Document 10981474
advertisement

INTERFACIAL PARTITIONING DURING SOLIDIFICATION
by
JAMES C. BAKER
B.S.
Michigan State University
(1965)
M.S.
Michigan State University
(1966)
Submitted
in Partial Fulfillment of the
Requirements
for the Degree of
DOCTOR OF PHILOSOPHY
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June, 1970
Signature of Author
Dep-tment of Metallurgy
May 14, 1970
af
Certified by
·Thesis Supervisor
.'
~ ~<
Accepted by
Chairman, DepartmentalY
Committee
on
Graduate Students
Archives
JUN
8
970
ii
INTERFACIAL PARTITIONING DURING SOLIDIFICATION
By
James C. Baker
Submitted to the Department of Metallurgy and Materials
May 14, 1970, in partial fulfillment of the requirement
degree of Doctor of Philosophy.
Science on
for the
ABSTRACT
It has been demonstrated experimentally that departures
from local equilibrium at the interface do occur during rapid
solidification of zinc-cadmium alloys.
The resulting solid
compositions are larger than the equilibrium solid compositions at
any temperature, including metastable equilibrium compositions
below the eutectic.
This implies the solute chemical potential
increases during the freezing process.
Non-equilibrium alloy
solidification theories of Jackson, Borisov, and Baralis cannot
predict an increase in solute chemical potential, and therefore are
invalid.
There is a group of theories that do predict an increase
in chemical potential during freezing.
This set of theories as
well as the above set can both be derived from irreversible
thermodynamics.
The two types of theories can best be understood
in the common framework of irreversible thermodynamics.
Within this
common framework contradictory predictions can be made from
theories starting with identical assumptions. Therefore, the
validity
of both groups of theories is in doubt, and irreversible
thermodynamics as currently applied is untrustworthy.
A kinetic theory is developed which indicates that if
solute atoms are adsorbed on the liquid-solid interface then their
chemical potential increases during the solidification process,
even at low interface velocities.
Thesis Supervisor:
John W. Cahn
Title: Professor of Physical Metallurgy
TABLE OF CONTENTS
Section
Page
Number
Number
ABSTRACT
ii
LIST OF ILLUSTRATIONS
iii
ACKNOWLEDGEMENT
GENERAL INTRODUCTION
1
1.
Introduction
1
2.
Continuous Interface Motion in Pure
Materials
2
3.
III
Equilibrium
Segregation at Phase
Boundaries
5
Driving Force for Interface Motion
in Binary Alloys
9
5.
Continuous Interface Motion in
Binary Alloys
16
6.
Scope and Objective
4.
II
vi
S
of Thesis
SOLUTE TRAPPING BY RAPID SOLIDIFICATION
20
31
1.
Introduction
31
2.
Experimental
33
3.
Discussion
34
4.
Appendix
40
THERMODYNAMIC ASPECTS OF SOLIDIFICATION
PROBLEMS
50
50
1.
Introduction
2.
The Assumption
of Local Equilibrium
54
3.
Steady-State Binary Solidification
56
Section
Number
IV
Page
Number
IRREVERSIBLE THERMODYNAMICS OF INTERFACE
PROCESSES
65
1.
Introduction
65
2.
General
65
3.
Solidification
4.
V
VI
Principles
Theories
73
A.
Borisov Theory
73
B.
C.
D.
E.
Jackson Theory
Baralis Theory
Aptekar-Kamenetskaya Theory
Jindal-Tiller Theory
74
75
76
77
Discussion
of Theories
78
AN ALTERNATE APPROACH TO INTERFACE
PARTITIONING DURING SOLIDIFICATION
81
1.
Introduction
81
2.
Diffusional
3.
Graphical
4.
Appendix
Solution
Representation
81
85
90
SUMMARY AND GENERAL CONCLUSION
100
1.
Summary
100
2.
Suggestions
3.
References
103
4.
Biographical Note
106
for Future Work
101
iii
LIST OF ILLUSTRATIONS
Figure
Number
I-1
Page
Number
Graphical method for obtaining the chemical
potentials from the molar free energy
curve.
I-2
Graphical
23
construction
for obtaining the
equilibrium chemical potentials and
compositions from the free energy curves
I-3
I-4
for a binary system.
24
Graphical method for obtaining the total
free energy change of
when a mole of
material of composition C is added to a
large amount of
at composition C.
25
Graphical
illustration
of free energy AG
per mole reacted for diffusionless
reactions
I-5
I-6
I-7
of composition C
from a to S.
26
When the ratios of the reacting components
coincides with those of one of the phase,
the general tangent-to-tangent rule for
obtaining free energy changes reduces to a
tangent-to-curve rule. Here Cr = C .
27
It is thermodynamically possible to form
solid of any composition between C s ( l) to
C s (2) from liquid of composition C.
28
The domain of all possible solid compositions that can form from various liquid
compositions
at a given temperature is
enclosed within the curve OABEP.
curves OE and EP represent
The
conditions of
equal chemical potential for solute and
solvent respectively. They are straight
lines for dilute solutions.
Their inter-,
section E is the equilibrium condition.
The line OB is for a diffusionless transformation, with B representing the T
condition.
I-8
o
Schematic free energy curves to illustrate
the evolution of the curve OABEP of
Figure I-7.
29
30
iv
Figure
Page
Number
Number
II-1
Portion of phase diagram showing
equilibrium
composition of solid and
liquid.
11-2
44
Portion of eutectic
phase diagram
illustrating equilibrium metastability
of solid and liquid.
45
II-3
Zinc-cadmium retrograde phase diagram.
46
II-4
Plot of unit cell volume of zinc-rich
solid at -196°C against weight per cent
cadmium of initial liquid.
47
II-5
Electron micrograph of Zn-3.5 w/o Cd
which was splat quenched to -196°C.
130,000X.
II-6
48
Electron micrograph
of replica of
Zn-3.5 w/o Cd which was cooled by the
piston and anvil technique.
8,000X.
III-1
49
When there is a spinodal in the miscibility
gap the stable-metastable phase diagram
lines end when the solidus meets the
spinodal. Further extensions of the
metastable liquidus and solidus line are
unstable and intersect at a point on the
T
III-2
0o
line.
62
The domain of possible
liquid interface
compositions for steady-state solidification of an alloy of composition C
at
three different temperatures. At steadystate Cs equals C.
As indicated b the
arrows upward fluctuations is solid composition reduce the solute content of the
liquid and downward fluctuations increase
it. Steady-state cannot be established
by a system at point F in temperature
region II.
maintained
Local equilibrium cannot be
in region III.
63
V
Figure
Number
III-3
IV-1
Page
Number
Liquidus, solidus and T o curves defining
the three regions of temperature of
Figure III-2.
64
In the double primed set the free energy
change per mole of solid formed AG is
apportioned among a solidification
reaction AGs for which the driving force
is A averaged among the atoms in the
liquid at the interface and a redistribu-
tion reaction in which the differences in
Ap act to bring about the composition
difference
V-1
(CL - C ).
L s
80
Graphical illustration of the interdiffusion coefficient D(x) and the solute
interaction energy E(x) as a function of
distance x through the interface.
The
value of the solute interaction energy at
V-2
the center of the boundary EB will be
allowed to vary from EB << EL to EB >> E S
in the analysis.
90
Partitioning at interface
of imposed growth rate.
91
as a function
-vi
ACKNOWLEDGEMENTS
The author wishes to express his sincere thanks
to Professor John W. Cahn for his peerless guidance
and continued encouragement throughout this investigation.
Helpful discussions with Professors K. C. Russell
and M. C. Flemings are greatly appreciated. Thanks is
also due to Dr. B. C. Giessen for his contribution of
time and splat cooling equipment.
Special thanks are due to Mrs. Janine Weins for
her assistance
in the electron microscopy
portion of the
study, and Dr. Ronald Heady for computer programming
assistance.
The author is deeply indebted to his wife, Bonnie,
for colossal patience and understanding throughout the
course of the work.
The financial support of the National Science
Foundation is gratefully appreciated.
1
Chapter
I
GENERAL INTRODUCTION
1.1
Introduction
The purpose of this chapter is to give the reader
the necessary background
and a framework for understanding
succeeding chapters. Consequently, a number of distinct
concepts need mentioning, and for this reason the various
sections of this chapter may appear to be somewhat
unrelated.
An attempt will be made throughout the chapter to
show that the same principles
which apply to interface
motion in solids, such as during recrystallization,
can be
used to describe and explain liquid-solid interface motion
during solidification. The next section concerns itself
with interface motion in pure materials during solidification and solid-solid transformations.
A couple of
theories of both types of transitions will be discussed.
Section
.5 will later extend these suppositions to
describe interface motion in binary systems. The key here
is to account for the additional variable of composition.
Sections
.3 and
.4 pertain to aspects of
interfaces which are vital prerequisites in understanding
boundary motion in binary alloys.
The last section gives
a perspective view of the remainder of the thesis.
2
1.2
Continuous Interface Motion in Pure Materials
Before attempting to understand binary interface
motion and the associated interface partitioning, a brief
treatise of interface migration in pure materials will be
given.
There are two general atomistic mechanisms of
crystal growth from the melt.
(a) The first is when the
interface advances normal to itself.
For this case the
nature of the interface is usually thought of as being
"rough" (1,2),or the driving force being sufficiently
large(3).
Wilson(4)
in 1899 was the first to propose a
kinetic theory to describe this type of freezing in pure
materials.
His prediction
V
=
is of the form
-MAG
(1.1)
where V is the growth rate, M is an interface mobility
which is dependent on temperature,
the molar free energy
driving free energy).
and AG is the change in
(equal but opposite in sign to the
This relationship
is still popular
today.
(b) The second mechanism
is when the interface
advances by lateral motion of steps or layers one or more
interatomic distances in height.
advance of the interface,
There is no normal
growth of an element of surface
advances only when a layer sweeps by.
A possible genera-
tion of such steps has been proposed by Frank(5) to occur
3
at screw dislocations.
It was later shown by Hillig and
Turnbull(6) that the growth rate by such a mechanism
V
(AG)
is
(1.2)
Interface motion by steps is believed to occur when the
liquid-solid interface is smooth(2), or when the driving
force is sufficiently small(3).
Regardless of the fact
many materials are believed to solidify by this mechanism,
the remainder of this thesis will be committed to a study
of normal interface advancement.
It will be called
"continuous interface motion."
The first theories of continuous interface motion in
solids were proposed by Turnbull(7)
and Mott(8) for
recrystallization. They derived rates of motion of highangle grain boundaries by the use of absolute reactionrate theory.
Turnbull bases his theory on the assumption
that the atoms traverse the interface individually.
final conclusion
V
=
-e
His
is
kT
AG
()exp
(-)
where e is the Naperian base,
a,
() aS
(-RT)
(1.3)
is the atomic jump
distance, h is Planck's constant, N is Avogadro's
AG is the molar free energy difference,
of activation, R is the universal
number,
AS a is the entropy
gas constant, Qa is the
activation energy and T is the absolute temperature in
Kelvin units.
4
If the structure of the interface is always assumed
not to change then equation
form of equation
(1.3) can be written in the
(1.1).
Mott's approach differs in that he assumes the
interface transport process is one where groups of atoms
of the parent grain melt and then solidify as a group onto
the daughter grain.
Mott's end result is of similar form
to that of Turnbull's
V
=
-e
(kT)
nAG
nLm
nLm
(h--)R-,exp(RT-)exp(-R-T-)
m
(1.4)
where n is the number of atoms in each group, Tm is the
melting temperature and Lm is the latent heat of fusion.
Experimental boundary migration rates by Aust and
Rutter(9), Rath and Hu(10), and Gordon and Vandermeer(ll)
show agreement with predictions of Turnbull's singleprocess theory, while Mott's group-process theory predicts
a velocity of several orders of magnitude higher.
A similar approach to Turnbull's has been proposed
specifically
for liquid to solid transformations
in pure
materials by Jackson and Chalmers(12). This analysis is
more detailed, though, because it considers geometric
factors and the probability
that an atom will be accommo-
dated by one of the phases at the interface.
The essential
feature of the Turnbull theory that interface motion is the
result of an individual atomic process is retained.
5
1.3
Equilibrium Segregation at Phase Boundaries
If theories similar to Turnbull's(7)
are tested for
materials without ultra-high purity large disagreements
between theory and experiment result. The pre-exponential
term is found to be much too small(ll).
Hence, impurities
or solute atoms influence interface motion significantly.
It is believed the solute retards the interface motion by
segregating to the interface.
For this reason it is of
interest to digress temporarily to allow one to comprehend
the interaction of a solute species with an interface.
Before the role of solute on moving boundaries
is
considered, an attempt will be made to convey that which
is known about segregation at stationary boundaries or
interfaces.
A means of characterizing equilibrium segregation at
boundaries will now be given.
If one assumes the struc-
ture of the interface is different than the bulk phase or
phases and hence the affinity for solute atoms is also
unequal, then it is plausible
to express the solute
chemical potential of the system in terms of a solute
interaction energy E(x) in dilute solution as
B
=
kTZnC(x) + E(x) + constant
The superscript B refers to the solute species.
(1.5)
The
distance x is in a direction normal to the interface.
equilibrium
( = constant),
At
C and E will be constant in
6
the bulk phase.
