. Sections were counterstained with 0.025% methylgreen in 0.1 M NaOAc, pH 4.0, dehydrated, and mounted with coverslips. Hematopoietic Progenitor Cell Analysis E12.5 fetal livers were dissected free and collected in Iscove's modified Dulbecco's medium (IMDM), 2% FCS, disaggregated by passage through 23 and 26 gauge needles, counted, and plated in duplicate in a-minimal essential medium supplemented with 0.9% methylcellulose, 30% FBS, 1%BSA, 2 mM glutamine, 0.1 mM 2-mercaptoethanol, 3 U/ml EPO and 2% pokeweed mitogen-stimulated murine spleen cell conditioned medium (Stem-Cell Technologies, Inc.). Colony formation was monitored at the appropriate times (CFU-E on days 2-3; BFU-E and CFUGM on days 6-8; CFU-GEMM on days 8-12), and were subsequently picked, applied to slides, stained and examined microscopically. Yolk sac progenitor assays were performed on E9.5 and E10.5 yolk sacs as described (Wong et al., 1986; Shivdasani et al., 1995) using the same medium as used for fetal liver colony assays. In vivo irradiation rescues were performed essentially as described (Till, 1961). 129/Sv E13.5 fetal livers were isolated and disaggregated as above, counted, and injected retro-orbitally into C57BL/6 female recipient mice which had been lethally irradiated with 1200 rads of gamma irradiation. Blood samples were collected two weeks following injection and thereafter at one month intervals. Peripheral blood smears were examined and GPI analysis was, also, performed to determine the percentage and timing of contribution by donor cells. Preparation of Blood Cell Fractions Whole blood was collected in anticoagulant (3.8% NaCitrate) via heart puncture and diluted with 1 volume of phosphate buffered saline. Samples were then layered onto an equal volume of FicollHypaque (Histopaque-1077, Sigma) and prepared according to the manufacturer's suggestion. Following centrifugation, fractions corresponding to platelets, monocytes, and red blood cells were 127 collected and processed as recommended. Final cell pellets were stored at -800C until they were used for GPI or hemoglobin (RBC's) analysis. GPI Assay The separation and detection of GPI isoforms was performed as described (Bradley, 1987). The tissues analyzed included: tail, bladder, colon, cecum, small intestine, stomach, pancreas, spleen, kidney, adrenal, liver (2 different lobes), atrium, ventricle, thymus, salivary gland, lung, eye, medulla oblongata, cerebrum, cerebellum, whole blood, RBC, lymphocytes and monocytes, platelets, and bone marrow. Hemoglobin assays were also performed on the RBC fractions to confirm purity and support the GPI data. This assay was performed as described (Pevny et al., 1991; Williams et al., 1994). Preparation of Mouse Embryonic Fibroblasts Mouse embryonic fibroblasts (MEFs) were prepared essentially as described (Bradley, 1987) and were maintained in DMEM supplemented with 10% heat inactivated fetal calf serum (Hyclone). Preparation of Embryonic Tissue Extracts Embryos were collected at E12.5 in phosphate buffered saline (PBS) and dissected free of yolk sac and placenta. Five different tissues were dissected out, frozen immediately in a dry ice/EtOH bath, and stored at -80 0C until further analysis. The tissues recovered included brain, liver, heart, lung, and carcass. Yolk sacs were also retained for genotyping by PCR analysis. Preparation of Cell Lysates For MEFs, cells from 3-5 pl00s were washed 3 times with cold PBS, scraped from the plates, and collected into PBS on ice. Cells were pelleted and resuspended in 1.2 ml of lysis buffer (50 mM Tris-HC1, pH 8.0, 150 mM NaC1, 5 mM MgC12, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 0.5 mM PMSF, 10 gpg/ml leupeptin, 10 ptg/ml aprotinin, and 10 gg/ml 128 pepstatin) using a 26 gauge needle. For E12.5 tissues, the tissues were immediately placed into 100 pl of lysis buffer and disaggregated by passage through 23 and 26 gauge needles. Extracts of equivalent genotypes were then pooled as follows: 13 for lung, heart, and liver, and 5 for brain and carcass. After this step, tissues and MEFs were treated identically. The lysates were incubated for 30 minutes at 40C on a rotator, cellular debris was pelleted, and the supernatant collected. Protein concentration was determined using the Bio-Rad detergent-compatible protein assay system and equivalent amounts of protein for each of the three genotypes (+/+, +/-, and -/-) were used in immunoprecipitations. Immunoprecipitation and Western Analysis Immunoprecipitations were performed in a final volume of 1 ml with 1-5 jIg/ml of Y13-259 antibody (Santa Cruz Biotechnology, Inc.) for 2 hours to overnight at 40 C on a rotator wheel. Protein G PLUS-Agarose (Santa Cruz Biotechnology, Inc.) was added and incubated for 2 hours at 40C. Immunoprecipitates were pelleted and washed as follows: 2 times with 1 ml of lysis buffer, 2 times with 1 ml of high salt solution (1 M NaC1, 10 mM Tris-HC1, pH 7.5, and 0.5% Triton X-100), and 2 times with 1 ml of KSCN wash (0.75 M KSCN, 10 mM Tris-HC1, pH 7.5, and 1% Titron X-100). Complexes were resuspended in protein sample buffer, separated on 15% SDS/PAGE gels, and transferred onto PVDF membranes using a semi-dry transfer system (Owl). Western blot analysis was performed using an enhanced chemiluminescence system (Amersham) according to the manufacturer's recommendations, with the following exceptions: blocking was done in PBS, 0.2% Tween-20, 5% non-fat dry milk; washes were done in PBS, 0.2% Tween-20; primary antibodies were incubated in blocking solution at 1 gg/ml for 2 hours at 25 0C or overnight at 40 C; and the secondary anti-mouse IgG antibody conjugated to horseradish peroxidase was incubated at a 1:7000 dilution in blocking solution for 2 hours at 25 0 C. The primary mouse monoclonal antibodies used were K-ras (F234), N-ras (F155), H-ras (F235) (all from Santa Cruz Biotechnology, Inc.), and pan ras (Ab2 = clone F111) (Oncogene Science). 129 Stripping of immunoblots was done by incubating the blots in 50 mM glycine, pH 2.5, 0.05% Tween-20 for 30 minutes at 600 C. 130 References Arai, K. I., Lee, F., Miyajima, A., Miyatake, S., Arai, N., and Yokota, T. (1990). Cytokines: coordinators of immune and inflammatory responses. Annu Rev Biochem 783-836. Barbacid, M. (1987). ras genes. Annu Rev Biochem 56, 779-827. Boguski, M. S., and McCormick, F. (1993). Proteins regulating Ras and its relatives. Nature 366, 643-54. Bollag, G., and McCormick, F. (1991). Regulators and effectors of ras proteins. Annu Rev Cell Biol 7, 601-32. Bos, J. L. (1988a). The ras gene family and human carcinogenesis. Mutat Res 195, 255-71. Bos, J. L. (1988b). Ras oncogenes in hematopoietic malignancies. Hematol Pathol 2, 55-63. Bos, J. L. (1989). ras oncogenes in human cancer: a review [published erratum appears in Cancer Res 1990 Feb 15;50(4):1352]. Cancer Res 49, 4682-9. Bourne, H. R., Sanders, D. A., and McCormick, F. (1990). conserved switch for diverse cell functions. Nature 348, 125-32. The GTPase superfamily: a Bradley, A. (1987). In Teratocarcinomas and Embryonic Stem Cells: A Practical Approach, E. J. Robertson, eds. (Oxford: IRL, Press), pp. 113-152. Capon, D. J., Seeburg, P. H., McGrath, J. P., Hayflick, J. S., Edman, U., Levinson, A. D., and Goeddel, D. V. (1983). Activation of Ki-ras2 gene in human colon and lung carcinomas by two different point mutations. Nature 304, 507-13. Chesa, P. G., Rettig, W. J., Melamed, M. R., Old, L. J., and Niman, H. L. (1987). Expression of p2lras in normal and malignant human tissues: lack of association with proliferation and malignancy. Proc Natl Acad Sci U S A 84, 3234-8. Daum, G., Eisenmann, T. I., Fries, H. W., Troppmair, J., and Rapp, U. R. (1994). The ins and outs of Raf kinases. Trends Biochem. Sci. 19, 474-480. Davis, R. J. (1994). MAPKs: new JNK expands the group. Trends Biochem Sci 19, 470-473. Downward, J. (1992a). Regulation of p2lras by GTPase activating proteins and guanine nucleotide exchange proteins. Curr Opin Genet Dev 2, 13-8. Downward, J. (1992b). Regulatory mechanisms for ras proteins. Bioessays 14, 177-84. Duffy, J. B., and Perrimon, N. (1996). Recent advances in understanding signal transduction pathways in worms and flies. Curr Opin Cell Biol 8, 231-238. Dzierzak, E., and Medvinsky, A. (1995). Mouse embryonic hematopoiesis. Trends Genet 11, 359-366. 131 Ellis, R. W., Defeo, D., Shih, T. Y., Gonda, M. A., Young, H. A., Tsuchida, N., Lowy, D. R., and Scolnick, E. M. (1981). The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nature 292, 506-11. Furth, M. E., Aldrich, T. H., and Cordon, C. C. (1987). proteins in normal human tissues. Oncogene 1, 47-58. Expression of ras proto-oncogene George, D. L., Scott, A. F., Trusko, S., Glick, B., Ford, E., and Dorney, D. J. (1985). Structure and expression of amplified cKi-ras gene sequences in Y1 mouse adrenal tumor cells. Embo J 4, 1199-203. Gibbs, J. B. (1991). Ras C-terminal processing enzymes--new drug targets? Cell 65, 1-4. Gibbs, J. B., Oliff, A., and Kohl, N. E. (1994). Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell 77, 175-8. Glomset, J. A., and Farnsworth, C. C. (1994). Role of protein modification reactions in programming interactions between ras-related GTPases and cell membranes. Annu Rev Cell Biol 10, 181-205. Gossler, A., Doetschman, T., Korn, R., Serfling, E., and Kemler, R. (1986). Transgenesis by means of blastocyst-derived embryonic stem cell lines. Proc Natl Acad Sci U S A 83, 9065-9. Hancock, J. F. (1993). Anti-Ras drugs come of age. Curr Biol 3, 770-772. Hancock, J. F., Cadwallader, K., Paterson, H., and Marshall, C. J. (1991). A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. Embo J 10, 4033-9. Hancock, J. F., Magee, A. I., Childs, J. E., and Marshall, C. J. (1989). All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167-77. Hancock, J. F., Paterson, H., and Marshall, C. J. (1990). A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p2lras to the plasma membrane. Cell 63, 133-9. James, G. L., Goldstein, J. L., and Brown, M. S. (1995). Polylysine and CVIM sequences of KRasB dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J Biol Chem 270, 6221-6226. Kataoka, T., Powers, S., McGill, C., Fasano, O., Strathern, J., Broach, J., and Wigler, M. (1984). Genetic analysis of yeast RAS 1 and RAS2 genes. Cell 37, 437-45. Kawamura, M., Kaibuchi, K., Kishi, K., and Takai, Y. (1993). Translocation of Ki-ras p2 1 between membrane and cytoplasm by smg GDS. Biochem Biophys Res Commun 190, 832-41. Kayne, P. S., and Sternberg, P. W. (1995). Ras pathways in Caenorhabditis elegans. Curr Opin Genet Dev 5, 38-43. 132 Khosravi, F. R., Cox, A. D., Kato, K., and Der, C. J. (1992). Protein prenylation: key to ras function and cancer intervention? Cell Growth Differ 3, 461-9. Khosravi, F. R., and Der, C. J. (1994). The Ras signal transduction pathway. Cancer Metastasis Rev 13, 67-89. Kinoshita, T., Yokota, T., Arai, K., and Miyajima, A. (1995). Suppression of apoptotic death in hematopoietic cells by signalling through the IL-3/GM-CSF receptors. Embo J 14, 266-275. Kohl, N. E., Omer, C. A., Conner, M. W., Anthony, N. J., Davide, J. P., deSolms, S. J., Giuliani, E. A., Gomez, R. P., Graham, S. L., Hamilton, K., and et, a. 1. (1995). Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med 1, 792-797. Laird, P. W., Zijderveld, A., Linders, K., Rudnicki, M. A., Jaenisch, R., and Berns, A. (1991). Simplified mammalian DNA isolation procedure. Nucleic Acids Res 19, 4293. Lemoine, N. R., Mayall, E. S., Wyllie, F. S., Williams, E. D., Goyns, M., Stringer, B., and Wynford, T. D. (1989). High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis. Oncogene 4, 159-64. Leon, J., Guerrero, I., and Pellicer, A. (1987). Differential expression of the ras gene family in mice. Mol Cell Biol 7, 1535-40. Lerner, E. C., Qian, Y., Blaskovich, M. A., Fossum, R. D., Vogt, A., Sun, J., Cox, A. D., Der, C. J., Hamilton, A. D., and Sebti, S. M. (1995a). Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem 270, 26802-26806. Lerner, E. C., Qian, Y., Hamilton, A. D., and Sebti, S. M. (1995b). Disruption of oncogenic KRas4B processing and signaling by a potent geranylgeranyltransferase I inhibitor. J Biol Chem 270, 26770-26773. Manne, V., Yan, N., Carboni, J. M., Tuomari, A. V., Ricca, C. S., Brown, J. G., Andahazy, M. L., Schmidt, R. J., Patel, D., Zahler, R., and et, a. 1. (1995). Bisubstrate inhibitors of farnesyltransferase: a novel class of specific inhibitors of ras transformed cells. Oncogene 10, 1763-1779. Mansour, S. L., Thomas, K. R., and Capecchi, M. R. (1988). Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to nonselectable genes. Nature 336, 348-52. Marshall, C. J. (1993). Protein prenylation: a mediator of protein-protein interactions. Science 259, 1865-6. McCormick, F. (1994). Activators and effectors of ras p21 proteins. Curr Opin Genet Dev 4, 71-6. 133 McGrath, J. P., Capon, D. J., Smith, D. H., Chen, E. Y., Seeburg, P. H., Goeddel, D. V., and Levinson, A. D. (1983). Structure and organization of the human Ki-ras proto-oncogene and a related processed pseudogene. Nature 304, 501-6. Mizuno, T., Kaibuchi, K., Yamamoto, T., Kawamura, M., Sakoda, T., Fujioka, H., Matsuura, Y., and Takai, Y. (1991). A stimulatory GDP/GTP exchange protein for smg p21 is active on the post-translationally processed form of c-Ki-ras p21 and rhoA p21. Proc Natl Acad Sci U S A 88, 6442-6. Mortensen, R. M., Conner, D. A., Chao, S., Geisterfer, L. A., and Seidman, J. G. (1992). Production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol 12, 2391-5. Muller, R., Slamon, D. J., Adamson, E. D., Tremblay, J. M., Muller, D., Cline, M. J., and Verma, I. M. (1983). Transcription of c-onc genes c-rasKi and c-fms during mouse development. Mol Cell Biol 3, 1062-9. Muller, R., Slamon, D. J., Tremblay, J. M., Cline, M. J., and Verma, I. M. (1982). Differential expression of cellular oncogenes during pre- and postnatal development of the mouse. Nature 299, 640-4. Nakanishi, H., Kaibuchi, K., Orita, S., Ueno, N., and Takai, Y. (1994). Different functions of Smg GDP dissociation stimulator and mammalian counterpart of yeast Cdc25. J. Biol. Chem. 269, 15085-91. Orita, S., Kaibuchi, K., Kuroda, S., Shimizu, K., Nakanishi, H., and Takai, Y. (1993). Comparison of kinetic properties between two mammalian ras p21 GDP/GTP exchange proteins, ras guanine nucleotide-releasing factor and smg GDP dissociation stimulation. J Biol Chem 268, 25542-6. Orkin, S. H. (1995). Hematopoiesis: how does it happen? Current Opinion in Cell Biology 7, 870-7. Pevny, L., Simon, M. C., Robertson, E., Klein, W. H., Tsai, S. F., D'Agati, V., Orkin, S. H., and Costantini, F. (1991). Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA- 1. Nature 349, 257-60. Powers, S., Kataoka, T., Fasano, O., Goldfarb, M., Strathern, J., Broach, J., and Wigler, M. (1984). Genes in S. cerevisiae encoding proteins with domains homologous to the mammalian ras proteins. Cell 36, 607-12. Reiss, Y., Goldstein, J. L., Seabra, M. C., Casey, P. J., and Brown, M. S. (1990). Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell 62, 81-8. Robb, L., Lyons, I., Li, R., Hartley, L., Kontgen, F., Harvey, R. P., Metcalf, D., and Begley, C. G. (1995). Absence of yolk sac hematopoiesis from mice with a targeted disruption of the scl gene. Proc Natl Acad Sci U S A 92, 7075-7079. 134 Satoh, T., Nakafuku, M., and Kaziro, Y. (1992). Function of Ras as a molecular switch in signal transduction. J Biol Chem 267, 24149-52. Shimizu, K., Birnbaum, D., Ruley, M. A., Fasano, O., Suard, Y., Edlund, L., Taparowsky, E., Goldfarb, M., and Wigler, M. (1983). Structure of the Ki-ras gene of the human lung carcinoma cell line Calu- 1. Nature 304, 497-500. Shivdasani, R. A., Mayer, E. L., and Orkin, S. H. (1995). Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature 373, 432-434. Sun, J., Qian, Y., Hamilton, A. D., and Sebti, S. M. (1995). Ras CAAX peptidomimetic FTI 276 selectively blocks tumor growth in nude mice of a human lung carcinoma with K-Ras mutation and p53 deletion. Cancer Res 55, 4243-4247. Takai, Y., Kaibuchi, K., Kikuchi, A., and Kawata, M. (1992). Small GTP-binding proteins. Int Rev Cytol 187-230. Till, J. E. a. M., E.A. (1961). Radiation Research. 14, 213. Towatari, M., May, G. E., Marais, R., Perkins, G. R., Marshall, C. J., Cowley, S., and Enver, T. (1995). Regulation of GATA-2 phosphorylation by mitogen-activated protein kinase and interleukin-3. J Biol Chem 270, 4101-4107. Tsai, F. Y., Keller, G., Kuo, F. C., Weiss, M., Chen, J., Rosenblatt, M., Alt, F. W., and Orkin, S. H. (1994). An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 371, 221-226. Tybulewicz, V. L., Crawford, C. E., Jackson, P. K., Bronson, R. T., and Mulligan, R. C. (1991). Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl protooncogene. Cell 65, 1153-63. Ulsh, L. S., and Shih, T. Y. (1984). Metabolic turnover of human c-rasH p21 protein of EJ bladder carcinoma and its normal cellular and viral homologs. Mol Cell Biol 4, 1647-52. Umanoff, H., Edelmann, W., Pellicer, A., and Kucherlapati, R. (1995). The murine N-ras gene is not essential for growth and development. Proc Natl Acad Sci U S A 92, 1709-1713. Warren, A. J., Colledge, W. H., Carlton, M. B., Evans, M. J., Smith, A. J., and Rabbitts, T. H. (1994). The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell 78, 45-57. Wassarman, D. A., Therrien, M., and Rubin, G. M. (1995). Drosophila. Curr Opin Genet Dev 5, 44-50. The Ras signaling pathway in Williams, B. O., Schmitt, E. M., Remington, L., Bronson, R. T., Albert, D. M., Weinberg, R. A., and Jacks, T. (1994). Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. Embo J 13, 4251-4259. 135 Williams, G. T., Smith, C. A., Spooncer, E., Dexter, T. M., and Taylor, D. R. (1990). Haemopoietic colony stimulating factors promote cell survival by suppressing apoptosis. Nature 343, 76-9. Willumsen, B. M., Papageorge, A. G., Kung, H. F., Bekesi, E., Robins, T., Johnsen, M., Vass, W. C., and Lowy, D. R. (1986). Mutational analysis of a ras catalytic domain. Mol Cell Biol 6, 2646-54. Wittenberg, C., and Reed, S. I. (1996). Plugging it in: signaling circuits and the yeast cell cycle. Curr Opin Cell Biol 8, 223-230. Wong, P. M., Chung, S. W., Chui, D. H., and Eaves, C. J. (1986). Properties of the earliest clonogenic hemopoietic precursors to appear in the developing murine yolk sac. Proc Natl Acad Sci USA 83, 3851-4. Yamauchi, N., Kiessling, A. A., and Cooper, G. M. (1994). The Ras/Raf signaling pathway is required for progression of mouse embryos through the two-cell stage. Mol Cell Biol 14, 66556662. 136 Chapter 3 Analysis of an Oncogenic Allele of K-ras in the Initiation and Progression of Tumorigenesis in the Mouse via a Novel Approach 137 Abstract Ras genes are critical in the regulation of normal and neoplastic cell growth. Mutational activation of Ras leads to altered growth control and occurs in -30% of all human cancers. K-ras is the most frequently mutated member in this gene family and is commonly associated with adenocarcinomas of the pancreas, colon, and lung. To elucidate further the role of K-ras in the initiation and progression of tumorigenesis, I have created a mouse strain carrying a spontaneously activatable allele of K-ras through a novel variation of the hit-and-run gene targeting strategy. ES cells carrying a disruption of one allele of the K-ras gene were generated via gene targeting by a single reciprocal recombination event. The resulting insertion allele was composed, in part, of an additional copy of exon 1 in which an activating mutation (Asp 12 ) had been introduced. Spontaneous resolution of this duplication via intrachromosomal recombination will produce an oncogenic allele of K-ras that differs from wild-type by only the introduced point mutations. This "run" step was performed in tissue culture, but in addition, an innovative approach was taken in which the resolution was allowed to occur in vivo. ES cell clones carrying the targeted insertion were used to generate germline chimeras and established mouse lines. Because every cell in the developing and adult mouse carried the insertion mutant allele, this recombination event was expected to occur broadly and predispose the mice to a wide range of tumor types. Indeed, mice carrying the mutant allele are highly susceptible to lung tumors and lymphoma, with an onset as early as 3 months of age. These animals should prove invaluable for testing various environmental and genetic factors that may cooperate with an oncogenic K-ras allele to promote tumorigenesis. In addition, they should provide an excellent model system for screening potential chemotherapeutic drugs designed to inhibit the transforming activity of oncogenic K-ras. 138 Introduction Over the past several years, a considerable amount of epidemiological and experimental evidence has implicated a pivotal function for oncogenic Ras mutants in the development of human cancers. First, oncogenic ras mutations have been detected in -30% of all human tumors (Bos, 1989; Khosravi and Der, 1994). Those tumor types most frequently associated with mutated ras alleles include hematological disorders of the myelomonocytic lineage as well as carcinomas. In contrast, oncogenic ras mutations are rare in tumors of neuroectodermal origin and more differentiated lymphoid malignancies, suggesting that ras does not play an important role in the development of these tumor types. Second, the biological differences between normal Ras proteins and their oncogenic counterparts have been extensively documented in in vitro cell culture systems. Depending upon the cell type, expression of oncogenic, but not normal, Ras proteins can either induce cellular proliferation and malignant transformation (e.g., rodent cell lines such as NIH/3T3 or Rat-i fibroblasts) or differentiation and growth inhibition (e.g., the PC12 pheochromocytoma cell line) (Barbacid, 1987). Third, mice engineered to harbor oncogenic alleles of ras, either through transgenic or retroviral infection approaches, display increased incidences of tumor formation (Adams and Cory, 1991). Finally, specific activating ras mutations are reproducibly and frequently associated with carcinogen- and radiation-induced tumors in rodent model systems (Guerrero and Pellicer, 1987; Mangues and Pellicer, 1992). However, ras mutation is not the only defect associated with the development of a particular tumor. Indeed, most cancers are a multistep process, resulting from the accumulation of independent genetic events (Weinberg, 1989; Vogelstein and Kinzler, 1993). Moreover, the progressive development of aneuploidy during tumorigenesis suggests that many of the aberrations observed in tumor cells may be a secondary consequence of the genomic instability that may occur subsequent to an already established malignancy. Therefore, the precise role that ras genes play in tumorigenesis has been difficult to evaluate. The evidence discussed above suggests that ras mutations play a causative role in neoplastic development rather than occurring as 139 a consequence of the process. Indeed, ras mutation has been implicated as an early event in the development of multiple tumor types (Barbacid, 1987; Bos, 1988; Vogelstein et al., 1988). Two of the best characterized systems for this analysis are the mouse skin carcinogenesis and human colorectal cancer models (Fearon and Vogelstein, 1990; Hennings et al., 1993; Yuspa, 1994). Because these tumors evolve through well-defined morphological stages, it has been possible to establish the order in which genetic alterations occur. In each system, ras mutations have been associated with both benign and malignant stages of the neoplasm at equally high frequencies (90% of skin papillomas and carcinomas; 50% of colonic adenomas and carcinomas), suggesting that ras activation occurs early in tumorigenesis. In skin carcinogenesis models, activation of the H-ras gene is thought to be an initiating event following treatment with a mutagen (e.g., DMBA) (Balmain et al., 1984; Quintanilla et al., 1986). These "initiated" cells lie dormant and, importantly, do not exhibit neoplastic properties unless they are induced to proliferate, either through treatment with a tumor promoter (e.g., TPA) or by a natural growth-promoting stimulus such as wounding (Brown et al., 1986). During promotion, the initiated cells are thought to possess a growth advantage, with their proliferation resulting in the formation of a benign papilloma. Over time, papillomas acquire additional genetic alterations and progress into malignant carcinomas of varying morphological stages (Bremner and Balmain, 1990). In colorectal cancer, alterations in both APC and K-ras have been implicated in its early stages. However, the initiating event appears to be mutation of the APC gene (Powell et al., 1992). Mutations in APC have been found in the majority of all colon cancers, including both sporadic and familial cases. Indeed, mutations in APC have been shown to be responsible for the familial adenomatous polyposis (FAP) syndrome in humans, with loss of heterozygosity at the APC locus occuring frequently in the development of adenomas (Groden et al., 1991; Nishisho et al., 1991; Powell et al., 1993; Levy et al., 1994). Further analysis of multiple benign lesions (including both aberrant crypt foci and polyps) has revealed that mutation of the APC locus is closely associated with dysplasia, defined by a disruption in normal morphology and considered to be a hallmark of malignant potential (Jen et al., 1994). In contrast, activation of K-ras is more frequently associated 140 with non-dysplastic lesions, in particular, aberrant crypt foci, which have an apparently limited potential to progress to larger tumors (Jen et al., 1994; Smith et al., 1994; Yamashita et al., 1995). Interestingly, only those dysplastic lesions that carried both APC and K-ras mutations were classifed as adenomas in these studies. Thus, these models provide evidence that ras oncogenes have an important role in both the initiation and progression of tumorigenesis. Importantly, however, activation of Ras is insufficient for full transformation and requires the presence of cooperating genetic events. In addition, the order in which these genetic events occur appears to have a significant impact on both tumor morphology and progression. We are interested in trying to create a better animal model with which to study the role of oncogenic Ras, K-ras in particular, in the initiation and progression of tumorigenesis. K-ras is the most frequently mutated member of the gene family, particularly in adenocarcinomas of the pancreas (-80%), colon (-50%), and lung (-25-50%) (Bos et al., 1987; Forrester et al., 1987; Vogelstein et al., 1988; Bos, 1989; Pellegata et al., 1994; Mills et al., 1995). These are three very common and clinically important tumor types, and an accurate animal model of the diseases would prove invaluable in the development of drugs for their treatment. In addition, I have recently demonstrated that K-ras has a unique role during normal mouse development (Chapter 2); thus, this may be true in tumor development as well. A number of different animal models have been developed to analyze the function of oncogenic Ras in the mouse. All of these models have been made through the use of transgenics and have a number of disadvantages associated with them (Adams and Cory, 1991). First, many of the existing transgenic strains have specifically looked at oncogenic H-ras. The best known of these, the OncomouseTM, carries an activated H-ras gene driven by the mouse mammary tumor virus (MMTV) promoter (Sinn et al., 1987). For the reasons listed above, it is important to look at activated K-ras in much more detail. This is underscored by the fact that the transforming ability of tumor cells harboring an activated allele of K-ras are harder to inhibit with existing chemotherapeutic agents than are tumor cells carrying oncogenic H-ras (James et al., 1995; Lerner et al., 1995a; Lerner et al., 1995b; Manne et al., 1995; Sun et al., 1995). A second problem 141 associated with the use of transgenes to deliver oncogenic Ras to the cell is their use of an artificial promoter to direct their expression to a specific tissue. Due to both the promoter strength and copy number of integrants, this results in an expression level of Ras that is much higher than the observed endogenous levels in both normal and tumor cells. This is important since the level of oncogenic Ras appears to modulate its transforming potency and is frequently auqmented late in tumor progression (Barbacid, 1987; Bos, 1988; Finney and Bishop, 1993). In addition, it is possible that drugs that would be effective against oncogenic Ras expressed under its normal physiological constraints may fail to work if the protein were expressed at artificially high levels. Thirdly, the mutant Ras protein is expressed in every cell of the affected tissue in the transgenic strain. This is in contrast to what normally occurs during tumorigenesis, in which a clone of cells harboring a mutation in ras would be surrounded by neighboring cells with normal Ras function. It is well known that the transformed properties of tumor cells can be affected through their interactions with neighboring cells. Indeed, normal cells can inhibit the transformed phenotype of ras-transfectedcells, but only when plated at a high cell density (Spandidos, 1986). Finally, the transgenic Ras strains have all targeted ras expression to a specific tissue, and thus the observed pathological consequences and tumor spectrums are limited with respect to each strain. In addition, the choice of promoter driving the transgene does not assure its expression in the appropriate target cell of the tissue, especially since the target cell of many tumors is not well defined. Since ras mutations are associated with a wide range of tumor types, an ideal animal model would show a broad tumor spectrum, each initiating from the correct target cell. I have attempted to address these and related issues by creating a mouse strain that overcomes all of the problems described above. Using a novel variation of the hit-and-run gene targeting strategy (Hasty et al., 1991), I have created a strain of mice carrying a spontaneously activatable allele of oncogenic K-ras in all cells of the animal. An intrachromosomal recombination event is necessary to activate this allele and is expected to occur at some stochastic frequency in all cells of the developing and adult mouse. Importantly, because the mutation is targeted to the K-ras locus, the oncogenic allele is expressed under its own promoter and positional 142 constraints. Mice carrying this latent allele are expected to be predisposed to a wide range of tumor types, and indeed, I show that these mice are highly prone to, at least, lung tumors and thymic lymphomas. 143 Results Generation of ES Cells Carrying a Spontaneously Activatable Allele of K-ras Figure 3.1B shows a schematic representation of the hit-and-run targeting procedure used to generate mouse embryonic stem (ES) cells carrying a spontaneously activatable allele of K-ras. This procedure consists of two steps of homologous recombination events that are normally facilitated by the presence of positive (hit step) and negative (run step) selection in vitro (Hasty et al., 1991; Hasty and Bradley, 1993). We have attempted a novel variation of this strategy in which the "run" step was allowed to occur spontaneously in the mouse. The insertion allele generated following the "hit" step is expected to be inactive and in order to restore a functional and oncogenic allele of K-ras, the intrachromosomal recombination event (run step) must occur. Due to the stochastic nature of this event occurring in the cells of the animal, I refer to this allele as the K-ras latent allele. In addition, I have also applied the standard negative selection in vitro to select for ES cells that have undergone the intrachromosomal recombination to generate a functional oncogenic allele of K-ras, and I, therefore, refer to this as the K-ras activated allele. Hereafter, the K-ras latent and activated alleles are referred to as K-rasD 12 -L and K-ras D12-A, respectively. In order to generate ES cells carrying an insertion allele that was applicable to study the "run" step both in vivo and in vitro, two different targeting constructs carrying an activating mutation in K-ras were designed. Figure 3.1A shows the mutations that were incorporated into each K-ras targeting vector. Through site-directed mutagenesis, I converted the glycine residue at codon 12 to an aspartic acid residue. This is a known oncogenic mutation in Ras and is commonly found in multiple tumor types in both humans and rodents (Guerrero and Pellicer, 1987; Bos, 1989; Mangues and Pellicer, 1992). Because this mutation was not identifiable with a restriction enzyme, a unique HindIII site was also introduced 20 nucleotides upstream from the activating mutation to facilitate screening for targeted events. However, unlike the Asp 12 mutation, this alteration did not result in an amino acid change at position 5. 