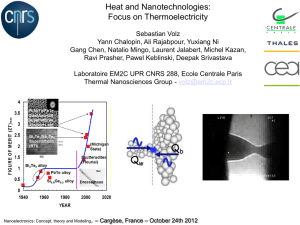
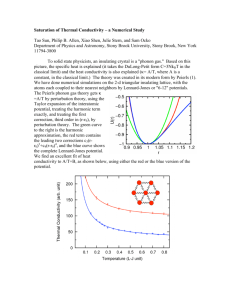
Exploring Heat Transfer at the Atomistic Level for
advertisement

Exploring Heat Transfer at the Atomistic Level for Thermal Energy Conversion and Management ARO-M" by MASSACHUSETTS INSTITUTE OF TECI-HNOLOGY Zhiting Tian AUG 15 2014 Submitted to the Department of Mechanical Engineering in partial fulfillment of the requirements for the degree of LIBRARIES Doctor of Philosophy in Mechanical Engineering at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY June 2014 Massachusetts Institute of Technology 2014. All rights reserved. Signature redacted Author................................ Department of Mechanical Engineering Signature redacted Certified by........... May 9, 2014 .............. Gang Chen Carl Richard Soderberg Professor of Power Engineering Department Head Thesis Supervisor Signature redactedr Accepted by......... David E. Hardt Ralph E. and Eloise F. Cross Professor of Mechanical Engineering Chairman, Department Committee on Graduate Students Exploring Heat Transfer at the Atomistic Level for Thermal Energy Conversion and Management by Zhiting Tian Submitted to the Department of Mechanical Engineering on May 9, 2014, in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Mechanical Engineering Abstract Heat transfer at the scales of atoms plays an important role in many applications such as thermoelectric energy conversion and thermal management of microelectronic devices. While nanoengineering offers unique opportunities to manipulate heat to our advantages, it also imposes challenges on the fundamental understanding of nanoscale heat transfer. As the characteristic lengths of the system size become comparable to the mean free paths of heat carriers, macroscopic theories based on heat diffusion are no longer valid due to size effects. Atomistic level simulation can provide powerful insights into the microscopic processes governing heat conduction, and is the focus of this thesis. In this thesis, we first introduce atomistic techniques to investigate phonon transport in bulk crystals. We start with normal mode analysis within the classical molecular dynamics framework to estimate the spectral phonon transport properties. Although it can provide the detailed phonon properties adequately, classical molecular dynamics with empirical potentials do not always yield accurate predictions. Then, we move to first-principles density functional theory (DFT) to compute mode-dependent phonon properties. Such simulations can well reproduce experimental values of phonon dispersion and thermal conductivity with no adjustable parameters, establishing confidence that such an approach can provide reliable information about the microscopic processes. These detailed calculations not only unveil which phonon modes are responsible for heat conduction in bulk crystals, but also expand our fundamental understanding of phonon transport, such as the importance of optical 3 phonons. Next, we study thermal transport across single and multiple interfaces via the atomistic Green's function method, especially the impact of interface roughness on phonon transmission across a single interface and coherent phonon transport in superlattices. Both the DFT and Green's function techniques provide fundamental parameters that then can be used to understand mesoscale transport. This paves the way for multiscale modeling from first-principles. Through these multiscale modeling efforts, we are able to obtain a comprehensive understanding of heat transfer from the atomistic to the macroscale, with important implications for energy applications. Complementary to the theoretical work, we measure the interface thermal conductance using ultrafast time-domain thermoreflectance experiments, examining thermal transport across solid-liquid interfaces modified by self-assembled monolayers. We find that an extra molecular layer can enhance the thermal transport across solid-liquid interfaces. In summary, theoretical, computational and theoretical approaches have been applied to study heat transfer at the atomistic level. The findings from this thesis have improved our fundamental understanding of phonon transport properties with important implications for energy applications and beyond, and build a foundation for multiscale simulation of phonon heat conduction at the mesoscale. Thesis Supervisor: Gang Chen Title: Carl Richard Soderberg Professor of Power Engineering Department Head 4 Dedication TO MY PARENTS For raising me to believe that girls can achieve anything TO MY HUSBAND For pushing me beyond my limits AND TO MY SON For inspiring me to dream big 5 6 Acknowledgements This thesis owes its completion to the support of numerous individuals. I would like to gratefully acknowledge their help here. First, I would like to thank my advisor, Professor Gang Chen. I am very fortunate to have Gang as my advisor. He has gone far above and far beyond my expectations of an advisor. Despite his super busy schedule, he always makes time for his students. He meets us individually as often as possible, provides comments on manuscripts within a day, spends huge amount of time teaching us how to deliver a good talk, and providing invaluable advice on job search. He called me almost every evening while I was deciding on my faculty job offers. Words cannot express my gratitude to him. I am particularly thankful to Gang for thinking on my feet while suggesting research topics to me, which makes my Ph.D. research a coherent story with a variety of skillsets. Meanwhile, he leaves enough room for me to independently develop my research. For example, when I wrote a proposal on my own and did experiments at Argonne National Lab, he was very supportive. Without him, I would not have been able to secure a faculty position straight from graduate school. Next, I would like to thank my thesis committee members, Millie Dresselhaus, Bora Mikic, Jeffery Grossman, and Evelyn Wang. They took significant amounts of time to give me advice on my research and career paths. They wrote reference letters for my faculty applications and gave me insightful advice on interview and offer decisions. I would like to thank people in the heat transfer committee who have greatly encouraged me. Special thanks go to Alan McGaughey at Carnegie Mellon University who kept an eye on my progress and offered to write reference letter for my job search. I would like to thank several female faculties, in addition to Millie and Evelyn, for setting up a great role model for me: particularly my mentor Dean Christina Ortiz at MIT, Jian Cao at Northwestern University, and Pamela Norris at University of Virginia. The interaction with them in person made me determined to pursue a career in academia. I would like to thank my lab mates in the NanoEngineering group. I am very lucky to work with a great group of people, the NanoEngineering group of MIT. My labmates were also a great resource, and I greatly benefitted from the communication and collaboration with them. In particular, I would like to Asegun Henry for his detailed 7 instruction on normal mode analysis, Sheng Shen, Austin Minnich and Amy Marconnet for their valuable suggestions and help on my job search, Keivan Esfarjani and Junichiro Shiomi for sharing with me their deep knowledge on phonons, Takuma Shiga and Jivtesh Garg for DFT calculations, Maria Luckyanova and Kimberlee Collins for teaching me how to conduct TDTR measurements, Tengfei Luo, Nuo Yang and Yann Chalopin for discussion on molecular dynamics simulations, Bo Qiu and Yuan Yang on other projects not covered in this thesis. Moreover, it has been a big family for me. They witnessed the important moments in my life and shared with me my happiness: getting married and having a baby. They prepared surprise party for my birthday which made me burst to tears. George Ni, Wei-Chun Hsu and Bolin Liao not only helped on my wedding but also help me move my home couple of times. I am very thankful to DOE S3TEC for supporting my research, Argonne National Lab for precious beam time and NSF Teragrid for supercomputer time. My experience at MIT has been made far richer by the terrific friends, especially through Tsinghua Alumni Association and Chinese Students and Scholars Association. Among those countless many, I would like to especially thank Jiexi Zhang and Tengfei Zheng for being my closest friends at MIT and supporting me all the time, especially throughout my pregnancy. I would like to thank Xian Li and Yu Jiang for being my very close friends since I came to the United States in 2007. Finally, above all else, I thank my family whose unwavering love carries me through all of life's adventures. No matter what happens, they are always there. My wonderful parents Yuzhong Tian and Tingrong Liao set the bar high for me and urged me to pursue my dream. My uncle Tingyuan Liao tried his best to support me choosing my path. My life has been more enjoyable accompanied my lovely cousin Shiying Liao. Last but not least, my husband Yan Zhou is the most amazing man I have ever met. His endless love and support makes me happy every day. He pushes me to reach higher. He sacrifices his job for me to accept my best offer. My son Austin Zirui Zhou is the most precious gift I ever have. He is my sunshine. I am extremely lucky to have both Yan and Austin. Having both of them in my life is the biggest achievement in my Ph.D. 8 Contents introduction...................................................................................................... 15 1.1 H eat Conduction by Phonons........................................................................ 15 1.2 Outline of the thesis ...................................................................................... 19 2. Spectral Phonon Properties of Ge Using Normal Mode Analysis ................... 2.1 Introduction.................................................................................................... 21 2.2 M ethodology ................................................................................................. 22 2.3 Results and D iscussion ................................................................................. 24 2.3.1 Phonon Lifetim es................................................................................... 24 2.3.2 Therm al Conductivity Validation........................................................... 25 2.3.3 Contribution from Different Phonon Modes........................................... 27 2.4 Conclusion..................................................................................................... 3. 21 29 Phonon Conduction in PbSe, PbTe and their alloys Using First-Principles Calculations..................................................................................................................31 3.1 Introduction.................................................................................................... 31 3.2 M ethodology ................................................................................................. 34 3.2.1 Harm onic Properties ............................................................................... 34 3.2.2 Anharm onic Properties........................................................................... 35 3.2.3 Lattice therm al conductivity ................................................................... 36 3.2.4 Alloy modeling ...................................................................................... 37 3.3 Results and D iscussion ................................................................................. 38 3.3.1 Com parison w ith experim ental results.................................................... 38 3.3.2 Com parison between PbSe and PbTe ..................................................... 41 3.3.3 The im portance of optical phonons........................................................ 44 3.3.4 The potential im pacts of nanostructuring ............................................... 45 3.3.5 The potential impacts of PbSe-PbTe Alloying ........................................ 47 3.4 4. Conclusion................................................................................................. 48 The Importance of Optical Phonons in Nanostructures ................................... 49 4.1 Introduction.................................................................................................... 9 49 5. 4.2 M ethodology ................................................................................................. 50 4.3 Results and D iscussion ................................................................................. 52 4.4 Conclusion ......................................................................................................... 57 Phonon Transmission across a Single Si/Ge Interface using the Green's Function M ethod .................................................................................................... ... - ....... 59 5.1 Introduction........................................................................................................59 5.2 M ethodology ................................................................................................. 62 5.3 Results and Discussion ................................................................................. 66 5.3.1. Rough interface with random distribution.................................................66 5.3.2. Rough interface with Gaussian distribution........................................... 5.4 Conclusion ...................................................................................................... 6. Phonon Transm ission across Si/Ge Superlattices ............................................. 73 77 79 6.1 Introduction.................................................................................................... 79 6.2 M ethodology................................................................................................. 81 6.3 Results and D iscussion ................................................................................. 83 6.4 Conclusion ...................................................................................................... 93 Solid-Liquid Interface Conductance Using Time-Domain Thermoreflectance 95 M easurem ents .............................................................................................................. 7. 7.1 Introduction.................................................................................................... 95 7.2 Sam ple Preparation and Experim ental Setup.................................................. 96 7.3 Results and D iscussion ................................................................................. 98 7.4 Conclusion ....................................................................................................... 8. 101 Sum m ary and Future Work................................................................................103 8.1 Sum m ary .......................................................................................................... 103 8.2 Future D irections ............................................................................................. 104 10 List of Figures Figure 2-1 Simulation cell of germanium showing two basis atoms per primitive cell. 10a x 10a x 10a is used as the simulation domain. 22 Figure 2-2 [100] Phonon dispersion of germanium from lattice dynamics. Two degenerate transverse acoustic branches, one longitudinal acoustic branch, one longitudinal optical branch and two degenerate transverse optical branches. ............................................................................. 23 Figure 2-3 From autocorrelation of normal mode energy to phonon lifetimes ........................................................................................................ . . 24 Figure 2-4 Phonon lifetimes of the transverse acoustic (TA), longitudinal acoustic (LA), transverse optical (TO) and longitudinal optical (LO) m odes in the [100] direction .......................................................... 25 Figure 2-5 Phonon density of states for each polarization: TAl, TA2, LA, TO 1, TO2 and LO .......................................................................... 26 Figure 2-6 Ensemble average of EMD simulations using Green-Kubo form ula............................................................................................ 27 Figure 2-7 Contributions from different crystallographic directions and different polarizations..................................................................... 28 Figure 2-8 Thermal conductivity accumulation with respect to the phonon m ean free paths.............................................................................. 28 Figure 3-1 Phonon dispersion for PbSe and PbTe: red lines: calculated results; black dots: experimental results .......................................... 39 Figure 3-2 Temperature dependent lattice thermal conductivity of PbSe and PbTe, red lines: calculated results; black crosses: experimental data..41 Figure 3-3 Frequency dependent phonon lifetimes of PbSe (squares) and PbTe (crosses) at 300 K: (a) TA, (b) LA, (c) TO, and (d) LO. ...... 42 Figure 3-4 Frequency dependent phonon group velocities of PbSe (squares) and PbTe (crosses) at 300 K: (a) TA, (b) LA, (c) TO and (d) LO........43 Figure 3-5 Thermal conductivity from different polarizations (TAl, TA2, LA, TOl, T02 and LO) versus temperature for PbSe and PbTe ......... 44 Figure 3-6 Temperature dependence of lattice thermal conductivity without acoustic-optical scattering: PbSe (black dashed line), PbTe (red dashed line) and with acoustic-optical scattering: PbSe (black solid line), PbTe (red solid line) ................................................................................. 45 Figure 3-7 Cumulative thermal conductivity with respect to phonon mean free path at 300 K for PbSe (red dashed line), PbTe (black solid line) 11 and PbTeo.5Seo.s (blue dotted line).................................................... 46 Figure 3-8 Calculated composition dependence of the lattice thermal conductivity in PbTe..xSex at 300 K (solid line) and 500 K (dashed line) 47 .............................................................................................................. Figure 4-1 Cumulative thermal conductivity with respect to MFPs at 277K from the 18 x 18 x18 k-mesh data; Inset (a)-Inset (b) Zoomed-in figures for MFP range of (a) 0-70nm and (b) 0-9nm, respectively......52 Figure 4-2 Thermal conductivity of silicon nanowires for d=37nm, 56nm and 11 5nm, lines: calculated results; crosses, squares and stars: experim ental results........................................................................ 53 Figure 4-3 Thermal conductivity from different polarizations versus temperature for d=20nm; (b) normalized optical phonon contributions to the total thermal conductivity versus temperature for d=1 Onm, 20nm, 55 100nm and lm m ............................................................................ Figure 4-4 (a) The thermal conductivity from different polarizations versus diameters at 277K, (b) the normalized optical phonon contributions to the total thermal conductivity vs. diameters at 100K, 177K, 277K, and 56 400K ..................................................................................................... Figure 5-1 The system is divided into three parts: left (L), center (C) and right (R). The left and right leads are semi-infinite crystal lattices. In the transverse direction, all the three regions have periodic boundary conditions imposed to represent the infinitely large lateral dimension. 63 .............................................................................................................. Figure 5-2 Total transmission function, transmittance and thermal conductance as a function of phonon frequency for an ideal Si/Ge interface (solid black line) and for a random rough Si/Ge interface (colored dashed or dotted lines): (a) Total transmission based on SW force constants; (b) Transmittance from Si to Ge based on SW force constants; (c) Thermal conductance based on SW force constants; (d) Total transmission based on DFT force constants; (e) Transmittance from Si to Ge based on DFT force constants; (f) Thermal conductance based on DFT force constants......................................................... 70 Figure 5-3 Phonon Density of States (DoS) of pure Si (Black solid line), pure Ge (red dashed line) and Si/Ge 1:1 mixture (green dotted line) using DFT force constants. ............................................................ 72 Figure 5-4 (a)Total transmission function, (b)transmittance, and (c) thermal conductance as a function of phonon frequency for an ideal Si/Ge interface (solid black line) and for a rough Si/Ge interface with a 12 Gaussian distribution (dashed blue lines) based on DFT force constants. Inset of (c): The number of Si atom in each layer for an ideal interface (solid black) and for a Gaussian rough interface (dashed blue)...........75 Figure 5-5 Thermal conductance ratio of a Gaussian rough interface to an ideal interface as a function of the mass ratio (lower x-axis) and the acoustic impedance ratio (upper x-axis) of the two materials using DFT force constants ................................................................................ 76 Figure 6-1 Schematic of the system setup: the left reservoir is pure Si, the right reservoir is pure Ge, the center region is the Si/Ge superlattice. 