But if the boundary is considered
to have
a finite thickness, then C and E may vary with position in
the boundary to maintain
= constant.
In this thesis EB
will designate the value of E at the center of the
boundary.
An understanding
of the solute interaction energy E
may be conceived by the following discussion.
Consider
the addition of a solute species to a pure material while
keeping the addition small to minimize solute-solute
interactions.
If there is an attractive or repulsive
force on the solute atoms in the vicinity of a boundary
then this force can be perceived
as minus the gradient of
The solute interaction energy E is
a potential energy.
this potential energy.
One would expect a higher concen-
tration of solute where the interaction energy is lower.
In equation
(1.5) the first term may be thought of as the
entropy contribution
For a given
to
and E the enthalpy contribution.
, such as at equilibrium,
the smaller the
solute interaction energy E the larger the composition C
will be.
At equilibrium, if one sets the solute chemical
potentials of the bulk phase a and of the boundary equal,
then the ratio of solute compositions
CB(eq)
C
C
(eq)
E
=
exp(
is given by
B
)
(1kT6a)
7
The subscript B refers to the boundary.
equation, it follows that if E
>
From this
EB then CB(eq) > C (eq)
and the solute is surface active (adsorption of solute).
Conversely, if EB > E
then solute interface desorption
exists at equilibrium.
Several methods of calculating the solute
interaction energy EB for large-angle grain boundaries in
single-phase systems have been reported(13,14).
one assumes that the disordered
But if
structure of a random
high-angle boundary resembles that of the liquid(15), then
a natural extension is to set EB = EL and CB(eq) = CL(eq)
for a given temperature.
For this case, equation
(1.6a)
may be rewritten as
=
EB
B - ESS
kTZn
CS(eq)
[c(eq
L(eq)
=
(1.6b)
kTZn[K(eq)]
where K(eq) is the equilibrium distribution coefficient
(not to be confused with k which is Boltzmann's
K(eq) can be determined
constant).
from the equilibrium phase diagram.
Turning our attention now to liquid-solid interfaces,
a plausible
assumption for allowing one to estimate EB and
the corresponding boundary segregation might be to:
assume the liquid-solid boundary to be disordered
similar to the liquid phase.
C B (eq)
Then E B
and
EL and
CL (eq).
One possible means of experimentally determining EB,
at least semiquantitatively,
for liquid-solid interfaces,
8
is to utilize the famous Gibbs(16) adsorption isotherm
equation
B
_a
=
(1.7)
=B T,P
or for when the solute composition is small
-
rB
-
Da )
RT ( YnCTP
p(1.8)
is the number of solute atoms adsorbed per unit inter-
face area, a is the surface free energy per unit area, and
B
p
and C are the solute chemical potential and composition
present in the system that are being adsorbed.
From this
equation it is seen that if raising the solute composition
or chemical potential causes a reduction in the work a
required to form a unit area of the surface, then rB is
If
positive and solute is adsorbed by the interface.
addition of solute increases a then the solute species
will be rejected at the interface
a is experimentally
rB can be found.
(desorption).
Hence, if
measured as a function of (znC) then
But
rB
is the number of solute atoms
adsorbed per unit interface and not a composition at a
particular position in the interface. Nevertheless, from
FB the
nature of the segregation can be determined
and
also CB(eq) and EB (at the center plane of the interface)
can be estimated if one assumes a particular E = E(x) or
C B (eq) = C(x) for the boundary.
This method of characterizing solute equilibrium
segregation at interfaces by an interaction energy has
9
been used by Cahn(17) to develop a theory for boundary
motion during recrystallization (see Section
.5).
Cahn's approach will be extended in Chapter V to enable
the partitioning
at moving interfaces to be expressed as a
function of growth velocity for binary alloy
solidification.
1.4
Driving Force for Interface Motion in
Binary Alloys
Before discussing interface motion in binary alloys
it is worthwhile
to comprehend
the thermodynamic
funda-
mentals which allow such a process to take place.
At
constant pressure for binary alloys the composition
variable in addition to the temperature must be taken into
account if the thermodynamics
is to be understood.
One
can then predict when such a process is energetically
possible and determine
if it does occur.
the driving force for the reaction
These driving forces will be illustrated
graphically.
In binary phases the chemical potentials
(A
and
B)
of A and B are definedCl18
i
-
i
(~G/3n)
(1.9)
T,P,n
as the rate of change in the Gibbs free energy G when an
i-th component is added at constant temperature and
pressure.
The two chemical potentials
are related by the
10
Gibbs-Duhem
equation, which for constant T and P is
(1
C)diA + Cd1B
-
=
(1.10)
0
where C is the mole fraction of B.
This equation permits
calculation of the chemical potential of one component
from a knowledge
of the other.
The molar Gibbs free
energy, Gm = G/(NA + NB), is related to the chemical
potentials
by
Gm(C)
=
(1 - C) PA(C)
and by use of the Gibbs-Duhem
A
-
_
= Gm
(C)
(1.11)
equation
C( m
C
(1.12)
B_
B
Equation
+ C
Gm +
(1.9) expresses
chemical potential.
(1 - C)(--)
the important property of the
It is the increase in free energy of
the entire system when an infinitesimal amount of one
component is added reversibly (per mole added). From this
property we have the important condition that for equilibrium the chemical potential must everywhere have the same
value.
Equations
(1.12) form the basis of the popular
graphical methods of tangents(19). They express the fact
that a tangent drawn to the molar free energy curve in
Figure 1.1 at the composition of interest intercepts the
C = 0 and C = 1 vertical axis at a value of Gm equal to
11
the chemical potential of the component. Hence a common
tangent among two or more phases implies equality of
chemical potentials and equilibrium (see Figure 1.2).
A
tangent to a Gm versus C curve also has an important meaning for reactions of a phase with composition change.
equation of a line tangent to Gm at C = C
Gm(C,C
)
=
(1 - C)A
If an infinitesimal
(C ) + C
is
(Ca)
(1.13)
amount of material of composition
C were to be added reversibly to
of composition C , then
Gm (C,C ) would be the change in free energy of
of material
The
added as in Figure 1.3.
composition of material added.
per mole
Here C is the
It may be quite different
from C , the composition of a, which is the value of C
where the line is tangent to the
free energy curve.
The
free energy change AG per mole reacted for diffusionless
reactions of composition C
from
to
is obtained
graphically in Figure 1.4 by reading the vertical distance
between tangents at C
(incidently the distance between
tangents for the case coincides with the distance between
curve to curve at C0).
Analytically
the free energy
changes per mole of B is
AG,, =
(l-CH
O)
0a
-
AC + C (B a
~0jB
The free energy change AG per mole
B
a
(1.14)
formed in a closed
system that transfer small amounts of components from a at
12
composition C
to
at a different
composition of C
is
attained graphically in a similar way as before by reading
the vertical distance between tangents at C
1.5).
This free energy change per mole
expressed if C
AG
=
is changed to C
(1 - C) (
-
formed can be
in equation
) + C
&
(see Figure
(1.14)
- hB
(1.15)
This substitution is necessary because the free energy
change desired is per mole
formed and 1 is no longer of
composition C.
To inquire if a solid can form from a single-phase
liquid of composition C
one draws a tangent to GL at C0
and sees if the free energy curve of any solid phase lies
below the tangent
(see Figure 1.6).
The composition range over which G
lies below the
tangent gives the range of composition of solid that can
form.
At temperatures
above the liquidus no solid can
form.
At the liquidus one solid just touches the tangent.
It is the only solid that can form and it must form with
the equilibrium composition. At lower temperatures an
increasing range of solid compositions can form.
To take advantage of the isothermal aspects of the
graphical methods this same range of solid compositions
can be presented
isothermally
by varying the liquid
composition CL.
At a given temperature the range of
thermodynamically possible solid compositions C
that can
13
form from a liquid of varying
compositions CL, is shown
in Figure 1.7 as an area enclosed by the curve where
AG = 0 (OABEP).
The right-hand most point on the curve E
depicts the equilibrium compositions. The tangent to the
liquid touches the solid only at one point
in Figure 1.8).
(see tangent 1
As the liquid composition moves to the
left (tangent 2) the range of possible solid compositions
at first increases and then decreases
again.
The maximum solid composition(C(T o ) in Figure 1.7,
tangent 3 in Figure 1.8) is the composition where the two
free-energy curves cross.
Only one liquid composition,
C(To), can give this solid.
If the liauid is either more
or less supersaturated this maximum solid composition
ceases to be possible.
Diffusionless
transformations,
shown as a line OB of slope 1 in Figure 1.7, can only
occur if the liquid composition
is below this crossover
composition(20). Following the useage in other transformations(21,22),
we shall call the condition of equal free
energies the T
condition and the composition C(To).
phase diagram T
solidus lines.
On a
forms a line between the liquidus and
The line marks the upper limit to the
liquid compositions and temperatures for diffusionless
transformations.
compositions
It also marks the upper limit to the
and temperatures
of solid that can form
isothermally from liquid of any composition.
14
interest are shown
Two other curves of thermodynamic
in Figure 1.7.
These are the lines where chemical poten-
tials of a component are equal in liquid and solid.
line
E, APB = 0; for line PE, AA
lines cross we have equilibrium.
For
Where the two
= 0.
These lines bound the
regions of the figure where a particular
component
solidifies with a decrease in chemical potential.
In the
triangular region OEP both components would experience a
decrease in chemical potential upon solidification. In
this joint composition range the line tangent to the solid
free energy versus composition curve lies everywhere below
the line tangent to the liquid.
Both components
independently experience a decrease in free energy upon
solidification. Outside the triangular region but within
the AG = 0 curve the tangent lines cross and although the
overall free energy decreases upon solidification, one of
the components experiences an increase in chemical potential.
Solidification
in this domain can only occur if the
two species do not solidify completely
one another.
independently
of
One species enters the solid with an increase
in its chemical potential because it is either passively
trapped by the advancing solidification front or because
it is a required participant
in an independent
solidifica-
tion reaction mechanism involving several species which
leads to an overall free energy decrease.
In either case we
will define "trapping" of a component to occur at an
15
interface when that species experiences an increase in
chemical potential there.
It is worthwhile
to describe the above conditions
quantitatively for one simple case, that of dilute solution
for both liquid and solid.
Then the chemical potentials
are given by Henry's Law for the minor component
B
=s B
B
L =BL
+ RT
n ys C
(1.16)
+
n yL CL
(1.17)
RT
where the B's and y's are related constants that depend on
temperatures and reference states. We may eliminate the
constants by noting that the chemical potentials are equal
at equilibrium when C s = C s (eq) and CL = C L (eq), then the
change in chemical potential across the solidification
front is
B
AP
B
PUs -
B
L
=
RT
n
C s C L (eq)
Cs (eq)
s
In terms of the distribution
(1.18)
~L
coefficient K at the inter-
face, defined as Cs/CL, and its
equilibrium value,
K(eq) = C s (eq)/CL (eq), this becomes
AP
B
=
RT
n
K/K(eq)]
(1.19)
The minor component experiences no change in chemical
potential when
Cs
=
K(eq) CL
(1.20)
16
This is a straight line OE with slope K(eq) through the
When K < K(eq), A B < 0.
origin in Figure 1.7.
This is
The area above line OE
the region below the line OE.
For the major component
corresponds to "solute trapping".
Raoult's Law hold and
CL (eq))
- C(1 - C
RT Zn (1 - C(eq)) (1 - C
5
L
A
A
=
(1.21)
The major component experiences no change in chemical
potential when
[Cs (eq) - Cs]
=
- C(eq)][CL(eq) - CL]
[1 - C(eq)/
........
.(1.22)
This is a straight line PE of slope [1 - C(eq)]/[l
- CL(e q ) ]
[Cs(eq), CL (eq)] in
= 1 through the equilibrium point
Figure 1.7.
The regions above and below the PE corresponds
respectively
to AiA
less than or greater than zero.
Solvent
trapping would occur below PE.
The AG = 0 curve is given by
(1 - C)
s
A
+ C
s
ASB
=
0
(1.23)
This curve must pass through the points 0, B, E, and P.
1.5
Continuous Interface Motion in Binary Alloys
In Section 1.2 it was shown by Equation
the response of an interface in pure materials
(1.1) that
(one
component systems) to the conditions at the interface can
17
be expressed by
V
=
-MAG
(1.1)
or in general, in terms of the variables V and T
f(V,T)
=
0
(1.24)
For binary systems the additional variable of
composition must be taken into account and two interface
response functions are needed to describe the interface
motion.
For an a+- transformation
fl (Ca ,CV,T)
=
0
f 2 (Ca ,CV,T)
=
0
where C a and C
the
and
these functions are
(1.25)
are the solute interface compositions of
phases.
Such response functions could equally
well be solved for the response, V and Ca, in terms of the
temperature T and
interface composition C.
V
=
gl(T,C)
Ca
=
g2 (T,C)
(1.26)
Such a form of the response functions are especially
valuable when considering a solid state transformation
under steady state conditions.
For this case C
equal to the overall solute composition C
is
of the system
and the temperature T is the imposed variable.
18
For steady state solidification te
desired response
functions are
since C
= C
T
=
hl(V,C
CL
=
h 2 (V,C s )
s
)
(1.27)
and the growth velocity V is now a
conveniently controllable parameter.
It should be noted
no matter which form of the response functions are chosen
they describe the same relationship
and are equivalent.