144 Figure 3.1 A. codon: 1 2 3 4 5 6 7 8 9 10 11 12 13 wild type: ATG ACT GAG TAT AAA CTT GTG GTG GTT GGA GCT GGT GGC activated: ATG ACT GAG TAT AAG CTT GTG GTG GTT GGA GCT GAT GGC I HindIl I G--D i Figure 3.1. Strategy for creating activatable alleles of K-ras. A. Genomic sequence comparison between the wild-type and mutant allele of K-ras from codons 1 to 13 of exon 1. Using site-directed mutagenesis, two alterations were made. Without changing the amino acid at position 5, a unique HindIII site was created to facilitate the screening of ES cell clones for targeted events. The second mutation converted Gly 1 2 to Asp and is oncogenic. B. Diagram represents the hit-and-run targeting approach that was used to introduce the activating mutation into K-ras. The initial targeting event occurs via a single reciprocal recombination between the K-ras gene and the insertion vector. Both insertion vectors were comprised of K-ras exon 1 and flanking sequences (denoted by M and N boxes) carrying the alterations described above (a ^ represents the HindIII mutation and an * represents the Asp 12 mutation). For the K-rasD12 -A allele, both the neo and tk selectable markers (as is shown) were included in the insertion vector outside of the region of homology. In contrast, the K-rasD1 2 -L allele targeting vector only contained the neo selectable marker. To direct integration, both vectors were linearized at a unique XhoI site contained within the region of homology. A correctly targeted cell will become G418-resistant (G418 r ) and, in the case of K-rasD12 -A, gancyclovir-sensitive (GANC s ) and will carry a duplication of the homologous sequences. Due to the insertion sequences, it is expected that this mutant allele will be non-functional. For the run step, either an intrachromatid recombination (depicted) or an unequal sister chromatid exchange within the duplicated sequences will result in the restoration of a functional K-ras allele. Depending upon which side of the mutation that the recombination occurs, either the desired mutation will be incorporated, as is shown, or a wild-type K-ras allele will be restored. For the K-rasD1 2 -A allele, these "pop-out" events were selected for by placing the cells into medium supplemented with gancyclovir. Cells which have correctly excised the selectable markers are G418 s , GANCr. In contrast, ES cells correctly targeted for the K-rasD12 -L allele were directly used to generate mice where the run step is expected to occur at some stochastic frequency in all cells of the animal. Figure 3.1 B. X XhoI Ex I G418rGANCS neo NTR I tk [ ,.,• m- Intrachromosomal Recombination Latent Allele = in vivo Activated Allele = in vitro G418sGANCr '''"~"'' toe I,In """":" Irr Al· ~ ~U-l LlrXL I~a ~I - The two targeting constructs are virtually identical except for the presence of the negative selectable marker, the tk gene, in the K-rasD12 -A allele vector. This marker was included to facilitate the identification of clones that had undergone the intrachromosomal recombination (run step) in vitro. However, since this process will occur in the mouse with the K-rasD12 -L allele, this marker was not necessary. In addition, this version of the tk gene causes sterility when expressed in the germ line of male mice, and therefore, was excluded from the K-rasD12 -L allele targeting vector. The genomic structure for each allele following a correctly targeted insertional recombination is diagrammed in Figure 3.2. As stated above, it is expected that these alleles of K-ras will be inactive due to the presence of the insertion sequences. Following introduction of the two different targeting vectors into 129/Sv D3 ES cells, the resulting G418 r clones were screened for targeted events by Southern blot analysis using a probe located 5' to the sequences present in the targeting vectors. This probe detects a 12.1 kilobase (kb) BamHI to KpnI fragment from wild-type DNA and an additional 9.1 kb BamHI mutant-specific fragment (Figure 3.2 and data not shown). As shown in Table 3.1, -20% of K-rasD12 -L and -40% of K-rasD12-A ES cell clones screened had acquired the desired sequences by homologous recombination as detected by the 5' probe and were, therefore, heterozygous for the mutant allele. To verify the presence of the HindIII mutation in the targeted clones, Southern blotting was performed using a 5' probe contained within the duplicated sequences. This analysis demonstrated that the HindIII mutation had integrated into none, one, or both of the duplicates for each allele (Figure 3.3). This variation in the integration pattern is expected and is associated with gap formation and repair, mismatch heteroduplex repair, and the branch migration of Holliday junctions during the initial insertional recombination event (Hasty and Bradley, 1993). As summarized in Table 3.1, all possible integration patterns were recovered with the K-rasD12 -L allele, whereas only three of the four possibilities were obtained with the K-rasD12 -A allele. Because the vectors were linearized 3' to the mutation, the highest expected pattern of integration would be one in which the mutation is present only in the 3' duplicate (i.e., -/+). Indeed, this was observed, with -50% of targeted clones for each allele exhibiting a -/+ (5'/3') integration pattern 147 Figure 3.2. Genomic configurations of the K-rasD12-L and K-rasD12 -A alleles following insertional recombination. Following integration into the K-ras locus, both alleles carry a duplication of exon 1 and the flanking sequences (represented by the shaded boxes) contained within the targeting vector. The extent of homology on either side of the mutations is 3.2 kb. The K-rasD12-L allele also carries a Pgk-1 promoter driven neo gene, which terminates with the Pgk-1 derived polyadenylation signal. In contrast, both the neo and tk selectable markers are present in the K-rasD12-A allele. A single Pgk-1 promoter was used to drive the expression of both genes. In between the two marker genes, the vector includes a 5' non-translated (NTR) fragment containing an internal ribosomal entry site, which allows efficient translation of the tk gene from the bicistronic message. In addition, two polyadenylation signals are included at the 3' end of the tk gene. In this configuration, both alleles should be non-functional. Based on the site of linearization within the homologous sequences, the expected pattern of integration would position the mutations in the 3' duplicate, as is shown. The positions of the probes used in this study are shown. Also represented are the expected fragments in targeted clones heterozygous for the insertion following digestion with BamHI (B) + KpnI (K) and detection with the 5' external probe. In addition, the expected fragments detected using the 5' internal probe following a HindIII (H)+ KpnI (K) are also depicted for all integration patterns and are the same for both mutant alleles. ^ indicates the targeted HindIII mutation and * represents the Asp 12 mutation. 148 eq L I$I x x 4I EP ~Cj ?r: fP EQ E9 ~1 ?C SC c~l W iiiii ::::: :·:·: r( iI x w e: ijijj :':': W x ·Q cy hi cr I I I 0d Table 3.1. Summary of Targeting Frequencies and Integration Patterns. Allele # G418 r clones screened # targeted clones Latent 229 45 12 2 24 2 (19.7%) (26.7%) (4.4%) (53.3%) (4.4%) # of targeted clones exhibiting each of the predicted integration patterns for HindIII -,+ +,+ -,+,- *5/45 (11.1%) contained >1 copy Activated 176 68 (38.6%) 25 (36.8%) 0 (0.0%) 30 (44.1%) 1 (1.5%) *10/68 (14.7%) contained >1 copy *2/68 (2.9%) were aberrant integrants The number of correctly targeted G418 r clones was determined by Southern blot analysis using a probe 5' to the region of homology. These clones were subsequently screened by Southern blotting using a 5' internal probe to determine the presence of the HindIII mutation in each duplicate. The integration patterns for each duplicate are depicted as 5', 3' with a + indicating the presence of the HindIII site. The highest expected integration pattern (-,+) was the most frequently represented event of the four. Some clones were found to have integrated more than one copy of the vector and their frequencies are also noted. 150 Activated Allele Latent Allele I 5' copy: 3' copy: wt+5'wt 5'mut 3'wt3'mut - + .. + + + + + I + + wowllrgeCg +)~ +·- t - + + Figure 3.3. Observed integration patterns in targeted K-ras clones. A representative subset of targeted clones from both alleles was digested with HindIII + KpnI and analyzed by Southern blotting using the 5' internal probe shown in Figure 3.2. The integration pattern for each clone is labelled above its respective lane, with a + sign indicating the presence of the HindIII mutation in the listed duplicate. The fragments for the wild-type allele as well as for each duplicate are identified to the left, with wt referring to a wild-type digestion pattern (i.e., no HindIII site) and mut. indicating that the HindIII site is present. The sizes of each of the fragments are as follows: wt and 5' wt copy are -10 kb, 5' mut copy is -8.9 kb, 3' wt copy is 7.2 kb, and the 3' mut copy is 6.1 kb. 151 for the HindIII mutation. Clones that had not incorporated the mutation into either duplicate (due to repair) were also frequently represented, whereas resolution of branch migration events in the 5' duplicate was more rare. This is not surprising, since the mutation was positioned 1.5 kb from the site of linearization, a distance that is sufficiently far enough away from the double strand break such that branch migration across the mutation will occur less frequently (Hasty and Bradley, 1993). In addition, all targeted clones were verified for legitimate recombination at their 3' ends as well as for the number of copies that had integrated into the locus (data not shown). All clones that were found to have integrated aberrantly or in multiple copies were not used in the subsequent run step. Genomic sequence analysis was performed to determine whether the Asp 12 mutation had co-integrated with the HindIII mutation. As shown in Figure 3.4, this analysis confirmed the presence of the HindIII A-to-G mutation at nucleotide 15 in those clones that had previously revealed its existence by Southern blotting. In addition, only those clones that had integrated a HindIII mutation into at least one of the duplicates also showed the presence of the G-to-A transition at nucleotide 35 (the nucleotide responsible for the Asp12 mutation). Moreover, based on the intensity of the nucleotide signals, the Asp 12 and HindIII mutations were always present in equal copy numbers (Figure 3.4 and data not shown). This suggests that the two mutations always co-integrated into the same duplicate(s). However, I can not distinguish between clones that had integrated one copy of each mutation into either the same or distinct duplicates. Creation of ES Cells Heterozygous for an Oncogenic Allele of K-ras Through Intrachromosomal Recombination of the K-rasD12 -A Allele in vitro. To create a functionally oncogenic allele of K-ras, an intrachromosomal recombination event must occur between the duplicated sequences that simultaneously excises the vector DNA containing the selectable markers. This can occur through either an intrachromatid recombination (depicted in Figure 3.1B) or an unequal sister-chromatid exchange. The ideal clone for negative selection is one that has the mutation present in both duplicates. Thus, irrespective of the position of the 152 I- Activated I Latent I g :12 ;->D :5', 3' HindIII wt DNA 113 G ATC GA TCGATC GATCGATCGATCGA TC Figure 3.4. The aspartic acid and HindIII mutations appear to co-integrate. Genomic DNAs from the clones shown in Figure 3.3 were sequenced to verify the presence of the activating mutation at codon 12. A subset of those clones is shown here. The integration pattern for the HindIII mutation is listed above each clone. Above that, the presence (+) or absence (-) of the oncogenic mutation is indicated as determined by sequence analysis. For those clones which had integrated the HindIII site in both duplicates, sequence analysis and band intensity appeared to indicate that the aspartic acid mutation had also integrated into both copies and is indicated by a ++ symbol. The sequence of wild-type (wt) ES cell DNA is shown for comparison. The position of each nucleotide alteration is indicated at the sides. 153 crossover, the mutation would be left in the chromosome following recombination. For the K-rasD12 -A allele, one clone (clone 194) was recovered with this structure. All other K-rasD12 -A clones that showed the presence of the Asp 12 mutation appeared to carry it in only one of the duplicates (presumably the 3' copy). Therefore, depending upon which side of the mutation that the cross-over occurs, either the mutation will remain in the chromosome or the wild-type allele will be restored. Since the vector contained 3.2 kb of homology on either side of the mutation (Figure 3.2), it was expected that both events would occur with an equal frequency. Importantly, irrespective of the initial integration pattern, the only alteration that would be remain in the genome following the recombination would be the desired HindIII and Asp 12 point mutations. To select for K-rasD12-A cells that had excised the DNA including the negative selectable marker, the six independent clones analyzed in Figure 3.3 were subjected to tk counter-selection. GANCr clones were isolated and examined for the presence of the "pop-out" event. As shown in Table 3.2, only 15% (14/94) of phenotypically GANC r clones had excised the neo and tk genes in the first round of screening. Unfortunately, the main limitation to this procedure is the spontaneously high frequency of the loss of sensitivity to the negative selection agent due to events other than homologous recombination. To reduce this background, DNAs were prepared and analyzed from only those clones that were both GANC r and G418s in the subsequent round of screening. Using this prescreening procedure, 100% (49/49) of such clones had correctly excised the selectable markers (Table 3.2). Of the 63 "pop-out" events obtained from both rounds of selection, 3 (-5%) clones were found to contain the desired HindIII mutation, whereas 95% had restored a wild-type allele (Table 3.2). As can be seen in Figure 3.5A, Southern blot analysis demonstrated that a 5' HindII site was retained (clones 194-A and -B) or created (clone 153-A) in the three "pop-out" clones following homologous recombination. Clone 153-B is representative of the many clones which had restored a wild-type allele. Further analysis using a probe located 5' to the duplicated sequences demonstrated that loss of the 3' HindIII site in clones 194-A and -B was not simply due to a mutation in the restriction site, but rather to an intrachromosomal recombination 154 Table 3.2. Summary of in vitro run step on K-rasD12-A allele. parental clone (5', 3') # GANCr # GANCr & G418s colonies expanded colonies # colonies screened # pop-out events allele restored in pop-out 12 G->D presence 17 1 15 1 wt ND 28 3 3 3 wt ND 5 (-,+) 17 0 17 0 NA NA 153 (-,+) 17 29 2 5 15 5 2 5 1-wt; 1-H3 wt no; no no 159 (-,+) 18 10 18 10 wt ND 62 36 35 35 wt ND 186 (-,+) 7 1 7 1 wt ND 194 (+,+) 22 0 22 0 NA NA 75 6 6 6 4-wt; 2-H3 no; yes 292 64 143 63 60-wt; 3-H3 2 3 (-,+) Totals: Six independently targeted clones with the above listed integration patterns for the HindIII mutation were placed into gancyclovir to select for clones which had popped-out the selectable markers. All GANC r clones were expanded and tested for their sensitivity to G418 as described in the Experimental Procedures. As predicted, Southern blot analysis demonstrated that only those clones which were both GANC r and G418s had removed the selectable markers. The nature of the restored allele, with respect to both the HindIII and aspartic acid mutations, was determined by Southern blot analysis as well as genomic sequencing and is summarized in the last two columns. NA = not applicable; ND = not determined. I 155 clonal step: Hit Run ="-I < •1 Hit Run run- I--hit hit 3' copy - -- hit 5' copy & run 3' copy -et wt/run oaf* 40 - hit 1 234 5678 -- 520 bp 520 bp- -- 406 bp 134 bp- Figure 3.5. Functional intrachromosomal recombination in the K-rasDI2 -A allele. A. Genomic DNAs from GANCr, G418s clones were digested with HindIII + KpnI and analyzed as described in Figure 3.3. Analysis shows that clones 153-A, 194-A, and 194-B have all undergone intrachromosomal recombination to create a mutated K-ras allele, whereas clone 153-B restored a wild-type allele. Clones 153 and 194 were the parental "hit" clones selected for GANCr. Clone 153 carries the HindIII mutation in the 3' duplicate, whereas clone 194 carries the mutation in both duplicates. Wild-type (wt) ES cell DNA is also shown for comparison. All fragments are identified to the side of each panel and are described in Figures 3.2 and 3.3. B. A parallel set of DNAs were digested with BamHI + KpnI and analyzed with the 5' external probe (Figure 3.2). Southern blotting shows that clones 153-A, 153-B, 194-A, and 194-B have all lost the 9.1 kb BamHI fragment specific to non-recombined, targeted clones. C. and D. PCR analysis was performed on a parallel set of DNAs as described in Experimental Procedures and shows the presence of the wild-type allele (520 bp fragment in both C. and D.) and the D 12 mutation (Figure C. 134 bp fragment in lanes 2, 5, 7, and 8) or the HindIII mutation (Figure D. 406 bp fi-agment in lanes 2, 3, 5, 7, and 8). Sample identity is as follows: lane 1 is wt ES cell DNA, lane 2 is clone 153, lane 3 is clone 153-A, lane 4 is clone 153-B, lane 5 is clone 194, lane 6 is clone 194-C2, lane 7 is clone 194-A, and lane 8 is clone 194-B. Clone 194-C is similar to clone 153-B in that it had restored a wild-type K-ras allele following drug selection. 