82 Figure 6-2 (a) Thermal conductivity of superlattices as a function of number of periods for smooth and rough superlattices at T=300K. (b) Transmittance as a function of frequency for superlattices (period =2a) with smooth interfaces; (c) Transmittance as a function of frequency for superlattices (period=2a) with rough interfaces........................86 Figure 6-3 Phonon dispersion of Si/Ge superlattice with period length = 2a in [100] direction............................................................................. 87 Figure 6-4 The total thermal conductivity, contribution from phonons with frequencies larger than the cutoff frequency, and not larger than the cutoff frequency at (a) T=20K, (b) 50K and (c) 300K....................89 Figure 6-5 Normalized thermal conductance per interface as a function of number of periods for rough-interfaced superlattices with period length 1 = 2a. The experimental value is extrapolated from the sample of period length 1 = 4.4nm and 100 periods.....................................90 Figure 6-6 Thermal conductivity of rough-interfaced superlattices as a function of superlattice length for period length 1 = a, 2a and 4a at 300K ............................................................................................... 93 Figure 7-1 Schematic of SAMs used in this study. (a) Hexenedithiol SHCH 2(CH 2) 4CH2SH; Undecanethiol (b) Hexanethiol CH3(CH 2)9CH2 SH; and CH3(CH 2) 4CH2SH; (d) (c) Hexadecanethiol CH3(CH2)1 4CH2 SH .......................................................................... 96 Figure 7-2 (a) Schematic of sample arrangement; (b) Phase data for samples without SAM and with hexanedithiol............................................ 98 Figure 7-3 Thermal conductance between Au and ethanol with and without SAMs from TDTR measurements at room temperature..................99 Figure 7-4 Contact angles, 0, of ethanol on Au surface modified by (a) hexanethiol and (b) hexanedithiol SAMs. ......................................... 13 100 14 1. Introduction 1.1 Heat Conduction by Phonons Emerging nanotechnological applications have necessitated reliable quantitative understanding of thermal transport. There are fundamental differences between heat transfer processes at the nanoscale and the macroscale due to quantum and classical size effects; for example, both the Fourier law for heat conduction and Planck's law for blackbody radiation break down in nanostructures. This makes modeling the nanoscale heat transfer challenging. In semiconductors and insulators, heat is carried primarily by vibrations in the crystal lattices known as phonons. A comprehensive understanding of phonon transport will facilitate design of innovative nanostructured materials and devices for thermal energy applications and beyond. Understanding heat conduction in crystalline solids started only when the quantum theory of lattice vibrations was developed and the phonon concept was established. The basic quantum of crystal lattice vibration is called a phonon. The phonon scattering originating from the anharmonic interatomic potential can be classified as either a normal process which conserves crystal momentum or an umklapp process which does not. It was recognized that the umklapp process creates a resistance to heat flow while the normal process only redistributes phonons. Peierls I extended the Boltzmann formulation for phonon transport taking into consideration only the umklapp process. Under the relaxation time approximation and assuming isotropic group velocity and phonon lifetime, the Boltzmann equation led to the kinetic theory expression for thermal conductivity as Kp= m. C(o) v(a)A(co)do = - C(C)V2(c)(c)do (1.1) where CQo) = hOD(o)dfBE / dT is the specific heat per unit frequency interval at frequency o and temperature T, D(w) is phonon density of states per unit volume 15 and per unit frequency interval, fBE is the Bose-Einstein distribution, V is the phonon group velocity, A = vr is the phonon mean free path (MFP) and r is the phonon lifetime. Callaway further modified the theory by accounting for both the normal and umklapp scattering processes, assuming that normal processes lead to a displaced Bose-Einstein distribution and indirectly affect the umklapp processes. The derived expression is similar to equation (1.1) except for a modification to phonon lifetimes. The quantities in equation (1.1) that determine the lattice thermal conductivity, however, are frequency dependent and difficult to obtain. Different approximations were made to perform the thermal conductivity integral. First, the Debye approximation that assumes a linear relation between the phonon frequency and the propagation wavevector is often used for the phonon group velocity and density of states, based on the fact that the Debye approximation has been very successfully used to explain the specific heat of crystalline materials. This left the phonon lifetime unknown. In the 1950s, Klemens 2 derived expressions using the quantum perturbation theory, i.e., Fermi's golden rule, for phonon lifetimes due to different scattering mechanisms. The expressions obtained are again based on the Debye approximation and contains unknown parameters. The experimental values of the thermal conductivity, speed of sound, and specific heat are often used to fit the unknown parameters in the Debye model and in the lifetimes. Such fittings usually work well at low temperatures. Deviations at high temperatures stimulated more refined models of dispersion, such as the one developed by Holland 3 that used two different linear dispersions to better represent the rapid flattening of transverse acoustic phonons in FCC crystals such as GaAs and Si. The shape of the temperature dependence of thermal conductivity is universal among crystalline solids. At high temperatures, the thermal conductivity usually decreases with increasing temperature as T-", with theoretically n=I although practically n=1 -1.5. This is because at high temperatures, phonon specific heat is a constant according to the Pettit-Delong law, and phonon energy, i.e. the number of phonons, increases linearly with temperature. Since the scattering rate is proportional to the number of phonons, the thermal conductivity decreases with increasing temperature. At low temperatures, the thermal conductivity is usually proportional to T3 . In this regime, phonon-phonon scattering is weak and phonon mean free paths (MFPs) are longer than the size of the sample. Phonons scatter more frequently with the boundaries and the phonon MFP is effectively equal to the sample size, and is independent of frequency. Thermal conductivity is thus proportional to the specific heat and hence the T3 behavior. This size effect was first discovered by De Haas and Biermasz 4, and explained by Casimir 5 (sometimes called the Casimir regime). Casimir assumed that 16 surfaces of samples are rough, and scatter phonons diffusely, although latter studies also investigated partially diffuse and partially specular surfaces 6. In between low and high temperatures, impurity scattering is usually important, and the peak value of the thermal conductivity depends sensitively on impurity concentrations. Although the approaches pioneered by Klemens were successful in explaining the trend of experimental observations, quantitative details of phonon transport may be far from what the past approaches described above predicted, especially in terms of the phonon lifetimes and MFPs. The reason that the Debye model together with Klemens' treatment on scattering can fit experimental data is because the thermal conductivity integral is quite forgiving. In fact, one can use different sets of parameters to fit the experimental data based on different approximations and scattering mechanisms. Extracting the exact phonon transport properties, especially phonon lifetimes and MFPs, thus remained unsolved. Since 1980s, efforts started on calculating thermal conductivity of crystals using molecular dynamics (MD) simulations 711. In classical MD, the approximate trajectories of each individual atom within the simulation domain are tracked based on an empirical interatomic potential and Newton's second law. Two prevailing methods used to study heat transport are equilibrium molecular dynamics (EMD) and nonequilibrium molecular dynamics (NEMD). EMD is suitable for transport properties, whereas NEMD simulates actual transport processes. EMD first obtains the history of individual particles in an equilibrium system, from which the transport properties are extracted on the basis of linear response theory. The thermal conductivity is calculated from the autocorrelation of the instantaneous heat flux through the Green-Kubo formula 1, 1. For NEMD, one can either impose a temperature difference to calculate the heat flux 14, the resulting temperature distribution The thermal conductivity is then 15, 16. or impose a heat flux to calculate determined by the Fourier law. The NEMD methods are relatively easy to implement and are usually faster than the EMD methods because the latter requires the calculation of the autocorrelation function, which can take a long time to decay. In addition, the EMD method may involve the artificial autocorrelation caused by the often-used periodic boundary condition. However, NEMD also suffers from several drawbacks. Firstly, the statistical foundation of NEMD is not as soundly established as that of EMD 1. Second, the finite simulation size in NEMD might be shorter than the MFP of some phonon modes, leading to artificial size effects with boundaries imposed at the heat reservoirs. Third, a large temperature difference is applied across a small simulation domain. 17 To extract the phonon lifetimes and MFPs, the phonon spectral energy density analysis or normal mode analysis 18-21 has been applied in combination with EMD. The key idea is to project the atomic displacements onto normal mode coordinates and to determine the phonon lifetimes by tracing the temporal amplitude decay of each mode or fitting the width of the spectral energy density peaks in the frequency domain. To apply this approach, one needs to utilize eigenvalues and eigenvectors from lattice dynamics calculations and to perform separate analysis from the traditional thermal conductivity calculations using MD. As a widely adopted simulation tool, MD simulations do not require any a priori knowledge of the phonon transport properties, are straightforward to implement, and automatically include the temperature-dependent anharmonicity. Yet MD is only rigorously applicable to solids above the Debye temperature and being entirely classical MD assumes each vibrational mode is equally excited. At low temperatures, quantum corrections for temperature-related effects are needed. The electronic contribution to thermal transport cannot be assessed in MD. Moreover, the empirical potentials used in classical MD can cause the thermal conductivity to deviate significantly from the experimental data 2 2 Conversely, first-principles calculations where the potential is obtained directly from the electron charge density via density functional theory (DFT) without any adjustable parameters provide the most reliable simple way of computing the lattice thermal conductivity. Ab inito MD simulations 23,24 use the forces computed on-the-fly by DFT and are computationally expensive, though. The alternative way is to extract the interatomic force constants from DFT calculations for limited atomic configurations and to then conduct further calculations. After DFT calculations, one can either obtain the force constants from reciprocal space based on density functional perturbation theory (DFPT) 25,26 or from real space calculations by fitting the force-displacement data in a supercell with a polynomial potential 27, 28. Despite the fact that the real space approach is simpler yet less precise, both approaches are accurate enough to reproduce the experimental results for the lattice thermal conductivity. Broido et al. 25 calculated the intrinsic lattice thermal conductivity of Si and Ge using the reciprocal space approach and obtained excellent agreement with experimental data. The reciprocal lattice approach was later applied to SixGei.x alloys and superlattices by Garg et aL. 29,30, and to PbSe, PbTe and PbTev.xSex by Tian et al. started the real space approach with Si half-Heusler 32, 28, 31. Esfarjani et al. and then extended the approach to PbTe 33 and GaAs 34. Once the harmonic and anharmonic force constants are obtained, one can either 18 perform MD simulations based on the developed Taylor expansion potential 32 or employ lattice dynamics calculations: first obtain the vibrational eigenmodes based on then compute the scattering rates of each mode the harmonic part of the potential, by treating the anharmonic potential as a perturbation using Fermi's golden rule, and solve the Boltzmann transport equation (BTE) to find the thermal conductivity 25,28-33 Although MD simulations based on fitted potentials have more flexibility to directly obtain the lattice thermal conductivity for complicated structures, lattice dynamics calculations can produce the detailed phonon transport properties without extra efforts because the thermal conductivities are the integrated quantities over the first Brillion zone using either the solution of BTE under the single-mode relaxation time approximation 28,29, 31-33,35 or based on an iterative solution of the integral BTE 25 1.2 Outline of the thesis This thesis focuses on phonon transport properties using emerging computational tools at the atomistic level to reveal the microscopic origin of the thermal conductivity of a solid. Chapters 2 and 3 deal with phonon transport properties in bulk materials, while Chapters 5 and 6 focus on phonon transmission across interfaces. Complementary to the theoretical work, experimental measurements are covered in Chapter 7. In Chapter 2, we introduce normal mode analysis which utilizes a combination of molecular dynamics and lattice dynamics to capture the spectral phonon transport properties of germanium. The contribution of different phonon polarizations and phonons with different mean free paths are estimated. In Chapter 3, we describe in detail first-principles calculations of phonon conduction in PbSe, PbTe and their alloys. The calculated phonon dispersion and thermal conductivity agree very well with experimental data. Mode-dependent phonon transport properties are extracted. The origin of the low thermal conductivity, the mean free path distribution and the importance of optical phonons are discussed. In Chapter 4, we examine the importance of optical phonons to thermal conductivity in nanostructures. Silicon nanowires are chosen as one example. In nanostructures, acoustic phonons with long mean free paths are strongly scattered at interfaces/boundaries, the optical phonons are much less influenced. This leads to a rebalance of the relative importance between acoustic and optical phonons. 19 In Chapter 5, we apply the Green's function method to study phonon transmission across single Si/Ge interfaces. We evaluate the effects of interface roughness and find that the roughness introduced by atomic mixing can enhance phonon transmission in a certain range through a smoother transition in the phonon vibrational spectrum. In Chapter 6, we apply the Green's function method to multiple Si/Ge interfaces, namely Si/Ge superlattices, and demonstrate that the thermal resistance cannot always be taken as a sum of individual interfaces. Coherent phonon transport is thus observed under certain conditions. In Chapter 7, we discuss about our experimental endeavors on thermal transport across a solid-liquid interface. The interface thermal conductance at a gold-ethanol interface modified by self-assembled monolayers is measured using time-domain thermoreflectance techniques. Finally, Chapter 8 lists potential future work and concludes the thesis. 20 2. Spectral Phonon Properties of Ge Using Normal Mode Analysis 2.1 Introduction Size effects are highly important in the micro/nanoscale regime where the phonon mean free paths become comparable to the device length and the thermal conductivity can decrease by several orders of magnitude. 17, 3640 To understand and manipulate thermal transport at these small length scales, knowledge of phonon mean free paths is required. However, the direct calculation of phonon mean free paths has been neglected for many years. The major challenge is to determine phonon lifetimes because most existing models2 are semi-empirical and a large uncertainty remains in fitting multiple parameters. To extract phonon lifetimes and MFPs, the phonon spectral energy density analysis or the normal mode analysis1 8 -21 have been developed. In the normal mode analysis, atomic vibrations from a MD trajectory are decomposed into vibrational eigenstates, or into so-called normal mode coordinates via lattice dynamics (LD) calculation. The lifetimes can then be extracted from the temporal decay of the normal mode energies. With knowledge of the phonon frequency, group velocity and lifetime of each mode, we can use the Boltzmann transport equation (BTE) to calculate the total thermal conductivity. Comparing these results with the thermal conductivity calculation based on the Green-Kubo formula, the methodology can be validated. More importantly, based on the mode-dependent lifetimes and mean free paths, we could fully detail the phonon contributions from different polarizations, frequencies and wavelengths. In this chapter, we perform normal mode analysis for germanium, one of the most important semiconductors, to fully detail the spectral dependence of the phonon transport properties in bulk germanium. The contributions of different phonon frequencies and polarizations to the thermal conductivity are discussed. 21 2.2 Methodology The detailed methodology of normal mode analysis can be found elsewhere.1'19 In short, the MD trajectory is transformed to normal mode coordinates via LD calculations. Equation (2.1) is used to calculate the normal mode amplitudes. A(I, p,t) = ( *(j,kp)-ii(jl,t) -exp(-ik -r(fl)) )11 (2.1) where e is the polarization vector obtained from LD calculations using GULP, and U' is the displacement of each atom recorded in EMD using LAMMPS . The normal mode energy is calculated using equation (2.2) E(kp,t) = -O2A(k, pt)- A*(k, p,t) + -A(k, p,t) - A*(k, p,t) 2 2 (2.2) The phonon lifetimes are then determined by equation (2.3)7' 18. f(E(k, p, t)&E(k, p,0))(23 _.(2.3) r~,p= 0 ) (&E2(k, p,0)) Detailed simulation procedures are described below. First, we use the StillingerWeber potential 42, 43 and construct a 10a*10a*10a simulation size with periodic boundary conditions in three dimensions as shown in Fig. 2-1. The system is maintained at 300K. The two basis atoms, denoted in different colors, are distinguished in order to perform the correct projection. Figure 2-1 Simulation cell of germanium showing two basis atoms per primitive cell. 10a x 10a x 10a is used as the simulation domain. 22 Second, phonon dispersion curves are obtained by LD calculations along three high symmetry lines [100], [110] and [111] within the First Brillouin Zone, although only [100] direction is shown in Fig. 2-2. There are six polarizations or phonon branches: two transverse acoustic (TA) branches, one longitudinal acoustic (LA) branch, two transverse optical (TO) branches and one longitudinal optical (LO) branch. The allowed wave vectors are discrete points due to the periodic boundary conditions applied over a finite number of unit cells. Third, we trace the evolution of each phonon mode in the EMD simulations. As shown in Fig. 2-3, from the autocorrelation of the normal mode energy, the frequency of this mode can be identified via the Fourier transform. Because energy autocorrelation is performed, the frequency thus obtained would be twice the mode frequency. We could compare this frequency with the eigenvalue we obtained from LD as a quick check whether this is the desired mode. Then, by fitting the peaks of the autocorrelation with an exponential function, the denominator of the power gives phonon lifetime of this mode. 12 .. .-7 10T 8- LO S6- 2- TA 0.5 1 k [2it/a] Figure 2-2 [100] Phonon dispersion of germanium from lattice dynamics. Two degenerate transverse acoustic branches, one longitudinal acoustic branch, one longitudinal optical branch and two degenerate transverse optical branches. 23 Autocorrelation of Normal Mode Energy 0.21 0.15 0) *--6.69THz Fourier Transform E <0 .5 0.1 0.05 N 0 z 0 0o S 0 2 50 100 150 -5 Time s k*1/ S0.8 5 10 15 20 Frequency fTHzI MD Simulation Data -Fitted Curve - E 0.6 xp(4 N! 0.4 cc E 05s 00 50 0 05. 1 1.5 2 100 Time rosl 150 200 Figure 2-3 From autocorrelation of normal mode energy to phonon lifetimes 2.3 Results and Discussion 2.3.1 Phonon Lifetimes We run five independent simulations at T=300K and then take the average. The [100] relaxation times are shown in Fig. 2-4. Similarly, we obtained the relaxation times for the [110] and [111] directions. The error bars denote the standard deviation from five runs. Acoustic relaxation times exhibit strong frequency dependence at the low frequencies. For low frequency modes, their lifetimes follow the (o-2 , consistent with Klemens' prediction. 2 For higher frequency acoustic modes and optical phonons, however, their lifetimes differ significantly from this trend. This reiterates the importance and necessity of extracting the detailed phonon properties. In addition, acoustic relaxation times are about one order of magnitude higher than optical relaxation times. 24 TA LA E 2 102 C 0 C 0 101 101 100 Frequency [THz] 10 0A TO CL 102 E L 10 C 0 101 C 0 101 7 10 9 8 11 Frequency [THz] Figure 2-4 Phonon lifetimes of the transverse acoustic (TA), longitudinal acoustic (LA), transverse optical (TO) and longitudinal optical (LO) modes in the [100] direction 2.3.2 Thermal Conductivity Validation The thermal conductivity is first retrieved using the BTE approach by integrating over the whole spectrum as expressed in equation (2.4), k "d "CP P 25 2.