From this view point it is readily evident that the
principles
of solidification
are equivalent
to those of
interface motion during solid state transitions, and basic
approaches
applicable to one should be satisfactory
for
the other.
For solidification,
due to the high diffusivities
the liquid and the usual comparatively
velocities,
in
small growth
it is customary to assume the liquid and solid
interface compositions are not substantially different
from equilibrium compositions. Such a condition is called
"local equilibrium"
this assumption
at the interface.
The significance of
is that the interface compositions
independent of growth velocity V and dependent oly
interface temperature T.
and solid compositions
are now
on
For any temperature the liquid
can be read from the liquidus and
solidus lines on the equilibrium
response functions are known.
phase diagram; hence the
19
If the above assumption
is not made two common
approaches have been used to determine the response
functions for the solidification process: Absolute
reaction rate theory and irreversible thermodynamics.
A theory for determining the response functions for
binary solidification using the absolute reaction rate
approach has been developed by Jackson(23).
It is an
extension of the work by Jackson and Chalmers(12)
component systems.
to two
Such an extension can easily be made
since an inherent assumption
of the previous work is that
the atoms traverse the phase boundary individually. Hence
the following convenient assumption of the Jackson theory
was a natural one:
the rate at which j atoms of each
species leave a phase and traverse the interface is proportional to the mole fraction of the species in that phase.
This assumption allows Jackson to develop an analysis which
lends itself
readily to physical interpretation. Details
of this theory will be given in the next chapter.
The more popular approach to the problem for binary
solidification
has been through the use of irreversible
thermodynamics. The starting point for these theories is
by assuming a relationship
similar to equation
(1.1) for
each atomic species, that is the flux of each species
across the phase boundary is linearly proportional
to the
gradient of its chemical potential. A thorough analysis of
these theories will be given in Chapter IV.
20
Since it is well known even traces of a second atomic
species retards interface motion during recrystallization,
Cahn(17) has formulated a theory to account for the interface drag of a second species on interface motion.
It is
assumed when the material is pure equation (1.1) is valid.
When a solute species is present its drag on the interface
is determined and in turn the resulting response functions
can be found.
The nature of the above solute effect
depends on the magnitude
of the solute interaction energy
EB relative to the interaction energy in the bulk phase.
These are the same equilibrium solute interaction energies
as found in equations
(1.5) and (1.6a).
An extension of
this Solute Drag Theory to binary solidification is given
in Chapter V.
The necessity of this alternate approach to
determine the interface partitioning during binary solidification was found to be needed since there are defects in
the previous two methods.
These deficiencies will be
covered in Chapter IV.
1.6
Scope and Objective
of Thesis
The purpose of this investigation
detailed examination
is to make a
of the response of a solid-liquid
interface to various conditions at the interface.
Specifically, this study has been made to gain insight
into the relationships between solute partitioning (solid
and liquid interface compositions), the interface
21
temperature, and the growth rate during binary alloy
solidification. Such relationships are called "response
functions."
A knowledge of the response functions is of utmost
importance in the field of solidification
since they are
needed boundary conditions to differential equations which
are capable of describing
Such a mathematical
the kinetics of solidification.
analysis in turn can help in
predicting and controlling cast alloy properties.
If it is assumed that the interface is highly mobile,
then even for small deviations from equilibrium the interface velocity is expected to be large.
For this case it
is expected that the solute partitioning
should be a
function of only the interface temperature and not
dependent on the growth rate.
interface compositions
Such a condition where the
are close to the equilibrium
compositions is called "local equilibrium." Since most
solidification
studies are carried out at low velocities
the condition of local equilibrium
has gained wide
acceptance as a valid form for the response functions.
first objective of this investigation
The
is to test the valid-
ity of local equilibrium under extreme solidification conditions.
In Chapter II it will be shown that compositions of a
resulting solid after freezing were found to be larger than
the stable or metastable
solidus composition at any tempera-
ture and therefore a departure
from local equilibrium
at the
22
liquid-solid interface occurred. This investigation was
performed by studying the zinc-rich solid phase in zinccadmium alloys after splat cooling.
Chapter III will then
concern itself with thermodynamics aspects of solidification when the solid composition lies in various regions of
the equilibrium phase diagram.
With the above experiments in mind, previous theories
on non-equilibrium
partitioning
With the aid of the experimental
are examined in Chapter IV.
results in the zinc-
cadmium work, serious deficiences have been found in each
of these theories.
Since most of these theories are
irreversible thermodynamic
in nature, special attention
is
given to this type of method and surprising conclusions are
drawn.
Chapter V will be concerned with attempting to
overcome the above shortcomings by using the solute drag
approach.
Before proceeding
to the next chapter it should be
noted that the major portion of Chapters II*, III**, and
IV** have been published.
J. C. Baker and J. W. Cahn, Acta. Met., 17, 575,
(1969) .
** J. C. Baker and J. W. Cahn, "Solidification,"
Soc. for Metals
(1970) .
Amer.
23
Cn
-4
4-i
-4c-J
G)J
Z
w9 '3N3
338
0 -1tV1
24
IIQZ
::L
I!
Zi
0n3
LI)
'I
4-J
(~~~)
C~~(
C
Cr ~
-
-
t
biJj-
W 9E' Agd3N3
33.
V1OA
,1
:tt
25
vL
,E
,2L
.-
Z
X
°
1o~~
C
o
.H
.
Gc *C
Z
Ik!3N3
333 A
N OVW
<
C
C-o
'
26
mr cQ
0
04.
0
0
-
o
o.~
0
E
0]0
0J4H
o
C-)
0
LiJ
II
C.-
.
0
1)
C
o-C
*'-1=
cn
,00
W
w9 ' AE)93N3 3-3DJ
:
27
cn~
M t
Cd
Cd
4JO
Cd
Cd-
Cd
Cd
Cd
Ci
2;
I
O4J
Q
Cd
Cd
Cd
o
c
c)
Cd
oi-
'I
0
Cd
0
Cd
Cd
0
Cd
Cd
Cd
CC
9d: N] 33
A D 8]3N]3
3J
V0<
a
HV-10
Cd
Cd
Cd
28
u0
I
i
v:
0
0
'0
E
©
0
ZJ
0 C
z Js
'~4 -
cn~~0
0 -
-
0<<CDL
)
~.
S4-
C
0~~~~Jr
-
'
~~~~~
-4
U>
00_
l
i)
0
Ad3N3 33WJJ VtO0J
,°
<
I
'-4E
29
iE
I
-
,
o
4-~ .;4
-
4-4
C
C~0(D-4-,
CUOC.
-~ CC
4_
r-:
tCH
-
aC
r
O
C.n
C.CCJ
~ Zn
C. -400
-
O0
0
- .
-
0;
2 ,.)~- 0
all0
5
~
W
0004-
02
C)
1-4
SO '30V8J31NI 1V NOIISOdVJOD clOS
~-'
0
4
Zn
X
02
*~
Z
)
0
C4-4
.0 C
-1) 0-- r0
o o~--a
C)
0
C C.) Zn
C)
C.iCr) Zn
Zn '4-r
0 H;
0> >Hr'
rLxU
0
j-~2
~$_,'4-j
: -oo rO
0
J-
"
4J
V
CC1-4 z 0 0 --~
0
CY
0
'. 4Jz0s
C.
U C
-z
I-
0
Z~T,~.-Q
C.
Zn-i
OC1);.)
o
. ,_~h
r1
c*1
-;C)0
m~~~1-
C) 4-1 Zn ,r
0A
C.)
Z
C.
4Zn
- -
30
-J
C0
0%>
C
cr
0
-4
4t
_
Li
CJ
v,
X_
oW
O<
-
U-
O
11
U)I
7a)
co
0
h a
4-4
1
!Dd3N3 338
dWV1OIN
31
Chapter II
SOLUTE TRAPPING BY RAPID SOLIDIFICATION
II.1
Introduction
Equilibrium between solid and liquid phases of metal
alloys implies that the position of the phase boundary is
fixed, i.e. the net rate of growth, V, is zero and that
both phases are of uniform composition, but the compositions
of the solid and melt are usually different.
brium compositions
These equili-
are dependent on temperature; the
possible compositions of solid and liquid correspond to two
lines on the phase diagram, the solidus, below which in
Figure 2.1 the solid is stable and the liquidus, above which
the liquid is stable.
If the liquid-solid
small rate
interface is allowed to move at a
(Vsmall), the solid and liquid phases need not be
of uniform composition.
But for all practical purposes, the
solid phase at the interface
can be assumed to be in equili-
brium with the liquid at the interface
CL(i) = CL(eq)).
(Cs(i) = C(eq)
and
In this thesis the above condition at the
liquid-solid interface will be referred to as "local
equilibrium."
For large growth rates it is questionable
condition of local equilibrium
if the
at the interface holds.
Consequently,
the objectives
chapter are:
Cl) to obtain high growth rates by the method
of the investigation of this
32
of splat quenching
and (2) to ascertain if there is a
detectable departure from local equilibrium at the interface for the case of large growth rates.
If we measured the temperature at the liquid-solid
interface during rapid solidification we would know the
equilibrium composition of solid from the phase diagram for
that temperature.
Then we could check the validity of local
equilibrium at the interface by comparing this equilibrium
composition with the actual composition of the resulting
solid.
But it is a major experimental
the temperature at the crystallization
problem to determine
front.
If this
temperature is unknown during the solidification, then for
most alloy systems any observed solute redistribution
be rationalized
can
from the assumption of local equilibrium at
the interface, since the composition of the solid can be
made to match a stable or metastable equilibrium solid
composition by choosing the appropriate solidification
temperature
above or below the eutectic as indicated in
Figure 2.2.
It is possible to avoid this problem of temperature
determination by selecting an alloy system with a retrograde
solidus (see Figure 2.3).
since a retrograde
The reasoning is as follows:
system has a maximum in the solidus
[Cs(eq max) > C(eq)],
if a composition of the actual solid
can be measured which is greater than the retrograde maximum
in the equilibrium
solid composition
Cs(i) > C(eq
max)], a
33
positive departure from local equilibrium at the interface
must have occurred
Hence, for this case
Cs(i) > Cs(eq)].
we can measure a departure from local equilibrium without
the temperature being known.
II.2
Experimental
The zinc-cadmium
investigation
system was chosen for this
(see Figure 2.3).
The solidus of the zinc-
rich end of this alloy phase diagram was reported to be
retrograde by Owen and Davies(24).
The retrograde
solidus
was confirmed by the present author using X-ray lattice
parameters to measure solid compositions at various temperatures.
High-purity
zinc (99.99%) and high-purity
(99.99%) were sealed in an argon atmosphere
melted together, and quenched
in water.
in
cadmium
vpyrextubing,
Portions of these
master alloys were then remelted and splat cooled to -1960 C.
The shock-tube method of splat quenching
al(25) was utilized.
of Duwez et
This technique employs a shock wave to
eject liquid metal through a small hole in a crucible,
atomizing the liquid in process.
The liquid globules
are
rapidly spread on a copper substrate.
0 C and
The resulting metal foils were kept at -196°
analyzed on an X-ray diffractometer.
Unit cell volumes
(0.866 a2c) of the zinc--rich phase were calculated
and
plotted versus weight per cent cadmium of the initial liquid
from zero to 5% (see Figure 2.4).
34
II.3
Discussion
The unit cell volume of the zinc-rich phase in Figure
2.4 can be considered
as a measure of the concentration
cadmium in solid solution.
of
Since we find that the unit
cell volume is linear with respect to the composition of
the initial liquid, we conclude that solid of compositions
up to 5 wt. % cadmium have formed.
The maximum composition
of equilibrium solid in accordance with the retrograde
phase diagram is 2.6 wt. % cadmium.
Consequently,
the solid
solubility has been increased beyond the maximum solidus
composition and a positive departure from local equilibrium
at the liquid-solid interface has occurred in the solid
composition, i.e. C(i)
> C(eq).
We are now in a position to compare the result of
increasing the actual solid composition beyond the
equilibrium composition of the solid with present theories.
There are two basic types of theories that are shown invalid
by this experiment; one by Jackson(23)
is based on reaction
rate theory, the second by Borisov(26)
is a thermodynamic
approach.
Let us first consider Jackson's kinetic description
of alloy solidification.
two assumptions:
It is developed on the basis of
(1) reaction rate theory can be applied
independently to each species, and (2) the rate at which j
atoms of each species leave a phase and traverse the
35
interface is proportional
in that phase.
to the mole fraction of the species
From these two assumptions Jackson attains
the equations for each atomic species
Vj
m
=
KJ C(i)
VD
F
=
K3
FLCD(i)
m
S
(2.1)
where Vm is the rate at which atoms of the j species cross
the interface to join the liquid, V
is the rate at which j
atoms cross the interface to join the solid, and
K j = AGjvNJV
j
exp(-QJ/RT)
(in the notation of the cited
paper) is a function of temperature but not of composition.
The net rate of growth of each species is equal to the
difference between V
Vj
=
From equations
and Vm and is given by the equation
VF -F V m
(2.1) and
(2.2)
(2.2) it can be shown Jackson's
analysis predicts a negative deviation in local equilibrium
for the solid at the interface, C(i)
< C(eq),
contrary to the results of this investigation.
as follows.