156 which had correctly excised the non-homologous vector sequences (source of the internal BamHI site) (Figure 3.5B). As discussed above, it was expected that -50% of all "pop-out" events from five of the clones and 100% from clone 194 would carry the desired mutation. However, as shown in Table 3.2, I observed a much lower frequency than this for both types of clones. This suggests that the majority of clones had gained resistance to the negative selection agent through mechanisms other than homologous recombination (e.g., chromosomal loss). Alternatively, the presence of a functional K-rasD12 allele may be detrimental to the growth and survival of the ES cell. To verify the presence of the Asp 12 mutation, I performed genomic sequence and PCR analysis on the three independent "pop-out" clones. Interestingly, only two of the three clones contained the activating mutation. Genomic sequencing confirmed the presence of both the A-toG and G-to-A transitions at nucleotides 15 and 35, respectively, in clones 194-A and 194-B. However, clone 153-A only carried the HindIII mutation at nucleotide 15 (Figure 3.6 and data not shown). This was demonstrated further by a sensitive PCR assay designed to detect each mutation. Clones 194-A and 194-B contained both the 406 bp HindIII-specific and the 134 bp Aspl 2 -specific PCR products in addition to the wild-type fragment (lanes 7 and 8, Figure 3.5C and D). In contrast, clone 153-A did not contain the Asp 12 -specific PCR product (lane 3, Figure 3.5 C and D). This suggests, therefore, that the Asp 12 mutation had either been repaired in clone 153-A or had been carried in a different duplicate (i.e. the 5' copy) than the HindIII mutation in the parental clone (153). The fact that I recovered two independent ES cell clones heterozygous for an activated allele of K-ras suggests that this oncogenic mutation is not detrimental to the ES cell. In support of this, no obvious defects in ES cell growth have been observed in either of these clones. However, I do not know if K-rasD12 is functionally expressed, as antibodies directed against this oncoprotein fail to work in our hands. Alternatively, it is possible that independent mutations may have occurred during the generation of these clones, enabling them to tolerate the expression of K-rasD 12 . In addition, I do not know at this time if the pluripotency of the K-rasD12 ES cells may 157 HindHI: 12 G->D: ++ ++ _ Hit __ Run __ Hit __ Run 12G-- GCI/A H2 A/G - GA TCGA TCGATCG AT C GATC G AT C Figure 3.6. Generation of ES cells carrying a functional oncogenic allele of K-ras. Genomic sequence analysis of the pop-out events shown in Figure 3.5. The presence of each mutation is listed above its respective clone. Either no (-), one (+), or two (++) copies of each mutation were found. The number of copies of the HindIII mutation was determined by Southern blot analysis and subsequently confirmed by sequencing. Copy number of the oncogenic mutation was inferred by sequence band intensity and direct comparison to the HindIII site alteration. Of the three pop-out events, clones 194-A (data not shown) and 194-B both carried a functional oncogenic allele of K-ras, whereas clone 153-A had lost the activating mutation. Clone 194-C had restored a wt allele based on Southern blot analysis and, as expected, did not show the presence of either mutation. For comparision, the sequence of wild-type (wt) ES cell DNA is also shown. Because this reaction was from a different gel, the mobility of the upper bands are different relative to the other samples. Therefore, the position of nucleotide 35 in the wt DNA sample is indicated by a dash. The position of the HindIII and Asp1 2 mutations in all other samples is indicated at the side. 158 be adversely affected and are currently trying to generate chimeras from each of these ES cell clones. Generation of Mice Carrying a Latent Oncogenic K-ras Allele, K-rasD 12 -L, and its Tumorigenic Consequences Four independent K-rasD12 -L heterozygous ES cell clones were selected for spontaneous recombination analysis in the mouse based on their integration patterns for the HindIII and Aspl2 mutations. Two of the clones had integrated the mutations into both duplicates, and therefore, should always generate a functional K-rasD12 allele following intrachromosomal recombination. The other two clones had integrated the HindIII mutation into the 5' duplicate, and the presence of the Asp 12 mutation was verified by sequence analysis (Figure 3.4 and data not shown). However, as discussed earlier, I can not say with certainty that it is the 5' duplicate that carried the mutation. The latter two clones were chosen to control for the possibility of K-rasD 12 expression from the downstream duplicate (e.g., due to a cryptic promoter site) prior to the recombination event. In addition, it is important to note, that all clones were maintained under positive selection (i.e., in the presence of G418) in vitro during clonal expansion in order to minimize subpopulations of cells spontaneously undergoing the intrachromosomal recombination prior to the in vivo analysis. Chimeric animals were created by injecting cells from the four independent K-rasD 12 -L clones into C57BL/6 blastocyst-stage embryos. These chimeras were then bred to C57BL/6 mice, and BL/6:129/Sv agouti offspring were genotyped to determine germline contribution. All four clones have produced chimeras that transmitted the K-rasD12-L allele through the germline (Figure 3.7). Because the PCR assay designed for genotyping only detected the presence of the insertion, I also performed the Aspl 2 -specific PCR analysis on F1 heterozygotes. This analysis confirmed the presence of the Asp 12 mutation in all twenty of the heterozygous offspring examined (data not shown). 159 * k** * 390 bp 220 bp - Figure 3.7. Germ line transmission of the K-rasD12-L allele. PCR analysis shows the presence of wild-type (220 bp) and mutant (390 bp) K-ras alleles in the DNA of Fl heterozygous offspring (indicated by an asterisk (*)). The other four animals were homozygous for the wild-type K-ras allele. Aspl2-specific PCR analysis was also performed (not shown) and confirmed the presence of the Asp 12 mutation in all heterozygous offspring. 160 12 It is expected that at some stochastic frequency, the duplicated sequences in the K-rasD -L allele will undergo intrachromosomal recombination to produce an activated allele of K-ras. The frequency and onset of this spontaneous event as well as the number of duplicates carrying the mutation will influence the end number of cells carrying a functional K-rasD12 allele. Because a large number of cells (in chimeras) or every cell (in germline Fl progeny) in the developing and adult mouse will carry the duplicated mutant allele, the somatic recombination event should occur broadly and predispose these mice to a wide range of tumor types. Because this study is currently ongoing, preliminary data on only a limited number of animals can be presented at this time. Starting at 3-4 months of age, a small number (6/40) of K-rasD12 -L chimeras and two Fl heterozygous animals have either died suddenly or exhibited signs of distress and were sacrificed for complete necropsy. These animals ranged in age from 84 to 220 days. Autopsy revealed the presence of obvious tumors in the lung and/or thymus in 75% and 50% of the animals, respectively. Tumor samples were saved for DNA analysis to determine the status of the intrachromosomal recombination event (currently in progress). Histopathology has only been perfomed on four of the animals at this time and confirmed the presence of 2 lung adenomas, 2 lung carcinoma, and 1 lymphoma (Figure 3.8). This is of great interest, given the high frequency of K-ras mutations associated with tumors of these tissues in both humans and rodents (Bos, 1989; Mangues and Pellicer, 1992). Importantly, these data suggest that this system is feasible for activating an oncogene in vivo. Based on a significant amount of in vitro and in vivo data, it is believed that an oncogenic allele of K-ras will provide the cell with a growth advantage, but will not be transforming in the absence of other cooperating mutational events (Barbacid, 1987). Thus, it is expected that these animals should have a reasonable lifespan. Indeed, most chimeras do not show any overt signs of illness at this time, with the oldest chimeras and Fl progeny being 8-9 months and 4 months of age, respectively. Because other mutational events are necessary for full transformation, I am currently in the process of breeding this mutation into other cancer-prone strains. In particular, due 161 to the frequent association of K-ras activation with mutation of the p53 and APC genes in several human tumor types, I am interested in examining the cooperative effects between these mutations. 162 Figure 3.8. Histopathology of tumors in K-rasD 12-L allele mice. A. Section showing the presence of both lymphoma (L) and a small adenoma (A) in the lung of a 3 month old chimera (100X magnification). B. Section showing the presence of a large adenoma (A) in the lung, which was surrounded by normal (N) lung tissue (100X). Importantly, this mouse exhibited multi-focal adenomas (not shown). C. Higher magnification (400X) view of the adenoma shown in panel B. D. Section through the lung demonstrating a clara cell carcinoma (100X). Clara cells normally function to secrete surfactin in the lung tissue. E. Different area of the carcinoma (C) depicted in panel D. This tumor was defined by the excess amount of surfactin present in the lung tissue and macrophages (pink staining cells indicated by black arrows). F. Higher magnification (400X) view of the carcinoma shown in panel E (400X). 163 Discussion Oncogenic ras mutations occur in -30% of all human tumors, with K-ras being the most frequently mutated member of this gene family. K-ras mutations are associated with three very common and clinically important tumor types, namely pancreatic, colon, and lung carcinomas (Bos, 1989). In order to develop drugs designed to inhibit tumorigenesis, an accurate animal model of the disease is invaluable. There are a number of problems associated with the current approaches available to analyze both oncogenes and tumor suppressor genes and their roles in the initiation and progression of tumorigenesis. We now demonstrate a novel method for activating an oncogenic allele of the K-ras gene in vivo, which may be applied to the generation of other improved animal models of cancer. Strategies For Analyzing Functional Mutations in the Mouse A number of different approaches have been utilized in the past to examine the effect of activated ras alleles on tumor development in vivo. Transgenic mice carrying activated ras genes have been created either through the use of retroviruses or direct microinjection of recombinant DNA into the pronucleus of fertilized eggs (Adams and Cory, 1991). Although an enormous amount of information has been gained through these approaches, the results must be interpreted with caution. As discussed earlier, there are a number of disadvantages associated with ras transgenes. Not only do they position the ras oncogene outside of its normal genomic context, but they also result in abnormally high levels of expression of the mutant protein due to either gene dosage (i.e., multiple integrants), the choice of promoter driving the activated ras allele, and/or position effects. This issue is important since the level of oncogenic Ras appears to modulate its transforming potency (see below). This problem can be overcome by targeting the expression of the desired mutation to the endogenous locus via homologous recombination. In the last decade, considerable technological advances in gene targeting approaches have provided a powerful tool for studying gene function in 165 vivo. Generally, these strategies require the introduction of a selectable drug marker into the targeted locus of mouse embryonic stem (ES) cells (Hasty and Bradley, 1993). The resulting targeted disruption has greatly facilitated the ability to study the effect of a null mutation on the whole animal and has been invaluable in studying the roles of tumor suppressor genes in tumorigenesis. However, this large scale alteration in gene structure is not desirable for creating subtle mutations in the mammalian genome for either fine-structure analysis or to mimic human genetic diseases resulting from small specific mutations (i.e., point mutations or small insertions and deletions). Recently, techniques designed to leave behind subtle genetic modifications, with very little or no alterations to the rest of the genome, have been developed. There are three documented methods designed for this purpose: the hit-and-run (or in-out) (Hasty et al., 1991; Valancius and Smithies, 1991), the double replacement (similar to tag-and-exchange) (Askew et al., 1993; Wu et al., 1994), and the combination of site-specific and homologous recombination (e.g., Cre/LoxP) (Kilby et al., 1993; Barinaga, 1994). One advantage of the first two approaches is that after the selectable marker has been removed, only the desired alteration remains in the genome. In contrast, the Cre/LoxP system leaves behind a small insertion consisting of the LoxP site following excision of the selectable marker. The main limition, however, of the first two approaches arises from their loss of sensitivity to the negative selectable marker as well as their sensitivity to the loss of the whole chromosome containing the targeted locus during the removal of selectable markers. Thus, as measured in vitro, the efficiency of the desired recombination event is lowered. In addition, the pluripotency of the targeted ES cells may be adversely affected by the exposure to two successive steps of selection. Prior to this work, these approaches had only been applied to ES cells in vitro. In fact, the double replacement strategy is only applicable in vitro. The drawback to this is that any subtle modification will be represented in every cell of the animal in established mouse lines. Thus, while this may be more representative of familially inherited mutations, it does not mimic sporadic forms of cancer. 