--dv (2.4) dE where C(v) =-= dT h v,. D(v) - df0 dT7, in which fo is the Bose-Einstein statistics and v is the group velocity for each mode which can be calculated from the derivative of the phonon dispersion. Note that D(v) is the polarization dependent density of states instead of the overall density of states. We calculated the polarization dependent density of states, as shown in Fig. 2-5, using lattice dynamics by sampling the whole first Brillouin zone with 50 x 50 x 50 k-points and we count the number of states in each frequency interval for each branch. The thermal conductivity obtained from the BTE approach is 171.5 W/m/K. 0.-02 .i) -- TA1 - ft-TA2 i -lLA 0.015 LO NI TO1 a) TO2 0.01 E 0 & --- E -a I tILI t! 0.005 0 ) 2 8 4 6 Frequency [THz] 1 10 Figure 2-5 Phonon density of states for each polarization: TAl, TA2, LA, TOl, T02 and LO To validate the results, Green-Kubo simulations12 44 based on linear response theory are performed. The thermal conductivity is computed via an autocorrelation of the heat flux using EMD. For each ensemble, we run 5 ns and calculate the auto-correlation of heat flux in 200ps. By taking ten ensemble averages, the thermal conductivity was found to be 175.5 W/m/K as shown in Fig. 2-6. 26 300 E 250200 - Ensemble Average 150- . 0 -100- 50- - .c 0 0 50 100 150 Time [ps] 200 250 Figure 2-6 Ensemble average of EMD simulations using Green-Kubo formula The agreement of the two approaches (171.5 W/m/K from BTE and 175.5 W/m/K from Green-Kubo formula) confirmed the reliability of the calculation procedures. However, both values are much higher than the experimental result of 60.2 W/m/K . due to the inaccurate phonon properties given by the Stillinger-Weber potential22 Although the absolute value is too high, it is still worth looking at the relative values and defining the contributions from different polarizations. 2.3.3 Contribution from Different Phonon Modes As shown in Fig. 2-7, the thermal conductivity in the [100] direction is largest while that in the [111] direction is smallest. Moreover, a major obstacle to the analytical study of phonon-phonon scattering has been the relative scaling of the contributions from different polarizations. Fortunately, our BTE approach could provide a clear picture of the contributions. The LA mode comprises about 40%, while the TAl and TA2 modes comprise about 50% and the LO comprises about 10%. With all the relaxation times, we could calculate the phonon mean free paths in the frequency domain. Thus, the accumulative contribution to the thermal conductivity from different phonon mean free paths can be determined as shown in Fig. 2-8. At room temperature, phonons with MFPs between 100 nm and 10 micron, comprise about 80% of the thermal conductivity. 27 [100] M[110] 0[111] 100 - a 50 - O Average E ------ 0 LA 50.00% TA1 TA2 TO1 LO L 40.70% ' ~ ~ T02 Average 22.40% 26.20% 9.45% 0.00% ---J- LA TA1 TA2 H- 0.58% 0.25% LO TO1 T02 Figure 2-7 Contributions from different crystallographic directions and different polarizations 1n" F 10 e Q 20 300K k 20 Ns 10 10-2 -02 Mea Fuue PaM (pm) Figure 2-8 Thermal conductivity accumulation with respect to the phonon mean free paths 28 2.4 Conclusion Normal mode analysis is applied to calculate phonon lifetimes while nonlinear dispersion and temperature dependent anharmonicity are included in the modeling. The spectral dependence of phonon transport prosperities was fully detailed. Contributions from different polarizations were provided based on the relaxation times extracted along different polarizations: LA ~ 40%, TA1+TA2 ~ 50%, LO -10%. Contributions from phonon mean free paths were exhibited: Phonons with mean free paths between 1 00nm and 10 micron comprise about 80% of the thermal conductivity. While these results provide detailed phonon transport properties, it should be mentioned that the empirical potential limits the accuracy of a quantitative prediction and thus the results should be taken more qualitatively. 29 30 3. Phonon Conduction in PbSe, PbTe and their alloys Using First-Principles Calculations 3.1 Introduction Although the normal mode analysis is able to provide mode-dependent properties within the classical molecular dynamics framework, the empirical potentials employed in these studies put question marks on the quantitative predictions. Since empirical potentials are fit to experimental properties of materials i.e., crystal structure, elastic constants, these potentials do not always yield accurate predictions for the thermal properties of a specific material. Therefore, there is no assurance that they would deliver an accurate microscopic picture such as for the mode-dependent phonon transport properties, as the germanium example shows in the previous chapter. The interatomic potential can be obtained directly from the electron charge density via density functional theory (DFT) without any adjustable parameters, thereby providing a most reliable way of computing the lattice thermal conductivity. However, ab inito MD simulations 23, 24 use the forces computed on-the-fly by DFT and are computationally expensive. The alternative way is to extract the interatomic force constants from DFT calculations for a limited number of atomic configurations and then to conduct further calculations. After DFT calculations, one can either obtain the force constants from reciprocal space calculations based on density functional perturbation theory (DFPT) 25, 26 or from real space calculations by fitting the force-displacement data in a supercell with a polynomial potential 21,28. Despite that, the real space approach is simpler yet less precise, and both approaches are accurate enough to reproduce the experimental results for the lattice thermal conductivity.25,28 31 Thermoelectric materials are of great interest for their potential in converting heat into electricity 45-49. The efficiency of thermoelectric power generators is determined by the dimensionless figure-of-merit zT ( zT = S 2 o.T / k , where S is the Seebeck 2 coefficient, a is the electrical conductivity, S a is the power factor and k is the thermal conductivity). First-principles calculations on some thermoelectric materials show that phonons have a wide mean free path (MFP) distribution, and hence 28 relatively large nanostructures can reduce their lattice thermal conductivity , 32, 49 Semiconducting lead chalcogenides, such as PbSe and PbTe, are attractive thermoelectric materials for intermediate temperature (600-800 K) applications47. Significant efforts have been made to enhance the zT value of PbTe 46-5. By introducing resonant states, TI doped p-type PbTe resulted in a high zT value of 1.5 at 773 K48. Non-resonant doping can also lead to zT-1.3 around 700 K in K or Na doped p-type PbTe 3 . Through band engineering to converge the valence bands, an extraordinary zT value of 1.8 at about 850 K was reported for doped PbTei.,Sex alloysso. Heremans et al.' observed an enhancement of the Seebeck coefficient in PbTe with nanograins. As the sister material of PbTe, PbSe has received much less attention although Se is more abundant and PbSe may offer an inexpensive alternative to PbTe especially for high temperature power generation. A recent calculation by Parker and Singh 56, predicted that heavily doped PbSe may reach zT ~ 2 at 1000 K due to the flattening of the valence band. The experiments5 7,5 8 later reported that the zT values could reach 1.2 and 1.3 at 850 K for heavily doped p-type and Al doped n-type PbSe, respectively. Past efforts in increasing the zT of PbTe and PbSe have mostly been based on improving the power factor S 2 .. Another approach to improve zT -is to reduce the lattice thermal conductivity without substantially sacrificing the electronic properties. Previous studies 5 9-6 1 demonstrated the effectiveness of the nanostructuring in suppressing the lattice thermal conductivity and thus improving zT. Most of the recent experimental studies on the strong reduction of the lattice thermal conductivity in nanostructured PbTe 54' 55 emphasized the importance of dislocations, nanoscale precipitates and strain while pointing out that the mere presence of nanostructuring cannot sufficiently increase the phonon scattering. He et al.5 2 found that not all nanostructures favorably scatter phonons. A necessary condition for the nanostructures to be effective in scattering phonons is to have their characteristic lengths, such as nanoparticle diameter and/or interparticle spacing, to be comparable or less than the MFP. Recent first principles calculations have shown that the MFP distribution is much narrower for PbTe3 3 , and thus, further characterizations of the distributions and the associated detailed heat conduction of lead chalcogenides are important for better materials' design. For example, the extracted MFPs from our 32 calculation can be combined with the Monte Carlo sampling of phonon free paths 62 to predict the thermal conductivity of the nanostructures of the lead chalcogenides. Besides nanostructuring, alloying may be another approach to reduce the lattice , thermal conductivity. Previous experimental and theoretical studies on Si-Ge alloys 29 63 have found a dramatic decrease in the lattice thermal conductivity from pure Si and Ge. There are still few reports on PbTe.xSe., and they only cover partial composition ranges (x<0.3). Based on the limited experimental data on the bulk PbSe-PbTe alloy6 4 p-type PbSe-PbTe alloy 0', 6s, and PbSe-PbTe nanodot superlattice66, the reduction in the lattice thermal conductivity is mild compared to that in Si-Ge alloys. The first principles calculation of the lattice thermal conductivity for PbSe-PbTe alloys over the whole composition range would allow us to better estimate the impacts of alloying. Despite the highly symmetric rock-salt structure of PbSe and PbTe, the lattice thermal conductivities reported in experiments were as low as 1.7-2.2 W/mK at 300 K4758 67-69. The first principles calculations are useful to gain insight into the low heat conduction, with the capability of accurately capturing the transport properties of each phonon mode, including the optical modes. In most bulk materials, the optical phonons are ignored for the lattice thermal conductivity calculation 70 . For instance, the optical phonons contribute only 5% of the lattice thermal conductivity in bulk silicon at room temperature 8,25,28,71. When the system size reaches the nanoscale, the optical phonons can contribute about 20% as discussed in Chapter 472. Another perspective to examine the importance of optical phonons is the acoustic-optical scattering, as described by Ward and Broido 73 . They removed the optical phonons in their calculations and observed an over three times increase in the lattice thermal conductivity for Si. The large anharmonicity of optical phonons was emphasized by the Oak Ridge group in PbTe to address the low thermal conductivity74. In this chapter, we explore the detailed phonon transport properties in PbSe and PbTe to gain more guidance for the thermoelectric applications of these materials systems. We first calculate the harmonic and anharmonic force constants from density functional perturbation theory (DFPT) calculations75-77. The anharmonic phonon lifetimes are then obtained based on Fermi's golden rule. The total lattice thermal conductivity is determined under the relaxation time approximation by summing up the contribution from each mode. Our results are validated by comparing them with the reported experimental data. We present a detailed analysis and we quantify contributions from different phonon modes to the thermal conductivity for both PbSe and PbTe, and discuss the importance of optical phonons and the potential impacts of 33 nanostructuring and alloying on further lattice thermal conductivity reduction in both materials systems. The results indicate that: 1) the optical phonons are important not only because they directly comprise over 20% of the lattice thermal conductivity, but also because they provide strong scattering channels for acoustic phonons, which is crucial for the low thermal conductivity; 2) nanostructures of less than -10 nm are needed to reduce the lattice thermal conductivity for pure PbSe and PbTe; 3) alloying should be a relatively effective way to reduce the lattice thermal conductivity. 3.2 Methodology Accurate interatomic force constants (IFCs) are crucial for the lattice thermal conductivity calculation. We adopt the DFPT approaches for both PbSe and PbTe. DFPT approaches have demonstrated unparalleled accuracy in reproducing the lattice thermal conductivity 25 , 29, 30 and are sufficiently computationally affordable for the simple rock-salt structure with only 2 atoms in the primitive cell. More specifically, in our work, both the harmonic and anharmonic IFCs are obtained based on DFPT calculations implemented in the Quantum Espresso package7 1. In the ground-state calculations, the newly developed norm conserving fully relativistic pseudopotentials which incorporate the spin-orbit interaction (SOI) effect appropriately are chosen under the local density approximation (LDA) for the electron exchange-correlation potential. Through a sensitivity study of the lattice thermal conductivity with SOI and without SOI, we find that for both PbSe and PbTe, the SOI effect is important and fully relativistic pseudopotentials are necessary. For example, the phonon lifetimes of all modes are noticeably larger with SOI, which results in a twice larger thermal conductivity with SOI than that without SOI at 300K. 3.2.1 Harmonic Properties The harmonic IFCs are obtained using the primitive cell calculation of 2 atoms. In the self-consistent calculation of electronic properties, a Monkhorst-Pack 10x 10x 10 mesh79 is used to sample the electronic states in the first Brillouin zone and an energy cutoff of 60 Ryd (-816 eV) is used for the plane-wave expansion to ensure the force convergence. In the following DFPT calculation, a Monkhorst-Pack 4 x 4 x 4 q-mesh is used to calculate the dynamical matrix at each q grid, which, through an inverse Fourier transform to real space, gives the harmonic IFCs. The harmonic IFCs allow computation of the dynamical matrix at any q point: 34 (3.1) ,,eiR D,,",(q)= VM,,m,, where D is the harmonic IFC, m is the atomic mass, R, of the unit cell is the translation vector ' , while q specifies the q th atom in the primitive cell, and a, fi are Cartesian components. The eigenvalues of the dynamical matrix yield the phonon frequencies and the phonon dispersion, from which the phonon group velocities can be calculated. 3.2.2 Anharmonic Properties There are two approaches to calculate the anharmonic IFCs in the reciprocal space. The results from both approaches are equivalent. One approach is based on 2n+1 theorem 26,8 0 which assumes that the third order IFCs can be obtained from the first order wave function. It is computationally effective since it does not involve the supercell calculation, but it is relatively complicated to implement. The other approach is to calculate the third order IFCs from the second order IFCs using a finite difference method, which is computationally more expensive but simpler to implement. We use the latter approach in this study. The third order derivatives are determined by taking the derivative of the second order IFCs through a central difference scheme as below: a2V q fya~v 1'17 Oi=~ q~. a2 V K P1 au/Xu7,,' U V 17 - au,",,7u ,,.8u ,,.. 2u1",, 2uo" 2uoa720 (3.2) where V is the interatomic potential. We first perform the IF point phonon calculation in a super cell to generate the harmonic IFCs for two different atomic configurations namely involving displacement of an atom along positive and negative Cartesian directions around the equilibrium position. All the required cubic IFCs are obtained by sequentially changing the displaced atom to be any of the atoms in the primitive cell. To ensure the accuracy of the cubic IFCs, we test three values for the displacements. We use a Monkhorst-Pack 4 x 4 x 4 mesh 79 to sample electronic states with the same energy cutoff of 60 Ryd (-816 eV). 35 The cubic IFCs are needed to compute the three-phonon scattering matrix elements, which measure the strength of the scattering events and are given by =/( V3 (qs,q's',q"s") i,"I-Rj. flyi,'-RI. 8NOc9(qs)co(q's')co(qofs x ,-afl 17 P17 P,,- 1117 e," (qs)e 17 (q's')e,,(q"s"t) 1 ? m,,m,,,m,,,, (3.3) where h is the Planck constant divided by 27r, N is the total number of modes in the first Brillouin zone, and s denotes the different polarizations. golden rule to the cubic Hamiltonian 81,82 the phonon lifetimes By applying Fermi's rqs due to the normal and umklapp three-phonon scattering processes can be expressed as V(qs, q's',q"s")12 = 21, = 7r x[2(nqs - nqs,,)S(a)(qs) + w(q's') -(qs")) qs s" qS +(1+nq's +nq',s)8(co(qs) - o)(q's') -(q's"))] (3.4) where nqs is the Bose-Einstein distribution function nqs = 1/(eh0p IkBT .1) The conservation of momentum requires q+q'+q"=G, where G is a reciprocal lattice vector, for which G =0 results in the normal processes and G 0 relates to the umklapp processes. The choices of q" are limited by the choices of q and q', and thus the summation involves only q'. 3.2.3 Lattice thermal conductivity We compute the lattice thermal conductivity approximation using the well-known formula 36 based on the relaxation time 3QK N=,(3.5)V 3QNo ,, 2Tq 'coqs aq aT where C2 is the volume of the unit cell and vqs is the amplitude of the group velocity. We use a 30 x 30 x 30 q-mesh within the first Brillouin zone to ensure convergence. Comparing the total calculated lattice thermal conductivity with the experimental data serves as a validation of our calculations. More importantly, decomposition of the total lattice thermal conductivity into each mode allows us to account for the contributions from phonons with different MFPs and polarizations, which provides insights into specific thermoelectric applications. The phonon MFP for each mode is defined as Aqs = vqs, (3.6) One way to quantify the contribution from phonons with various MFPs is to evaluate the thermal conductivity accumulation with respect to MFPs 18, 83 . By summing the thermal conductivity contribution coming from modes with MFPs up to A, the cumulative thermal conductivity can be determined as follows: 1 (A)N= 3 A <A vqsAqhoqs, (3.7) (3.7) To separate the contribution among the different polarizations, we simply sum the thermal conductivity of the modes for each polarization s as 1 2nqs y ,qshoqs K,= 3QNI 3.2.4 Alloy modeling 37 (3.8) To take into account alloy effects, we use the virtual crystal approach, first introduced by Abeles 63, where the disordered crystal is replaced with an ordered one with an average lattice parameter, atomic mass and a set of force constants which vary according to the composition. The mass disorder and anharmonicity are both treated as perturbations. Garg et al.29 has applied this approach to Si-Ge alloys using the force constants from DFPT and reached excellent with the experimental data by following this approach. The effective phonon scattering rate is defined as the sum of the scattering rates due to mass disorder and anharmonicity: 1 qs -= 1 Iqs While the anharmonic phonon lifetimes vr,,- (9 1 + (3.9) qs are calculated in the same way as the pure cases except for different input parameters, the harmonic phonon lifetimes due to mass disorder is given by8: = qs ;r 2N S( q, q's9 - q)sg where e is the polarization vector, and g 2(-)= f(a-) and 2 (.) s(a)eS(a) f(a)[1 -m 12 (a) / m (-)]2, (3.10) in which m, (a) are the concentration and the atomic mass of ith isotope of the a atom. 3.3 Results and Discussion 3.3.1 Comparison with experimental results 38 PbSe r-I 4 0 a) U) 0 0 0 G K) L G X( 5 PbTe 0 3 0 00 a) 0r a) I-. LL 1< . 0 G K X G L Figure 3-1 Phonon dispersion for PbSe and PbTe: red lines: calculated results; black dots: experimental results from neutron scattering Figure 3-1 shows the phonon dispersion relations of PbSe and PbTe along the high symmetry directions within the first Brillouin zone of the primitive cell with two 39 atoms. There are six polarizations: two transverse acoustic (TA), one longitudinal acoustic (LA), two transverse optical (TO) and one longitudinal optical(LO) modes. The disperion of PbSe agrees reasonably well with the experimental results 8. The splitting of the LO and TO branches at r point, which depends on the Born effective charges and dielectric constants, agrees with that in the experiment. The dispersion of PbTe matches well with the experiments86 except for the LO branch. The discrepancy for the TO-LO splitting at the F point comes from the difference in the Born effective charges. By setting the Born effective charge to the value obtained in the experiment (6.5 e ), the dispersion meets the experimental data while all other modes and the total thermal conductivity changes by less than 1%. It has also been claimed in previous work 32 , 33 that the inclusion of the LO-TO splitting has only negligible effects. For better comparison in terms of the actual frequency range, we use the tuned Born effective charge for the latter discussions. Although the frequency of the TO mode at zone center matches perfectly with the experimental value measured at room temperature, some uncerntainties exist in the calculation of this specific mode. As found in previous studies, the TO mode at the Gamma point is soft and directly relates to the ferroelectric ground state 87, 88 ' . The ferroelectric mode is difficult to calculate accurately due to its strong temperature and volume dependences, and different pseudopotentials and lattice constants lead to different frequencies 87-89 . However, since we focus on the integrated properties of all the phonon modes, the discrepancy of a single mode or of few modes near the zone center with very small or even zero group velocity does not make any noticeable change to our conclusions because they hardly carry any heat. The dispersion relations of PbSe and PbTe are similar but do not scale with the total primitive cell mass ratio because they have one element Pb in common. Although the frequencies of the optical modes of PbTe drop significantly compared to those of PbSe, the differences between the acoustic modes, especially the TA modes, are much smaller. 