For species B at equilibrium V=
which is
The proof is
0 and equation
(2.2) becomes
VB
F
=
=
VVB
m
(2.3)
36
Combining equations
(2.3) and
Km C S (eq)
=
m
(2.1)
B
KF
F CL (err)
VB is positive for solidification
equation
(2.4)
and is given by
(2.2)
= VB
VvB =V
Combining equations
K
C
L
F
and substituting for K
(2.5)
(i)
(2.5) gives
-
K
B
m C S (i)
from equation
m
VB
VBm
(2.1) and
BB
V
-
B CL(i)
=C(eq)K
L
F
[
()
(2.6)
(2.4)
CS (i)
CS(eq)
(2.7)
For solidification we must have a decrease in free
energy, hence it can be easily shown any deviation
local equilibrium in the liquid composition
from
is such that
the liquid composition at the interface is less than the
liquidus composition
CL(i)
Equations
<
CL (eq)
(2.8)
(2.7) and (2.8) yield
C S (i)
<
C s (eq)
(2.9)
Hence, Jackson's theory predicts the wrong direction for the
deviation from local equilibrium
and the theory is invalid.
37
We now turn to Borisov's theory, which makes the
thermodynamic assumption for a binary system that the
chemical potentials of both species must decrease during
solidification,
i.e. the values of the chemical potentials
at the interface must satisfy the following condition:
A (
0
<
i)
USL
(2.10)
PB(i)
Equations
-
<
BL(i)
0
(2.10) will now be shown to be inconsistent
with the experimental results, C(i)
> C(eq),
hence making
Borisov's theory also incorrect.
At equilibrium for species B
B
1L(eq)
=
ps(eq)
(211
(2.11)
Except at critical unmixing points and spinodals we have
the following condition for each species in any stable or
metastable phase
, (i)/C (i)
>
0
(2.12)
For solidification with a decrease in free energy equation
(2.8) holds true and when combined with equation
L(eq)
>
vL(i)
From our experimental result, C(i) > Cs(eq), and
equation
(2.12)
(2.12) gives
(2.13)
38
US(i)
Combining equations
vS (eq)
(2.14)
(2.11), (2.13), and (2.14)
_
-B(i)
S
Therefore,
>
L
the assumption
0
>
p(i)
of equations
(2.15)
(2.10) and Borisov's
theory are invalid.
It should be pointed out that equations
(2.10) are
not necessary thermodynamic conditions for solidification.
The thermodynamic
transition.
requirement is AG < 0 for the overall
This permits A
>
0 for one of the species.
It is now of interest to see why theories like
Jackson's and Borisov's fail to agree with our experimental
result.
Both types of theories make the inherent assumption
that some spontaneous activity is required of each species
during solidification.
the solid are inactive.
The atoms that do not traverse to
Thus each species must lower its
free energy on solidification. Experimentally we find that
the cadmium atoms experience
potential.
an increase in chemical
This means that if the cadmium atoms could act
independently
they would attempt to avoid the solid.
the time the cadmium's potential
passive.
During
is being raised it must be
The cadmium is trapped in the solid by the
solidification
front and if it could have been active
would have remained in the liquid.
it
39
There is one atomistic theory which has in it the
basic passivity of the solute and which therefore predicts
the correct deviation in solid composition from local
equilibrium.
This is Chernov's
of surface active solute atoms.
theory(27)
These solute atoms seek
the interface and are subsequently
of the crystal grows.
for the trapping
buried as the next layer
It is during the burying that they
are passive and their chemical potential
is raised.
If
they can diffuse they attempt to remain in the surface.
The author believes Chernov predicts the correct
departure because it is inherent in his beginning assumptions and not a product of the solution to his diffusion
equation. The disputed assumption is:
the initial composi-
tion of the new solid layer is identical to the interface
layer from which it formed.
Any later change in composition
of this solid is then due to diffusion
in the solid.
Hence,
for a surface active solute species this total initial
capture of solute atoms is independent of the layer's
lateral growth and neglects
solute escape by interface
diffusion ahead of the moving layer
(the author feels this
may be an important effect especially at small layer growths).
An analysis believed to be more realistic than the one
above is given in Chapter V.
It also predicts a positive
deviation in solid composition from local equilibrium when
the solute atoms are surface active.
40
II.4
Appendix
A.
Verification
of the Retrograde
Solidus in
Zinc-Cadmium
The validity of the conclusion of this experiment
requires that the solidus including its metastable extension
below the eutectic never exceeds the composition value at
the retrograde maximum.
But theoretical and experimental
tests were performed to check that the solidus indeed has a
maximum, and the metastable extension was shown theoretically to continue to decrease monotonically.
The various equilibrium solidus compositions were
calculated by utilizing available thermodynamic data from
the literature(28)
and the following expression derived by
Thurmond and Struthers(29).
kn
(A +
LB)--Bs
+HL)
C (eq)
(AHf
]
C
(e q )
CL
L (eq)
=
T
RT
AB
AfA2B
-
-RT
(2.16)
Both the liquid and solid solutions are assumed to be
regular solutions.
AHf and ASf are the heat of fusion and
entropy of fusion at the melting point of solute B, AH- and
-B
AHL
s
are the differential
heat of solutions of the solute in
the solid and liquid phases.
For regular solutions the
B
above three AH's and ASf are constant.
side in the Zn-Cd system they are(28):
For the zinc-rich
41
AHcd
Afif~
153
~cal.-C
f
= 201530
cal.
=
~~
~~~~~~L
Cd
ASf = 2.58 cal/°.
TCs
=
6904 cal.
Using CL(eq) values from the phase diagram, Cs(eq)
values have been calculated
at the temperature where the
experimentally determined phase diagram predicts a
maximum
(600°K) and at the eutectic temperature
The two calculated
solidus compositions
600°K) and 1.23 at/o (at 539°K).
0 K).
(539°
are 1.30 at/o
(at
Hence, these two
composition values predict a retrograde
solidus, and are
in close agreement with the equilibrium hase diagram
values(24) of 1.39 at/o and 1.20 at/o.
Let us now study equation
(2.16) in detail for the
Whenever
Zn-Cd system.
-B
AHs
the right-hand
B
-B
(2.17)
>> (AHf + AH)
side of equation
(2.16) is always negative
and increases in absolute magnitude as the temperature T
decreases.
The above implies
n[Cs(ea)/CL(eq)] + -A and
Cs(eq)
0 as the temperature T tends toward zero degrees
Kelvin.
For the zinc-cadmium
system expression
(2.17) is
obeyed strongly. So strongly that heat capacity differences
cannot produce appreciable
changes in it.
Hence, below the
eutectic temperature the metastable equilibrium solidus
compositions will continue to decrease with decreasing
temperature.
42
For a maximum to occur in the stable portion of
AHs and ETmA - T (eutectic)
a solidus, it is desired that -B
be large.
For systems where AHS < (AHf + AHL) the
solidus cannot have a retrograde maximum at any temperature.
The maximum in the solidus was checked experimentally
by X-ray lattice parameter measurements.
First, alloys of
3.5 w/o Cd were heated into the two phase liquid-solid
phase diagram region and were allowed to equilibrate at
the supposed temperature where C 5 (eq) is a maximum and at
a lower temperature.
Both specimens were quenched in
water and examined by X-ray lattice parameter measurements
at -196°C.
It was found that the specimen with the
higher equilibration
temperature had a larger unit cell
volume at -196°C and hence was of higher composition.
This
in turn supports the retrograde nature of the solidus.
B.
Verification
Solid Phase
of Presence of Only a Single
The second precaution
was to examine the various
splat cooled alloys by transmission electron microscopy.
The test was to determine
if only the zinc-rich
phase in
Figure II.3 was present as indicated by X-ray analysis,
or if the cadmium-rich
amounts.
phase was also present in small
If the s-phase is the only phase
alloy compositions
resent when
greater than C (eq) maximum are splat
s
cooled, then all the cadmium is in solution in the ~ phase
and C s > C (eq).
s '
This is what was found by the
43
transmission electron microscopy (see Figure II.5).
Only
one phase appears to be present and the grain boundaries
are clean.
If a phase particles
invalidate the experimental
But the actual finding of no
had been found, it does not
conclusion
that C s > Cs(eq).
phase present does support
the conclusion that the composition of cadmium in solution
is greater than the equilibrium composition.
One would expect if the cooling rate was decreased
sufficiently, the system would tend toward equilibrium
during the freezing process and form both
and a phases.
The necessary segregation needed for the formation of
these two phases most likely would occur by dendritic
solidification.
Figure II.6 is a micrograph of a
replica of the surface of a Zn-3.5 w/o Cd alloy cooled
by the piston and anvil technique(30).
appears to consist of
particles.
The specimen
dendrites and a interdendritic
The rate of cooling in the piston and anvil
technique(30)
is believed to be 105(°C/sec), where as
the splat cooling methods
leads to a cooling rate of
approximately 107(°C/sec).
44
e
._
©
0
.-4
F -CT
.
o
E
©n
r~
0
0
0Hu
--
-4
r
3 n 1v
3d
3i
45
cJ
(J
o
-j
o~~ ©
0~~~~~
u ,
C'4
0~~~
I-~~~~~~~~~~
3 n
v d3d vq31
0
a _
46
0
0
C~~~~~~C.1
(~~~~~~)
~
0~~~
z~~~~
s
2~~~~
a~~~~~a
C
Q
lpIC
_I
C
0',I
t~
D
U~~~~~l
_
CC~~~~~~~~~l
;~~~~~~~~~~~~~
,0
cO
E
B3
W LJVW13 dV3-L
N
47
30.40
0
o
C0(0
I
<r
30.30 D
-_
0
-J
-J
0w-
2.00
1.00
WEIGHT
Figure II-4.
3.00
4.00
% CADMIUM OF INITIAL
5.00
LIQUID
Plot of unit cell volume of zinc-rich solid at
-196°C against weight per cent cadmium of
initial liquid.
48
Figure II-5.
Electron micrograph of Zn -3.5w/o Cd which was splat
quenched to -196°C.
130,000X@
-
49
rC'
.
-.4e
L..
'
r
S
l ~ br
c
t
,
Y
a
V
f
_
t.
v~-
8i,-O X
.t
'- ,,'f
9~ ~ ~C ,r ~i ~ (~~Y
-
Figure II-6.
t
Electron micrograph of replica of Zn -3.5w/o Cd which
was cooled by the piston
and anvil
technique.
8 ,0OOX.
50
Chapter III
THERMODYNAMIC ASPECTS OF SOLIDIFICATION PROBLEMS
III.1
Introduction
The mathematical
analysis of an N-component
solidification problem with one solid phase consists of
setting up 2N differential
(2N-2) for interdiffusion
equations;
2 for heat flow and
in the two phases.
In addition
the initial conditions and the boundary conditions must
be given.
Of these, (2N+2) boundary conditions pertain
The reason this number of
to the liquid-solid
interface.
boundary conditions
are needed is that the interface
not at a fixed position and additional
is
conditions are
needed to specify the interface velocity. The interface
boundary conditions are:
(1) Continuity
of temperature.
It is believed
that the heat transfer across the interface is sufficiently
rapid that even in the most extreme heat fluxes temperature remains continuous across the interface.
(2) Conservation
of heat.
This condition includes
a term for the latent heat of fusion L
and thus involves
the velocity V
Ks(VT.n)sm - KL (VT .n)L
=
L V
(3.1)
51
(3) N conservation of mass equations; one for each
component
Ds (VC.n)s - DL(VC.n)L
=
(Cs - CL )V
(3.2)
These also contain the velocity.
(4) N "response" functions which describe the
response of the interface to the conditions at the
interface. These would give N quantities, e.g. the
velocity of the interface and the composition of the solid,
in terms of the instantaneous
temperature,
conditions; the interface
the composition of the liquid, and the orienta-
tion and defect structure of the interface. This section
will consider the thermodynamic aspects of these response
functions.
For a single component system if we ignore the last
three factors, such a response function would be
f(V,
AT)
=
0
(3.3)
or equivalently when solved for either V or AT
V
=
g(AT)
(3.4)
AT
=
h(V)
All three equations express the same relationship
should be fully equivalent.
and
If we impose a velocity
note the interface temperature and then in another
and
52
experiment apply this AT we should observe that the system
responds with the same V.
For a binary system the two response functions
would be
Cs, V, T)
=
0
f 2 (CL, Cs, V, T)
=
0
fl(CL
(3.5)
which might be solved for the response, V and C,
in terms
of the interface temperature T and CL
V
=
gl(T, CL)
(3.6)
CS
=
g 2 (T, CL)
Such a response function could equally well be expressed in
terms of the compositions that would be found for a given T
and V.
CS
=
m 1 (T, V)
CL
=
m 2 (T, V)
(3.7)
As we shall see this form is convenient
from a thermodynamic
point of view. Another way of expressing the respone
functions would be to express the temperature T and CL in
terms of a given V and C
53
T
=
h1 (V, C)
CL
=
h 2 (V, Cs)
(3.8)
This is a valuable
form for steady state solidification.
These four ways of expressing the response function are
fully equivalent
to each other and an experimental determina-
tion of one form should be adequate for reexpression
the other forms.
in all
Because they are boundary conditions to
differential equations one form or another may be convenient
for a particular
experimental
geometry but for a particular
system the relations should be universal.
Thermodynamics places only general restriction on the
response functions.
It does not specify them.