166 In contrast, the Cre/LoxP system has provided a very powerful genetic tool with which to create subtle mutations via a site-specific recombinase. Importantly, this method has the potential to create the desired alteration in a tissue/cell-specific manner by directing Cre expression, and thus excision, to a specific cell type or lineage (Barinaga, 1994). However, despite the high efficiency of Cre recombinase-mediated excision, this method is severely limited by the availability of transgenic mice expressing the Cre recombinase under the appropriate tissue/cell-specific promoter. This is important since the target cell of the initiating (and subsequent) mutation in many cancers is not well defined. Therefore, finding the appropriate promoter to direct the expression of the desired modification is not necessarily straightforward. In addition, all cells within the affected tissue will express the mutation. Thus, while this may increase the target cell population for subsequent cooperating events, it does not mimic spontaneous tumorigenesis, nor does it allow for the potentially inhibitory interaction with normal neighboring cells. Finally, since its expression will be directed to a specific tissue, only a limited tumor spectrum would be presented. Overall, despite being a powerful strategy for creating subtle mutations in a more controllable fashion, it has many of the same disadvantages associated with transgenics for animal cancer models. We have now demonstrated a variation on the hit-and-run targeting procedure in vivo. This approach overcomes many of the disadvantages associated with these other systems, especially for studying tumor development. Importantly, the stochastic process of generating the mutation in somatic cells is more representative of many of the genetic hits that occur during the initiation and progression of tumorigenesis. In addition, this strategy obviates the need for a specific promoter to target expression of the mutation to the correct target cell. In theory, every cell in a mouse carrying a latent version of the mutated allele has the potential to generate the alteration through an intrachromosomal recombination. The end number of cells expressing a functional mutation will be determined by the number of duplicates carrying the mutation, the position of the mutation relative to the lengths of the arms of homology, and the frequency and onset of the functional recombination event. For example, functional recombination in a pluripotent stem cell or early in 167 development could result in the clonal expansion of the mutated allele into potentially many different cell lineages, assuming the differentiating capacity of the cell is not affected. Thus, a large number of cells/cell types in the whole animal may carry the mutation. In contrast, only a limited number of cells will carry the functional mutation if the recombination occurs later in the adult mouse. Therefore, depending upon the frequency and onset of the activation, this model has the capacity to represent both familial and sporadic forms of cancer. Furthermore, this approach allows expression of the mutated allele to be controlled by its endogenous promoter and positional constraints. With respect to Ras, all previous approaches utilizing transgenes have implemented promoters that result in a significantly higher than normal level of activated Ras expression. This becomes an issue because the transforming potency of Ras appears to be modulated by its level of expression. Although point mutations within ras are gainof-function and ectopic overexpression permits transformation even in the presence of a normal allele (Tabin and Weinberg, 1985), their proposed dominance has been questioned (Capon et al., 1983; Santos et al., 1984; Craig and Sager, 1985; Geiser et al., 1986). Analysis of ras genes in tumor cells has revealed that in many cases the expression of the mutant allele is frequently augmented by gene amplification or other mechanisms (Barbacid, 1987; Bos, 1988; Cohen and Levinson, 1988; Finney and Bishop, 1993). In particular, the ratio between the mutant and the normal allele is oftentimes increased during progression (Bos et al., 1987; Bremner and Balmain, 1990). In addition, loss of the normal ras allele has been observed in human tumors and frequently occurs in mouse skin tumor progression (Yokota et al., 1986; Bremner and Balmain, 1990). However, the significance of this latter finding is not known and may simply reflect the coincident deletion of a linked gene (e.g., a tumor suppressor gene) important for tumor progression. Recently, Finney and Bishop directly addressed this issue of dominance. Utilizing a modified hit-and-run strategy, they were able to introduce an activating mutation (H-rasE12 ) into the endogenous H-ras gene in Ratl fibroblasts (Finney and Bishop, 1993). Importantly, heterozygous cells expressing the mutant and normal alleles at equal levels were neither 168 transformed nor tumorigenic. Instead, they were predisposed to neoplastic transformation at a low frequency. Interestingly, all spontaneously transformed cell lines retained the normal H-ras allele and greater than 90% of the lines had amplified the mutant allele. In support of this observation, analysis of clones carrying a single copy of the mutant H-ras allele driven by the MLV-LTR (parental "hit" step clones) revealed that mutant H-ras expression was 20 times greater in these cells than in cells where its expression was driven from its natural promoter, and as a result, they were highly transformed and tumorigenic. Together, these results imply that a single point mutation in H-ras is insufficient for neoplastic transformation, even of already established cell lines. Thus, in order to render a cell transformed, at least one additional event must occur, of which increasing the expression of a mutant ras allele may be one. Since our approach allows the oncogenic allele of K-ras to be expressed under the normal physiological control of its native promoter, any augmentation in the expression of the allele would be a consequence of natural events occurring during tumorigenesis. The relevance of this may become important in the development of drugs designed to inhibit Ras-transformed cells. It is possible that chemotherapeutic drugs that would be effective against a mutant Ras protein controlled under its natural constraints would fail to work as effectively if the protein were expressed at artificially high levels. Therefore, having an accurate animal model of the disease is invaluable. This is underscored by the fact that many of the compounds that have proven effective in cell-free or cell culture systems are often times found to be ineffective or toxic when tested in vivo. Examining the Multistep Nature of Cancer in the Mouse By using the novel approach described here, every cell in the animal will carry the spontaneously activatable allele. Therefore, it is expected that these animals will be predisposed to a broad spectrum of tumor types with variability in the onset of presentation. I have now demonstrated that this approach is feasible for activating an oncogene in vivo. In addition, more than one tumor type was represented in these animals, suggesting that more than one cell type is capable of 169 carrying out the recombination event. At this time, K-rasD12 -L animals appear to be highly susceptible to lung tumors and thymic lymphomas. This is not surprising, given the frequent association of K-ras mutations in these tumor types in mouse carcinogenesis models (Guerrero and Pellicer, 1987; Mangues and Pellicer, 1992). Furthermore, the onset of tumorigenesis in these animals appears to occur within a broad window of time. The earliest presentation occurred at -3 months of age; however, at this time, chimeras with greater than 95% contribution (based on coat color) by K-rasD12 -L ES cells have survived out to 8.5 months of age with no overt abnormalities. In addition to activating Ras, additional genetic events must occur in order for a cell to be fully transformed. Thus, in those K-rasD12 -L animals exhibiting an increased lifespan, it may be possible to uncover the cooperating genetic events associated with a particular tumor type. Over the past decade, it has become clear that cancer is a genetic disease, arising from the accumulation of several mutations, most of which are somatic in nature (Vogelstein and Kinzler, 1993). Because each cancer appears to be clonal in origin, it is likely that they represent sequential mutations of growth-regulatory genes in a single cell and its descendants. In addition, some tumors, such as those of the colon, evolve through a series of well-defined morphological stages, thus making it possible to establish an order in which mutations occur in that tumor type (Fearon and Vogelstein, 1990). Many of the alterations in colorectal cancer have now been identified and include mutations of the APC, ras,DCC, and p53 genes as well as others. The APCMin (hereafter referred to as Min) mouse has provided an excellent model system with which to study the early events in intestinal neoplasia. The Min mouse strain was generated in a chemical mutagenesis screen and was subsequently shown to have a nonsense mutation in the APC gene (Moser et al., 1990; Su et al., 1992). Min mice develop a severe and early onset of polyposis, with remarkable similarities to the presentation of FAP in humans. However, the distribution of tumors along the intestine differs between humans and mice. Whereas humans exhibit the majority of lesions in the colon and rectum, Min mice display numerous adenomas in both the small and large intestines, with highest concentrations present in the small bowel (Moser et al., 1990; Kim et al., 1993). In addition, the formation of adenomas in Min mice appears to be dependent upon the loss of the 170 wild-type APC allele (Levy et al., 1994; Luongo et al., 1994; Oshima et al., 1995). Interestingly, adenomas from APC mutant mice do not appear to be associated with activating mutations in K-ras (R. Smits and R. Fodde, personal communication), and their progression to carcinoma is strain dependent (i.e., due to allelic modifiers) (Moser et al., 1992). The reason(s) for these differences between the mouse and human models of intestinal neoplasia is presently unknown. Other investigators have tried to examine this multistep nature of intestinal neoplastic progression through the use of transgenic mice. Transgenic mouse lines expressing either SV40 TAg, an oncogenic form of K-ras (Val 12), or a dominant negative p53 mutant (Ala 143) were created and analyzed singly, in combination, or in association with the APCMin mutation (Kim et al., 1993). Whereas expression of K-rasV 12 alone resulted in no detectable abnormalities, it did cooperate with SV40 TAg to produce marked proliferative and dysplastic changes in the intestinal epithelium. However, this dysplasia failed to progress to form adenomas despite continued expression of the transgenes. Importantly, when K-rasV12 and APCMin mutations were analyzed in combination, no synergistic effect was observed. One possible explanation for the lack of cooperation between the Min and K-rasV12 mutations may be due to inappropriate target cell expression of K-rasV 12 . The expression of all of the transgenes in this study were directed to post-mitotic enterocytes. In contrast, initiation of tumorigenesis in Min mice typically occurs in the intestinal crypts (location of stem cells) rather than in villus-associated epithelial cells (Moser et al., 1992). Thus, cooperation between Min and oncogenic alleles of K-ras may require appropriately targeted expression of K-ras to the crypt stem cells or their immediate descendants. Indeed, D'abaco et al. recently demonstrated that co-expression of a defective APC allele and an activated allele of H-ras was sufficient to transform normal colonic epithelial cells and render them tumorigenic (D'Abaco et al., 1996). Making use of the Immorto mouse and Immorto-Min mouse hybrids, they were able to derive colonic epithelial cell lines and examine the effect of an overexpressed, activated H-ras allele on tumor progression. The Immorto mouse is a transgenic mouse carrying a temperature-sensitive allele of SV40 TAg under the control of an interferon-y inducible promoter. Introduction of oncogenic H-ras into both cell lines allowed 171 them to continually proliferate in the absence of functional SV40 TAg, unlike the parental cell lines that require SV40 TAg to proliferate and survive. Thus, an activated allele of H-ras was able to replace the immortalization function of SV40 TAg. Moreover, the loss of one functional APC allele and co-expression of an oncogenic allele of H-ras was sufficient to fully transform mouse colonic epithelial cells. Interestingly, tumorigenesis appeared to proceed without the loss or mutation of the wild-type APC allele as well. However, despite showing a cooperative effect between the APCMin and activated H-ras mutations, the above result must be interpreted with caution. First, human colorectal cancer is associated with K-ras mutation, not H-ras (Bos et al., 1987; Vogelstein et al., 1988). Because I have shown that K-ras has a unique role in normal mouse development, it is possible that it may also have a unique role in the formation of specific tumor types. Therefore, it would be important to know if activation of K-ras has a similar cooperative effect in this system. In addition, this effect was observed in the presence of abnormally high levels of oncogenic H-ras. Finally, it would interesting to know if these two mutations actually cooperated in vivo in either the progression of intestinal neoplasia or its distributional presentation in the intestinal tract. K-rasD12 -L animals should provide a valuable tool for examining this and related issues. Due to the frequent association of K-ras mutations with alterations in p53 in several human tumor types (Vogelstein et al., 1988; Mitsudomi et al., 1992; Pellegata et al., 1994), and APC in human colorectal cancer (Vogelstein et al., 1988; Jen et al., 1994), I are currently in the process of crossing the K-rasD12-L allele into p53 deficient and APCMin mouse strains. It will be of great interest to determine what the cooperating effects, if any, that these mutations will have, particularly with respect to the initiation and progression of neoplasms in the intestine/colon, pancreas, and lung. Moreover, a novel cooperative role between these genes in tumor development may be uncovered. In addition, it will be important to examine possible cooperating effects between the K-rasD12 -L allele and mutations in other growth regulatory genes, such as the Rb and Nfl tumor suppressor genes. Lastly, this novel approach for mutational activation may be invaluable in the generation of other improved animal cancer models. 172 Experimental Procedures Construction of Targeting Vectors The K-ras genomic clone used to construct both targeting vectors was previously described in Chapter 2. For site directed mutagenesis, a 1.9 kb SalI-XhoI fragment containing exon 1 sequences was isolated from pK-ras-3' and subcloned into pGEM-7Z+ (Promega). Mutagenesis was then performed to create pK-ras-ExlD 12 using the Amersham Scultptor kit. The sequence of the oligo used to create the HindIII and D 12 mutations was 5'-ATGACTGAGTATAAGCTIGTG GTGGTTGGAGCTGATGGCGTAGGC-3', with the two nucleotide alterations indicated in bold face type. All clones were sequenced to insure that only the desired mutations were incorporated into exon 1. Next, p-3'-K-rasD12 was created via a three piece ligation with the following fragments: a 1.9 kb SalI-XhoI fragment from pK-ras-ExlD12, a 1.7 kb XhoI-XbaI fragment from pK-ras-3', and a 3.0 kb SalI-XhoI fragment from pKSII+ (Stratagene). Two different vectors were created to contain the desired selectable markers for each allele. For the latent allele backbone, a 1.8 kb XhoI-SacI fragment from pPGKRN containing a wild-type neo gene was subcloned into a 5.5 kb XhoI-XbaI fragment from pPNT (Tybulewicz et al., 1991) to create pPRNT. Next, pKSII+-Rneo was created by subcloning a 1.9 kb EcoRI-Acc65I neo fragment from pPRNT into EcoRI-Acc65I digested pKSII + . For the activated allele, pKSII+Rneo:tk was constructed by inserting a 4.0 kb EcoRI-NsiI fragment from pPGKneoNTRtkpA (Wu et al., 1994) into the EcoRV site of pKSII+AXhoI. pKSII+AXhoI was created by deleting sequences from the Apal to Clal sites in pKSII + for the purpose of removing the XhoI site and was necessary in order to create a unique XhoI site in the final targeting vector for linearization. The final targeting constructs were then created via three piece ligations. A 2.8 kb NotISall fragment from pK-ras-5' and a 3.6 kb SalI-XbaI fragment from p-3'-K-rasD12 were ligated together with either a 4.8 kb XbaI-NotI fragment from pKSII+-Rneo to create pK-rasD12-L , or a 7 kb XbaI-NotI fragment from pKSII+-Rneo:tk to create pK-rasD12-A. Exon 1 sequences in the final targeting vectors were subsequently confirmed by sequencing. 173 Electroporation, Selection of ES Cell Clones, and Southern Blot Analysis D3 ES cells (Gossler et al., 1986) were cultured and electroporated as described in Chapter 2. The ES medium was supplemented with G418 at 200 gg/ml (active weight) 48 hours after electroporation. After 9-10 days of selection, colonies were picked and expanded. Genomic DNAs were isolated as described (Laird et al., 1991). To identify targeted clones, DNAs were digested with BamHI + KpnI, resolved on 0.8% agarose gels, and Southern blot analysis was performed as described in Chapter 2 using a 5' external probe. Targeted clones were analyzed further using Southern blotting to determine the integration pattern and copy number. Confirmation of the D 12 mutation was done by sequence analysis as described below. All clones were maintained under positive selection (i.e., G418) prior to the "run" step in order to minimize mixed populations due to pop-out events during clonal expansion. Revertants of the K-rasD12 -A allele were isolated essentially as described (Hasty and Bradley, 1993). Clonal lines were placed into ES medium supplemented with 2 gpM gancyclovir 24-48 hours after plating and selection was continued for 8-10 days. Due to the high frequency of clones which become GANC r via mechanisms other than intrachromosomal recombination, a prescreening procedure was used (Wu et al., 1994). Individual GANC r clones were isolated and grown to confluence. Following trypsinization, cells from each clone were divided as follows: one half was frozen down and one quarter was replated in ES medium containing either gancyclovir (I) or G418 (II). Cells which grew in the first set of plates (I) but not in the second set of plates (II) were GANCr, G418 s and, therefore, reflected the loss of both selectable markers. DNA was prepared from those cells as before and subsequently screened by Southern blotting to determine the nature of the pop-out event. Clones retaining the HindII mutation were sequenced to verify the presence of the D 12 mutation. 