40 PbSe, 2 4- 2 0 -0 - 4-, PbTe 0 L- 0 200 400 600 Temperature [KI 800 0 200 400 600 Temperature [1 800 Figure 3-2 Temperature dependent lattice thermal conductivity of PbSe and PbTe, red lines: calculated results; black crosses: experimental data. We compare the calculated lattice thermal conductivities with experimental results in figure 3-2. For both PbSe and PbTe, the calculations achieve decent agreement with experimental values 68, 69 . The small discrepancies of PbSe between 300 K and 400 K might come from the impurity or defect scattering in the experimental sample, which becomes inferior to three-phonon scattering at higher temperatures. Above 400 K, the calculated results lie on top of the experimental data. The agreement for PbTe over the whole temperature range is excellent. The good agreement bears out the accuracy of our approach, and the validity of the relaxation time approximation, and supports our following discussions. 3.3.2 Comparison between PbSe and PbTe The calculated lattice thermal conductivity of PbSe is 11% higher than that of PbTe at temperatures of 300 K-700 K. The atomic masses of Pb, Se and Te are 207.2, 78.96 and 127.6, respectively. Se is about 40% lighter than Te, but due to the heavy mass of Pb, the mass difference for PbTe and PbSe is only 17%. At a first glance, the mass difference seems to fully explain the thermal conductivity difference. Yet how the mass difference actually leads to the variance in different quantities is far from a simple deduction, as we will show below. 41 3 10 3 C) 10 10 TA -2 T'0) -2 2_ CL) 0 E :r_ E JD 10 L - 2 10 Frequency[THz] 101 10 (A PbSe PbTe 10 0 -(b) 10 10 - (a) 10 10-1 10 LA -____ 10 Frequency[THz] LO 0~ a- E 10 - E 1U 10 S0 -- - ---- (d) 101 0 a S 01 100 10 Frequency[THz] Frequency[THz] Figure 3-3 Frequency dependent phonon lifetimes of PbSe (squares) and PbTe (crosses) at 300 K: (a) TA, (b) LA, (c) TO, and (d) LO. We show the phonon lifetimes in figure 3-3. In the low frequency range, the lifetimes of the acoustic modes exhibit a co- dependence, in agreement with Klemens' prediction90 . The trends of the lifetimes with respect to frequencies are similar for PbSe and PbTe. For most of the TA modes, the lifetimes of PbSe are substantially larger than those of PbTe, while for the LA and optical modes, the lifetimes of PbSe are not necessarily higher. This is a nontrivial observation since the anharmonicity of PbSe is normally expected to be larger due to the larger average Griineisen parameter reported from experiments 67. For the optical modes, the lifetimes of PbTe are obviously larger. 42 300 0 -- TA- -- 40C 0 -- E 200 0 E 0 05 0 30C 0 x PbSe PbTe 20C 0 100 0 10C 0 1 Frequency[T Hz] 0 2 ------- 40C 0 0 2 Frequency[T Hz] 4 2 4 Frequency[T Hz] 6 300 E 200 E 30OC 0 0@ 0 10 (c) 2 3 Frequency[THz] 4 Figure 3-4 Frequency dependent phonon group velocities of PbSe (squares) and PbTe (crosses) at 300 K: (a) TA, (b) LA, (c) TO and (d) LO. With heavier mass, PbTe was anticipated to have smaller group velocities in general. Nevertheless, figure 3-4 shows that for the TA modes, the group velocities of PbSe and PbTe are almost the same because of the closely matched acoustic dispersions. Noticeably, these TA modes are fairly soft with maximum value around 2000 m/s. In terms of the LA modes, the group velocities of PbSe are moderately higher. Between 1 THz and 2 THz, several TO modes of PbTe possess exceptionally high group velocities (>3500 m/s) and even higher than the TO modes of PbSe. For the LO modes, the group velocities of PbTe are perceptibly smaller than those of PbSe. 43 %PbSe TA1 PbTe ---TA2 LA E 0.6 E 0.6- 0.5 0.5 ---- TO2 0.4 LO 0 U 0.3- : 0.3 0 0.2 -i 0.2 F -FU E - E 0.1 00 600 500 400 Temperature[K] .1 700 ------ - 00 400 500 600 700 Temperature[K] Figure 3-5 Thermal conductivity from different polarizations (TAl, TA2, LA, TOl, T02 and LO) versus temperature for PbSe and PbTe Integrating the transport properties over the entire first Brillouin zone, we can obtain the polarization dependent thermal conductivities as shown in figure 3-5. Remarkably, over a wide temperature range of 300 K to 700 K, the three acoustic branches for PbTe contribute equally and three optical branches contribute almost evenly to the thermal conductivity of PbTe. In the case of PbSe, by contrast, the contribution among acoustic and among optical modes are all distinguishable. Considering all the differences in phonon frequencies, lifetimes, and group velocities, it is impossible to identify the decisive one source of the differences between PbTe and PbSe, despite the simple mass difference argument. 3.3.3 The importance of optical phonons The normalized optical phonon contributions can be calculated by adding TO and LO modes together. For the whole temperature range considered (300 K-700 K), the contributions of optical phonons remain about 25% for PbSe and 22% for PbTe. These findings are rather surprising especially considering the simple rocksalt crystal structures of these two materials and the fact that only half of the modes are optical phonons. Our calculations demonstrate that optical phonons are not always negligible even in simple crystalline bulk materials. 44 without acoustic-optical scattering E 1 _,010 PbSe PbTe with acoustic-optical scattering E PbSe 10 PbTe 100 200 300 400 500 600 700 800 Temperature [K] Figure 3-6 Temperature dependence of lattice thermal conductivity without acoustic-optical scattering: PbSe (black dashed line), PbTe (red dashed line) and with acoustic-optical scattering: PbSe (black solid line), PbTe (red solid line). Moreover, optical phonons provide important scattering channels for acoustic phonons and are essential for the low thermal conductivity of PbSe and PbTe. By removing the acoustic-optical scattering, the thermal conductivity of PbSe/PbTe increases dramatically by a factor of six/five over the entire temperature range investigated here (300 K to 700 K) as shown in figure 3-6. This difference is about twice larger than that of Si73 . Due to the softening of the optical phonons, the longitudinal acoustic and transverse optical phonons are strongly coupled, as observed in PbTe by Delaire et al.74 in the experiment, and by Shiga et al.3 3 in the calculation, and help lower the lattice thermal conductivity. 3.3.4 The potential impacts of nanostructuring 45 2. E 2 PbTe 0 1 5 E SAK-PbSe Te 1PbSe-V 05 E 0 .57 --, .- . ___- 101 100 Phonon Mean Free Path[nm] 10 102 Figure 3-7 Cumulative thermal conductivity with respect to phonon mean free path at 300 K for PbSe (red dashed line), PbTe (black solid line) and PbTeo.5 Seo 5 (blue dotted line) The calculated cumulative thermal conductivity with respect to phonon mean free paths (MFPs) at 500 K is shown in figure 3-7. The total accumulation for PbSe keeps increasing as the MFPs increase while the accumulation for PbTe gradually approaches a plateau after the MFPs reach 10 nm. Phonons with MFPs smaller than 10 nm comprise around 80% of the lattice thermal conductivity for PbSe and about 90% for PbTe. In other words, even if the interface backscattered all the ballistic phonons, the nanostructuring with length scale 10 nm would only potentially reduce the thermal conductivity by 20% for PbSe and 10% for PbTe at the most. Therefore, to significantly reduce the lattice thermal conductivity in these materials, nanostructures with characteristic length smaller than 10 nm are required. Therefore, smaller scale inhomogeneities and alloying might be more effective in reducing the lattice thermal conductivity. 46 3.3.5 The potential impacts of PbSe-PbTe Alloying 2.5 L L- -E 300K 500K 2 -a 1.5 0 E a) 1- 0.5' 0 0.2 0.4 0.6 PbTe ixSex 0.8 1 Figure 3-8 Calculated composition dependence of the lattice thermal conductivity in PbTei.xSex at 300 K (solid line) and 500 K (dashed line) We plot the lattice thermal conductivity of different composition of PbTei-xSex alloy in figure 3-8. At x = 0.5, we obtain a maximum decrease in k of 30% (1.46 W/mK) compared to the average lattice thermal conductivity of PbSe and PbTe (2.1 W/mK) at 300 K. There is no sharp decrease feature in the dilute alloy limit as reported in the Si-Ge alloy 29 due to the small difference in acoustic impedance between PbSe and PbTe. As the temperature increases, the phonon-phonon scattering becomes dominant, and the influence from alloy scattering becomes less important. Therefore, comparing 300 K with 500 K, the reduction of lattice thermal conductivity is greater at 500 K. The mean free path accumulation of PbTeo.5 Seo.5 is plotted in figure 3-7. The phonons with high frequencies and short mean free paths are strongly scattered by mass disorder, while the phonons with small frequencies and long mean free paths are much less influenced. This leads to a redistribution among different mean free paths and consequently a shift in the accumulation curve results. Since the accumulation 47 curve of PbTeo.5 Seo.5 is considerably more flat above 10 nm, similar to PbSe and PbTe, nanostructuring on alloys could not push down lattice thermal conductivity by a significant amount. Taking into account the practical difficulty in introducing nanostructures at the scale of 10 nm and the potential reduction in the lattice thermal conductivity, the simple alloying approach is more promising in reducing the lattice thermal conductivity because experimentally grain growth in these materials is a problem with annealing. 3.4 Conclusion We perform first-principles calculations to detail the spectral phonon transport properties of PbSe and PbTe. We first extract harmonic and anharmonic force constants from density functional perturbation theory calculations within a supercell. We then extract the phonon lifetimes based on Fermi's golden rule and we then compute the thermal conductivity under the relaxation time approximation. The total lattice thermal conductivities quantitatively agree with the experimental results. Comparison of mode-dependence properties between PbSe and PbTe suggests that the transport properties of these two sister materials are similar in principle but different in specifics. The optical phonons not only directly contribute a considerable amount to the total lattice thermal conductivity of bulk PbSe and PbTe but also serve as important scattering channels for acoustic phonons. Both PbSe and PbTe possess very low lattice thermal conductivities, which is attractive for thermoelectric applications. Nanostructuring, however, would be difficult to further reduce the lattice thermal conductivity unless the characteristic lengths of the nanostructures could be reduced and maintained to much less than 10 nm. Alloying, on the other hand, has advantages over nanostructuring in reducing the lattice thermal conductivity. The parallel studies of these two materials provide insights into the phonon properties and may help design better thermoelectric materials. 48 4. The Importance of Optical Phonons in Nanostructures 4.1 Introduction It is generally understood that optical phonon contributions to the thermal conductivity k are small and negligible in bulk materials because of their short lifetimes and low group velocities. Several recent theoretical efforts 18 ,2 5,71 that fully detail the spectral phonon transport properties of bulk silicon (Si) all concluded that the contribution of optical phonons to k is around 5% at room temperature, regardless of the method used. Chapter 3, however, highlights the importance of optical phonons for bulk PbSe and PbTe mainly due to the softening of their TO modes and calls for attention to optical phonons in certain bulk materials. The importance of optical phonons in nanostructures, on the other hand, is more universal. When the system size decreases, the contributions of optical phonons to heat conduction become increasingly important. While acoustic phonons are strongly scattered at boundaries and interfaces, optical phonons have short mean free paths (MFPs) and are scattered much more strongly inside the nanostructures than at the boundaries. Such a difference in scattering leads to a rebalance of the relative importance of optical phonons and acoustic phonons to the thermal conductivity of nanostructures. In this chapter, we examine this shift using Si as a test case, because several puzzling experimental results for Si nanowires 36 ' 91-93 have yet to be explained satisfactorily, despite several theoretical and computational studies94-99. Based on first principles calculations, we first examine the cumulative contributions to the thermal conductivity in bulk Si by phonons with different MFPs and polarizations, namely longitudinal acoustic (LA), transverse acoustic (TA), longitudinal optical (LO) and transverse optical (TO). We then model the thermal conductivity of Si nanowires based on the spectral properties in bulk Si and evaluate the contributions of optical 49 phonons as a function of nanowire diameter over a wide temperature range. Our modeling results show that around room temperature optical phonon contributions can increase to 18% when the nanowire diameter is reduced to 20 nm. 4.2 Methodology The detailed methodology and calculation procedures for bulk Si are presented 27,32 . In short, electronic structure calculations based on density functional elsewhere theory were applied to extract interatomic force constants via the direct displacement method.2 7 The cubic anharmonic force constants lead to three-phonon lifetimes. The phonon lifetimes rk,p due to the normal and umklapp three-phonon scattering processes have been calculated for each polarization p and each k point sampled in the first Brillouin zone based on the scattering rate determined from application of Fermi's golden rule. The thermal conductivity is then computed from the relaxation time approximation using the well-known formula (4.1) 1~~k v2_ kP = ho~T~hA;p 3 QNk t, kT K~ where 2 is the volume of the unit cell and nk,p is the Bose-Einstein distribution function. The phonon MFP for each mode is defined as (4.2) Ak~p = Vk~prk~p By sorting the thermal conductivity contribution of each mode according to increasing MFPs18, 83, the polarization dependent and total cumulative thermal conductivity can be determined by equation (4.3a) and (4.3b), respectively, as follows: K,(1A) = hCo kp<A 1(A)= vpAk)wk k 3QNk K(A) = EZKP(A) P 50 _ a (4.3a) (b (4.3b) As the system size decreases, it is often found that a large thermal conductivity reduction occurs as the nanometer regime is approached. 40' 83, 100-104 Past studies suggest that this reduction is due mainly to increased boundary scattering. The effective phonon lifetimes including boundary scattering can be estimated by adding the scattering rates due to anharmonic and boundary processes: 1 k,p 1I 1 +1I p-p B Tkp Tk,p (44) where for nanowires with diameter d, the Casimir limit gives 1 v (4.5) d kp which assumes purely diffuse scattering at the boundary. Alternatively, by solving Boltzmann's equation for an infinite wire, Sondheimer also arrived at a similar result, and generalized it to the case of boundaries with scattering continuously going from specular to diffuse. 28 51 4.3 Results and Discussion 160 r- (a) E LOTA1to 140 2 10120 A 5-- - 4-' 100 20 0 0.4 :3 0 - 0 80 TA2 40 60 LA 60 TO1 p. .0.2- TA2 T02 40 LO - 0 I- - 20 H 0 2 6 4 A [nm] 0 100 .m... rX -- 8 TA1 ItLA . U Total I -A[nm],b 0.4 (b) -' E -c f 10 102 1 10 3 10 4 Mean Free Path A [nm] Figure 4-1 Cumulative thermal conductivity with respect to MFPs at 277K from the 18 x 18 x 18 k-mesh data; Inset (a)-Inset (b) Zoomed-in figures for MFP range of (a) 0-70nm and (b) 0-9nm, respectively The detailed cumulative contributions to the thermal conductivity by phonons of different MFPs and polarizations at 277K are shown in Fig. 4-1. Note the slope Inset (a) in change in the thermal conductivity when the phonon MFPs are -30nm. Fig. 4-1 shows that this sharp increase is due to the rapid growth in contributions from the TA phonons. Contributions to the total thermal conductivity for phonons with a MFP less than -30 nm are mainly due to LA and optical phonons. Although TO phonons have very short MFPs (less than 9 nm), they are dominant contributors (inset (b)) to the thermal conductivity in this MFP range due to their large density of states (DOS). 52 Fig. 4-1 also leads to additional insights on the contributions of acoustic modes to the thermal conductivity. Below 45 nm, LA modes contribute more to the thermal conductivity than TA modes. These small MFP modes correspond to phonons near the first Brillouin zone edge where the group velocities of TA modes are lower than those of LA modes. The sharp increase of the TA2 accumulation between 40nm and I00nm is due to larger DOS of TA2 modes. 60 E 7 50- /~* ~1l5nm 40I -o 30 a 56nm 0 & a) X / X* 100 0 37nm -- - - - - - -- - - -... . 20 : 50 100 200 250 150 Temperature [K] 300 350 Figure 4-2 Thermal conductivity of silicon nanowires for d=37nm, 56nm and 115nm, lines: calculated results; crosses, squares and stars: experimental results Considering the properties of different polarizations in bulk Si presented above, we investigate how the contributions change in the context of nanowires. To validate our model, we first compare the thermal conductivities of nanowires with experimental results. To best represent the experimental sample, we include isotope scattering and add the scattering rate according to Matthiessen's rule as below: 53 1 1 TkpTP+T Tk 4 1 B k,p + 1 and 1 = Aco4 k,p +TITi (4.6) k~p Tkp s' is analytically determined from the isotope concentration . where A = 1.32 x 10 k,p + Using Matthiessen's rule to include boundary scattering is an approximation and there are other studies solving BTE directly 0 . Without any fitting parameters, we have obtained decent agreement with experimental data3 6 for d=l 15nm, 56nm and 37nm as shown in Fig. 4-2. The good agreement between experiment and theory supports the following discussions based on the boundary scattering effect. We do not include results for the nanowire of 22nm since we could not explain the experimental results after taking into account the optical phonon contributions. 7- (a) 67 E TA2 5 > 4-- E 2- TO 1&T02 ; 0~ 0 200 800 600 400 Temperature [K] 54 1000 C 0.25 -b- o 1 Onm 0.2 0 0.1 00nm ~ | - 1 ~ ~ C 0.15 -- 1mm 0.0 0 0 200 600 400 Temperature [K] 800 1000 Figure 4-3 Thermal conductivity from different polarizations versus temperature for d=20nm; (b) normalized optical phonon contributions to the total thermal conductivity versus temperature for d= 1 Onm, 20nm, 1 00nm and 1mm. The temperature dependence of the relative phonon contributions from the different polarizations is shown in Fig. 4-3(a) for the d=20nm nanowire. At temperatures below 100K, the three acoustic thermal conductivity contributions exhibit a T 3 dependence, the same as the temperature dependence of the specific heat, due to the dominance of boundary scattering. The two TA modes grow more rapidly than the LA modes since the specific heat of the TA modes rises more rapidly due to their lower frequency and higher DOS. The LO modes contributions are noteworthy over most of the temperature range considered. The TO modes, however, make a negligible contribution for this temperature range. Adding LO and TO modes together, the normalized optical phonon contributions are shown in Fig. 4-3(b) for different nanowire diameters. As the diameter decreases, the optical phonon contributions relative to the acoustic phonons become larger as expected. Between OK and 300K, the optical phonon contributions increase due to the increase in their specific heat. 55 ) - 3025 20- 20 TA2 - TA1 15- LA 10 LO --- 5 TO1 &T02 0 wan 0 50 200 100 150 Diameter d [nm] aw 250 0.25 M 300 (b)~ 0.2- 0.15 \ 400K 277K-- 0.1 0.05 . -L t --_r , ,I 0L 0 1OOK - 50 -100 177K - 150 200 Diameter d [nm] 250 300 Figure 4-4 (a) The thermal conductivity from different polarizations versus diameters at 277K, (b) the normalized optical phonon contributions to the total thermal conductivity vs. diameters at 100K, 177K, 277K, and 400K Fig. 4-4(a) depicts the thermal conductivities from different polarizations for Si nanowires of different diameters, varying from 5nm to 300nm, at 277K. Acoustic phonon contributions increase with increasing diameter in the plotted diameter range (5-300nm), while the optical phonon contributions saturate around 100 nm due to 56 their lower MFP values arising from three phonon scattering processes. Note that the LO modes contribute much more significantly than the TO modes. The normalized contributions with the total thermal conductivities at different temperatures are shown in Fig. 4(b). At 277K, the optical phonon contributions grow from below 10% to 21% when d decreases from 300nm to 5nm. It suggests that optical phonons can have significant impact on the thermal conductivity in nanostructures, especially at temperatures on the order of the Debye temperature or higher. 