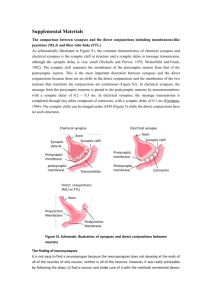
Coordination between Presynaptic and Postsynaptic Properties
Measured at Single Terminals in Hippocampal Cultures
by
Jonathan G. Murnick
M.Phil. Physiology
University of Cambridge, 1996
A.B. Chemistry
Princeton University, 1994
Submitted to the Department of Electrical Engineering and Computer Science in Partial
Fulfillment of the Requirements for the Degree of
Doctor of Philosophy
at the
Massachusetts Institute of Technology
September 2003
C 2003 Massachusetts Institute of Technology
All rights reserved
Signature of Author: . ..............................
Department
Certified by:
. ..... ...........................................................
lectrical Engineering and Computer Science
August 29, 2003
.......................
Guosong Liu
Associate. Professor of Neurobiology
T4hesisj$hervisor
Accepted by: ....................
Arthur C. Smith
Chair, Committee on Graduate Students
Department of Electrical Engineering and Computer Science
MASSACHUSETTS INSTITUTE
OF TECHNOLOGY
OCT 1 5 2003
BAFKER
Coordination between Presynaptic and Postsynaptic Properties
Measured at Single Terminals in Hippocampal Cultures
by
Jonathan G. Murnick
Submitted to the Department of Electrical Engineering and Computer Science on
August 29, 2003 in Partial Fulfillment of the Requirements for the Degree of Doctor of
Philosophy in Electrical Engineering and Computer Science
Abstract
I examined the spatial scale at which pre-synaptic activity interacts with synaptic strength
and developmental events in the post-synaptic cell. I performed this work in primary
hippocampal cultures of postnatal Sprague-Dawley rats after 8-10 days in vitro. I
measured electrophysiologically the functional addition of GluR2 and NR2A subunits to
AMPA and NMDA receptors, respectively, as well as the absolute strength of AMPAand NMDA-mediated currents. I examined both spontaneous quantal events and local
responses using glutamate iontophoresis at putative single-synaptic sites. I monitored
pre-synaptic strength via vesicle staining with FM dyes. I found an inverse correlation
between pre-synaptic and post-synaptic strength at individual synapses: Intensity of
presynaptic FM-staining was inversely correlated with AMPA current magnitude
measured by local iontophoretic stimulation. I also found a negative correlation at
individual puncta between FM-staining intensity and decay time of iontophoreticallyevoked NMDA decay current. Presumably, this correlation reflects a higher proportion
of NR2A subunits at presynaptically more active synapses. I propose that homeostatic
feedback mechanisms may operate at a subecellular level to maintain excitatory synaptic
input at a single synapse or within a dendritic branch.
In order to stimulate individual synaptic sites, I used a novel iontophoresis system,
presented here. The system incorporates a 0.1 ptm electrode tip for local stimulation,
combined with fast capacitance compensation to achieve high-speed application from a
high-resistance tip. Ejection of fluorescent dye from the electrode shows that transmitter
can be limited to the width of a single synapse and to a time scale similar to an
The speed and localization of transmitter is confirmed by
endogenous event.
iontophoretically stimulating single labeled synapses in cultured hippocampal neurons
held under voltage clamp. The amount of transmitter ejected is linear and reproducible
over a physiologically relevant range, making this technique useful for examining
receptor kinetics and receptor insertion/removal. The system should be capable of
delivering any charged neurotransmitter.
Thesis Supervisor: Guosong Liu
Title: Associate Professor of Neurobiology
- 2 -
CHAPTER 1 INTRODUCTION ........................................................................
4
CHAPTER 2 BACKGROUND...........................................................................
7
Techniques for Probing Single Synapses .................................................................................
Homeostatic Regulation of Synaptic Strength......................................................................
Activity-mediated Change in Receptor Subtype ...................................................................
7
10
11
CHAPTER 3 IONTOPHORESIS SYSTEM PERFORMANCE .............
13
.........
Description of System ..........................................................................................
---------------................
Speed of Release ...........................................................................................
... --------------.............
Spatial Localization ...............................................................................-----.
Control of Transmitter Concentration .................................................................................
13
15
18
20
CHAPTER 4 DEVELOPMENTAL CHANGES AT SYNAPSES IN CULTURE .. 24
Presynaptic Function: Vesicle Turnover .........................................................................
Postsynaptic Properties: GluR2 Insertion .............................................................................
Postsynaptic Function: AMPA Conductance ........................................................................
....
Postsynaptic Properties: Other.......................................................................
Possible Pre/Post Interactions.......................................................................................37
CHAPTER 5 PRE/POST SYNAPTIC INTERACTIONS AT INDIVIDUAL
SYNAPSES......................................................................................................
24
29
32
34
39
Overview of Technique..................................................................... ..... .......... 39
41
Inverse Correlation between Pre- and Post-synaptic Strengths ...........................................
Pre/Post Synaptic Coordination Originates Locally within Cells.........................................43
45
Interactions within a Local Dendritic Neighborhood..........................................................
NR2A..................47
of
fraction
postsynaptic
higher
a
have
terminals
presynaptic
Stronger
Presence of GluR2 is not Coordinated with Presynaptic Strength.......................................48
49
Effect of Activity Block on Pre/Post Relationships ...............................................................
CHAPTER 6 DISCUSSION..............................................................................
51
Iontophoresis Technique ............................................................................
Pre- and Post-synaptic Properties ..................................................................................
Opportunities for Further Study.............................................................................................62
51
55
CHAPTER 7 METHODS..................................................................................
65
Hippocampal Cell Culture and Patch Clamp Recording....................................................
Fluorescence Data Acquisition...............................................................................................
65
66
CHAPTER 8 REFERENCES ..........................................................................
68
APPENDIX: CELL CULTURE PROTOCOL ...................................................
72
- 3 -
Chapter 1 Introduction
Although the microscopic structure of the brain has been studied for over a hundred
years, the brain's fundamental computational unit-the smallest component that takes
two or more inputs and produces a non-linear output used for subsequent computationsstill is unknown. Conventional wisdom holds that this computational unit is the neuron.
In the most basic formulation, a single neuron can receive hundreds to thousands of
excitatory and inhibitory inputs to its extensive dendritic tree. The cell soma sums these
inputs and, if some threshold is reached, fires an action potential. This "integrate-andfire" behavior is based on known physiology of active electrical processes within neurons
and is widely used as a model for exploring the function of neural circuits.
Many variations of and extensions to integrate-and-fire have been proposed, such as
the weighting of inputs based on their timing or their distance from the soma. Still, the
widely held assumption is that the basic computation within the brain occurs when a
neuron fires or does not fire an action potential.
Recent studies have suggested that the most basic computations in the brain are
performed subcellularly-at the level of one dendritic branch or even among a few
individual synapses.
Several authors have argued that action potentials can and do
originate within dendrites under physiological conditions, as well as within the soma.
These action potentials may not simply amplify synaptic inputs, but also help create
integrate-and-fire circuits within individual dendritic branches (Poirazi et al., 2003).
- 4 -
Since no one has yet deciphered the neural code, it is impossible to say definitively
what information a neural circuit derives from a particular firing pattern. Therefore, one
cannot be sure when and where a computation is being performed.
However, it is
possible to infer something about the functional significance of a structure by examining
how it is regulated endogenously.
In particular for this thesis, I look at activity-
dependent regulation of synaptic strength and properties, a long-established principle of
neural functioning.
Several recent studies have established a principle of activity-
mediated homeostatic regulation of synaptic strength. Other work has demonstrated the
importance of neural activity in regulating the insertion of GluR2 and NR2A subunits
into AMPA and NMDA receptors, respectively.
Most of these studies have been carried out by modifying activity in entire cell cultures
or brain slices, or in an entire brain region in vivo (e.g. eye-opening). A few authors have
shown that homeostatic regulation of synaptic strength can occur at the level of a single
neuron. For example, lowering activity levels of just a few neurons in a cell culture leads
to a selective increase in the synaptic input strength to the cells with lower activity.
However, if independent neural computations are carried out on a subcellular scale, it
would be reasonable to expect that activity-dependent regulation of synaptic properties
also occurs at a subcellular level. No study to date has looked at the effects of activity on
such a local scale.
In this work, I look at the relationship between presynaptic and postsynaptic strength at
single terminals.
I also examine the effect of presynaptic activity in regulating
postsynaptic receptor subtypes. I find that presynaptic strength and both postsynaptic
strength and receptor properties are indeed coordinated at individual synapses.
- 5 -
In order to measure synaptic strength and postsynaptic properties at individual
terminals, I needed to develop and refine techniques for single-synapse investigation. I
rely heavily on glutamate iontophoresis for direct postsynaptic stimulation of putative
synaptic sites.
I show that a properly designed iontophoresis electrode can deliver
glutamate with a time course similar to endogenous events and a spatial localization
small enough to stimulate only a single synaptic terminal.
- 6 -
Chapter 2 Background
Techniques for Probing Single Synapses
The strength of a synaptic connection is determined presynaptically by release
probability and concentration of neurotransmitter and postsynaptically by the number and
properties of receptors.
Recent advances in the understanding of glutamate receptor
regulation trafficking during development and plasticity have shown that both the number
and properties of AMPA and NMDA receptors are present in different amounts at
different times and with different degrees of localization during development (Pickard et
al., 2000; Sans et al., 2000; Stocca and Vicini, 1998; Tovar and Westbrook, 1999; Zhu et
al., 2000). Similar diversity is thought to occur during plasticity events such as LTP and
LTD.
This highly dynamic picture demands tools to measure instantaneously the
presence and functionality of postsynaptic receptors.
Many studies have fruitfully examined these changes by examining mEPSC's or
evoked responses.
While these studies have led to an increased understanding of
glutamate receptor behavior, it has been difficult to separate clearly pre-synaptic from
post-synaptic mediated phenomena. Additionally, synaptic heterogeneity prevents highlevel analysis of synaptic function (Hessler et al., 1993; Murthy et al., 1997; Rosenmund
et al., 1993).
Finally, these approaches cannot directly address synapses receptors on the
postsynaptic cell that may not lie directly under a pre-synaptic site (Cottrell et al., 2000).
- 7 -
Other studies have visualized post-synaptic receptors directly using monoclonal
antibody staining (Rao et al., 1998) or fluorescent fusion proteins (Zhu et al., 2000).
While these techniques can distinguish changes in postsynaptic receptor quantity and
localization from changes in presynaptic release, they cannot do so instantaneously.
Further, even when they are sensitive enough to distinguish surface from subsurface
receptors, they cannot speak to the functionality of those receptors.
The number and properties of functional postsynaptic receptors can be assessed
directly by their responses to local application of transmitter. To address questions of
postsynaptic receptor identity, functionality, and localization in dynamic living cells,
there is a need for techniques that can deliver neurotransmitter directly to receptors with
precise spatial and temporal control. An ideal technique for single-synaptic stimulation
should mimic the fast time course and highly local spatial distribution of neurotransmitter
released from a synaptic vesicle.
Current techniques for stimulating an isolated
postsynaptic site include excised dendritic patch, laser uncaging, iontophoresis, and
caged glutamate.
An excised patch can be moved quickly in and out of different
solutions within 0.3ms (Fleck et al., 1996; Geiger et al., 1995; Jonas et al., 1994; Jonas
and Sakmann, 1992; Sommer et al., 1990; Tong and Jahr, 1994), enabling brief
stimulation. It is also possible to select a section of dendrite containing a high synaptic
density, thus ensuring stimulation localized to one or few synapses. Still, the synapse is
removed from its normal surroundings, which may have undesirable or unclear effects for
an experiment (e.g. rundown) (Rosenmund et al., 1995b).
Laser-released caged neurotransmitter allows the study of synapses without disrupting
a cell's physical integrity, facilitating direct comparison of responses at different areas of
- 8 -
a dendrite (Callaway and Katz, 1993; Parpura and Haydon, 1999b; Pettit et al., 1997;
Wang and Augustine, 1995). However, this technique has a stimulation rise-time about
an order of magnitude slower than endogenous stimulation, which can greatly alter the
responses of certain receptors (Kullmann, 1999; Trussell et al., 1988). It is also difficult
to limit the radius of uncaging beyond about 20ptm, which is too large a region to confine
stimulation to a single synapse (Callaway and Katz, 1993; Pettit et al., 1997; Wang and
Augustine, 1995). Chemical two-photon uncaging has made some improvement to the
spatial localization of this technique (Pettit et al., 1997), but its time course is still slow
compared to endogenous release.
One recent report has demonstrated two-photon
excitation uncaging for AMPA receptor stimulation with near-endogenous space and time
resolution (Matsuzaki et al., 2001). However, this approach is very costly and cannot
easily be extended to other molecules besides glutamate.
There is still a need for a technique of synaptic stimulation that is fast, local, and usable
on intact cells.
High speed and localized application of neurotransmitter through
iontophoresis may provide a solution to these problems. While many previous studies
have described iontophoresis for application of neurotransmitter (e.g., Cash and Yuste,
1999; for review see Curtis, 1964; Hicks, 1984), these applications are generally too slow
and broad to mimic the temporal and spatial profile of endogenously released
transmitters.
Trussel, et al. described an application of glutamate iontophoresis with
speed and localization approaching endogenous stimulation (Trussell et al., 1988).
However, they suffered from inconsistencies between electrodes and used large holding
currents, which limit the linearity and reproducibility of the technique.
- 9 -
In the following chapter, I demonstrate a novel iontophoresis technique generating a
highly localized release zone with a diameter of approximately 0.1 tm. This is more than
an order of magnitude smaller than any previously reported for iontophoresis, and it
allows stimulation at single-synapse resolution, even after accounting for diffusion of
transmitter away from the release site. Such a small electrode tip necessarily creates a
high-resistance stimulation system, which poses challenges for achieving fast application.
However, a commercially available, specialized amplifier with fast capacitance
compensation enables responses rivaling endogenous vesicle release, with a linear
response and high reproducibility.
Homeostatic Regulation of Synaptic Strength
Several recent studies have established a principle of activity-mediated homeostatic
regulation of synaptic strength (Davis and Bezprozvanny, 2001; Liu and Tsien, 1995;
Turrigiano et al., 1998). As excitability of a neural network increases, the strength of
excitatory synaptic connections decreases to maintain a constant baseline firing rate.