174 Genomic Sequence Analysis PCR was used to amplify K-ras exon 1 sequences. The 5' primer (5'-GGGTAGGTGTTGGGAT AGCTGTCGACAAGC-3') was located in intron 0, and the 3' primer (5'-CCTITACAAGCGCA CGCAGACTGTAGAGC-3') was located in intron 1. The 520 bp fragment was phenolchloroform extracted, purified in a Microcon 100 microconcentrator (Amicon), and then sequenced directly (U.S. Biochemical). The primer used for sequence analysis was 5'- TCTTGTGTGAGACATG-3', located immediately 5' to exon 1. Generation of Chimeras C57BL/6 blastocyst-stage embryos were injected with 10-15 K-rasD12 -L ES cells or 8-10 K-rasD12 -A ES cells and then transferred to pseudopregnant Swiss Webster females for further development, essentially as described (Bradley, 1987). Chimeric mice were mated to C57BL/6 and 129/Sv animals and F1 agouti offspring were genotyped. Germline transmission of the mutant allele was detected by either Southern blot or PCR analysis of tail DNA obtained at weaning. PCR Analysis Tail DNA was processed as described (Laird et al., 1991) and analyzed by PCR. All PCR conditions were exactly as those described in Chapter 2, except for the choice of primer pairs. K-rasD12-L primers and their final concentrations used for genotyping were as follows: 0.6 [pM Lat. 5'WT (5'-TGCACAGCTTAGTGAGACCC-3'), 0.4 pM Lat. 3'WT (5'-GACTGCTCTCTTT CACCTCC-3'), and 0.2 p.M Lat. 3'MUT (5'-GGAGCAAAGCTGCTATTGGC-3'). Lat. 5'WT + Lat. 3'WT yields a 220 bp fragment, whereas Lat. 5'WT + Lat. 3'MUT produces a 390 bp mutantspecific fragment. For K-rasD12 -A allele PCR, the following primers and their final concentrations were used to detect the presence of the D 12 mutation: 0.6 p.M 5'K-raslO (5'-AGGGTAGGTGTT GGGATAGC-3'), 0.6 pgM Act. K-ras 3'11 (5'-CCTTTACAAGCGCACGCAGAC-3'), and 0.4 gM Act. K-ras 5'H3 (5'-GCTTGTGGTGGTTGGAGCTGA-3'). 175 To detect the presence of the HindIII mutation, the first two primers were retained and Act. K-ras 5'H3 was replaced with its exact complement, Act. K-ras 3'H3 (5'-TCAGCTCCAACCACCACAAGC-3'). These two primers contain the D 12 and HindIII nucleotide alterations at their 5' and 3' ends, thus making it possible to exploit the destability of the specific nucleotide change at the 3' end for specificity. 5'K-raslO + Act. K-ras 3'11 yields a 520 bp wt fragment, Act. K-ras 5'H3 + Act. K-ras 3'11 produces a 134 bp D 12 -specific fragment, and 5'K-raslO + Act. K-ras 3'H3 yields a 406 bp HindlIl-specific fragment. Histological Analysis All animals showing obvious tumors or other signs of distress were sacrificed and subjected to full necropsy. For histological analysis, all tissues and tumors were fixed in either Bouin's fixative or 10% neutral buffered formalin, dehydrated in graded solutions of alcohol, embedded in paraffin, sectioned at 4-6 gLm and stained with hematoxylin and eosin (H & E). 176 References Adams, J. M., and Cory, S. (1991). Transgenic models of tumor development. Science 254, 1161-7. Askew, G. R., Doetschman, T., and Lingrel, J. B. (1993). Site-directed point mutations in embryonic stem cells: a gene-targeting tag-and-exchange strategy. Mol Cell Biol 13, 4115-24. Balmain, A., Ramsden, M., Bowden, G. T., and Smith, J. (1984). Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature 307, 658-60. Barbacid, M. (1987). ras genes. Annu Rev Biochem 56, 779-827. Barinaga, M. (1994). Knockout mice: round two [news; comment]. Science 265, 26-8. Bos, J. L. (1988). The ras gene family and human carcinogenesis. Mutat Res 195, 255-71. Bos, J. L. (1989). ras oncogenes in human cancer: a review [published erratum appears in Cancer Res 1990 Feb 15;50(4):1352]. Cancer Res 49, 4682-9. Bos, J. L., Fearon, E. R., Hamilton, S. R., Verlaan, de, V. M., van, B. J., van, der, Eb, Aj, and Vogelstein, B. (1987). Prevalence of ras gene mutations in human colorectal cancers. Nature 327, 293-7. Bradley, A. (1987). In Teratocarcinomas and Embryonic Stem Cells: A Practical Approach, E. J. Robertson, eds. (Oxford: IRL, Press), pp. 113-152. Bremner, R., and Balmain, A. (1990). Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell 61, 407-17. Brown, K., Quintanilla, M., Ramsden, M., Kerr, I. B., Young, S., and Balmain, A. (1986). v-ras genes from Harvey and BALB murine sarcoma viruses can act as initiators of two-stage mouse skin carcinogenesis. Cell 46, 447-56. Capon, D. J., Seeburg, P. H., McGrath, J. P., Hayflick, J. S., Edman, U., Levinson, A. D., and Goeddel, D. V. (1983). Activation of Ki-ras2 gene in human colon and lung carcinomas by two different point mutations. Nature 304, 507-13. Cohen, J. B., and Levinson, A. D. (1988). A point mutation in the last intron responsible for increased expression and transforming activity of the c-Ha-ras oncogene. Nature 334, 119-24. Craig, R. W., and Sager, R. (1985). Suppression of tumorigenicity in hybrids of normal and oncogene- transformed CHEF cells. Proc Natl Acad Sci U S A 82, 2062-6. D'Abaco, G. M., Whitehead, R. H., and Burgess, A. W. (1996). Synergy between Apc M in and an activated ras mutation is sufficient to induce colon carcinomas. Mol. Cell. Biol. 16, 884-891. 177 Fearon, E. R., and Vogelstein, B. (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759-67. Finney, R. E., and Bishop, J. M. (1993). Predisposition to neoplastic transformation caused by gene replacement of H-rasl. Science 260, 1524-7. Forrester, K., Almoguera, C., Han, K., Grizzle, W. E., and Perucho, M. (1987). Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature 327, 298-303. Geiser, A. G., Der, C. J., Marshall, C. J., and Stanbridge, E. J. (1986). Suppression of tumorigenicity with continued expression of the c-Ha-ras oncogene in EJ bladder carcinomahuman fibroblast hybrid cells. Proc Natl Acad Sci U S A 83, 5209-13. Gossler, A., Doetschman, T., Korn, R., Serfling, E., and Kemler, R. (1986). Transgenesis by means of blastocyst-derived embryonic stem cell lines. Proc Natl Acad Sci U S A 83, 9065-9. Groden, J., Thliveris, A., Samowitz, W., Carlson, M., Gelbert, L., Albertsen, H., Joslyn, G., Stevens, J., Spirio, L., Robertson, M., and et, a. 1. (1991). Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66, 589-600. Guerrero, I., and Pellicer, A. (1987). Mutational activation of oncogenes in animal model systems of carcinogenesis. Mutat Res 185, 293-308. Hasty, P., and Bradley, A. (1993). Gene targeting vectors for mammalian cells. Targeting: A Practical Approach, A. L. Joyner, eds. (Oxford: IRL Press), pp. 1-31. In Gene Hasty, P., Ramirez, S. R., Krumlauf, R., and Bradley, A. (1991). Introduction of a subtle mutation into the Hox-2.6 locus in embryonic stem cells [published erratum appears in Nature 1991 Sep 5;353(6339):94]. Nature 350, 243-6. Hennings, H., Glick, A. B., Greenhalgh, D. A., Morgan, D. L., Strickland, J. E., Tennenbaum, T., and Yuspa, S. H. (1993). Critical aspects of initiation, promotion, and progression in multistage epidermal carcinogenesis. Proc Soc Exp Biol Med 202, 1-8. James, G. L., Goldstein, J. L., and Brown, M. S. (1995). Polylysine and CVIM sequences of KRasB dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J Biol Chem 270, 6221-6226. Jen, J., Powell, S. M., Papadopoulos, N., Smith, K. J., Hamilton, S. R., Vogelstein, B., and Kinzler, K. W. (1994). Molecular determinants of dysplasia in colorectal lesions. Cancer Res 54, 5523-5526. Khosravi, F. R., and Der, C. J. (1994). The Ras signal transduction pathway. Cancer Metastasis Rev 13, 67-89. Kilby, N. J., Snaith, M. R., and Murray, J. A. (1993). genome engineering. Trends Genet 9, 413-21. 178 Site-specific recombinases: tools for Kim, S. H., Roth, K. A., Moser, A. R., and Gordon, J. I. (1993). Transgenic mouse models that explore the multistep hypothesis of intestinal neoplasia. J Cell Biol 123, 877-93. Laird, P. W., Zijderveld, A., Linders, K., Rudnicki, M. A., Jaenisch, R., and Berns, A. (1991). Simplified mammalian DNA isolation procedure. Nucleic Acids Res 19, 4293. Lerner, E. C., Qian, Y., Blaskovich, M. A., Fossum, R. D., Vogt, A., Sun, J., Cox, A. D., Der, C. J., Hamilton, A. D., and Sebti, S. M. (1995a). Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem 270, 26802-26806. Lerner, E. C., Qian, Y., Hamilton, A. D., and Sebti, S. M. (1995b). Disruption of oncogenic KRas4B processing and signaling by a potent geranylgeranyltransferase I inhibitor. J Biol Chem 270, 26770-26773. Levy, D. B., Smith, K. J., Beazer, B. Y., Hamilton, S. R., Vogelstein, B., and Kinzler, K. W. (1994). Inactivation of both APC alleles in human and mouse tumors. Cancer Res 54, 5953-8. Luongo, C., Moser, A. R., Gledhill, S., and Dove, W. F. (1994). Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res 54, 5947-52. Mangues, R., and Pellicer, A. (1992). ras activation in experimental carcinogenesis. Semin Cancer Biol 3, 229-39. Manne, V., Yan, N., Carboni, J. M., Tuomari, A. V., Ricca, C. S., Brown, J. G., Andahazy, M. L., Schmidt, R. J., Patel, D., Zahler, R., and et, a. 1. (1995). Bisubstrate inhibitors of farnesyltransferase: a novel class of specific inhibitors of ras transformed cells. Oncogene 10, 1763-1779. Mills, N. E., Fishman, C. L., Rom, W. N., Dubin, N., and Jacobson, D. R. (1995). Increased prevalence of K-ras oncogene mutations in lung adenocarcinoma. Cancer Res 55, 1444-1447. Mitsudomi, T., Steinberg, S. M., Nau, M. M., Carbone, D., D'Amico, D., Bodner, S., Oie, H. K., Linnoila, R. I., Mulshine, J. L., Minna, J. D., and et, a. 1. (1992). p53 gene mutations in nonsmall-cell lung cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene 7, 171-80. Moser, A. R., Dove, W. F., Roth, K. A., and Gordon, J. I. (1992). The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol 116, 1517-26. Moser, A. R., Pitot, H. C., and Dove, W. F. (1990). A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247, 322-4. Nishisho, I., Nakamura, Y., Miyoshi, Y., Miki, Y., Ando, H., Horii, A., Koyama, K., Utsunomiya, J., Baba, S., and Hedge, P. (1991). Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 253, 665-9. 179 Oshima, M., Oshima, H., Kitagawa, K., Kobayashi, M., Itakura, C., and Taketo, M. (1995). Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A 92, 4482-4486. Pellegata, N. S., Sessa, F., Renault, B., Bonato, M., Leone, B. E., Solcia, E., and Ranzani, G. N. (1994). K-ras and p53 gene mutations in pancreatic cancer: ductal and nonductal tumors progress through different genetic lesions. Cancer Res 54, 1556-60. Powell, S. M., Petersen, G. M., Krush, A. J., Booker, S., Jen, J., Giardiello, F. M., Hamilton, S. R., Vogelstein, B., and Kinzler, K. W. (1993). Molecular diagnosis of familial adenomatous polyposis [see comments]. N Engl J Med 329, 1982-7. Powell, S. M., Zilz, N., Beazer, B. Y., Bryan, T. M., Hamilton, S. R., Thibodeau, S. N., Vogelstein, B., and Kinzler, K. W. (1992). APC mutations occur early during colorectal tumorigenesis. Nature 359, 235-7. Quintanilla, M., Brown, K., Ramsden, M., and Balmain, A. (1986). Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 322, 78-80. Santos, E., Martin, Z. D., Reddy, E. P., Pierotti, M. A., Della, P. G., and Barbacid, M. (1984). Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science 223, 661-4. Sinn, E., Muller, W., Pattengale, P., Tepler, I., Wallace, R., and Leder, P. (1987). Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 49, 465-75. Smith, A. J., Stem, H. S., Penner, M., Hay, K., Mitri, A., Bapat, B. V., and Gallinger, S. (1994). Somatic APC and K-ras codon 12 mutations in aberrant crypt foci from human colons. Cancer Res 54, 5527-5530. Spandidos, D. A. (1986). The human T24 Ha-rasl oncogene: a study of the effects of overexpression of the mutated ras gene product in rodent cells. Anticancer Res 6, 259-62. Su, L. K., Kinzler, K. W., Vogelstein, B., Preisinger, A. C., Moser, A. R., Luongo, C., Gould, K. A., and Dove, W. F. (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene [published erratum appears in Science 1992 May 22;256(5060): 1114]. Science 256, 668-70. Sun, J., Qian, Y., Hamilton, A. D., and Sebti, S. M. (1995). Ras CAAX peptidomimetic FTI 276 selectively blocks tumor growth in nude mice of a human lung carcinoma with K-Ras mutation and p53 deletion. Cancer Res 55, 4243-4247. Tabin, C. J., and Weinberg, R. A. (1985). Analysis of viral and somatic activations of the cHaras gene. J Virol 53, 260-5. Tybulewicz, V. L., Crawford, C. E., Jackson, P. K., Bronson, R. T., and Mulligan, R. C. (1991). Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl protooncogene. Cell 65, 1153-63. 180 Valancius, V., and Smithies, 0. (1991). Testing an "in-out" targeting procedure for making subtle genomic modifications in mouse embryonic stem cells. Mol Cell Biol 11, 1402-8. Vogelstein, B., Fearon, E. R., Hamilton, S. R., Kern, S. E., Preisinger, A. C., Leppert, M., Nakamura, Y., White, R., Smits, A. M., and Bos, J. L. (1988). Genetic alterations during colorectal-tumor development. N Engl J Med 319, 525-32. Vogelstein, B., and Kinzler, K. W. (1993). The multistep nature of cancer. Trends Genet 9, 138-41. Weinberg, R. A. (1989). Oncogenes and Multistep Carcinogenesis. In Oncogenes and the Molecular Origins of Cancer, eds. Cold Spring Harbor Laboratory Press), pp. 307-326. Wu, H., Liu, X., and Jaenisch, R. (1994). Double replacement: strategy for efficient introduction of subtle mutations into the murine Colla-1 gene by homologous recombination in embryonic stem cells. Proc Natl Acad Sci U S A 91, 2819-23. Yamashita, N., Minamoto, T., Ochiai, A., Onda, M., and Esumi, H. (1995). Frequent and characteristic K-ras activation in aberrant crypt foci of colon. Is there preference among K-ras mutants for malignant progression? Cancer 1527-1533. Yokota, J., Tsunetsugu, Y. Y., Battifora, H., Le, F. C., and Cline, M. J. (1986). Alterations of myc, myb, and rasHa proto-oncogenes in cancers are frequent and show clinical correlation. Science 231, 261-5. Yuspa, S. H. (1994). The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis--thirty-third G. H. A. Clowes Memorial Award Lecture. Cancer Res 54, 1178-89. 181 Chapter 4 Overview and Future Directions 182 This thesis focused on trying to understand the role(s) that ras family members play in the normal growth and development of the mouse as well as in the initiation and progression of tumorigenesis. Unveiling the function that ras oncogenes serve in basic cellular growth and differentiation will further our knowledge on how its disregulation can lead to neoplastic development. ras genes (and other members of the signaling cascade) are highly conserved throughout evolution, underscoring their fundamental role in growth control of the cell. However, ever since the identification of these genes in the mammalian genome, no one had been able to demonstrate a distinct role for any of the three members of the ras gene family. I have now demonstrated a unique and vital function of K-ras in mouse embryogenesis. In the future, it will be of great importance to determine why K-ras function is unique. I do not know if the requirement for K-ras reflects a unique function of the gene product not shared by N-ras or H-ras or simply differences in the expression patterns of the family members. A more precise analysis of the expression of ras genes must be done in order to determine if a critical cell type exclusively expresses K-ras. In particular, this analysis should be directed to the fetal liver due to the defects associated with this tissue in K-ras-1- embryos. Alternatively, K-ras could be relaying a unique signal transduction cascade. The differences in K-ras4B processing and its specific regulation by the smg-GDS exchange factor suggests that K-ras may be brought into association with a distinct subset of effectors, thereby elliciting a unique function in cell signaling. By generating mutant alleles specific for each K-ras isoform, one could determine the requirements and distinct functions that may be associated with each isoform. The K-ras-deficient embryos and ES cell clones are a valuable source for isolating specific cell populations devoid of K-ras function. Therefore, biochemical characterization of these cells may allow identification of a distinct signaling pathway(s) emanating from K-ras in response to specific growth/survival factors. This will not only be of value in understanding the role of K-ras in normal cell growth and differentiation, but may also offer another potential (and perhaps more specific) target for drug therapy. Hematopoietic defects and significant cell death were observed in both K-ras-1 - embryos and compound mutants of N- and K-ras. Given the finding that Ras 183 mediates a survival signal downstream of certain cytokines, it will be important to dissect the function that each Ras member serves in relaying such signals. The availability of mice deficient in specific ras genes now makes this type of analysis feasible. I was able to show partial overlap in function between N- and K-ras in early murine development. Thus, a more thorough analysis of various combinations of