4.4 Conclusion In summary, we have used the relaxation times determined from first principles derived force constants to calculate the thermal conductivity of bulk Si and Si nanowires. Detailed analysis of the respective contributions shows that optical phonons comprise up to 20% of the total thermal conductivity in Si nanowires around room temperature, despite conventional contributions are usually negligible. wisdom which suggests that their This finding brings to light the importance of optical phonon contributions to heat conduction in nanostructures. Although Si is taken as the model material, we expect that similar behavior should exist in many other materials. 57 58 5. Phonon Transmission across a Single Si/Ge Interface using the Green's Function Method 5.1 Introduction The reduced lattice thermal conductivity observed in many nanostructured materials has significant implications for applications from thermoelectric energy conversion to microelectronics thermal management. The Boltzmann transport equation (BTE) can be used to accurately model the phonon transport in nanostructures if the input parameters, such as the phonon mean free paths and interfacial transmission, can be properly represented. In recent years, excellent progress has been made in computing the mode-dependent phonon mean free paths in bulk materials using first-principles approaches 28, 32,33,106 as covered in Chapter 3. In contrast, research on phonon transmission across interfaces is still limited and prior first-principles studies of phonon interfacial transport are rather scarce. First-principles based approaches have been recently applied to nanotubes107108 ; however, their applications to interfaces between bulk 3D materials are significantly more demanding due to the large number of transverse wavevectors required. Interface roughness due to atomic disorder and defects commonly occurs at interfaces during material synthesis. A thorough understanding of the influence of interface roughness on phonon transport is crucial for surface engineering and improved device design. It is generally accepted that interface roughness is a very important driving mechanism for thermal conductivity reduction in different nanostructures such as nanowires and superlattices. However, it is not clear how interface roughness affects interfacial phonon transmission. et al.1 09 Using a lattice Green's function formalism, Fagas found that the phonon transmittance is strongly dependent on phonon 59 frequency and the disorder correlation length by varying the atomic masses in a two-dimensional disordered atomic layer. Following the same approach, Zhao and Freund" 0 studied the phonon scattering at a rough interface induced by atomic mixing between two FCC lattices, and found that the transmittance is insensitive to the roughness parameters. Using molecular dynamics (MD) simulations, Sun and Murthy"' focused on the transmittance change as the roughness thickness was increased. For long wavelength phonons, they concluded that the transmittance is For mid-range wavelength phonons, the independent of roughness thickness. transmittance is reduced as roughness thickness increases but eventually saturates to become independent of the roughness. Nevertheless, the above studies have not drawn a comparison between the ideal and rough interface, and furthermore, the conclusions were derived from empirical potentials. 112 dynamics model, Kechrakos Using a simplified lattice found that the interface conductance can be enhanced by as much as a factor of three for highly mismatched materials. The calculation only included one monolayer roughness and one branch mode. Stevens et al.113 observed that interface mixing improved thermal transport by nearly a factor of 2 through non-equilibrium molecular dynamics (NEMD) simulations. using NEMD, English et al. 114 Most recently, found that by sandwiching an intermediate layer between two dissimilar materials, the interfacial thermal conductance enhanced compared to that of the two dissimilar materials. unable to unveil any information about the can be NEMD, however, is mode-dependent Additionally, an empirical potential was used in their simulations. transmission. The behavior of different phonon modes at a rough interface using reliably accurate force constants would be preferable, and as we will show in this paper, results can differ by up to 50% depending on the choice of the force field. Phonon interface transmittance is critical in determining the interfacial thermal resistance. Phonon interface transmittance models have yet to reliably predict experimental observations. There are two widely used models for the phonon transmittance at an interface: the acoustic mismatch model (AMM) 115 and the diffuse mismatch model (DMM)1 16. As a continuum model, the AMM assumes that phonons undergo specular reflection or transmission at the interface. This model is valid in the long-wavelength limit, where due to their small details compared to the incident phonon wavelength, interfaces are seen as sharp. The DMM, on the other hand, assumes not only purely diffuse scattering at the interface, but also an equivalence between phonon reflectance from one side to the transmittance from the other side. This model, as opposed to AMM is valid for very rough or dirty interfaces and short wavelength phonons. Neither AMM nor DMM consistently predict interface thermal boundary resistance. 60 Using molecular dynamics (MD)", 17-2, wave-packets can be created and the phonon transmittance can be obtained by tracking the energy transmitted and reflected after encountering an interface. Although easy to implement, it is computationally expensive since one separate MD simulation is needed for every incoming phonon mode, although using the multiple phonon wave packets reduces computational intensity' 1. Additionally, MD simulations cannot capture wide angles of incidence because it requires a large lateral size that is difficult to achieve. Linear lattice dynamics (LD) calculations 122-125 have been performed to extract the mode-dependent phonon transmittance by solving the reflected and transmitted wave functions subject to boundary conditions. However, this method can be difficult to implement for complex atomic structures. As an alternative and more straighforward approach, Green's function methods dedicated to solve for the response from a point source perturbation are employed to compute the phonon transmission function that can be easily related to transmittance as described in Sec. II. The Green's function approach has been described thoroughly for ' transmission function calculations in electron transport by Datta 126 . Mingo et al.12177 128 applied the approach to deal with phonon transport within an elastic scattering domain in nanowires and referred to this method as the atomistic Green's function (AGF) method. Later, Zhang et al.1 29 extended the method to phonon transport in 3D structures. They calculated the phonon transmission across the Si-Ge interface using an empirical interatomic potential and investigated the strain effect on interfacial transport. A general formulation and full derivation have been detailed by ' Zhang et al. 26 and Mingo' 30 31 1 . Several other studies utilize the same framework 0 7 108, 132-134 including the only first-principles based calculations with the AGF method in 1D structures 10'108. Here we incorporate the first-principles force constants into AGF and demonstrate the importance of using accurate force constants. Without any fitting to experimental data, the force constants from first-principles calculations demonstrated the ability to accurately reproduce the lattice thermal conductivity of bulk materials 25 28 32 33 , , , ,106. These force constants can also improve the quantitative prediction for interfacial phonon transport. In this Chapter, we employ the AGF method to study the interface roughness stemming from atomic mixing between Si and Ge interfaces. Although thermal conductivity reduction in nanostructured materials can usually be described by phonon scattering due to interface roughness, we show how a Green's function method in conjunction with the Laudauer formalism suggests that interface roughness induced by atomic mixing can increase phonon transmission and interfacial thermal conductance. This is the first attempt to incorporate first-principles force constants derived from ab initio density functional theory (DFT) into Green's function calculation for infinitely large 3D crystal structure. 61 We also demonstrate the importance of accurate force constants by comparing the phonon transmission and thermal conductance using force constants obtained from the semi-empirical Stillinger and Weber (SW) potential, and first-principles DFT calculations. 5.2 Methodology 2 The detailed methodology of AGF has been presented elsewhere ' 2 . In short, the system is partitioned into three regions: the left lead, the central region (also known as the scattering region) and the right lead, as shown in Fig. 5-1. The advantage of the Green's function approach lies in its ability to replace the infinite leads by finite leads with self-energies 126. The self-energy 1a describes the effect of the lead a on the central block and is defined as (5.1) Ea =#CagaOCa where a stands for left (L) or right (R), C stands for center; the # 's are the harmonic force constant matrices divided by their corresponding atomic masses: #aa means onsite force constants of a block in lead a, #' is the two neighboring blocks within lead a and #a means the the hopping matrices between complex conjugate of # ; g is the surface Green's function defined by: = 2 _ a + ]' (5.2) The surface Green's function corresponds to the uncoupled semi-infinite system and 135 The coupled Green's function for the - L - is solved iteratively using a fast algorithm3. central region is expressed as: GR = [C2_ 62 R (5.3) where the superscript R stands for retarded, o is phonon frequency, and $c represents the onsite force constants of the central region. * ** 000 ** 0 * LO ******* 00 0 0 0 OCO e 0 00000 0 0 oo ORO 0000000 e 0 0 0 0 Figure 5-1 The system is divided into three parts: left (L), center (C) and right (R). The left and right leads are semi-infinite crystal lattices. In the transverse direction, all the three regions have periodic boundary conditions imposed to represent the infinitely large lateral dimension. To tackle the infinitely large size in the transverse direction, a Fourier transform is performed parallel to the interface to decouple the infinite degrees of freedom into independent transverse wavevectors, k, assuming ideal translational invariance. We can then treat them as independent one dimensional chains with different transverse As the phonon frequency and transverse momentum are conserved wavectors. across the interface, the transmission function, E(c,k), as a function of these parameters is given as a trace over the Green's function of the center and coupling terms between the leads and the center: E(o, k) =Tr[FL ( where Ia =i[ER _ EA] ,)GR k)R(co, k )G^( )] describes the rate at which phonons enter and exit the leads. The retarded Green's function, GR, and retarded self-energy, ER , are the Hermitian conjugate of the advanced Green's function, GA , and advanced self-energy, respectively. (5.4) IA, The total transmission at a given frequency is simply the sum of the transmission function of different transverse wavevectors normalized by the total number of transverse k points: E(I/, k ) E(c)=1/N k, 63 . While the phonon frequency and transverse wavevector are conserved, mode conversion is allowed and the longitudinal wavevector can change. In other words, the phonons can elastically scatter into different directions at rough interfaces. The thermal conductance per unit area, -, based on the total transmission function, E(co), is calculated using Landauer's formula136 I 1 "0 af(oj, T) TE(co)dco c-(T)=- x - f h C s 21 0 aT (5.5a) f is the Bose-Einstein distribution and s is the cross-sectional area of the simulation cell perpendicular to the direction of the heat flow direction. Note that this definition yields a finite thermal conductance in the limit of an identical material because the temperature drop, AT, used to derive equation (5.5a) is between the reservoir temperatures, T, and T2, instead of the temperature drop across the interface. In other words, equation (5.5a) is the formula corresponding to a where two-probe setup where the thermometer probes the bulk phonons incident on the interface'1. If a thermometer probes the temperature drop right across the interface (this 17 corresponds to a four-probe setup), equation (5.5a) needs to be modified ' 137 Despite the highly nonequilibrium distribution near the interface, we can define two equivalent equilibrium temperatures, Tei and Te2, as proposed by Chen . The equivalent equilibrium temperature corresponds to the final equilibrium temperature of these phonons if we assume they adiabatically approach equilibrium. Then we could use the Bose-Einstein distribution as a function of the equivalent equilibrium temperature to represent the local energy density. On the other hand, we can express the local energy density as a summation of the phonons emitted from both ends with the reservoir temperatures. By equating the two approaches, we obtain the relation between the equivalent equilibrium temperature and the heat reservoir temperature as Te = T, + (T 2 - T 1)a/(2a1 ) and Te 2 = T2 - (T 2 - T1)a/(2a 2). Finally, we reach a modified expression for the thermal conductance as -I(T) = a(T) x 1- where o, and 1 ( a(T)+ (T)) -2 (T) 2 a-1 (T) (5.5b) -2 are the "thermal conductance" of pure material 1 and pure 64 material 2 using equation (5.5a), respectively, with E(w) equaling the number of phonon bands at the frequency co. For a pure material, equation (5.5b) gives infinite thermal conductance as there is no temperature drop across the virtual interface. In the limit of low conductance (a- <<-i, a-<<a 2 ), equation 5.5a and 5.5b reach the same value as the denominator approaches 1. In the following discussion (Sec. III), equation 5.5b is applied. The transmittance can be related to the transmission function as 'T 2 (0)= (5.6) T21()= (CO) 2(CO) where r,(c2) is the transmittance from material 1 to material 2, while r2 () transmittance from material 2 to material 1. Transmittance describes the fraction of the incident phonons of frequency o that is transmitted. between zero and unity. is the Consequently, its value lies The transmission function, on the other hand, can exceed unity because it describes the number of modes transmitted at a specific frequency. The maximum value of the transmission function at a certain frequency would be the total number of phonon modes available at that frequency. Although the transmission function from either side is identical, the requirement of detailed balance requires the transmittance to have a directional dependence. In this study, we first construct an ideal Si/Ge interface as shown in Fig. 5-1 with Si on the left of the interface and Ge on the right of the interface, using the lattice constant of Si. a=5.43 A Lattice constants for the SW potential and DFT potential for Si are and a=5.3976 A, respectively. The transverse direction of all the three regions is set to be 3a x 3a, which has converged by comparing to the results of the 6a x 6a simulation size. transverse directions. Periodic boundary conditions are imposed in the The longitudinal length of the central region is 2a, which equals the largest thickness of the rough region investigated in this study. For simplicity, we use the force constants obtained from Si throughout the system since those of Ge are very similar in magnitude. The major factor affecting the phonons of Si and Ge are their very different masses. 28.0855 and 72.63 respectively. 41 and DFT, LAMMPS The atomic masses for Si and Ge are To obtain the force constants from the SW potential and Quantum Espresso 7 8 are used to record the force and displacement data, respectively. For our DFT calculation, we use the local density approximation of Perdew and Zunger 138 with a cutoff energy of 40 Ryd and 4 x 4 x 4 k-points for a 2 x 2 x 2 supercell of 64 atoms. 65 By fitting the general expression of the Taylor expansion of the interatomic potential to the set of force-displacements obtained from different atomic configurations , we extract the harmonic force constants that are input into our transmission calculation. We take exactly the same parameters as Esfarjani et al.28 used where they obtained excellent agreement with experimental data for the phonon dispersion and thermal conductivity of Si. This gives us confidence in the DFT force constants and corresponding phonon properties. The harmonic force constants that determine the phonon are essential for the transmission and thermal conductance. To calculate the total transmission, the number of transverse k points within the Brillouin zone is chosen to be 10 x 10 to ensure convergence. A similar frequencies and eigenvectors procedure has been followed for rough interfaces except for the system setup that obtains the force constants. For rough interfaces, the atoms in the interface region are assigned to one of the two atomic masses according to some probability (uniform or Gaussian), constrained by the thickness of the rough region, and then the effective 4 were obtained by dividing the Si force constants by the newly assigned masses. Lattice mismatch between Si and Ge, i.e. strain effects, and As observed by the NEMD anharmonicity are not included in this study. force constants simulations 139, anharmonic effects were not important for temperatures lower than 500 K. To first validate our methodology, we compare our calculated thermal conductance of an ideal Si/Ge interface using the SW potential and equation (5.5a) with available data in the literature. 2 Our result yields 2.8 x 108 W/m K at 300 K, which is close to 124 3.1 x 108 W/m 2 K from the lattice dynamics calculation by Zhao and Freund , and (3.2 0.2) x 108 W/m 2 K McGaughey13 9 . from the NEMD calculation by Landry and We can then focus on the discussion on rough interfaces using equation (5.5b). 5.3 Results and Discussion 5.3.1. Rough interface with random distribution To create random atomic mixing, we select a certain number of layers (2, 4, 6, and 8) in the central region and randomly shuffle the atoms within these layers. Three independent configurations are constructed for each roughness thickness and calculations are conducted for each configuration. The average value is plotted for each thickness of the rough region. The total transmission function, transmittance and 66 thermal conductance are plotted in Fig. 5-2. The total transmission function, transmittance and thermal conductance of the ideal interface are plotted in Fig. 5-2 as a reference. 14 rS 1 2C 0 C., C ... \ 1 0-- Ideal --- 2-layer Ran dom 4-layer Ran dom --- 6-layer Ran dom -8-layer Ran dom (a) Rough Rough Rough Rough 8 0 cc 4. p 2 0 0 100 300 200 Frequency[cm-1] 67 400 500 & 09 0017 [>iI9jne-iedwe 00C ON~ 001 ij6no~j wopue~j Jee-q q6no~j wopue~j JeAel-9 0 0 -..- q~no>.j wopue~j jael-t... C) Lfbfo~j wopue>j JeAel-Z.. 0 CL --- - -- p CD - - am ow - - - 0 W" - son- ~ -- -I on ago ts10 . X 0 '09 EL-wo]Aouenbij ooC o0z 00t, 00M MAS ?o0 -~ 4 q1 o~ wpe~ j~eopu~j e~e-gI q~n~j j~elt,, q~noj wpuej wou qKo~ CD) Iaeleei (q) G) L0Oc i -- ~1 'p 6'0 **:~ ~1 - 12 DFT - 10 0 C) 8- 0 6- C: 4 (d) Ideal --- 2-layer Random ----- 4-layer Random 6-layer Random --- 8-layer Random Rough Rough Rough Rough 20 0 100 300 200 Frequency[cm-1] 400 500 0.9 (e) Ideal Rough --- 2-layer Random ..... 4-layer Random Rough 0.8 --- 6-layer Random Rough -8-layer Random Rough 0.7a) C E CO 0.6 0.50.4 A 0.3 0.2 0.1 DFT 0 100 300 200 Frequency[cm-1] 69 400 500 x 108 L f 3 DFT 2 .5- 00 c\E 1 2 0 -Ideal Random Rough 4-layer Random Rough -- 6-layer Random Rough - -8-layer Random Rough 0-'(.) f---2-layer 0 0 .5 0 100 200 300 Temperature[K] 400 5 )0 Figure 5-2 Total transmission function, transmittance and thermal conductance as a function of phonon frequency for an ideal Si/Ge interface (solid black line) and for a random rough Si/Ge interface (colored dashed or dotted lines): (a) Total transmission based on SW force constants; (b) Transmittance from Si to Ge based on SW force constants; (c) Thermal conductance based on SW force constants; (d) Total transmission based on DFT force constants; (e) Transmittance from Si to Ge based on DFT force constants; (f) Thermal conductance based on DFT force constants. One counter-intuitive finding, arguably the most important highlight, from Fig. 5-2 is that the phonon transmission across a rough Si/Ge interface can be higher than the ideal Si/Ge interface for certain frequencies, contributing to a larger thermal conductance at certain roughness thicknesses. In the low frequency limit, the long wave-length phonons do not sense the interface roughness and propagate through the interface as if they are traveling across an ideal sharp interface. Due to its short length scale, atomic roughness has negligible influence on the long-wavelength phonons. In the high frequency limit, the transmission is zero because there are no 70 available states on the Ge side. The most interesting phenomena are observed for the phonons with mid-range frequencies, where the atomic roughness could play a role in enhancing the transmission. The roughness softens the abrupt change of the acoustic impedance at the interface and facilitates phonon propagation. Surface roughness can also allow phonons with large incidence angles, which would otherwise be internally reflected at the interface, to be transmitted. More specifically, this can be understood by investigating the phonon density of states (DoS) of the two materials where incident and outgoing phonons are contained, and the interfacial region where reflection and transmission happens. As shown in Fig. 5-3, the phonon DoS of pure Si and Ge are quite different, while the Si/Ge mixture has an intermediate DoS which serves to bridge the gap between Si and Ge. Therefore, phonons that originally cannot propagate across the Si/Ge interface can now be transmitted via new elastic scattering channels created in the Si/Ge mixture. Accordingly, the phonon transmission and transmittance are boosted in the 200 to 300 /cm frequency range where the overlap of the two DoS is enhanced. This frequency range corresponds to the top of the TA branches close to the zone boundary, where the typical phonon wavelength is a few lattice constants at the most. Although one configuration of a Si/Ge mixture is used in Fig. 5-3, it can represent the trend for general Si/Ge mixtures at the interface since the atomic ratio of all the configurations involved in our calculation is 1:1 with the only difference being the atomic positions. been well-known that interface roughness photons 140-143 and electrons 144-147. In fact, it has can increase the transmittance of For phonons, interface roughness leads to a reduction in thermal conductivity in nanowires 3,91,12 because of back scattering and in superlattices'48-150 due to the loss of coherence. But for an individual interface, interface roughness is able to increase transmittance. This has not received much attention before. 