Similarly, as excitability goes down, excitatory synaptic connection strength increases to
compensate.
In cortical cultures, artificially altering excitability has been shown to
induce compensatory changes in both AMPA and NMDA receptor number, as well as
presynaptic release probability at excitatory terminals (Murthy et al., 2001; Watt et al.,
2000).
Although the majority of this work has involved perturbations to an entire
culture dish, a couple of elegant studies have been able to isolate the effect to single cells.
Although it did not involve any exogenous perturbations, one early study showed an
inverse relationship between number of inputs to a cultured neuron and the strength of
those inputs (Liu and Tsien, 1995). More recent work has used transfection techniques to
-10-
selectively reduce the excitability of isolated cultured neurons (Burrone et al., 2002).
That study demonstrated an increase in synaptic input to the quieted cells. To date, no
study has tried to examine homeostasis at a subcellular level, as I do here.
Activity-mediatedChange in Receptor Subtype
Addition of GIuR2 to synaptic receptors
At early stages of development, AMPA-type receptors are thought to consist of
heterotetramers of GluRl and GluR4 subunits.
As development progresses, GluR2
subunits are added to the AMPA receptor structure, producing tetramers composed of
GluRl and GluR2, or possibly GluR1, GluR2 and GluR4 (Pickard et al., 2000). The
addition of GluR2 converts AMPA receptors from calcium-permeable to calciumimpermeable. Since calcium concentration is a potent intracellular signal, particularly at
synapses, this change in permeability likely has important functional consequences. Each
of these changes in postsynaptic receptors has been shown to be activity-dependent, in
that blocking neural activity leads to slowing or abolishment of the developmental switch
(Barria and Malinow, 2002; Kumar et al., 2002; Liu and Cull-Candy, 2002; Zhu et al.,
2000). However, it is not known if neural activity independently triggers each of these
three changes. Possibly, activity triggers one postsynaptic change, which then triggers
others in a cascade; or, one developmental switch may be permissive for another.
Switch from NR2B to NR2A subunits
Hippocampal NMDA receptors are composed of two subunits: NR1 and either NR2B
or NR2A.
Early in development, only NRl/NR2B receptors are present in the
hippocampus and other brain areas. As development progresses, the fraction of NMDA
receptors composed of NR1/NR2A increases (Tovar and Westbrook, 1999). Eventually,
- 11 -
NR2A-containing and NR2B-containing NMDA receptors are present in roughly equal
numbers, and the NR2A fraction stabilizes. NR2B-containing receptors have a channelopen time about twice as long as NR2A-containing receptors, and they therefore pass
more calcium into the cell when opened (Brimecombe et al., 1997).
Given the
importance of calcium as a molecular signal, one would predict significant functional
consequences of this switch. Indeed, knockout mice overexpressing NR2B in adulthood
perform better on memory tasks than their wild-type counterparts (Tang et al.).
Importantly, the insertion of NR2A is known to depend on neural activity, and it can be
regulated either up or down by changes in activity (Quinlan et al., 1999; Yoshii et al.,
2003).
-12-
Chapter 3 lontophoresis System Performance
Deleted:
Description of System
Mic'*elec
I used an MVCS 02 (NPI Electronic. Tamm, Germany) high-speed iontophoresis
amplifier to control transmitter release through the iontophoresis electrode. Although the
MVCS 02 headstage is designed to hold a glass electrode directly, I found that
configuration to cause excessive drift of the electrode tip, probably due to thermal
fluctuations within the headstage.
electrode
Instead I held the glass electrode with a standard
holder (Warner Instruments MEW-F15T), which I connected to the
Grounding was achieved with a pellet
iontophoresis headstage with a 3-inch wire.
attached to the patch clamp headstage. The patch clamp amplifier and iontophoresis
amplifier grounds were both connected to a common ground in the instrument rack; no
separate bath ground was used for the iontophoresis.
{Deleted:
I fabricated
iontophoresis microelectrodes
from quart
glass
capillary tubes
Deleted:
(O.D. = 1.0mm, I.D. = 0.7mm, Sutter Instrument Co.) pulled in a single, stage, with a
horizontal pipette puller (Sutter Instrument Co. P-2000)
These electrodes have a tip
opening of approximately 0.1 [tm, based on electron microscopy performed by the puller
manufacturer.
When filled with 150mM glutamatic acid (pH adjusted to 7.0 with
NaOH), they had a resistance of approximately I OOMO. Although the results described
here were collected using quartz microelectrodes, I have also successfully fabricated
13
-
Deleted:
electrodes using aluminosilicate glass, which can be shaped at lower temperatures.
However, I have also found that using small-diameter capillary glass (O.D. = 1.0mm, as
opposed to 1.2mm or 1.5mm) is critical for electrode consistency.
Occasionally, glutamate leaked slightly from the electrode tips, as visualized by
increased noise in a patch recording when placed near a synapse. However, these leaks
could be eliminated with 1-2nA of positive holding current. Electrodes with a resistance
of less than 80MQ tended to have larger leaks that were not controllable with holding
SDeleted
current. _I placed the electrode tip, within 1.0tm, and ideally within 0.5pm, of a synapse
to avoid the slowing and broadening of transmitter flux associated with diffusion (Figure
3.2A). In certain experiments, the iontophoresis electrode additionally contained 5mM
Oregon Green dye (Molecular Probes).
With resistances in the hundreds of MO, the system's time constant r =R C became an
obstacle to fast stimulation.
I measured a time constant of approximately 200 ins,
implying that the electrode capacitance was approximately 2 nF. To mimic endogenous
release, glutamate should be released in a single pulse lasting no longer than ims;
unfortunately, this type of pulse would be low-pass filtered by the electrode so as to
practically disappear. To help minimize this problem, I decreased R and C as much as
possible. I decreased R by pulling electrodes with a short shank, as a long, thin shank
increases electrode resistance. I also decreased C by lowering the extracellular solution
level as much as possible without affecting the cells' viability; this modification
Deletec
decreases C, since largely the submerged portion of the electrode contributes to
capacitance. To compensate for the remaining RC filtering, the MVCS 02 is equipped
with positive-feedback capacitance compensation in the stimulation current. Figure 3.1 A
-
14-
illustrates the improvement to the ejection response achieved with capacitance
compensation.
To properly adjust the capacitance compensation, I applied a -lOnA square-wave pulse
at 20Hz to the iontophoresis electrode after filling it and immersing the tip in the bath. I
monitored the tip voltage on an oscilloscope while adjusting the compensation so that the
voltage also described a square wave.
This method also gave us a measure of the
electrode resistance via the relationship V = IR. Typically the initial electrode resistance
was > 500MQ, possibly due to microscopic air bubbles in the tip.
At this point, I
"zapped" the electrode once or twice with a -250nA pulse for 5-10s, after which the
resistance would have a stable value around 100MQ.
I reapplied a square wave
periodically during the course of a recording session to monitor R and adjusted the
compensation if necessary, but it typically varied very little over the course of several
hours.
Speed of Release
Endogenous synaptic transmission is characterized by fast release of transmitter from a
vesicle, which is located across the synaptic cleft from the postsynaptic receptors.
Ideally, I would like the iontophoretic pulse to reproduce, as accurately as possible, the
fast and highly localized properties of vesicular release. To assess the speed of ejection, I
added Oregon Green fluorescent dye to the iontophoresis electrode, in addition to
glutamate.
Oregon Green was chosen because it has physical properties close to
glutamate's: It is a small molecule with a single negative charge at physiological pH. It
also has a fluorescence emission that is not pH-sensitive, so any pH changes between the
electrode interior and the bath solution should not produce artifacts. The dye has a higher
-
15-
molecular weight than glutamate (glutamate M.W. = 164; Oregon Green M.W. = 332), so
it is expected to diffuse more slowly in the bath after ejection. The glutamate ejected
from the tip will not be as spatially localized as the dye. Still an increase in molecular
weight by a factor of 2 would decrease the diffusion constant by a factor of
Deleted
approximately_1A which should minimally affect the results.
A
B
5ms
-
lontophoresis
-
mEPSCs
5 ms
1ms
C
...
50
D
mEPSCs
-
lontophoresis
mns
--
lontophoresis
mEPSCs
20 ms
10 ms
Figure 3.1. Temporal resolution of iontophoretic application. A Fluorescence intensity over time following
a ims / -50nA iontophoretic ejection of glutamate and Oregon Green, measured at the tip of the electrode
(black trace). The gray trace shows the same current pulse without capacitance compensation. B, C, D
Comparable time-course of iontophoretic and synaptic AMPA and NMDA receptor currents. B AMPA
receptor current, locally evoked by a 1 ms iontophoretic application of glutamate (-50 nA, solid, average of
4 consecutive traces) to an isolated FM1-43 labeled synapse, overlaid with two amplitude-matched
mEPSCs recorded from the same cell (dashed). C NMDA receptor current waveforms (solid; average of 4
consecutive traces; recorded in 0 Mg2+ and 5pM NBQX), locally evoked as in B, overlaid with an NMDAonly mEPSC waveform recorded from the same cell (-50 nA, dashed; average of 100 events, not the same
cell as in B). Inset: Same waveforms scaled to better compare the activation kinetics of both receptor
currents. D Both AMPA and NMDA receptor components, locally evoked by a 1 ms iontophoretic
glutamate application (-25 nA, black, average of 4 consecutive traces; recorded in 0 Mg2+, different cell
from B,C) to an isolated FM 1-43 labeled synapse, overlaid with two amplitude-matched mEPSCs recorded
from the same cell (gray).
-16-
Deleted:
I examined the dye concentration during a ims iontophoretic pulse at a point directly in
front of the electrode tip. As shown in Figure 3. 1A, the dye concentration rises sharply
and linearly for the duration of the constant-current pulse, reaching its peak 1.1 ms after
the ejection begins.
After the pulse is complete, the response drops quickly as dye
diffuses away from the tip, falling by a factor of e by 1.4 ms after the peak.
When this system was used to deliver glutamate to activate glutamate receptors at
a postsynaptic site labeled with FM 1-43, both AMPA and NMDA receptor mediated
responses could be detected (Figure 3.lB&C}.
To determine whether this glutamate
transient is sufficiently fast to mimic the activation of AM PA receptor during endogenous
Deleted:
synaptic
transmission,
I compared,
spontaneous
AMPA-mediated
mEPSCs
to
Figure 2B
Deleted:
iontophoretically evoked EPSCs of similar amplitudefrom the same cell (Figure 3.1B}.
Note that the rise times of the iontophoretic event is similar to the mEPSCs (20%-80%
Deleted:
rise ionto.: 0.65 ins, mEPSC avg.: 0.75 is). _Jjie falling phases of the AMPA currents
Deleted:
are similar, as well (80%-20% decay ionto.: 8.2 ms; mEPSC avg.: 5.8 ms).
Deleted:
An analogous comparison can be made for the time course of NMIA-receptor-
synaptic
cu
technique c
an endogen
mediated responses (Figure 3.1CL Endogenous and iontophoretic EPSCs match both on
Deleted:
the rising and falling phases (20%-80% rise ionto.: 7.1 ms, mEPSC avg.: 6.8 ms; 80%-
Deleted:
NMDAcui
20% decay ionto.: 138 ms, mEPSC avg.: 124 ms). -The matching decay phases of the
Deleted:
NMDArec
endogenous
and
iontophoretic
responses
suggest
that
the
ejected
transmitter
concentration drops quickly enough that it does not lead to additional or prolonged
currents, compared to endogenous events. Since modest changes in the time course of
neurotransmitter concentration can lead to large changes in relative AMPA and NMDA
-17-
application
endogenou:
receptor activation (Renger et al., 2001), I also compared endogenous to iontophoretic
events recorded in 0 Mg>.
Both a fast-decaying AMPA component and a slowly-
decaying NMDA component of EPSCs were present under these conditions (Figure
3.1D).
Both components of mEPSCs and iontophoretic EPSCs are again similar,
showing that the iontophoretic glutamate concentration time course is near enough to
endogenous to mimic the proportion of AMPA:NMDA receptor activation.
Spatial Localization
I assessed the spatial spread of dye after ejection at several different ejection currents to
determine the spatial concentration profile of glutamate. If the technique is to be useful
to study isolated synapses, it must be able to stimulate synaptic receptors without
activating receptors at nearby synapses.
Figure 3.2B shows the peak intensity after
ejection along a line drawn perpendicular to the tip at a distance of 0.5pm. This is where
a dendrite would normally lie in relation to the tip during stimulation of a synapse.
Typical currents used for synaptic stimulation are 100nA and smaller. As is evident from
the figure, the dye concentration is 80% less than the peak concentration at a distance of
about 0.5 pm from the tip.
-18-
A
B
25 nA
50 nA
100 nA
200 nA
-
sca
line--
-6
m
0--
3
-3
6
Distance (pm)
D
C
Figure 3.2. Spatial resolution of the iontophoresis application. A High magnification confocal image of the
tip of an iontophoretic electrode apposed to an isolated FM1-43 labeled synapse. B Maximum dye
fluorescence after iontophoretic ejection of dye/glutamate mixture at varying ejection currents.
Fluorescence was measured with a microscope line scan along a line perpendicular to and 0.5 ptm away
from the pipette. This is the typical location of a dendrite during iontophoretic stimulation of a synaptic
site. Fluorescence is in arbitrary units. C AMPA-mediated response at a series of locations separated by
1.Opm. White dots (along dendrite) indicate locations of ejection, and yellow dots (connected by line)
indicate strength of AMPA response. The two white traces show representative raw responses at
corresponding locations, directly over the FM-labeled site, and 8pm away. D NMDA-mediated responses
(in 5jiM NBQX) at 0.7pm intervals along a path parallel to the dendrite. White traces show responses
directly over the FM-labeled site, and 4.2 pm away.
Figure 3.2C shows the AMPA current in response to a 50nA pulse of glutamate for
-Deleted:
different positions of the iontophoresis tip with respect to an FM-labeled synaptic site
The response is strongest with the tip directly over the synapse and drops off quickly as
-19-
the tip is moved away in 0.5pm increments.
At a distance of 1.5pm, there is no
discernible current, indicating that it is possible to resolve AMPA-mediated responses
from synapses separated by at least 1.5pm. Figure 3.2D shows the same experiment in
conditions allowing for the observation of NMDA-receptor-mediated current.