the three mutant alleles (H-, N-, and K-ras) would reveal the extent of functional overlap between various family members and help define the threshold level of activity that is required for carrying out vital functions. Due to the lethality of K-ras-1- embryos and the limited contribution that could be achieved by K-ras-1 - ES cells in chimeras, it was difficult to assess what the requirement for K-ras was in the adult animal. Therefore, the generation of tissue/cell specific mutant alleles would allow for a more defined analysis of what cells, if any, require K-ras function in the adult animal. Alternatively, the novel strategy that I developed could, in theory, be applied as a method for cell/tissue specific "knock-in". Because the insertion allele is expected to be inactive, breeding the mutation to homozygosity would result in embryonic lethality. Therefore, the generation of viable offspring from heterozygous intercrosses would select for those animals that restore K-ras function through intrachromosomal recombination in the critical cell type(s) of genotypically homozygous mutant animals. Thus, extensive expression analysis on these mice could reveal which cells require K-ras function for the completion of embryogenesis and later in adult life. The success of this approach would depend, in part, on the frequency and onset of the activating recombination. Defining the requirement for K-ras in adult tissues is important, given the enormous amount of research by pharmaceutical companies devoted to developing anti-cancer drugs based on the inhibition of oncogenic or normal Ras function. Our results suggest that drugs that block Ras function non-specifically would be highly toxic. In fact, the recent finding that K-ras4B (the predominant isoform) is geranylgeranylated provides a strong explanation for the non-toxic effect of farnesyl-transferase inhibitors in vivo, as geranylgeranylation is highly resistant to their 184 inhibitory effect. Thus, it would seem more plausable to develop drugs designed to inhibit specific members of the Ras family rather than overall Ras activity. On this note, the creation of the K-rasD12 L allele mouse strain should prove invaluable in the development of drugs designed to specifically inhibit tumor cells carrying an oncogenic allele of K-ras. In the future, it will be necessary to define the mechanism(s) of activation of the K-rasD12 L allele to confirm its association with the observed tumor phenotypes. Southern blotting of tumor DNAs will be instrumental in this analysis and will help establish whether activation can occur in the absence of intrachromosomal recombination. Moreover, this analysis can address the importance of allelic amplification during tumor progression. In addition, it will be very interesting to characterize the lesions that are presented in older animals. For example, is there an even broader spectrum of pathological consequences associated with oncogenic K-ras in the mouse? How limited are the tumors with respect to their progression? And most importantly, how does this allele cooperate, if at all, with other tumor suppressor mutants in the initiation and progression of tumorigenesis? Having established the tumor phenotypes associated with the K-rasD12 L allele, I will be able to test the efficacy of various anti-Ras chemotherapeutic agents on tumor regression as well as tumor formation. All too often, drugs that show promise in cell culture or cell-free systems prove ineffective or toxic when tested in vivo. Therefore, the availability of mice which more accurately reflect the natural tumorgenic process may have a significant impact on these studies. I believe that this novel method of oncogene activation may be applied to other genes to generate additional improved animal models of cancer. For example, the creation of mice deficient in tumor suppressor genes has greatly extended our knowledge on the function of these genes, not only in tumorigenesis, but in development as well. However, mice heterozygous for the mutations don't always present the same phenotype that is associated with their familial inheritance in humans. This may partly be explained through species-specific differences in either the sensitivity to the mutation(s) or the rate-limiting cooperative events in the appropriate target cell. Due to the high susceptibility of lethal tumor types in some of these mutant strains, their lifespan is 185 significantly shortened. Thus, the potential for cooperating events to occur is severely limited and may, therefore, not allow for presentation of the familial phenotype. It is tempting to speculate that through the combination of more carefully targeted mutations in the mouse, we will be able to better recapitulate human cancer models and define the relevant aberrations associated with each cancer. All of this in hope of creating better chemotherapeutic treatments designed to specifically inhibit the neoplastic processes associated with each type of cancer. 186 Appendix 1 The PDGF Inducible Factor SIF is Related to the Interferon Activated p91 Transcription Factor This chapter represents some of my work while in Dr. Brent Cochran's lab. It was submitted for publication, although it was never published. Abstract Transcriptional regulation of the c-fos proto-oncogene is controlled by an array of regulatory elements and transcription factors. One of these elements is a conserved regulatory sequence which binds to a growth factor inducible DNA-binding factor termed SIF (c-sis/PDGF inducible factor). This element can confer PDGF responsiveness onto the c-fos promoter independently of the serum response element (SRE). Here we show that the SIF DNA binding complex reacts with antibodies against the 91 kD subunit of the interferon-stimulated transcription factor ISGF3. In addition, we show that interferon-y induces SIF DNA binding activity and that the binding specificity of SIF is closely related to that of the interferon-y activated factors. These results suggest that growth factors and interferons activate common signal transduction pathways. 188 Introduction/Results/Discussion c-fos transcription is transiently activated by a wide variety of extracellular stimuli (Verma and Sassone-Corsi, 1987). We have previously described a DNA binding factor whose binding activity is induced by v-sis conditioned media (SCM) and PDGF, but not by phorbol esters (Hayes et al., 1987; Wagner et al., 1990). This factor binds to a conserved sequence (the sis/PDGF inducible element SIE) just upstream of the serum response element (SRE) of the c-fos promoter. In addition to PDGF, SIF binding activity is induced by a variety of growth factors including EGF (Sadowski and Gilman, 1993), growth hormone (Meyer et al., 1993), CSF-1, and insulin (unpublished results). The SIE is the only c-fos promoter element whose in vivo footprint changes upon c-fos activation (Herrera et al., 1989). In cultured cells, the SIE and SRE are each sufficient to confer transcriptional activity onto a truncated c-fos promoter in response to PDGF (Wagner et al., 1990). However, studies in transgenic mice analyzing point mutations in either element indicate that both are required for in vivo c-fos expression (T. Curran, personal communication). UV cross-linking experiments identify an approximately 91 kD protein that interacts specifically with the SIF DNA binding site (Figure 1.1). In crude extracts of quiescent Balb/c-3T3 cells, several proteins become crosslinked to a 3 2 P-labeled SIE probe in unstimulated cells. An additional band of approximately 91kD is crosslinked in the extracts from v-sis conditioned media (SCM) treated Balb/c-3T3 cells. Moreover, crosslinking of this 91 kD protein can be specifically reduced by inclusion of unlabelled SIE oligonucleotides in the binding reaction. From this data and from purification and sedimentation analysis (data not shown), it can be concluded that a protein of approximately 91 kD in molecular weight is the principal DNA binding component of SIF. Recently, a 91 kD protein has been identified as a component of the interferon stimulated transcription factor, ISGF3 (Fu, 1992; Schindler et al., 1992a). Interferon induces the tyrosine phosphorylation and DNA binding activity of this transcription factor (Schindler et al., 1992b; 189 4( CO cC, ct, cO' 92- 68- ! 45- 31- a aU Figure 1.1. Specific crosslinking of a 91 kD protein to the SIE in extracts of v-sis conditioned media treated cells. Extracts from quiescent (Q) or v-sis conditioned medium (SCM) treated Balb/c-3T3 cells were crosslinked to an SIE DNA probe and electrophoresed through SDS-PAGE gels and exposed to X-ray film. In the last lane, the crosslinking was done in the presence of 0.5 pmol unlabelled SIE oligonucleotide. The positions of the molecular weight markers are indicated along with the position of an induced specific band. 190 Shuai et al., 1992). Because of the similarities in molecular weight, inducible DNA binding, and the fact that some genes are induced by both PDGF and interferon (Garcia-Blanco et al., 1989), we investigated whether ISGF3 and SIF share any common components. To determine whether any of the SIE complexes are immunologically related to the IFN induced complexes, antibodies to the 91 and 113 kD components of ISGF3 were pre-incubated with extracts from PDGF-treated cells and analyzed by mobility-shift assays (Figure 1.2). Antisera to the 91 kD protein efficiently bound to and supershifted all of the PDGF-induced complexes. However, antisera to the 113 kD protein or sera from a non-immunized rabbit did not react with the SIF complexes. The antisera to p91 is directed against a C-terminal peptide of this protein and does not recognize the 84 kD protein produced by the same gene (Igarashi et al., 1993). Inclusion of excess peptide antigen in the binding reaction eliminated the supershift (data not shown). An antibody directed against a part of p91 that includes the SH2 domain reacts with selected, but not all complexes that form specifically with an SIE oligonucleotide (data not shown). Thus, the epitopes which this antisera recognizes are either occluded in some of these complexes or the protein recognized by the C-terminal antibody in this complex is closely related, but not identical to the ISGF3 p91. There are two classes of interferon responsive regulatory elements. One is termed the ISRE and is primarily responsive to interferons ca and P3 (Friedman and Stark, 1985; Levy et al., 1988). Subsequently, it has been found that there are also regulatory elements specifically responsive to IFN-y (Lew et al., 1991). The p91 ISGF3 protein is found in the IFN-a stimulated ISRE binding complex, but does not determine binding specificity at this site (Veals et al., 1992). However, in response to stimulation by interferon-y, inducible binding is seen to IFN-y responsive elements (IGRE), and this binding activity may be mediated directly by p91 (Shuai et al., 1992; Pearse et al., 1993). Recently, a consensus sequence has been derived for the minimal IFN-y activation sequences (Pearse et al., 1993). Known SIF DNA binding elements closely resemble this consensus. 191 Balb/c-3T3 C,Q'Oj , 'o W a sY Figure 1.2. Antisera to the ISGF3 p91 supershifts SIF complexes. Nuclear extracts from Balb/c-3T3 cells were used in mobility shift assays with the SIE probe after pre-incubation with either antibodies against the p91 or p113 proteins of ISGF3 or from normal non-immune rabbit sera (NRS). All extracts were made from PDGF treated cells except the lane marked Q for quiescent cell extracts. 192 A comparison of IFN-y responsive elements and the c-fos SIE is shown in Table 1.1. The sequence similarity between these two elements has led us to ask whether IFN-y can induce binding to the SIE and whether SIE and IFN-y responsive elements can cross compete with one another for binding to SIF and IFN-y activated factors. Results of these experiments are shown in Figure 1.3. U937 cells were treated for thirty minutes with IFN-y and extracts from these cells were analyzed by mobility shift assay using a probe derived from the IFN-y response region (GRR) of the high affinity Fc receptor for IgG gene (FcyRI). A complex is induced by IFN-y in these cells which has previously been shown to be composed of at least in part the p91 protein (Wilson and Finbloom, 1992; Pearse et al., 1993). Both the wild type SIE found in c-fos and a high affinity SIE oligonucleotide can compete for binding to this complex in the mobility shift assay, whereas a non-binding point mutant of the SIE fails to compete for this band (Wagner et al., 1990). The band is competed with an unlabelled homologous GRR oligonucleotide and an oligonucleotide derived from the FcyRI which contains primarily the minimal IFN-y responsive element of the GRR. It is also competed by the IFN-y responsive element of the Ly6E gene. The interferon-cx, 03 responsive ISRE does not compete for this binding. Interestingly, the IGRE elements from the GBP and MIG genes also fail to compete for this binding, or do so only very weakly. A similar set of competition experiments was performed using the wild type c-fos SIE as a probe with extracts from PDGF treated HepG2 cells containing exogenous PDGF receptors (Valius and Kazlauskas, 1993). An essentially identical competition was seen in this experiment. In other experiments (not shown), we have found that IFN-y induces binding to the high affinity SIE site in both U937 cells and A431 cells. In more quantitative competition assays (not shown), the high affinity SIE oligonucleotide also proves to be a high affinity competitor for 91 kD binding to the GRR probe. The order of affinity for binding from either extracts of IFN-y or PDGF treated cells is the high affinity SIE followed by the wild type c-fos SIE and the GRR and then the Ly6E site (SIE + > SIE,GRR > Ly6E). The IFN-y responsive elements from the GBP and MIG genes, as well as the consensus ISRE, show little or no competition. 193 Table 1.1. Comparison of the sequences of the c-fos SIE and interferon responsive elements. IFN-y consensus TTCCNNNAA A human c-fos SIE AGTTCCCGTCAATCCC SIE+ GRR core GRR long CATTTCCCGTAAATCCCT TATTTCCCAGAAAAGGAA GCATGTTTCAAGGATTTGAGATGTATTTCCCAGAAAAGG Ly6E ATAITTCCTGTAAGTGCAT MIG GBP ISRE consensus TGGTTTCACATCCCTTACTATAAA TACTTTCAGTTTCATATTACTCTAAA AGTTTCACTTTTCC TT C The consensus of IFN-y responsive elements is from Pearse et al. (Pearse et al., 1993). SIE is the sis/PDGF inducible element from the c-fos gene. SIE + is a high affinity SIF binding site (Wagner et al., 1990). GRR and Ly6E are the core IFN-y responsive elements from the FcyRI and Ly6E genes (Pearse et al., 1993). MIG is an element from the monokine induced by interferon-y gene (Wright and Farber, 1991). The GRR long element is the optimal IFN-y response element from the FcyRI gene (Pearse et al., 1993). GBP is the interferon responsive element from the guanylate binding protein gene (Lew et al., 1991). The ISRE consensus is from Levy et al. (Levy et al., 1988). Sequences with homology to the consensus IFN-y responsive element are underlined. 194 A GRR-I probe L . , U937 cells SIF probe HEPG2 cells . NM m mm O Figure 1.3. The c-fos SIE and IFN-y responsive elements cross compete for binding to IFN-y and PDGF induced complexes. Mobility shifts assays were performed with extracts from IFN-y treated U937 cells or from PDGF-BB treated HepG2 cells that express the PDGF3 receptor (Valius and Kazlauskas, 1993). The GRR oligonucleotide was used as a probe for the U937 cell extracts and the c-fos wild type SIE was used as a probe for the HepG2 cell extracts. Where indicated, competitor oligonucleotides were added to the binding reactions. SIF + is the high affinity SIF binding site m67. SIF is the wildtype c-fos SIE, and SIF* is the nonbinding mutant SIE m56. These sequences are given in Wagner et al. (Wagner et al., 1990). The ISRE oligonucleotide is derived from the ISG-54 gene (Levy et al., 1988) and is 5'-AATTTCACTTT CTAGTTTCACTTICCCTT TTGT-3' and 5'-AATTACAAAAGGGAAAGTGAAACTAGAAAGTGA-3'. The sequences of the other competitor oligonucleotides are as indicated in Table 1.1 except that they are double stranded with overhanging HindIII termini for labelling. 