71 0.09 -Si- 0.08 - --- Ge Si/Ge mixture I" 0.07 0.06S0.05- - 0.04 0.03- 0.020.01- 0 100 400 300 200 500 600 Frequency[cm-1] Figure 5-3 Phonon Density of States (DoS) of pure Si (Black solid line), pure Ge (red dashed line) and Si/Ge 1:1 mixture (green dotted line) using DFT force constants. For the 2-layer rough configuration, SW predicts a -20% increase in the thermal conductance at 300K (Fig. 5-2(c)), while DFT predicts a -30% increase, compared to Empirical potentials can qualitatively capture the trend, but are unable to quantitatively predict the difference. As the thickness of the rough region increases, the transmission does not keep increasing, which is consistent with earlier observations"' 14 There are two competing factors: 1) overlapping perfect interfaces (Fig. 5-2(f)). DoS which increases transmission; 2) diffuse scattering at the rough interface which reduces transmission. As observed in the SW case (Fig. 5-2(a)), the 2-layer rough Above a thickness of two layers, diffuse scattering becomes the more significant mechanism that affects thermal conductance. In the DFT case (Fig. 5-2(d)), however, the 4-layer rough configuration configuration gives the highest transmission. gives the highest transmission at a frequency of around 120 cm~ and 2-layer roughness gives highest transmission between 230 cm~1 and 300 cm~I, which leads to fairly close thermal conductance between the 2-layer rough configuration and the This finding cannot be 4-layer rough configuration as shown in Fig. 5-2(f). represented by the calculation using SW prediction partly because their phonon bandwidths are different from DFT. Compared to the ideal interface, the thermal conductance is larger when the rough region is thinner than 6 layers using the SW force constants and up to 8 layers using the DFT force constants. This discrepancy reiterates the necessity of adopting the DFT force constants to provide precise 72 guidance in practical applications. constants results are presented. In the following discussion, only the DFT force As thickness increases even further, the thermal conductance decreases below that of the ideal interface. understood by considering the limiting case. increases to infinity, diffuse This can be easily As the thickness of rough region scattering becomes dominant and the thermal conductance should approach the alloy limit. 5.3.2. Rough interface with Gaussian distribution To mimic atomic diffusion at an interface, we also create an atomic profile for one type to obey a half Gaussian distribution as shown in the Fig. 5-4(c) inset. The phonon transmission, phonon transmittance and thermal conductance are plotted in Fig. 5-4. constants. A significant increase in phonon transmission is observed using DFT force At 300K, there is 32.6% increase for a 6 layer roughness thickness. For the same roughness thickness, the Gaussian distribution shows more enhanced transmission compared to the random roughness distribution. Comparison with experimental data is difficult since there is no experimental data on a single Si/Ge interface. On the other hand, several experiments had reported reduced thermal conductivity on Si/Ge superlattices 148 149. If we assume that the measured thermal conductivity is due to interfacial resistances only, as one would expect in the very thin limit when phonon transport is completely incoherent15 1 and yet ballistic through individual layers of the superlattice, the extrapolated thermal conductance is 2 x 101 W/m 2 K 148 (period = 3 nm) and 1.8 x 10 W/m 2 K 149 (period = 4.4 nm) at 300 K. Both the extrapolated values are close but about one order of magnitude larger than our calculated value of 2 x 108 W/m 2 K for an ideal interface and 2.8 x 108 W/m 2 K for a Gaussian rough interface based on DFT force constants. The higher than predicted value is actually consistent with recent experimental observation152 that long wavelength phonons maintain their coherence in thermal transport in superlattices, and hence lead to a higher conductance value than that of a single interface as we calculated. 73 (a) Ideal --- 6-layer Gaussia i Rough 1 0 r 0 C, 0 C,) 6-i it E 2- 0 0 100 200 300 Frequency[cm-1] 400 1 A 0.8 500 (b) -Ideal -- 6-Iayi er Gaussian Rough I I 0.6 CD A 1 0.4 10 00 FrqecIm1 30 0.2 0O 0 400 74 500 x10 -Ideal - - -layer (c) Gaussian Rough - --- - 3 2.51 2 20 1.5 0 0$ ~0 0 -nterface ;;Z 15. 40'- 10 1 .E 5. z0 0.5 -4-3-2-1 1 2 34 0 Figure Layer Number C 5-4 (a)Total 100 200 300 Terrperature[K] transmission function, 400 (b)transmittance, and 500 (c) thermal conductance as a function of phonon frequency for an ideal Si/Ge interface (solid black line) and for a rough Si/Ge interface with a Gaussian distribution (dashed blue lines) based on DFT force constants. Inset of (c): The number of Si atom in each layer for an ideal interface (solid black) and for a Gaussian rough interface (dashed blue). 75 Z221 1.41 .41 2 2.45 2.83 3.16 8 10 1.35 1.3- S1.2 $ 1.25\ b 1.15 1.1 Emmmmmm0. 1.05 2 4 6 m 2/m 1 Figure 5-5 Thermal conductance ratio of a Gaussian rough interface to an ideal interface as a function of the mass ratio (lower x-axis) and the acoustic impedance ratio (upper x-axis) of the two materials using DFT force constants To explore the generality of the transmission enhancement between different materials, we keep the Gaussian rough configuration and vary the mass of the atoms on the Ge sites from 1.25 times that of Si to 10 times that of Si, corresponding to acoustic mismatch values from 1 to 3.16. The thermal conductance ratio of a Gaussian interface over an ideal interface is plotted in Fig. 5-5 as a function of the mass/acoustic impedance ratio of the two materials on both sides of the interface. Since the roughness is caused by the mass difference, when the mass ratio is 1, there is no atomic mixing and no roughness at the interface. As the mass ratio increases, the phonon dispersions of the two materials begin to differ from each other and the roughness favors phonon propagation via graded acoustic impedances at the interface. The thermal conductance ratio reaches its maximum at 2.586, which happens to be the mass ratio of Si to Ge. As the mass ratio increases even further, the phonon dispersions of two materials fall further apart from each other and it becomes less effective to bridge the large gap through the effects of roughness. Therefore, the thermal conductance ratio drops and flattens out with increasing mass ratio. Nevertheless, the thermal conductance ratio is kept over unity up to a mass ratio of 10 76 and will stay above unity in the infinite mass mismatch limit since such an interface provides a smooth transition for intermediate frequency phonons to transmit thermal energy across the interface. Although there are variations in the extent to which surface roughness increases thermal conductance, the enhancement generally holds. 5.4 Conclusion In summary, we apply the atomistic Green's function method to calculate the phonon transmission across an ideal and rough Si/Ge interface. The atomistic roughness can increase the phonon transmission across two dissimilar materials if the roughness thickness and profile are properly controlled, contrary to the commonly held notion that rougheness reduces transmission. This effect is more pronounced if the acoustic mismatch between the two materials is moderately large. new design considerations for interface engineering. This finding elucidates As our contribution to the AGF framework, we incorporate the first-principles force constants determined from DFT into the AGF method for phonon transport in an infinitely large 3D structure. The comparison between the results from SW force constants and those from DFT force constants demonstrates that DFT force constants are necessary in reliable predictions. Since interface transmission is crucial for bridging the calculation of pure materials to nanocomposites, we can now integrate the interfacial transmission and the bulk mean free paths, both calculated from first-principles DFT, to accurately model heat transport in complex nanostructured materials. 77 78 6. Phonon Transmission across Si/Ge Superlattices 6.1 Introduction As mentioned in Chapter 5, the calculated thermal conductance across a single Si/Ge interface is one order of magnitude lower than the extrapolated value from experimental data on Si/Ge superlattices. This suggests that phonon transport across multiple interfaces is not a simple summation of individual interfaces and coherent phonon transport is expected to exist in Si/Ge superlattices. In this chapter, we investigate phonon transmission across Si/Ge superlattices using the Green's function method with first-principles force constants derived from ab initio density functional theory. The thermal properties of semiconductor superlattices have been under intense investigation due to their potential uses in thermoelectric energy conversion37,39,153 and optoelectronic devices. 154 The thermal conductivity of superlattices can be even lower than their alloy counterparts.148, 15-157 Although diffuse scattering at interfaces is responsible for the remarkable thermal conductivity reduction,51, 158 coherent phonon transport has been experimentally observed in GaAs/AlAs superlattices1 59 and for perovskite oxides 60. To further reduce the thermal conductivity for thermoelectric applications, it is crucial to understand and control the different phonon transport modes in superlattices. Phonon heat conduction in superlattices can be attributed to incoherent and coherent phonon modes. Coherent modes preserve their phase as they propagate through multiple interfaces. For these phonons, Bloch mode extends through the whole structure and the superlattices can be treated as a homogeneous material with its own unit cell. If interfaces destroy the superposition of waves, due to roughness or other structures, phonon modes lose their phase information and their transport is incoherent. For these modes, superlattices act as a composite made of a 79 stack of two alternating materials. Previous theoretical studies on superlattices have focused on changing the periodicity. Most common theories developed to understand phonon transport in superlattices fall into one of two pictures: the incoherent particle picture which is rooted in solving the Boltzmann transport equation,151 , 161 and the coherent wave picture where lattice dynamics calculations were employed. 162-164 Either picture could fully explain the experimentally observed thermal conductivity trend as a function of period length in both in-plane and cross-plane directions, though. 39 A combination of both pictures is desired. Lattice dynamics based on damped wavefunctions was used to predict a 165 66 minimal in the thermal conductivity of superlattices in the cross-plane direction. ,1 More recently, a perturbation method based on the Fermi golden rule 30, 159, 167 was developed but the method may have limitations on treating interface scattering, as strong scattering may not be captured by perturbation. One alternative approach is to use molecular dynamics simulations,1 50"6 11 which do not assume the nature of phonon transport but on the other hand are classical in nature. Yet the empirical potentials involved in molecular dynamics limit the accuracy and it is difficult to explore the detailed phonon mode behavior. The Green's function method has been applied to study phonon transport across single and multiple Si/Ge interfaces. For single Si/Ge interfaces, effects of strain 129, lattice mismatch' 6 9 and interface roughness"o, 170 on phonon transmission have been investigated. Green's function study on Si/Ge superlattices, however, is scarce. Zhang et. aL.129 briefly discussed the impact of the number of interfaces on the overall thermal resistance across multiple Si/Ge interfaces while the transmission function was not detailed. In this study, we use the Green's function method to investigate the coherent phonon transport across Si/Ge superlattices. First-principles force constants have been incorporated as our previous work on single Si/Ge interfaces 170 In this chapter, we calculate the phonon transmission and corresponding thermal conductivity of Si/Ge superlattices with varying interface roughnesses. By keeping the period thickness fixed while changing the number of periods, we show that interface roughness partially destroys coherent phonon transport, especially at high temperatures. The competition between the low-frequency coherent modes and high-frequency incoherent modes leads to an optimum period length for the minimum thermal conductivity. To destroy the coherence of the low frequency modes, a scattering length scale on the order of period length is required. This finding is useful to guide the design of superlattices to reach even lower value of the thermal conductivity. 80 6.2 Methodology We follow the same atomistic Green's function method1 27, 170 single Si/Ge interface. 129 as we applied for a The only difference is that, in this study, we use the Si/Ge superlattices as the center region as shown in Fig. 6-1. As a brief overview, we employ the force constant, #c, from ab initio density functional theory into the Green's function to determine the transmission function. The retarded Green's function is given by GR where GR =[)2 _CO 1 C -YL (C)-YR (OT is the retarded Green's function, 0 is the phonon frequency, represents the onsite force constants of the center region and the self-energy 6.1) #c E, describes the effect of the lead a on the center block. The transmission function, E(o), is given as a trace over the Green's function of the center region and the coupling terms between the leads and the center: E(c) = Tr[FL (w)GR ()R (o)G A (c)] where = -a] .describes (6.2) the rate at which phonons enter and exit the leads. In these calculations transverse periodic boundary conditions are assumed and the above formulas hold for every single transverse momentum, over which a final summation needs to be performed in order to obtain the total transmission. The interface transmittance is then defined as r(W) = - () (6.3) ) .pure(( In our system setup, we use Em pure (to) = ESi (to) The 2-probe thermal conductance per unit area, a, based on the total transmission function, E(eo), is calculated using Landauer's formula 136 81 a(T) = s hc af(co, T) E()dco I 1 2;r 0 T (6.4a) where f is the Bose-Einstein distribution function and s is the cross-sectional area of the simulation cell perpendicular to the direction of heat flow. The 4-probe conductance can then be written as1 70 - 1 ( (T) + 2 o-,(T) (6.4b) ) -'(T) = o-(T) x c 2 (T) Although the difference of thermal conductance between 2-probe formula and 4-probe formula becomes small as the number of interfaces increases, we use 4-probe formula throughout this study to be consistent with our previous calculation of single interface and experiments. The thermal conductivity of a sample length L is defined to be L times the 4-probe conductance: (6.5) k ='L s k sts4 "4 G Figure 6-1 Schematic of the system setup: the left reservoir is pure Si, the right reservoir is pure Ge, the center region is the Si/Ge superlattice. The calculations in this paper do not include phonon-phonon scattering. According to experimental' 4 8' 155 and modeling 30 SO, 151 results on Si/Ge superlattices, anharmonic effects are not important for temperatures below 500K. The anharmonicity would become important when the phonon mean free path due to anharmonicity becomes smaller than the superlattices length L. In the harmonic regime, specular scattering leads to coherent wave effects171-1 73 while diffuse scattering could destroy coherence. 82 6.3 Results and Discussion For incoherent transport, the interfaces behave like a series of thermal resistors and the effective thermal conductivity becomes independent of the number of periods. For coherent transport, the thermal resistance keeps constant with respect to the number of periods and the thermal conductivity increases linearly with an increasing number of periods. The thermal conductivities for smooth- and rough-interfaced superlattices are shown in Fig. 6-2a. The thermal conductivity of smooth-interfaced superlattices demonstrates a linear increase with respect to the number of periods, indicating coherent transport at 300K although ultimately anharmonicity limits the number of periods over which transport is coherent. The thermal conductivity of rough-interfaced superlattices increases more slowly than linear, indicating partially coherent and partially incoherent transport. Another noticeable point is that roughness increases the thermal conductivity of small-period superlattices, in contrast to conventional wisdom. This is because atomic roughness generates a smoother change of density of states between layers 170 We then compare the transmittance across smooth-interfaced superlattices (Fig. 6-2b) with a rough-interfaced one (Fig. 6-2c). When the number of periods equals 1, it reverts to the single Si/Ge interface as we investigated before 17 0 , which we include as a reference. What we are mainly interested in here are multiple interfaces. For a number of periods > 1, there are clearly two frequency regimes: the low frequency regime and the higher frequency regime separated by the vertical lines at 55.6 cm-. The low frequency regime is defined as the region where the transmittance does not change as the number of periods is increased. This indicates that low frequency, long wavelength phonons pass through the entirety of the superlattices as if it is a homogeneous medium. They form passing bands and transport phonon waves coherently. The low frequency regime is the same for both smooth and rough superlattices. In the rough case, the constancy of the transmittance versus the number of periods for low-frequency phonons is due to the fact that such phonons have wavelengths larger than the roughness scale and thus see an effectively homogeneous interface of atoms with an effective mass intermediate between Si and Ge. As such, they do not get scattered by the roughness at the interface and thus, similar to ideal interfaces, their transmittance does not change with the number of periods. In the higher frequency regime, the transmittance for smooth-interfaced superlattices no longer changes as the number of periods becomes larger than 5, suggesting the formation of minibands. Because the superlattice eigenstates are formed from the constructive interference between all the multiple reflected waves, the wave needs to 83 go a few periods away and be reflected back for a few times in order to get a coherent eigenstate of the superlattice. In contrast, the transmittance for rough-interfaced superlattices keeps dropping due to more diffuse scattering at the interfaces. In other words, roughness destroys coherence especially for higher frequency modes. 2 (a) E 1.5 J 3 -o 1 E 0.5- -u-* Smooth Interfaces -- +- Rough Interfaces r- 0 0 5 10 15 Number of Periods 84 20 1 I' -- ---- 3 pd ....... 5 pd ..-- 10 pd 20 pd 0.8r a) C 1 pd 0.6 E 0.4 ' C 0.20 (b) 0 100 200 Frequency [cm-1] 300 400 1 -- 1 d ---- 3 pd ..-. 5 pd ----- 10 pd 20 pd 0.8 L a) CU) 0.6 \ EW, 0.4- I'I....' - 0.2 0 100 200 Frequency [cm~ 1] 85 300 400 Figure 6-2 (a) Thermal conductivity of superlattices as a function of the number of periods for smooth and rough superlattices at T=300K. (b) Transmittance as a function of frequency for superlattices (period =2a) with smooth interfaces; (c) Transmittance as a function of frequency for superlattices (period=2a) with rough interfaces. To unveil the cutoff frequency, wcutoff, of the low frequency regime, we plot the phonon dispersion of the SiGe superlattice with period length 1 = 2a in [100] direction (Fig. 6-3). The zone boundary frequency of the lowest acoustic branch is 55.6 cm 1 . It is intriguing that the cutoff frequency is the lowest acoustic phonon branch at the folded Brillouin zone edge. Although some of the higher frequency phonons have a long wavelength in the folded zone representation, they are unable to maintain their coherence. Therefore, phonon wavelengths of higher frequency modes in the folded zone do not matter. We expect that this argument generally holds for different materials. As the period length increases, the first Brillouin zone becomes shorter since the edge of the folded zone is proportional to the inverse of the period length. Thus, the cutoff frequency of the totally coherent regime is determined by the reduced first Brillouin zone, or the period length. It is, therefore, difficult to destroy the coherence of the low frequency modes unless scattering length scale comparable to the period length can be introduced. 