The
NMDA receptor, with its slower kinetics, can act like an integrator of low transmitter
levels (Dub6 and Liu, 1999; Kullmann et al., 1999; Lester et al., 1990).
It is more
sensitive than the AMPA receptor to low transmitter levels and correspondingly shows a
{Deleted
measurable response at a greater tip distance,
Control of Transmitter Concentration
One of the most powerful features of the iontophoresis technique is the ability to
precisely control the amount of glutamate applied to the synapse. Figure 3.3A shows that
the amount of dye, and therefore glutamate, ejected is linear over the range of 25-200nA.
Thus, a linear change in the ejection current translates into a proportional change in the
quantity of neurotransmitter ejected. At higher currents, the iontophoresis device cannot
achieve the necessary driving force through the high-resistance electrode.
In our
hippocampal culture system, 200nA is more than sufficient to saturate the response of a
single synapse, so this presents no problem. However, NPI Electronic produces a highervoltage device that can presumably extend this linear range to higher ejection currents, if
the experimental situation were to demand it. Figure 3.3B shows that the apparatus is
linear with respect to ejection time, as well. At a constant ejection current, the amount of
dye ejected is proportional to the ejection time. Since I examined times up to 8 ins,
diffusion of dye away from the tip could occur to a significant extent during the course of
ejection. For these long ejection times, peak dye concentration was therefore no longer
-20-
proportional to ejection time. Instead, I measured integrated dye fluorescence over 30 ms
during and after ejection, which measured the total amount of dye during the response
that flowed past a point directly in front of the tip.
A
C
3000-
12-
a)
1.0/LI
0.5-
2000.
0.6-
E
1000.
CU
~0
Q)
pA
A
7 S0
0.2-
0-
0
or
0
B
0.0
0
100
2.5
300
200
5 7.510
50
25
rr
75100
so0
Current (nA)
Current (nA)
a)
N
20,00020
C)
a)'
OA-
1.2s
0
z
15,000-
L..
1.00.8-
0
10,000-
0.6-
y
0.4CU
5,000-
0-
t
0.2
100 nwm
B,-.
0
1
2
3
4
5
6
7
8 9
Pulse length (ins)
10
25
50
75 100
250
Current (nA)
Figure 3.3 Linearity of iontophoresis ejection. A Peak fluorescence intensity during the first 2 ms after
ejection, at a point 0.5rn in front of the pipette, from raw traces in Figure 3.2B. Fluorescence intensity is
in arbitrary units at several ejection currents ranging from -25 to -400 nA, applied for 1 ms. Dashed line is
a least-squares fit to the points between -25nA and -200nA. B Integrated fluorescence at a single point in
front of the tip, with an ejection current of -8nA and ejection times ranging from 0.25 to 8 ms. Dashed line
is a least-squares fit to all points. Inset shows raw traces. C Dose-response curve for AMPA-mediated
responses. Inset shows raw traces. Note the log scale. D Dose-response curve for NMDA-mediated
responses (in 5 pM NBQX). Inset shows raw traces.
With the ability to control transmitter concentration, it is possible to measure kinetic
properties of synaptic receptors without removing them from the rest of the cell. Doseresponse curves for iontophoretic glutamate application to AMPA and NMDA receptors
are shown in Figure 3.3C&D. Note that the AMPA and NMDA receptors have a similar
-21-
EC50 , which cannot be predicted from Kd values for the two receptor types. Particularly
in the case of the NMDA receptor, the EC 50 is controlled by the interaction between the
receptor's kinetic parameters and the fast glutamate transient (Dube and Liu, 1999).
Therefore, to understand the response to endogenous transmission, it is important to use a
technique such as iontophoresis that can apply glutamate with a similar time course to
vesicle release.
The iontophoresis system can release pulses of glutamate at rates of up to 100Hz, and
the pulse sizes are reliably identical at rates up to 10Hz (Figure 3.4A). At rates above
10Hz, I have found that there can be interaction between pulses, such that a second pulse
is consistently larger or smaller than the initial pulse. The repeatability of release over
Deletec
short time scales lends itself to the study of fast receptor desensitization (Figure 3.4B),
The consistency of iontophoretic release is useful for measurement of many other
receptor properties. For example, Figure 3.4C shows the response of a single synapse to
two different transmitter concentrations over a period of 10 minutes. Since the amount of
glutamate applied is constant from trial to trial, the variation in the response is likely due
to stochastic properties of channel opening. One could perform noise analyses on these
data to calculate parameters such as number of channels per synapse and opening
probability (Sigworth, 1981).
-22
-
Multiple
B0
A
C
0
50 me
1
0
im
i
Time (min)
4
50 pA
100 ms
Figure 3.4 Reproducibility of iontophoresis ejection. A Dye fluorescence directly in front of the pipette
during two successive ejections of -32 nA, separated by 100 ms. B Stability of the response to stimulation
of an isolated FM-labeled site to repeated -50 nA applications of glutamate at 0.2 Hz. C Desensitization of
AMPA receptors, shown with paired pulses of -100 nA glutamate at varying time intervals (20, 40, 80,
160, 320 ms). Top: control conditions; Bottom: in the presence of 50 ptm cyclothiazide, which blocks
desensitization of AMPA receptors.
-23-
Chapter 4 Developmental Changes at Synapses in
Culture
Presynaptic Function: Vesicle Turnover
I assessed presynaptic release by staining with the fluorescent dye FM.
FM dye
specifically stains endocytotic vesicles and is commonly used to label and monitor
neurotransmitter vesicles at synapses (Cochilla et al., 1999). In the presence of dye, cells
are stimulated to release vesicles.
After their release at presynaptic terminals,
neurotransmitter vesicles are quickly taken up again via endocytosis, along with any
bound FM dye. The cells are then washed in dye-free solution containing tetrodotoxin
(TTX), which blocks all action potentials and-therefore-most vesicle release.
dye-labeled terminals are then stable in the presence of TTX for many hours.
-24-
The
stained
vesicle
releasable
vesicle
wash
TTX
high-K+
FM 4-64
endocytosis
ex ocytosis
-~
Figure 4.1 Cartoon outlining the process of FM staining of vesicles. After (Ryan et al., 1993).
Two stimulation protocols are commonly used to stain synapses with FM: lowintensity stimulation for staining only the docked or "readily-releasable" vesicles; or
high-intensity stimulation for staining the entire recycling vesicle pool (Murthy et al.,
2001; Murthy et al., 1997). Low-intensity stimulation consists of a few to a few dozen
electrical pulses, causing an equivalent number of action potentials in the cultured
neurons. This protocol stains each synapse in proportion to its individual probability of
release (P)-the probability per action potential that the terminal releases a vesicle
(Dobrunz and Stevens, 1997).
High-intensity stimulation is performed with high-
frequency electrical pulses or by immersing the neurons in a depolarizing ionic solution
(as I do in this study) (Cochilla et al., 1999). Although I am interested in the Pr at each
synapse, the high-intensity protocol stains the entire vesicle pool and is not a direct
measure of Pr. High-intensity stimulation has two key advantages for my experiments,
however: 1) The stained puncta are several times brighter than with low-intensity
stimulation, which is helpful for positioning the iontophoresis electrode to stimulate
-25-
individual synapses.
2) High-intensity stimulation can be done via solution changes,
while low-intensity stimulation requires electrodes in close proximity to the cell culture.
These stimulation electrodes obstruct access for the voltage-clamp and iontophoresis
electrodes that I need for single-synapse recording, increasing the practical difficulties of
the experiments. Fortunately, the release probability, the docked vesicle pool size, and
the full recycling vesicle pool size have been shown to be in direct proportion to one
another at hippocampal synapses (Murthy et al., 1997; Murthy and Stevens, 1999). A
key assumption throughout this thesis, then, is that the FM intensity at an individual
punctum is a reliable indicator of P,.
-26-
D
A
25.000
E 20.000
:t
C.
15.000
--
10.000
~--TTX
Control
5.000
0.000
8DIV
10DIV
15DIV
70000
60000T
50000
z 40000
E 2 30000
20000
10000
0
F
15DIV
1ODIV
8DIV
1.800
1600S1.4001.200
-4 control
1.000
E 0.800TT
0.600
0.0.400
0.200
0.000
8DV
10DIV
15DW/
Figure 4.2 Developmental Increase in FM Staining. A, B, C Typical hippocampal cell cultures stained
with FM 1-43 dye to label individual synaptic puncta. A 8 DIV B 10 DIV C 15 DIV D Developmental
increase in FM puncta density, in control conditions and in the presence of TTX (applied starting from 7
DIV) E Developmental increase in FM intensity per punctum F Mean area of puncta, quantified with FM
staining, in control conditions and TTX (applied from 7 DIV)
Functional maturation of synapses occurs primarily between 8 and 15 days in vitro
(DIV) in hippocampal cultures.
The number of synapses with vesicles undergoing
endocytosis and the number of vesicles per synapse increases rapidly during this period.
Figure 4.2 shows the corresponding increases both in puncta density and intensity per
punctum.
The most marked increase is in the density of mature synapses, which
-27-
increases more than fivefold. Normalizing for the length of dendrite controls for the fact
that total dendrite length is increasing steadily as the culture matures. Figure 4.2E also
shows that the mean FM intensity per punctum also increases during development. Since
I assume FM intensity is proportional to Pr, each postsynaptic site is stimulated more and
more frequently with glutamate as the neurons mature. Considering also the increasing
density of puncta, it is evident that the amount of excitatory input to a given length of
dendritic branch increases rapidly during development.
I also monitored the mean puncta size through development (Figure 4.2F). In contrast
to density and intensity, size is relatively constant. Mean puncta size is constant from 1015DIV, when the largest increase in density occurs. This is consistent with previous
work indicating that synapse size is constant through development. The modest increase
from 8-10DIV may be an artifact of the increasing intensity.
As individual puncta
become brighter overall, their dimmer edges may become visible above background
resulting in apparently larger puncta.
Figure 4.2D&F also illustrate the effect on presynaptic maturation of blocking activity.
I used tetrodotoxin (TTX) to block spontaneous electrical activity in the cultures starting
at 7 DIV. At 10 DIV and 15 DIV, I measured puncta density and mean area. Mean
punctum area after TTX treatment is identical to control conditions. The density also
increases rapidly, with a time course similar to control conditions. These data show that
density does not quite increase to control levels after eight days of TTX treatment
(1 5DIV), although this difference is not statistically significant. Together, these results
suggest that activity is not necessary for presynaptic maturation.
-28-
It is a welcome observation that vesicle release matures similarly in the presence and
absence of activity, as it makes subsequent experiments easier to interpret.
I am
interested in the coordination of pre- and postsynaptic maturation by neural activityspecifically excitatory activity at individual synapses mediated by release of glutamate
vesicles. If a subsequent experiment shows that blocking activity prevents or delays a
postsynaptic developmental event, I can be sure that I am observing a failure of pre and
postsynaptic coordination.
Conversely, if blocking activity had blocked presynaptic
maturation, a failure of postsynaptic development could be interpreted either as a failure
of coordination or as a coordinated failure of the prc/postsynaptic unit.
PostsynapticProperties: GIuR2 Insertion
The postsynaptic developemental change that I studied most extensively was the
insertion of GluR2 subunits into AMPA receptors.
examining the rectification of AMPA-mediated events.
I monitored this insertion by
AMPA receptors without the
GluR2 subunit are inwardly rectifying-they conduct inward (negative) current much
better than outward. AMPA receptors that do contain GluR2 are non-rectifying-they
have a linear I-V relationship. Typically, the degree of rectification is quantified with a
rectification index (RI), derived from the ratio of peak AMPA current at a positive
holding potential to peak AMPA current at a negative holding potential (Kumar et al.,
2002). I use +40 mV and -60 mV holding potentials here. Since the AMPA receptor
channel has a reversal potential of approximately 0 mV in the recording conditions used
here, the maximum possible ratio-achieved when the I-V curve is perfectly linear-is
about 0.66 (40/60). I define RI here as [1
-
-
(I+40/-60) / 0.66] An RI of zero indicates that
29
-
all of the AMPA receptors contributing to a conductance contain the GluR2 subunit. As
the fraction of AMPA receptors without GluR2 increases, the RI increases toward one.
A
B
1.00 - C)
+40mV
-
0.80-
0
0.60
cc
C
0.40-
-4---- control
-m-r X
.9
-60mV
12
-0.20-
10-13DIV
8DIV
10
15DIV
pL
200 ms
0
C
E
60-
40-
-A--
20-
1.000
_-
40
4
~1
20
-
0.800
I
-KY>
-T--
~2
0.600
-20
-
-40
-20
-60k
0
p
A
4
I
-
---2
10Q10
-80
-40
-20
0
20
40
80
80
-20
m-
0
my
--
0.400
C
i
80 -40
-80
I
--
0.200
0.000
-0.200
7
6D[\/
7D[\/
BDKI
10DIV
150r'/
Figure 4.3. Developmental time course of GluR2 receptor insertion. The amount of GluR2 present at
synapses was assessed using the ratio of conductances at +40mV and -60mV. Complete rectification
corresponds a rectification index (RI) of 1, while no rectification is an RI of 0. A Rectification in whole
cells was quantified by the RI of the mean mEPSC at +40 and -60mV. Sample traces shown are from a 15
DIV cell. B Between 8-15 DIV, when mEPSC's could be recorded, there was essentially no rectification,
indicating the presence of GluR2 at nearly every synapse, both in control conditions and after treatment
with TTX from 7 DIV. C,D Rectification could be quantified earlier at single terminals using iontophoretic
stimulation while holding the cell at positive and negative holding potentials. C Sample traces and I-V
curve for a typical synapse at 6 DIV. D Sample traces and I-V curve for a typical synapse at 10 DIV.
E Summary data showing that the majority of change in RI occurs before 8 DIV.
I first assessed rectification by comparing the mean amplitudes of mEPSC's recorded
at -60mV and +40mV. I used the ratio of these amplitudes to determine RI for a cell
-30-
(Figure 4.3A). A comparison of cells ranging in age from 8-15 DIV shows surprisingly
little change in RI (Figure 4.3B). From 10-15 DIV, the RI is approximately equal to
zero, indicating that GluR2 is present in all or nearly all AMPA receptors. Even at 8
DIV, RI is equal to 0.13, indicating that GluR2 is present in most AMPA receptors.