For the
single component it specifies that the sign of V be related
to the sign of AT.
For the binary system it specifies only
that for a given T, solidification
(V > 0) requires that C
5
and CL lie inside or on the boundary of the curve OABEP of
Figure 1.7. For purposes of mathematical analysis such a
general restriction is insufficiently precise. We have to
know more about the response functions.
Because these response functions are boundary
conditions to a solidification
problem it is difficult to
perform experiments that isolate them sufficiently clearly.
Ideally interface compositions, temperature, and velocity
should be measured directly.
Even in the single component
54
system the interface temperature is often inferred from
bath temperatures with or without a heat flow correction.
In multicomponent
systems as we shall discuss, equilibrium
at the interface is usually assumed(31) without even an
attempt at experimental verification.
III.2
The Assumption of Local Equilibrium
If we assume that the boundary is so mobile that V
is large for any deviation
from equilibrium,
functions become the conditions
the response
for local equilibrium,
of which there also are N in number.
Hence, for one
component
AT
=
0
for all V
and for two components
(3.9)
the compositions at the interface
are
Cs
=
C s (eq)
=
m 1 (T, 0)
(3.10)
CL
=
C L (eq)
=
m 2 (T, 0)
for all V.
The assumption is valid whenever the deviation from
equilibrium,
expressed as AT, C s - C s (eq), or CL - CL(eq)
is small compared to the total temperature and composition
ranges in the problem.
Although
appears in these conditions,
vation conditions
can proceed.
the velocity V no longer
it still appears in the conser-
(2) and (3), and the mathematical
analysis
55
The popularity
of local equilibrium
as an
assumption rests on its expected widespread validity for
most solidification problems which involve rather low
interface velocities,
and the fact that it gives a form
to the response function that permits one to begin calculation of solidification
of the assumption
problems.
The test of the validity
could come from two kinds of experiments:
the simultaneous measurement
or checking the applicability
of all the interface variables
of predictions of calculations
made using the assumptions. The former test has never been
made and the latter test is most usually made qualitatively.
Qualitative predictions from the assumption can only
show consistency or inconsistency.
For instance we can, by
using the assumption of local equilibrium, predict an
enhancement of solid solubility in a eutectic beyond the
maximum phase diagram value simply by invoking the assumption that the eutectic has been suppressed and solidifica-
tion proceeds on metastable liquidus and solidus extensions
below the eutectic.
(Figure 3.1)
However unless we
simultaneously measure the interface temperature, we will
not know whether we produced a deviation from local
equilibrium or actually suppressed the eutectic reaction.
Thus even the enhanced solubility produced by rapid
solidification is by itself inadequate to prove that a
significant deviation from local equilibrium was produced
because the metastable solidus usually continues to
56
increase in composition.
In the Zn-Cd system which has a
retrograde solidus, the equilibrium solid solubilitv has
a true maximum and the metastable
lower compositions.
extension continues to
The maximum was exceeded by rapid
solidification indicating that under extreme conditions
the significant deviations from local equilibrium can
be produced as shown in Chapter II.
It is interesting
to note another case where
local
equilibrium ceases to exist during solidification. Even
though the equilibrium
shape of a crystal may be faceted,
when originated during solidification the faceting is
usually a result of the growth kinetics.
Since such
shapes are not solutions to the differential
condition of local equilibrium
hold.
For instance, Glicksman
equations, the
at the interface does not
and Vold(32)
found faceting
during the freezing of Bi at small velocities,
but rounding
of the interface during melting at small growth rates;
this indicatesan
inconsistency with the condition of
local equilibrium
at the interface.
III.3
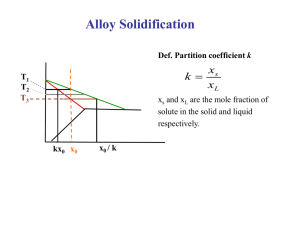
Steady-State Binary Solidification
Of the various experimental and theoretical methods
of dealing with solidification the steady-state planefront condition is an exceptionally
useful one.
We impose
a velocity V on the system and assume that after an initial
57-
transient the temperature and composition profiles and
the position of the interface will move with the
velocity V.
solid C
C
Under these conditions the composition of the
must be equal to the alloy's overall composition
0
C
Because C
= C
"diffusionless"
=
(3.11)
C
the process has some aspects of
solidification,
that there will be a diffusional
but it is highly likely
layer ahead of the inter-
face and the composition of liquid at the interface CL will
differ from C .
Under steady-state conditions this layer
remains unchanged with time and may be hard to detect.
It
will have a thickness of order DL/V, or less than 11 when
V exceeds 1 cm/sec.(33).
A study of steady-state solidification thus permits
us to fix V and C
and if we determine experimentally
the
value of T and CL at the interface we would be evaluating
the response functions. The functions although evaluated
by a steady-state experiment should be universally valid
for other geometries
state experiment
in that system.
If in a non-steady
the interface found itself with the same
value of T and CL as found in the steady-state experiment,
it should respond with the same V and C s .
Figure 3.2 shows Figure 1.7 redrawn for purposes
of steady-state
solidification.
The horizontal
line
58
represents the alloy composition
that is equal to C s .
The possible values of CL are those that lie on the
horizontal line but within the curve OABEP appropriate
The OABEP curves are given for the
for that temperature.
various temperatures, each representing a temperature
lying within three distinct regions of the phase diagram
given in Figure 3.3. Let us now examine steady-state
solidification
of an alloy of composition C
= Cs and
investigate the possibilities and the phenomena that
we would encounter
in the three temperature regions.
<
Region
I
CL(eq);
To
< T
TL(C
);
Point B below C 0
From the steady-state
Cs > C(To).
condition C
= C s implies
As can be seen in Figures 3.2 and 1.8 steady-
state solidification
cannot occur in this region, because
AG > 0 for all possible values of CL .
state solidification
Only non-steady-
can occur in this region(20) with a
solid composition in the allowed range of Figure 3.2,
C
s
< C .
0
The rejection of solute enriches the liquid with
a likely drop in solidification
temperature or a breakdown
of the plane front(34).
Region
II
(eq) < C<
CCseq
0
C(T 0o);
T o (Co0 ) < T < T O
Co lies between B and E
59
In this region steady-state
solidification
is
possible provided that the composition of liquid remains
in the range depicted in Figure 3.2.
The solid formed
is metastable with respect to partial remelting.
There is possible diffusional instability in such
solidification as indicated by the down arrow in Figure
3.2.
If momentarily the composition of the solid drops
below C,
conservation of mass requires that the liquid
be enriched in the minor component.
fluctuation from the horizontal
result in a shift to the right
Thus a downward
line (C
s
= C ) should
0
(CL increasing).
If a
system finds itself at a point F in the vicinity of EII
it cannot reestablish steady-state
(climbing to the
horizontal line) without decreasing CL.
But because the
system is at F, the solid that is forming is below the
average composition C0 and the excess is rejected into
the liquid making a reduction in CL unlikely.
state cannot be reestablished
Thus steady-
at this temperature
and
since we impose a velocity on the system the plane front
interface again either breaks up or lags back to a lower
temperature.
Steady-state solidification
in region II is thus
thermodynamically possible, but has difficulty in starting
from equilibrium and could be diffusionally
also leads to a metastable
partially remelt.
unstable.
solid, which might in turn
It
60
Regardless
of whether we have steady-state
the solute experiences
an increase in chemical potential
upon solidification
upon~~~~~ when C
occurs.
or not
> C (eq) and solute traDping
soldfcto
hnC
As we shall see in Chapter IV, this type of
solidification is prohibited by various theories.
The
experimental finding of Zn-Cd in Chapter II indicates
that solidification
when C
> C (eq) does occur.
5
Region
III
C
< C
(eq);
5
T < T ;
C
lies below
E
Like region II steady-state growth of the solid is
thermodynamically
possible in this region.
region II the resulting solid is stable.
But unlike
In addition the
diffusional problem appears to be stable(22).
If the
system starts at point EII I , C s will be greater than C o
and the liquid will be depleted of excess solute.
the system spontaneously
Thus
leaves the point EII I and will
settle somewhere on the horizontal
line C
5
= C
0
in Figure
3.2 for steady-state solidification.
Solute trapping occurs to the left of the intersection of line C0 with line OE.
transformation
The diffusionless
(line OB) requires solute trapping and
hence is not predicted by theories that forbid solute
trapping.
To the right of line OE no solute trapping is
required, but to the right of PE solvent trapping begins.
The equilibrium liquid concentration
lies outside the
61
domain of the possible compositions in all regions except
when
T = T.
Local equilibrium. When the velocity V is very
small but finite we must come close to the condition of
local equilibrium.
This means T
close to the point E.
T s and the system is
It is important to point out that
the immediate vicinity of the point E could place the
system either in region II or in region III.
of solute trapping,
Conditions
solvent trapping, or no trapping at
all are also infinitesimally
close to the point E.
Therefore one cannot argue a priori that because the
system must approach E at low velocities that it comes
to rest in any of these domains.
62
U)
I;
3
G1)
1)
0
C
-4
0
-
trL.J ~Z
4
V-U) C)
.-S
,,.4
z-4 U )
0
3)
) 4- -U UC
U)
F-
_-
LV
,4JC
C
'
-
X r.
*
,.,
CJ
--
<
4L C
3)
0 0U U
-n
t
--4
flv3dv3i
3 Wn
0
C:
-X
C
n
4
63
0
0.0
0-
4-
'"
.,4
;0
,ZCJ0.
-z
O
0
O
. CJ
~*-
'
J-J
0
O 0 4 C:
-4
0 0-'
Htu .O
0
,p .- 4'0 r_.
~
E
i.
Og
,-
- v
'D
L
C0
LS
7.2°
QU_
-
0 5~
-w
4) 4
0X
0
o1
0
t<
0
_
*~
0
rP;
-JS -4
0)
o0 4 4
0
0;
_
a.
O
0
0E*A
4
-0 0
00
4>
a.40
-J
oU.
-j
~
0
-
0-
,,
0-
0
'.00"-V'
0
°©D
QH
-L
~0
QDo
0
I
i.4
(N
0
0
3OV
.3INIiV NOIIISOdIOD (IlOS
z 0:
0
Ja
0
'0 _
0f)
.
0
o
0
4-_
0> Vf
64
CJ N,
el. ,Dr
4
Oc:
-,
0
L-
C:
C
El
-j
,,
U
o-
.I
-
o
n-
-4
0
1
t~13
3n llV id v 3 i
65
Chapter IV
IRREVERSIBLE THEP4ODYNAMICS
OF INTERFACE PROCESSES
IV. 1 Introduction
In this chapter we will describe methods of applying
irreversible thermodynamics to interface processes during
solidification and obtaining the interface response functions.
The means of choosing the fluxes and thermodyna-
mic forces for the various interface reactions will be
examined. Four current theories using this approach will
then be analyzed and compared with existing experimental
results.
Special attention will be focused on the
following most often used assumptions and principles:
the assumptions of independent
reactions at the inter-
face (no coupling of processes),
the Onsager reciprocal
relationships, and the principle of minimum entropy
production.
The section will be concluded by summarizing
what has been learned about the validity of various
aspects of the irreversible
thermodynamic theories and
where the field now stands.
IV.2 General Principles
The heart of irreversible thermodynamicsC35,36,37)
is that it proposes much needed relationships between
rates of reactions and thermodynamic quantities. When
applying it to the interface processes during
66
non-equilibrium solidification, the usual assumption is
that the fluxes, Jir across the interface and the conjugate
thermodynamic
forces, X
are related linearly.
In other
J
words a phenomenological
J.
1
=
relation of the form
EL..X.
is assumed for the interface reactions.
coefficients, Lij,
determined
(4.1)
.J13JJ
J
are phenomenological
for any given temperature.
The kinetic
quantities
to be
If there are only
two processes occuring at the interface then equation
(4.1) becomes
J1
LlX1
+
L21X1
+
=
L12X2
(4.2)
J2
In equation
=
L22X2
(4.2) the coefficients Lll and L22 relate each
flux to its conjugate force.
The cross coefficients,
L1 2
and L2 1 , give the coupling interaction of the two
processes.
If the processes
are independent, L 1 2 and L21
are zero and
J11
=
LllX1
L11X1
(4.3)
J2
=
L 2 2 X2
There is, as we shall see, considerable freedom about
the choice one may make for the independent fluxes and
forces. Generally each flux-force pair is chosen such that
67
its product is a separate term satisfying the entropy
production relationshipS38}
Ta
where
=
J1X1 + J2X2
(4.4)
- is the rate of entropy production.
For example, a
proper choice of flux-force pairs should not lead to a
cross-term J1 X2 .
Because T
cannot be negative, if we
define our flux-force pairs to satisfy equation (4.4), then
the L matrix of equation
(4.2) is required by thermodynamics
to be positive definite(39)
(or positive indefinite
if there
are infinitely large kinetic barriers to some of the
reactions).
This is
L
1>
11
-
0
>
0
LL
1 2L 2 1
<
L22
(4.5)
L
L
LllL22
For isothermal processes Ta = - dG/dt and for binary
solidification
at steady state
Ta
=
-pV[CsA
+(l
C)Ap
(4.6)
We will define two sets of flux-force pairs that
satisfy equation
(4.4) for the interface process in binary
systems. Either of these are therefore equally valid
variables for developing
face processes.
a theory of irreversible
inter-
Both have been used with some modification.