86 500 400 E200 100 0 X Figure 6-3 Phonon dispersion of Si/Ge superlattice with period length 2a in [100] direction We then explore the temperature dependence of coherent and incoherent phonon transport. This temperature dependence comes only from phonon occupation or heat capacity. At all temperatures, phonons with frequencies smaller than the cutoff frequency yield a linearly increasing thermal conductivity as a function of number of periods as shown in Fig. 6-4. To illustrate this effect, we choose temperatures of 20, 50 and 300K which correspond to frequencies of 13.9, 34.7 and 208.1 cm~respectively. These are to be compared with the cutoff frequency of 55.6 cm- . At low temperatures, only low frequency modes are excited, thus the phonon transport is mostly coherent. As the temperature increases, more and more high frequency modes are excited and incoherent phonon transport plays a more and more important role. Correspondingly, we observe that the thermal conductivity increases more slowly than linear and becomes flatter as the number of periods increases. 87 0.1 E - (a) 0.08 L__ _ T=20K -+-Total --&-) > OCUtoff 0.06- .- .. 4-a 0 0..<= o cutoff 0.04- E g......- . 0.02 0- -------------- 5 0 0.4 20 15 10 Number of Periods (b) T=50K ,.0.35 0.3L 0.25. - 0.2' 0 o - Total --e-- (0 > (Oof ,'ctf 0.15 -U 0 cutoff ..--- 0.05 0 5 15 10 Number of Periods 88 20 (C) T=300K 0 .80.6--0 0.4- -E Total --- O > 0) cutoff E0 cutoff p0.2 0 5 15 10 Number of Periods 20 Figure 6-4 The total thermal conductivity, contribution from phonons with frequencies larger than the cutoff frequency, and not larger than the cutoff frequency at (a) T=20K, (b) 50K and (c) 300K. In our previous paper on single Si/Ge interfaces 170, a we found that the conductance at single Si/Ge interface is an order of magnitude lower than the extracted experimental thermal conductance from Si/Ge superlattices assuming the thermal resistance only happens at the interfaces. We predicted that the discrepancy comes from the long-wavelength phonons which maintain their coherence. Now with coherent transport, the calculated thermal conductance per interface increases with the number of periods and matches well with experiments (Fig. 6-5). Although the size of the superlattices is much smaller than that of the experimental sample due to computational limitations, we can at least see that the trend is consistent. It states that the thermal conductance is not intrinsic to the interface, but depends on what exists on both sides of the interface. 89 Extrapolated Experimental Value (9 _1.5~ -E -U (0U C 0 E I- 0 10 20 30 Number of Periods 40 Figure 6-5 Normalized thermal conductance per interface as a function of number of periods for rough-interfaced superlattices with period length 1 = 2a . The experimental value is extrapolated from the sample of period length 1 = 4.4nm and 100 periods. To destroy coherence in rough superlattices for the purpose of reducing the thermal conductivity, there are two competing effects as the period length increases: 1) the low frequency regime with totally coherent transport shrinks, which is beneficial; and 2) the interface density decreases and the importance of interface roughness decreases, which is detrimental. We plot the thermal conductivity as a function of superlattice length for period length I = a, 2a and 4a, respectively in Fig. 6-6. At the same superlattice length, superlattices with period length 1 = 2a = 1nm possess minimum thermal conductivity. The crossover from the coherent to incoherent regime is naturally included in our formulation, and we thus observe the minimum thermal conductivity of superlattices 60' 166' 167 under the atomistic Green's function framework. We then introduce a simple model below to identify the dependence of the thermal 90 conductivity on period length. We write the thermal conductivity as a sum over the contribution of acoustic (the three low-lying folded acoustic phonons) and optical (rest of the bands) phonons: k = kac + kop = cutoff f( C (&v)D(c)v(&v)A(co)d(A + fwmax Cj,(w)D(cv)v(co)A(w )dw cutoff (6.6) where &max represents the highest phonon frequency. For acoustic phonons, we 2 assume that the specific heat C,(o) = kB, that the density of states D (CO) = A&) (A is a coefficient which can be determined from the Debye approximation or lattice dynamics calculations), and v(cv) = c (average speed of sound of acoustic modes), and A(w) = L (in the absence of anharmonicity, scattering occurs at the sample boundaries). Then, Acv since f''toff 0 2 AW 2 kB cLdcd = fOC'f dc = 3 -cell and Qceul k CL (6.7) = 1 Atrans, with Atrans being the area of the unit cell in the transverse direction. For optical phonons, we use C,(c) = hcv , kac = D(c) = D (average density of states in a given volume per unit frequency interval), v(cv) = c0 p (average speed of sound for optical modes, which is much smaller than the speed of sound) and A(co) = 12 1-p 1 where p accounts for the probability of pure specular scattering at each interface, and we have assumed that the thickness of each medium is equal to half the superlattice period. kop hcvL)Dcop '=f"" 8T cutoff 'a' If the further assumption of -I- kBT i 2 1-p d ~:- -Ocell kL cop niL 2 1-p (6.8) «1 is made. Here N is the total number of atoms in a period. Therefore, the overall conductivity (neglecting anharmonicity) would be a sum of 91 k = 3 1Atrans kc L + ' 1cell kEcop 1+p 2 1-p (6.9) Equation (6.9) has a minimum at an optimum period length of l 'opt where t2 M3cellcL(1-p) (3N-3)Atranscop(1+p) fDcelloc~L-p) NOAtranscop(1p) (6.10) cello and No are the volume and number of atoms in a unit cell, respectively. It is noteworthy that the optimum period length depends on the superlattice length because the relative contribution from coherent and incoherent phonons would vary as the superlattices length changes. When designing superlattices to reduce the thermal conductivity, the optimum period length would be desirable. For SiGe superlattices considered in this work, No = 8, -celo = a3 , Atrans = 3a x 3a and a = 0.54nm. We assume c = 5400m/s (speed of sound for germanium), cop = 100m/s, and p = 0.5. This leads to lopt = 0.82nm at L = Snm, lopt = 1.16nm at L = 10nm, and lopt = 1.64nm at L = 20nm. All the optimum period lengths are close to 2a = 1.08nm, which is consistent with our calculations using the atomistic Green's function method. In strongly anharmonic materials, however, kop would be independent of 1 and there is no minimum thermal conductivity. 92 . 1 0.9 00 0 0.8 - 0 0 0 00 00 0t 0t 00 0.7 -.- Period: =a a) =2a Period: =4a - 25 20 15 10 5 Length [nm] Figure 6-6 Thermal conductivity of rough-interfaced superlattices as a function of superlattice length for a period length 1 = a, 2a and 4a at 300K. 6.4 Conclusion In summary, we apply the atomistic Green's function method to calculate the phonon transmission across Si/Ge superlattices. We focus our discussion on coherent vs. incoherent phonon transport in superlattices. We show examples of totally coherent phonon transport in smooth-interfaced superlattices, and partially coherent and partially incoherent demonstrate that phonon the in transport contribution from rough-interfaced coherent superlattices. phonons We decreases in rough-interfaced superlattices as the temperature increases. To obtain lowest thermal conductivity, there is an optimum length resulting from the competition between coherence of low-frequency phonons and incoherence of high frequency phonons caused by interface scattering when anharmonicity is negligible. Our theoretical study complements earlier experiments, providing guidance for the design of superlattices. 93 94 7. Solid-Liquid Interface Conductance Using Time-Domain Thermoreflectance Measurements 7.1 Introduction Interfacial thermal conductance has been a subject of fundamental and practical interest for many years. Usually, extra molecular layers at an interface add to the total thermal resistance network and reduce the thermal conductance, especially for solid-solid interfaces17 4 . Chemical functionalization, however, has significant influence on the interface thermal conductance between solid-solid interfaces via the interfacial bonding mechanism 175, 176 Recent studies have shown that covalent . chemical bonding at solid-solid interfaces using self-assembled monolayers (SAMs) can improve the interfacial thermal conductance 77 . Compared to solid-solid interfaces, the thermal conductance across solid-liquid interfaces has received limited attention. Better understanding of solid-liquid interfacial transport is important for different applications such cancer as treatment nanoparticles1 78, solar thermal heating 17 9 based on thermal therapeutics and 80 8 2 , colloids and nanofluids1 -1 . Experiments on thermal conductance of solid-liquid interfaces typically employ suspensions of metal nanorods in water or organic solvents 183-187. Although planar solid-liquid interfaces modified with experimentally studied' 88 and hydrophilic hydrophobic SAMs have been , there have been no controlled studies of solid-liquid interfaces with SAMs and without SAMs. In this chapter, we systematically study the thermal conductance dependence on the functional end groups using the same class of SAM, as well as the dependence on 95 We show that the addition of the extra SAM layer between the planar chain length. Au and ethanol enhances the thermal transport as SAM serves as a transitional layer. This shares the same idea as discussed in Chapter 5 that a smoother transition favors phonon transport across an interface. Specifically, we find that increasing the chain length does not adversely impact the interfacial thermal conductance, while different functional end groups enhance the thermal conductance to different extents. 7.2 Sample Preparation and Experimental Setup Alkanethiol and alkanedithiol SAMs are formed on a gold surface using the standard wet chemical preparation method8 9 . Specifically, Au coated glass slides (purchased from Phasis Sirl) are immersed in a dilute (-2 mM) ethanolic solution of the thiols (purchased from Sigma-Aldrich) for 18-24 hours at room temperature. Excess thiol molecules not bonded to the surfaces are removed by cleaning with ethanol and a nitrogen gun. Molecular schematics of the four different SAMs grown for this study are shown in Fig. 7-1. All the SAMs have an oil-like chain (-(CH 2),-) as a molecular backbone, a head group containing a sulfur atom which can strongly bond to the Au surface, and a terminal end group. Hexanedithiol and hexanethiol have the same alkane chain length but a different functional group (thiol group -SH vs methyl group -CH 3). In contrast, hexanethiol, undecanethiol and hexadecanethiol have the same end group (-CH 3) but different alkane chain lengths. -(CH2) n- (a) Figure 7-1 (b) Schematic SHCH 2(CH 2)4 CH2SH; of (b) (d) (c) SAMs study. (a) Hexenedithiol CH3(CH 2) 4CH2SH; (c) Undecanethiol used Hexanethiol in this CH 3(CH 2)9CH 2SH; and (d) Hexadecanethiol CH3(CH 2)1 4CH2SH For characterization of the thermal interface conductance between the Au and ethanol, the Au slides (with and without SAMs) are placed in contact with half of a demountable cuvette with a 1 mm thick channel, which is then filled with ethanol, as 96 shown in Fig. 7-2(a). The Au film, on which the SAMs are grown, serves as the transducer layer for time-domain thermoreflectance (TDTR) measurements of the interfacial thermal conductance. The detailed TDTR methodology can be found elsewhere190 ' 191. In brief, pump laser pulses (~150 fs pulse-width, 80.7 MHz repetition rate) with a spot diameter of 60 im pass through the glass slide and are absorbed by the 100 nm Au film, heating the sample. Optically time-delayed probe pulses coaxial with the pump beam with a diameter of 12 gm measure the temperature decay after the pump pulses through the change in reflectivity. The amplitude of the pump pulse train is modulated at 9 MHz to allow for lock-in detection of the thermoreflectance response. The sample properties, including the interface thermal conductance, impact the cooling curve and are extracted by fitting the data with a diffusive heat transfer model 190. Typical phase data are shown in Fig. 7-2(b). (a) Glass A -SAM Ethanol 97 (b) -40k - with Hexanedithiol 5 -45 Cu -50 without SAM -55 500 1000 2000 1500 2500 3000 3500 Delay (ps) Figure 7-2 (a) Schematic of sample arrangement; (b) Phase data for samples without SAM and with hexanedithiol. 7.3 Results and Discussion To measure the Au-ethanol interface thermal conductance, we first calibrate the thermal properties of the glass slides and ethanol using a clean gold-coated glass slide. The thermal conductivities of glass and ethanol are found to be 1.25 W/m/K and 0.17 W/m/K, respectively, and the measured thermal conductance between glass and Au is 51 MW/m 2/K, consistent with textbook values. These values are kept constant and in all the subsequent fittings we only fit the thermal conductance between Au and ethanol for the samples with SAMs. The thermal conductance between Au and ethanol at room temperature are shown in Fig. 7-3. The error bars represent the standard deviation between 4-5 different locations on the same sample. The error bars are smaller for the sample without SAMs, due to the greater uniformity of the bare Au surface. The spot-to-spot variation may be indicative of non-uniform SAM coverage due to the polycrystalline nature of the Au surface. 98 120 100I 801 E 60T 40- gL 20- Without SAMs Hexanethiol Hexanedithiol Undecanethiol Hexadecanethiol Figure 7-3 Thermal conductance between Au and ethanol with and without SAMs from TDTR measurements at room temperature. Counterintuitively, the interface conductance between Au and ethanol is improved by the existence of an extra molecular layer in all cases, as shown in Fig. 7-3. While hexanedithiol improves the thermal conductance by a factor of -5, hexanthiol, undecanethiol and hexadecanethiol all improve it by only a factor of-2. For all SAMs studied, this improvement is likely due to the strong chemical bond formed between Au and SAMs and between SAMs and ethanol. At one end of the oily chain, all the SAMs have one thiol group covalently bonded to the gold surface. At the other end, hexanedithiol has a thiol group exposed to the ethanol, while hexanethiol, undecanethiol and hexadecanethiol have a methyl functional group exposed to the ethanol. Since ethanol itself contains both a polar group (-OH) and a nonpolar methyl group, both the thiol end group and the methyl end group could form stronger chemical bonds with ethanol than the bare gold surface in contact with ethanol. In other words, the SAMs serve as a bridge between Au and ethanol and facilitate thermal transport. While the thermal conductance of the hexanedithiol SAM significantly outperforms the hexanethiol SAM, they differ only by the functional end group. We measure the advancing contact angle at three different locations on each sample by using a contact angle goniometer. The measured contact angles of ethanol on hexanethiol, undecanethiol and hexadecanethiol are 0 = 340 4', 0 = 350 30 and 0 = 410 2* respectively, while the contact angle on hexanedithiol is too small to be measured, as shown in Fig. 7-4. This implies the stronger bonding between hexanedithiol and 99 ethanol, which likely leads to the higher thermal conductance than the sample with hexanethiol1 9 2 . These results agree with earlier experiments, which showed that hydrophilic SAMs produced higher interface conductances than hydrophobic SAMs for interfaces between water and metal 8 8 . In addition to changing the bonding strength, the SAM layers may impact the acoustic mismatch between solid and liquid. Specifically, SAM surfaces which demonstrate smaller contact angles may reduce the acoustic mismatch between the gold and ethanol, by making the interface more solid-like '93 Figure 7-4 Contact angles, 0, of ethanol on Au surface modified by (a) hexanethiol and (b) hexanedithiol SAMs. We use optical spectroscopic ellipsometry to estimate the SAM thickness with a Cauchy model. The measured thicknesses for hexanethiol, undecanethiol and hexadecanethiol are 0.943 0.057 nm, 1.389 0.054 nm and 2.284+0.061 100 nm . 19 4 respectively, in good agreement with previous measurements on hexadecanethiol Moreover, the measured thickness increases almost linearly as the number of carbon atoms increases from 6, to 11, to 16. The tilt angles are 0*-35'. Despite the difference in chain length and film thickness, there is no observable difference in the thermal conductance for hexanethiol, undecanethiol and hexadecanethiol. This indicates that phonon transport is ballistic along the alkane chain, as well as that the phonon vibrational spectra match regardless of the chain length investigated in this work. This is consistent with earlier experiments 174, 195 and simulations 196, 197 for solid-SAM-solid interfaces. 7.4 Conclusion In summary, we use TDTR measurements to study the thermal conductance between Au and ethanol with various interfacial SAMs. We show that the SAMs enhance the thermal transport from Au to ethanol. The interfacial thermal conductance is insensitive to the length of the alkane chain length, but strongly dependent on the functional group. Our results shed lights on strategies to further tune the interfacial conductance for practical applications. 101 102 8. Summary and Future Work 8.1 Summary This thesis explored nanoscale heat transfer using both atomistic simulations and ultrafast laser measurements. The detailed calculations expand our fundamental understanding of the transport processes and provide guidance for practical materials design to meet different needs. The first portion of the thesis focuses on phonon transport properties in bulk materials while the second portion focuses on phonon transport across interfaces. Chapter 2 describes how to obtain spectral phonon transport properties in germanium. Using a combination of molecular dynamics and lattice dynamics, we can extract the information of phonon lifetimes by tracing the temporal amplitude decay of each mode. With the knowledge of group velocity from phonon dispersion, we can obtain phonon mean free paths and which phonons carry most heat. The empirical potential employed, however, leads to a large discrepancy on the total thermal conductivity between calculation and experiment. A better potential is thus needed. Chapter 3 introduces in detail how to obtain phonon properties in PbSe, PbTe and their alloys using first-principles calculations. Density functional theory is used to compute the electronic band structure and derive more accurate force constants. The excellent agreements with experimental values for the phonon dispersion and thermal conductivity demonstrate the predictive power of this approach. The phonon properties including lifetimes, group velocities and mean free paths are presented. In addition, the importance of optical phonons is emphasized, which is crucial for the low thermal conductivity of these two materials and their alloys. Chapter 4 discusses the importance of optical phonons in silicon nanowires. Since the long mean free path acoustic phonons get strongly scattered at interfaces/boundaries, optical phonons are much less influenced. This leads to a redistribution of the relative importance between acoustic and optical phonons. Chapter 5 investigates phonon transmission across a single Si/Ge interface using the Green's function method. Phonon transmission can be enhanced by interface 103 roughness introduced by atomic mixing. This can attribute to a smoother transition in the phonon vibrational spectrum. Chapter 6 looks into coherent phonon transport in Si/Ge superlattices using the Green's function method. Interface roughness can destroy coherence of high frequency phonon modes. To destroy coherence of low frequency modes, a scattering length scale on the order of a period length is needed. Chapter 7 examines the influence of chemical bonding on the solid-liquid interface conductance using time-domain thermoreflectance measurements. Self-assembled monolayers are used to modify the gold-ethanol interface. Enhanced thermal conductance is observed compared to the gold-ethanol interface without self-assembled monolayers. Certain self-assembled monolayers are more effective than others. These atomistic level studies not only improve our fundamental understanding of nanoscale thermal transport but also pave the way for multiscale simulation from first-principles. 