Therefore, if there is a developmental increase in GluR2 fraction in this culture system, it
must occur primarily before 8 DIV. It was difficult to obtain reliable data on rectification
before 8 DIV using mEPSC's. Although recordings from cultures younger than 10 DIV
contain numerous NMDA-only "silent" mEPSC's, AMPA-mediated mEPSC's are rare.
AMPA-mediated mEPSC's in 8 DIV cultures occur at a rate of approximately two per
minute, so even measuring mEPSC rectification at that age required exceptionally long
data records.
To measure RI at younger ages, I used glutamate iontophoresis to apply
neurotransmitter directly to synapses.
With this technique, I could step the holding
potential from -60 to +40mV while stimulating with glutamate, thereby measuring the RI
at a single synapse.
Figure 4.3 shows the results of stimulating several synapses in
several cells at different developmental ages, starting at 6 DIV. It is evident that the
youngest cultures do display substantial AMPA rectification, indicating a lack of GluR2.
These data show that GluR2 does increase developmentally in these cultures; the
majority of GluR2 incorporation occurs between 6-8 DIV. The process is substantially
complete before 10 DIV, before the largest changes in presynaptic release measured by
FM.
Before I observed this result, one plausible theory was that maturation of
presynaptic vesicle release allows the postsynaptic cell to sense activity, leading to
changes in receptor subtype. This theory seems to be less likely, since GluR2 seems to
-31-
be present at most synapses expressing mEPSC's at ages before much FM staining is
visible.
As a further test of whether GluR2 insertion might be driven by activity, I
examined mEPSC rectification in cells treated with TTX to block activity. After treating
with TTX from 7 DIV onward, GluR2 insertion appears to proceed normally (Figure
4.3B). From 10-15 DIV GluR2 is present at maximal levels, identical to the control
conditions.
PostsynapticFunction: AMPA Conductance
I also looked for any developmental changes in the amplitude of AMPA-mediated
currents, to see if they might occur on a similar time scale to changes in presynaptic
release. I followed the mean AMPA mEPSC amplitude from 8-15 DIV (Figure 4.4). It
increases consistently, approximately doubling over this time period. This is the same
time period over which I found using FM dye that presynaptic release increases most
rapidly. Thus, presynaptic release and postsynaptic AMPA strength are two properties
that might be coordinately regulated, perhaps in an activity-mediated process.
The
remainder of this thesis is concerned largely with dissecting the features of this
relationship.
A
-
B
25.000 - --
.
--..............................................
70.000
20.000
60.000
15.000
10.000 -
____________________
~2.0
S5.000 -
0.000
control
--
E
-
8DIV
10DIV
15DIV
----------
50.000
40.000
--
~30.000
L.< 20.000
10.000
0.000
8DIV
10DIV
15DIV
Figure 4.4 A Average AMPA-mediated mEPSC size measured from 8-15 DIV, showing a doubling in
mean event size. Treatment with TTX from 7 DIV causes a further increase. B Response to a fixed
glutamate pulse averaged over several synapses in several cells from 8-15 DIV. Note the small decrease in
mean AMPA conductance.
-32-
This developmental increase in mEPSC size could be due to an increase in the
transmitter released presynaptically, an increase in the postsynaptic AMPA conductance,
or some combination of the two. I attempted to distinguish these possibilities using
iontophoresis to stimulate directly the postsynaptic receptors.
By looking at the
postsynaptic current in response to a fixed amount of glutamate, I could assess any
changes in postsynaptic conductance specifically. Figure 4.4 shows the response to a
fixed -32nA, 0.5ms glutamate pulse averaged over a number of synapses from several
cells at 8, 10, or 15 DIV, the developmental period when mEPSC's are known to increase
in magnitude. Interestingly, the average AMPA current measured postsynaptically with
iontophoresis does not change significantly over this time period, and it may even
decrease slightly. Thus, the developmental increase in mEPSC size is due to an increase
in presynaptic quantal strength.
It is important to note that this increase in presynaptic strength is distinct from the
presynaptic increase in vesicle pool size visualized with FM and described above. The
increase in presynaptic strength visualized with FM is presumed to reflect an increased
P, i.e. the synapse is stronger because vesicles are released more frequently in response
to action potentials. However, mEPSC's are thought to represent single vesicle release
events, so a presynaptically-mediated increase in mEPSC size involves an increased
amount of transmitter release per vesicle.
Blocking activity with TTX starting at 7 DIV causes an increase in mEPSC amplitude
measured 3 days later and a further increase after 8 days (Figure 4.4). This increase is
consistent with homeostatic regulation of excitability that has been described recently in
-33-
several systems.
As network activity is reduced, the strength of excitatory synapses
increases to try to compensate for the lack of excitatory input.
PostsynapticProperties: Other
When I began this work, I examined two other postsynaptic developmental changes to
see if they happened with a similar time course to presynaptic maturation: insertion of
NR2A subunits into NMDA receptors, and the flop/flip transition in functional AMPA
receptors. Both of these transitions proved difficult to characterize in the culture system
for various technical reasons described below.
The interaction between presynaptic
strength and postsynaptic AMPA amplitude proved to be a fertile area for further
exploration, so I did not pursue the NR2A orflop/flip transitions.
A classic developmental change in pyramidal neurons is an increase in the fraction of
NR2A subunits in NMDA receptors. These subunits endow the NMDA receptor with
faster decay kinetics, so it passes less calcium into the postsynaptic cell with each
opening event. I tried to identify the addition of NR2A at synapses by examining the
sensitivity of mEPSC's to ifenprodil, a drug which selectively blocks NMDA receptors
containing the NR2B subunit. Unfortunately, under the 0 Mg 2 + recording conditions I
was using in order to detect NMDA currents in mEPSC's, all NMDA receptors appeared
to be in a partially desensitized state. Ifenprodil, as well as presumably blocking NR2Bcontaining
receptors
selectively,
also seemed
to remove the
global baseline
densensitization. This combination of effects produced results that I could not interpret
quantitatively. One potential workaround for this problem, had I chosen to pursue it,
would be to use additional pharmacologic agents known to block NMDA desensitization.
-34-
As an alternative to ifenprodil sensitivity, it is also theoretically possible to identify the
contribution of NR2A subunits to NMDA-mediated currents directly from the faster
decay kinetics. In practice, this is often difficult because the conditions used to record
NMDA-mediated mEPSC's also unmask random NMDA channel openings throughout
the cell, greatly increasing background noise. NMDA-mediated mEPSC's are themselves
small and noisy (due to the intrinsic stochastic variations in NMDA channel gating), so
even finding mEPSC's in the current recording is a challenge, let alone extracting their
decay times. Iontophoretic application of glutamate can get around some of these issues
by evoking an event with a known start time, and by allowing repetitive stimulation of
the same receptors to average out stochastic variation. I use this technique successfully
to measure NMDA decay time at individual synapses (see next chapter), but I have not
systematically characterized the developmental time course of NR2A addition in the
culture system.
The final postsynaptic change I attempted to follow was the transition of AMPA
receptor subunits from the flop to flip forms. This change occurs via a posttranslational
modification of the AMPA subunit mRNA transcript, before the protein is synthesized.
This modification is known to occur more frequently as development progresses.
Functionally, it subtly changes the kinetics of the receptor and may also affect the
regulation of insertion and removal of AMPA receptors into the cell membrane. The
flop/flip transition has not been well-studied physiologically in intact cells, perhaps
partially due to the experimental difficulties in doing so.
The standard method for
differentiating the flop and flip states is to use the drug cyclothiazide (CTZ), which
selectively blocks desensitization of receptors containing flip subunits.
-35-
After treating
with CTZ, the decay of AMPA mediated currents changes from a single exponential to a
sum of 2-3 exponentials. Looking for a developmental increase in flip-type subunits
amounts to looking for a developmental change in the parameters of these exponentials,
which I found difficult to quantify. An additional problem with these experiments is that,
once applied, CTZ takes at least thirty minutes to wash out, severely limiting the number
of experiments that can be done with each culture coverslip. Once it was obvious that the
effects of CTZ did not change dramatically over development, I chose not to spend any
more patience or culture resources in this experimental direction.
70000
60000
50000
40000
E
30000
a
_
_
_
_
_
_
_
_
_
_
_
10000
0
1.00
.3 0.80
-08
0.600.40
-0.20
20.00
-0.20-
CL
20000
18.000
16.000-
U)
14.000
1 2.000
10.000
E
8.000
6.000
.
C
4.000
2.000-
* 0.00080.000
-__
_
_
_
_
_
_
70.000
7C
.60.0005 0.000*
<
4
. 00-
4 0 0
301*000
20.000 -_
_
_
_
_
_
_
_
_
_
_
_
_
_
_
_
_
_
_
_
_
=10.000 - ___________________
S0.000
_
- ____________________
8DIV
1ODIV
15DIV
Figure 4.5 Correspondences between pre and postsynaptic developmental events. This figure aligns data
from previous figures to highlight relationships between the developmental time courses.
-36-
Possible Pre/Post Interactions
In Figure 4.5, I align the developmental timecourses reported in the previous sections.
This chart can suggest which properties might or might not be coordinated between the
pre and postsynaptic sides of the synapse. Specifically, I would look for postsynaptic
developmental changes that happen over the same time scale as presynaptic maturation. I
intentionally chose the period 8-15 DIV, since this is the time frame when presynaptic
terminals in culture are known to rapidly increase their Pr. This fact is confirmed by the
observed increase in FM staining intensity.
The next plot to consider is the insertion of GluR2 subunits to AMPA receptors,
quantified by the RI. Although this process is known to be activity-dependent in several
systems, it does not here appear to depend on mature presynaptic vesicle release.
Therefore I do not expect to find any relationship between presynaptic strength and
GluR2 content at individual terminals.
The final two plots are the most intriguing and deserving of closer study. I have shown
a developmental increase in average mEPSC amplitude, which reflects properties of both
pre and postsynaptic sides of the terminal.
The iontophoresis response, reflecting
postsynaptic strength only, is constant or decreases slightly over this developmental
period, implying that the presynaptic side must increase to produce an overall increase in
mEPSC amplitude.
The mEPSC amplitude reflects quantal event strength, which is
independent of Pr, so I tentatively assume that there is an increase in quantal glutamate
content over development.
In the next chapter, I examine the relationships between
presynaptic strength-quantified by FM intensity, reflecting Pr-and postsynaptic
glutamate currents. It is important to keep in mind that while Pr is a very important
component of presynaptic strength, it is likely not the only component.
-37-
Other factors,
such as quantal glutamate content, may also contribute, but I do not have the
experimental tools here to measure those factors at single terminals.
-38-
Chapter 5 Pre/Post Synaptic Interactions at Individual
Synapses
Overview of Technique
The experiments described in this chapter all involve correlating the presynaptic
strength at an individual terminal with the postsynaptic electrophysiological properties at
the same or nearby terminals.
Figure 5.1 summarizes the basic techniques I used to
measure these values. Briefly, a culture coverslip was initially stained with FM 4-64 dye
using high-K+ stimulation to visualize synaptic puncta.
Then, a morphologically
pyramidal neuron was selected for whole cell patch clamp based on its transmitted light
image. Next, I patched the cell and held it under whole-cell voltage clamp using a patch
solution containing Alexa 488 dye. The dye quickly diffuses to fill the dendritic tree and
allows visualization of branches up to a few hundred microns from the soma. At this
point, I saved a high-resolution fluorescence overview image showing both the dendritic
tree and synaptic puncta.
I chose one of the puncta to stimulate and brought the
iontophoresis electrode directly to that punctum.
I stimulated that terminal with
glutamate while altering the holding potential of the voltage clamp to measure various
synaptic properties, as I describe elsewhere. For each punctum that I stimulated, I saved
for later reference another image of the electrode positioned adjacent to that punctum. I
repeated this process with as many different puncta as I could until I lost the patch or
broke the iontophoresis electrode by crashing it into the bottom of the chamber. Later, I
-39-
analyzed offline the high-resolution overview image using custom-written scripts in
Matlab to identify and measure the intensity of individual puncta. By comparing this
image with the reference images of the iontophoresis electrode next to each stimulated
punctum, I could accurately identify the stimulated puncta and correlate their intensities
with their measured electrophysiological properties. The initial high-resolution overview
image was used for all FM intensity measurements, because the FM dye bleaches quickly
with repeated imaging.
A
D
C
B
E
glutamate
nidlw -19nA
20
5 rns
Figure 5.1 Summary of Techniques. A Sample experimental preparation: FM 4-64 dye (red) stains
individual terminals, while a single patched neuron is filled with Alexa 488 dye (green) to visualize its
dendritic tree structure. B Higher magnification image (white box in (A)). The yellow arrow marks the
terminal chosen for stimulation with iontophoresis. C Image in (B) after processing with image
enhancement and segmentation algorithms. D The glutamate iontophoresis electrode is brought to within 1
mm of a terminal. (See scale bar in (B).) E A negative current pulse is applied to the iontophoresis
electrode to eject a fixed amount of glutamate. The blue current is recorded in the voltage-clamped
postsynaptic neuron. It is a typical response mediated by AMPA receptors. Unless otherwise noted, a -32
nA pulse applied for 0.5 ms is used throughout this work.
-40-
Inverse Correlation between Pre- and Post-synaptic Strengths
The first properties I examined at single terminals were simply pre and postsynaptic
strength. Presynaptic strength was quantified by FM intensity, and postsynaptic strength
was measured as the amplitude of the current response to a fixed iontophoretic pulse of
glutamate (-32 nA, 0.5 ms). I found a clear negative or inverse correlation between
The data fit an
presynaptic and postsynaptic strength on cells at 8 DIV (Figure 5.2).
inverse correlation best (IAMPA vs. FM1), and an inverse relationship is consistent with an
inverse relationship found between synaptic strength and synapse density in individual
cells.
However, a linear correlation (IAMPA vs. -FM) is nearly as strong and also
statistically significant, and the other correlations I find at single synapses in this work
are more clearly linear. In any case, it is evident that a stronger presynaptic punctum
implies a weaker postsynaptic AMPA current.
A
80*
.60
5
8 DIV
+
B
80
10 DIV
.6
404
20.