195 Thus, the IFN-y responsive elements can be divided into two groups -- those that compete for SIF binding and those which do not. One sequence difference between these classes of elements is that those that compete have the sequence TTCC and those that do not compete begin with the sequence TTAC. This sequence is located in the part of the SIE where SIF binding is disrupted by DNA methylation (Hayes et al., 1987). The second difference between these elements is that the GBP and MIG genes contain composite elements with nearby or overlapping ISREs to the IFN-y consensus element (Lew et al., 1991; Wright and Farber, 1991). It may be that interactions between proteins binding this site and proteins binding the ISRE slightly alter the affinity of p91 for these IFN-y responsive elements. Alternatively, a distinct interferon stimulated complex may bind to these sites. In any case, IFN-y induces a binding activity that is capable of binding to the SIE. This complex has a sequence specificity similar to complexes induced by PDGF. Both of these complexes appear to contain the ISGF3-91 kD or closely related protein. However, the SIE binding complexes induced by PDGF and interferon-y are likely to be distinct. That is, the mobility shift band pattern observed on the high affinity SIE differs slightly between extracts of IFN-y treated cells and from those of PDGF treated cells (data not shown). To confirm further that the 91 kD protein which complexes with the SIE is closely related to the 91 kD protein that is part of the ISGF3 complex, we have investigated complex formation on the SIE using extracts of mutant cells which have specific deficiencies in the response to interferons. One of these mutant cells is specifically deficient in expression of mRNAs which encode the 91 and 84 kD proteins of ISGF3 (McKendry et al., 1991; Muller et al., 1993) (G.R. Stark and I.M. Kerr, personal communication). Treatment of the parental 2fTGH cell line with IFN-y induces a complex which binds the high affinity SIE (Figure 1.4). This complex is supershifted by the anti-C-terminal p91 antisera and is specifically competed by unlabelled SIE (data not shown). In the cell line (U3A) which is defective in the expression of the gene encoding the 91 kD protein, no induction can be detected, although a faint constitutive complex is unaffected. In contrast, induction of SIE binding is not affected in the U2A cell line. U2A cells are defective for p48 ISGF3, which mediates binding to the ISRE (Stark and Kerr, personal 196 U2A U3A 2FTGH C) 4~'y '44p SMS S* -I -r hr ~ EIN OW 0nm Mn, d @W UUIIIWI Figure 1.4. Inducible binding to the SIE is diminished in cell lines deficient in ISGF3-p91. Extracts of the 2fTGH cell line and the mutant derivatives U2A and U3A were used in mobility shift assays with the the high affinity SIE probe. In each case, quiescent cells were treated with IFN-a or y or with PDGF-BB. 197 communication). Thus, expression of the p91 gene is required for IFN-y induced complex formation on the SIE. The parental 2fTGH cells do not show a response to PDGF, so it cannot be determined what the effect of eliminating p91 expression is on PDGF inducible complex formation. The growth factor inducible DNA binding complex SIF is thus related to the IFN-y stimulated p91 by size, binding specificity, and immunoreactivity. It is likely that the 91 kD protein of ISGF3 is a component of SIF, although it is possible that the PDGF induced complex contains a closely related protein instead. Moreover, on tris/glycine gels as many as four distinct bands can be observed with the high affinity SIE probe after PDGF stimulation. The multiple complexes formed on the SIE observed on mobility shift gels after induction by IFN-y or PDGF may be due to differences in phosphorylation, the ability to complex with other proteins, as well as binding by other proteins that could be closely related to p91. p91 has previously been shown to be phosphorylated on tyrosine in response to interferon (Schindler et al., 1992b; Shuai et al., 1992). By genetic complementation it has been found that the Jak2 tyrosine kinase is required for the response to IFN-y (Watling et al., 1993). Jak2 is a cytoplasmic tyrosine kinase with two putative tyrosine kinase domains (Harpur et al., 1992). Jak2 has also been found to be tightly associated with the growth hormone receptor in pre-adipocytes and growth hormone induces SIF DNA binding (Argetsinger et al., 1993; Meyer et al., 1993). Thus, the JAK family kinases are candidates for being the ISGF3 and/or SIF kinases although this has not been directly demonstrated. Since IFN-y can induce transient c-fos expression (Wan et al., 1988) and the SIE can mediate c-fos induction by PDGF, it is likely that a pathway involving JAK family kinases will be activated by PDGF and other growth factors as well. We are currently investigating this possibility. 198 Experimental Procedures Maintenance of Cell Lines Balb/c-3T3 cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS). HepG2/PDGF-O3R cells were maintained in DMEM supplemented with 10% FBS and G418 at 500 p.g/ml. U937 cells were grown in suspension in RPMI-193 supplemented with 10% FBS and 2mM L-glutamine. 2fTGH and mutant derivate cell lines (U2A and U3A) were maintained in DMEM supplemented with 10% FBS and 2mM L-glutamine in 10% C02. Prepartion of Nuclear Extracts Cell lines were grown to confluence and subsequently serum starved as follows: Balb/c-3T3, 2fTGH, U2A and U3A cell lines for 48 hours in 0.5% FBS, and HepG2 cells in DMEM alone for 72 hours. U937 cells were grown to high density only prior to stimulation. Cells were then stimulated with either PDGF-BB (25 ng/ml, R&D systems) or IFN-a or y (200 U/ml, recombinant human, Genzyme) for 30' at 37 0 C. Cells were rinsed three times with ice-cold phosphate-buffered saline (PBS). PBS containing 1 mM Na3VO4 and 5 mM NaF was added to each plate, and the cells were scraped from the dish and pelleted at 1500 rpm for 10 min at 40 C. Cells were resuspended in the same buffer and pelleted as before. Then, the pellet was resuspended in 0.8 ml ice-cold hypotonic buffer, transferred to microfuge tubes, and allowed to swell on ice for 15-30 min. The lysate was vortexed vigorously for I min and the nuclei pelleted (14,000 g for 30 s). The nuclear pellets were resuspended in 100-150 gl of high salt buffer and rotated at 40 C for 30 min. The extracted proteins were separated from residual nuclei (14,000 g for 20 min) and the supernatant was quick frozen in a dry ice/methanol bath. The buffer compositions were as follows: 1) hypotonic buffer: 10 mM Hepes (pH 7.9), 10 mM KCI, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 1 mM Na4P207, 20 mM NaF, 1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulphonyl fluoride (PMSF), 0.125 gM okadaic acid, and 1 g.g/ml each of leupeptin, antipain, pepstatin, and chymostatin; 2) high salt buffer: 20 199 mM Hepes (pH 7.9), 420 mM NaC1, 1.5 mM MgC12, 25% glycerol, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 1 mM Na4P207, 20 mM NaF, 1 mM DTT, 0.5 mM PMSF, 0.125 IM okadaic acid, and 1 gg/ml each of leupeptin, antipain, pepstatin, and chymostatin. For mobilityshift assays, nuclear extracts (12 gg) were incubated with or without antisera in binding buffer for 1 h on ice, after which 3 2 P-endlabelled oligonucleotide m67-SIE probe (Wagner et al., 1990) (-30,000 c.p.m., ~5 fmol) was added and the reactions were incubated for 30 min at 30 0 C. Final binding reactions (20 pl) contained: 14 mM Hepes, pH 7.9, 85 mM NaC1, 10 mM KC1, 0.3 mM MgC12,1.25 mM DTT, 0.25 mM EDTA, 0.2 mM EGTA, 15% glycerol, 50 gg/ml poly(dIdC)-poly(dI-dC), and 250 pg/ml acetylated bovine serum albumin (BSA). Binding reactions were analyzed on 5% polyacrylamide gels (39:1 acrylamide:bis) containing 2.5% glycerol in 1X TGE (50 mM Tris, 380 mM glycine, and 2 mM EDTA) buffer at room temperature. For U937 nuclear extracts, mobility-shift reactions contained 150 gg/ml of poly(dI-dC)-poly(dI-dC) and were analyzed on 5% polyacrylamide gels (39:1 acrylamide:bis) containing 2.5% glycerol in 0.5X TBE (45 mM Tris, 45 mM borate, and 1 mM EDTA) buffer at room temperature. Unlabelled competitor oligonucleotides were added to the reactions at 100X molar excess. Crosslinking of Nuclear Extracts Balb/c-3T3 cells were rendered quiescent as described above and treated for 30 min with v-sis conditioned medium (SCM). Nuclear extracts were prepared as described (Wagner et al., 1990). The probe was made from a c-fos SIE oligonucleotide (5'-AGCTTCAGTTCCCGTCAATCAAG CT-3') cloned into the SmaI site of mpl8. A body labeled BrdU substituted probe was prepared and used for UV crosslinking according to the protocol described by Chodosh et al. (Chodosh et al., 1986). 200 Acknowledgments We thank Jessica Schwartz, Christy Carter-Su, George Stark, and Ian Kerr for comunication of unpublished results and comments on the manuscript. We are also grateful to George Stark, Ian Kerr, and Andrius Kazlauskas for the provision of the cell lines used in this study. This work was supported by NIH grant P01-CA42063 to B.H.C. 201 References Argetsinger, L. S., Campbell, G. S., Yang, X., Witthuhn, B. A., Silvennoinen, O., Ihle, J. N., and Carter-Su, C. (1993). Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell 74, 237-44. Chodosh, L. A., Carthew, R. W., and Sharp, P. A. (1986). A single polypeptide possesses the binding and transcription activities of the adenovirus major late transcription factor. Mol Cell Biol 6, 4723-33. Friedman, R. L., and Stark, G. R. (1985). alpha-Interferon-induced transcription of HLA and metallothionein genes containing homologous upstream sequences. Nature 314, 637-9. Fu, X. Y. (1992). A transcription factor with SH2 and SH3 domains is directly activated by an interferon alpha-induced cytoplasmic protein tyrosine kinase(s). Cell 70, 323-35. Garcia-Blanco, M. A., Lengyel, P., Morrison, E., Brownlee, C., Stiles, C. D., Rutherford, M., Hannigan, G., and Williams, B. R. (1989). Regulation of 2',5'-oligoadenylate synthetase gene expression by interferons and platelet-derived growth factor. Mol Cell Biol 9, 1060-8. Harpur, A. G., Andres, A. C., Ziemiecki, A., Aston, R. R., and Wilks, A. F. (1992). JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene 7, 1347-53. Hayes, T. E., Kitchen, A. M., and Cochran, B. H. (1987). A rapidly inducible DNA-binding activity which binds upstream of the c- fos proto-oncogene. J Cell Physiol Suppl 5, 63-8. Herrera, R. E., Shaw, P. E., and Nordheim, A. (1989). Occupation of the c-fos serum response element in vivo by a multi- protein complex is unaltered by growth factor induction. Nature 340, 68-70. Igarashi, K., David, M., Larner, A. C., and Finbloom, D. S. (1993). In vitro activation of a transcription factor by gamma interferon requires a membrane-associated tyrosine kinase and is mimicked by vanadate. Mol Cell Biol 13, 3984-9. Levy, D. E., Kessler, D. S., Pine, R., Reich, N., and Darnell, J., Jr. (1988). Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev 2, 383-93. Lew, D. J., Decker, T., Strehlow, I., and Darnell, J. E. (1991). Overlapping elements in the guanylate-binding protein gene promoter mediate transcriptional induction by alpha and gamma interferons. Mol Cell Biol 11, 182-91. McKendry, R., John, J., Flavell, D., Muller, M., Kerr, I. M., and Stark, G. R. (1991). Highfrequency mutagenesis of human cells and characterization of a mutant unresponsive to both alpha and gamma interferons. Proc Natl Acad Sci U S A 88, 11455-9. Meyer, D. J., Stephenson, E. W., Johnson, L., Cochran, B. H., and Schwartz, J. (1993). The serum response element can mediate induction of c-fos by growth hormone. Proc Natl Acad Sci USA 90, 6721-5. 202 Muller, M., Laxton, C., Briscoe, J., Schindler, C., Improta, T., Darnell, J. J., Stark, G. R., and Kerr, I. M. (1993). Complementation of a mutant cell line: central role of the 91 kDa polypeptide of ISGF3 in the interferon-alpha and -gamma signal transduction pathways. Embo J 12, 4221-8. Pearse, R. N., Feinman, R., Shuai, K., Darnell, J., Jr., and Ravetch, J. V. (1993). Interferon gamma-induced transcription of the high-affinity Fc receptor for IgG requires assembly of a complex that includes the 91-kDa subunit of transcription factor ISGF3. Proc Natl Acad Sci U S A 90, 4314-8. Sadowski, H. B., and Gilman, M. Z. (1993). Cell-free activation of a DNA-binding protein by epidermal growth factor. Nature 362, 79-83. Schindler, C., Fu, X. Y., Improta, T., Aebersold, R., and Darnell, J., Jr. (1992a). Proteins of transcription factor ISGF-3: one gene encodes the 91-and 84- kDa ISGF-3 proteins that are activated by interferon alpha. Proc Natl Acad Sci U S A 89, 7836-9. Schindler, C., Shuai, K., Prezioso, V. R., and Darnell, J., Jr. (1992b). Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor [see comments]. Science 257, 809-13. Shuai, K., Schindler, C., Prezioso, V. R., and Darnell, J., Jr. (1992). Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science 258, 180812. Valius, M., and Kazlauskas, A. (1993). Phospholipase C-gamma 1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor's mitogenic signal. Cell 73, 321-34. Veals, S. A., Schindler, C., Leonard, D., Fu, X. Y., Aebersold, R., Darnell, J., Jr., and Levy, D. E. (1992). Subunit of an alpha-interferon-responsive transcription factor is related to interferon regulatory factor and Myb families of DNA-binding proteins. Mol Cell Biol 12, 3315-24. Verma, I. M., and Sassone-Corsi, P. (1987). regulation. Cell 51, 513-4. Proto-oncogene fos: complex but versatile Wagner, B. J., Hayes, T. E., Hoban, C. J., and Cochran, B. H. (1990). The SIF binding element confers sis/PDGF inducibility onto the c-fos promoter. Embo J 9, 4477-84. Wan, Y. J., Levi, B. Z., and Ozato, K. (1988). Induction of c-fos gene expression by interferons. J Interferon Res 8, 105-12. Watling, D., Guschin, D., Muller, M., Silvennoinen, O., Witthuhn, B. A., Quelle, F. W., Rogers, N. C., Schindler, C., Stark, G. R., Ihle, J. N., and et, a. (1993). Complementation by the protein tyrosine kinase JAK2 of a mutant cell line defective in the interferon-gamma signal transduction pathway [see comments]. Nature 366, 166-70. Wilson, K. C., and Finbloom, D. S. (1992). Interferon gamma rapidly induces in human monocytes a DNA-binding factor that recognizes the gamma response region within the promoter of the gene for the high-affinity Fc gamma receptor. Proc Natl Acad Sci U S A 89, 11964-8. 203 Wright, T. M., and Farber, J. M. (1991). 5' regulatory region of a novel cytokine gene mediates selective activation by interferon gamma. J Exp Med 173, 417-22. 204 CURRICULUM VITAE Leisa Johnson Home Address: Work Address: 73 5th St. Apt. 2 Cambridge, MA 02141 (617)-497-2378 MIT-CCR E17-518 77 Mass. Ave Cambridge, MA 02139 (617)-253-0264 Date of Birth: January 19, 1967 Education: 1989 B.A. Hastings College Chemistry Hastings, NE 1996 Ph.D. Massachusetts Institute of Technology Biology Thesis advisor: Tyler Jacks Cambridge, MA 1988 University of Arizona Cancer Center Advisor: Roger L. Miesfeld Undergraduate summer research position Tucson, AZ 1989 Eppley Institute for Research in Cancer and Allied Diseases Advisor: Jill Pelling Undergraduate research position Omaha, NE 1990-1993 Massachusetts Institute of Technology Graduate student in the laboratory of Brent Cochran who moved to Tufts Medical School in 1993 Boston, MA 1993-1996 Massachusetts Institute of Technology Graduate student in the laboratory of Tyler Jacks Boston, MA Research Experience: Awards: 1985 Valedictorian Academic All-American Academic Scholarship Athletic (volleyball and track) Scholarship 205 1987 Physics Scholarship 1989 summa cum laude Chemical Rubber Plant Co. Outstanding Senior Chemist Teaching Experience: 1988 Organic Chemistry - lab assistant Nurses Organic Chemistry - teaching assistant Hastings College 1991 Molecular Biology, 7.08 - teaching assistant Massachusetts Institute of Technology 1992 Introductory Biology Lab, 7.02 - teaching assistant and director of developmental lab section Massachusetts Institute of Technology Bibliography: Original Reports: 1. Meyer, D.J., Stephenson, E.W., Johnson, L., Cochran, B.H., and Schwartz, J. The serum response element can mediate induction of c-fos by growth hormone. Proc Natl Acad Sci USA 1993; 90: 6721-6725. 2. Johnson, L., Greenbaum, D., Mercer, K., Murphy, E., Schmitt, E., Bronson, R.T., Umanoff, H., Edelmann, W., Kucherlapati, R., and Jacks, T. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Submitted. 3. Johnson, L., Greenbaum, D., Bronson, R.T., and Jacks, T. Analysis of an oncogenic allele of K-ras in the initiation and progression of tumorigenesis in the mouse via a novel approach. In preparation. Abstracts: 1. Johnson, L., Reddy, P.S., and Cochran, B.H. Regulation and characterization of the SIS/PDGF inducible factor. Eighth Annual Meeting on Oncogenes; June 1992. 2. Johnson, L., Reddy, P.S., and Cochran, B.H. Regulation and characterization of the SIS/PDGF inducible factor. Kestone Symposia on Molecular and Cellular Biology, Transcription: Factors, Regulation, and Differentiation; January 1993. 3. Johnson, L., Fu, X.Y., and Cochran, B.H. A component of the SIS/PDGF inducible factor reacts with an antibody against the 91kD subunit of ISGF3. Ninth Annual Meeting on Oncogenes; June 1993. 206 4. Johnson, L., Greenbaum, D., Schmitt, E., Murphy, L., Bronson, R.T., Umanoff, H., Kucherlapati, R., and Jacks, T. Targeted mutation of the K-ras gene in the mouse. Mouse Molecular Genetics; 1995. 207