8.2 Future Directions One natural extension of this thesis work is to pursue multiscale modeling of thermal transport. Understanding of heat conduction in nano to meso-scale structures from multiscale thermal modeling could guide the practical materials design for applications from thermoelectrics to the thermal management of electronic systems. Using the extracted mode-dependent phonon bulk mean free paths and interfacial transmission, both from first-principles approaches, and by integrating them into the Boltzmann transport equation or Monte Carlo simulation, we can predict the thermal conductivity of various nanocomposites. Furthermore, developing an improved effective medium model based on the knowledge of multiscale simulation would be useful for practical simulations. The other direction would be look further into the thermal transport of soft materials. On the fundamental side, mechanisms of thermal transport in soft materials are of great interest. Significant progress has been made in understanding thermal transport properties in crystalline solids. The knowledge of thermal transport in soft materials, however, is presently falling far behind. While the periodicity in crystals with well-defined vibrational or phonon modes has been actively studied, the lack of 104 periodicity in soft materials imposes a big challenge onto the fundamental description of the vibration modes, let alone their properties. The vibrational modes are no longer pure plane-waves, except in the low-frequency limit. The transport mechanisms are still under debate. For amorphous materials, fracton transport was proposed and this approach attributes the increase in thermal conductivity above the plateau, in part, to anharmonic fracton hopping. 198 Yet Freeman and Anderson discussed that it was not clear which type of excitation transport heat above the plateau.1 99 Cahill and Pohl argued that their experimental data did not support the fracton theory. 200 Using normal mode analysis, Larkin and McGaughey 201 showed that the lifetimes of amorphous materials show little frequency dependence and a significant number of modes fall below the loffe-Regel limit. More studies along this line would be useful to gain a comprehensive understanding. In addition, thermal transport across interfaces between hard and soft materials needs more investigation. It would be interesting to study how the vibrational modes couple to each other at the interface and how much one can tune the coupling to engineer the interface conductance. On the applications side, a deep understanding of thermal transport in soft materials and soft/hard interfaces promises applications ranging from photothermal nanotheuraputics to battery thermal management. 105 106 Biography 1 R. Peierls, Annalen Der Physik 3, 1055 (1929). 2 P. G. Klemens, Solid State Physics-Advances in Research and Applications 7, 5 1(1958). M. G. Holland, Physical Review 132, 2461 (1963). W. J. de Haas and T. Biermasz, Physica 5, 619 (1938). H. B. G. Casimir, Physica 5, 495 (1938). 6 J. M. Ziman, Electrons and phonons; the theory of transportphenomena in 3 4 solids (Clarendon Press, Oxford, 1960). 7 8 9 10 11 12 13 A. J. C. Ladd, B. Moran, and W. G. Hoover, Physical Review B 34, 5058 (1986). S. G. Volz and G. Chen, Physical Review B 61, 2651 (2000). J. Li, L. Porter, and S. Yip, Journal of Nuclear Materials 255, 139 (1998). J. W. Che, T. Cagin, W. Q. Deng, and W. A. Goddard, Journal of Chemical Physics 113, 6888 (2000). C. Oligschleger and J. C. Schon, Physical Review B 59, 4125 (1999). M. S. Green, Journal of Chemical Physics 22, 398 (1954). R. Kubo, M. Yokota, and S. Nakajima, Journal of the Physical Society of Japan 12, 1203 (1957). 14 W. G. A. Hoover, W.T., in Theoretical Chemistry: Advances and Perspectives, edited by H. Eyring and D. Henderson (Academic Press, New York, 1975), Vol. 1, p. 1. 15 S. Kotake and S. Wakuri, JSME International Journal Series B-Fluids and 16 Thermal Engineering 37, 103 (1994). T. Ikeshoji and B. Hafskjold, Molecular Physics 81, 251 (1994). 17 G. Chen, Nanoscale energy transportand conversion : a paralleltreatment of electrons, molecules, phonons, and photons (Oxford University Press, New York, 2005). 18 19 20 21 A. S. Henry and G. Chen, Journal of Computational and Theoretical Nanoscience 5, 141 (2008). A. J. H. McGaughey and M. Kaviany, Physical Review B 69, 094303 (2004). J. E. Turney, E. S. Landry, A. J. H. McGaughey, and C. H. Amon, Physical Review B 79, 064301 (2009). B. Qiu, H. Bao, G. Q. Zhang, Y Wu, and X. L. Ruan, Computational Materials 107 Science 53, 278 (2012). 22 D. A. Broido, A. Ward, and N. Mingo, Physical Review B 72, 014308 (2005). 23 M. E. Tuckerman, Journal of Physics-Condensed Matter 14, R1297 (2002). H. Kim and M. Kaviany, Physical Review B 86, 045213 (2012). D. Broido, M. Malorny, G. Birner, N. Mingo, and D. Stewart, Applied Physics 24 25 28 Letters 91, 231922 (2007). G. Deinzer, G. Birner, and D. Strauch, Physical Review B 67, 144304 (2003). K. Esfarjani and H. Stokes, Physical Review B 77, 144112 (2008). K. Esfarjani, G. Chen, and H. Stokes, Physical Review B 84, 085204 (2011). 29 J. Garg, N. Bonini, B. Kozinsky, and N. Marzari, Physical Review Letters 106, 30 045901 (2011). J. Garg, N. Bonini, and N. Marzari, Nano Letters 11, 5135 (2011). Z. T. Tian, J. Garg, K. Esfarjani, T. Shiga, J. Shiomi, and G. Chen, Physical 26 27 31 32 Review B 85, 184303 (2012). J. Shiomi, K. Esfarjani, and G. Chen, Physical Review B 84, 104302 (2011). 33 T. Shiga, J. Shiomi, J. Ma, 0. Delaire, T. Radzynski, A. Lusakowski, K. Esfarjani, and G. Chen, Physical Review B 85, 155203 (2012). 34 T. Luo, J. Garg, J. Shiomi, K. Esfarjani, and G. Chen, Europhysics Letters 101, 35 16001 (2013). A. J. Minnich, J. A. Johnson, A. J. Schmidt, K. Esfarjani, M. S. Dresselhaus, K. A. Nelson, and G. Chen, Physical Review Letters 107, 095901 (2011). 36 37 D. Y. Li, Y. Y. Wu, P. Kim, L. Shi, P. D. Yang, and A. Majumdar, Applied Physics Letters 83, 2934 (2003). M. S. Dresselhaus, G. Chen, M. Y Tang, R. G. Yang, H. Lee, D. Z. Wang, Z. F. 38 A. J. Minnich, M. S. Dresselhaus, Z. F. Ren, and G. Chen, Energy & Ren, J. P. Fleurial, and P. Gogna, Advanced Materials 19, 1043 (2007). Environmental Science 2, 466 (2009). 39 40 Z. T. Tian, S. Lee, and G. Chen, Journal of Heat Transfer 135, 061605 (2013). J. Chen, G. Zhang, and B. W. Li, Nano Letters 10, 3978 (2010). 41 S. Plimpton, Journal of Computational Physics 117, 1 (1995). 42 J. Zi, K. M. Zhang, and X. D. Xie, Physical Review B 41, 12915 (1990). 43 44 F. H. Stillinger and T. A. Weber, Physical Review B 31, 5262 (1985). R. Kubo, Journal of the Physical Society of Japan 12(1957). 45 H. J. Goldsmid, Introduction to Thermoelectricity (Springer, 2010). 46 G. J. Snyder and E. S. Toberer, Nature Materials 7, 105 (2008). 47 J. R. Sootsman, D. Y Chung, and M. G. Kanatzidis, Angewandte Chemie-International Edition 48, 8616 (2009). 48 J. P. Heremans, V. Jovovic, E. S. Toberer, A. Saramat, K. Kurosaki, A. 108 Charoenphakdee, S. Yamanaka, and G. J. Snyder, Science 321, 554 (2008). 49 M. Zebarjadi, K. Esfarjani, M. S. Dresselhaus, Z. F. Ren, and G. Chen, Energy & Environmental Science 5, 5147 (2012). 50 51 Y. Z. Pei, X. Y. Shi, A. LaLonde, H. Wang, L. D. Chen, and G. J. Snyder, Nature 473, 66 (2011). J. P. Heremans, C. M. Thrush, and D. T. Morelli, Physical Review B 70, 115334 (2004). Q. He, J. Androulakis, M. G. Kanatzidis, and V. P. Dravid, Nano Letters 12, 52 J. 53 343 (2012). J. Androulakis, I. Todorov, D. Y. Chung, S. Ballikaya, G. Y. Wang, C. Uher, 54 and M. Kanatzidis, Physical Review B 82, 115209 (2010). J. Q. He, J. R. Sootsman, S. N. Girard, J. C. Zheng, J. G. Wen, Y. M. Zhu, M. G. Kanatzidis, and V. P. Dravid, Journal of the American Chemical Society 132, 8669 (2010). 55 J. Q. He, S. N. Girard, M. G. Kanatzidis, and V. P. Dravid, Advanced Functional Materials 20, 764 (2010). 56 57 D. Parker and D. J. Singh, Physical Review B 82, 035204 (2010). Q. Y. Zhang, H. Wang, W. S. Liu, H. Z. Wang, B. Yu, Q. Zhang, Z. T. Tian, G. Ni, S. Lee, K. Esfarjani, G. Chen, and Z. F. Ren, Energy & Environmental 58 59 Science 5, 5246 (2012). H. Wang, Y. Z. Pei, A. D. LaLonde, and G. J. Snyder, Advanced Materials 23, 1366 (2011). B. Poudel, Q. Hao, Y. Ma, Y. C. Lan, A. Minnich, B. Yu, X. A. Yan, D. Z. Wang, A. Muto, D. Vashaee, X. Y Chen, J. M. Liu, M. S. Dresselhaus, G. 60 61 62 63 64 Chen, and Z. F. Ren, Science 320, 634 (2008). G. Joshi, X. Yan, H. Z. Wang, W. S. Liu, G. Chen, and Z. F. Ren, Advanced Energy Materials 1, 643 (2011). X. A. Yan, G. Joshi, W. S. Liu, Y C. Lan, H. Wang, S. Lee, J. W. Simonson, S. J. Poon, T. M. Tritt, G. Chen, and Z. F. Ren, Nano Letters 11, 556 (2011). A. J. H. McGaughey and A. Jain, Applied Physics Letters 100, 061911 (2012). B. Abeles, Physical Review 131, 1906 (1963). A. F. Ioffe, S. V. Airapetiants, A. V. Ioffe, N. V. Kolomoetz, and L. S. Stilbans, Doklady Akademii Nauk Sssr 106, 981 (1956). 65 I. Kudman, Journal of Materials Science 7, 1027 (1972). 66 Y. K. Koh, C. J. Vineis, S. D. Calawa, M. P. Walsh, and D. G. Cahill, Applied Physics Letters 94, 153101 (2009). 67 Y. I. Ravich, B. A. Efimova, and I. A. Smirnov, Semiconducting Lead Chalcogenides (Plenum, New York, 1970). 109 68 A. A. El-Sharkawy, A. M. Abou El-Azm, M. I. Kenawy, A. S. Hillal, and H. M. Abu-Basha, International Journal of Thermophysics 4, 9 (1983). 69 70 E. D. Devyatkova and I. A. Smirnov, Sov. Phys. Solid State 3(1962). T. Beechem, J. C. Duda, P. E. Hopkins, and P. M. Norris, Applied Physics Letters 97, 061907 (2010). 71 D. P. Sellan, J. E. Turney, A. J. H. McGaughey, and C. H. Amon, Journal of 72 Applied Physics 108, 113524 (2010). Z. T. Tian, K. Esfarjani, J. Shiomi, A. S. Henry, and G. Chen, Applied Physics 7 Letters 99, 053122 (2011). A. Ward and D. A. Broido, Physical Review B 81, 085205 (2010). 0. Delaire, J. Ma, K. Marty, A. F. May, M. A. McGuire, M. H. Du, D. J. Singh, 74 A. Podlesnyak, G. Ehlers, M. D. Lumsden, and B. C. Sales, Nature Materials 10, 614 (2011). 75 S. Baroni, P. Giannozzi, and A. Testa, Physical Review Letters 58, 1861 (1987). 76 A. Debernardi, S. Baroni, and E. Molinari, Physical Review Letters 75, 1819 (1995). X. Gonze, Physical Review A 52, 1086 (1995). 78 P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. KokaUj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. Seitsonen, A. Smogunov, P. Umari, and R. Wentzcovitch, Journal of Physics-Condensed Matter 21, ARTN 395502 (2009). 79 80 81 H. J. Monkhorst and J. D. Pack, Physical Review B 13, 5188 (1976). X. Gonze and J. P. Vigneron, Physical Review B 39, 13120 (1989). A. A. Maradudin and A. E. Fein, Physical Review 128, 2589 (1962). 82 R. A. Cowley, Reports on Progress in Physics 31, 123 (1968). 83 C. Dames and G. Chen, in Thermoelectrics handbook: macro to nano edited by Rowe, David Michael (CRC/Taylor & Francis, Boca Raton, 2006). 85 S. Tamura, Physical Review B 27, 858 (1983). P. R. Vijaraghavan, S. K. Sinha, and P. K. Iyengar, Proc.Nuc.Phys.and Solid State Symp. 16(1973). 86 W. Cochran, R. A. Cowley, G. Dolling, and M. M. Elcombe, Proc. R. Soc. 84 87 Lond. A 293, 8 (1966). J. M. An, A. Subedi, and D. J. Singh, Solid State Communications 148, 417 (2008). 110 88 0. Kilian, G. Allan, and L. Wirtz, Physical Review B 80, 245208 (2009). 89 Y. Zhang, X. Z. Ke, C. F. Chen, J. Yang, and P. R. C. Kent, Physical Review B 80, 024304 (2009). 90 P. G. Klemens, Proceedings of the Royal Society of London Series a-Mathematical and Physical Sciences 208, 108 (1951). 91 92 93 A. I. Boukai, Y Bunimovich, J. Tahir-Kheli, J. K. Yu, W. A. Goddard, and J. R. Heath, Nature 451, 168 (2008). A. I. Hochbaum, R. K. Chen, R. D. Delgado, W. J. Liang, E. C. Garnett, M. Najarian, A. Majumdar, and P. D. Yang, Nature 451, 163 (2008). K. Hippalgaonkar, B. Huang, R. Chen, K. Sawyer, P. Ercius, and A. Majumdar, Nano Letters 10, 4341 (2010). 94 95 96 97 S. G. Volz and G. Chen, Applied Physics Letters 75, 2056 (1999). N. Mingo, L. Yang, D. Li, and A. Majumdar, Nano Letters 3, 1713 (2003). P. G. Murphy and J. E. Moore, Physical Review B 76, 155313 (2007). I. Ponomareva, D. Srivastava, and M. Menon, Nano Letters 7, 1155 (2007). 99 R. Chen, A. I. Hochbaum, P. Murphy, J. Moore, P. D. Yang, and A. Majumdar, Physical Review Letters 101, 105501 (2008). D. Donadio and G. Galli, Physical Review Letters 102, 195901 (2009). 100 G. Chen, T. Zeng, T. Borca-Tasciuc, and D. Song, Materials Science and 98 Engineering a-Structural Materials Properties Microstructure and Processing 101 102 103 104 105 292, 155 (2000). D. G. Cahill, W. K. Ford, K. E. Goodson, G. D. Mahan, A. Majumdar, H. J. Maris, R. Merlin, and Phillpot, Sr., Journal of Applied Physics 93, 793 (2003). C. Dames and G. Chen, Journal of Applied Physics 95, 682 (2004). L. H. Liang and B. W. Li, Physical Review B 73, 153303 (2006). S. K. Bux, R. G. Blair, P. K. Gogna, H. Lee, G. Chen, M. S. Dresselhaus, R. B. Kaner, and J. P. Fleurial, Advanced Functional Materials 19, 2445 (2009). Y. F. Chen, D. Y Li, J. R. Lukes, and A. Majumdar, Journal of Heat Transfer-Transactions of the Asme 127, 1129 (2005). 106 Z. Tian, J. Garg, K. Esfarjani, T. Shiga, J. Shiomi, and G. Chen, Phys. Rev. B 85, ARTN 184303 (2012). 107 N. Mingo, D. Stewart, D. Broido, and D. Srivastava, Physical Review B 77, 033418 (2008). 108 109 D. Stewart, I. Savic, and N. Mingo, Nano Letters 9, 81 (2009). G. Fagas, A. Kozorezov, C. Lambert, J. Wigmore, A. Peacock, A. Poelaert, and 110 R. den Hartog, Physical Review B 60, 6459 (1999). H. Zhao and J. Freund, Journal of Applied Physics 105, 013515 (2009). II L. Sun and J. Murthy, Journal of Heat Transfer-Transactions of the ASME 132, 111 102403 (2010). 113 D. Kechrakos, Journal of Physics-Condensed Matter 3, 1443 (1991). R. J. Stevens, L. V. Zhigilei, and P. M. Norris, International Journal of Heat 114 and Mass Transfer 50, 3977 (2007). T. English, J. Duda, J. Smoyer, D. Jordan, P. Norris, and L. Zhigilei, Physical 112 117 Review B 85, ARTN 035438 (2012). W. Little, Canadian Journal of Physics 37, 334 (1959). E. Swartz and R. Pohl, Reviews of Modern Physics 61, 605 (1989). P. Schelling, S. Phillpot, and P. Keblinski, Applied Physics Letters 80, 2484 118 (2002). C. Kimmer, S. Aubry, A. Skye, and P. Schelling, Physical Review B 75, ARTN 115 116 119 120 144105 (2007). P. Schelling and S. Phillpot, Journal of Applied Physics 93, 5377 (2003). Z. Tian, S. Kim, Y. Sun, and B. White, Proceedings of the ASME InterPACK 125 Conference 2009, Vol 1, 607 (2009). N. Zuckerman and J. Lukes, Physical Review B 77, 094302 (2008). D. Young and H. Maris, Physical Review B 40, 3685 (1989). S. Pettersson and G. Mahan, Physical Review B 42, 7386 (1990). H. Zhao and J. Freund, Journal of Applied Physics 97, 024903 (2005). J. Wang and J. Wang, Physical Review B 74, 054303 (2006). 126 S. Datta, Electronic Transport in Mesoscopic Systems (Cambridge University 127 Press, Cambridge, UK, 1997). N. Mingo and L. Yang, Physical Review B 68, ARTN 245406 (2003). N. Mingo and L. Yang, Physical Review B 70, ARTN 249901 (2004). 121 122 123 124 128 129 W. Zhang, T. Fisher, and N. Mingo, Journal of Heat Transfer-Transactions of the ASME 129, 483 (2007). 130 W. Zhang, T. Fisher, and N. Mingo, Numerical Heat Transfer Part B-Fundamentals 51, 333 (2007). 131 S. Volz, Thermal Nanosystems and Nanomaterials 118, 1 (2009). 132 A. Dhar and D. Roy, Journal of Statistical Physics 125, 805 (2006). 133 P. Hopkins, P. Norris, M. Tsegaye, and A. Ghosh, Journal of Applied Physics 134 106, 063503 (2009). X. Li and R. Yang, Journal of Physics-Condensed Matter 24, 155302 (2012). 135 M. Sancho, J. Sancho, and J. Rubio, Journal of Physics F-Metal Physics 15, 136 137 851 (1985). R. Landauer, Philosophical Magazine 21, 863 (1970). J. Katerberg, C. Reynolds, and A. Anderson, Physical Review B 16, 673 (1977). 112 138 139 140 141 J. P. Perdew and A. Zunger, Physical Review B 23, 5048 (1981). E. S. Landry and A. J. H. McGaughey, Physical Review B 80, 165304 (2009). Y. Kanamori, M. Sasaki, and K. Hane, Optics Letters 24, 1422 (1999). Y. F. Huang, S. Chattopadhyay, Y. J. Jen, C. Y Peng, T. A. Liu, Y. K. Hsu, C. L. 142 Pan, H. C. Lo, C. H. Hsu, Y. H. Chang, C. S. Lee, K. H. Chen, and L. C. Chen, Nature Nanotechnology 2, 770 (2007). K. Choi, S. H. Park, Y M. Song, Y T. Lee, C. K. Hwangbo, H. Yang, and H. S. 143 Lee, Advanced Materials 22, 3713 (2010). K. C. Park, H. J. Choi, C. H. Chang, R. E. Cohen, G. H. McKinley, and G. 144 Barbastathis, Acs Nano 6, 3789 (2012). K. Tai, L. Yang, Y Wang, J. Wynn, and A. Cho, Applied Physics Letters 56, 2496 (1990). 145 M. Peters, B. Thibeault, D. Young, J. Scott, F. Peters, A. Gossard, and L. Coldren, Applied Physics Letters 63, 3411 (1993). 146 M. Hong, J. Mannaerts, J. Hong, R. Fischer, K. Tai, J. Kwo, J. Vandenberg, Y. 147 Wang, and J. Gamelin, Journal of Crystal Growth 111, 1071 (1991). S. Chalmers, K. Lear, and K. Killeen, Applied Physics Letters 62, 1585 (1993). 148 S. M. Lee, D. G. Cahill, and R. Venkatasubramanian, Applied Physics Letters 149 70, 2957 (1997). T. Borca-Tasciuc, W. L. Liu, J. L. Liu, T. F. Zeng, D. W. Song, C. D. Moore, G. Chen, K. L. Wang, M. S. Goorsky, T. Radetic, R. Gronsky, T. Koga, and M. S. Dresselhaus, Superlattices Microstruct. 28(2000). 150 151 152 E. S. Landry and A. J. H. McGaughey, Physical Review B 79, 075316 (2009). G. Chen, Physical Review B 57, 14958 (1998). M. N. Luckyanova, Garg, Jivtesh, Esfarjani, Keivan, Jandl, Adam, Bulsara, Mayank T., Schmidt, Aaron J., Minnich, Austin J., Chen, Shuo, Dresselhaus, Mildred S., Ren, Zhifeng, Fitzgerald, Eugene A., and Chen, Gang, 338, 936 (2012). 153 G. Chen, M. S. Dresselhaus, G. Dresselhaus, J. P. Fleurial, and T. Caillat, International Materials Reviews 48, 45 (2003). 154 C. L. Tien and G. Chen, Journal of Heat Transfer 116, 799 (1994). T. Borca-Tasciuc, W. L. Liu, J. L. Liu, T. F. Zeng, D. W. Song, C. D. Moore, G. Chen, K. L. Wang, M. S. Goorsky, T. Radetic, R. Gronsky, T. Koga, and M. S. 155 Dresselhaus, Superlattices and Microstructures 28, 199 (2000). 156 W. S. Capinski, H. J. Maris, T. Ruf, M. Cardona, K. Ploog, and D. S. Katzer, Physical Review B 59, 8105 (1999). 157 J. C. Caylor, K. Coonley, J. Stuart, T. Colpitts, and R. Venkatasubramanian, 113 Applied Physics Letters 87, 023105 (2005). 158 159 G. Chen, Journal of Heat Transfer-Transactions of the Asme 124, 320 (2002). M. N. Luckyanova, J. Garg, K. Esfarjani, A. Jandl, M. T. Bulsara, A. J. Schmidt, A. J. Minnich, S. Chen, M. S. Dresselhaus, Z. F. Ren, E. A. Fitzgerald, and G. Chen, Science 338, 936 (2012). 160 J. Ravichandran, A. K. Yadav, R. Cheaito, P. B. Rossen, A. Soukiassian, S. J. Suresha, J. C. Duda, B. M. Foley, C.-H. L. Ye Zhu, A. W. Lichtenberger, J. E. Moore, D. A. Muller, D. G. Schlom, P. E. Hopkins, A. Majumdar, R. Ramesh, and M. A. Zurbuchen, Nature Materials, 168 (2013). 161 162 163 164 165 166 167 168 169 170 171 172 173 174 G. Chen and M. Neagu, Applied Physics Letters 71, 2761 (1997). S. Y Ren and J. D. Dow, Physical Review B 25, 3750 (1982). P. Hyldgaard and G. D. Mahan, Physical Review B 56, 10754 (1997). D. A. Broido and T. L. Reinecke, Physical Review B 70, 081310 (2004). B. Yang and G. Chen, Physical Review B 67, 195311 (2003). M. V. Simkin and G. D. Mahan, Physical Review Letters 84, 927 (2000). J. Garg and G. Chen, Physical Review B 87, 140302 (2013). S. Volz, J. B. Saulnier, G. Chen, and P. Beauchamp, High Temperatures-High Pressures 32, 709 (2000). X. B. Li and R. G. Yang, Physical Review B 86, 054305 (2012). Z. T. Tian, K. Esfarjani, and G. Chen, Physical Review B 86, 235304 (2012). P. E. Hopkins and J. R. Serrano, Physical Review B 80, 201408 (2009). L. Hu, L. F. Zhang, M. Hu, J. S. Wang, B. W. Li, and P. Keblinski, Physical Review B 81, 235427 (2010). Z. T. Tian, B. E. White, and Y. Sun, Applied Physics Letters 96, 263113 (2010). R. Y Wang, R. A. Segalman, and A. Majumdar, Applied Physics Letters 89, 173113 (2006). 175 176 177 P. E. Hopkins, M. Baraket, E. V. Barnat, T. E. Beechem, S. P. Kearney, J. C. Duda, J. T. Robinson, and S. G. Walton, Nano Letters 12, 590 (2012). K. C. Collins, S. Chen, and G. Chen, Applied Physics Letters 97, 083102 (2010). M. D. Losego, M. E. Grady, N. R. Sottos, D. G. Cahill, and P. V. Braun, Nature Materials 11, 502 (2012). 178 S. Lal, S. E. Clare, and N. J. Halas, Accounts of Chemical Research 41, 1842 (2008). 179 180 o. Neumann, A. S. Urban, J. Day, S. Lal, P. Nordlander, and N. J. Halas, Acs Nano 7, 42 (2013). J. W. Gao, R. T. Zheng, H. Ohtani, D. S. Zhu, and G. Chen, Nano Letters 9, 114 181 4128 (2009). R. T. Zheng, J. W. Gao, J. J. Wang, S. P. Feng, H. Ohtani, J. B. Wang, and G. Chen, Nano Letters 12, 188 (2012). 182 R. Prasher, P. E. Phelan, and P. Bhattacharya, Nano Letters 6, 1529 (2006). 183 J. Y. Huang, J. Park, W. Wang, C. J. Murphy, and D. G. Cahill, Acs Nano 7, 184 185 186 589 (2013). J. Park, J. Y Huang, W. Wang, C. J. Murphy, and D. G. Cahill, Journal of Physical Chemistry C 116, 26335 (2012). A. J. Schmidt, J. D. Alper, M. Chiesa, G. Chen, S. K. Das, and K. Hamad-Schifferli, Journal of Physical Chemistry C 112, 13320 (2008). 0. M. Wilson, X. Y Hu, D. G. Cahill, and P. V. Braun, Physical Review B 66, 224301 (2002). 187 Z. B. Ge, D. G. Cahill, and P. V. Braun, Journal of Physical Chemistry B 108, 188 Z. B. Ge, D. G. Cahill, and P. V. Braun, Physical Review Letters 96, 186101 18870 (2004). 189 190 191 192 (2006). J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo, and G. M. Whitesides, Chemical Reviews 105, 1103 (2005). A. Schmidt, M. Chiesa, X. Y. Chen, and G. Chen, Review of Scientific Instruments 79, 064902 (2008). H. K. Lyeo and D. G. Cahill, Physical Review B 73, 144301 (2006). D. D. Gandhi, M. Lane, Y Zhou, A. P. Singh, S. Nayak, U. Tisch, M. Eizenberg, and G. Ramanath, Nature 447, 299 (2007). 193 S. Murad and I. K. Puri, Applied Physics Letters 92, 133105 (2008). 194 F. Bordi, M. Prato, 0. Cavalleri, C. Cametti, M. Canepa, and A. Gliozzi, Journal of Physical Chemistry B 108, 20263 (2004). 195 Z. H. Wang, J. A. Carter, A. Lagutchev, Y. K. Koh, N. H. Seong, D. G. Cahill, and D. D. Dlott, Science 317, 787 (2007). 196 T. F. Luo and J. R. Lloyd, Journal of Heat Transfer-Transactions of the Asme 132, 032401 (2010). 197 J. C. Duda, C. B. Saltonstall, P. M. Norris, and P. E. Hopkins, Journal of Chemical Physics 134, 094704 (2011). 198 S. Alexander, 0. Entinwohlman, and R. Orbach, Physical Review B 34, 2726 (1986). 199 J. J. Freeman and A. C. Anderson, Physical Review B 34, 5684 (1986). 200 D. G. Cahill and R. 0. Pohl, Physical Review B 35, 4067 (1987). J. M. Larkin and A. J. H. McGaughey, Journal of Applied Physics 114, 023507 201 (2013). 115