20
00
5
10
FM Intensity (arbitrary unli9 04
00
5
10
FM Intensity (arbitrary unqq 0 4
Figure 5.2 Pre and Postsynaptic Strength are Inversely Correlated. A A plot of AMPA current vs. FM
intensity shows that they are inversely related at 8 DIV, i.e. brighter FM-staining at the presynaptic
punctum is associated with a weaker AMPA response at the corresponding postsynaptic site. (R = 0.66, p
= 0.005, n = 16 synpases from 6 cells). Each point represents one synapse. B At 10 DIV, there is still a
negative correlation between AMPA current and FM intensity, although it is not as strong (R = -0.33, p =
0.35, n = 10 synapses from 3 cells).
-41-
I also examined pre and postsynaptic strength on some synapses of 10 DIV cells.
There is a negative correlation apparent here too (definitely linear), although it is not as
strong as at 8 DIV and not statistically significant for the number of synapses I measured.
The key difference between the 8 DIV and 10 DIV data is that the older cells have none
of the very strongest postsynaptic terminals.
A
B
15
V8 DIV
9 10DIV
0~
+40mV
.6-a
'NMD A
C
10k
U)
1....
I-
0
0
5
z
0
0
20 pA
40 ms
E
2
4
6
FM Intensity (arbitrary unitV
8
104
7
7
-60mV
'AMPA
AMPA:NMDA
Current Ratio
6
5
4
3
2
1
c)
+ 8DIV
+ 10 DIV-
+
++
4
6
2
FM Intensity (arbitrary
8
4
Figure 5.3 A Both AMPA- and NMDA-mediated currents can be measured at each terminal. At a holding
potential of +40 mV, the Mg2+ block is removed, and both AMPA and NMDA currents are seen. The
NMDA current is measured as the peak current between 30-60 ms after the glutamate pulse. B There is no
obvious correlation between FM intensity and NMDA current at individual synapses. C The ratio of
AMPA:NMDA current at each synapses is strongly negatively correlated with FM intensity. (R = -0.78, p
= 0.0001, n = 18 synapses from 7 cells).
-42-
I measured the strength of NMDA currents at individual synapses by looking at the
response to an iontophoretic glutamate pulse while holding the voltage at +40 mV to
remove the Mg 2 + block from NMDA channels (Figure 5.3).
Although the AMPA
component is also present, it decays faster than the NMDA component. I quantify the
NMDA conductance by measuring the peak current between 30-60 ms after the
glutamate pulse, when AMPA current has already decayed.
There is no apparent relationship between presynaptic FM intensity and postsynaptic
NMDA current, either at 8 DIV or 10 DIV (Figure 5.3). Interestingly, however, plotting
the AMPA:NMDA current ratio versus FM intensity produces a very strong negative
correlation. Additionally, the 10 DIV synapses follow the same relationship as 8 DIV
synapses, even though the 10 DIV synapses were much more weakly correlated when
considering AMPA current alone. It is not immediately clear why "normalizing" the
AMPA current by the NMDA current produces a tighter correlation with FM intensity,
although I suggest some possible reasons in the Discussion chapter.
Pre/Post Synaptic Coordination Originates Locallywithin Cells
The data presented so far provide evidence that presynaptic and postsynaptic strengths
are negatively correlated. However, they do not necessarily show that their coordinate
regulation occurs at a single-synapse scale. Without breaking down the data cell by cell,
it is possible that the regulation occurs on a cell-wide scale, or even a culture-wide scale.
For example, one cell could have all strong presynaptic inputs and all weak postsynaptic
currents, while another cell could have all weak presynaptic inputs and strong
postsynaptic currents.
To conclude that coordination of synaptic strength occurs on a
-43
-
subcellular scale, it necessary to observe a negative correlation between pre and
postsynaptic strength within individual cells (Figure 5.4A).
D
A
0
z
5
LOCAL
REGULATION
CELL-WIDE
REGULATIO
0
0
C r~
5
FM 104
B
10 pAj_
20 ms
Figure 5.4 Pre/Post Synaptic Coordination Originates Locally within Cells. A Schemata showing two
possible modes of coordination of pre- and postsynaptic strength. left: Regulation occurs on a cell-wide
level. In this case, there are some cells in the culture with both bright presynaptic FM puncta and weak
AMPA responses, and other cells with weak presynaptic FM and strong AMPA responses. right:
Regulation at a subcellular level. Individual cells each have both strong and weak FM puncta and
cooresponding weak and strong AMPA responses. B-D Example cell showing local regulation of pre and
postsynaptic strength. B Patched cell filled with Alexa 488 and stained with FM 4-64. The white box
outlines the inset shown in (C). C Magnified view showing four puncta that were stimulated with
iontophoresis, along with the corresponding responses at -60 mV and +40 mV. Note that the weakest
current response is at the brightest FM-stained punctum in the upper right, while the strongest current
response is at the dimmest FM punctum in the upper left. D AMPA:NMDA ratio plotted versus FM
intensity for these four puncta, showing the negative correlation within this cell.
This regulation does indeed occur within individual cells, as can be observed in one
example cell in Figure 5.4. A negative correlation between AMPA:NMDA ratio and FM
intensity is observed among the four synapses recorded from that cell. Summary data in
shows that all cells from which I recorded at least two synapses exhibit a similar negative
-44-
correlation.
I conclude that the inverse coordination of pre and postsynaptic strength
occurs, if not at the level of individual synapses, then at least at a subcellular spatial
scale.
-61
CU
-
4
z2
a<0
0
6
8
2
4
FM Intensity (arbitrary uni s 0
Figure 5.5 Summary data from all cells recorded, showing that FM and AMPA:NMDA ratio are
coordinated within each cell. Synapses recorded from the same cell are shown in the same color. A line is
fit to the points for each cell from which at least two synapses were recorded (n = 5 cells).
Interactions within a Local Dendritic Neighborhood
Given the idea that the dendritic branch may be the fundamental neural computational
unit, I was particularly interested in seeing whether homeostatic regulation might occur
on the scale of a local dendritic branch. There is no a priorireason why the coordination
I have described at single synapses might not reflect an inverse relationship between
postsynaptic conductance and presynaptic input within a small local neighborhood. The
cells I studied at 8-10 DIV have a low density of mature inputs compared with adult
cells, and in many cases a "local neighborhood" could conceivably consist of just the one
punctum I stimulated and one or two others.
-45
-
A
B
80-
0
6
CU
C1 60-
.
,
40
00
C
2
z
20-
< 0I
0
1
2
3
FM Intensity (arbitrary units) 105
0.5
1
1.5
2
2.5
FM Intensity (arbitrary units) 10 5
D
0.7
-0.6
0.6
-0.65
0
CU
0
C
0
0.5-
-0.7
0.4-
0
U
0
0.3-
U.2
0
I
5
10
15
20
X (1 am)
-0.75
0.8
-0
5
10
15
20
X (p
1 m)
Figure 5.6 Effects on correlations of considering neighborhood presynaptic strength. A AMPA
conductance to -32nA, 0.5ms glutamate pulse plotted versus total FM intensity in a 7.5 mm neighborhood.
Compare . B AMPA:NMDA ratio plotted versus intensity in a 7.5 mm neighborhood. Compare Figure
4.3C C Effect on correlation of varying effect of neighbors with a smoothly varying exponential weighting
factor, space constant 1. Effect on inverse correlation shown in Figure 4.2A. D Similar effect of varying
space constant on correlation shown in 4.3C.
If the regulation does occur within a neighborhood, I would expect that including
neighboring puncta when calculating presynaptic input strength should increase the
correlations I measure. I first considered the effect of including the sum total of intensity
of presynaptic puncta onto a dendrite within a distance of 7.5 pm of the punctum I
stimulated.
(I chose 7.5 pm, because I was initially thinking about a mechanism
involving diffusion of Ca2+ within the dendrite, and this distance was my estimate of the
upper limit of the Ca2+ diffusion distance.) I looked specifically at two of the strongest
-46-
correlations that I observed at single terminals: between IAMPA and 1/FM in 8 DIV cells,
and between
IAMPA/INMDA
and -FM in 8 DIV and 10 DIV cells combined. Figure 4.5
shows that including the entire FM input intensity within 7.5
tm only lessens the
correlation in both cases.
Building on the idea of a diffusible transmitter within the dendrite, I also considered a
more smoothly varying model. In this model, postsynaptic strength at a synapse is most
strongly influenced by the presynaptic input at that same terminal, but neighboring
terminals also have an effect that decreases with distance. I examined a model in which
the effect of a neighboring terminal decreases exponentially with distance (Figure 5.6).
For each of the two correlations above, I considered exponential space constants from 120 ptm.
For both correlations, increasing the space constant-thereby increasing the
impact of neighbors in calculating presynaptic strength-always reduced the degree of
correlation between pre and postsynaptic strength. The correlation was always strongest
when considering only the presynaptic strength of the single stimulated punctum and
neglecting its neighbors.
Stronger presynapticterminals have a higher postsynaptic
fraction of NR2A
Even though it was difficult to measure using whole-cell mEPSC's, I was expecting to
see a change in the amount of NR2A subunit present at NMDA receptors during the
developmental period I was studying.
For reasons explained in the previous chapter, it
was somewhat easier to measure clear NMDA currents using iontophoresis than by
observing mEPSC's. I took advantage of this fact to measure the decay time constant of
NMDA currents at individual stimulated terminals. For many stimulated terminals, I was
able to fit a decaying exponential to the NMDA current and extract the time constant
-47-
(Figure 5.7). Since NMDA receptors containing NR2A subunits decay faster than those
with NR2B, a faster decay time should indicate a greater fraction of NR2A.
Plotting NMDA decay t versus presynaptic strength shows a very strong negative
correlation at 10 DIV synapses. Presumably, this represents increased NR2A subunit
insertion at synapses with more active presynaptic terminals. I also observed a negative
correlation at 8 DIV synapses, although quite a bit weaker. Possibly, the lack of effect at
8 DIV is due to a relative lack of NR2A anywhere in the cell at that age. Studies in rat
hippocampus in vivo have shown that NR2A mRNA does not significantly increase after
birth until 10 days of age (Ritter et al., 2002).
A
+4OmV
-r=124ms
B 300
4ms
300v
1
200.
200
100-
100
-6OmVi-.
z
z
0
2
6
FM Intensity (arbitrary unit'
1,
8
'
2
6
8
FM Intensity (arbitrary units) 104
Figure 5.7 Stronger presynaptic terminals have a higher postsynaptic fraction of NR2A. A The degree of
NR2A subunit incorporation into NMDA receptors can be quantified by examining the decay time constant
of NMDA currents. This panel illustrates how a decaying exponential can be fit to the NMDA response at
+40mV in order to measure the decay time constant (t). B The decay time constant is strongly negatively
correlated with the FM intensity at single synapses at 10 DIV. Presumably this reflects greater insertion of
NR2A subunits into NMDA receptors at more active presynaptic terminals. (R = -1.00, p < 0.0001, n = 6
synapses from 2 cells) C There is a similar negative correlation at 8 DIV, although it is not as strong. (R
-0.43, p = 0.29, n = 8 synapses from 5 cells)
Presence of GIuR2 is not Coordinated with Presynaptic Strength
I also checked for any correlation between presynaptic strength and the presence of
GluR2 postsynaptically using the RI. As expected from the developmental time course
described in the previous chapter, there was no correlation evident (Figure 5.8). The
majority of synapses at both 8 DIV and 10 DIV exhibited very little rectification,
suggesting that GluR2 was already incorporated into most receptors at every synapse.
-48-
I
*8
x
x 0.8
-C
DIV
10DIV
0.6
0
cu 0.4
a 0.2
0+
0
2
4
6
FM Intensity (arbitrary uni
8
04
Figure 5.8 GluR2 insertion at single terminals. Measurement of the I-V relationship at individual synapses
at 8-10 DIV shows that most synapses are non-rectifying, and there is no correlation with FM intensity.
Effect of ActivityBlock on Pre/Post Relationships
As a further check that the relationships I observed at single synapses indeed reflect the
effects of presynaptic vesicle release on the postsynaptic terminal, I blocked most vesicle
release by blocking action potential activity with TTX. If the correlations are dependent
on vesicle release, this treatment should weaken or abolish them. I treated with TTX
from 6-8 DIV and recorded single synapses at 8 DIV.
I looked at the negative correlations between IAMPA/INMDA and FM and between rNMDA
and FM. Both of these correlations are not evident at 8 DIV after 2 days' TTX treatment
(Figure 5.9). In the case of AMPA:NMDA current ratio, I interpret this result as clear
evidence that the negative correlation described in Figure 5.9 does depend on actionpotential-mediated vesicle release. This is further support for the theory that postsynaptic
strength is downregulated homeostatically in response to an increase in presynaptic
vesicle release.
The lack of a relationship between NMDA decay and FM intensity at 8 DIV after TTX
suggests that this correlation is activity-dependent as well.
-49-
A
B
0
*
BDIV
I
10 DIV
0
cc
U
U
TTX
U
10i
lWb
*
0
.*
U
z
5
5
Em
U
U.
CL
OL
0
Cn
U
2
4
6
8
FM Intensity (arbitrary units) 10 4
,
8 DIV
4
2
0
D
300
U
U
8
6
FM Intensity (arbitrary units 104
3 n
0 TTX
10 DIV
E
E
200
c1
200
1
0
0
0
a
00
100
0
< 100
z
z
0
8
6
2
4
FM Intensity (arbitrary units) 104
0
I
2
I
4
8
6
FM Intensity (arbitrary unitsc,)
10
Figure 5.9 Pre/Post synaptic coordination is dependent on neural activity. I treated cultures at 6 DIV with
TTX to block all action potential activity and measured single synaptic properties at 8 DIV, after 2 days of
treatment. A, B Control and TTX-treated cultures, showing that AMPA:NMDA ratio is no longer
correlated with FM intensity after TTX treatment. (TTX: n = 12 synapses from 5 cells) C, D Control and
TTX-treated cultures, showing that NMDA decay is uncorrelated with FM intensity after TTX treatment.
(TTX: n = 7 synapses from 5 cells)
-50-
Chapter 6 Discussion
lontophoresisTechnique
Similarities between lontophoretic and Endogenous Responses
The measured EPSC traces evoked with iontophoresis appear nearly identical to
spontaneous mEPSC's of the same amplitudes. As well as can be ascertained by my
patch clamp recordings, these two modes of stimulation are functionally identical.
Nevertheless, there are important differences in the neurotransmitter concentration profile
in iontophoretic versus endogenous release.