68
We can choose the J's to be the net fluxes across the
interface of the two species during steady-state growth
=
J15
= =A
pV(
- C)
(4.7)
= B
=
2
=
VC s
and the forces to be the chemical potential
changes across
the interface
x 1 'X =
-AUA
(4.8)
X'
where Ap
2i
i
= (is
2
-
-AB
=
i
iL). Alternatively we might choose J
to
flux without regard to composition
be a total solidification
change and J2 to be what has been called a redistribution
flux
J1
J,~= PV
1
J2 w
=
(4.9)
pV(Cs - CL )
Then we must choose
XI" Xi=
-(1
- CL)
L
AA
-
C L~ A
(4.10)
X2
"
=
AU
-
AB
2
X1
is the free energy change if components of the liquid
add to the solid in the ratio in which they are in the
liquid at the interface.
It might be considered a driving
force averaged according to the interface composition of
69
the liquid.
The first reaction by itself (J2 " = 0)
would lead to solidification without a composition change.
X 2 " is the driving force for the composition change across
the interface.
and J2describe
Together J
any solidifi-
cation reaction.
The reader may verify that both sets satisfy equation
(4.4) algebraically.
For the single primed set it is easily
demonstrated graphically as well.
For the double primed
set, Figure 4.1 shows how the molar free energy change is
apportioned among the flux-force pairs.
AGI is the free
energy change due to the interface process when a mole
is the free energy change
solidifies at steady-state.
AG
due to the first flux-force
air and the difference
in the
slopes of the tangents times the composition difference
is
the free energy change due to the second flux-force pair.
Two ways of finding the independent processes have been
given and for each process the proper driving force has been
found.
is tied
In the first set the fluxes of each secies
to the thermodynamic
driving force for that species.
the second the overall solidification
overall driving force which
In
rate is tied to an
is the average of the driving
force experienced by the atoms in the liquid, and the extent
of redistribution
species.
is tied to the driving forces between the
Either set is as complete as the other.
They
both give identical free energy changes.
A well-known condition in irreversible thermodynamics
is the Onsager reciprocal
relationships(40).
It states
70
that L12 = L21.
We invite the reader to assume the
relationship holds true in one of the primed sets and then
substitute
into the other set.
that the Onsager relationship
set.
It is often stated(41)
It is immediately apparent
does not hold in the other
that if the flux-force pairs
are chosen to satisfy equation (4.4), the Onsager relationships are automatically
criticized(42)
guaranteed.
This has been
and the present example shows that this
criticism is valid.
The contradiction
the reader will have
found proves the Onsager relationships are not guaranteed
by choosing our flux-force pairs to satisfy equation (4.4).
It is also tempting to assume that the fluxes are
independent and set L12 = L21 = 0.
However, in the examples
above it is sobering to consider that if we make this
assumption when using one set of flux-force pairs, the
independence does not hold in the other set.
For example
if we assume
J1
=
Lll X1
(4.11)
J2
22 2
then
X
(1
CL)X1
+ CLX2
(4.12)
2
2
1
Or solving for X's,
X1' - X"
1
X2
X
CLX
2
= XI + (l
X
CL)X
22
1
C4.131
71
And substitution
Tilt = J
I =
T,
T2
L
leads to
LX
'1 !"2X
2 XI
/']X
(4.14)
IT-:
c)z,
( ' :s-
-c7 z ) 2'
'
Now not only would these fluxes be coupled, but the offIf
to the diagonal.
diagonal terms are comparable
L
L2
=
122
0 implies
L21
0 implies
=
0,
and
=
or L2
22
either
=
L
=
0
0.
In addition we note that again
12
r
21
What this proves is that if the first set of fluxforce pairs are assumed independent,
the second set
cannot be, and must appear coupled.
We cannot assert a
priori that because the flux-force pairs are chosen from
equation
(4.4) that they are uncoupled.
is no contribution
Even though there
from J 1 X2 or J2X1 to T
guarantee that there is not a contribution
or vice versa.
we have no
to J1 from X 2
In fact the example shows the coupling can
be quite large in one set when it was assumed absent in
the other.
Thus a perfectly
becomes very complicated
flux-force pair.
simple flux-force relationship
wahen expressed in terms of another
It must be stressed that in going from
the primed set to the double primed set no new physical
72
assumptions were made.
The two sets of equations express
the same physical behavior in different but compatible
language, in that any experimental
be described in either set.
outcome can equally well
The errors are introduced when
someone makes a theoretical assertion about the L's.
Which of these two sets of flux-force pairs is the
correct choice is not at issue.
describing
the same phenomenon.
Both are capable of
Of importance however is
if we wish to assume that coupling between fluxes is weak
and set cross terms equal to zero we must be careful.
Compatability with equation (4.4) is not a sufficient
condition to insure the chosen fluxes and forces are
independent,
and does not guarantee that the Onsager's
Reciprocal Relations
(L1 2 = L21 ) hold for the coupling terms.
Another principle that is commonly invoked is that at
steady-state there is minimum entropy productionC43,44,45).
This can be used as a principle
for obtaining relations
among the L's but has often been criticized(42,46)
theoretical grounds as invalid.
on
We shall show below that
it leads to a result that contradicts solidification
experiments.
Quite apart from these considerations,
the assumption
of linearity may not be valid when applied to the interface
response.
Several mechanisms of solidification, nucleation
and lateral growth of new layers, and the spiral dislocation ramp would not lead to linear laws.
73
IV.3 Solidification Theories
The four irreversible
thermodynamic
theories to be
discussed now are by Borisov(47), Baralis(43), AptekarKamenetskaya(48), and Jindal-Tiller(49). The theories
differ in the way the flux-force
Another theory by Jackson(23)
pairs are chosen.
is of interest and will be
discussed although it is not an irreversible thermodynamic
theory. With minor differences the first two theories use
the primed flux-force set, while the second two use the
double primed set.
Since additional assumptions are made
about the L's, it is apparent the validity for both groups
of theories is questionable. We will now consider the
theories individually.
1.
Borisov Theory
Borisov assumes the net fluxes of each atomic species
traversing the interface is independent
JA
=
LAAXA
(4.15)
JB
=
LBBXB
Since the above flux-force
set is the primed set mentioned
previously
J
A
pVCl - Co0~~
) =
-LAA
AA CJs
L
(4.16)
JB
B
pVC0
-
L
BB
CB,
B
s
B)
L
74
By equations
quantities.
(4.5) LAA and LBB are always positive
And since pV(l - C
and pVC° are positive
for solidification, Borisov's theory yields the following
condition:
A
A
B
B
s
L
Ds - HL
<0
<0
(4.17)
<
0
This limits the allowed processes
to the triangle OEP of
Figure 1.7 and implies no trapping of either atomic species
is possible during solidification. For uncoupled reactions
each species must traverse the interface only when it is
thermodynamically favorable. This prediction is contrary
to the Zn-Cd experimental result of solute trapping in
Chapter II.
2.
Jackson Theory
Unlike the other four theories, the Jackson theory
is not an irreversible thermodynamic
description of alloy solidification.
theory, but a kinetic
It is of interest
since his resulting relationships have been put into the
thermodynamic language of Borisov's.
theory is seen to be equivalent
When doing so, the
to Borisov's(50).
Jackson
develops the theory on the basis of two assumptions:
(i) reaction rate theory can be applied independently
for
each species during solidification to determine the net
flux of each type of atomic species across the interface,
and (ii) the rate at which each atomic species leave a phase anJ
73
IV.3 Solidification Theories
The four irreversible
thermodynamic
discussed now are by Borisov(47),
theories to be
Baralis(43), Aptekar-
Kamenetskaya(48), and Jindal-Tiller(49). The theories
pairs are chosen.
differ in the way the flux-force
Another theory by Jackson(23)
is of interest and will be
discussed although it is not an irreversible thermodynamic
theory.
With minor differences
the first two theories use
the primed flux-force set, while the second two use the
double primed set.
Since additional assumptions are made
about the L's, it is apparent the validity for both groups
of theories is questionable.
We will now consider the
theories individually.
1.
Borisov Theory
Borisov assumes the net fluxes of each atomic species
traversing the interface is independent
JA
=
LAAXA
(4.15)
JB
=
LBBXB
Since the above flux-force
set is the primed set mentioned
previously
J
JB
=
=
pVl
- Co )
~A
0
pVCo
=
-LAAs A
AA
B
BB(-
s
B
L)
AL
- 3~
(4.16)
(4.16)
75
traverse the interface is proportional
to the mole
fraction of the species in that phase.
Assumption
necessarily
leads to equation
(i)
4.17) and limits the theory
to the triangle OEP of Figure 1.7.
3.
Baralis Theory
Like Borisov, Baralis assumes the primed flux-force
pairs, but he doesn't assume independent fluxes; i.e.,
PV1
JA=
A = pV(1
-
C0 )
C
o
=
- AA(NA
IA
-LA
s -_'ALL-
LAB(s_B EL)
_
)
LAB
(4.18)
and
J
= pVC0 =
-
LBA (A
A)
L
(B
_pB)
Equations (4.18) are general and allow for coupling, which
in turn permits the possibility
of one of the species
chemical potentials to increase upon solidification.
He assumes Onsager's Relationship,
LAB = LBA, for the
coupling coefficients. In addition he assumes that the
rate of entropy production
growth.
is a minimum for steady-state
In applying the latter assumption he arrives at
the conclusion that
A
A
s -
B
s
-
<
DL
B
L
<
0
Hence, this analysis also limits the processes
to the
triangle OEP of Figure 1.7 and is contradicted
by the
Cd-Zn experiment.
Even though the theory allows for
76
interaction of the two atomic-fluxes
the principle
of
minimum entropy production" restricts the coupling and
forbids the possibility of trapping.
4.
Aptekar-Kamenetskaya Theory
The interface processes assumed in this theory are
equivalent
to the double primed flux-force
set, that is a
total solidification reaction and redistribution reaction
which are independent. The total solidification flux-force
relationship
is of a slightly different form, though, than
for the double primed set, because of the dissimilarity
other assumptions.
in
This treatment does not assume steady-
state growth, but assumes the diffusivities
and solid phases are infinite
in the liquid
(this implies the unlikely
circumstance of the two phases having uniform composition
at any instant of time).
The above condition for most
growth rates leads to a variation of liquid and solid
compositions at the interface with solidification time,
which in turn corresponds to a driving force across the
interface that is a function of time.
As a result, there
is a subtle distinction between the two expressions
driving force for the total solidification
of
reaction.
It
also should be added that this treatment is not a complete
theory since the number of unknown quantities exceeds the
number of equations.
Regardless
of the above serious
shortcomings, Aptekar and Kamenetskaya do predict the
possibility
of solute trapping,
s-
_L
> 0.
77
5.
Jindal-Tiller Theory
In principle,
this theory also assumes the double
primed set, although the only reactions considered in
detail are the redistribution reactions
JJB R
=
RV(C5
=
pV(CL
A
J
-
C
-
L
=
B
L-
s
A
A
L(Ps
(4.19)
B
B
R
)
L(4.19)
A
_L
(4.20)
B
aA
If LR
and LR
are chosen to be the interdiffusion kinetic
R R~~~
coefficients, then Lcfe
= LA and combining equations (4.19)
R
R
and (4.20) yield the double primed redistribution relation
JR
=
pV(Cs - CL)
where LR = LB/2.
shows that AA
Comparison
=
LR(APA
-
AB)
of equations
(4.21)
(4.19) and (4.20)
and APB always have opposite signs.
Thus
this theory restricts the process to lie outside the
triangle OEP but inside the curve OABEP where trapping
always occurs.
It should be noted that the expression
for the total
solidification flux-force pair should be solved simultaneously
with equations (4.19) and (4.20), otherwise the various values
of the growth rate V may be unrealistic.
Also it is not
certain that equation (4.4) is satisfied since no expression
for the total solidification
reaction was given.
A major
deficiency of this theory is that Cs/CL does not equal K(eq)
78
when V = 0, but
(2Y7K(eq)-
(See their Figure 6.)
1).
A limiting condition for all correct theories should be
Cs/CL = K(eq) when V = 0.
IV.4 Discussion
of Theories
It has been shown earlier from classical thermodynamics
that for solidification
the compositions of the solid and
liquid at the interface must be inside the OABEP curve of
Figure 1.7.
In this section it has been shown that
irreversible thermodynamics is unreliable in attempts to
restrict the two compositions further.
Theories using
similar assumptions can arrive at contrary results.
For
example, Borisov assumes his flux-force pairs are uncoupled
and finds trapping to be impossible, where as Jindal and
Tiller make the same assumption for their flux-force pairs
and find trapping always occurs.
the two approaches
The basic difference
is that they choose different
in
coordinate
systems for the atomic fluxes, JA and JB' which are proportional to AA
and AB.
Borisov uses the one moving with the
interface which gives a constant flux of material for
steady-state for the minor component
B
=
VPCs
'
while Jindal and Tiller chooses a substantial or laboratory
coordinate system imbedded in the liquid which differs by
VpCL
=
VQCC
- CL)
79
For the usual case where C
> Cs the two JB 's have
opposite signs and, if uncoupled,
in 4
B
lead to opposite signs
.