First, the speed of release from the
iontophoresis tip (typically Ims) may be slower than release from a synaptic vesicle,
which may occur in as little as 0.2 ms (Clements, 1996; Clements et al., 1992; Eccles and
Jaeger, 1958). Second, endogenous transmitter is released directly into the cleft, while
iontophoretically ejected glutamate must diffuse into the cleft from just outside. Again,
this may slow the time-to-peak-concentration of iontophoretically released glutamate
compared with endogenous release.
Still, the important issue is not whether iontophoresis is as fast as endogenous release,
but whether it is as fast on a time scale that is relevant to receptor binding. During the
rise of the current, changes in concentration of transmitter are essentially low-pass
filtered with a time constant equal to that of the rate-limiting step in channel opening.
For the AMPA receptor, this step is thought to be glutamate binding, which occurs with a
time constant of approximately 0.1 ms at a typical saturating cleft glutamate
-51 -
concentration of 10OOpM (Clements, 1996). For the NMDA receptor, the rate-limiting
step likely is channel opening, with a time constant of 3-6 ms. Given its fast binding
kinetics, the AMPA receptor theoretically could detect the different rise times of the
transmitter concentration in iontophoresis and endogenous release; I may simply not be
able to visualize the differences when I low-pass filter patch recordings at 1 Hz.
Conversely, the recording setup does not filter NMDA-mediated responses at any lower
frequency than I would expect from NMDA receptor kinetics. I can thus be certain that
the rise time of iontophoretically-evoked NMDA EPSC's is as fast as endogenous
stimulation.
The falling phases of AMPA- and NMDA-mediated currents are both slower than their
respective rising phases. The time course of current decay is determined by a mix of
factors, including channel closing and desensitization (Rosenmund et al., 1995a). Both
iontophoretically and endogenously released transmitter leave the synaptic cleft via
diffusion and glutamate uptake processes.
The diffusion is likely faster in the
endogenous case, because there is zero transmitter concentration immediately outside the
cleft. In the iontophoretic case, there is a non-zero concentration directly outside the
cleft, leading to slower diffusion.
How much slower depends on unknown factors,
particularly on any differences in diffusion constants inside versus outside the cleft.
Whatever the size of this difference, it is not large enough to alter the shape of AMPA
and NMDA current decay.
However, this does not rule out that the glutamate time
course may have more subtle effects that are not directly reflected in individual EPSC's,
and that these effects may differ between endogenous and iontophoretic transmitter
delivery.
-52-
Single-synapse Stimulation in Practice
Although the results described in this work show that the iontophoresis technique is
capable of stimulation at high spatial and temporal resolution, when I perform
experiments I still use a few quick procedures to insure the system is working properly
and that I am recording synaptic currents.
After selecting an FM-labeled spot to
stimulate in a patched cell, I bring the iontophoresis electrode as close as possible to it. I
try not to place the tip visibly inside the punctum to reduce the risk of puncturing the preor postsynaptic cell. When manipulating the electrode, I keep it about 5pm above the
surface of the culture dish and lower it as a final step, also to reduce the risk of damaging
cells.
I then increase the holding current in 0.5nA increments until further increases
cause no more reduction in the noise in the patch recording. If the total holding current
required is greater than 3nA, there is usually an uncontrollable leak, so I replace the
electrode and start over-however, this almost never happens if R > IOOMQ as described
in the Methods section. Finally, I provide a test pulse of about -50nA for Ims and
observe the response.
Synaptic currents are most readily distinguished from
extrasynaptic responses by their fast rise time of 2ms or less. Typically, I then move the
elctrode 1-2pim away from the FM punctum, and the response to a stimulus is markedly
smaller and slower.
After this confirmation, I return the electrode to the punctum
confident that I am stimulating a local synaptic zone, and I proceed with an experiment.
Comparison with Caged Glutamate
Caged glutamate released by laser stimulation is another technique that has been used
to release transmitter locally near a synapse in a living cell. For single-photon excitation
uncaging, the localization of transmitter release is determined by the size of the laser light
spot. The spot radius has been reported as low as 1-3 tm, in the plane perpendicular to
-53-
the laser beam, although the localization is substantially worse along the beam's axis
(Parpura and Haydon, 1999a; Parpura and Haydon, 1999b; Pettit et al., 1997). After the
initial release of caged molecules, diffusion further spreads the transmitter, making the
radius of neurotransmitter excitation larger than the spot size. In contrast, iontophoretic
transmitter is released from a 0.1 pm tip, and very little diffuses beyond 1.5 tm. Another
advantage of iontophoresis is in its rapid time course. With single-photon laser uncaging,
reported stimulation times vary from 5-300 ins, in order to uncage enough molecules for
a maximal reponse. The iontophoresis technique more closely simulates endogenous
transmission time course, which may be important in various experimental scenariossuch as situations where it is important to limit entry of calcium ions to an amount that
would occur during an endogenous event.
Very recently, spatial localization of 0.5-1.1im and a stimulation time of 50pis has
been reported using two-photon excitation with the compound MNI-glutamate
(Matsuzaki et al., 2001). While it is not clear whether this technique can release enough
glutamate to saturate synaptic AMPA receptors, it does represent a substantial
improvement over previous single-photon excitation techniques. Like the iontophoresis
technique described here, it can provide glutamate stimulation with near-endogenous
space and time localization. However, it does require an expensive two-photon laser
system and, presently, custom synthesis of MNI-glutamate.
Unlike iontophoresis, it
cannot be extended to other receptor agonists and antagonists without substantial further
research and development of two-photon caged compounds.
For some experimental priorities, laser uncaging has clear advantages over the
iontophoresis technique described here. Most importantly, it is relatively easy to specify
-54-
the locus of transmitter release in three dimensions, simply by moving the focus of the
laser beam.
In contrast, the iontophoresis electrode tip must be positioned carefully
adjacent to a labeled synapse. In our cell culture preparation, this can be accomplished
usually within one minute, which is not a problem when studying a single synapse.
However, when an experimenter wishes to stimulate multiple synapses successively, or
even to repeatedly move between them, it becomes very difficult to rapidly reposition the
tip manually.
To help overcome these problems, another member of our research group has written
software for computer control of the tip with a Sutter M-285 motorized micromanipulator
(Murnick et al., 2002). With this combination, it is possible quickly and accurately to
move to any point in the microscope field with a click of the mouse.
He has also
programmed the manipulator to move repeatedly and accurately between several
synapses. Finally, the manipulator can be instructed to move up 10 pim or 20 ptm, then
move to a new point into the plane of the image, and then descend again to the proper
depth; all these movements can be accomplished in under 300 ms. This sequence of
motions may make it possible to use this technique to stimulate labeled synapses within a
slice preparation, as well.
Pre- and Post-synaptic Properties
Summary of Results
1) The amplitude of AMPA mEPSC events increases during synaptic maturation from
8-15 DIV, while the AMPA conductance postsynaptically measured at individual
synaptic sites is constant or declines slightly.
quantal size increases as synapses mature.
-55-
This suggests that presynaptic vesicle
2) I found an inverse correlation between presynaptic strength (FM intensity) and
postsynaptic strength (AMPA current in response to glutamate iontophoresis) at single
synapses. The correlation becomes stronger when the AMPA current is "normalized" by
the NMDA current at the synapse.
3) This inverse or negative correlation between pre and postsynaptic strength is
regulated at a subcellular level. Simple tests to determine if the relevant presynaptic
strength might include a local dendritic neighborhood did not find any evidence for a
neighborhood effect. The only factor I measured influencing the postsynaptic strength at
a terminal was the presynaptic strength at the same terminal.
4) There is a strong negative correlation between presynaptic strength and postsynaptic
NMDA decay time constant at single synapses at 10 DIV. Presumably this reflects the
greater presence of NR2A subunits at more active synapses.
5) I did not observe a correlation between pre and postsynaptic strength when
measuring at 8 DIV after the culture was treated with TTX for 48 hrs. This suggests that
activity-dependent vesicle release is necessary to coordinate pre and postsynaptic effects
at single synapses.
6) I found no correlation between FM intensity and the presence of GluR2 subunits in
AMPA receptors at single synapses. This was consistent with my measurements of the
time course of GluR2 insertion during development, suggesting that the majority of
GluR2 is present before presynaptic terminals mature.
Measurement of Probability of Release
In this work, I have used FM staining intensity as a proxy for presynaptic strength. I
have argued above that FM intensity at a synapse is a reasonable indicator of P, the
-56-
probability of a terminal's releasing a vesicle upon the arrival of an action potential.
Note particularly that Pr is proportional to N, the number of readily releasable vesicles at
a terminal. This implies that the mean release probability for each individual vesicle at a
terminal does not vary from synapse to synapse. Indeed, the individual vesicle exocytotic
probability has been estimated at 0.057 for a range of synapses in cultured hippocampal
neurons (Murthy et al., 2001). In other words, to increase Pr, a terminal will increase the
number of docked vesicles, rather than increasing the exocytosis probability of each
individual docked vesicle.
Another question is whether Pr is adequately reflects presynaptic strength. Although a
higher Pr means the postsynaptic receptors see more glutamate per action potential, over
time, the amount of glutamate seen by a synapse also depends on the number of action
potentials reaching each presynaptic terminal.
For instance a terminal with a higher
intrinsic firing rate might also tend to have a larger releasable pool. Were this the case,
the effects I measure in this work would be amplified: What I consider a strong
synapse
one with a high Pr -would
in fact be even stronger because of a high
presynaptic firing rate. One the other hand, if pool size is inversely proportional to firing
rate, it would serve to even out the presynaptic strength over all synapses: This would
reduce or eliminate variation in presynaptic strength, thereby weakening all the
correlations I have measure here. Finally, pool size and firing rate may be unrelated
within the cultured neurons.
I know of no study that has looked at the relationship
between intrinsic firing rate or excitability and vesicle pool size within individual
hippocampal cells. Although such a study could present major technical challenges, it
would be a valuable experiment to try.
57-
Homeostasis at Single Terminals
One obvious interpretation of the finding that pre and postsynaptic strength are
inversely correlated is that there may be some sort of homeostatic regulation occurring at
the individual terminal. On the one hand, this is a very satisfying interpretation that is
consistent with much recent work on homeostasis. It seems clear that neural networks,
and likely individual cells, increase or decrease the strength of their inputs in order to
compensate for a lack of stimulation or for overstimulation.
From a computational
standpoint, this could keep a neuron-or a branch of dendrite-in its working activity
range, neither firing continuously nor shut down completely. My results could indicate
that this homeostatic regulation is occurring at an exquisitely fine scale: balancing the
strength of individual synapses.
On the other hand, strict homeostatic regulation at individual terminals could have the
effect of undoing most learning in the brain. If every synapse were constantly reverting
to some baseline synaptic strength, information could not be stored as patterns of
different synaptic strengths, removing one vast potential substrate for memory. Still, the
situation is not so dire, for several reasons. One is the influence of differing time scales.
Long-term potentiation (LTP), a well-studied learning paradigm involving changes in
synaptic strength, occurs on a time scale of seconds to minutes and reaches maximal
effect within a couple of hours. Homeostatic changes, in contrast, happen on a time scale
of hours to days, and may take a week to reach maximal effect. The intervening time
leaves opportunity for the neuron to make other changes in response to LTP, such as
forming new synapses, before it homeostatically "fades."
Secondly, synaptic strength is not the only parameter that can be regulated at a single
synapse.
I have shown here that the amount of NR2A subunits present in NMDA
-58-
receptors may also be regulated, possibly independently of synaptic strength. Even if
synaptic strength always reverts to some baseline after an initial change, changes in these
other synaptic parameters may persist.
A7
6.
CV=0.55
5.
4
B
7
C 7.
60
CV=0.89
5
0
2
4
CV=0.47
5
FM Intensity (normalized)
C
03
03
2
2.
. D
1
0
2
4
Pre x Post (normalized)
1
6
5.
0
CV = 0.61
2
4
Pre x Post (normalized)
54033
2
0
2
4
AMPA Current (normalized)
Figure 6.1 Effect of single-synapse homeostasis on dynamic range. A Histogram of presynaptic strength of
16 representative synapses used in this work, measured by FM intensity. D Histogram of postsynaptic
strength for the same 16 synapses, measured as AMPA current amplitude with iontophoretic glutamate.
B Theoretical distribution of the product of pre- and post-synaptic strength, assuming random pairings of
pre- and post-synaptic terminals. Note that the variation in the product, quantified by CV, is larger than the
variation of either the pre or post side alone. C Actual distribution of products of pre- and post-synaptic
strength. Note that the inverse relationship between pre- and post-synaptic strength reduces, but does not
eliminate, variability in responses across synaptic junctions.
Third, while I do show an inverse correlation between pre and postsynaptic strength,
they are not correlated 100%. One can approximate total synaptic strength as the product
of presynaptic and postsynaptic strengths.
Interestingly, among the synapses that I
studied, the variability in presynaptic strength alone was similar to the variability in
postsynaptic strength alone (see Figure 6.1).
If pre- and post-synaptic terminals were
matched randomly, the variability of the product would be higher than either the pre or
post alone. However, the inverse correlation that exists between pre- and post-synaptic
-59-
strength limits the variability in the product. The fact that there is an inverse correlation
reduces the dynamic range of the synapse, but it does not eliminate it. Potentially,
homeostasis at single terminals may cost some dynamic range, but confer benefits on the
system such as global stability or improved signal-to-noise ratio.
Significance of the AMPA:NMDA Ratio
As I noted above, it is not immediately obvious why the AMPA:NMDA ratio should be
more tightly negative correlated with FM intensity than AMPA strength alone, although
it quite clearly is. I have three possible explanations: one technical and two biological.
The technical explanation is that the effect of taking the AMPA:NMDA ratio may be to
literally normalize for the variability in amount of glutamate released from different
iontophoresis tips.
I have studied carefully the amount of substrate released for a
constant current pulse from the iontophoresis electrode, and it is highly reproducible. I
did not, however, check that this amount was constant from week to week.
The
iontophoresis electrode is fabricated using a specialized commercial device that uses a
laser to melt quartz glass and solenoids to pull out the electrodes. It too is designed to
fabricate highly reproducible electrodes, but the laser power and alignment could vary
with time and ambient temperature.
The iontophoresis amplifier stimulates with a
constant current pulse, and each electrode is individually compensated for capacitance, so
I doubt that there could be much variation in glutamate release between tips. Still, I
cannot know for sure. It is possible that NMDA conductance is relatively constant from
synapse to synapse, and that it therefore acts as some sort of normalization of the
glutamate pulse for measuring AMPA conductance.
-
60 -
A different, biological explanation for the significance of the AMPA:NMDA ratio is
that the homeostatic effects on AMPA strength may be driven by Ca2 + entry. Ca
entry
at synapses is known to be involved in both synaptic potentiation and depression. A
synapse with more NMDA conductance may give rise to wider variation in Ca2 entry
postsynaptically for the same variation in presynaptic strength. If the changes in AMPA
conductance are Ca2+-mediated, a wider variation in Ca2 entry could result in a greater
range of possible AMPA conductances.
Dividing by NMDA conductance effectively
normalizes for the size of the dynamic range.
A third explanation is that there are multiple competing local influences on AMPAmediated synaptic strength. There may be an influence that is independent of presynaptic
strength that regulates synaptic AMPA and NMDA receptors up or down together (Watt
et al., 2000) (see Figure 6.2).
The inverse relationship described here between
presynaptic strength and postsynaptic AMPA strength may be superimposed on that
process. Taking the AMPA:NMDA ratio would control for the effects of that process.
Most of the variation that remained would then be due to the homeostatic regulation that I
am interested in.
-61-
20
S8 DIV
DIV
+10
1510
: 5
z++
0
0
20
40
60
AMPA Current (pA)
80
Figure 6.2 Postsynaptic AMPA and NMDA are correlated at individual synapses.
showing a positive correlation between AMPA and NMDA currents at each terminal.
Iontophoresis data
Opportunitiesfor Further Study
The present work describes an improved means to sample the kinetic properties of
clusters of ligand-gated receptors in situ in intact cells and without disrupting the
intracellular signaling that can modulate these receptors. It provides local fast delivery of
neurotransmitters that approximate synaptic transmission. This approach can be used to
investigate gating and modulation of ionotropic receptors at synapses and those localized
outside synapses.
With a good reporter assay (e.g., intracellular calcium fluxes) this
approach could be extended to the investigation of other receptors such as those coupled
to G-proteins. When coupled with a controller system capable of precisely manipulating
the iontophoresis electrode, this technique allows for analysis of the distribution of
receptors and receptor properties along the dendritic surface. The high spatial resolution
of the technique provides opportunities to answer these questions to a high level of detail.
Finally, the ability to use iontophoresis in intact, living cells enables the study of
receptor dynamics as well. For expample, since most synapses contain hundreds of
glutamate receptors (Cottrell et al., 2000), the variation associated with random channel
-62-
opening is relatively small compared with their mean amplitude (CV = 0.1). Therefore,
average responses to a fixed concentration of glutamate should be determined largely by
the number of functional receptors at the studied postsynaptic site.
This offers a new
opportunity to monitor the dynamic nature of AMPA receptor insertion and removal at a
given synapse.
I have shown that the modified iontophoresis technique I have described here can be
used to stimulate receptors clustered at single synaptic sites quickly and locally, and that
it produces responses similar to presynaptic release events. This provides investigators
with a unique tool to investigate native ligand-gated channels "in synapsis".
My results concerning the single-synapse homeostasis of synaptic strength and
presynaptic influence on NMDA subtype can lead to a wealth of further studies. An
important question to ask is the mechanism of these processes.
These single-synapse
interactions may involve feed-forward and/or retrograde messengers.
If the regulation
involves transcriptional-level signals from the nucleus, one can ask how the effects are
localized to independent single terminals.
Additional further work includes confirmatory experiments using modalities other than
iontophoresis.
Quantitative immunostaining of postsynaptic receptors, combined with
FM-staining of pre-synaptic terminals could potentially confirm these results.
Other
modalities might also be able to explore the relationships described here in older cultures,
where synaptic densities are high enough that single-synapse iontophoresis is impossible.
It might also be possible to extend the results to more intact preparations, such as brain
slice.
-63-
Finally, it will be interesting to explore the theoretical and computational implications
of both single-synapse homeostasis and local regulation of NMDA receptor subtype
potentially implying local regulation of plasticity rules.
-64-
Chapter 7 Methods
Hippocampal Cell Culture and Patch Clamp Recording
Primary cultures of CAl-enriched hippocampal neurons were prepared from neonatal
rats (P1) as described in the Appendix. The age of the cultures used in this study ranged
from 8 to 28 days in vitro (DIV). For glutamate receptor recordings, the composition of
the whole-cell recording electrode solution was (in mM): CsMeSO 3, 125, HEPES 10,
NaCl 8, CaCl 2 1, EGTA 10, MgATP 2, and NaGTP 0.3, and was adjusted to pH 7.25
with CsOH. (For the data in Figure 3.1D only, the recording electrode solution was, in
mM: K-gluconate 120, KCl 10, HEPES 10, NaCl 8, CaCl 2 0.06, EGTA 0.6, MgATP 2,
NaGTP 0.3.)
When AMPA-current rectification was being studied, 100tM spermine
was included in the patch solution to prevent washout of intracellular polyamines.
Alexa488 was also included in some patches for visualization of the dendritic tree.
Deleted:
The composition of the extracellular solution was (in mM): NaCl 128, KCl 3, CaCl 2
2.6, MgCl 2 1.3, glucose 30, glycine 0.005, and HEPES 25 (adjusted to pH 7.4 with
NaOH). Tetrodotoxin (TTX, 0.5 pM; Oretek Inc., Lake Oswego, OR) and picrotoxin
(50pM, Sigma, St Louis, MO) were added on the day of the experiment to block action
potentials and spontaneous inhibitory synaptic events.
The composition of the FM1-43 (Molecular Probes, Eugene, OR) staining solution was
(in mM): KCl 90, NaCl 39, Glucose 30, HEPES 25, CaCl 2 2, MgCl 2 1, NBQX (6-nitro-7-
-65-
sulphamoylbenzo[f]quinoxaline-2,3-dione)
0.005,
AP-5
(DL-2-amino-5-
phosphonovalerate) 0.1, and FM 1-43 0.01 (adjusted to pH 7.4 with NaOH).
The
composition of the FM4-64 (Molecular Probes, Eugene, OR) staining solution was (in
mM): KCl 90, NaCl 39, Glucose 30, HEPES 25, CaCl 2 2, MgCl 2 1, kynurenic acid and
FM 4-64 0.015 (adjusted to pH 7.4 with NaOH). Recordings were made with a 200B or
a 700A integrating patch clamp amplifier (Axon Instruments, Foster City, CA) with a
1 kHz low-pass filter. Data were digitized at 10 kHz using a Digidata 1200B A/D
converter (Axon Instruments). Access resistance was monitored online and was typically
<20MQ. AP-5 and NBQX were purchased from Tocris Cookson (Ballwin, MO).
{eIetee
Following a one-piinute incubation in the FM solution and a >5 min wash, neurons were
visualized under a confocal microscope (Olympus Fluoview) using a 40x planachromat
water immersion objective (1.15 NA).
Fluorescence Data Acquisition
I assessed various iontophoresis properties by observing the ejection of Oregon Green
dye from the electrode under the confocal microscope. Fluorescence data were recorded
using Fluoview software (Olympus). Further processing was done using custom scripts
written for Matlab (MathWorks, Natick, MA). Data showing peak intensity along a line
(Figure 3.2) were recorded using the Fluoview line scan function. Points along the line
were sampled at a spacing of 0.077pm, and one complete line was scanned every 1.8ms.
I did not synchronize the scan with the iontophoresis pulse, so the scan did not always
coincide with the peak dye concentration (i.e. the time immediately following ejection).
Therefore, for each line scan shown, I recorded 100 scans and took an average of the five
most intense traces. The resulting trace was smoothed with a 0.2 im Gaussian filter.
-66
-
Figures showing the fluorescence over time at a single point were recorded by directing
the laser to scan and record a planar area, while the Fluoview parameters file was altered
to disable the scanning motors. Data were sampled at 500kHz and filtered with a 20 ts
boxcar filter. Each record shown is an average of 5-10 traces, which were aligned using
Deleted:
Deleted:
a least squares fit of the traces to each other
67
Chapter 8 References
Barria, A., and Malinow, R. (2002). Subunit-specific NMDA receptor trafficking to
synapses. Neuron 35, 345-353.
Brimecombe, J. C., Boeckman, F. A., and Aizenman, E. (1997). Functional consequences
of NR2 subunit composition in single recombinant N-methyl-D-aspartate receptors. Proc
Natl Acad Sci U S A 94, 11019-11024.
Burrone, J., O'Byrne, M., and Murthy, V. N. (2002). Multiple forms of synaptic plasticity
triggered by selective suppression of activity in individual neurons. Nature 420, 414-418.
Callaway, E. M., and Katz, L. C. (1993). Photostimulation using caged glutamate reveals
functional circuitry in living brain slices. Proc Natl Acad Sci U S A 90, 7661-7665.
Cash, S., and Yuste, R. (1999). Linear summation of excitatory inputs by CA1 pyramidal
neurons. Neuron 22, 383-394.
Clements, J. D. (1996). Transmitter timecourse in the synaptic cleft: its role in central
synaptic function. Trends Neurosci 19, 163-171.
Clements, J. D., Lester, R. A., Tong, G., Jahr, C. E., and Westbrook, G. L. (1992). The
time course of glutamate in the synaptic cleft. Science 258, 1498-1501.
Cochilla, A. J., Angleson, J. K., and Betz, W. J. (1999). Monitoring secretory membrane
with FM1-43 fluorescence. Annu Rev Neurosci 22, 1-10.
Cottrell, J. R., Dub6, G. R., Egles, C., and Liu, G. (2000). Distribution, Density and
Clustering of Functional Glutamate Receptors Before and After Synaptogenesis in
Hippocampal Neurons. J Neurophysiol 84, 1573-1587.
Curtis, D. R. (1964). Microelectrophoresis. In Physical Techniques in Biological
Research, W. L. Nastuk, ed. (New York, Academic Press), pp. 144-190.
Davis, G. W., and Bezprozvanny, I. (2001). Maintaining the stability of neural function: a
homeostatic hypothesis. Annu Rev Physiol 63, 847-869.
Dobrunz, L. E., and Stevens, C. F. (1997). Heterogeneity of release probability,
facilitation, and depletion at central synapses. Neuron 18, 995-1008.
Dub6, G. R., and Liu, G. (1999). AMPA and NMDA receptors display similar affinity
during rapid synaptic-like glutamate applications. Soc Neurosci Abstr 25, 399.395.
Eccles, J. C., and Jaeger, J. C. (1958). The relationship between the mode of operation
and the dimension of the junctional regions at synapses and motor-end-organs. Proc R
Soc Lond B Biol Sci 148, 38-56.
-68-
Fleck, M. W., Bahring, R., Patneau, D. K., and Mayer, M. L. (1996). AMPA receptor
heterogeneity in rat hippocampal neurons revealed by differential sensitivity to
cyclothiazide. J Neurophysiol 75, 2322-2333.
Geiger, J. R., Melcher, T., Koh, D. S., Sakmann, B., Seeburg, P. H., Jonas, P., and
Monyer, H. (1995). Relative abundance of subunit mRNAs determines gating and Ca 2 +
permeability of AMPA receptors in principal neurons and interneurons in rat CNS.
Neuron 15, 193-204.
Hessler, N. A., Shirke, A. M., and Malinow, R. (1993). The probability of transmitter
release at a mammalian central synapse. Nature 366, 569-572.
Hicks, T. P. (1984). The history and development of microiontophoresis in experimental
neurobiology. Prog Neurobiol 22, 185-240.
Jonas, P., Racca, C., Sakmann, B., Seeburg, P. H., and Monyer, H. (1994). Differences in
Ca2 permeability of AMPA-type glutamate receptor channels in neocortical neurons
caused by differential GluR-B subunit expression. Neuron 12, 1281-1289.
Jonas, P., and Sakmann, B. (1992). Glutamate receptor channels in isolated patches from
CAI and CA3 pyramidal cells of rat hippocampal slices. J Physiol (Lond) 455, 143-171.
Kullmann, D. M. (1999). Synaptic and extrasynaptic roles of glutamate in the mammalian
hippocampus. Acta Physiol Scand 166, 79-83.
Kullmann, D. M., Min, M. Y., Asztely, F., and Rusakov, D. A. (1999). Extracellular
glutamate diffusion determines the occupancy of glutamate receptors at CAl synapses in
the hippocampus. Philos Trans R Soc Lond B Biol Sci 354, 395-402.
Kumar, S. S., Bacci, A., Kharazia, V., and Huguenard, J. R. (2002). A developmental
switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci 22,
3005-3015.
Lester, R. A., Clements, J. D., Westbrook, G. L., and Jahr, C. E. (1990). Channel kinetics
determine the time course of NMDA receptor-mediated synaptic currents. Nature 346,
565-567.
Liu, G., and Tsien, R. W. (1995). Properties of synaptic transmission at single
hippocampal synaptic boutons. Nature 375, 404-408.
Liu, S. J., and Cull-Candy, S. G. (2002). Activity-dependent change in AMPA receptor
properties in cerebellar stellate cells. J Neurosci 22, 3881-3889.
Matsuzaki, M., Ellis-Davies, G. C. R., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H.
(2001). Dendritic spine geometry is critical for AMPA receptor expression in
hippocampal CAl pyramidal neurons. Nature Neurosci 4, 1086-1092.
Murnick, J. G., Dube, G., Krupa, B., and Liu, G. (2002). High-resolution iontophoresis
for single-synapse stimulation. J Neurosci Methods 116, 65-75.
Murthy, V. N., Schikorski, T., Stevens, C. F., and Zhu, Y. (2001). Inactivity produces
increases in neurotransmitter release and synapse size. Neuron 32, 673-682.
Murthy, V. N., Sejnowski, T. J., and Stevens, C. F. (1997). Heterogeneous release
properties of visualized individual hippocampal synapses. Neuron 18, 599-612.
-
69
-
Murthy, V. N., and Stevens, C. F. (1999). Reversal of synaptic vesicle docking at central
synapses. Nat Neurosci 2, 503-507.
Parpura, V., and Haydon, P. G. (1999a). "Uncaging" using optical fibers to deliver UV
light directly to the sample. Croat Med J 40, 340-345.