We should also point out that all four irreversible
thermodynamic theories have serious deficiencies. New
principles
are required in this field or tighter restric-
tions for applying the old principles
are necessary.
most of all, there is a need for experiments
But
to study the
deviations from local equilibrium and coupling effects.
80
f:n[:4_
I
~
~~~~~~~~~~1
I,
.
_'J %
-
I~)
S.Cu
C-_
.
.
~ ~ ~I--
-
<:[
__.~~i
A,, 0_
W3N3 T3d.
V<:OW
A,9813N3 33diA 8V-I0
r-
o
Uz.4
2J
0'.-°
2- 0
· 0 ,,, L..
0s
Cr
UJ,-r
J
C)
_,
04-i
C 0C2
l
'£
v~ .--.J.
5
C.
81
Chapter V
AN ALTERNATE APPROACH TO INTERFACE
PARTITIONING DURING SOLIDIFICATION
V.1
Introduction
In the previous chapter it was found that the method
of irreversible thermodynamics
as applied to the study of
interface processes during solidification leads to values
of interface distribution coefficients that were
questionable. The present chapter is devoted to overcoming
this shortcoming by using a different approach to the
problem.
The method of Solute Interface Drag will be
applied.
Specifically,
an attempt to solve for the liquid
and solid interface compositions
for an imposed and
specified velocity is made when the moving interface is
represented by a potential well in motion.
It is assumed
in regard to the solute species, that the interface at a
given temperature can be characterized by a solute interaction energy E(x) and a diffusion coefficient D(x).
Both
E(x) and D(x) are functions of an arbitrarily chosen plane
in the interface.
V.2
Diffusional Solution
Let us assume dilute solution everywhere, then the
chemical potential of the solute species is given by
equation
(I.5)
=
kT.nC(x) + E(x) + constant
(5 . 1)
82
where E for the liquid and solid are related by
.Cs (eq)
(EL - E s)
kTZn
=
CL
(5.2)
(e q )
Now define the interdiffusion coefficient as
D(x)
=
- JkT/ (d
-
(5.3)
2
where J is the flow of atoms per cm.
per sec. measured
a co-ordinate system fixed in the liquid phase.
changes are neglected.
Combining equations
in
Volume
(5.1) and (5.3)
the flux becomes
-J j =k
DC kT
iD
Dx
=
D ax
C + DC
kT DE
x
(5.4)
(54)
The first term on the right-hand side of this equation is
the well known fickian flux, and the second term is the
flux resulting from the interfacial potential gradient
9E/Dx. However, a fixed co-ordinate system relative to the
interface is mathematically
desired and for this new
co-ordinate system the flux relationship becomes
-J
=
D -C + DCkT
ax + VC
(5.5)
With regard to this new co-ordinate system, the conservation
equation becomes
aC
a~ =
a
D --x
+
2[V
aD
D
[Iv+ x + kT
DE
]
C
x
C
+
kT
D
x
E +
x
+
32E
a -]
......
. (5.6)
83
This relationship
at any instant in time must be
satisfied at each point in the system, the composition C
being a function of both position x and time t.
It has to
be solved for given initial and boundary conditions.
Relative to the interface fixed co-ordinate system and
under steady-state conditions, the composition everywhere
remains unchanged with respect to time.
DC/3t = 0 and J = constant, and equation
Therefore
(5.6) may be
expressed as
0
=
_
_
x (-D
C
DC
~E
kC
E
DC
-
VC)
-
x
(5.8)
(J)
This is a necessary result for the satisfaction
state conditions where J is a constant.
of steady-
Integration from
-a to x yields
o
x Dj
=
f
a-a
x
dx
f
=
-00
dJ
=
J(x)
- J(--)
(5.9)
-00
or
0
[-D
=
~C -
DC DE
kT
Dx-
VC] + VC
(5.10)
Rewriting the above equation
DC + [
ax
kT
DE + V] C
x
D-
V
D Co
(5.11)
It is readily seen that the above equation is a linear
differential equation of the first order and can be solved
in general terms without determining separately homogeneous
84
and particular
solutions.
~x
The form
+ P(x)y
=
of the equation is
Q(x)
(5.12)
which can be solved by using a standard integrating factor
(51)
I.F.
=
ex[/fP(x)dx]
The solution of equations o
(5.13)
the form (5.12) is obtained by
the following integration:
Y
1
=
(I.F.)
x=x
f
(I.F.)Q(x)dx
Hence, the solution of equation
C (x)
=VC
exp{
0
(5.14)
(5.11) becomes
E~~~~~~~~~x)
x-[k
D (n)
E(x)
~~~ _~~~kT
V xi' dn ) }
o
(§) +
dn
c-o
X
o
.. (5.15)
The above relationship
(5.11) , and is completely
in equations
is the exact solution to equation
general as long as the assumptions
(5.1) and (5.3) hold.
comosition everywhere
for any imposed velocity at steady-
state for arbitrary E(x) and D(x).
of equation
(5.15) are difficult
be easily shown from equation
interface compositions
C
It describes the
The limiting conditions
to determine.
But it can
(5.1) that the ratio of the
of the solid and liquid is given by
(i)
(L5.16?
C L (i)
=
exp
L
E
- E
_skT
) = K(eq)
(5.16)
]
85
when V = 0.
This is a necessary
any valid theory.
that equation
limiting condition for
Later in the section it will be shown
5.15) reduces to condition (5.16) when
V = 0 and to another limiting condition
C (i)
C(i)
i(5.17)
1
5
CL
-
when V +
V.3
Graphical Representation
As pointed out above, equation
composition profile at steady-state
and D(x).
This relationship
(5.15) describes the
for an arbitrary E(x)
then not only yields the
desired interface compositions but also the compositions
everywhere else.
on understanding
Nevertheless this section will focus only
the nature of the interface partitioning.
To represent graphically some of the properties of equation
(5.15), D(x) and E(x) are assumed to be functions of x as
shown in Figure 5.1.
For such a model, equation
(5.15) can
be reduced to yield the following expression for the
interface partitioning:
CL (i)
C=
CL (i)
(i)
CsC(i)
-is
SV(
CO
B
exp(
ES - EL
kT ) {exp(
D
f
S)
V ) exp(-5D
L
+
s
kT
EL
1
s
EBEL
] [
[exp
k -- ~~ S][
~ DVE xD(E
L ~~)ep]~
D (L
1+
LL
][exp kT
l
-- )exp(-)l
- kLT
V
_ ex-
e
1 + (V) (.kT
.........(5.18)
86
as shown in the appendix.
In terms of the equilibrium
distribution coefficient, Keq),
CL i)
CLCi)
Cs i)
CO
0i1
s
equation
5.18) becomes
F1 -X-Y +
_
KCeq) -
-Y
(-X)
K(ec)
+ ("~~~~~~~~~~~An)e ) KBC)-e
KB(eq)
)
1+
e- Y
(eq))]
(eq)(Keq))C(l-K
1
lnK (
+
1+
Y
(5.19)
.
where KB(eq), X, and Y are defined as in the appendix.
the case where Keq)
For
is a constant this kinetic result
(equation 5.19) is only dependent on the imposed velocity
and not on the interface temperatures.
The graphical representation
typical K(eq) = 10
-1
of equation
is given in Figure 5.2.
(5.19) for a
The various
curves are for different assumed values of KB(eq) or (EL-EB).
Curves 1 and 2 show the nature of the dependence of the
interface partitioning on the growth velocity when the
solute species is surface desorbed at equilibrium. Curves 5
and 6 are for the case when the solute species is surface
active
(surface adsorption at equilibrium).
Curves 3 and 4
show the partition when the equilibrium interface composition
is equal to the equilibrium solid and liquid compositions.
Let us now consider in detail the case when the solute
atoms are surface inactive
deviations
curves 1 and 2).
from local equilibrium
that is for a given steady-state
The initial
are negative, K < KCeq),
solid composition the liquid
interface composition increases with growth velocity.
Since
87
EB ?B EL
L the solute species attempt to remain in the liquid
and avoid the moving interface; this leads to an increase
in the liquid interface composition as the velocity
increases because the diffusion distances in the liquid
relative to the interface per unit time are decreasing with
increasing interface velocity.
K < K(eq), the solidification
lie in the single-phase
One can then conclude since
must take place when C
and T
solid region on the equilibrium
phase diagram to allow CL(i)
< CL(eq) which is a thermo-
dynamic requirement.
At higher growth velocities we see curves 1 and 2 pass
through a maximum and then log (CLCi)/C S ) converges
continuously to zero.
The reason for this behavior is that
the solute species has increasing difficulty remaining
in
the liquid by diffusing ahead of the interface at these
higher steady state velocities.
zero the transformation
When log (CL(i)/Cs) becomes
may be considered as diffusionless.
From the above reasoning one can conclude that if the
diffusivity in the liquid were larger than for the case in
Figure 5.2, the maximum in curves 1 and 2 would be shifted
to a larger velocity.
The same is true for the velocity
needed for a diffusionless
transition.
It is also of interest to analyze curves 1 and 2 in
terms of solute trapping, K > KCeq).
From Figure 5.2 it is
necessary for log Y > 0 for solute trapping to occur, or
102
cm/sec.
V > V>
10 cm/sec.
Such large
may be
be physically
physically
Such
large velocities
velocities may
88
unrealistic and hence solute trapping is most likely
impossible when EB > EL.
Turning now to the case where the solute atoms are
surface activez EB < EL and curves 5 and 6, it is observed
that any deviation from local equilibrium is positive.
Hence solidification
in the two phased region, C
> Cs(eq),
is possible and solute trapping will always result no
matter in what region of the phase diagram the solidification
occurs.
This behavior is rationalized because during
freezing the solute atoms rush to the interface from the
liquid when EB < EL and then are captured in solid such that
K > K(eq) even though EB < E S
The reason for this is
because the diffusivity of the interface is changing from DL
on the liquid side to a smaller value, D,
on the solid side.
Such a phenomena also allows it to be kinetically possible
to have C(i)
> CL(i) at large velocities, but which are
smaller than those necessary
for a diffusionless
transformation.
Similar to the above, the intermediate cases, curves 3
and 4, result in positive departures
at high velocities
from local equilibrium
because of the difference
across the interface.
But C(i)
in diffusivity
> CL(i) is not possible
because the solute species does not rush to the interface as
in the previous case.
The solute species in the liquid is
more or less not affected by the interface potential since
ES
S -< E B
B
-L
EL
89
As noted in Section
.3 of the first chapter, the
most plausible assumption concerning the nature of the
interface is to assume it to be disordered
the liquid phase.
Then EB
and similar to
= EL and the partitioning
represented by curve 4 of Figure 5.2.
is
For this probable
case solute trapping will always occur during steady-state
solidification and CL Ci) will monotonically decrease from
C0/K(eq) to C
0
o
0
with increasing
growth velocity.
shows the condition of local equilibrium
Curve 4
at the interface
to be a good assumption for velocities
of millimeters per
second or smaller.
becomes diffusion-
The transformation
less with velocities larger than centimeters per second.
It should be kept in mind that these estimates are for the
particular model developed in the appendix.
90
V.4
Appendix
In this section equation
(5,15) will be integrated
for the
case where D(x) and E(x) are assumed to be functions of x as shown
in Figure 5.1.
The origin of the x-axis is taken as the solid side
of the interface.
E = EB.
The center of the interface x =
is where
The liquid side of the interface is at x = 2,
The
interdiffusion coefficient is assumed to change from D S to D L at
x -
L
Equation
(5.15) will be integrated
from -
to
2
since
the interface liquid composition is desired for determining the
interface partitioning. Equation (5.15) is now rewritten.
D
73f
The second integral in equation
LTvb&.,I
J
'
0
Xv
RA
4o
eT--ITvI
J
9 Df)
-r.- C.Pug,C 0
z
(5.15)
(5.15) is broken up into the sum
of the following three integrals.
N
D(f)
('VJP
P(r)
91
I-
E(f) +
I- vrEGRAL
COA1s1DRA't6
kT
) D(r
Q
SrfTld&R4eL
a
IT~v
are CGIR
Feeon F/G
/RE
V_
F
=) O
At
£r)
kT
'F
D(
V-i
D -Ds
E= Es
f
yg Jn
JoDf~n
REWR/T//4
-I t6AL
=-v
-ot
Ds
0
O
0
-T
'rPc.AL
+2k
d F
Ds
92
AMAJ6/I& VAg/,S
LE OPI1JTEGATO/
c/ f
dGDS
o
lT v
ArL&A
L0
hr
/
A1E7RGAL
0=
7-U,,em
AOW TA
-rAi
ed
V
(S.zo)
kT
7RiL
v at
df
Dgv
0
Feo FUR EEV-1
E-E=-
-FCp0:~x
T
~
Es -4E)
f
A/voD V
x
-
)
D(-)
V
f
of S
fl/
I,v rRgq/-( =
ioI
Ds
,'E
-
£
I-r6,RT-EL
0
(E,-FE
S
z
k
Z0L
df
94 VJ T
-I
DS
/-f/
Vkr-
s
d
1r
~~~~~k
_ZV-Nr R
)
93
rT
+VkTk
Os
/C -Ea)] -9
_7
0
Ty FY ns EeP
a
s-!S
/O
_tsq -re Gg
p.A
r: GAL
R
=
E
0i
9jL
(5.2/)
Dsj
@eq
A/0 Ev,
Sww64r,& -T&
Ip g-6L
2S
r
Ir-t 6 AL(9 -
LF
A, 7
o/(
-I)
J
FRo
mF/1Ue V-1
D=DL
E(f) =E s
Amb
F
V
.O
0
dL
V(n.)
f-S
7-
S
- EI-i6)
v S
-FoR f 2S
LoR
01L
Y>d
94
()
RE,/TS-IG6r,I-EGRAL
2S
D4+
IM&RA
04_L
l/
I NrEGRALO
7-
E
r
+
,D!