Parpura, V., and Haydon, P. G. (1999b). UV photolysis using a micromanipulated optical
fiber to deliver UV energy directly to the sample. J Neurosci Methods 87, 25-34.
Pettit, D. L., Wang, S. S., Gee, K. R., and Augustine, G. J. (1997). Chemical two-photon
uncaging: a novel approach to mapping glutamate receptors. Neuron 19, 465-471.
Pickard, L., Noel, J., Henley, J. M., Collingridge, G. L., and Molnar, E. (2000).
Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA
receptor subunit composition in living hippocampal neurons. J Neurosci 20, 7922-7931.
Poirazi, P., Brannon, T., and Mel, B. W. (2003). Pyramidal neuron as two-layer neural
network. Neuron 37, 989-999.
Quinlan, E. M., Olstein, D. H., and Bear, M. F. (1999). Bidirectional, experiencedependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat
visual cortex during postnatal development. Proc Natl Acad Sci U S A 96, 12876-12880.
Rao, A., Kim, E., Sheng, M., and Craig, A. M. (1998). Heterogeneity in the molecular
composition of excitatory postsynaptic sites during development of hippocampal neurons
in culture. J Neurosci 18, 1217-1229.
Renger, J. J., Egles, C., and Liu, G. (2001). A developmental switch in neurotransmitter
flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron 29, 469484.
Ritter, L. M., Vazquez, D. M., and Meador-Woodruff, J. H. (2002). Ontogeny of
ionotropic glutamate receptor subunit expression in the rat hippocampus. Brain Res Dev
Brain Res 139, 227-236.
Rosenmund, C., Clements, J. D., and Westbrook, G. L. (1993). Nonuniform probability
of glutamate release at a hippocampal synapse. Science 262, 754-757.
Rosenmund, C., Feltz, A., and Westbrook, G. L. (1995a). Calcium-dependent inactivation
of synaptic NMDA receptors in hippocampal neurons. J Neurophysiol 73, 427-430.
Rosenmund, C., Feltz, A., and Westbrook, G. L. (1995b). Synaptic NMDA receptor
channels have a low open probability. J Neurosci 15, 2788-2795.
Ryan, T. A., Reuter, H., Wendland, B., Schweizer, F. E., Tsien, R. W., and Smith, S. J.
(1993). The kinetics of synaptic vesicle recycling measured at single presynaptic boutons.
Neuron 11, 713-724.
Sans, N. A., Montcouquiol, M. E., and Raymond, J. (2000). Postnatal developmental
changes in AMPA and NMDA receptors in the rat vestibular nuclei. Brain Res Dev Brain
Res 123, 41-52.
Sigworth, F. J. (1981). Interpreting power spectra from nonstationary membrane current
fluctuations. Biophys J 35, 289-300.
-
70 -
Sommer, B., Keinanen, K., Verdoorn, T. A., Wisden, W., Burnashev, N., Herb, A.,
Kohler, M., Takagi, T., Sakmann, B., and Seeburg, P. H. (1990). Flip and flop: a cellspecific functional switch in glutamate-operated channels of the CNS. Science 249, 15801585.
Stocca, G., and Vicini, S. (1998). Increased contribution of NR2A subunit to synaptic
NMDA receptors in developing rat cortical neurons. J Physiol 507, 13-24.
Tang, Y. P., Shimizu, E., Dube, G. R., Rampon, C., Kerchner, G. A., Zhuo, M., Liu, G.,
and Tsien, J. Z. (1999). Genetic enhancement of learning and memory in mice. Nature
401, 63-69.
Tong, G., and Jahr, C. E. (1994). Multivesicular release from excitatory synapses of
cultured hippocampal neurons. Neuron 12, 51-59.
Tovar, K. R., and Westbrook, G. L. (1999). The incorporation of NMDA receptors with a
distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci 19,
4180-4188.
Trussell, L. 0., Thio, L. L., Zorumski, C. F., and Fischbach, G. D. (1988). Rapid
desensitization of glutamate receptors in vertebrate central neurons. Proc Natl Acad Sci U
S A 85, 4562-4566.
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998).
Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892896.
Wang, S. S., and Augustine, G. J. (1995). Confocal imaging and local photolysis of caged
compounds: dual probes of synaptic function. Neuron 15, 755-760.
Watt, A. J., van Rossum, M. C., MacLeod, K. M., Nelson, S. B., and Turrigiano, G. G.
(2000). Activity coregulates quantal AMPA and NMDA currents at neocortical synapses.
Neuron 26, 659-670.
Yoshii, A., Sheng, M. H., and Constantine-Paton, M. (2003). Eye opening induces a rapid
dendritic localization of PSD-95 in central visual neurons. Proc Natl Acad Sci U S A 100,
1334-1339.
Zhu, J. J., Esteban, J. A., Hayashi, Y., and Malinow, R. (2000). Postnatal synaptic
potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat
Neurosci 3, 1098-1106.
-71-
Appendix: Cell Culture Protocol
Solutions:
Basic Medium: MEM, Gibco 51200-038 (No phenol red)
>
>
>
>
500 ml of MEM
2.5 g glucose
100 mg NaHCO3
50 mg transferrin, Calbiochem 616420
filter-sterilize. Store at 4'C.
Platin2 Medium: On the day of use take 89 ml of the above MEM/Basic Medium and add:
> 30 mg glutamine [1 ml 0.2 M glutamine stock/ 100 ml medium, store glutamine at -20 0 C]
> 2.5 mg insulin Sigma 15500 [0.2 ml of insulin stock (12.5 mg insulin/1 ml 10 mM HCl)/
100 ml medium, store insulin at -20'C]
> 10 ml fetal bovine serum (FBS) [if not done, heat inactivate serum at 57 C for 30 min.,
stored at 4'C]
Filter-sterilize. For 15 plates, equilibrate about 35 ml of medium in incubator (37*C, 5% CO 2 )
Feedin2 medium: Make fresh weekly and keep on hand for feeding.
> 97 ml of Basic Medium
> 1 ml of glutamine stock
> 2 ml of B-27 stock [Gibco 17504-044, 50X concentration]
> 300 pl of ARA-C stock [1 mM ARA-C, cytosine arabinoside, Sigma C6645, sterile]
Filter-sterilize.
Hank's Salt Solution: Hank's balanced salt solution (HBSS) without calcium or magnesium
(Sigma H2387)
> 350 mg/L NaHCO3 (4 mM)
> 1.3 g/L HEPES (5 mM)
pH solution to 7.3-7.4 and filter-sterilize. Wrap the lid with parafilm to help maintain pH of
solutions.
-
72 -
HBSS/20% FBS.
Make up 50 ml at a time on the day of the dissociation.
Digestion Solution:
>
400 mg/50ml
137 mM NaCl
18 mg/50 ml
5 mM KCl
50 mg/50 ml
S7 mM Na2HPO4
S25 mM HEPES 150 mg/50 ml
pH to 7.2 and store at 4'C. Solution may be sterilized for storage purposes but will also be
sterilized at time of use.
Dissociation Solution
>
Add 150 mg of MgSO2*(7H20) to 50 ml of HBSS [12 mM].
DNase and Trypsin Stocks
>
DNase type IV, Sigma D5025 1 mg Dnase/50 jLl H20 stored at -20'C.
>
Trypsin type XI, Sigma T1005 10 mg trypsin/200 pl H20 stored at -20'C.
All solutions should be at 4'C, i.e. on ice, during the procedure, except plating medium,
which should be at 37*C.
Materials Preparation
Coverslips:
Circular glass coverslips, 12 mm size, No. 0 thickness, Carolina Biological Supply P-763-3009.
Using sonication, wash coverslips in xylene for ~30 min. Repeat wash with 100%
ethanol (3x). Rinse at least 5 times with distilled de-ionized water. Separate coverslips manually
and allow them to air-dry before transferring to a glass petri dish and autoclaving (dry cycle 20
min sterilize/60 min dry).
Generally, use 15 dishes [2 coverslips/35 mm dish] for each prep [3 rat pups/prep]. Using
sterile technique, transfer coverslips to dishes and dot with ~0. 1 ml diluted Matrigel
[Collaborative Research Inc., Bedford, MA]. Aliquotes of Matrigel (-50 tl) are kept at -20'C.
-73-
Thaw on ice and dilute 1:100 in sterile, cold MEM. If the temperature of Matrigel increases
above 4'C, it will "polymerize". Incubate coverslips with Matrigel at 37 0 C while proceeding
with cultures. Just before use, aspirate Matrigel gently and plate cells directly onto coverslips.
Siliconized Pasteur Pipettes:
Coat the inside of Pasteur pipettes by drawing up a solution of 0.2% silicon oil/ether
(Aldrich 14,615-3). Rinse out several times with dd-water. Air-dry, plug with cotton, and
autoclave on dry cycle.
Odds and Ends:
Autoclaved, not plugged, 9 inch Pasteur pipettes.
Dissecting instruments in excellent condition, sterilized by soaking in 70% ETOH for >30 min..
Animals
Neonatal Sprague-Dawley rats, 1-2 days old; use 4 pups/preparation. The younger the animals
are the fewer glia in the prep.
Dissection
1.
Decapitate animal and remove the brain into a 60 mm petri dish containing ice-cold
HBSS/20% FBS. Complete dissection before proceeding to next animal.
2.
Hemi-dissect brain, removing brainstem and thalamus to expose hippocampus. Remove
hippocampus leaving some of the subiculum attached. Tug and pull as little as possible.
Always cut, not crush, tissue.
3.
Transfer hippocampi to a second 60 mi dish of cold HBSS/20% FBS. Use the back end
of a Pasteur pipette or a cut plastic transfer-pipette for the gentlest handling. Using
tweezers, peel-of all adhering membranes and blood vessels. You can cut away the
fimbriae with a scalpel or scissors. Be careful not to stab the hippocampi while
proceeding.
4.
Transfer cleaned hippocampi to a fresh dish of cold HBSS/20% FBS. Unroll the dentate
gyrus. Use the subiculum to pin down the tissue while unrolling and cutting off the
dentate gyrus with scissors. Also cut away the hippocampi from the remaining
subiculum then transfer dissected hippocampi to a third 60 mm dish containing cold
HBSS/20% FCS.
-74-
5.
Cut hippocampi end to end, into about ten thin slices and store on ice until all dissections
are completed.
6.
Using a plastic pipette, under sterile conditions, transfer the sliced hippocampal tissue to
a 15 ml polypropylene (not polycarbonate) conical tube. Let tissue settle and discard
supernatant.
7.
Wash lx with 10 ml of HBSS/20% FBS, and 3x with 10 ml of HBSS alone.
Digestion
8.
Add one aliquot (10 mg) of trypsin and one aliquot of DNase (1 mg) to 2 ml of digestion
solution and sterile filter directly onto the last pellet. Incubate for 5 minutes at 37'C,
occasionally shaking gently. Discard solution and stop digestion by adding about 10 ml
of HBSS/20% FBS.
9.
Wash Ix with 10 ml of HBSS/20% FBS, and 1x with 10 ml of HBSS alone.
Dissociation
10. Add one aliquot of DNase (1 mg) to 2 ml of dissociation solution and sterile filter onto
last pellet as above. Fire-polish two sterile, siliconized/plugged Pasteur pipettes, making
bore size of tips successively smaller. Too small tips will lyse the cells, too large tips
will not dissociate cell aggregates. Mechanically dissociate cells by gently triturating,
avoid bubbles, and always pipette onto the wall of the tube. Allow clumps of
undissociated tissue to settle for 2 minutes then transfer supernatant to another tube. This
separates cells from tissue particles.
11. Add 3 ml HBSS/20% FBS to the cell suspension (5 ml total), and centrifuge for 10
minutes at 1,000 rpm, 4'C.
12. Discard supernatant and resuspend pellet in 1.8 ml of pre-equilibrated plating medium.
Place a small drop (~0. 1 ml) on each coverslip, using all the cell suspension. Incubate for
1-2 hours to allow the cells to attach. The first short cell processes should be visible by
that time. Add 2 ml of pre-equilibrated plating medium to each dish and incubate for 48
hours.
13. After 2 days change the plating medium to selection medium by replacing 1 ml of old
medium with 1 ml of fresh pre-equilibrated feeding medium. For the first change of
medium, you will need to add 6 tM ARA-C, twice the normal concentration. By
replacing only half the medium the ARA-C concentration is diluted in half. All
subsequent feedings are done with the addition of 3 tM ARA-C.
-75
14. Feed cells three times a week. Try to replace lml of the old medium with 1 ml of fresh
pre-equilibrated medium. However, always keep the neurons covered with medium as
they are extremely sensitive and will die if exposed even briefly.
-
76 -
Acknowledgments
Thanks to all the members of my committee for guidance in my thesis
research and writing. Special thanks to Guosong Liu, my thesis advisor,
for his tireless efforts to teach me. Every current and former member of
the Liu lab has helped me on many occasions. I especially want to thank
Nathan Wilson and Gilles Dub6 for the extensive amounts of time they
have devoted to aiding me. I also owe a deep debt to Bing Li, our lab
technician, for consistent and expert preparation of the cell cultures that
were the substrate for my work.
Completing a Ph.D. requires personal support as well. For that I thank
most of all my wife, Simona Murnick, my father, Daniel Murnick, and my
friend and colleague, Sayan Mukherjee.
My work was supported by the Riken-M.I.T. Neuroscience Center and
NIH grant NS37342 to Guosong Liu. I was also supported by an NDSEG
fellowship from the Department of Defense, an NIH MSTP grant through
Harvard Medical School, and NIMH training grant MH 15761.
Biographical Note
Jonathan Mumick was born in 1972 and grew up in New Jersey. He
attended Phillips Andover Academy from 1986-1990. He did his
undergraduate studies at Princeton from 1990-1994, from where he
graduated with a degree in chemistry. He received a Churchill Fellowship
to study at Cambridge University in Cambridge, England. While at
Cambridge in 1994-1995, he studied electrophysiology of toad rod
photoreceptors under the guidance of Professor Trevor Lamb. In 1995, he
began the combined M.D.-Ph.D. program at Harvard Medical School. As
part of that program, he began his doctoral work in 1997 in M.I.T's
Department of Electrical Engineering and Computer Science. He expects
to complete his medical degree in 2005 and plans to pursue a career in
academic medicine.
-77-