,
LT
k -v
]
kjL-s-VT
I
I
2-L-6E4.
S
+
D-L
- ,-VAT
8
frt
J{
B·Vs
(s.22)
+v
ReWA/rI/A
Eau rvo,w(5S.1) I
Ao gCYLlr
Qt Tc- 72siZ mNr7GR6 1 S
7is
A-r(x- S) .
(2$) = CoVf
E.
7T rTC:)
I°I'Jl
DE'
A,F
s_
QVCo Et.
Ds
D
(. 23)
95
0! Lr
0
r
]
7
r
:3
(D
(5. 2z)
0-3=6'.{a Urr)®~r7-T
hE
C(2-)=
fi
1
•2a1L63
Co n 8/A/Vv G & 0 U 7-/a A1
IQ1 = C',
V-tf
a£2,= Co2&7r&
(5. 2 0)j 5.Z 3)
EL
k 7-
D4
DL/
Q
(S 2 Y)
-/- -2!
(s- z)
96
C
-_
Z
ONS/OER
c0M
I/IvJA Ec r9 -/Dvs55Z/)%(S 2
aE~b-E
l k7
/
4Df)
iS-Ea
kT7
24)
g
-__
D 5-
(s. 27)
1
S -,Fs-
,I,-T-
t:s
CO1/SoER
Q
/Jr/O
5(
Co /, AJ/ 6 E4
.3C=C
I
EL-E
0,6 rRqlV F6?V47'JdAJ(E/ 8
Co
C~ )
-' '
"
i
(5.2
)
(5.28)
-
D4
-r) - -f~A~
~-S
j
(S-. 2 4) (5-,27), f (s- 2 a
WC
OF 7EYPRvEl/vUSS7EC -/
a
Coon 131AIII& 67IPV4 / 0 AJ (7. L
S)
C__)
IstD
-
! -.,-
J
_
=
4
2(2 ),(Y 2 3)
.
s7
DLJ
.
"
'
£tutE -E
'
k
- )
'gq-,EL
)(-}
/
1('
-t# - VI
+f
97
FIo
/nv
Co
VE6/V
K (q
Chv aiPSSD
e
CF
7~~-,n
(s- /i)
cuA raM
NCE-
E
=-e
_ Cs (eq)
cl _
-
4-
7'-',
CL (eq)
C8y (e )
-
'64 - 'LO
k7
CL (eq-)
Ds
DL
C
()
Cs ()
)- /+
(e
c (4D =
1eq)
/¢(
C =s~
(
Is A sslEo
O~c 7/
IA
U
,O
(-X-Y)
I
I k(e)
(k3 (e)
rs
T/sv /5 7-,
1
44P(-X
I,
t
PL o - r7 D /I
T-A-AT Ds = /0- DL
s -' / L R e-
V - R ) Ke)
(eq)
a
x
- ~~iiY
q
t6(f
/,/4 u?c V -2 727
_-v 7
= K) Avo
/o 7-A 7-/o N
1'We)
R
(5.19)
98
_
___
___
____
o;
o
ce ~
0
Co
cr-
G)
-A
0)
I
g0
O 0W> =
o4-Z
0)4J0
0
C
_cl
'--JC
0
0)
_
-
tJ
-Q
;>o
O
---- LU
-
U)
L~J
C
O4-
_
r
-
oR '-'
crr
_
_
0
,'L
W
_
0
-J
J~~~~u
--
-7~~~~
_
O
_
Xr
_
'H
0) 0
C) C >' 0)
(1)
0- 0)C
CO4-
,n
U)
V
o-4 4
W
0
CO
vq}
0
4-}
Ll
Q)
0
0)
0
CO
0)
-fc
0)
r
LUJ
_
_I
I
I
99
+
N
Co
4J-
a)
u)
C.)
0+
co
1
-1
Cd
m
' coln
L
ou)
MT C
4-
I
O
0J
0
H
CQJ
I
'H
+
+
+
..-
+
--.
-!)lo
+
al E090-
0
I
I
C
100
Chapter VI
SUMMARY AND GENERAL CONCLUSION
VI.1
Summary
We have seen that classical thermodynamics
is useful
and rigorous in determining free energy differences between
various states of a system, thus indicating if certain
changes of state are possible or not.
Also thermodynamics
is approximate but very useful when the condition of local
can be applied.
equilibrium
solidification
But for the case of binary
it has been shown in Chapter II that the
condition of local equilibrium
always hold.
at the interface does not
And when the condition of local equilibrium
at the interface is not valid for binary solidification,
thermodynamics
restricts the domain of the possible but it
may still be so large that thermodynamics
is inadequate in
predicting what will happen.
The need to specify what will occur for situations
similar to the above case has led to the development of
the Theory of Irreversible Thermodynamics.
The author has
shown that this theory as currently applied is not
rigorous or trustworthy, especially when applied to the
interface processes during solidification. It predicts
interface response functions which are of questionable
validity.
It is unreliable
in attempts to restrict the
liquid and solid interface compositions
further than
101
classical thermodynamics
has done as illustrated in
Figure 1.7.
Due to the shortcomings
in the ability of
irreversible thermodynamics to predict correctly the
interface response functions during solidification, the
author has choosen an alternate method to the solution of
(Chapter V).
the problem
By applying Solute Drag Theory,
an expression has been found which relates the liquid and
solid interface compositions to the imposed growth
velocity.
The analysis predicts
solute trapping will
always occur if the solute is surface active at the
liquid-solid interface; otherwise it most likely will not
occur.
The prediction of solute trapping during binary
solidification is experimentally verified for Zn-Cd
alloys in Chapter II.
VI.2
Suggestions for Future Work
The previous chapter concerned itself with deriving
an expression for the interface partitioning during binary
alloy solidification as a function of imposed steady-state
velocity.
But to describe
"completely" the response of
the interface in terms of the conditions at the interface
another relationship involving the interface temperature
T is needed.
For a binary system two response functions
are needed for a complete description. The author can
think of a possible approach for determining
this necessary
102
second relationship, but what may be first needed is
additional experiments to study the deviations
local equilibrium,
from
and to show whether or not there is
interaction between various interface rocesses. An
excellent experiment would be one which determines the
exact direction of deviations
in CL(i) and C (i) in
L
Figure 1.7 from their equilibrium values.
~s
Such an
experiment would give much needed information on the
nature of the interface processes;
this would hel
tightening restrictions for applying old
in developing new ones.
in
rinciples or
Hence, better guidance would
be available for formulating an expression
second response function.
for the
103
VI.3
References
1.
K. . Jackson, "Liquid Metals and Solidification<
(1958).
p. 174, Amer. Soc. for Metals, Cleveland
Acta
7, 148
Met.,
(1959).
2.
K. A. Jackson,
3.
J. W. Cahn, Acta Met.,
4.
H. A. Wilson, Camb. Phil. Soc., 10, 25 (1898-1900).
5.
F. C. Frank, Discussions
No.
6.
5, p. 149
(1949);
8, 554 (1960).
of the Faraday Society,
Phil.
Mag.,
41, 200
(1950).
W. B. Hillig and D. Turnbull, J. Chem. Phys., 24,
194
(1956).
7.
D. Turnbull, Trans. AIME, 191, 661 (1951).
8.
N. F. Mott,
9.
K. T. Aust and J. W. Rutter, Trans. AIME, 215, 119
(1959),
Proc.
and ibid.
Phys.
218,
Soc.,
682
60, 391
(1948).
(1960).
10.
B. B. Rath and H. Hu, Trans. AIME, 245, 1577 (1969).
11.
P. Gordon and R. A. Vandermeer, "Recrystallization,
Grain Growth and Textures," p. 205, Amer. Soc. for
Metals (1965).
12.
K. A. Jackson and B. Chalmers, Can.
473
. Phys., 34,
(1956).
13.
P. Niessen, Ph.D. Thesis, Univ. of Toronto
14.
D. G. Cole, P. Feltham, and E. Gilliam, Proc. Phys.
Soc., 67, 131 (1954).
15.
P. Niessen
and W. C. Winecrard, J. Inst.
(1964).
Metals,
88,
300 (1963-64)
16.
J. W. Gibbs, "On the Equilibrium of Heterogeneous
Substances," Vol. 1, p. 219, Collected Works,
Longsmans, Green and Co., New York (1928).
17.
J. W. Cahn,
18.
L. S. Darken and R. W. Gurry, "Physical Chemistry
of Metals," p. 215, Mc Graw-Hill Book Co., New York
(1953).
Acta
Met.,
10, 789
(1962).
104
18; p. 240.
19.
Ref.
20.
H. Biloni and B. Chalmers,
233, 373
Trans. AIE,
(1965).
21.
22.
L. Kaufman and Morris Cohen, Prog. Metal
Pergamon Press, 7, 165 (1958).
Phys.,
D. A. Karlyn, J. W. Cahn, and Morris Cohen, Trans.
(1967).
197
245,
AIME,
683
36,
(1958).
K. A. Jackson,
24.
E. A. Owen and D. A. Davies, Brit. J. Appl. Phys.,
16, 1291
Can.
J. Phys.,
23.
(1965).
and R. H. Willens,
Trans.
AIMIE, 227,
362
25.
Pol Duwez
(1963).
26.
V. T. Borisov, Soviet Phys.-Dok,
27.
A. A. Chernov, "Growth of Crystals," Vol. 3, p. 35,
Consultants Bureau, New York (1962).
28.
J. R. Brown, J. Inst. Metals, 83, 49, 1954; R. W. Bohl
and V. D. Hildebrandt, J. Am. Chem. Soc., 79, 2711
7, 50 (1962).
(1957).
29.
C. D. Thurmond and J. D. Struthers,
57, 831 (1963).
J. Phys.
30.
Pol Duwez, Trans. ASM, 60, 607 (1967).
31.
Principles of Solidification,"
B. Chalmers,
John Wiley and Sons, New York (1964).
32.
M. E. Glicksman and C. L. Vold, Acta Met.,
Chem.,
. 129,
15, 1409
(1967).
33.
Ref.
31;
p.
34.
W. W. Mullins and R. F. Sekerka, J. Al.
35, 444
35.
133.
Phys.,
(1964).
S. R. de Groot
"Non-Equilibrium
and P. Mazur,
Thermodynamics," North-Holland Publishing Co.,
Amsterdam
36.
(1962).
I. Prigogine,
"Thermodynamics
of Irreversible
Processes," Interscience Publishers, New York and
London
37.
(1961).
E. S. M4achlin, Trans.
AiME,
197,
437
(1953).
105
38.
K. G. Denbigh, "The Thermodynamics of the Steady
State," . 29, John Wiley and Sons Inc., London
(1958).
36; p. 46.
39.
Ref.
40.
L. Onsager,
(1931).
41.
Ref.
42.
B. D. Coleman and C. Truesdell,
Phys.
Rev.,
37,
405
(1931);
38,
2265
35; p. 61-64.
33, 28
J. Chem. Phys.,
(1960).
43.
G. Baralis,
J. Cryst.
44.
Ref.
76.
45.
J. S. Kirkaldy,
46.
J. W. Cahn and W. W. Mullins, "Decomposition of
Austenite by Diffusional Processes," p. 123,
36;
p.
Can.
Growth,
J. Phys.,
3-4,
627
(1968).
37,
739
(1959).
Interscience Publishers, New York and London (1962).
47.
Ref.
26.
48.
I. L. Aptekar and D. S. Kamenetskaya,
Metalloved,
14, 358 (1962).
49.
B. K. Jindal
4632 (1968).
50.
V. T. Borisov, "Crystallization Processes," p.
Consultants Bureau, New York (1966).
51.
W. T. Martin and E. Reissner,
Equations,"
and W. A. Tiller,
J. Chem.
(1964).
Met.
Phys.,
i.
49,
69,
"Elementary Differential
p. 60, Addison-Wesley
Reading, Mass.
Fiz.
Pub.
Co.
Inc.,
106
VI.4
Biographical Note
The author, James C. Baker, was born in Flint,
Michigan on May 24, 1942.
He graduated from Flint
Beecher High School in June, 1960.
He entered Michigan
State University in September, 1961 and graduated with
a B.S. in Metallurgical
While an undergraduate
engineering
Engineering in June, 1965.
he received membership
honoraries.
to two
He was the winner of a
Wheelabrator Graduate Fellowship in 1965-66, and
received his M.S. in Metallurgical
University
in June, 1966.
as an instructor
Candidate
at the
During 1965-66 he also served
in the Metallurgy
In September,
Engineering
Department.
1966 the author entered M.I.T. as a
for the Degree of Doctor of Philosophy
Materials Science.
in
During his tenure he held a National
Science Foundation Graduate Traineeship at the Institute.
Following the completion of his studies, the author
will join the Homer Research Laboratories of Bethlehem
Steel Corporation in Bethlehem, Pennsylvania.